
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoejection–recapture of the Ca2+ cation studied by time resolved spectroscopy and TDDFT calculations: the case study of an azacrown–iridium(III) complex†
Clément
Guerrin
 a,
Laure
De Thieulloy
c,
Julien
Dubois
a,
Clément
Barois
b,
Aurélie
Perrier
cd,
Isabelle
Leray
b,
Cedric
Mongin
*b and
Stephane
Aloise
*a
a,
Laure
De Thieulloy
c,
Julien
Dubois
a,
Clément
Barois
b,
Aurélie
Perrier
cd,
Isabelle
Leray
b,
Cedric
Mongin
*b and
Stephane
Aloise
*a
aUniv. Lille, CNRS, UMR 8516 – LASIRe – Laboratoire Avancé de Spectroscopie pour les Intéractions la Réactivité et l’Environnement, F-59000 Lille, France. E-mail: stephane.aloise@univ-lille.fr
bUniversité Paris-Saclay, ENS Paris-Saclay, CNRS, PPSM, Gif-sur-Yvette 91190, France
cChimie ParisTech, PSL Research University, CNRS, Institute of Chemistry for Life and Health Sciences (i-CLeHS), F-75005 Paris, France
dLaboratoire Interdisciplinaire des Energies de Demain, Université Paris Cité, CNRS, LIED, UMR, 8236 Paris, France
First published on 25th July 2024
Abstract
In this study, we examined the photophysical properties of an azacrown–iridium(III) complex while focusing on its interactions with calcium ions (Ca2+). We explored the dynamic processes within the complex combining time-dependent density functional theory (TDDFT) calculations and time-resolved spectroscopies. In the presence of Ca2+, the complex exhibits significant shifts in absorption and emission profiles, from 494 nm to 375 nm, aligning with theoretical predictions. Notably, we observed the ultrafast photo-ejection of Ca2+ within 70 femtoseconds, followed by its recapture in 250 nanoseconds, revealing a 10-million-fold timescale difference between the two phenomena. These behaviors confirm the established photophysical properties of polypyridyl iridium(III) complexes and their intrinsic sensitivity to their surrounding environment. Our comprehensive kinetic analysis highlights the azacrown moiety's competitive binding and photo-release capabilities, suggesting its potential for practical sensing applications. The versatile properties of these iridium(III) complexes offer promising prospects for their application as stimuli-responsive materials and in advanced optoelectronic devices, targeted imaging, and biomedical ion sensors and delivery systems.
1. Introduction
Complexation and precise spatiotemporal release of bioactive compounds are becoming increasingly vital in the fields of supramolecular chemistry, life science, and nanomedicine.1 A significant focus has been directed towards the development of chemical cages for metal ions, particularly calcium ions (Ca2+), due to their biological significance.2 In the pursuit of achieving precise control over bioactive cations, the use of light is appealing because of its non-invasive nature and its ability to be delivered with high spatial and temporal precision. Among all the possible chemical strategies, one solution involves using photo-induced charge transfer (PCT) molecules combined with a complexing agent, typically an aza-crown compound: PCT induces positive polarization of the heteroatom of the macrocycle, leading to coulombic repulsion between the heteroatom and the metal cation initially present in the cavity. Such a system has been proposed using various compounds, including azacrown-substituted [(bpy)Re(CO)3L]+ complexes3–5 and azacrown-substituted merocyanine.6–9 In 2016, we investigated a novel system, azacrown-substituted pyridinum betaine (PyB-Aza) by combining stationary spectroscopy, ultrafast absorption spectroscopy and DFT calculations. Focusing on PyB-Aza, the first aim was to assess the competitive complexation of the imidazole bridge vs. the macrocycle. In acetonitrile, it was found by absorption and emission that the imidazole moiety binds efficiently through lateral electrostatic interaction to form a (1,1) metal–ligand complex with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 6.8. From a photochemical point of view, the rate of the photo-release process, assessed by tracking the transient stimulated emission band was found to be less than 200 fs, the fastest photorelease characteristic time reported so far. Furthermore, it has been shown that this rate of photorelease is proportional to the complexation constant. Finally, no evidence was found for a photoinduced translocation from an imidazole bridge to crown ether. Currently, our group is interested in exploring novel chemical strategies.
K = 6.8. From a photochemical point of view, the rate of the photo-release process, assessed by tracking the transient stimulated emission band was found to be less than 200 fs, the fastest photorelease characteristic time reported so far. Furthermore, it has been shown that this rate of photorelease is proportional to the complexation constant. Finally, no evidence was found for a photoinduced translocation from an imidazole bridge to crown ether. Currently, our group is interested in exploring novel chemical strategies.
The study of iridium(III) complexes’ photophysics represents a cutting-edge topic within the field of photoluminescent materials.10–13 These complexes, renowned for their exceptional luminescent characteristics, are pivotal contenders for a diverse range of applications, spanning optoelectronic devices to bioimaging agents.14–16 Upon further examination, iridium(III) complexes can be classified into two primary categories: cyclometalated complexes and coordination complexes, including polypyridyl complexes. Cyclometalated iridium(III) compounds present at least one characteristic carbon–iridium bond and exhibit strong absorption in the visible region arising from intense metal-to-ligand charge transfer (MLCT) singlet state transition and high emissivity due to their lowest energy excited states being a blend of strong 3MLCT and triplet ligand-centered (3LC) π–π* states, rather than metal centered (MC) non-radiative d–d states.11,12 Among phosphorescent compounds, cyclometalated iridium(III) complexes emit from triplet excited states at room temperature with exceptional quantum yields up to unity and radiative rates in the microsecond regime.17
In contrast, polypyridyl complexes often exhibit ligand-to-ligand charge transfer (LLCT) and intra-ligand charge transfer (ILCT), which influence their photophysical behavior and result in weaker or even no MLCT transitions.12,18–21 Consequently, these complexes are often less emissive and display weaker phosphorescence at room temperature. However, due to the significant alteration of the polarization in the excited state, they are highly susceptible to minor alterations in their immediate surroundings, rendering them promising candidates for ion chelation and photo-ejection monitoring.20,22
This work will concentrate on the photophysical properties of polypyridyl complexes, with a particular emphasis on heteroleptic bisterpyridine iridium(III) complexes. These complexes are distinguished by their molar extinction coefficients, which are typically lower than those of cyclometalated complexes. Thus, the oscillator strength of the transitions is a critical parameter, influencing their photophysical properties.
The absorption and emission properties of these complexes are largely influenced by the nature of the ligands. The strong absorption bands around 450 to 500 nm, corresponding to the 1ILCT state, and the emission band above 500 nm, arising from the intersystem crossing (ISC) to the 3ILCT, are sensitive to changes in solvents and pH.11,12 This makes them excellent candidates for use in sensors and other applications where changes in environmental conditions need to be monitored. They also exhibit emission in the visible region involving the low-lying 3ILCT state. Furthermore, strong wavelength-dependent photoluminescence has been reported, as well as dual phosphorescence.23,24 The latter phenomenon is associated with the formation of a specific ion pair, which results in the iridium center and a counter ion, particularly PF6−, being in close proximity in organic solvents such as acetonitrile.23 This leads to a decrease in the excited state energy, which in turn causes the emission from the 3ILCT to shift towards the red end of the spectrum. The emission of the ion pair is similar to that observed in the solid state, where the crystalline structure allows for the specific interaction to occur. This study has explored the photophysics of these systems using time-resolved spectroscopies, characterizing very short 1ILCT lifetimes (approximately 100 fs),25 dynamics dependent on substituents,26 and solvent-dependent ISC 1ILCT → 3ILCT (70 fs–3 ps).27 It is also noteworthy that the use of iridium(III) cyclometalated complexes substituted with metallic cation chelating moieties has been previously investigated. This research has demonstrated a notable alteration of the phosphorescent properties, both in the absence of a metallic cation (weak 3ILCT emissive states) and in the presence of a metallic cation (strong 3LLCT/3MLCT emissive state).28
In this paper, we are investigating a new class of iridium(III) terpyridine complex, molecule 1 (Scheme 1), that comprises an azacrown moiety dedicated to trap and photo-release calcium cations. This molecule has been studied using a combination of stationary and ultrafast spectroscopies together with (TD-)DFT calculations.
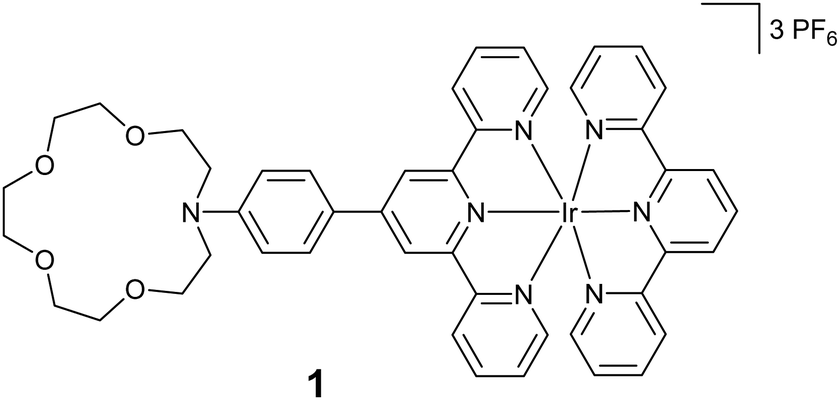 | ||
| Scheme 1 Structure of molecule 1. | ||
2. Results and discussion
2.1. Synthesis
The synthesis of molecule 1 is outlined in Scheme 2. Terpyridine 4 was synthesized from 2 and 3 according to a method from the literature via the Paal Knorr reaction in 70% yield.29 The synthesis of the iridium complex was achieved using a two-step method described by Collin et al.30 IrAzaTpyCl3 was obtained in 40% yield by a reaction of IrCl3 and 4 in ethylene glycol at 160 °C. The formation of the final complex was achieved by heating complex 5 in the presence of 6, followed by anion exchange with PF6−. The purities of the complexes were carefully checked by 1H NMR spectroscopy and high-resolution mass spectrometry.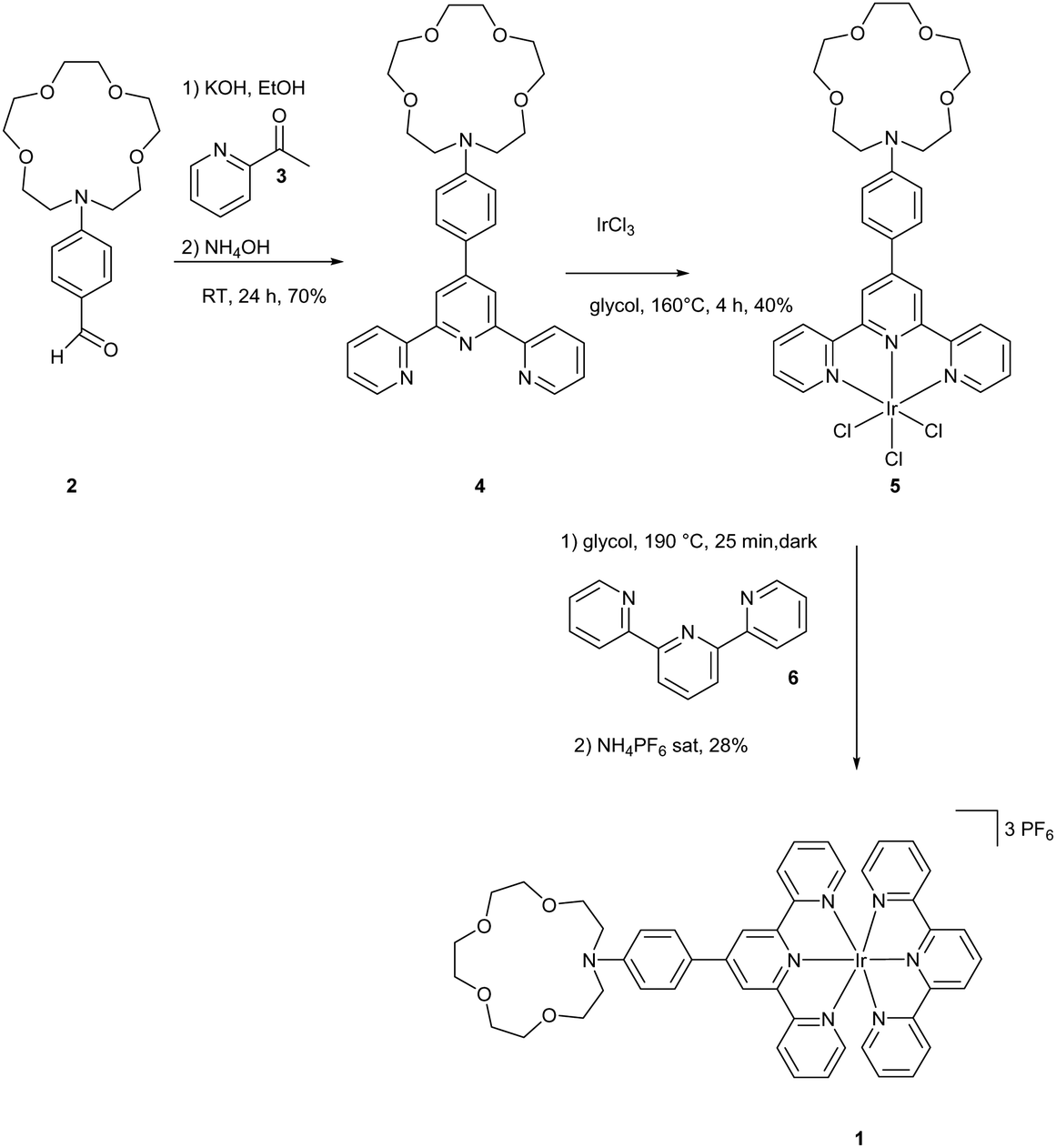 | ||
| Scheme 2 Synthesis of the iridium complex 1. | ||
2.2. Photophysical framework and complexation scheme
K = 2.4 ± 0.002 (K = 250) is assigned to a (1:1) stoichiometry complex as evidenced by the JOB plot presented in Fig. S1 in the ESI.† Note that this latter value is in accordance with various aza-crown–Ca2+ complexes found in the literature for PCT based fluorescent sensors inducing cation photorelease.31,32
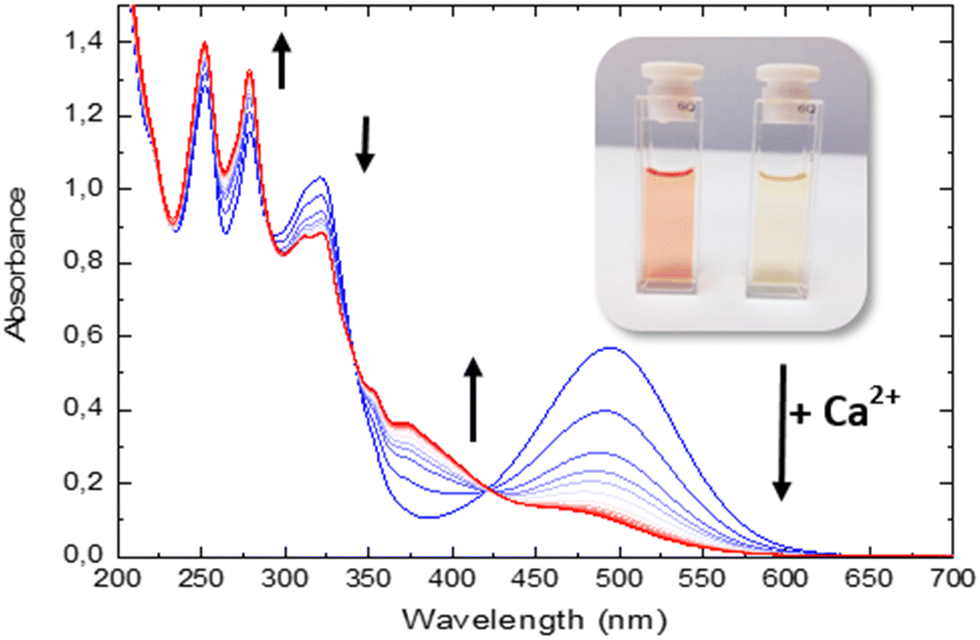 | ||
| Fig. 1 Absorption spectra of 1 in CH3CN (28 μM) upon successive addition of calcium perchlorate solution from 0 to 0.29 M. The inset shows a picture of the solution of 1 before (blue) and after adding a large excess of Ca2+ ion (red). | ||
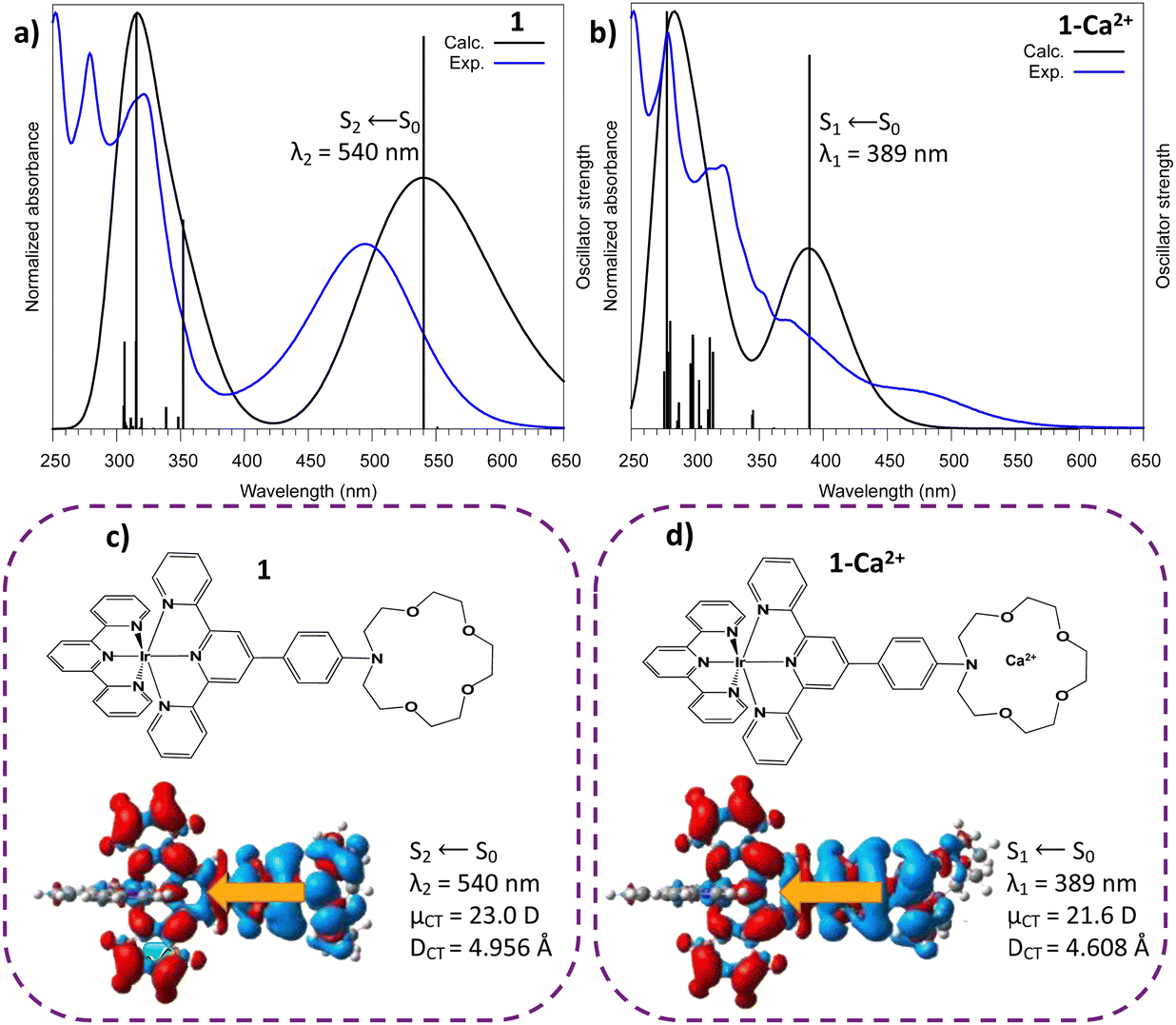 | ||
Fig. 2 Theoretical excitation energies and convoluted absorption spectra (convolution: FWHM = 0.25 eV) (a) for 1 and (b) the 1–Ca2+ complex. The variations of the electronic density upon excitation (Δρ(![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) ), red: gain of electronic density, blue: depletion of electronic density) are shown for the most important transition related to the calculated bands peaking at 540 nm for 1 (c) and at 389 nm for 1–Ca2+ (d). The charge transfer dipole moment ), red: gain of electronic density, blue: depletion of electronic density) are shown for the most important transition related to the calculated bands peaking at 540 nm for 1 (c) and at 389 nm for 1–Ca2+ (d). The charge transfer dipole moment ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) CT is also represented (golden arrow) and the charge transfer parameters are given. CT is also represented (golden arrow) and the charge transfer parameters are given. | ||
For 1–Ca2+ (with the cation inside the crown ether), the maximum absorption band is now related to the S1 electronic state which arises from a similar aza → tpy ILCT transition with CT descriptor values close to those of molecule 1 (μCT = 21.6 D, DCT = 4.6 Å). These descriptors suggest that 1–Ca2+ is a good candidate for cation photoejection since there is a depletion of electronic density on the azacrown complexation site after excitation. In addition to this, we considered the evolution of the distances of Ca2+ in the azacrown upon relaxation on the first singlet excited state (Table S1, ESI†). One can notice a pre-ejection process of Ca2+ from the azacrown with (i) a distance between the aza-nitrogen and Ca2+ stretched by more than 1 Å and (ii) an average distance between the oxygens and Ca2+ increased by 0.05 Å.
Finally, as anticipated with transient laser experiments, for molecule 1, an excitation near 350 nm coincides with the S6 ← S0 transition which presents an ILCT character (calculated value: 352 nm; see Fig. 2 and Fig. S2, Table S2, ESI†). In contrast, for complex 1–Ca2+, there is no transition with a strong oscillator strength in this region (Fig. 2 and Table S3, ESI†). In summary, according to theoretical predictions, UV excitation of 1–Ca2+ will only induce a S1 ← S0 electronic excitation and an instantaneous Aza → tpy ILCT process, leaving a positive charge on the crown nitrogen, inducing the pre-ejection of Ca2+ into the bulk. The corresponding ILCT state of molecule 1, namely S2, should then be populated.
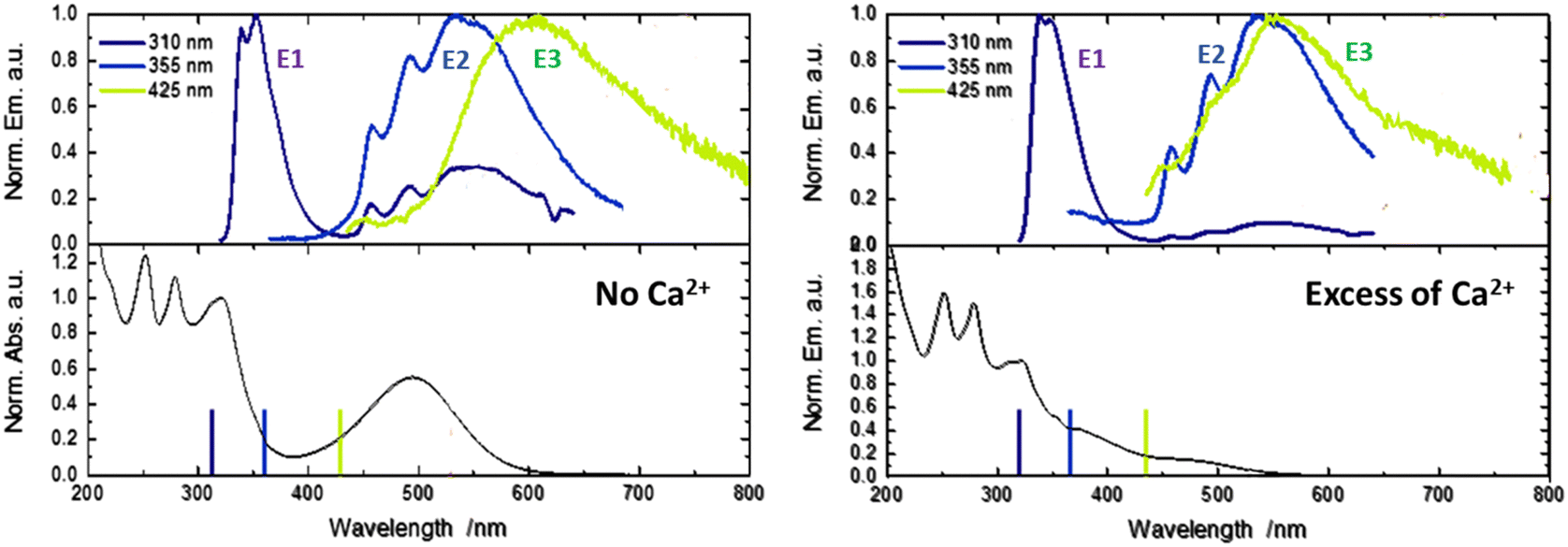 | ||
| Fig. 3 Absorption and emission spectra upon 310, 355 and 425 nm excitation for 1 in CH3CN (left) and 1–Ca2+ (excess of Ca2+) in CH3CN (right) (10 μM). The E3 band was missing after addition of calcium perchlorate. | ||
By comparison with the literature, with its typical vibronic structure, the E2 band is assigned at first glance to the triplet phosphorescence originating from the 3LLCT state for homoleptic iridium complexes.24 However, in our case, for molecule 1, TDDFT calculations provide evidence that T1(3ILCT) is the lowest in energy while the triplet T2(3LLCT) is quasi degenerated with both the singlet state populated after photo-ejection, namely the S2(ILCT) and S1(LLCT) states (Fig. S2, ESI†). Therefore, the E2 band could be attributed to delayed emission involving LLCT states. Finally, the unusual E3 band that arises for 425 nm light excitation is due to ion-pair formation between the metallic center Ir3+ and the counter anion (PF6)−.23 With anticipation, this latter state can be important to rationalize the upcoming transient spectroscopy experiments.
2.3. Ultrafast dynamics and cation ejection
In order to depict the full picture of molecule 1 with and without Ca2+ cations, we will focus on transient spectroscopy with laser excitation near 350 nm to ensure that triplet states are surely populated knowing that cation ejection can occur at singlet or triplet manifolds. Two set-ups were employed corresponding to two distinct ranges – femtosecond/picosecond and nanosecond/microsecond regimes. The overall results are reported in a Jablonski diagram at the end of the article.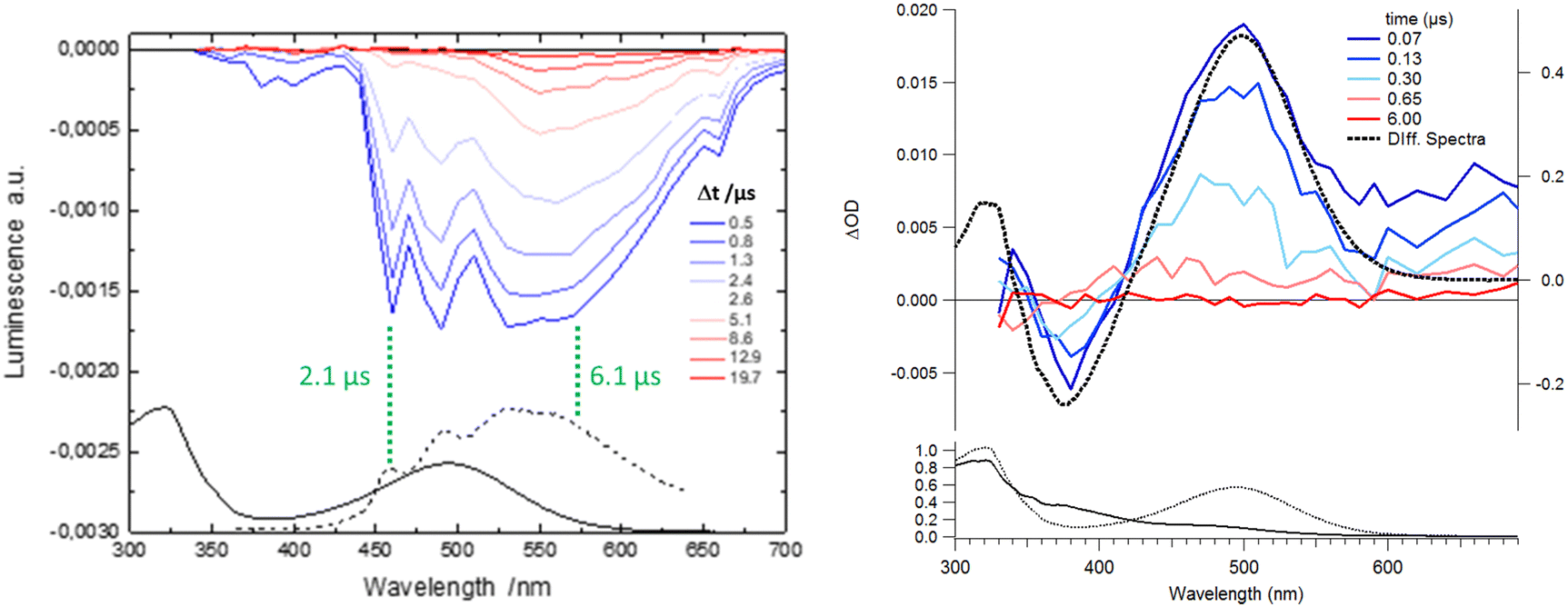 | ||
| Fig. 4 (left) Transient emission traces obtained with laser flash photolysis in luminescent mode for 1 in CH3CN (200 μM) with 355 nm laser excitation. Bi-exponential fits have been performed at two chosen wavelengths (green dashed line) and the predominant characteristic time is indicated. (right) Transient absorption for 1–Ca2+ (excess of Ca2+) in CH3CN with 355 nm laser excitation. The kinetics at a selected wavelength are better fitted with a mono-exponential function. | ||
According to the strategy of Moore et al., the recapture of the Ca2+ cation can be highlighted with laser flash photolysis with a regular absorption configuration. The transient data have to be compared with the difference in the stationary spectra with and without excess of the cation (the absorption curve with the Ca2+ cation is subtracted from the absorption curve without Ca2+).4,31 Both μ-second TA data after 355 nm laser excitation and difference spectra are presented in Fig. 4. Clearly, the transient band perfectly matches the difference spectra, providing evidence that this band is associated with the photoproduct, namely molecule 1 in its excited states following Ca2+ photo-release. The kinetics at 500 nm is fitted with a mono-exponential function with a 250 ns characteristic time that represents the mean time for the Ca2+ recapture.
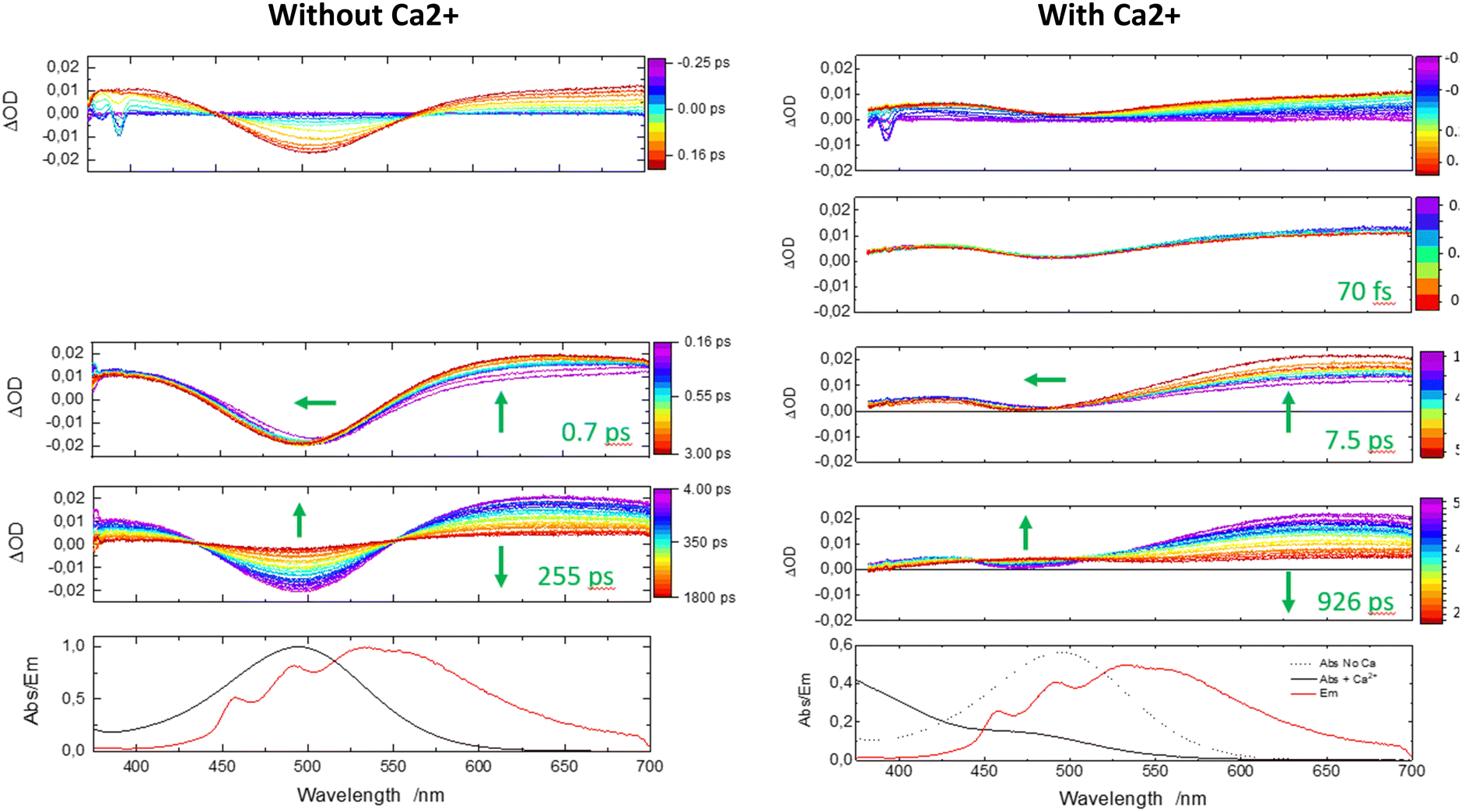 | ||
| Fig. 5 Femtosecond TA for a solution of 1 in CH3CN without (left) and with excess (right) of Ca2+ cations under 350 nm laser excitation presented in different temporal windows from −0.25 ps to 2000 ps. Global fitting has been undertaken at selected wavelengths (max of transient bands, bleaching bands…) and the characteristic times are indicated. The first panel represents the rise of the TA signal. The last panels are stationary absorption and emission curves for the 350 nm excitation. | ||
Now, one can analyze the femtosecond transient data taking into account the presence of the Ca2+ ion. In comparison, the first panel shows immediately a similar rise in positive transient bands related to S1(ILCT) (originating from 1–Ca2+ species) except that the negative bleaching band is strongly reduced in accordance with stationary spectroscopy. Then, a novel discrete process, a slight decrease of intensity at the red spectral edge, is reported on a second temporal window [0.45; 0.9 ps] with a characteristic time of 70 fs. Because the complexation constant is somewhat weak (K = 250), one does not expect a drastic decrease of the excited state absorption, and for that reason, we assign that process to the photo-release of the Ca2+ cation. That assignment is consistent with the 100 fs photo-release reported for betaine pyridinium molecules.34 The following temporal window [1; 50 ps] logically displays the expected result, that is to say, ISC from S1(ILCT) to T1(ILCT) but with a characteristic time of 7.5 ps. This drastic increase by a factor of 10 compared to molecule 1 alone can be rationalized by an electrostatic effect induced by the “loose complex”, i.e. Ca2+ is still in the neighboring molecule.8 Indeed, the energy levels (not computed by TDDFT) should be influenced by the presence of the cation. Following the analogy with the previous case, the next temporal window [50; 2000 ps] concerns the decay to the ground state, T1(ILCT) → S0. The characteristic lifetime of 926 ps is also multiplied by a factor of 3 rationalized in terms of the energy gap law for the ISC processes (Fig. 6).35
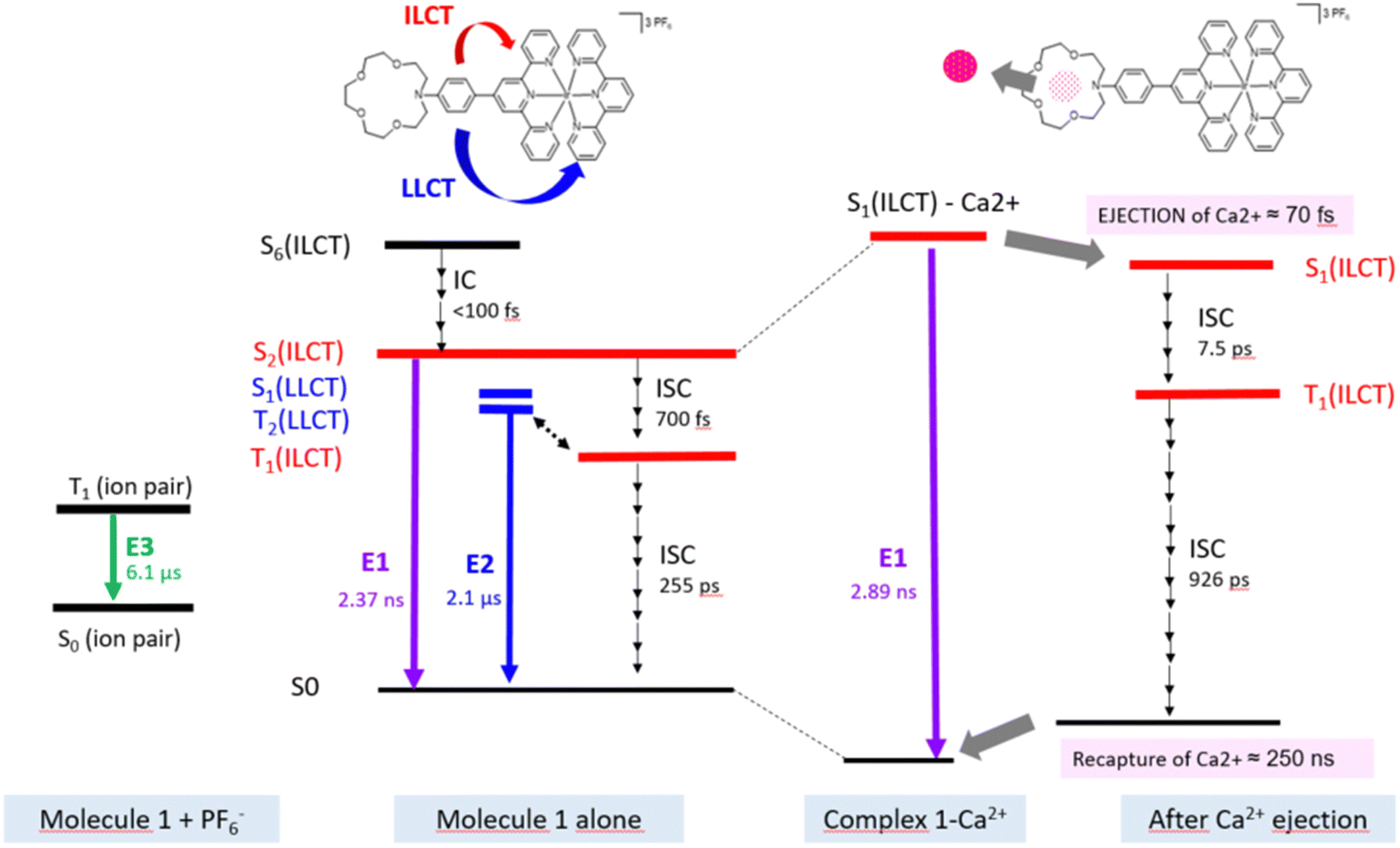 | ||
| Fig. 6 Proposed Jablonski diagram for molecule 1 and complex 1–Ca2+ according to our experiment and theoretical calculations. | ||
3. Conclusion
The results of our investigation on the photophysical properties of an iridium(III) polypyridyl heteroleptic complex, namely an azacrown–iridium(III) complex, have elucidated several crucial aspects of its response to photoexcitation and interactions with Ca2+ ions. The integration of TD-DFT calculations and time-resolved spectroscopies has allowed us to characterize the intricate dynamics governing this complex. Our results indicate that the introduction of Ca2+ significantly alters the absorption and emission profiles, with the complex displaying a notable shift from 494 nm to 375 nm upon metal binding, consistent with the calculated transitions. In addition, the theoretical results as a whole are very consistent with the spectra obtained from experimental studies, thus demonstrating the relevance of this method in the selection of a molecule to be synthesized. Furthermore, we documented the photo-ejection of Ca2+, occurring at a remarkable 70 fs, and its subsequent recapture at 250 ns, and these phenomena aligned with existing literature on ultrafast cation dynamics in comparable systems. These results are among the few to show both photo-ejection and recapture of the cation and feature a unique 10 million time factor between the two processes, allowing temporal isolation of the two phenomena and potential applications in fields where ion control, both temporal and spatial, is critical.Comparisons with earlier studies show that this complex exhibits typical behaviors of polypyridyl iridium(III) complexes, such as strong intra-ligand charge transfer (ILCT) and ligand-to-ligand charge transfer electronic transitions as well as sensitivity to environmental changes through, for example, ion-pair formation. These findings are consistent with established knowledge of the photophysics of iridium complexes, particularly regarding the influence of ligand environments on charge transfer states and emission properties. The distinction in our results lies in the detailed kinetic analysis and the exploration of interaction dynamics, which have not been thoroughly examined in prior studies. This filled a significant gap, particularly in understanding the excited states' landscape and their role in facilitating efficient ion transfer processes. The competitive binding and photo-release capabilities of the azacrown moiety in our system are also corroborated by previous reports on similar aza-crown compounds, cementing our understanding of their potential in sensor, biolabeling or optoelectronic applications.
Looking ahead, the versatile photophysical properties of these complexes present exciting opportunities for the development of advanced biosensors, molecular machines and stimuli-responsive materials. The precise control over cation binding and release demonstrated here could be leveraged to design sophisticated ion sensors and delivery systems with potential for applications in biomedical fields. Additionally, further exploration into the fine-tuning of ligand environments and photophysical responses assisted by preliminary TD-DFT calculations may yield new insights into the manipulation of charge transfer processes for various technological applications. The promising results showcased in this study lay the groundwork for future developments in the use of iridium(III) complexes for excited state cation transfer for innovative applications in dynamic and responsive material systems.
4. Experimental
4.1. Synthesis
HRMS have been obtained by the mass spectrometry service from the Institut de Chimie des Substances Naturelles (ICSN), at the research center from Gif sur Yvette.
4.2. Calculations
All the calculations were performed using the Gaussian 16 package36 relying on (TD-)DFT to determine the ground-state (GS, S0) and excited-state (ES, Sn and Tn) properties, respectively. We have used (i) the 6-311+G(d) basis set for the H, C, N, O, and Ca atoms, and (ii) the LANL2DZ37 relativistic pseudopotential and the corresponding basis set for Ir. The geometry optimizations and the calculation of the excited state properties have been carried out with the PBE038 hybrid exchange–correlation functional. Previous works have demonstrated the performances of global hybrid functionals39 and, more specifically, PBE0, to investigate the nature, dynamics, and reactivity of the (charge transfer) excited states of iridium(III) complexes.40,41We calculated and analyzed frequency vibrations to guarantee that we obtained true minima on the potential energy surfaces. For all TD-DFT calculations, 25 excited states are considered. The “stick spectra” are convoluted with a Gaussian function (full width at half maximum FWHM = 0.25 eV). The computed vertical excitation wavelengths λ with the highest oscillator strength f are compared to the experimental maximum absorption wavelength. One should note that these values do not necessarily coincide, and a more refined computational model should consider the vibronic structure of the electronic absorption spectra.
All calculations were performed by considering the polarizable continuum model42 (IEF-PCM) in the equilibrium limit using the linear-response (LR)PCM scheme to quantify the impact of the acetonitrile solvent (ACN).
The Breit–Pauli spin–orbit matrix elements (SOCMEs) are calculated using PySOC,43 a program interface based on MolSOC44 and implemented within the MOMAP package.45 At that stage, the level of calculation is exactly the same as the one considered in the TD-DFT calculations (solvent included).
To analyze the CT character of the different excited states, we have used three different descriptors. First, the variation of electronic density Δρ() is calculated on a grid of points using the cubegen utility provided by the Gaussian package:
| Δρ() = ρES() − ρGS() |
) and ρES() are the electron density of the ground state and the considered excited states, respectively.
Secondly, the density derived indexes namely the DCT index and the norm of the dipole moment corresponding to the CT observed upon excitation μCT are computed using the DctViaCube program.46
4.3. Spectroscopies
The stationary absorption spectra were measured with a CARY 100bio absorption spectrometer while emission spectra were recorded with a Fluoromax3 spectrometer. The TCSPC were measured with FT200 apparatus (PicoQuant) using femtosecond laser pulses at 310 nm.Our nanosecond transient absorption spectroscopy setup has been described elsewhere.47 Briefly, a third harmonic pulse (355 nm) of a Nd:YAG laser with ca. 1 mJ output energy and 5 ns pulse width was used as the excitation light and a pulsed Xe lamp was utilized as the probe light. Transient absorption spectra were obtained from the transient absorption decays recorded at various wavelengths by sampling the absorbance changes for different given delay-times. Note that a “luminescent” mode is available by switching off the Xe lamp.
Femtosecond time-resolved experiments were performed using a pump–probe spectrometer48 based on a Ti:sapphire laser system (Coherent Mira-900-D oscillator and Libra-S regenerative amplifier) delivering 800 nm, 1 mJ and 90 fs pulses with a repetition rate of 1 kHz. The pump pulses adjusted and tuned at 320 nm were generated by frequency quadrupling the output of a Quantronix Palitra OPA pumped at 800 nm and the energy at the sample was about 2 μJ (0.2 mJ cm−2). Note that, we chose to excite the molecule far from the maximum of absorption localized near 270 nm to avoid a short-time 2-photon absorption signal coming from the solvent. The probe pulses were obtained by focusing 1 μJ, 800 nm pulses into a 1 mm thick CaF2 plate to generate a white light continuum. The pump–probe polarization configuration was set at a magic angle (54.7°). Transient absorbance was obtained by comparing the signal and reference spectra with and without pump pulses for different delay-times. The delay-time between the pump and probe was varied up to 0.5 ns using an optical delay line. Sample solutions (∼10−3–10−5 mol dm−3, OD = 1 at the pump wavelength) were circulated in a cell equipped with a 200 μm thick CaF2 entrance window and a 2 mm optical path. All the transient spectra presented in this paper are GVD corrected according to the typical extrapolation method.49 The characteristic times deduced from kinetics were obtained using a global fitting approach, and the data being fitted with the results of a multiexponential function convolved with a Gaussian pulse mimicking the pump–probe cross-correlation function (FWHM ∼ 180 fs).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the support of the French Agence Nationale de la Recherche (ANR), under grant ANR-18-CE29-0014 (project ExStaCaT). This work was granted access to the HPC resources of IDRIS under the allocations 2021-A0010810135 and 2023-AD010814608 made by GENCI (“Grand Equipement National de Calcul Intensif”).References
- C. Li, Y. Wu, Y. Zhu, J. Yan, S. Liu, J. Xu, S. Fa, T. Yan, D. Zhu, Y. Yan and J. Liu, Molecular Motor-Driven Light-Controlled Logic-Gated K+ Channel for Cancer Cell Apoptosis, Adv. Mater., 2024, 2312352, DOI:10.1002/adma.202312352.
- G. Bonsignore, S. Martinotti and E. Ranzato, Wound Repair and Ca2+ Signalling Interplay: The Role of Ca2+ Channels in Skin, Cells, 2024, 13(6), 491, DOI:10.3390/cells13060491.
- I. K. Lednev, T.-Q. Ye, R. E. Hester and J. N. Moore, Photocontrol of Cation Complexation with a Benzothiazolium Styryl Azacrown Ether Dye: Spectroscopic Studies on Picosecond and Kilosecond Time Scales, J. Phys. Chem. A, 1997, 101(27), 4966–4972, DOI:10.1021/jp970685y.
- I. K. Lednev, R. E. Hester and J. N. Moore, Direct Observation of Photocontrolled Ion Release: A Nanosecond Time-Resolved Spectroscopic Study of a Benzothiazolium Styryl Azacrown Ether Dye Complexed with Barium, J. Phys. Chem. A, 1997, 101(40), 7371–7378, DOI:10.1021/jp972360n.
- J. D. Lewis and J. N. Moore, Cation Sensors Containing a (Bpy)Re(CO)(3) Group Linked to an Azacrown Ether via an Alkenyl or Alkynyl Spacer: Synthesis, Characterisation, and Complexation with Metal Cations in Solution, Dalton Trans., 2004, 1376–1385, 10.1039/b401092b.
- C. Ley, F. Lacombat, P. Plaza, M. M. Martin, I. Leray and B. Valeur, Femtosecond to Subnanosecond Multistep Calcium Photoejection from a Crown Ether-Linked Merocyanine, ChemPhysChem, 2009, 10(1), 276–281, DOI:10.1002/cphc.200800612.
- M. M. Martin, P. Plaza, N. D. Hung, Y. H. Meyer, J. Bourson and B. Valeur, Photoejection of cations from complexes with a crown-ether-linked merocyanine evidenced by ultrafast spectroscopy, Chem. Phys. Lett., 1993, 202(5), 425–430, DOI:10.1016/0009-2614(93)90065-9.
- M. M. Martin, P. Plaza, Y. H. Meyer, L. Begin, J. Bourson and B. Valeur, A New Concept of Photogeneration of Cations: Evidence for Photoejection of Ca(2+) and Li (+) from Complexes with a Crown-Ether-Linked Merocyanine by Picosecond Spectroscopy, J. Fluoresc., 1994, 4(4), 271, DOI:10.1007/bf01881437.
- M. M. Martin, P. Plaza and Y. H. Meyer, Ultrafast intramolecular charge-transfer in the merocyanine dye DCM, Chem. Phys., 1995, 192(3), 367–377 CrossRef CAS.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Rational Design of Phosphorescent Iridium(III) Complexes for Emission Color Tunability and Their Applications in OLEDs, Coord. Chem. Rev., 2018, 374, 55–92, DOI:10.1016/j.ccr.2018.06.014.
- J. C. Deaton and F. N. Castellano, Archetypal Iridium(III) Compounds for Optoelectronic and Photonic Applications, Iridium(III) in Optoelectronic and Photonics Applications, John Wiley & Sons, Ltd, 2017, pp. 1–69 Search PubMed.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Photochemistry and Photophysics of Coordination Compounds: Iridium, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin, Heidelberg, 2007, pp. 143–203 Search PubMed.
- E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, From Ruthenium(II) to Iridium(III): 15 Years of Triads Based on Bis-Terpyridine Complexes, Chem. Soc. Rev., 2004, 33(3), 147–155, 10.1039/B308983E.
- Y. Zhang and J. Qiao, Near-Infrared Emitting Iridium Complexes: Molecular Design, Photophysical Properties, and Related Applications, iScience, 2021, 24(8), 102858, DOI:10.1016/j.isci.2021.102858.
- H. Shi, Y. Wang, S. Lin, J. Lou and Q. Zhang, Recent Development and Application of Cyclometalated Iridium(III) Complexes as Chemical and Biological Probes, Dalton Trans., 2021, 50(19), 6410–6417, 10.1039/D1DT00592H.
- F. Monti, A. Baschieri, L. Sambri and N. Armaroli, Excited-State Engineering in Heteroleptic Ionic Iridium(III) Complexes, Acc. Chem. Res., 2021, 54(6), 1492, DOI:10.1021/acs.accounts.0c00825.
- Y. You and S. Y. Park, Phosphorescent Iridium(III) Complexes: Toward High Phosphorescence Quantum Efficiency through Ligand Control, Dalton Trans., 2009, 1267–1282, 10.1039/B812281D.
- L. Flamigni, B. Ventura, F. Barigelletti, E. Baranoff, J.-P. Collin and J.-P. Sauvage, Luminescent Iridium(III)-Terpyridine Complexes – Interplay of Ligand Centred and Charge Transfer States, Eur. J. Inorg. Chem., 2005,(7), 1312–1318, DOI:10.1002/ejic.200400801.
- J.-P. Collin, I. M. Dixon, J.-P. Sauvage, J. A. G. Williams, F. Barigelletti and L. Flamigni, Synthesis and Photophysical Properties of Iridium(III) Bisterpyridine and Its Homologues: A Family of Complexes with a Long-Lived Excited State, J. Am. Chem. Soc., 1999, 121(21), 5009–5016, DOI:10.1021/ja9833669.
- M. Licini and J. A. G. Williams, Iridium(III) Bis-Terpyridine Complexes Displaying Long-Lived pH Sensitive Luminescence, Chem. Commun., 1999, 1943–1944, 10.1039/A906203C.
- B. Liu, S. Monro, Z. Li, M. A. Jabed, D. Ramirez, C. G. Cameron, K. Colón, J. Roque, S. Kilina, J. Tian, S. A. McFarland and W. Sun, A New Class of Homoleptic and Heteroleptic Bis(Terpyridine) Iridium(III) Complexes with Strong Photodynamic Therapy Effects, ACS Appl. Bio Mater., 2019, 2(7), 2964–2977, DOI:10.1021/acsabm.9b00312.
- K. J. Arm, W. Leslie and J. A. G. Williams, Synthesis and pH-Sensitive Luminescence of Bis-Terpyridyl Iridium(III) Complexes Incorporating Pendent Pyridyl Groups, Inorg. Chim. Acta, 2006, 359(4), 1222–1232, DOI:10.1016/j.ica.2005.09.021.
- W. Ito, S. Hattori, M. Kondo, H. Sakagami, O. Kobayashi, T. Ishimoto and K. Shinozaki, Dual Emission from an Iridium(III) Complex/Counter Anion Ion Pair, Dalton Trans., 2021, 50(5), 1887–1894, 10.1039/D1DT00021G.
- Y.-S. Yeh, Y.-M. Cheng, P.-T. Chou, G.-H. Lee, C.-H. Yang, Y. Chi, C.-F. Shu and C.-H. Wang, A New Family of Homoleptic Ir(III) Complexes: Tris-Pyridyl Azolate Derivatives with Dual Phosphorescence, ChemPhysChem, 2006, 7(11), 2294–2297, DOI:10.1002/cphc.200600461.
- K. Yamanaka, K. Sato, S. Sato, S. Nozawa, S. Lee, R. Fukaya, H. Fukuzawa, D. You, S. Saito, T. Takanashi, T. Katayama, T. Togashi, T. Nonaka, K. Dohmae, S. Adachi, K. Ueda, M. Yabashi, T. Morikawa and R. Asahi, Ultrafast Charge-Transfer Dynamics in a Visible-Light-Excited Iridium(III) Terpyridine 2-Phenylpyridine Complex Studied by Femtosecond X-Ray Absorption Spectroscopy, J. Photochem. Photobiol., A, 2023, 435, 114267, DOI:10.1016/j.jphotochem.2022.114267.
- R. Bevernaegie, L. Marcélis, A. Moreno-Betancourt, B. Laramée-Milette, G. S. Hanan, F. Loiseau, M. Sliwa and B. Elias, Ultrafast Charge Transfer Excited State Dynamics in Trifluoromethyl-Substituted Iridium(Iii) Complexes, Phys. Chem. Chem. Phys., 2018, 20(43), 27256–27260, 10.1039/C8CP04265A.
- Z. Kuang, X. Wang, Z. Wang, G. He, Q. Guo, L. He and A. Xia, Phosphorescent Cationic Iridium(III) Complexes with 1,3,4-Oxadiazole Cyclometalating Ligands: Solvent-Dependent Excited-State Dynamics, Chin. J. Chem. Phys., 2017, 30(3), 259–267, DOI:10.1063/1674-0068/30/cjcp1703058.
- N. Zhao, Y.-H. Wu, H.-M. Wen, X. Zhang and Z.-N. Chen, Conversion from ILCT to LLCT/MLCT Excited State by Heavy Metal Ion Binding in Iridium(III) Complexes with Functionalized 2,2′-Bipyridyl Ligands, Organometallics, 2009, 28(19), 5603–5611, DOI:10.1021/om900334f.
- U. Resch-Genger, Y. Q. Li, J. L. Bricks, V. Kharlanov and W. Rettig, Bifunctional Charge Transfer Operated Fluorescent Probes with Acceptor and Donor Receptors. 1. Biphenyl-Type Sensor Molecules with Protonation-Induced Anti-Energy Gap Rule Behavior, J. Phys. Chem. A, 2006, 110(38), 10956–10971 CrossRef CAS PubMed.
- J.-P. Collin, I. M. Dixon, J.-P. Sauvage, J. A. G. Williams, F. Barigelletti and L. Flamigni, Synthesis and Photophysical Properties of Iridium(III) Bisterpyridine and Its Homologues: A Family of Complexes with a Long-Lived Excited State, J. Am. Chem. Soc., 1999, 121(21), 5009–5016, DOI:10.1021/ja9833669.
- J. D. Lewis and J. N. Moore, Cation Sensors Containing a (Bpy)Re(CO)(3) Group Linked to an Azacrown Ether via an Alkenyl or Alkynyl Spacer: Synthesis, Characterisation, and Complexation with Metal Cations in Solution, Dalton Trans., 2004, 1376–1385 RSC.
- P. Plaza, I. Leray, P. Changenet-Barret, M. M. Martin and B. Valeur, Reversible Bulk Photorelease of Strontium Ion from a Crown Ether-Linked Merocyanine, ChemPhysChem, 2002, 3(8), 668–674 CrossRef CAS PubMed.
- C. H. Ryu, M. Kim, C. Sohn, J. H. Hong, S. Im, S. Y. Kim and K. M. Lee, Monodentate Benzo[d]Imidazole-Based Iridium(III) Complexes and Their Dual Fluorescent and Phosphorescent Emissions, Bull. Korean Chem. Soc., 2020, 41(2), 176–183, DOI:10.1002/bkcs.11949.
- S. Aloise, Z. Pawlowska, O. Poizat, G. Buntinx, A. Perrier, F. Maurel, K. Ohkawa, A. Kimoto and J. Abe, Excited-State Dynamics of Thiophene Substituted Betaine Pyridinium Compounds, Phys. Chem. Chem. Phys., 2014, 16(4), 1460–1468, 10.1039/c3cp53614a.
- N. J. Turro, Modern Molecular Photochemistry, The Benjamin/Cummings Publishing Co., Inc., Menlo Park, California, 1978 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Williams Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, 2016 Search PubMed.
- W. R. Wadt and P. J. Hay, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi, J. Chem. Phys., 1985, 82(1), 284–298, DOI:10.1063/1.448800.
- C. Adamo, M. Cossi and V. Barone, An Accurate Density Functional Method for the Study of Magnetic Properties: The PBE0 Model, Theochem, 1999, 493, 145–157 CrossRef CAS.
- S. Mai, M. F. S. J. Menger, M. Marazzi, D. L. Stolba, A. Monari and L. González, Competing Ultrafast Photoinduced Electron Transfer and Intersystem Crossing of [Re(CO)3(Dmp)(His124)(Trp122)]+ in Pseudomonas Aeruginosa Azurin: A Nonadiabatic Dynamics Study, Theor. Chem. Acc., 2020, 139(3), 65, DOI:10.1007/s00214-020-2555-6.
- T. Liu, B.-H. Xia, Q.-C. Zheng, X. Zhou, Q.-J. Pan and H.-X. Zhang, DFT/TD-DFT Investigation on Ir(III) Complexes with N-Heterocyclic Carbene Ligands: Geometries, Electronic Structures, Absorption, and Phosphorescence Properties, J. Comput. Chem., 2010, 31(3), 628–638, DOI:10.1002/jcc.21360.
- I. Ciofini, P. P. Lainé, F. Bedioui and C. Adamo, Photoinduced Intramolecular Electron Transfer in Ruthenium and Osmium Polyads: Insights from Theory, J. Am. Chem. Soc., 2004, 126(34), 10763–10777, DOI:10.1021/ja0482278.
- B. Mennucci, Polarizable Continuum Model, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404, DOI:10.1002/wcms.1086.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, Evaluation of Spin-Orbit Couplings with Linear-Response Time-Dependent Density Functional Methods, J. Chem. Theory Comput., 2017, 13(2), 515–524, DOI:10.1021/acs.jctc.6b00915.
- S. G. Chiodo and M. Leopoldini, MolSOC: A Spin–Orbit Coupling Code, Comput. Phys. Commun., 2014, 185(2), 676–683, DOI:10.1016/j.cpc.2013.10.014.
- Z. Shuai, Thermal Vibration Correlation Function Formalism for Molecular Excited State Decay Rates, Chin. J. Chem., 2020, 38(11), 1223–1232, DOI:10.1002/cjoc.202000226.
- T. Le Bahers, C. Adamo and I. Ciofini, A Qualitative Index of Spatial Extent in Charge-Transfer Excitations, J. Chem. Theory Comput., 2011, 7(8), 2498–2506, DOI:10.1021/ct200308m.
- G. Buntinx, O. Poizat and N. Leygue, Transient absorption and resonance Raman investigation of the photochemical reactivity of 2,2’-bipyrimidine in solution, J. Phys. Chem., 1995, 99(8), 2343–2352, DOI:10.1021/j100008a016.
- B. Moine, J. Rehault, S. Aloise, J. C. Micheau, C. Moustrou, A. Samat, O. Poizat and G. Buntinx, Transient Absorption Studies of the Photochromic Behavior of 3H-Naphtho[2,1-b]Pyrans Linked to Thiophene Oligomers via an Acetylenic Junction, J. Phys. Chem. A, 2008, 112(21), 4719–4726, DOI:10.1024/jp711865j.
- T. Nakayama, Y. Amijima, K. Ibuki and K. Hamanoue, Construction of a Subpicosecond Double-Beam Laser Photolysis System Utilizing a Femtosecond Ti: Sapphire Oscillator and Three Ti: Sapphire Amplifiers (a Regenerative Amplifier and Two Double Passed Linear Amplifiers), and Measurements of the Transient Absorption Spectra by a Pump-Probe Method, Rev. Sci. Instrum., 1997, 68(12), 4364–4371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02274b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |