
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Navigating epoxidation complexity: building a data science toolbox to design vanadium catalysts†
José
Ferraz-Caetano
 *a,
Filipe
Teixeira
b and
M. Natália D. S.
Cordeiro
*a
*a,
Filipe
Teixeira
b and
M. Natália D. S.
Cordeiro
*a
aLAQV-REQUIMTE – Department of Chemistry and Biochemistry – Faculty of Sciences, University of Porto – Rua do Campo Alegre, s/n, Porto 4169-007, Portugal. E-mail: jose.caetano@fc.up.pt; ncordeir@fc.up.pt
bCQUM – Centre of Chemistry, University of Minho, Campus de Gualtar, Braga 4710-057, Portugal
First published on 26th February 2024
Abstract
This communication presents a novel approach to set up a machine learning-ready database for epoxidation reactions, focusing on vanadium catalysts. Utilising data driven analysis, we identified key reaction yield trends through chemical descriptors, providing insights for catalyst design and reaction optimisation.
The catalytic epoxidation of small alkenes and allylic alcohols (ESA) represents a critical process in industrial chemistry.1 It yields various fine chemicals, encompassing a wide range of applications from the synthesis of complex molecules to the production of high-value pharmaceuticals.2 Central to this process is the role of vanadium catalysts, known for their efficiency and selectivity in facilitating these reactions.3,4 Given their unique ability to transfer oxygen atoms onto substrates, vanadium complexes have emerged as a preferred choice in various epoxidation processes.5,6 The versatility of vanadium-based catalysts, particularly in their oxidation states and ligand interactions, has led to significant advancements in the field, enabling the production of epoxides with high yield and specificity.7 This has not only enhanced industrial processes efficiency but also contributed to the development of eco-friendly practices, allowing the use of sustainable substrates.
Despite the recognised potential of vanadium catalysts in ESA reactions, significant challenges in process optimisation still persist.1 Chemists have relied on trial-and-error strategies to optimise these reactions, but this approach involves systematically varying reaction parameters one-at-a-time. While it can eventually lead to optimised conditions, this method is time-consuming, resource-intensive, and often impractical. It is the complexity of these reactions, combined with a myriad of factors including catalyst structure, reaction conditions, and substrate properties, that poses a significant hurdle in ESA optimisation.
To effectively handle these multiple reaction variables, data science strategies emerge as an alternative over classic methods.8 With the integration of these techniques with machine learning (ML) predictive algorithms, they tackle traditional approaches’ inefficiencies through the exploration of complex chemical parameters.9 Unlike sequential explorations, these algorithms can simultaneously analyse multiple variables, rapidly identifying patterns and relationships within extensive datasets. This capability is crucial in uncovering intricate interactions between chemical descriptors, efficiently navigating towards optimal reaction conditions, yield maximisation, cost reduction, and environmental impact.10,11 A key benefit of employing data science in chemical reaction optimisation is the reduction in experimental workload. Indeed, the advent of big data has brought forth various tools capable of transcribing chemical reaction information into extensive sets of computer-readable data, and even predict complex chemical properties.12 These data-driven tools are equipped to handle large volumes of raw chemical information in real-time, extracting insights towards cost-saving experimental planning.
Although current computational chemistry has many ML tools at its disposal, there is commonly one key element missing: a comprehensive digital database of chemical reactions.13,14 The scarcity of digital computational databases for chemical reactions are attributed to two main challenges. First, due to the nature of chemical data itself, with a multitude of variables influencing each reaction. Capturing this complexity in a digital format requires systematic data structuring, each with its unique set of parameters and outcomes. And secondly, due to the labour-intensive data collection process for such chemical databases. Much of the data resides in scientific literature, often in unstructured formats such as text, tables and images. And extracting and digitising this data can be a daunting task, requiring significant effort and expertise. But even if we are able to address both these challenges, there is still the issue of data quality. Chemical data from different sources can vary in terms of accuracy and detail, which may comprise a holistic landscape of the chemical reaction data. This requires rigorous quality control and validation, which adds even more complexity to database development.
In this communication, we present a comprehensive methodology to build a ML-ready database of vanadium-catalysed ESA reactions. By explaining how it encodes pivotal chemical information, we present a database used for ML modelling, which can be used to identify key reaction yield trends, describe reaction feature importance and other molecular insights. Our intent is to layout a blueprint for ML database building for forthcoming studies, using ESA reactions as our case-study. We thus finish this communication with a descriptive example on how to obtain one of the many insights provided by this database.
The cornerstone of our project was the development of a framework that would tackle current database challenges. In our case, all molecular structures were meticulously transformed into 3D representations and SMILES (Simplified Molecular Input Line Entry System) strings. This conversion was crucial for enabling computational models to process the molecular data effectively. Additionally, experimental variables were digitised and represented numerically, ensuring that the data was not only comprehensive but also uniformly structured for computational purposes. This step addressed the challenge of translating complex chemical data from diverse sources into a standardised, machine-readable format. Our database development also used open-source software to encode a multitude of variables for each reaction. This approach allowed to capture a wide array of information, ranging from molecular properties to reaction conditions. By leveraging open-source tools, we ensured that our database was not only robust in its data representation but also with high data quality. We achieved this by harmonising experimental input, such as quantities of reactants, types of solvents used, and reaction temperatures, ensuring consistent and comparable data across different experiments. Additionally, we employed generalisable chemical descriptors, which provided a standardised way of describing chemical entities and reactions. These are crucial for building predictive models also applicable to a broader range of chemical reactions. This universality facilitates the creation of a broad playbook for reaction optimisation, transcending the specificities of individual reaction systems.
Aiming at a data-driven model to optimise epoxidation reactions, our dataset integrates a diverse array of chemical descriptors, encompassing experimental conditions, solvent properties, and molecular features, to create a robust foundation for predictive modelling. We outline the following steps for model building:
Data compilation: The initial phase involved the meticulous collection of ![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif)
![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) using vanadium catalysts. This data was sourced from an extensive review of existing literature from 16 bibliographical references, experimental records, and established chemical databases. The gathered information included details on homogeneous catalyst reactions, including catalyst structures, substrates, ligands, oxidants, and experimental conditions such as temperature, yield, enantiomeric excess (EE), and reaction time. These parameters included the condition that epoxidation reactions occur at temperatures above 195 K, the use of a single type of oxidant, and the restriction to only one solvent per reaction. The reactions compiled in this database were specifically chosen based on several critical parameters to maintain a manageable level of chemical complexity.
using vanadium catalysts. This data was sourced from an extensive review of existing literature from 16 bibliographical references, experimental records, and established chemical databases. The gathered information included details on homogeneous catalyst reactions, including catalyst structures, substrates, ligands, oxidants, and experimental conditions such as temperature, yield, enantiomeric excess (EE), and reaction time. These parameters included the condition that epoxidation reactions occur at temperatures above 195 K, the use of a single type of oxidant, and the restriction to only one solvent per reaction. The reactions compiled in this database were specifically chosen based on several critical parameters to maintain a manageable level of chemical complexity.
In total, the database encompasses 273 distinct reactions, featuring five different vanadium-catalyst scaffolds: vanadyl(IV) sulphate – [VOSO4], vanadyl acetylacetonate-salen – [![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif)
![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) (salen)], vanadyl isopropoxide – [VO(OiPr)3], vanadyl acetylacetonate – [VO(acac)2], and vanadyl dichloride-salen – [VCl2(salen)]. The choice of these specific catalysts was driven by their varied and representative nature, ensuring a comprehensive coverage of possible reaction scenarios. Each component of the reactions – catalysts, ligands, substrates, oxidants, and solvents – was meticulously represented in a 3D structure using MarvinSketch. This step was crucial for converting these components into unique SMILES strings, thereby standardising the data format and facilitating computational analysis. This was complemented by additional experimental data extracted from the literature, encompassing EE, solvent and oxidant type. Source code and detailed database are presented in the ESI.†
(salen)], vanadyl isopropoxide – [VO(OiPr)3], vanadyl acetylacetonate – [VO(acac)2], and vanadyl dichloride-salen – [VCl2(salen)]. The choice of these specific catalysts was driven by their varied and representative nature, ensuring a comprehensive coverage of possible reaction scenarios. Each component of the reactions – catalysts, ligands, substrates, oxidants, and solvents – was meticulously represented in a 3D structure using MarvinSketch. This step was crucial for converting these components into unique SMILES strings, thereby standardising the data format and facilitating computational analysis. This was complemented by additional experimental data extracted from the literature, encompassing EE, solvent and oxidant type. Source code and detailed database are presented in the ESI.†
Descriptor generation: The heart of the dataset lies in the integration of a comprehensive set of descriptors, meticulously chosen to capture the nuances of epoxidation reactions. These descriptors fall into three primary categories: (i) Experimental reaction descriptors: These include quantitative measures such as substrate and catalyst quantities, solution volumes, reaction times and temperature. They provide a direct insight into the experimental setup and conditions under which the epoxidation reactions were conducted. (ii) Solvent characteristics: Given the critical role of solvents in reaction dynamics, descriptors such as solvent optimal frequency, hydrogen bond acidity and basicity, surface tension, dielectric constant, aromaticity, and electronic halo were included. These parameters were calculated using the Minnesota Solvent Descriptor Database, offering a deeper understanding of the solvent's reaction influence. (iii) Molecular descriptors: A comprehensive range of molecular descriptors was employed to capture the chemical nature of the catalysts, substrates, ligands, and solvents. These were calculated by the open-source RDKit software‡, using each SMILES string as input, and are assorted into different chemical groups: volume surface area (VSA), electronic and structural descriptors. RDKit excels as the best software for descriptor generation due to its comprehensive range of accurate and reliable chemical descriptors, coupled with seamless integration with major Python ML libraries. In the ESI,† we provide a complete list of all descriptors used.
Data processing and standardisation: To ensure consistency and reliability, the collected data underwent rigorous processing, including the normalization of units, handling of missing values, and standardisation of formats. The final dataset comprised more than 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 data entries, while the targets were specific reaction outcomes such as yield and EE.
000 data entries, while the targets were specific reaction outcomes such as yield and EE.
Once the dataset is built, the next stage is defining the data analysis strategy. Using this database, we will give a simple example of running a routine for identifying key chemical features in designing efficient vanadium-based catalysts. This includes Pearson and Spearman correlation coefficients, principal component analysis, and ML algorithms like random forests and gradient boosting for feature importance evaluation. The approach begins with a comprehensive data collection phase, ensuring a robust dataset that captures a wide array of variables potentially influencing yields. Subsequent data pre-processing and cleaning ensure the accuracy and reliability of the analysis. The strategy is twofold: first, we evaluate correlation coefficients from key descriptors correlated with high epoxidation yields. Then, we amass these chemical descriptors, related to each reaction's catalyst, to pinpoint structural insights in the experimental design of novel catalysts, aiming to optimise reaction processes.
In Fig. 1, we present reaction yield distribution in the ESA database. By calculating correlation coefficients between each descriptor and the reaction yield, we can prioritize those descriptors that demonstrate the strongest relationships, either positive or negative. This is especially valuable in dealing with complex datasets where manual pattern inspection is impractical. Correlation metrics offer a quantitative basis to streamline the selection process, enabling scientists to focus on the most promising features, as descriptors with high positive correlation coefficients might be key for yield optimisation. It also filters out descriptors with negligible correlation, leading to more interpretable results and effective experimentation.
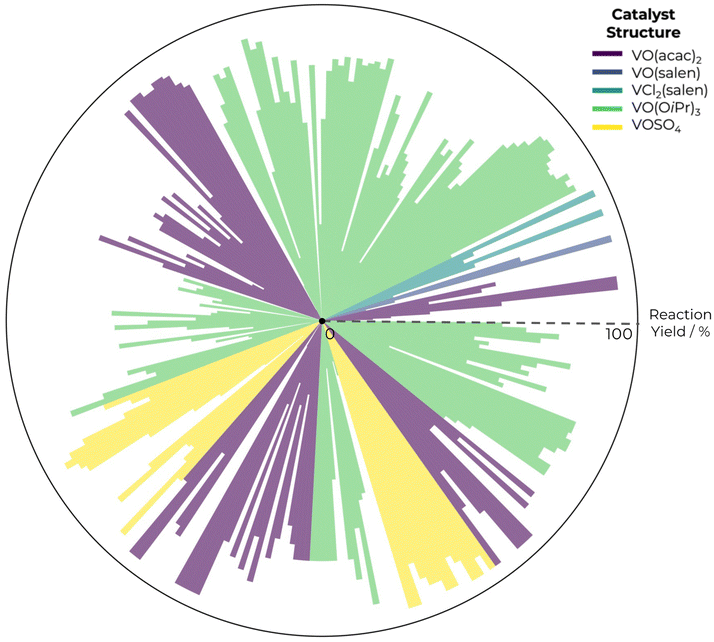 | ||
| Fig. 1 Graphical radar plot for reaction yield distribution in the ESA database. | ||
Fig. 2 depicts the correlation importance with ESA reaction yield with different descriptor types and detailed results are present in the ESI.† Catalyst descriptors are accountable for 25.3% of correlation importance, as catalyst's ligands are accountable to 15.3%. Also, reaction descriptors (namely quantities, temperature, time) are interestingly connected with ESA yields amongst different chemical properties with 32.8% overall correlation.
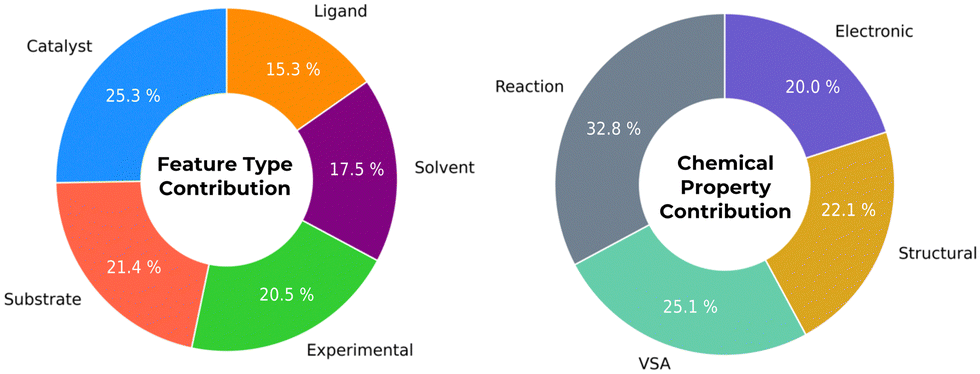 | ||
| Fig. 2 Average contributions from different database descriptor groups based on ESA yield successful predictions. | ||
It is possible to identify which catalyst structural and electronic features are more relevant. For example, ligand feature EState-VSA5 emerges as an interesting factor, accounting with 20% of the ESA yield correlation. This value indicates that this descriptor has a significant contribution in assessing the best ligand structures in the outcome of ESA reactions. EState-VSA5, combines electronic and topological information within a specific van der Waals surface area (VSA) – in an EState between 1.54 and 1.81 – offering a nuanced understanding of molecular behaviour. This descriptor captures the subtle interplay between ligand electronic properties and molecular size, which are crucial in vanadium catalytic activity. In the context of epoxidation, where the efficiency and selectivity of the catalyst are paramount, such insights enables chemists to fine-tune molecular structures to achieve desired reaction outcomes. By paying close attention to this descriptor, researchers can better navigate the complex landscape of catalyst optimization, making informed decisions that balance the intricate factors influencing epoxidation reactions.
In Fig. 3, we present an illustrative example of a catalyst structure represented in the model's database, with a specific focus on the key hotspots associated with the EState-VSA5 descriptor. This figure serves as a visual guide, highlighting areas within the catalyst's ligand structure that are crucial for optimizing its performance in ESA reactions. The hotspots, marked distinctly on the catalyst structure, correspond to regions where the EState-VSA5 descriptor exerts significant influence. These areas are indicative of where electronic and topological properties converge, offering potential sites for chemical modification.
 | ||
| Fig. 3 Graphical depiction of ligand hotspots sites for a VOSO4 catalyst scaffold for ESA epoxidation, suggested for fine-tuning optimisation using the EState-VSA7 descriptor. | ||
The visualisation of these hotspots is not only instrumental in identifying pivotal aspects of the catalyst's structure, but also in suggesting strategic points for chemical fine-tuning. By targeting these key areas, experimentalists can modify the catalyst to enhance its efficiency and selectivity in epoxidation reactions.
This approach exemplifies the versatility of combining insights from various descriptors to inform catalyst design. By translating abstract descriptor values into tangible structural features, it provides a roadmap for chemists to experimentally test and validate the impact of specific modifications. This integration of computational predictions with experimental strategies is pivotal in advancing the field of catalysis, bridging the gap between theory and real-world applications.
We have outlined a comprehensive blueprint for setting up a ML-readable database, tailored for vanadium-catalysed ESA reactions. The meticulous process of compiling, digitising, and standardising a vast array of chemical data culminated in a robust database, primed for ML applications. The deployment of an array of chemical descriptors within this database was instrumental in data-driven analytics to correlate reaction yields with chemical descriptors. Encompassing a wide spectrum of molecular and reaction parameters, these descriptors have demonstrated their prowess in identifying key features that significantly impact the reaction outcomes.
The analysis of this ESA database led to the potential identification of key ligand hotspots – areas within the ligand structure that are pivotal in determining the efficiency and selectivity of the epoxidation reactions. These hotspots, illuminated through various chemical descriptors, offer valuable guidance for chemists. By focusing on these areas, researchers can strategically tune vanadium catalysts, enhancing their performance and potentially leading to advancements in catalyst design. In essence, this communication presents a viable pathway for harnessing the power of data science in chemistry, while also highlighting the transformative impact of data-centred models in unravelling complex chemical reactions.
Author contributions
Conceptualisation, J. F. C., F. T. and M. N. D. S. C.; writing – original draft preparation and editing, J. F. C.; writing – review, F. T. and M. N. D. S. C.; supervision, F. T. and M. N. D. S. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Fundação para a Ciência e Tecnologia (FCT/MCTES) support to LAQV-REQUIMTE (UIDP/50006/2020). J. F. C.'s PhD Fellowship is supported by a doctoral Grant (SFRH/BD/151159/2021) financed by the FCT, with funds from the Portuguese State and the European Union Budget, through the Social European Fund and Programa Por_Centro, under the MIT Portugal Program.Notes and references
- J. Ferraz-Caetano, F. Teixeira and M. N. D. S. Cordeiro, Int. J. Mol. Sci., 2023, 24, 12299 CrossRef CAS.
- S. Meninno and A. Lattanzi, ACS Organic Inorganic Au, 2022, 2, 289–305 CrossRef CAS.
- C. D. Nunes, P. D. Vaz, V. Félix, L. F. Veiros, T. Moniz, M. Rangel, S. Realista, A. C. Mourato and M. J. Calhorda, Dalton Trans., 2015, 44, 5125–5138 RSC.
- A. Lattanzi, S. Piccirillo and A. Scettri, Eur. J. Org. Chem., 2005, 1669–1674 CrossRef CAS.
- K. B. Sharpless, J. M. Townsend and D. R. Williams, J. Am. Chem. Soc., 1972, 94, 295–296 CrossRef CAS.
- M. L. Kuznetsov and J. C. Pessoa, Dalton Trans., 2009, 5460–5468, 10.1039/B902424G.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- E. Shim, A. Tewari, T. Cernak and P. M. Zimmerman, J. Chem. Inf. Model., 2023, 63, 3659–3668 CrossRef CAS PubMed.
- K. Takahashi, J. Ohyama, S. Nishimura, J. Fujima, L. Takahashi, T. Uno and T. Taniike, Chem. Commun., 2023, 59, 2222–2238 RSC.
- S. V. Johansson, H. G. Svensson, E. Bjerrum, A. Schliep, M. H. Chehreghani, C. Tyrchan and O. Engkvist, Mol. Inf., 2022, 41, e2200043 CrossRef PubMed.
- E. Mosqueira-Rey, E. Hernández-Pereira, D. Alonso-Ríos, J. Bobes-Bascarán and Á. Fernández-Leal, Artificial Intelligence Review, 2023, 56, 3005–3054 CrossRef.
- J. Ferraz-Caetano, F. Teixeira and M. N. D. S. Cordeiro, J. Chem. Inf. Model., 2023 DOI:10.1021/acs.jcim.3c00544.
- L. Alzubaidi, J. Bai, A. Al-Sabaawi, J. Santamaría, A. S. Albahri, B. S. N. Al-dabbagh, M. A. Fadhel, M. Manoufali, J. Zhang, A. H. Al-Timemy, Y. Duan, A. Abdullah, L. Farhan, Y. Lu, A. Gupta, F. Albu, A. Abbosh and Y. Gu, J. Big Data, 2023, 10, 46 CrossRef.
- T. Taniike and K. Takahashi, Nat. Catal., 2023, 6, 108–111 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Various software, complete descriptor list, source of the developed code and full statistical results are available in the SI and at our online repository. See DOI: https://doi.org/10.1039/d3nj05784d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |