
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexanuclear {ZnII4FeIII2} and {ZnII4CrIII2} complexes from the use of potentially tetradentate NOO′O′′ Schiff-base ligands†‡
Konstantinos N.
Pantelis
a,
Sotiris G.
Skiadas
 a,
Zoi G.
Lada
b,
Rodolphe
Clérac
c,
Yiannis
Sanakis
*d,
Pierre
Dechambenoit
*c and
Spyros P.
Perlepes
*ab
a,
Zoi G.
Lada
b,
Rodolphe
Clérac
c,
Yiannis
Sanakis
*d,
Pierre
Dechambenoit
*c and
Spyros P.
Perlepes
*ab
aDepartment of Chemistry, University of Patras, Patras 26504, Greece. E-mail: perlepes@upatras.gr
bInstitute of Chemical Engineering Sciences (ICE-HT), Foundation for Research and Technology-Hellas (FORTH), Platani, P.O. Box 144, Patras 26504, Greece
cUniv. Bordeaux, CNRS, CRPP, UMR 5031, F-33600 Pessac, France. E-mail: pierre.dechambenoit@u-bordeaux.fr
dInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, Aghia Paraskevi Attikis 15310, Greece. E-mail: i.sanakis@inn.demokritos.gr
First published on 31st May 2024
Abstract
The use of N-(2-carboxyphenyl)salicylideneimine (saphHCOOH) and N-(4-chloro-carboxyphenyl)salicylideneimine (4ClsaphHCOOH) for the synthesis of hexanuclear {ZnII4MIII2} (M = Cr, Fe) complexes is described. [Zn4Fe2(saphCOO)6(NO3)2(EtOH)2] (1), [Zn4Cr2(saphCOO)6(NO3)2(H2O)2] (2) and [Zn4Fe2(4ClsaphCOO)6(NO3)2(EtOH)2] (3), as 4CH2Cl2·2EtOH (1, 3) and 4MeCN·2EtOH (2) solvates, have been isolated and their structures have been determined by single-crystal X-ray crystallography. The three complexes are centrosymmetric and almost isostructural. The metal topology can be described as two isosceles triangles which are linked through phenolate oxygen atoms of two 3.2111 (Harris notation) saphCOO2−/4ClsaphCOO2− ligands, each of which bridges exclusively three ZnII atoms. Four 2.1111 ligands are also incorporated in the molecules, each bridging one MIII center and one ZnII ion. The former is chelated with the Ophenolate, Nimine and one Ocarboxylate being the donor atoms (thus forming two 6-membered chelating rings with a common MIII–N edge), while the latter is bound to the other carboxylate oxygen. The coordination spheres are of the types {FeO4N2}/{ZnO4N}/{ZnO5} for 1 and 3, and {CrO4N2}/{ZnO4N}/{ZnO4} for 2. Complexes 1–3 are the first heterometallic complexes (with any metals) based on saphHCOOH and 4ClsaphHCOOH, thus illustrating their ability for the preparation of mixed-metal molecular species. Interesting H-bonding patterns are present in the crystal structures. The IR and Raman spectra of the solid complexes are discussed in terms of the coordination modes of the ligands involved, while molar conductivity data and UV/VIS spectra in CH2Cl2 are also reported and interpreted. The δ and ΔEQ57Fe-Mössbauer parameters of 1 and 3 at 300 and 80 K indicate isolated high-spin FeIII centers. Variable-temperature (1.8–300 K) magnetic susceptibility data for 1–3 suggest a very weak exchange interaction between the MIII ions in agreement with their long distances (∼9 Å) in the molecules.
Introduction
The field of mixed-metal, atom- or ion-based materials is receiving1–3 great attention of researchers working in solid-state chemistry and condensed-matter physics, for example in the area of multiferroics.2 Molecular inorganic chemists are also interested in synthesizing and characterizing mixed-metal compounds. This general topic is stimulated by its relevance to several areas of bioinorganic chemistry,4,5 molecular nanomagnetism,6,7 catalysis,8 metallosupramolecular chemistry,9 porous complexes,10 quantum technology11 and development of precursors for multifunctional materials.12 Despite the recent explosive growth on complexes containing two different transition-metal ions,13,14 such complexes are significantly less reported in comparison to the plethora of 3d–4f species.15–20 With this in mind, we have initiated a project to examine the synthesis, structures and properties of heterometallic 3d–3d′ complexes.From a synthetic inorganic chemistry viewpoint, methods must be developed to combine two different 3d-metal ions. For some 3d-metal ion pairs, e.g. FeIII–NiII and FeIII–CuII, strategies can be used based on the HSAB model.15,21 For other pairs, e.g. the pairs CrIII–ZnII and FeIII–ZnII of the present work, this is not an easy task and often simply combining sources of the two different metal ions leads to complexes containing only one of them; thus success is many times results from several “try and see” exercises. It is obvious that this “self-assembly” process to prepare 3d–3d′ metal complexes is heavily relied on the choice of the organic ligand.22
Polydentate O,N-based Schiff bases are candidates for this goal. Even 160 years after their first synthesis by the Italian-German Hugo Schiff (1834–1915), these azomethine (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) or imine (
N–) or imine (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CN–) groups-containing molecules continue to surprise inorganic chemists because of their close relevance to a number of interdisciplinary research fields.23–25 Because of their logical modular synthesis that allows strict control over the nature of donor atoms, denticity, chelating and/or bridging function, as well as their adjustable electronic and steric features, polydentate Schiff bases have played a great role in the development of coordination chemistry after the second world war,26 being also important for the preparation of heterometallic complexes.23–26 In a broader sense, formation of azomethines/imines is one of the types of reactions which define dynamic covalent chemistry (DCC), an approach which is used in the synthesis of complicated molecules and extended structures because of the inherent “proof-reading” and “error-checking” associated with these reversible processes.27,28
CN–) groups-containing molecules continue to surprise inorganic chemists because of their close relevance to a number of interdisciplinary research fields.23–25 Because of their logical modular synthesis that allows strict control over the nature of donor atoms, denticity, chelating and/or bridging function, as well as their adjustable electronic and steric features, polydentate Schiff bases have played a great role in the development of coordination chemistry after the second world war,26 being also important for the preparation of heterometallic complexes.23–26 In a broader sense, formation of azomethines/imines is one of the types of reactions which define dynamic covalent chemistry (DCC), an approach which is used in the synthesis of complicated molecules and extended structures because of the inherent “proof-reading” and “error-checking” associated with these reversible processes.27,28
An interesting group of the family of tetradentate Schiff-base ligands consists of molecules derived from the condensation of salicylaldehyde (or its derivatives) with 2-aminobenzoic acid (or its derivatives). Two members of this group have been employed in this work, namely N-(2-carboxylphenyl)salicylideneimine [the IUPAC name is {((2-hydroxyphenyl)methylidene)amino}benzoic acid] abbreviated as saphHCOOH, and N-(4-chloro-carboxylphenyl)salicylideneimine [the IUPAC name is 4-chloro-{((2-hyroxyphenyl)methylidene)amino}benzoic acid] abbreviated as 4ClsaphHCOOH; the two H atoms in the abbreviations denote the number of the ionizable protons. The structural formulas of the free neutral ligands are illustrated in Scheme 1. These molecules contain an ONO′O′′ donor set; deprotonation of the –COOH group or both the –COOH and –OH groups may favor coordination of the resulting anionic ligands to two or more same or different metal ions, thus resulting in dimers, polynuclear coordination complexes or polymers. Somewhat to our surprise, the coordination chemistry of saphHCOO− and saphCOO2− has been exclusively limited to homometallic complexes (selected literature is given in ref. 29–33); no heterometallic compounds have been structurally characterized. The metal chemistry of 4ClsaphHCOOH is almost completely unexplored (by contrast with its 5-chloro analogue for which interesting homometallic complexes have been reported),34,35 the only structurally characterized complex being [Cu(4ClsaphCOO)(phen)], where phen is the chelating ancillary ligand 1,10-phenanthroline.36,37
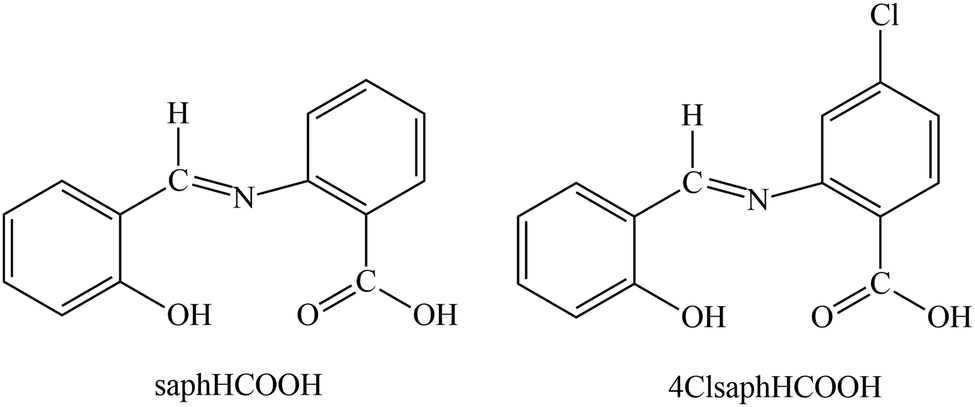 | ||
| Scheme 1 The structural formula of the two potentially tetradentate ONO′O′′ Schiff-base ligands used in the present work and their abbreviations. The single-crystal X-ray structure of saphHCOOH38 reveals that the compound exists in the solid state as an intermediate form between the phenol-imine (illustrated in this scheme) and quinoid tautomers. | ||
In this work, we describe our efforts to prepare ZnII–FeIII and ZnII–CrIII compounds based on saphHCOOH and 4ClsaphHCOOH. ZnII–FeIII complexes are interesting to the scientific community because: (i) the heterodinuclear {ZnIIFeIII} unit has been recognized at the active sites of purple acid phosphatase and human calcineurin that facilitate the hydrolysis of phosphodiesters into phosphomonoesters, and several complexes have been reported as structural and functional models of these enzymes;39,40 (ii) the ternary zinc ferrite, ZnIIFeIII2O4, has been recognized as one of the promising lithium ion batteries anode substitutes41 and molecular analogues of this mixed-metal oxide are highly desirable; (iii) 3D porous frameworks containing ZnII and FeIII exhibit high CO2/N2 and CO2/CH4 selectivity;42 (iv) formal reduction of FeIII complexes containing redox-active pincer ligands leads to “metalloligands” of FeII which allow the construction of iron–zinc complexes with interesting redox and magnetic properties;43 and (v) ZnII is an ideal diamagnetic substitute for CoII in polynuclear CoII/FeIII complexes allowing for the in-depth understanding of the iron(III)⋯iron(III) magnetic interactions in such complexes. ZnII–CrIII complexes attract the interest of the inorganic chemistry community because – among others –: (i) diamagnetic ZnII “metalloligands” can be used in the study of the presence of long-range CrIII⋯CrIII exchange interactions even if the distance between the paramagnetic metal ions is as long as ∼7 Å;44 (ii) the simultaneous presence of the two metal ions have resulted in a new trinodal metal–organic material platform comprised of simple and inexpensive triangular, tetrahedral and trigonal-prismatic molecular building blocks,45 leading to molecular materials with chemical stability (organic solvents, water and bases) and high uptake of CO2 and H2. These properties are desirable in the context of the cost/performance ratio that is needed to develop metal–organic materials for applications; (iii) heterometallic Anderson-type {ZnII5CrIII2} “wheels” help in the in-depth study of substitutional disorder in the positions of the CrIII and ZnII ions around the outside ring;46 (iv) heterometallic rings based on generic {(ZnIIxCrIIIy)} formulas (x, y are integer numbers) exhibit exciting structural and physical properties, and they have been proposed for various applications47 including quantum information processing48 and magnetic refrigeration;49 and (v) doping of cyclic {ZnIICrIII7} molecular nanomagnets into diamagnetic and isostructural hosts allows pulsed X-band EPR studies (including relaxation and nutation experiments on the S = 3/2 ground state), the doping without scrambling of the CrIII and ZnII ions being aided by the kinetic inertness of the former.50
This work can also be considered as a continuation of the interest of our groups in various aspects of the coordination chemistry of N,O-based Schiff bases.22,30,33,51–55
Experimental section
Materials and instrumentation
All manipulations were performed under aerobic conditions using reagents and solvents (Sigma-Aldrich) as received. The free ligands saphHCOOH29,32,56 and 4ClsaphHCOOH36,37,57 were synthesized as described in the literature; their purity was checked by 1H NMR spectroscopy in DMSO-d6. C, H and N microanalyses were conducted by the Instrumental Analysis Laboratory of the University of Patras using a Carlo-Erba analyzer, model EA 1108. FT-IR spectra (4000–400 cm−1) were recorded using a PerkinElmer 16PC spectrometer with samples prepared as KBr (spectroscopic grade) pellets under pressure. Raman spectra were recorded an a T-64000 Jobin Yvon-Horiba micro-Raman setup utilized in a single-spectrograph configuration. The excitation wavelength was 514.5 nm emitted from a DPSS laser (Cobolt Fandango TMISO Laser, Norfolk, UK). The laser power on the samples was 1 mW. The backscattered radiation was collected from a single configuration of the monochromator after passing through an appropriate edge filter (LP02-633RU-25, Laser2000 Ltd, Huntingdon, Cambridgeshire, UK). The calibration of the instrument was achieved via the standard peak position of Si at 520.5 cm−1. The spectral resolution was 5 cm−1. UV/Vis spectra in solution were recorded on a Hitachi U-300 spectrometer. Conductivity measurements in CH2Cl2 were carried out at room temperature (22–24 °C) with a Metrohm-Herisau E-527 bridge and a cell of standard design; the concentration of the solutions was ∼10−3 M. 1H NMR spectra in DMSO-d6 (to check the purity of the free ligands) were run on a 600.13-MHz Bruker Avance DPX spectrometer. 57Fe-Mössbauer spectra from powdered dried samples of the iron(III)-containing complexes were recorded using a constant-acceleration conventional spectrometer with a source of 57Co (Rh matrix). Spectra in the 80–300 K range were obtained using a Janis cryostat. Isomer shift values (δ) are reported relative to the Fe foil at 293 K. Magnetic susceptibility and magnetization measurements were performed on a Quantum Design SQUID magnetometer MPMS-XL at temperatures between 1.8 and 300 K, and dc magnetic fields ranging from −7 to +7 T. The data were collected on polycrystalline samples suspended in mineral oil and introduced in a sealed polyethylene bag. Prior to the experiments, the field-dependent magnetization was measured at 100 K on each sample in order to detect the presence of any bulk ferromagnetic impurities; paramagnetic or diamagnetic materials should exhibit a strictly linear dependence of magnetization that extrapolates to zero at zero dc field. The samples appeared to be free of any ferromagnetic impurities. The magnetic susceptibilities were corrected for the oil, the sample holder, and the intrinsic diamagnetic contributions.Synthetic details
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). To the resulting yellow suspension was added solid Fe(NO3)·9H2O (0.040 g, 0.10 mmol). A dark red solution was obtained which was stirred for 1 h and then layered with Et2O (5 mL). X-ray quality brown crystals of the product were precipitated within 11 d. The crystals were collected by filtration, washed with Et2O (2 × 2 mL) and dried in a vacuum desiccator, first over silica gel and later over P4O10. The yield was ca. 60% (based on the ligand available). The sample was analyzed satisfactorily as lattice solvent-free (1). Anal. calcd for C88H66N8O26Zn4Fe2:C 52.20; H 3.29; N 5.53%. Found: C 52.46; H 3.27; N 5.39%. IR (KBr, cm−1): 3422wb, 2925w, 1591s, 1544s, 1490m, 1469sh, 1447m, 1384s, 1295m, 1228w, 1185m, 1153m, 1126w, 1095w, 1040w, 923w, 875m, 850w, 795m, 760m, 714m, 687w, 648w, 604w, 560w, 523w, 461w, 417w. Selected Raman peaks (cm−1): 1614s, 1587s, 1534m, 1458w, 1410w, 1361w, 1340m, 1310w, 1272w, 1251w, 1186m, 1164w, 613s, 563m. UV/Vis (CH2Cl2, nm): 230sb, 258sh, 285sh, 380mb, 405mb, 510wb, 585wb. ΛM (CH2Cl2, 10−3 M, 23 °C) = 4 S cm2 mol−1. 57Fe-Mössbauer (mm s−1): δ = ∼0.35 and ΔEQ = ∼0.6 at room temperature.
:1 v/v). To the resulting yellow suspension was added solid Cr(NO3)3·9H2O (0.040 g, 0.10 mmol). An olive green solution was obtained, which was stirred under reflux for 2 h. No noticeable color change took place. The solution was layered with Et2O (4 mL) and reddish green X-ray quality crystals of the product were precipitated in a period of 5 d. The crystals were collected by filtration, washed with Et2O and dried in a vacuum desiccator, first over silica gel and later over anhydrous CaCl2. The yield was ca. 50% (based on the ligand available). The sample was analyzed satisfactorily as lattice solvent-free, i.e. as 2. Anal. calcd for C84H58N8O26Zn4Cr2:C 51.44; H 2.99; N 5.71%. Found: C 51.78; H 2.87; N 5.87%. IR (KBr, cm−1): 3482mb, 1594s, 1552m, 1467sh, 1449m, 1384s, 1293m, 1230w, 1188w, 1152w, 1126w, 1095w, 1042w, 926w, 878w, 857w, 797w, 761m, 714w, 690w, 616w, 538w, 462w, 441w. Selected Raman peaks (cm−1): 1630m, 1587s, 1545m, 1473m, 1442s, 1308w, 1275w, 1186m, 1156w, 624m, 461w. UV/Vis (CH2Cl2, nm): 225mb, 240mb, 280wb, 325wb, 420wb, 522wb, 583wb. ΛM (CH2Cl2, 10−3 M, 23 °C) = 2 S cm2 mol−1.
:1 v/v). To the resulting yellow suspension was added solid Fe(NO3)·9H2O (0.040 g, 0.10 mmol). A dark red solution was obtained which was stirred for 1 h and then layered with Et2O (5 mL). X-ray quality brown crystals of the product were precipitated within 11 d. The crystals were collected by filtration, washed with Et2O (2 × 2 mL) and dried in a vacuum desiccator, first over silica gel and later over P4O10. The yield was ca. 60% (based on the ligand available). The sample was analyzed satisfactorily as lattice solvent-free (1). Anal. calcd for C88H66N8O26Zn4Fe2:C 52.20; H 3.29; N 5.53%. Found: C 52.46; H 3.27; N 5.39%. IR (KBr, cm−1): 3422wb, 2925w, 1591s, 1544s, 1490m, 1469sh, 1447m, 1384s, 1295m, 1228w, 1185m, 1153m, 1126w, 1095w, 1040w, 923w, 875m, 850w, 795m, 760m, 714m, 687w, 648w, 604w, 560w, 523w, 461w, 417w. Selected Raman peaks (cm−1): 1614s, 1587s, 1534m, 1458w, 1410w, 1361w, 1340m, 1310w, 1272w, 1251w, 1186m, 1164w, 613s, 563m. UV/Vis (CH2Cl2, nm): 230sb, 258sh, 285sh, 380mb, 405mb, 510wb, 585wb. ΛM (CH2Cl2, 10−3 M, 23 °C) = 4 S cm2 mol−1. 57Fe-Mössbauer (mm s−1): δ = ∼0.35 and ΔEQ = ∼0.6 at room temperature.
:1 v/v). To the resulting yellow suspension was added solid Cr(NO3)3·9H2O (0.040 g, 0.10 mmol). An olive green solution was obtained, which was stirred under reflux for 2 h. No noticeable color change took place. The solution was layered with Et2O (4 mL) and reddish green X-ray quality crystals of the product were precipitated in a period of 5 d. The crystals were collected by filtration, washed with Et2O and dried in a vacuum desiccator, first over silica gel and later over anhydrous CaCl2. The yield was ca. 50% (based on the ligand available). The sample was analyzed satisfactorily as lattice solvent-free, i.e. as 2. Anal. calcd for C84H58N8O26Zn4Cr2:C 51.44; H 2.99; N 5.71%. Found: C 51.78; H 2.87; N 5.87%. IR (KBr, cm−1): 3482mb, 1594s, 1552m, 1467sh, 1449m, 1384s, 1293m, 1230w, 1188w, 1152w, 1126w, 1095w, 1042w, 926w, 878w, 857w, 797w, 761m, 714w, 690w, 616w, 538w, 462w, 441w. Selected Raman peaks (cm−1): 1630m, 1587s, 1545m, 1473m, 1442s, 1308w, 1275w, 1186m, 1156w, 624m, 461w. UV/Vis (CH2Cl2, nm): 225mb, 240mb, 280wb, 325wb, 420wb, 522wb, 583wb. ΛM (CH2Cl2, 10−3 M, 23 °C) = 2 S cm2 mol−1.
Single-crystal X-ray crystallography
Crystals of the complexes were taken directly from the mother liquor. Crystallographic data were collected with a Bruker APEX II Quasar diffractometer, equipped with a graphite monochromator centered on the path of Mo Kα radiation. The crystals were coated with CargilleTM NHV immersion oil and mounted on a fiber loop, followed by data collection at 120 K. The program SAINT was used to integrate the data, which were therefore corrected using SADABS.58 The structures were solved using SHELXT59 and refined by a full-matrix least-squares technique on F2 using SHELXL-2019.60 All non-H atoms were refined with anisotropic displacement parameters, whereas H atoms were assigned to ideal positions and refined isotropically using a suitable riding model, except those on the O atom of coordinated EtOH or H2O molecules, which were located on the difference density map and introduced using DFIX constraints.The crystals of 1·4CH2Cl2·2EtOH and 3·4CH2Cl2·2EtOH are small and weakly diffracting. Therefore, the data were cut at 0.90 and 1.00 Å, respectively, as there is no diffraction above and Rint becomes larger. The result is quite low θmax values and data/parameters ratios.
The CIF files have been deposited at the Cambridge Crystallographic Data Center as supplementary publication with the deposition numbers 2316071–2316073.‡ Crystallographic data are summarized in Table S1 (ESI‡).
Results and discussion
Synthetic comments
As stated in “Introduction”, our goal was to prepare heterometallic ZnII–FeIII and ZnII–CrIII complexes based on saphHCOOH and 4ClsaphHCOOH. Several synthetic parameters were investigated, such as the source of the two metal ions (nitrate, acetate, halide, perchlorate), the solvent, the concentration, the molar ratio of the reactants, the absence/presence of external bases (LiOH, NaOMe, Et3N), the reaction time, the temperature, the pressure (i.e. solvothermal techniques) and the crystallization techniques, before arriving at the optimized processes described in the Experimental section. The best choice of the metal sources for obtaining single crystals of the products were Zn(O2CMe)2·2H2O, and the nitrate “salts” of FeIII and CrIII. The ideal solvents proved to be EtOH/CH2Cl2 for the ZnII/FeIII complexes and MeCN/EtOH for the ZnII/CrIII compound. Elevated temperatures (i.e. refluxing conditions) were necessary to overcome the kinetic inertness of the 3d3 CrIII ion. Assuming that the hexanuclear complexes are the only products from their respective reaction mixtures, the formation of 1–3 can be summarized be eqn (1) and (2), where LH2 is saphHCOOH or 4ClsaphHCOOH, and S is CH2Cl2 for 1, 3 and MeCN for 2: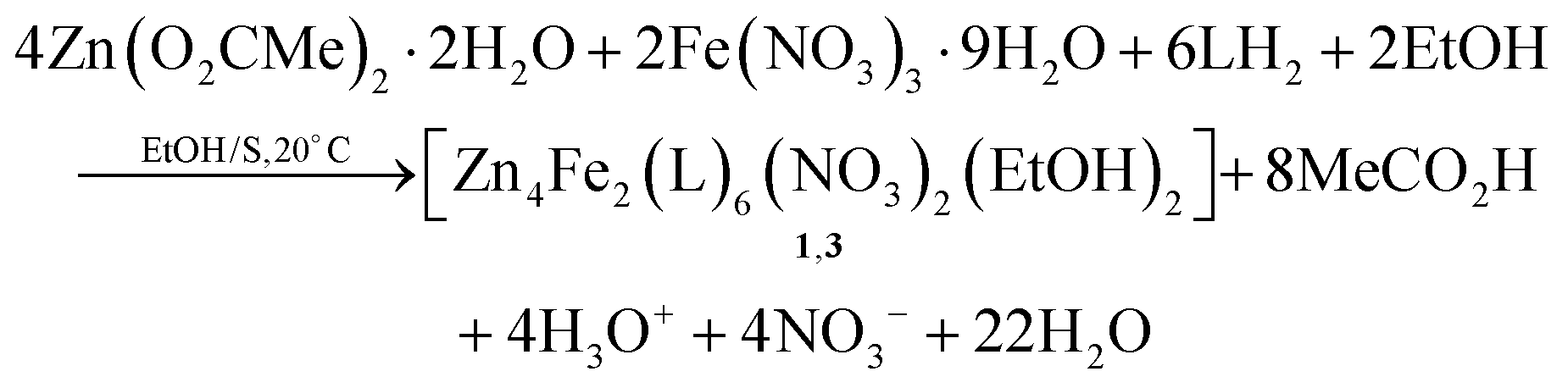 | (1) |
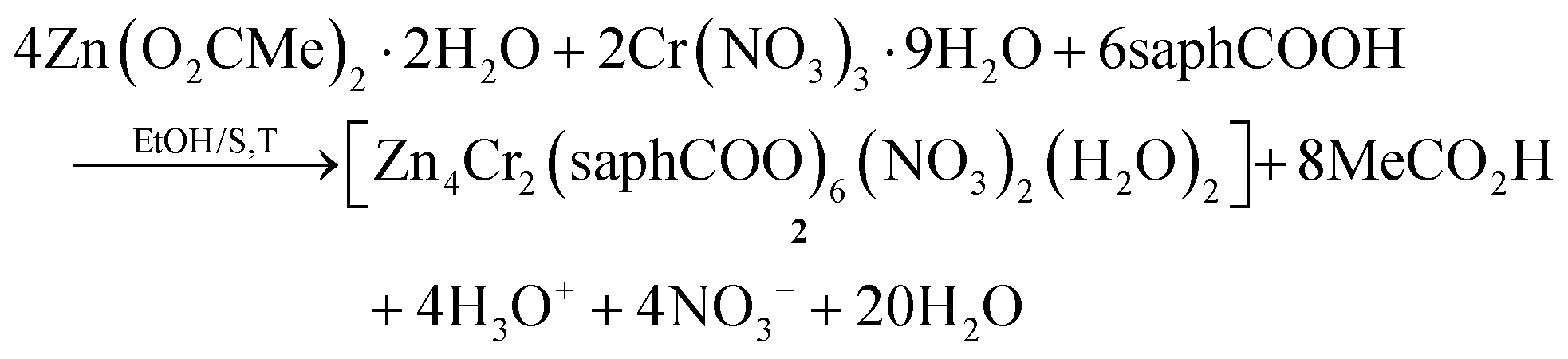 | (2) |
Few synthetic points deserve comments: (i) small changes in the reactants’ ratio do not affect the product identity. (ii) Heterometallic complexes containing the singly deprotonated saphHCOO− and 4ClsaphHCOO− ligands could not be prepared. (iii) A procedure similar to that described for 2, but using 4ClsaphHCOOH instead of saphHCOOH, gave a green powder whose analytical data are consistent with the formulation [Zn4Cr2(4ClsaphCOO)6(NO3)2(H2O)2]·xsolvents; in addition, the IR spectrum of this powder is similar to that of 2. However, since we could not obtain single crystals of this product, its characterization was not pursued further; and (iv) looking at the stoichiometric chemical eqn (1) and (2) we see that dilute HNO3 (strong acid) is also produced, which might decompose 1–3, e.g. by singly protonating the coordinated dianionic ligands. We believe that the released protons are neutralized by the MeCO2− ions that are present in excess in the reaction mixture. The stoichiometric MeCO2−:saphHCOOH/4ClsaphHCOOH molar ratio indicated by eqn (1) and (2) is 8:6, whereas the experimental one is 2:1 (i.e. 12:6); this means that the acetates are in excess acting as additional effective proton acceptors. In support of this viewpoint, the complexes can not be isolated using an 1:1.5 Zn(O2CMe)2·2H2O:LH2 (i.e. MeCO2−:LH2 = 4:6) ratio. Thus, an alternative description of the actual processes, perhaps more correct, can be represented by eqn (3); in this equation M is Fe or Cr, LH2 is saphHCOOH (for 1 and 2) or 4ClsaphHCOOH (in the case of 3) and n = 30. If this is correct, FeIII and CrIII are in deficiency in the reaction mixtures and this is consistent with the moderate yields (ca. 50%) of the reactions.
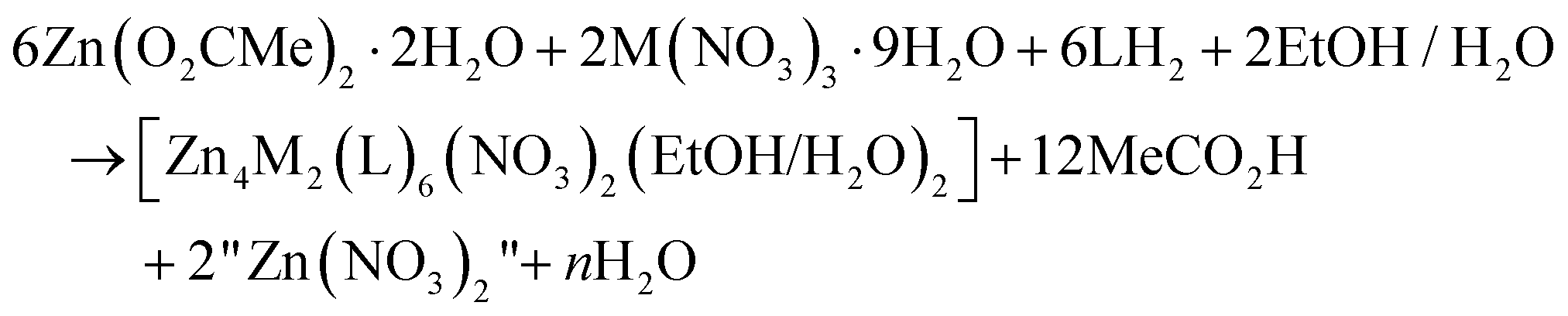 | (3) |
Description of structures
The structures of the three complexes have been determined by single-crystal X-ray crystallography. Selected interatomic distances and bond angles are listed in Tables 1 and 2. Various structural plots are shown in Fig. 1–7 and Fig. S1–S8 (ESI‡). Further details can be found in the deposited CIFs. The molecular structures of the three complexes are very similar and thus only the structure of 1·4CH2Cl2·2EtOH will be described in detail.| 1·4CH2Cl2·2EtOHa | 3·4CH2Cl2·2EtOHb | ||||||
|---|---|---|---|---|---|---|---|
| a Symmetry operation used to generate equivalent atoms: (′) −x, −y − 1, −z − 1. b Symmetry operation used to generate equivalent atoms: (′) −x + 1, −y + 1, −z + 2. | |||||||
| Interatomic distances | |||||||
| Zn1⋯Zn1′ | 3.118(2) | Zn2–O9 | 1.924(6) | Zn1⋯Zn1′ | 3.095(2) | Zn2–O9 | 1.927(4) |
| Zn2⋯Zn2′ | 11.349(2) | Zn2–O6 | 1.946(6) | Zn2⋯Zn2′ | 11.336(2) | Zn2–O6 | 1.967(4) |
| Fe1⋯Fe1′ | 9.022(2) | Zn2–O13 | 2.025(6) | Fe1⋯Fe1′ | 9.111(2) | Zn2–O13 | 2.055(5) |
| Fe1⋯Zn1 | 4.505(2) | Zn2–O10 | 2.040(7) | Fe1⋯Zn1 | 4.468(2) | Zn2–O10 | 2.025(4) |
| Fe1⋯Zn2 | 4.547(2) | Zn2⋯O11 | 2.566(6) | Fe1⋯Zn2 | 4.470(2) | Zn2⋯O11 | 2.558(6) |
| Fe1⋯Zn2′ | 9.189(2) | Fe1–O1 | 1.900(6) | Fe1⋯Zn2′ | 9.262(3) | Fe1–O1 | 1.903(4) |
| Zn1⋯Zn2′ | 6.745(2) | Fe1–O5 | 1.992(6) | Zn1⋯Zn2′ | 6.732(2) | Fe1–O5 | 1.990(4) |
| Zn1–O7 | 1.988(6) | Fe1–N1 | 2.140(8) | Zn1–O7 | 1.998(4) | Fe1–N1 | 2.124(5) |
| Zn1–N3 | 2.070(7) | Fe1–N2 | 2.181(8) | Zn1–N3 | 2.072(5) | Fe1–N2 | 2.221(5) |
| Bond angles | |||||||
| O7–Zn1–O3′ | 120.1(2) | O11⋯Zn2–O10 | 54.4(3) | O7–Zn1–O3′ | 114.7(2) | O11⋯Zn2–O10 | 54.5(3) |
| O7–Zn1–O7′ | 79.7(2) | O11⋯Zn2–O6 | 91.5(2) | O7–Zn1–O7′ | 81.1(2) | O11⋯Zn2–O6 | 91.0(3) |
| O7–Zn1–O8 | 140.9(2) | O11⋯Zn2–O13 | 143.2(3) | O7–Zn1–O8 | 142.3(2) | O11⋯Zn2–O13 | 146.1(3) |
| O7′–Zn1–N3 | 167.1(2) | Zn1–O7–Zn1′ | 100.3(2) | O7′–Zn1–N3 | 168.3(2) | Zn1–O7–Zn1′ | 98.1(2) |
| O13–Zn2–O10 | 89.2(3) | O4–Fe1–O2 | 158.1(2) | O13–Zn2–O10 | 92.1(2) | O4–Fe1–O2 | 155.7(2) |
| O9–Zn2–O10 | 111.1(3) | O5–Fe1–N1 | 172.0(3) | O9–Zn2–O10 | 113.7(2) | O5–Fe1–N1 | 168.4(2) |
| O9–Zn2–O13 | 102.1(3) | O1–Fe1–N2 | 168.3(3) | O9–Zn2–O13 | 103.0(2) | O1–Fe1–N2 | 166.2(2) |
| O11⋯Zn2–O9 | 88.4(3) | N1–Fe1–N2 | 103.1(3) | O11⋯Zn2–O9 | 87.5(3) | N1–Fe1–N2 | 105.3(2) |
| a Symmetry operation used to generate equivalent atoms: (′) −x + 1, −y + 1, −z + 1. | |||
|---|---|---|---|
| Interatomic distances | |||
| Zn1⋯Cr1 | 4.545(2) | Zn1–O6 | 1.957(2) |
| Zn1⋯Cr1′ | 9.063(2) | Zn1–O13 | 1.977(2) |
| Zn1⋯Zn2 | 4.804(2) | Zn1–O9 | 1.960(2) |
| Zn1⋯Zn2′ | 6.633(2) | Zn2–O7 | 2.013(2) |
| Zn1⋯Zn1′ | 11.175(1) | Zn2–O3′ | 2.006(2) |
| Zn2⋯Cr1 | 4.478(2) | Zn2–N3 | 2.048(2) |
| Zn2⋯Cr1′ | 4.993(2) | Cr1–O4 | 1.940(2) |
| Zn2⋯Zn2′ | 3.044(1) | Cr1–O2 | 1.993(2) |
| Cr1⋯Cr1′ | 8.984(2) | Cr1–N2 | 2.066(2) |
| Bond angles | |||
| O6–Zn1–O9 | 130.5(1) | O7–Zn2–O8 | 144.9(1) |
| O6–Zn1–O13 | 117.3(1) | O3′–Zn2–O7 | 117.5(1) |
| O6–Zn1–O10 | 102.5(1) | O3′–Zn2–N3 | 98.0(1) |
| O9–Zn1–O13 | 98.7(1) | O3′–Zn2–O7′ | 96.8(1) |
| O9–Zn1–O10 | 98.7(1) | O4–Cr1–O2 | 170.4(1) |
| O13–Zn1–O10 | 105.8(1) | O5–Cr1–N1 | 176.3(1) |
| N3–Zn2–O7′ | 165.2(1) | O1–Cr1–N2 | 171.9(1) |
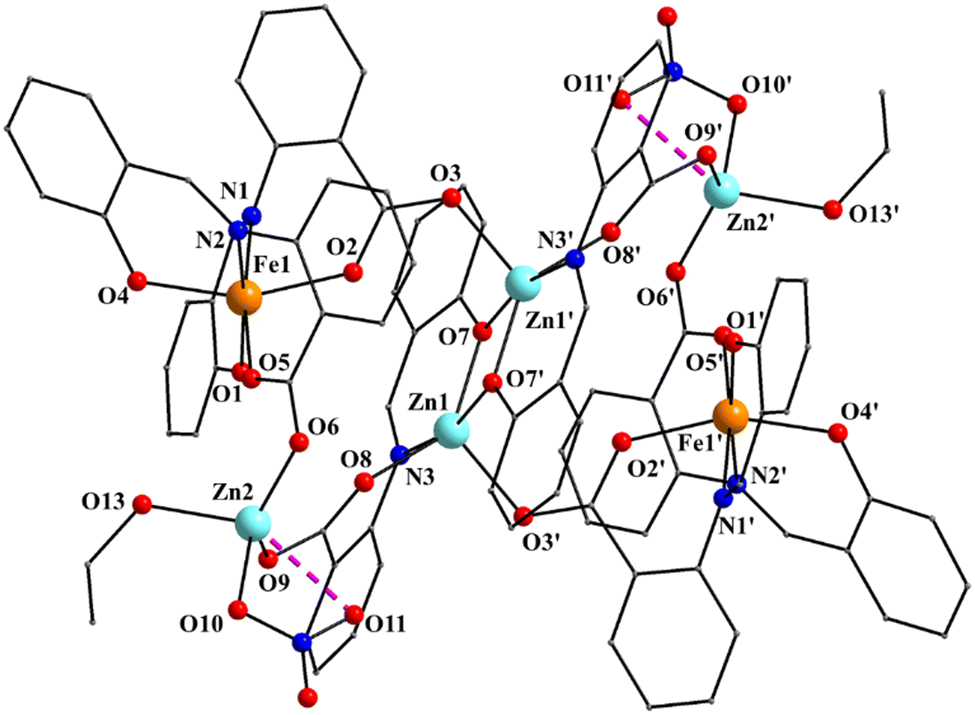 | ||
| Fig. 1 Partially labeled plot of the molecule [Zn4Fe2(saphCOO)6(NO3)2(EtOH)2] that is present in the crystal structure of 1·4CH2Cl2·2EtOH. The dashed lines indicate a weakly bonding interaction. Symmetry operation used to generate equivalent atoms: (′) −x, −y − 1, −z − 1. | ||
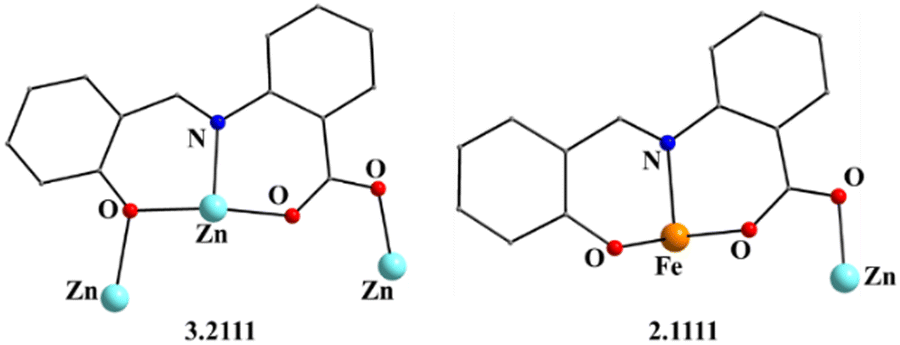 | ||
| Fig. 2 The coordination modes (using Harris notation) of the saphCOO2− ligands in complex 1·4CH2Cl2·2EtOH; four ligands adopt the 2.1111 ligation mode and two the 3.2111 one. | ||
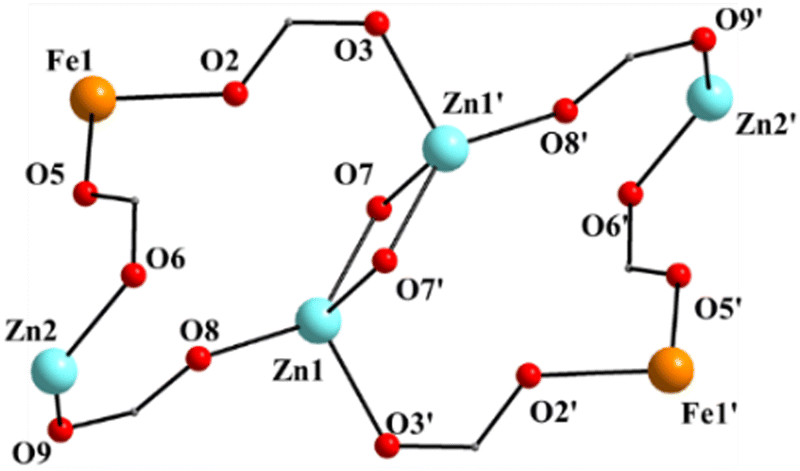 | ||
| Fig. 3 The labeled core of 1; the primes (′) are used for atoms generated by the symmetry operation −x, −y − 1, −z − 1. | ||
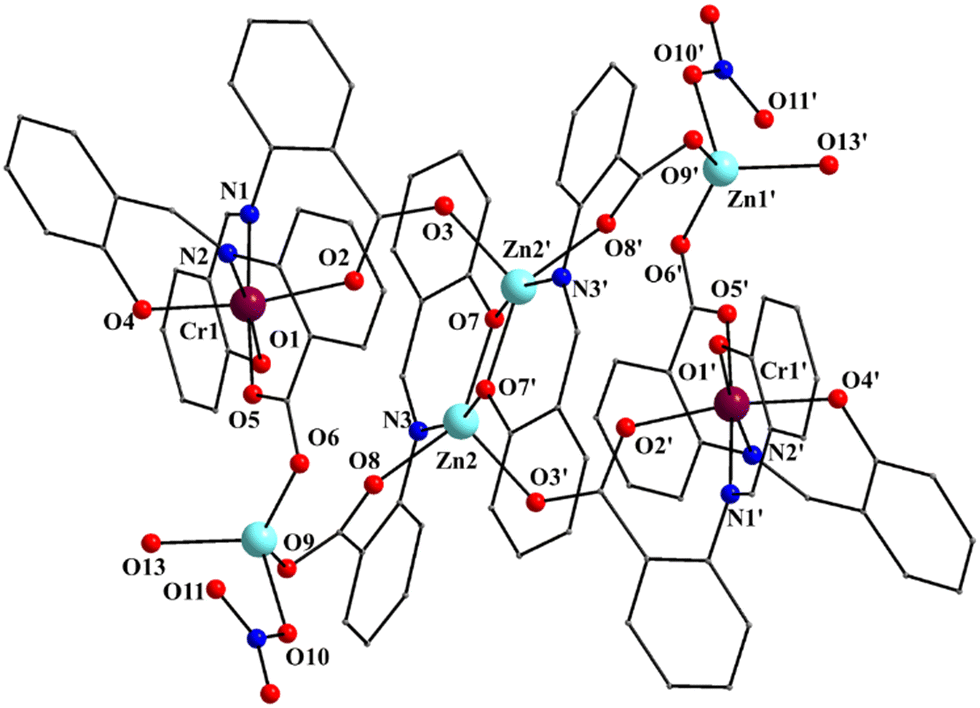 | ||
| Fig. 4 Partially labeled plot of the molecule [Zn4Cr2(saphCOO)6(NO3)2(H2O)2] that is present in the crystal structure of 2·4MeCN·2EtOH. The atom labeling scheme is similar to that adopted for 1 and 3, the only difference being that the central ZnII atoms of the planar rhombus are labeled as Zn2/Zn2′ and the outer ones as Zn1/Zn1′ in 2, whereas the inverse applies for 1 and 3 (Fig. 1 and Fig. S1, ESI‡). Symmetry operation used to generate equivalent atoms: (′) −x + 1, −y + 1, −z + 1. | ||
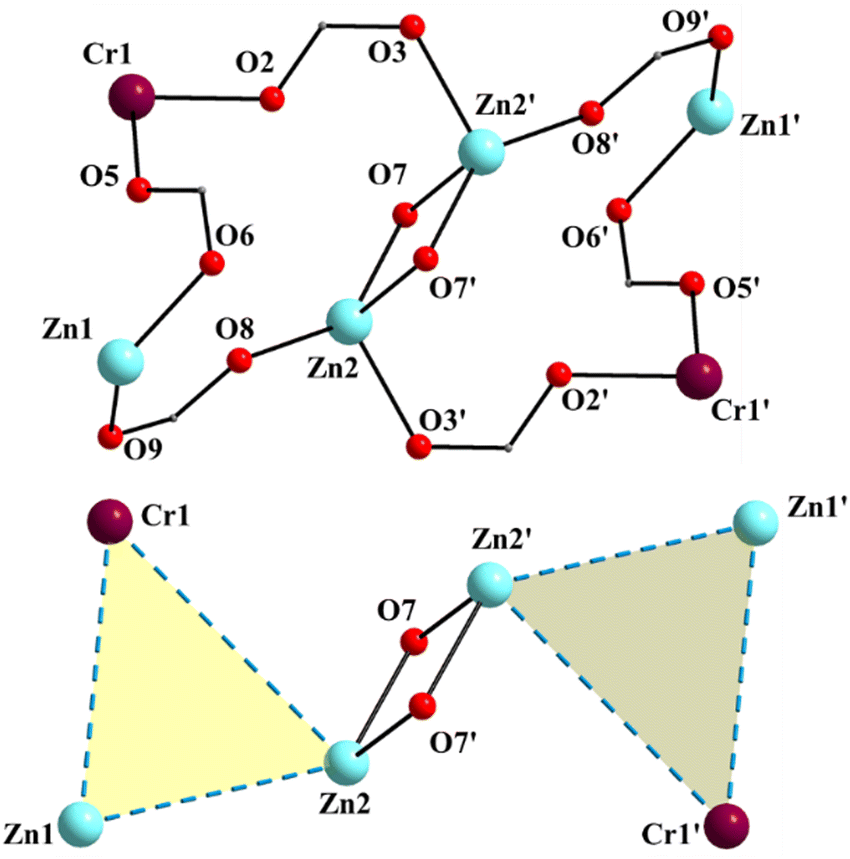 | ||
| Fig. 5 The labeled core of 2 (up) and a view of the metal topology (also including the phenolate monoatomic bridging oxygen atoms) of the molecule (down). The primes are used for atoms generated by the symmetry operation −x + 1, −y + 1, −z + 1. | ||
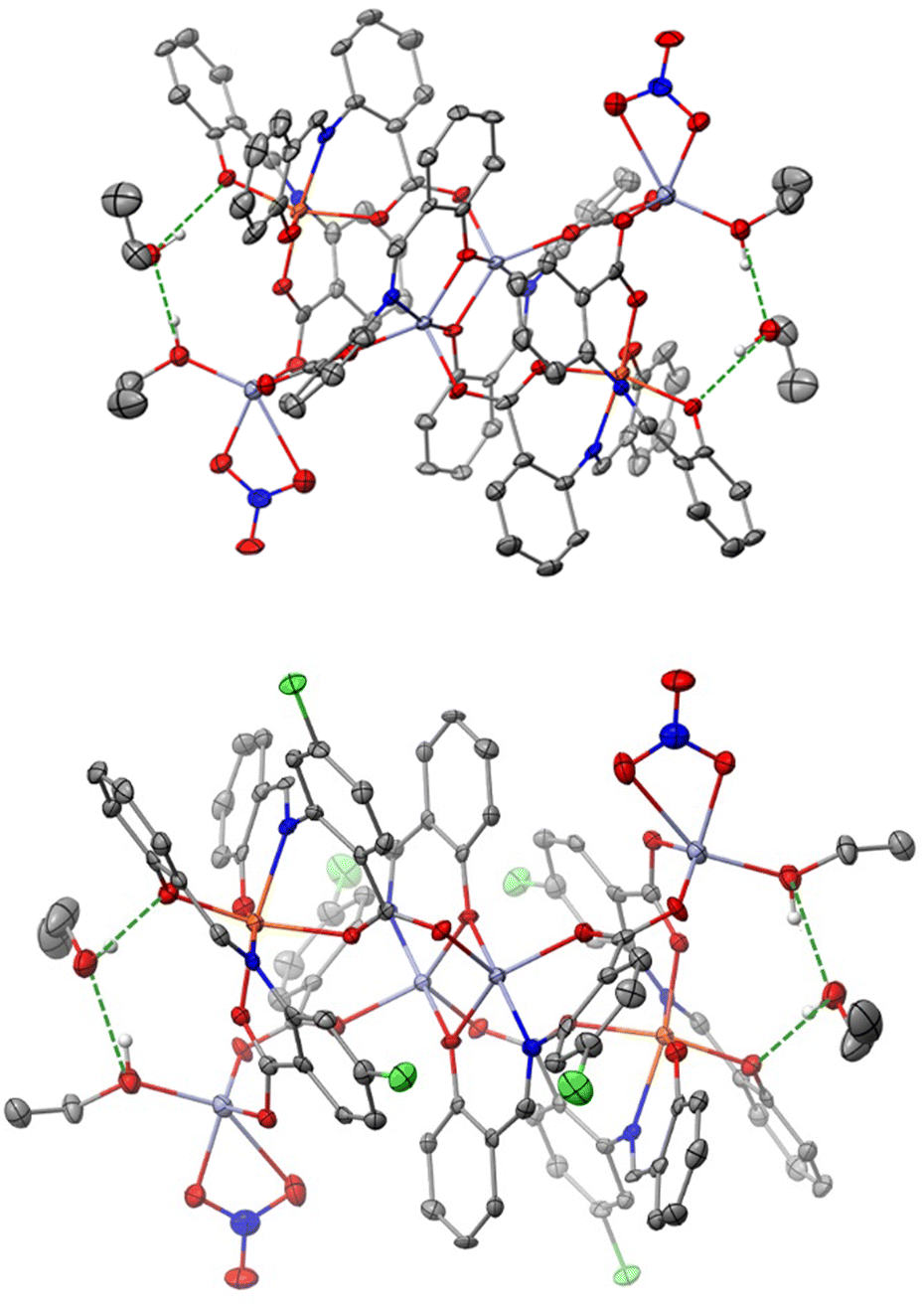 | ||
| Fig. 6 ORTEP-type views of the complexes 1·4CH2Cl2·2EtOH (top) and 3·4CH2Cl2·2EtOH (bottom), and the two hydrogen bonded lattice EtOH molecules in the crystal at 120 K. Thermal ellipsoids are depicted at a 50% probability level. Lattice CH2Cl2 molecules and H atoms are omitted for clarity, except those involved in H-bonding. H bonds are illustrated with dashed green lines. C, grey; H, white; N, blue; O, red; Cl, light green, Fe, orange; Zn, light blue. | ||
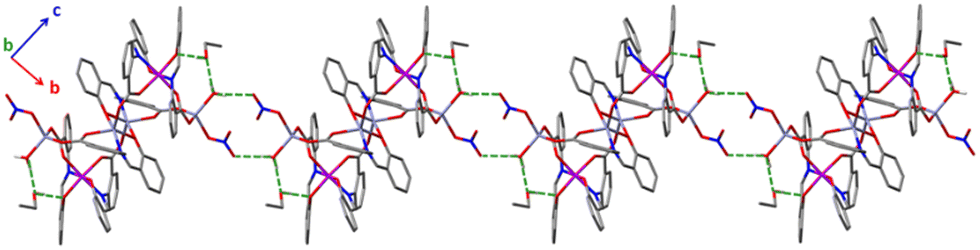 | ||
| Fig. 7 Stick representation of the H-bonded molecules 2 forming an 1D H-bonded network. H atoms (except those involved in H bonds) and lattice MeCN molecules are omitted for clarity. H bonds are depicted in dashed green lines. C, grey; H, white; N, blue; O, red; Cr, purple; Zn, light blue. | ||
Complex 1·4CH2Cl2·2EtOH crystallizes in the monoclinic space group P21/c. Its structure consists of hexanuclear [Zn4Fe2(saphCOO)6(NO3)2(EtOH)2] molecules, and lattice CH2Cl2 and EtOH molecules in an 1:4:2 ratio; the latter two will be not described here, but later in the context of the supramolecular interactions. The hexanuclear molecule possesses a crystallographically imposed inversion center, which is located at the midpoint of the Zn1⋯Zn1′ vector. The metal topology can be described as two isosceles triangles (Zn1Zn2Fe1 and Zn1′Zn2′Fe1′) which are linked through symmetry-related phenolate oxygen atoms (O7, O7′) of two 3.2111 (Harris notation61) saphCOO2− ligands, which bridge exclusively three ZnII atoms (Zn1, Zn1′, Zn2 and their symmetry equivalents). The Zn1O7Zn1′O7′ rhombus is perfectly planar (due to symmetry); its sides are 1.988(6) and 2.071(5) Å, its angles are 79.7(2) and 100.3(2)°, while the diagonal is 3.118(2) Å. The bridging of Fe1 and Zn2 is achieved by the syn, anti carboxylate group (O5, O6) of a 2.1111 saphCOO2− ligand. Zn1 and Zn2 are connected through the syn,anti carboxylate group (O8, O9) of the above mentioned 3.2111 ligand. Zn1′ and Fe1 are linked through the syn, anti carboxylate group (O2, O3) of a second 2.1111 saphCOO2− ligand. There is no direct linking between Zn1 and Fe1 through monoatomic or triatomic bridges. The phenolate oxygen atoms of the 2.1111 ligands (O4, O1) are terminally ligated to Fe1. Thus, there are four 2.1111 and two 3.2111 saphCOO2− groups in the whole molecule (Fig. 2). A terminal EtOH molecule (O13) and a highly anisobidentate chelating nitrate ion (O10, O11) complete 5-coordination at Zn2. Considering the latter, the anisobidentate chelating character is indicated by the Zn2–Onitrate bond lengths [2.040(7), 2.566(6) Å] which are significantly different; thus, the O11 could be considered as semicoordinated and its bond to Zn2 is drawn with a dashed line in Fig. 1. From Fig. 1 and the above discussion, it is clear that the coordination spheres are of the {Fe1O4N2}, {Zn1O4N} and {Zn2O5} types. Considering the triatomic carboxylate bridges as parts of the core, this is {Zn4Fe2(μ-OR)2(η1:η1:μ-O2CR′)4(η1:η1:μ-O2CR′′)2}6+, where R, R′ and R′′ are parts of the saphCOO2− ligands.
The Zn–O/N bond lengths are unremarkable,42,62 while the FeIII–O/N bond distances are typical for high-spin iron(III) centers in an octahedral environment.42,43 The FeIII coordination geometry is distorted octahedral, with the trans angles being in the range 158.1(2)–172.0(3)°. The Zn1 (and its symmetry equivalent) coordination geometry may be described as highly distorted square pyramidal, with the carboxylate atom O3′ occupying the apical position. Analysis of the shape-determining angles using the approach of Anderson, Reedijk et al.63 yields a value for the trigonality index, τ, of 0.45 for this metal ion (τ = 0 and 1.0 for ideal square pyramidal and trigonal bipyramidal geometry, respectively). Although the τ value for Zn2 is significantly smaller (0.19) than that for Zn1, its geometry also appears highly distorted; it may be described as square pyramidal with the nitrate oxygen O10 at the apex of the pyramid. Reasons for the distortion are the small basal plane angles [143.2(3) and 131.3(3)°], significantly less than the ideal value of 180°, and the small bite angle of ∼55° of the anisobidentate chelating NO3−.
Complex 3·4CH2Cl2·2EtOH is isostructural with 1·4CH2Cl2·2EtOH, but not isomorphous (Table S1, ESI‡). As it is obvious from Table 1 and Fig. 1, Fig. S1 (ESI‡), the centrosymmetric molecules of 1 and 3 are very similar (for convenience, an analogous atom labeling scheme has been adopted). Again the 4ClsaphCOO2− ligands adopt similar coordination modes (Fig. S2 and Fig. 2, ESI‡). As in 1, the nitrate groups are anisobidentate in 3, the coordination spheres are of the {Fe1O4N2}, {Zn1O4N} and {Zn2O5} types, and the core is again {Zn4Fe2(μ-OR)2(η1:η1:μ-O2CR′)4(η1:η1:μ-O2CR′′)2}6+. Thus, the Cl atom in the position 4 of the aminobenzoic aromatic ring has no influence on the molecular structure.
Complex 2·4MeCN·2EtOH crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Its structure consists of centrosymmetric hexanuclear [Zn4Cr2(saphCOO)6(NO3)2(H2O)2], and lattice MeCN and EtOH molecules in an 1:4:2 ratio. The molecule of 2 has a rather similar structure to that of 1, with the replacement of the FeIII atoms in the latter by the CrIII atoms in the former (Fig. 1–5 and Fig. S3, ESI‡). There are two differences in the molecular structures of the two complexes. First, in 2 the outer ZnII atoms (Zn1/Zn1′ in Fig. 4) are bound to two aqua ligands (the corresponding terminal ligands are EtOH molecules in 1). And second, the terminal nitrato ligands are clearly monodentate [the Zn1⋯O11/Zn1′⋯O11′ distance is ∼2.78 Å. i.e. non-bonding] in 2 as opposed to 1 in which these groups are anisobidentate; as a result the coordination geometry at Zn1 may be better described as distorted tetrahedral, the donor atom – Zn1/Zn1′ – donor atom angles being in the 98.7(1)–130.5(1) range (Table 2). The Cr–N/O bond lengths are typical for octahedral CrIII centers.44,46,64,65
. Its structure consists of centrosymmetric hexanuclear [Zn4Cr2(saphCOO)6(NO3)2(H2O)2], and lattice MeCN and EtOH molecules in an 1:4:2 ratio. The molecule of 2 has a rather similar structure to that of 1, with the replacement of the FeIII atoms in the latter by the CrIII atoms in the former (Fig. 1–5 and Fig. S3, ESI‡). There are two differences in the molecular structures of the two complexes. First, in 2 the outer ZnII atoms (Zn1/Zn1′ in Fig. 4) are bound to two aqua ligands (the corresponding terminal ligands are EtOH molecules in 1). And second, the terminal nitrato ligands are clearly monodentate [the Zn1⋯O11/Zn1′⋯O11′ distance is ∼2.78 Å. i.e. non-bonding] in 2 as opposed to 1 in which these groups are anisobidentate; as a result the coordination geometry at Zn1 may be better described as distorted tetrahedral, the donor atom – Zn1/Zn1′ – donor atom angles being in the 98.7(1)–130.5(1) range (Table 2). The Cr–N/O bond lengths are typical for octahedral CrIII centers.44,46,64,65
We now discuss the intermolecular interactions in the three structures. In 1·4CH2Cl2·2EtOH and 3·4CH2Cl2·2EtOH, each polynuclear molecule is H-bonded to lattice EtOH molecules, both as donor and as acceptor (Fig. 6 and Fig. S4, ESI‡). The intermolecular distances are d(Olatticeethanol⋯Ophenolate) = 2.730(3) Å and d(Olatticeethanol⋯Ocoordinatedethanol) = 2.687(4) Å in 1·4CH2Cl2·2EtOH, and d(Olatticeethanol⋯Ophenolate) = 2.719(3) Å and d(Ocoordinatedethanol⋯Olatticeethanol) = 2.698(3) Å in 3·4CH2Cl2·2EtOH. Even though the two compounds are not isomorphous, the packing is similar with four dichloromethane molecules per hexanuclear complex filling the void space (Fig. S4 and S5, ESI‡).
In 2·4MeCN·2EtOH, each polynuclear molecule is H-bonded to two lattice EtOH molecules, both as donor and as acceptor (Fig. 7 and Fig. S6, ESI‡), with intermolecular distances of d(Olatticeethanol⋯Ophenolate) = 2.668(3) Å and d(Ocoordinatedwater⋯Olatticeethanol) = 2.644(3) Å. The coordinated H2O molecule is also H-bonded to a “free” oxygen atom of the coordinated nitrato group of a neighboring hexanuclear molecule [d(O⋯O) = 2.775(3) Å], forming an 1D H-bonded network in the crystal (Fig. 7). Four lattice MeCN molecules per complex are also present and fill the void space. The shortest intermolecular metal⋯metal distances are 12.175 Å (1·4CH2Cl2·2EtOH), 7.733 Å (3·4CH2Cl2·2EtOH) and 7.629 Å (2·4MeCN·2EtOH).
Space-filling diagrams of 1 and 3 are shown in Fig. S7 and S8 (ESI‡), respectively. The outer C⋯C distances in the molecules are ca. 1.9 nm.
Complexes 1 and 3 join a rather small family of structurally characterized metal complexes containing the saphCOO2− and saphHCOO− ligands (selected compounds are described in ref. 29–33 and 66–69); most of them possess the doubly deprotonated ligand. As mentioned in “Introduction”, these are the first heterometallic complexes based on these ligands. On the contrary, 2 is only the second characterized complex with any form of 4ClsaphHCOOH as ligand, the first being [Cu(4ClsaphCOO)(phen)].36,37 For convenience, the to-date crystallographically established coordination modes of the two ligands are illustrated in Fig. 8.
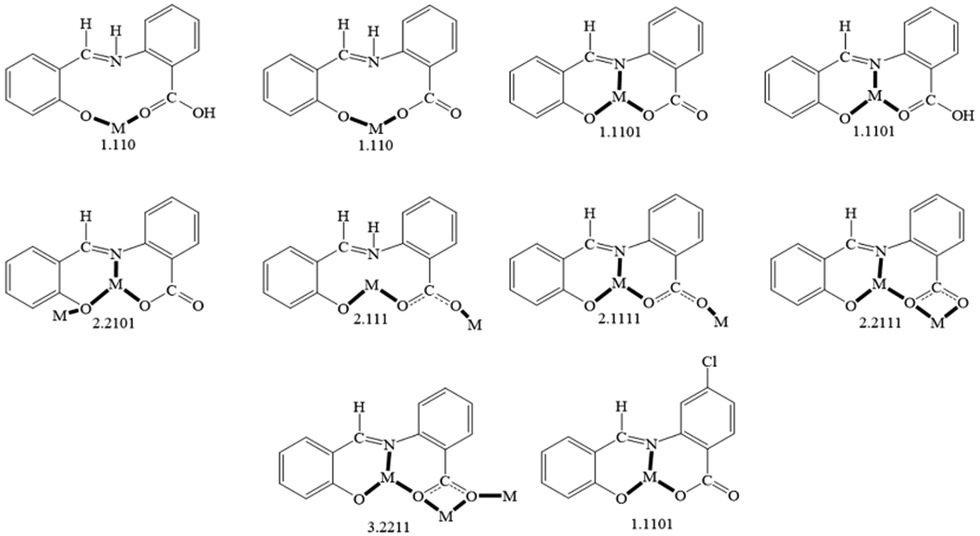 | ||
| Fig. 8 The to-date crystallographically observed coordination modes of saphHCOOH, saphHCOO−, saphCOO2− and 4ClsaphCOO2−, and the Harris notation that describes these modes; complexes containing the neutral or the singly deprotonated 4ClsaphHCOOH ligand have not yet been reported. The new ligation mode 3.2111 of the present work (Fig. 2 and Fig. S3, ESI‡) is not included in the figure. Coordination bonds have been drawn with bold lines. | ||
The ligation mode 3.2111 (Fig. 2 and Fig. S3, ESI‡) has not been observed before. The 2.1111 mode (Fig. 2 and 8) has been established only in few complexes of saphCOO2−,66–69 whereas this mode is new for 4ClsaphCOO2−.
Complexes 1, 3 and 2 are members of families that have heterometallic ZnII–FeIII (selected literature is given in ref. 39–43, 62 and 64) and ZnII–CrIII (for example, see ref. 44–47, 64, 65 and 70) complexes, respectively. The nuclearity {ZnII4FeIII2} has been observed once,62 whereas the nuclearity {ZnII4CrIII2} was unknown prior to this work.
IR, Raman and UV/Vis spectra
The three complexes were characterized by vibrational and solution UV/Vis spectroscopies on well dried (i.e. as lattice solvent-free) samples (Fig. 9 and Fig. S9–S13, ESI‡).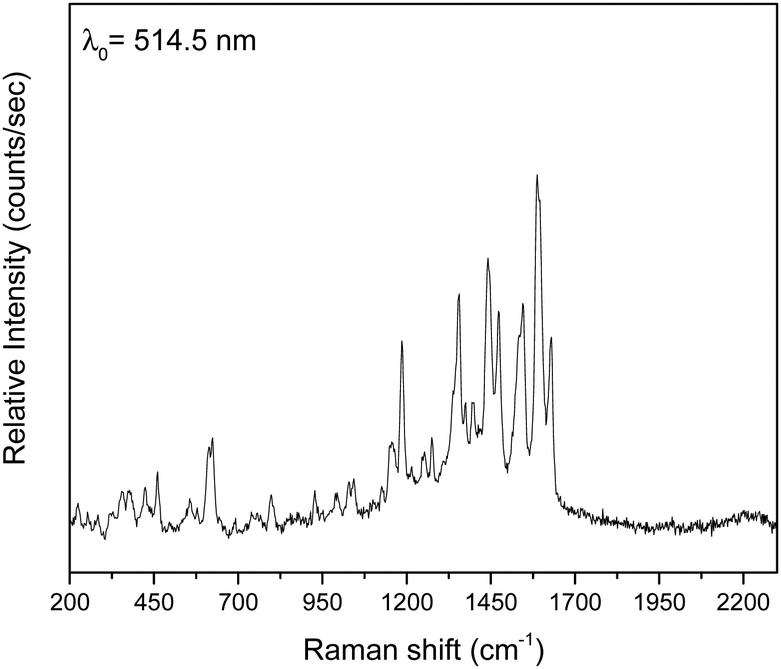 | ||
| Fig. 9 The Raman spectrum of a dried sample of 2 in the 2300–200 cm−1 region. | ||
The IR and Raman spectra of the complexes are very similar in accordance with their similar structures. The weak-to-medium intensity broad IR band at 3482–3420 cm−1 is assigned to the ν(OH) vibration of the coordinated EtOH (1, 3) and H2O (2) molecules; the broadness of the band suggests H bonding.51 As expected, this mode is hardly seen in the Raman spectra. The IR spectra exhibit a strong band at ∼1595 cm−1 which is assigned to the ν(CN) vibration of the Schiff-base linkage,51,52 most probably overlapping with an aromatic stretch. This spectral mode appears as a strong peak at ∼1615 cm−1 in 1 and 3, and as a medium-intensity peak at 1630 cm−1 (2) in the Raman spectra.71 The IR spectra exhibit a medium to strong band at ca. 1385 cm−1, characteristic of the ν3(E′)[νd(NO)] mode of the planar D3h ionic NO3−;72 however, such nitrates are not present in the crystal structure of the complexes. The appearance of this band indicates that the nitrato ligands (or an amount of them) in the samples are replaced by bromides that are present in the KBr matrix (used for the preparation of pellets), and so ionic nitrates (KNO3) are produced;73 it is well known that this replacement is facilitated by the pressure applied. In accordance with this explanation, the peak at ∼1385 cm−1 is always absent in the Raman spectra of the complexes. In these spectra, the peaks at 1458–1442 and 1317–1308 cm−1 are assigned to the ν5(B2)[νas(NO2)] and ν1(A1)[νs(NO2)] modes, respectively, of the nitrate group assuming C2v symmetry; their separation is small (∼150 cm−1) indicating monodentate nitrates.72 Thus, from the Raman spectroscopy viewpoint the nitrato ligands appear monodentate, suggesting that the anisobidentate chelating character in 1 and 3 is not reflected in the spectra. A larger separation (∼200 cm−1) of the two highest wavenumber stretching nitrogen–oxygen is expected for “normal” bidentate chelating nitrato ligands.72 The fact that the ∼1450 and ∼1300 cm−1 bands are present in the IR spectra is an indication that there is an amount of coordinated nitrate group still present in the samples and only a percentage of the whole NO3− content has been transformed to ionic nitrates. The location of the carboxylate stretching bands in the expected 1600–1300 cm−1 region72 is difficult due to the appearance of aromatic and nitrate stretching vibrations in this region. The IR bands at 1550–1540 and ca. 1380 cm−1 are tentatively assigned72,74 to the νas(CO2) and νs(CO2) vibrations respectively, the latter overlapping with the ionic nitrate stretching vibration ν3(E′)[vide supra]. Only in the spectrum of 3 (Fig. S11, ESI‡), the νs(CO2) band (at 1401 cm−1) is well separated from the nitrate band. The large νas(CO2)–νs(CO2) difference (∼160 cm−1) is indicative of the bidentate bridging ligation of the carboxylate groups of saphCOO2− and 4ClsaphCOO2−.72,74 The νas(CO2) and νs(CO2) Raman peaks appear at ca. 1540 and ca. 1275 cm−1, respectively, with the latter not seen clearly in the spectrum of 3; again the large separation suggests bidentate bridging behavior.72
The UV/Vis spectra of 1–3 were recorded in CH2Cl2 (Fig. S13, ESI‡). The molar conductivities, ΛM, are negligible (1–4 S cm2 mol−1), suggesting the absence of ionic species in solution.75 This fact, in combination with the non-donor properties of CH2Cl2, might indicate that the molecular structures of the complexes are retained in solution. In the spectra of 1 and 3, the bands below 300 nm are due to transitions of the aromatic ring, the longest wavelength maximum also having a –CHN– transition character.76 The 380, 405 and 510 nm bands are assigned to ligand-to-metal charge transfer (LMCT) transitions,77i.e. from ligands′ π orbitals to the t2g orbitals of iron(III). Because of the oxidizing power of FeIII, LMCT bands often obscure the very low intensity d–d transitions. The weak band at ca. 585 nm may be due to the 6A1g → 4T2g transition77 in a high-spin 3d5 octahedral system. The interpretation of the electronic spectrum of 2 is not easy. The bands at 225 and 240 nm are assigned77 to transition within the π system of the aromatic rings, the longest wavelength maximum possibly also having a –CHN– transition character.76 Since Cr(III) is neither a good reducing nor a good oxidizing agent, charge transfer bands tend not to obscure the three spin-allowed transitions. The bands at 280, 420 and 522 nm are attributed77 to the 4A2g → 4T1g (P), 4A2g → 4T1g (F) and 4A2g → 4T2g d–d transitions, respectively, in an octahedral 3d3 crystal field. The possibility the 280 nm band to have a mixed –CHN/d–d character can not be ruled out. The band at 325 nm can be tentatively assigned to a LMCT transition, while that at 583 nm to the spin-forbidden 4A2g → 2T2g, 2Eg transition.77 The longest wavelength spin-allowed d–d transition at 522 nm gives directly the eg–t2g gap (10 Dq) which is ∼19150 cm−1, typical for octahedral CrIII complexes in a mixed N/O environment.77
57Fe Mössbauer spectra
Zero-field Mössbauer spectra of powdered samples of well dried 1 and 3 were recorded at 300 and 80 K. Spectra are presented in Fig. 10 and Fig. S14 (ESI‡). At 300 K, the spectrum of 1 consists of a relative broad doublet with an apparent isomer shift value (δ) of ∼0.35 mm s−1 with an estimated quadruple splitting parameter, ΔEQ, of ∼0.6 mm s−1. The δ value is consistent with a high-spin FeIII ion in an octahedral environment comprising N/O-donors.78 A more accurate determination of ΔEQ is hindered by the broadness of the peak. The broadness of the spectrum is attributed to relaxation effects and this is often observed in isolated high-spin ferric ions in the solid state.79 In comparison to 3, the 300 K spectrum of 1 exhibits already a broad background. At 80 K, the doublet becomes broader and the broad background increases. We attribute this behavior to spin relaxation effects.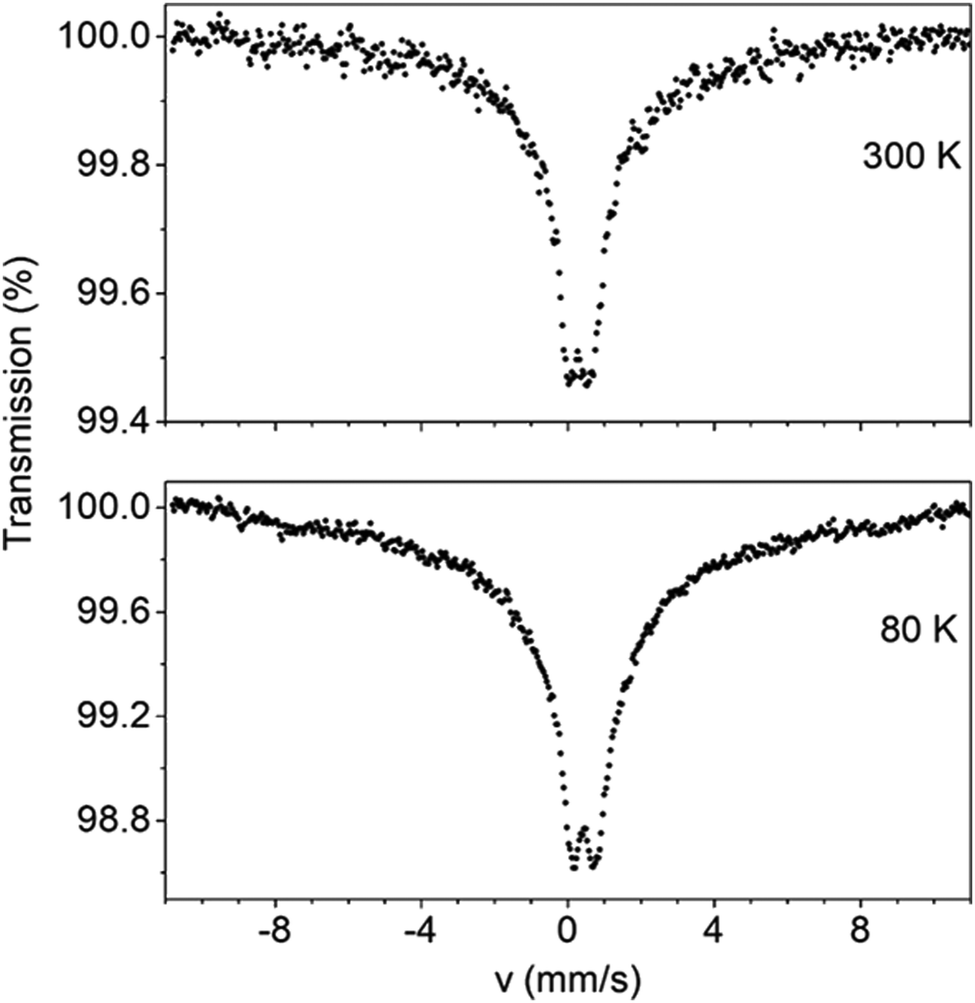 | ||
| Fig. 10 Mössbauer spectra of powdered samples of dried complex 1 recorded at 300 and 80 K in zero applied field. | ||
The Mössbauer behavior of 3 is similar to that of 1. At room temperature, the spectrum consists of a broad absorption peak corresponding to a δ value of 0.38 mm s−1. Again this value is consistent with a high-spin FeIII ion possessing a mixed O/N-donor coordination sphere.78 The ΔEQ value is estimated to be ∼0.5 mm s−1. At 80 K, the central peak becomes broader. At the same time, a broad background develops having the behavior of an unresolved magnetic sextet. This is consistent (like the broadness of the spectrum of 179) with magnetic relaxation effects. As the temperature decreases, the spin–lattice relaxation rate decreases leading to the conversion of the Mössbauer spectrum from a doublet to sextet.
Magnetic properties in brief
Variable-temperature (1.8–300 K) dc magnetic susceptibility studies were performed on powdered microcrystalline complexes of the three complexes at 0.1 T. Data are shown in Fig. 11, 12 and Fig. S15–S18 (ESI‡). The crystals were filtered directly from mother liquor and immediately prepared for measurements. The χT value of 1·4CH2Cl2·2EtOH at room temperature is 9.8 cm3 K mol−1, identical with the expected value (g = 2.12) for two non-interacting high-spin FeIII centers (χ is the molar magnetic susceptibility). The value of the χT product remains practically constant in the 300–20 K range and then decreases reaching a value of 8.9 cm3 K mol−1 at 1.85 K. This behavior is indicative of a very weak (if any) antiferromagnetic exchange interaction between the paramagnetic metal spins. The data have been subjected to a least-squares fit to the expression80 derived from the spin-Hamiltonian Ĥ = −2J(Ŝ1·Ŝ2), with S1 = S2 = 5/2. The calculated value is J/kB = −0.03(1) K (with g = 2.12(5)). Magnetization data at 1.85 K are approaching saturation at 7 T with a value of 11.0μB for two independent S = 5/2 ions and a g value of 2.10(5). Data for complex 3·4CH2Cl2·2EtOH are similar with a room-temperature χT value of 9.1 cm3 K mol−1 (g = 2.04(5)) and J/kB = −0.06(1) K.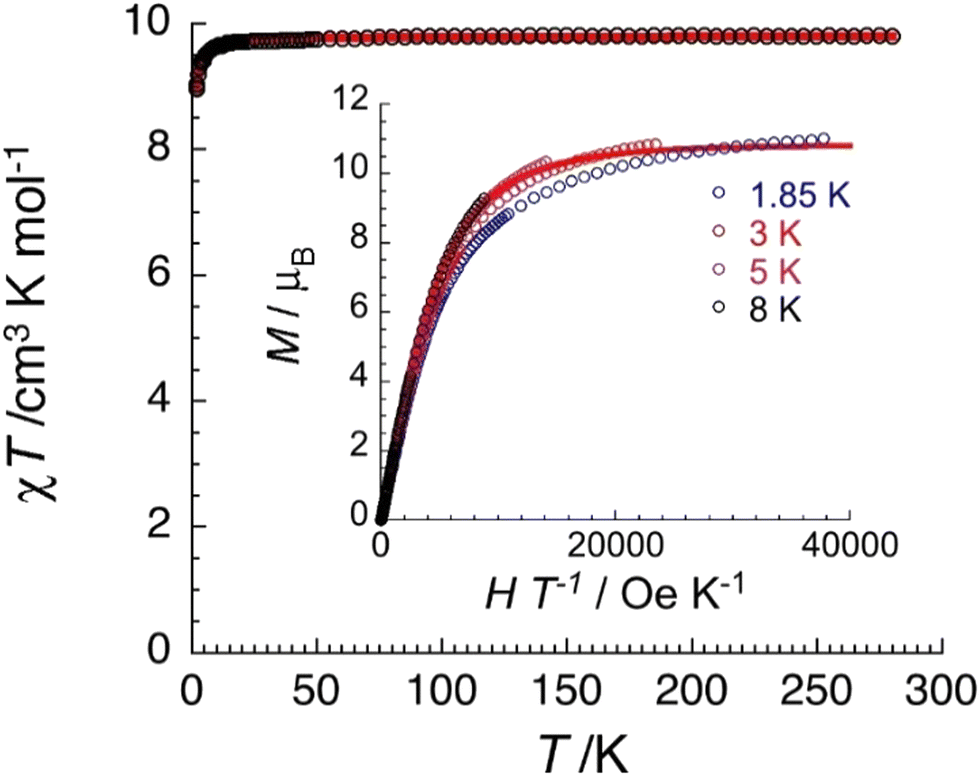 | ||
| Fig. 11 Temperature dependence of the χT product (where χ is the molar magnetic susceptibility that equals M/H per complex, and T the temperature) collected in an applied dc magnetic field (H) of 0.1 T for 1·4CH2Cl2·2EtOH. The solid red line is the best fit of the experimental data to an Heisenberg SFe = 5/2 spin dimer model as discussed in the text. Inset: Field dependence of the magnetization (M) for 1·4CH2Cl2·2EtOH below 8 K plotted as M vs. H/T. The solid red line is the best fit of the experimental data to two S = 5/2 Brillouin functions as discussed in the text. | ||
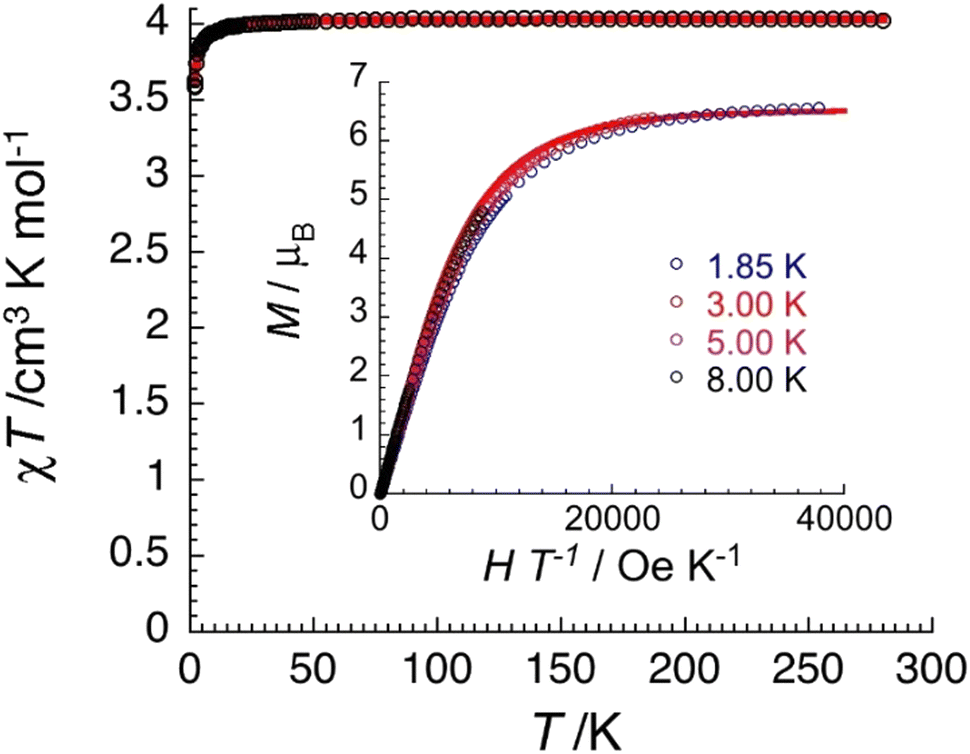 | ||
| Fig. 12 Temperature dependence of the χT product (where χ is the molar magnetic susceptibility that equals M/H per complex, and T the temperature) collected in an applied dc magnetic field (H) of 0.1 T for 2·4MeCN·2EtOH. The solid red line is the best fit of the experimental data to an Heisenberg SCr = 3/2 spin dimer model as discussed in the text. Inset: field dependence of the magnetization (M) for 2·4MeCN·2EtOH below 8 K plotted as M vs. H/T. The solid red line is the best fit of the experimental data to two S = 3/2 Brillouin functions as discussed in the text. | ||
The χT value of 2·4MeCN·2EtOH at room temperature is 4.0 cm3 K mol−1, close to the expected value (g = 2.06) for two non-interacting S = 3/2 CrIII centers. The value of the χT product remains constant in the 300–15 K range and then decreases reaching a value of 3.6 cm3 K mol−1 at 1.85 K. This behavior suggests a very weak antiferromagnetic exchange interaction between the CrIII spins. The data were fitted to the expression derived from the HDVV spin-Hamiltonian Ĥ = −2J(Ŝ1·Ŝ2) giving a J/kB value of −0.16(1) K (with g = 2.07(5)). Magnetization data have reached saturation at 7 T with a value of 6.6μB for two independent S = 3/2 ions with g = 2.20(5).
The negligible magnetic coupling for the complexes was expected considering the long (>7.5 Å, vide supra) intra- and intermolecular distances between the paramagnetic metal ions. However, cases have been reported44 in {ZnIICrIII2} complexes, where appreciable spin coupling between terminal CrIII ions separated by ca. 7 Å, is operated.
Conclusions and perspectives
The chemical message of this work is the ability of saphHCOOH and 4ClsaphHCOOH to form heterometallic 3d–3d′ complexes. Complexes 1–3 are the first heterometallic species based on these ligands. Their donor-atom system is ideal for both tridentate chelating Ophenolate, Nimine, Ocarboxylate and bridging behavior; candidate bridging atoms are the “free” (i.e. not coordinated) carboxylate oxygen, and both the coordinated phenolate and carboxylate atoms. A priori prediction for the preference for heterometallic or homometallic species is not possible. More judicious choice of the different metal ions is necessary to enforce the formation of heterometallic complexes. From the inorganic chemistry viewpoint, the complexes have interesting molecular structures, metal topologies, and enrich the groups of ZnII–FeIII and ZnII–CrIII compounds; moreover, the {ZnII4CrIII2} nuclearity is new, the {ZnII4FeIII2} one has been characterized for the second time and the 3.2111 coordination mode of saphCOO2−/4ClsaphCOO2− is observed for the first time. Given the long distance between the paramagnetic FeIII and CrIII ions, the magnetic exchange interactions are extremely weak and of antiferromagnetic nature.With knowledge and experience obtained in the present study, our efforts are directed, among others, to: (1) the preparation of heterometallic 3d–3d′ and 3d–4f complexes of the singly deprotonated (saphHCOO−, 4ClsaphHCOO−) ligands; (2) the replacement of ZnII with the other metal ions of Group 12 (CdII, HgII) to see if this change affects the chemical and structural identity of the products; (3) the use of two different paramagnetic metal ions, e.g. the pairs NiII–FeIII, NiII–CrIII, CuII–FeIII, CuII–CrIII, CuII–LnIII, NiII–LnIII (Ln = trivalent lanthanoid), to bring them at short distances with the goal to obtain new polynuclear complexes with interesting magnetic properties; (4) the replacement of FeIII in 1 and 3 with diamagnetic trivalent metals (e.g. GaIII and InIII) or LnIII ions aiming at the isolation of new species with photoluminescence properties based on the organic ligands or the 4f element, respectively; and (5) the incorporation of electron-releasing or electron-withdrawing (except Cl used in this work) non-donor substituents on one or both the aromatic rings, and study of their heterometallic coordination chemistry to investigate the effect of substituent(s) on the electronic and structural characteristics of the products.
Author contributions
Konstantinos N. Pantelis: data curation, formal analysis, investigation, methodology. Sotiris G. Skiadas: data curation, formal analysis, investigation. Zoi G. Lada: data curation, formal analysis, investigation. Rodolphe Clérac: data curation, formal analysis, methodology, software, editing – original draft. Yiannis Sanakis: data curation, formal analysis, methodology, software, writting – original draft. Pierre Dechambenoit: conceptualization, investigation, methodology, software, writing – original draft. Spyros P. Perlepes: conceptualization, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Head of the Laboratory of Applied Molecular Spectroscopy, Research Director George A. Voyiatzis (ICE-HT/FORTH), for the access to the Raman facilities. R. C. and P. D. thank the University of Bordeaux, the Région Nouvelle Aquitaine, Quantum Matter Bordeaux (QMBx), the Centre National de la Recherche Scientifique (CNRS), and the Association Francaise de Magnétisme Moléculaire.References
- M. G. Kanatzidis, R. Pöttgen and W. Jeitschko, Angew. Chem., Int. Ed., 2005, 44, 6996–7023 CrossRef CAS PubMed.
- S.-W. Cheong and M. Mostovoy, Nat. Mater., 2007, 6, 13–20 CrossRef CAS PubMed.
- N. Liu, C. Homann, S. Morfin, M. S. Kesanakurti, N. D. Calvert, A. J. Shuhendler, T. Al and E. Hemmer, Nanoscale, 2023, 15, 19546–19556 RSC.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- S. Mukherjee, J. A. Stull, J. Yano, T. C. Stamatatos, K. Pringouri, T. A. Stich, K. A. Abboud, R. D. Britt, V. K. Yachandra and G. Christou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2257–2262 CrossRef CAS PubMed.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420–3437 CrossRef CAS PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- J. L. Atwood and J. M. Lehn, Comprehensive Supramolecular Chemistry, Pergamon, Oxford, UK, 1996 Search PubMed.
- M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- D. Aguilà, L. A. Barrios, V. Velasco, O. Roubeau, A. Repollés, P. J. Alonso, J. Sesé, S. J. Teat, F. Luis and G. Aromí, J. Am. Chem. Soc., 2014, 136, 14215–14222 CrossRef PubMed.
- N. C. Anastasiadis, C. D. Polyzou, G. E. Kostakis, V. Bekiari, Y. Lan, S. P. Perlepes, K. F. Konidaris and A. K. Powell, Dalton Trans., 2015, 44, 19791–19795 RSC.
- D. Aguilà, Y. Prado, E. S. Koumousi, C. Mathonière and R. Clérac, Chem. Soc. Rev., 2016, 45, 203–224 RSC.
- T. Iwazumi, A. Kusumoto, R. Takeshita, D. Kobayashi, H. Tokoro, K. Nakagawa and S.-I. Ohkoshi, J. Electron Spectrosc. Relat. Phenom., 2024, 271, 147421 CrossRef CAS.
- A recent review from our group: Z. G. Lada, C. D. Polyzou, V. Nika, T. C. Stamatatos, K. F. Konidaris and S. P. Perlepes, Inorg. Chim. Acta, 2022, 539, 120954 CrossRef CAS.
- K. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289–290, 74–122 CrossRef CAS.
- L. Rosado Piquer and E. C. Sañudo, Dalton Trans., 2015, 44, 8771–8780 RSC.
- J. W. Sharples and D. Collison, Coord. Chem. Rev., 2014, 260, 1–20 CrossRef CAS PubMed.
- H.-S. Wang, K. Zhang, Y. Song and Z.-Q. Pan, Inorg. Chim. Acta, 2021, 521, 120318 CrossRef CAS.
- A. Dey, J. Acharya and V. Chandrasekhar, Chem. – Asian J., 2019, 14, 4433–4453 CrossRef CAS PubMed.
- C. D. Polyzou, C. G. Efthymiou, A. Escuer, L. Cunha-Silva, C. Papatriantafyllopoulou and S. P. Perlepes, Pure Appl. Chem., 2013, 85, 315–327 CrossRef CAS.
- Z. G. Lada, E. Katsoulakou and S. P. Perlepes, in Single-Molecule Magnets, ed. M. Hołyńska, Wiley-VCH, Weinheim, Germany, 2019, pp. 282–286 Search PubMed.
- For a review from our group, see: S. T. Tsantis, D. I. Tzimopoulos, M. Holynska and S. P. Perlepes, Int. J. Mol. Sci., 2020, 21, 555 CrossRef CAS PubMed.
- L. Rigamonti, A. Forni, S. Righetto and A. Pasini, Dalton Trans., 2019, 48, 11217–11234 RSC.
- J. Long, Front. Chem, 2019, 7, 63 CrossRef CAS PubMed.
- R. Molina-Hernández and A. Mederos, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Amsterdam, The Netherlands, 2004, pp. 411–446 Search PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- I. C. Lazzarini, A. V. Funes, L. Carrella, L. Sorace, E. Rentschler and P. Alborés, Eur. J. Inorg. Chem., 2014, 2561–2568 CrossRef CAS.
- N. C. Anastasiadis, N. Lalioti, A. Terzis, V. Psycharis, C. P. Raptopoulou and S. P. Perlepes, Inorg. Chem. Commun., 2015, 51, 118–121 CrossRef CAS.
- H. Ke, G.-F. Xu, Y.-N. Guo, P. Gamez, C. M. Beavers, S. J. Teat and J. Tang, Chem. Commun., 2010, 46, 6057–6059 RSC.
- I. C. Lazzarini, L. Carrella, E. Rentschler and P. Alborés, Inorg. Chim. Acta, 2016, 453, 692–696 CrossRef CAS.
- N. C. Anastasiadis, C. M. Granadeiro, J. Mayans, C. P. Raptopoulou, V. Bekiari, L. Cunha-Silva, V. Psycharis, A. Escuer, S. S. Balula, K. F. Konidaris and S. P. Perlepes, Inorg. Chem., 2019, 58, 9581–9585 CrossRef CAS PubMed.
- E. C. Mazarakioti, J. Regier, L. Cunha-Silva, W. Wernsdorfer, M. Pilkington, J. Tang and T. C. Stamatatos, Inorg. Chem., 2017, 56, 3568–3578 CrossRef CAS PubMed.
- A. A. Athanasopoulou, M. Pilkington, C. P. Raptopoulou, A. Escuer and T. C. Stamatatos, Chem. Commun., 2014, 50, 14942–14945 RSC.
- X. Li, C.-F. Bi, Y.-H. Fan, X. Zhang, X.-D. Wei and X.-M. Meng, Transition Met. Chem., 2014, 39, 577–584 CrossRef CAS.
- X. Li, D. Zhang, Z. Liu, Y. Xu and D. Wang, Inorg. Chim. Acta, 2018, 471, 280–289 CrossRef CAS.
- A. G. J. Ligtenbarg, R. Hage, A. Meetsma and B. L. Feringa, J. Chem. Soc., Perkin Trans. 2, 1999, 807–812 RSC.
- H. Machinaga, K. Matsufuji, M. Ohba, M. Kodera and H. Okawa, Chem. Lett., 2002, 716–717 CrossRef CAS.
- P. Biswas, M. Ghosh, S. K. Dutta, U. Flörke and K. Nag, Inorg. Chem., 2006, 45, 4830–4844 CrossRef CAS PubMed.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999–15008 RSC.
- A. Hazra, S. Bonakala, S. K. Reddy, S. Balasubramanian and T. K. Maji, Inorg. Chem., 2013, 52, 11385–11397 CrossRef CAS PubMed.
- J. L. Wong, R. F. Higgins, I. Bhowmick, D. X. Cao, G. Szigethy, J. W. Ziller, M. P. Shores and A. F. Heyduk, Chem. Sci., 2016, 7, 1594–1599 RSC.
- D. Burdinski, E. Bill, F. Birkelbach, K. Wieghardt and P. Chaudhuri, Inorg. Chem., 2001, 40, 1160–1166 CrossRef CAS PubMed.
- A. Schoedel, A. J. Cairns, Y. Belmabkhout, L. Wojtas, M. Mohamed, Z. Zhang, D. M. Proserpio, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2013, 52, 2902–2905 CrossRef CAS PubMed.
- H. W. L. Fraser, G. S. Nichol, D. Uhrín, U. G. Nielsen, M. Evangelisti, J. Schnack and E. K. Brechin, Dalton Trans., 2018, 47, 11834–11842 RSC.
- R. Alotaibi, E. Little, J. M. Fowler, A. Brookfield, R. W. Adams, A. Achari, G. A. Timco, G. F. S. Whitehead, N. F. Chilton, R. R. Nair, D. Collison and R. E. P. Winpenny, Inorg. Chem., 2021, 60, 15675–15685 CrossRef CAS PubMed.
- F. Troiani, A. Ghirri, M. Affronte, S. Carretta, P. Santini, G. Amoretti, S. Piligkos, G. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2005, 94, 207208 CrossRef CAS PubMed.
- M. Affronte, A. Ghirri, S. Carretta, G. Amoretti, S. Piligkos, G. A. Timco and R. E. P. Winpenny, Appl. Phys. Lett., 2004, 84, 3468–3470 CrossRef CAS.
- F. Moro, D. Kaminski, F. Tuna, G. F. S. Whitehead, G. A. Timco, D. Collison, R. E. P. Winpenny, A. Ardavan and E. J. L. McInnes, Chem. Commun., 2014, 50, 91–93 RSC.
- S. T. Tsantis, V. Bekiari, C. P. Raptopoulou, D. I. Tzimopoulos, V. Psycharis and S. P. Perlepes, Polyhedron, 2018, 152, 172–178 CrossRef CAS.
- N. C. Anastasiadis, D. A. Kalofolias, A. Philippidis, S. Tzani, C. P. Raptopoulou, V. Psycharis, C. J. Milios, A. Escuer and S. P. Perlepes, Dalton Trans., 2015, 44, 10200–10209 RSC.
- K. I. Alexopoulou, A. Terzis, C. P. Raptopoulou, V. Psycharis, A. Escuer and S. P. Perlepes, Inorg. Chem., 2015, 54, 5615–5617 CrossRef CAS PubMed.
- D. Maniaki, I. Mylonas-Margaritis, J. Mayans, A. Savvidou, C. P. Raptopoulou, V. Bekiari, V. Psycharis, A. Escuer and S. P. Perlepes, Dalton Trans., 2018, 47, 11859–11872 RSC.
- I. Mylonas-Margaritis, Z. G. Lada, A. A. Kitos, D. Maniaki, K. Skordi, A. J. Tasiopoulos, V. Bekiari, A. Escuer, J. Mayans, V. Nastopoulos, E. G. Bakalbassis, D. Papaioannou and S. P. Perlepes, Dalton Trans., 2023, 52, 8332–8343 RSC.
- A. D. Westland and M. T. H. Tarafder, Inorg. Chem., 1981, 20, 3992–3995 CrossRef CAS.
- Z. H. Abd El-Wahab, Spectrochim. Acta, Part A, 2007, 67, 25–38 CrossRef PubMed.
- G. M. Sheldrick, SADABS, ver. 2.03, Bruker Analytical X-Ray Systems, Madison, WI, USA, 2000 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349–2356 RSC.
- I. A. Lutsenko, M. A. Kiskin, N. N. Efimov, E. A. Ugolkova, Y. V. Maksimov, V. K. Imshennik, A. S. Goloveshkin, A. V. Khoroshilov, A. S. Lytvynenko, A. A. Sidorov and I. L. Eremenko, Polyhedron, 2017, 137, 165–175 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. Sanz, H. M. O'Connor, V. Martí-Centelles, P. Comar, M. B. Pitak, S. J. Coles, G. Lorusso, E. Palacios, M. Evangelisti, A. Baldansuren, N. F. Chilton, H. Weihe, E. J. L. McInnes, P. J. Lusby, S. Piligkos and E. K. Brechin, Chem. Sci., 2017, 8, 5526–5535 RSC.
- V. V. Semenaka, O. V. Nesterova, V. N. Kokozay, V. V. Dyakonenko, R. I. Zubatyuk, O. V. Shishkin, R. Boča, J. Jezierska and A. Ozarowski, Inorg. Chem., 2010, 49, 5460–5471 CrossRef CAS PubMed.
- W.-Z. Ju, L. Shi, K. Chen and J.-Y. Xue, Acta Crystallogr., Sect. C: Struct. Chem., 2005, 61, m1427–m1428 CAS.
- J. Wang, F. L. Bei, X. J. Yang, L. D. Lu and X. Wang, J. Mol. Struct., 2002, 643, 129–133 CrossRef CAS.
- S. S. Tonde, A. S. Kumbhar, S. B. Padhye and R. J. Butcher, J. Inorg. Biochem., 2006, 100, 51–57 CrossRef CAS PubMed.
- R. H. Laye and E. C. Sañudo, Inorg. Chim. Acta, 2009, 362, 2205–2212 CrossRef CAS.
- F. K. Larsen, J. Overgaard, S. Parsons, E. Rentschler, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 5978–5981 CrossRef CAS PubMed.
- F. R. Dollish, W. G. Fateley and F. F. Bentley, Characteristic Raman Frequencies of Organic Compounds, Wiley, New York, USA, 1974, pp. 110, 134–137 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, USA, 4th edn, 1986, pp. 121–124, 231–233, 254–257 Search PubMed.
- G. J. Kleywegt, W. G. R. Wiesmeijer, G. J. Van Driel, W. L. Driessen, J. Reedijk and J. H. Noordik, J. Chem. Soc., Dalton Trans., 1985, 2177–2184 RSC.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- C. N. R. Rao, Ultra-Violet and Visible Spectroscopy, Butterworths, London, UK, 1967, 2nd edn, pp. 20–33, 58–73 Search PubMed.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, The Netherlands, 2nd edn, 1984, pp. 329–332, 417–420, 450, 452, 453 Search PubMed.
- M. Savva, D. I. Alexandropoulos, M. Pissas, S. P. Perlepes, C. Papatriantafyllopoulou, Y. Sanakis and A. J. Tasiopoulos, Dalton Trans., 2023, 52, 6997–7008 RSC.
- N. N. Greenwood and T. C. Gibbs, Mössbauer Spectroscopy, Chapman and Hall, London, UK, 1971, pp. 1–660 Search PubMed.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 203–283 CrossRef.
Footnotes |
| † Dedicated to Professor Mark Turnbull on the occasion of his retirement; a great inorganic chemist and magnetochemist, a precious friend. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data (Table S1), structural plots (Fig. S1–S8), various spectra (Fig. S9–S14) and magnetic measurements plots (Fig. S15–S18). CCDC 2316071–2316073. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj02030h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |