
Synthesis and spectroscopic properties of carotenoid bis-phenylhydrazone astaxanthin: extending conjugation to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N group†
N group†
Emrah
Özcan
 *a,
Gürkan
Keşan
a,
Pavel
Chábera
b,
Radek
Litvín
c and
Tomáš
Polívka
a
*a,
Gürkan
Keşan
a,
Pavel
Chábera
b,
Radek
Litvín
c and
Tomáš
Polívka
a
aDepartment of Physics, Faculty of Science, University of South Bohemia, Branišovská 1760, České Budějovice 370 05, Czech Republic. E-mail: ozcane00@prf.jcu.cz
bDivision of Chemical Physics, Department of Chemistry, Lund University, Box 142, Lund SE-221 00, Sweden
cDepartment of Chemistry, Faculty of Science, University of South Bohemia, Branišovská 1760, České Budějovice 370 05, Czech Republic
First published on 1st July 2024
Abstract
We report on the synthesis and detailed spectroscopic characterization of bis-phenylhydrazone astaxanthin (BPH-Asx), a derivative of astaxanthin (Asx), in which the conjugated carbonyl group of Asx is replaced by a conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. BPH-Asx was successfully synthesized and characterized using various spectroscopic techniques, revealing subtle changes in absorption spectra and significant alterations in excited-state dynamics compared to Asx. The results reveal a shortened S1 lifetime, 1.4 ps for BPH-Asx compared to 5 ps for Asx, indicating a significant impact on its excited-state dynamics. Since no polarity-induced effect was observed for BPH-Asx, the changes induced by the conjugated CN group are due to prolongation of effective conjugation. Moreover, the identification of a distinctive S* signal with a 3 ps lifetime in BPH-Asx underscores the relation between effective conjugation and presence of the S* signal that is not detected in Asx.
N bond. BPH-Asx was successfully synthesized and characterized using various spectroscopic techniques, revealing subtle changes in absorption spectra and significant alterations in excited-state dynamics compared to Asx. The results reveal a shortened S1 lifetime, 1.4 ps for BPH-Asx compared to 5 ps for Asx, indicating a significant impact on its excited-state dynamics. Since no polarity-induced effect was observed for BPH-Asx, the changes induced by the conjugated CN group are due to prolongation of effective conjugation. Moreover, the identification of a distinctive S* signal with a 3 ps lifetime in BPH-Asx underscores the relation between effective conjugation and presence of the S* signal that is not detected in Asx.
Introduction
Carotenoids are a class of natural pigments with extraordinary photoprotective1 and light-harvesting2,3 abilities in biological systems. Moreover, their rich excited-state dynamics4 as well as capacity to engage in various functions, such as quenching of singlet-excited states of chlorophylls,5–8 chlorophyll triplets,9,10 and antioxidation achieved by scavenging singlet oxygen or other reactive oxygen species,11–16 emphasize their importance in both biology and chemistry. Due to their important roles in light-induced processes, photophysical and photochemical properties of carotenoids have been frequently studied; however, owing to the complicated excited-state structure and dynamics, gaps in understanding the carotenoid photophysics still remain.According to the basic description of the excited states of all carotenoids, two states are highlighted: strongly absorbing S2 state and lower-lying S1 state, to which a one-photon transition from the ground state (S0) is forbidden due to the multiply-excited character.17 Thus, the lowest energy one-photon transition from the ground state occurs to the S2 state, which relaxes to the S1 state whose properties are readily monitored via its characteristic S1–Sn band in transient absorption spectra.18,19 This general picture is valid for all carotenoids and their conjugated CC bond chain structures, common in all carotenoids, primarily determine their photophysical properties.17
Beyond this three-state model (S0, S1 and S2), other states have been identified. In keto-carotenoids, featuring a conjugated CO bond in their structure, an intramolecular charge transfer (ICT) state coupled to the dark S1 state is stabilized in a polar environment, resulting in a state commonly referred as the S1/ICT.20,21 The presence of the ICT state is easily detected by characteristic bands in the transient absorption spectrum. The ICT-like transition is red-shifted from the S1–Sn band for most keto-carotenoids. The amplitude ratio between the S1–Sn and ICT-like bands serves as a measure of degree of charge transfer character of the coupled S1/ICT state.20,21 Numerous investigations have demonstrated that the degree of charge transfer character is proportional to solvent polarity but the conjugation length and position of the conjugated keto group are also crucial factors. The ICT signal increases for short keto-carotenoids having a single keto group positioned asymmetrically while the charge transfer nature of the S1/ICT state is minimized for long ones with two keto groups positioned symmetrically.20,22,23 This is for example the case of astaxanthin (Asx), one of the widely studied keto-carotenoids, which has two symmetric conjugated keto (CO) groups at the terminal rings, resulting in only a weak ICT signal detected in a broad range of solvents.24–26
Another state known as S* has been identified in carotenoids with long conjugation. The S* signal is typically demonstrated as a distinct blue shoulder at the S1–Sn band, and its lifetime is longer than that of the S1 state.27 The origin of the S* signal is still a matter of debate. The first detection of the S* signal assigned it to a hot ground state,28 but this assignment was later challenged and the S* signal was instead attributed to a separate excited state.27 Since then, numerous studies favoring either ground state29,30 or excited state31,32 hypothesis have been reported. Yet, it seems that both hot ground state and excited state contribute to the S* signal, with particular contribution depending on conjugation length: while for short conjugation (N < 11) the excited state contribution dominates, a hot ground state is the key source of the S* signal for long carotenoids.33
Since excited state dynamics of carotenoids depends on the structure of the conjugated system, synthetic carotenoids with various modifications of the conjugated chain helped to understand some aspects of carotenoid photophysics. These modifications often focused on synthesis of carotenoids with a conjugation length longer than that of natural carotenoids. Such approach, involving a synthesis of β-carotene analogs with 15 or even 19 CC bonds (in contrast to 11 in natural β-carotene), led to the first observation of the S* signal.28 Series of synthetic carotenoids with varying conjugation lengths later helped to understand excited-state dynamics of β-carotene,34 zeaxanthin35 or keto-carotenoids peridinin36 and fucoxanthin.37
Besides synthesis of carotenoid series with the same structure but different conjugation lengths, chemical modifications of carotenoids have also targeted various functional groups involved in conjugation in order to test their role in excited-state dynamics. To this end, alterations of the allene group of peridinin38 or fucoxanthin39 were used to test the effect of the allene group on ICT state of these keto-carotenoids. Similarly, symmetry of peridinin was modified by ‘moving’ the lactone ring along the main conjugated chain.40 Many synthetic carotenoids have been introduced in the past few decades including those having non-natural atoms such as sulfur, nitrogen, or phosphorus in their structure.41 Yet, excited state dynamics of these carotenoids has not been studied, apart from two exceptions featuring nitrogen atoms in their structure. First, ultrafast dynamics of all-trans-7′,7′-dicyano-7′-apo-β-carotene,42 demonstrating strong effect of the cyano groups on excited state lifetime. Second, astaxanthin (Asx) esterified by the amino acid lysine, synthesized to make Asx water soluble, was subjected to ultrafast transient absorption spectroscopy.43
Asx is a subject of chemical modification also in this study. It has two oxygen atoms on each cyclohexene rings in the form of a keto (CO) and hydroxyl (C–OH) group. These groups at terminal rings offer a possibility of chemical/structural modification of Asx, synthesizing novel carotenoids.44–46 Since such modification of Asx, especially that modifying the keto group, is expected to change the photophysical properties, it is an ideal tool to explore features of excited states of the Asx. More specifically, by the synthesis of novel carotenoids (Scheme 1), we can obtain astaxanthin whose conjugated CO group, the expected generator of the ICT state, is modified to some other functional group. Such modification could figure out whether a carbonyl group is needed for observation of spectroscopic features related to the ICT state.
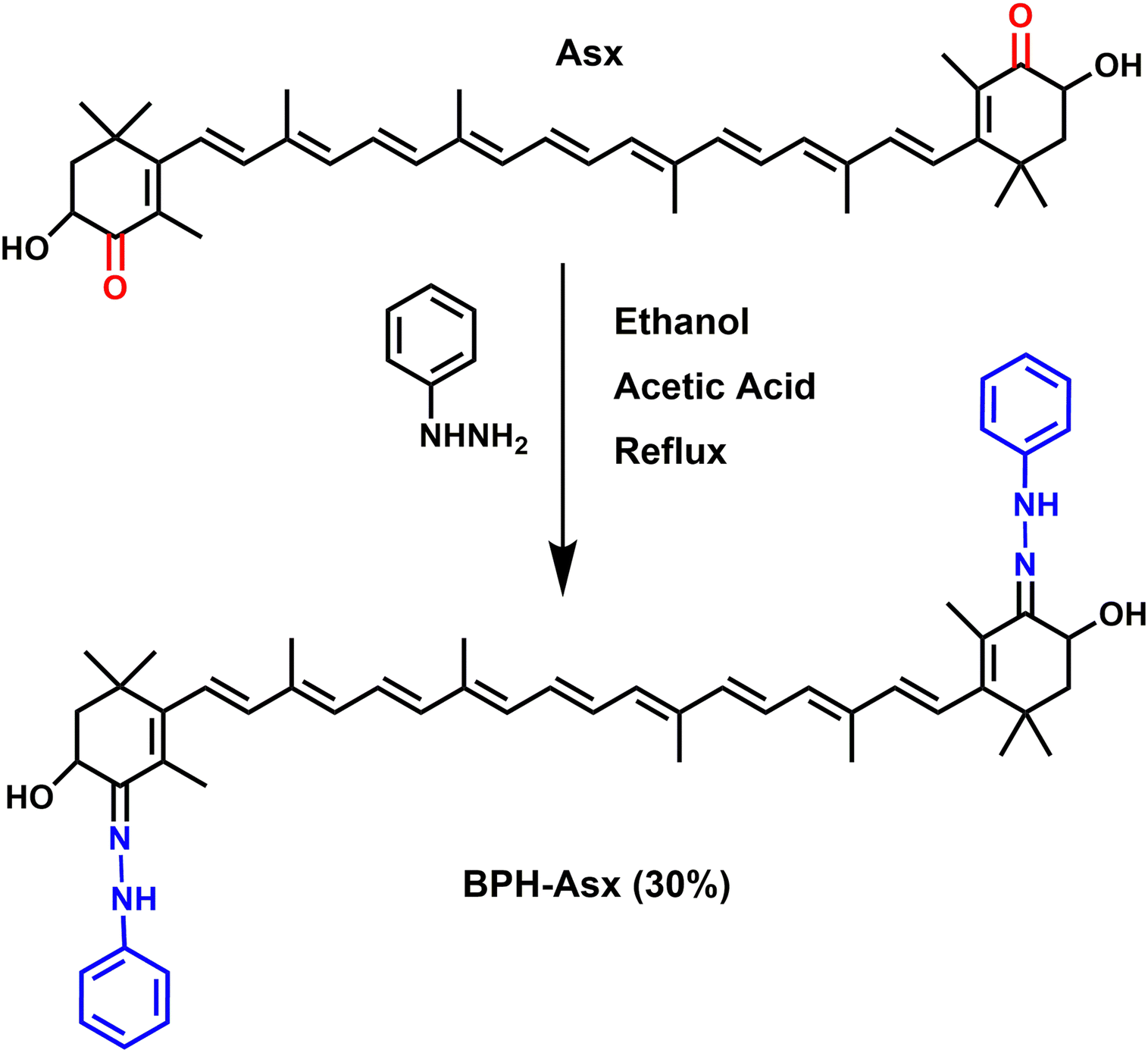 | ||
| Scheme 1 Synthetic pathway of bis-phenylhydrazone astaxanthin (BPH-Asx). | ||
Here, we report on synthesis of a novel astaxanthin derivative named bis-phenylhydrazone astaxanthin (BPH-Asx) which instead of conjugated CO group has CN bond in conjugation. We have conducted a detailed study of its photophysical properties that were investigated by ultrafast time-resolved transient absorption spectroscopy. The data showed that Asx could be successfully modified, resulting in stable BPH-Asx, which has significantly different spectroscopic properties compared to Asx.
Experimental section
Materials and methods
Astaxanthin (Sigma Aldrich, ≥97%, HPLC), phenylhydrazine (Merck, ≥97%, for synthesis), ethanol (Penta, ≥97%, GC), glacial acetic acid (Lachner, 99.8 G.R.), acetonitrile (ACN, Fluka, ≥99.9%, HPLC), benzene (Fluka, ≥99.5%, GC), methanol (Merck, ≥99.9%, HPLC), and dichloromethane (DCM, Merck, ≥99.9%, GC) were used as obtained without further purification. Reactions were monitored by thin layer chromatography using Merck TLC Silica gel 60 with DCM/methanol (1/1, v/v) as the eluent. MALDI-TOF mass spectra were acquired in linear modes with average of 50 shots on a Bruker Daltonics Microflex mass spectrometer equipped with a nitrogen UV-Laser operating at 337 nm. ESI-MS detection was conducted on a Bruker QqTOF compact instrument operated using Compass Control 4.0 software (Bruker Daltonics, Germany). Compass DataAnalysis 4.4 (Build 200.55.2969) (Bruker Daltonics, Germany) software was used for data processing. NMR spectra (1H and 13C NMR) were recorded for all compounds in CDCI3 by a Varian INOVA 500 MHz spectrometer using TMS as internal reference. High-performance liquid chromatography (HPLC) was performed using a Waters Alliance HPLC system with a PDA 2998 detector (Waters, USA). The compounds were injected in methanol and separated on a reverse phase Nova-Pak C18 column (3.9 × 300 mm, 4 μm, silica-based, end-capped; Waters, USA) using a linear gradient elution. A tertiary solvent system used was as follows:47 solvent A (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 methanol: 0.5 M ammonium acetate (aq., pH 7.2 v/v)), solvent B (90:10 acetonitrile:water), solvent C (100% ethyl acetate). The flow rate was 1 mL min−1. Absorption spectra of the samples were measured in a 10-mm path length quartz cuvette using UV-vis spectrometer (Shimadzu UV-2600). Fluorescence spectra were measured in 3 × 3 mm quartz cells using Horiba Fluorolog-3 spectrometer, using Xe arc lamp, double monochromators and a photomultiplier detector at right angle detection geometry.
:20 methanol: 0.5 M ammonium acetate (aq., pH 7.2 v/v)), solvent B (90:10 acetonitrile:water), solvent C (100% ethyl acetate). The flow rate was 1 mL min−1. Absorption spectra of the samples were measured in a 10-mm path length quartz cuvette using UV-vis spectrometer (Shimadzu UV-2600). Fluorescence spectra were measured in 3 × 3 mm quartz cells using Horiba Fluorolog-3 spectrometer, using Xe arc lamp, double monochromators and a photomultiplier detector at right angle detection geometry.
Synthesis of bis-phenylhydrazone astaxanthin (BPH-Asx)
Astaxanthin 10 mg (0.017 mmol), excess phenylhydrazine 7.34 mg (0.068 mmol) and a few drops of glacial acetic acid in ethanol (25 mL) were heated at reflux overnight. The resulting precipitate was filtered and washed with cold ethanol (100 mL). As the TLC of the solid part was clear, no further separation was applied. BPH-Asx was obtained as an orange-red color powder (4 mg, 30%). MALDI TOF (m/z) (Fig. S1, ESI†) calc. 776.50, found: 776.998 [M + H]+; ESI-MS (Fig. S1, ESI†): C52H63N4O2 [M+] calculated 775.4945, found: 775.4936 m/z. 1H NMR (Fig. S2, ESI;† 500 MHz, CDCl3) δ 9.61 (s, 2H, NH, j), 7.24 (4H, Ar–CH, h), 7.12 (d, J = 8.0 Hz, 4H, Ar–CH, h), 6.84 (t, J = 7.6 Hz, 2H, Ar–CH, g), 6.66 (t, J = 14.9 Hz, 4H, alkene H, f), 6.42 (d, J = 14.9 Hz, 2H, alkene H, f), 6.29–6.22 (m, 8H, alkene H, f), 5.30 (s, 2H, CH, e) 4.83 (s, 2H, OH, d), 2.10 (s, 4H, CH2, c), 1.99–2.00 (s, 12H, CH3, b), 1.13–1.25 (s, 18H, CH3, a) ppm. 13C NMR (Fig. S3, ESI;† 125 MHz, CDCl3) δ 150.3, 138.4, 137.5, 136.7, 135.1, 130.3, 129.6, 129.1, 128.5, 128.3, 126.8, 124.3, 114.4, 68.5, 53.8, 30.8, 30.2, 27.5, 26.4, 21.9 ppm. FT-IR (Fig. S4, ESI;† ATR, cm−1) v = 3300 cm−1 (NH), v = 1750 cm−1 (CN), v = 1251 cm−1 (C–N), v = 1500–1600 cm−1 (CO).
Transient absorption spectroscopy
Transient absorption (TA) spectroscopy was measured using an in-house build setup, based on a Solstice ACE (Spectra Physics) laser amplifier system that produces ∼60 fs pulses at a central wavelength of 796 nm at 4 kHz repetition rate. The amplifier output is divided into two parts that each pump an optical parametric amplifier (TOPAS-C, Light Conversion). One generates the pump beam while the other produces a NIR beam (1360 nm) that is focused onto a 3 mm CaF2 crystal to generate a supercontinuum probe beam. The delay between pump and probe pulses is introduced by a computer-controlled delay stage (Aerotech, 10 ns) placed in the probe beam path. After supercontinuum generation the probe pulses are split into two parts: the former being focused to ∼100 μm spot size and overlapping with the pump pulse in the sample volume, and the latter serving as a reference. After passing the sample the probe beam is collimated again and relayed onto the entrance aperture of a prism spectrograph. Both beams are then dispersed onto a double photodiode array, each holding 512 elements (Pascher Instruments). The intensity of excitation pulses was set to 230 μW, yielding excitation density of 6.5 × 1013 photons per pulse per cm2. Mutual polarization between pump and probe beams was set to the magic angle (54.7°) by placing a Berek compensator in the pump beam. Time-resolution of the setup after dispersion correction is estimated to be ≤100 fs. The measured samples, placed in a 1-mm pathlength optical cuvette, were translated after each scan to avoid photodegradation. To check for stability of each sample steady-state absorption spectra were measured before and after experiments.Data analysis
The resulting spectro-temporal data sets were analyzed by a global fitting software (CarpetView, Light Conversion, Lithuania). It was assumed that the excited system evolves irreversibly and sequentially to visualize the excited-state dynamics. Each component of the sequential scheme illustrates an individual excited-state species, and the spectral profile of each species is called the evolution-associated difference spectrum (EADS). The same software was used for chirp-correction of the spectra.Results
Synthesis and structural characterization
BPH-Asx was successfully prepared by using the procedure described in literature.48 The synthesis procedure of BPH-Asx is detailed in Scheme 1. The BPH-Asx was synthesized by the condensation reaction of commercially available Asx and phenylhydrazine in ethanol and in the presence of a catalytic amount of glacial acetic acid. After reaction reflux overnight, solid BPH-Asx was obtained with a reasonable yield (30%) by just washing with cold EtOH/MeOH to remove excess/unreacted phenylhydrazine. The BPH-Asx obtained without further purification appeared as an orange-red solid. The successful condensation of Asx with phenylhydrazine to produce the desired target compound, BPH-Asx, was confirmed by using various spectroscopic techniques.The molecular ion peak of the BPH-Asx was determined by the MALDI-TOF mass spectrometer as 776.998 m/z, which is in good agreement with the predicted structure, and further confirmed by ESI-MS (Fig. S1, ESI†). The novel BHP-Asx structure was further supported by comparing its 1H NMR spectroscopic data with that of commercially available Asx (Fig. S2, ESI†). The proposed chemical structure for BPH-Asx were confirmed by analysis of both aromatic and aliphatic protons. Specifically, in the case of BPH-Asx, additional resonances of aromatic benzene protons at 7.24 and 7.12 (10H, g and h), along with a new resonance of NH (2H, j) proton at 9.61 ppm, were identified. The presence of these additional characteristic protons strongly supports the structural confirmation of BPH-Asx. In the 13C NMR spectrum, the aromatic carbons of the BHP-Asx were marked between 150.3 and 114.4 and the aliphatic carbons signals were seen between the 68.5 and 21.9 ppm regions of the spectra (Fig. S3, ESI†). FT-IR spectroscopy is a useful and effective method for investigating structural changes in molecules and we compare the FT-IR spectra of BPH-Asx and Asx in Fig. S4 (ESI†). In the FT-IR spectrum of BPH-Asx, distinctive peaks were observed at 3300 cm−1 corresponding to the NH stretching vibrations, 1750 cm−1 indicate of the CN stretching vibrations, and 1251 cm−1 were attributed to the C–N stretching vibrations. Notably, in comparison to astaxanthin, BPH-Asx exhibited these new characteristic peaks and the absence of the strong peak at 1660 cm−1 associated with the CO stretching vibrations. These spectral structural differences between BPH-Asx and Asx further confirm the proposed structure and alterations of functional groups.
Asx and its modified derivative BPH-Asx were analyzed by HPLC for further characterization (Fig. S5, ESI†). The chromatogram of Asx consists of a single broad peak at 11.1 min, identified as all-transAsx.49 The chromatogram of BPH-Asx exhibits five peaks, occurring at longer retention times compared to Asx. The highest peak at 12.9 minutes (81%) was identified as BPH-Asx. Two minor peaks at 13.9 minutes (6%), 14.1 minutes (6%) showed absorbance bands around 380 nm (Fig. S6, ESI†) and likely correspond to cis-isomers of BPH-Asx.49 Presence of small amount of some unidentified Asx-related species was indicated by a minor peak at 12.2 minutes (3%). The last minor peak at 15.6 minutes (4%) showed significantly red-shifted absorbance (Fig. S6, ESI†) indicating significantly longer effective conjugation in the ground state. This could be caused by the s-trans configuration of the terminal rings making the whole conjugation linear including the two CN groups (Scheme 1).
Steady-state and transient absorption spectroscopy
Absorption spectra of Asx and BPH-Asx in acetonitrile (ACN) and dichloromethane (DCM) are depicted in Fig. 1. The absorption maximum of Asx is at 476 nm in ACN with a single broad peak, reflecting the S0–S2 transition, in agreement with previous reports on Asx in this solvent.25 The absorption maximum of Asx red shifts with increasing solvent polarizability, peaking at 486 nm in DCM. For BPH-Asx, absorption spectra in both solvents exhibit a small red shift compared to Asx: the absorption maxima are at 483 and 488 nm in ACN and DCM, respectively. A slightly broader distribution of the S0–S2 transition energies is observed for BPH-Asx, but overall, the comparison of absorption spectra suggests that the transition dipole moment associated with the S0–S2 transition undergoes only modest changes upon change from Asx to BPH-Asx. The properties of the S0–S2 transition are further confirmed by fluorescence spectra shown in Fig. S7 (ESI†). The weak fluorescence originates from the S2 state in agreement with data reported on Asx earlier.50 The solvent induced red-shift of fluorescence spectrum is larger for Asx than for BPH-Asx, mirroring the behavior of absorption spectra. | ||
| Fig. 1 Steady state absorption spectra of Asx and BPH-Asx in acetonitrile and dichloromethane. | ||
Transient absorption spectra at different time delays following excitation are shown in Fig. 2. The excitation wavelength was at 500 nm for Asx and BPH-Asx in both solvents, intended to excite the molecules just below the maximum of the S0–S2 transition. The data provide characteristic carotenoid transient absorption spectra, comprising ground state bleaching and excited state absorption (ESA) attributed to the S1–Sn transition for both Asx and BPH-Asx. The S1–Sn band of Asx in ACN, peaking at 629 nm, is consistent with previous studies,25 fully forming within the first picosecond. In DCM, the S1–Sn peak shifts to 642 nm (Fig. 2a and b). In addition to the dominant S1–Sn transition, a weak band in the 700–800 nm spectral region indicates the presence of an ICT state. The ICT signal is weak for Asx due to its symmetrically positioned conjugated CO groups that minimize the charge transfer character of the coupled S1/ICT state as has been also demonstrated for other carbonyl carotenoids.22 The 800–900 nm spectral region exhibits an additional ESA band at early delays (0.15 ps), which is linked to the ESA from the initially excited S2 state.51
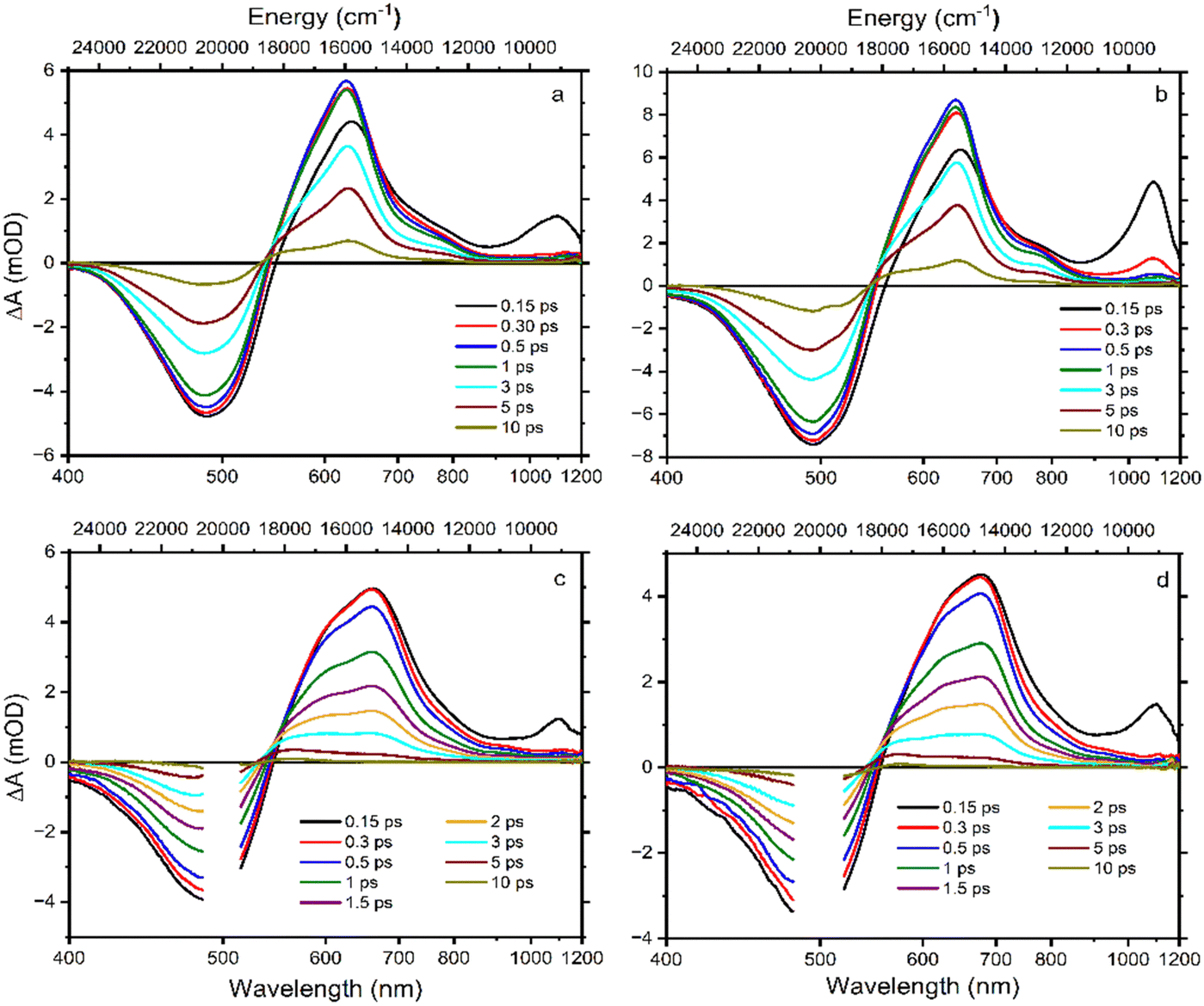 | ||
| Fig. 2 Transient absorption spectra of Asx in ACN (a), Asx in DCM (b), BPH-Asx in ACN (c), and BPH-Asx in DCM (d). The delay times after 500 nm excitation are indicated in each panel. The color-coding is the same in all panels. | ||
Although the general features observed in the transient absorption spectra of both carotenoids look similar, the modification of the conjugated chain from Asx to BPH-Asx induces some changes in the transient absorption spectra. As expected, both the ground state bleaching and S1–Sn ESA exhibit a red shift, reflecting the observed difference in the ground state absorption spectra. The S1–Sn bands of BPH-Asx have maxima at 642 nm and 681 nm in ACN and DCM, respectively (Fig. 2c and d). Besides the changes in the S1–Sn band, the ESA signal associated with the S2–Sn band of BPH-Asx at 0.15 ps after excitation did not show any shift but had less amplitude compared to Asx. The signal associated with the ICT state is much less pronounced in BPH-Asx as it nearly disappears in both solvents. However, this can be partly due to a broader S1–Sn band of BPH-Asx, resulting in a weak ICT band hidden under the dominant S1–Sn transition, which extends to 800 nm for BPH-Asx.
To visualize these changes, Fig. 3a compares normalized transient absorption spectra at 1 ps after excitation of both compounds in both solvents. The decrease in the magnitude of the ICT band, along with the red shift observed when going from Asx to BPH-Asx, is evident. Excited state dynamics was monitored by kinetics measured at the maximum of the S1–Sn band (Fig. 3b). The decay is significantly faster for BPH-Asx compared to Asx, while there is no change for both compounds with respect to solvent polarity.
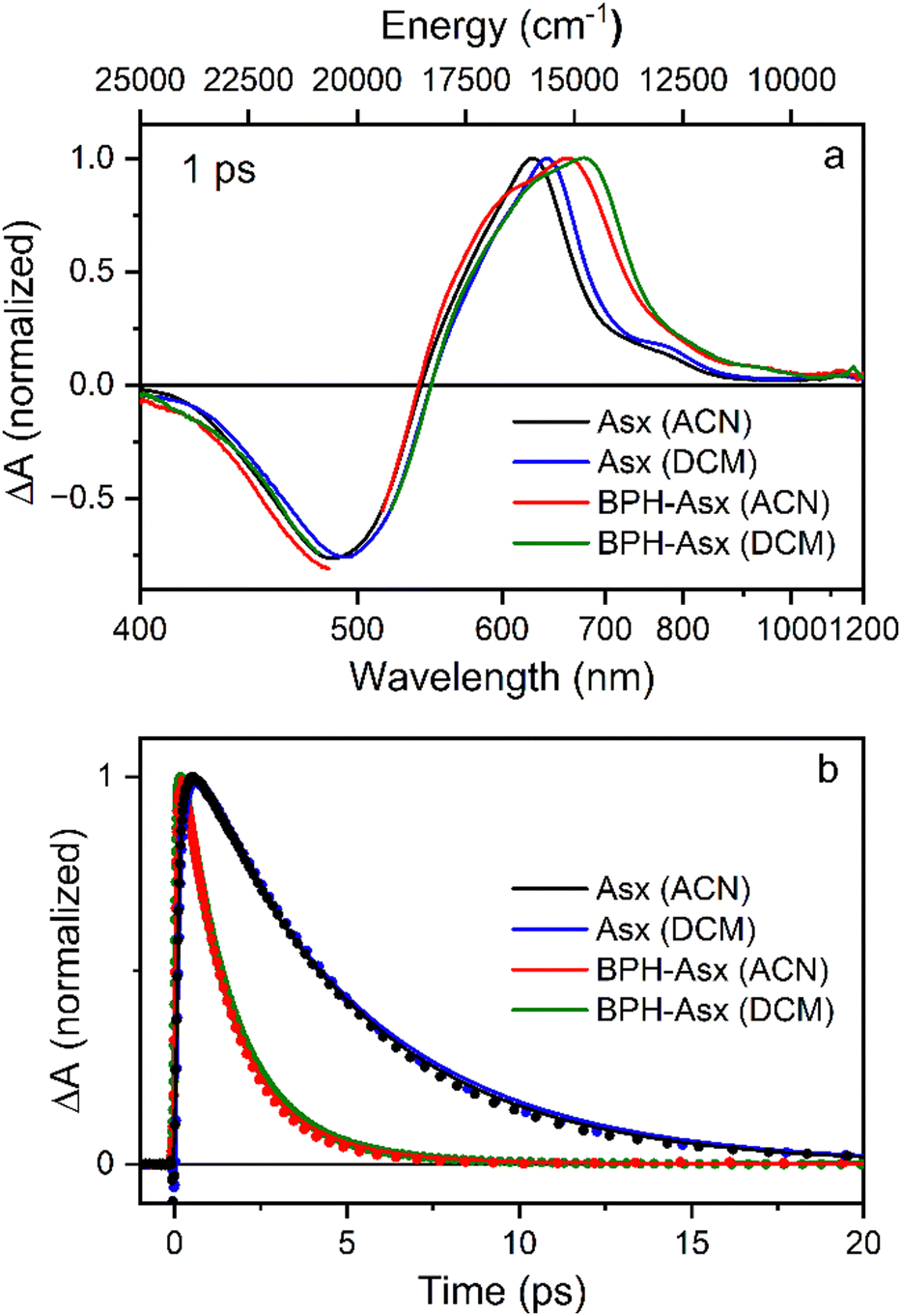 | ||
| Fig. 3 (a) Normalized transient absorption spectra of Asx and BPH-Asx in both solvents. The spectra were measured at 1 ps after excitation at 500 nm for all compounds/solvents. (b) Normalized kinetics measured at the S1–Sn maximum for each sample. The solid lines represent fits obtained from global fitting analysis. | ||
Excited state lifetimes were determined using a global fitting analysis, and the results are summarized in Fig. 4. For both compounds, three decay components are sufficient to obtain good fits. The first EADS for Asx in both solvents show features typical of the excited Asx S2 state, and its lifetime is at sub-100 fs time scale, reaching the limit of our time resolution. The second EADS has already typical features of the S1–Sn band except the increased amplitude at the low energy side of the band, which is characteristic of a hot S1/ICT state.52
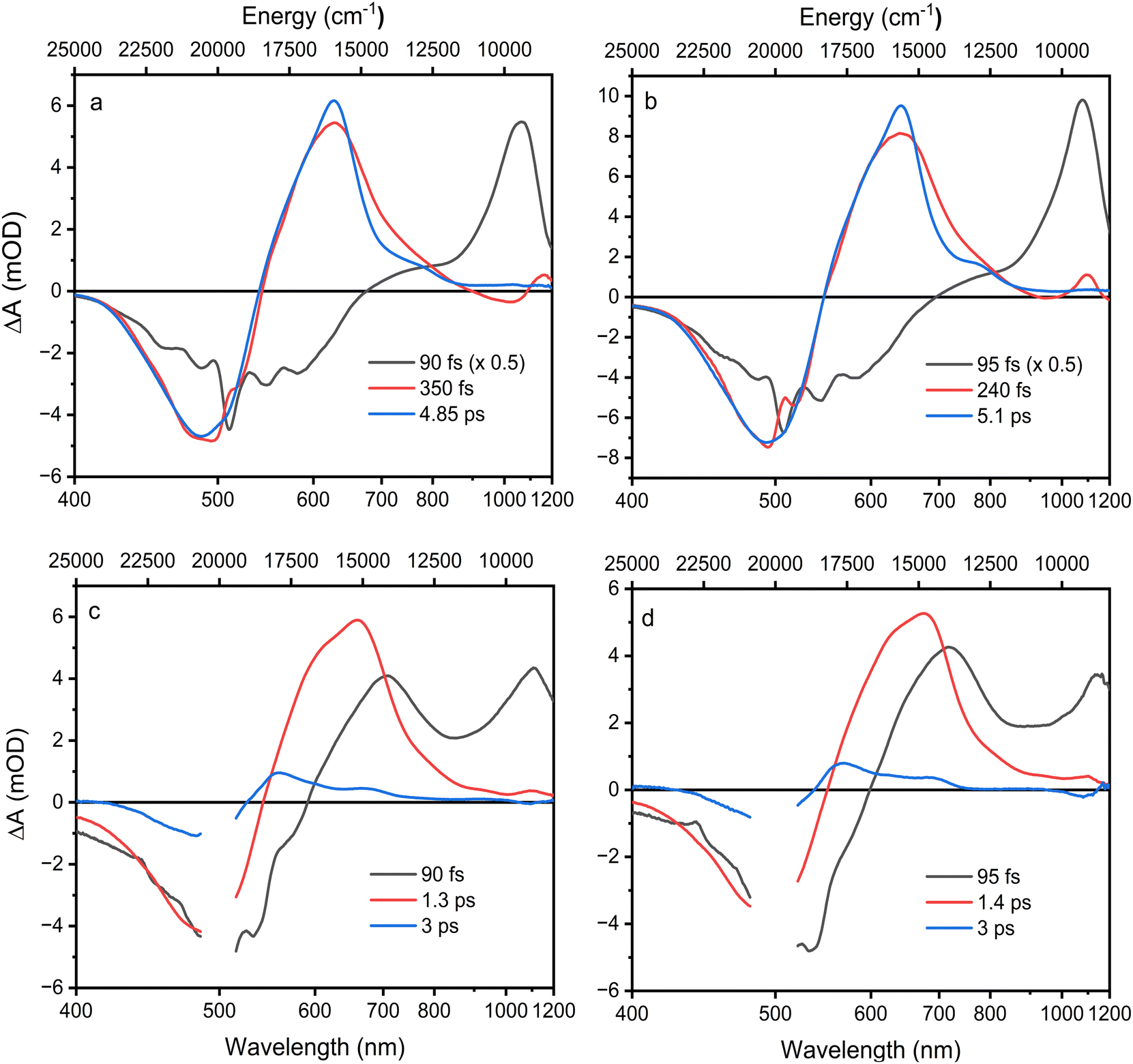 | ||
| Fig. 4 EADS obtained from global fitting of Asx in ACN (a), Asx in DCM (b), BPH-Asx in ACN (c), and BPH-Asx in DCM (d). | ||
Decay of the hot S1/ICT state occurs within a few hundred femtoseconds, differing slightly between ACN (350 fs) and DCM (240 fs) and yields EADS of the relaxed S1/ICT state. This EADS exhibits the characteristic profile of the relaxed S1/ICT state and decays with a time constant of 4.85 ps (ACN) and 5 ps (DCM). This pattern of the excited state dynamics of Asx in ACN agrees with the results obtained earlier.25,52
For BPH-Asx, three decay components provide a good fit, but the individual EADS differ from those obtained for Asx. The first EADS with sub-100 fs lifetime clearly contains features associated with both S2 state (the ESA signal peaking around 1100 nm) and the hot S1/ICT state. This implies that S2 and hot S1/ICT decays are both very short and occur on a comparable time scale, preventing separation of their contributions with our time resolution. Then, the second EADS corresponds to the relaxed S1/ICT state, which has a lifetime of 1.3 ps in ACN and 1.4 ps in DCM. In contrast to Asx, however, global fitting of BPH-Asx reveals EADS with a lifetime longer than the S1/ICT state. This EADS (blue in Fig. 4c and d) has a spectral shape characteristic of an S* signal, which is typically indicated by a distinct blue shoulder at the S1–Sn band, with a longer lifetime compared to the S1 state.27 Here, the S* EADS has a lifetime of 3 ps.
To further visualize the difference, we compared kinetics measured at the peak maxima of S1/ICT and S* bands for both Asx and BPH-Asx (Fig. 5). The kinetics clearly demonstrate that while the kinetics of Asx are identical, BPH-Asx exhibits a slower decay of the S* signal compared to the S1/ICT decay in both ACN and DCM, as expected.
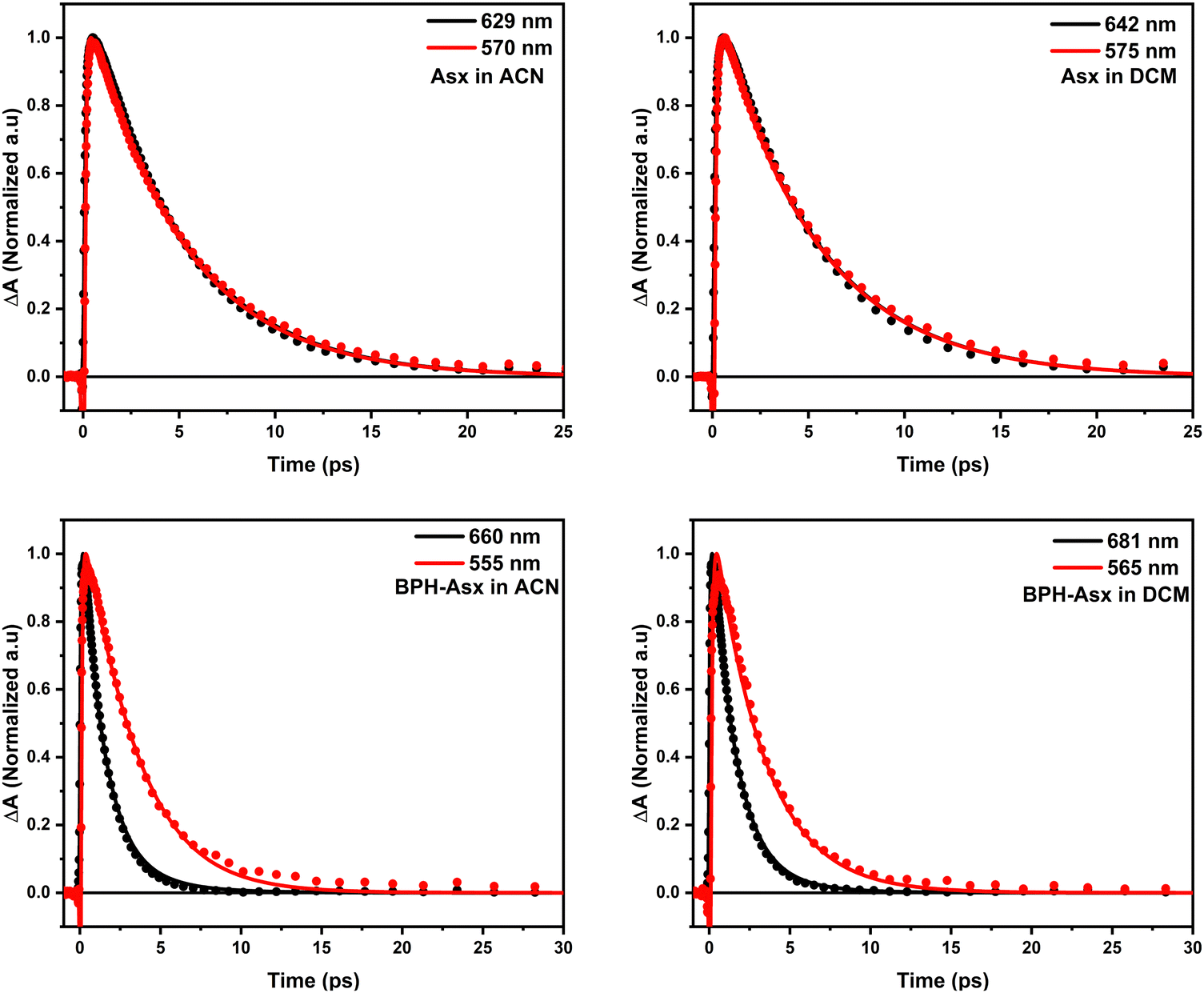 | ||
| Fig. 5 Comparison of normalized kinetics measured at the S1 (black) and S* (red) maximum bands of Asx and BPH-Asx in polar and non-polar solvents. The lines represent fits obtained from global fitting analysis. | ||
Discussion
A straightforward chemical reaction of Asx with phenylhydrazine and acetic acid in ethanol produced stable BPH-Asx with ∼30% yield, demonstrating a successful synthesis of a first carotenoid with a conjugated CN group. Formally, structure of the conjugated system of BPH-Asx is the same as for Asx except the CO groups of Asx, located symmetrically at both terminal rings, are replaced by CN groups in BPH-Asx. This allows to study the effect of replacement of the CO group by the CN group on spectroscopic properties directly. The presence of keto-groups in Asx generates a featureless absorption spectrum, most likely due to enhanced conformational disorder in the ground state produced by a broad distribution of end ring torsions. The relevant carotenoid without these conjugated keto-groups (zeaxanthin) has absorption spectrum with clearly resolved vibrational bands, proving the importance of the keto-groups in forming the featureless absorption spectrum. The same apparently happens when the keto groups are replaced by the CN group: the overall shape of the main absorption band is very similar, though the absorption spectrum of BPH-Asx is slightly broader, suggesting further enhancement of conformational disorder. Furthermore, absorption maximum of BPH-Asx is red-shifted by a few nanometers which is the first indication of changes of spectroscopic properties induced by the conjugated CN group. Solvent polarity has minimal effect on absorption spectrum of BPH-Asx, which is in striking contrast to the dicyano-apo-carotene, another carotenoid featuring nitrogen atoms in conjugation.42
While the changes in absorption spectra are rather subtle, more pronounced changes occur in excited state dynamics. The red shift of BPH-Asx is enhanced in transient absorption spectra reflecting the S1–Sn band, but the most significant effect of the conjugated CN group is on the S1/ICT lifetime. Even though the ICT band amplitude is not enhanced in BPH-Asx, the S1/ICT lifetime drops from ∼5 ps (Asx) to ∼1.4 ps (BPH-Asx). Since the S1/ICT lifetimes are essentially identical in DCM and ACN (Fig. 4), the shortening can hardly be associated with solvent polarity. To verify this, we have measured additional data for both carotenoids in non-polar benzene (Fig. S8 and S9, ESI†). The data clearly show that further decrease of solvent polarity does not have any effect on spectroscopic properties. The S1/ICT lifetime is the same in benzene and ACN, proving that the observed shortening of the S1/ICT lifetime is not due to solvent polarity, but it rather indicates a prolongation of effective conjugation length for BPH-Asx.
The effective conjugation length can be determined by comparison of the S1/ICT lifetime with that of carotenes.53 The S1/ICT lifetime of Asx, ∼5 ps, is comparable to that of the linear lycopene with N = 11. Shortening of the S1/ICT lifetime to 1.4 ps (BPH-Asx) suggests effective conjugation of ∼13. This implies that the conjugated CN groups contribute to the total conjugation length significantly more than the CO groups. Another support for explanation of the observed changes in spectroscopic properties solely by prolongation of the effective conjugation is detection of the S* signal exclusively in BPH-Asx. The blue shoulder (S*) of the S1–Sn band decaying slower than the main S1–Sn band (Fig. 5) is a feature reported exclusively for carotenoids with N > 11.33 Though the origin of this signal is not completely clear, recent studies have showed that for these long carotenoids the S* signal is likely due to a hot ground state populated by fast decay of the S1 state.33 Thus, the distinct lifetimes of the S* and S1/ICT signals, 3 and 1.4 ps provide further support for explanation of the differences between spectroscopic properties of Asx and BPH-Asx by prolongation of the effective conjugation.
To identify possible origin of the proposed prolongation of effective conjugation, we have examined both molecules by calculations using density functional theory. The structures were optimized using the B3LYP level of theory with 6-31g(d,p) basis set. Since the prolongation of the effective conjugation in carotenoids with conjugation extended to terminal rings is often associated with twisting of the end rings resulting in planarization of the conjugated system,53 we have focused on dihedral angles between the terminal rings and the main conjugation chain. For the relaxed ground state structures of Asx and BPH-Asxin vacuo we have obtained values of −38.5° and −43.7° (see Fig. S10 for relaxed structures, ESI†). These values are close to those reported for Asx earlier54 and indicate that planarization of BPH-Asx cannot be the reason for prolongation of the conjugation length. Thus, a different mechanism must operate here, likely related to the properties of the conjugated CN group that is further connected to other, non-conjugated part of the phenylhydrazone group. Even though non-conjugated groups usually do not contribute to the effective conjugation length, for some keto-carotenoids, electron distribution in the excited state has been affected by non-conjugated groups, resulting in a change of excited-state properties.55 It is likely that comparable mechanism works also here as there is only mild change (small red-shift of absorption spectrum) of spectroscopic properties in the ground state, but significant change occurs for the lowest excited state, somehow mimicking the behaviour reported in ref. 54.
Longer effective conjugation length of BPH-Asx and no effect of solvent polarity on its excited states is in striking contrast with another carotenoid involving a nitrogen atom in conjugated system, dicyano-apo-carotene, which exhibits a strong dependence of excited state properties on solvent polarity as its S1 lifetime varies by an order of magnitude from 11.7 ps in 3-methylpentane to 1.9 ps in ACN.42 This is due to a significant charge transfer character of the excited state, because dicyano-apo-carotene has two C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups but both located at the same side of the molecule, generating large asymmetry in the electron distribution along the conjugated chain. BPH-Asx in contrast, has two CN groups positioned symmetrically at ends of the conjugated chain, preventing asymmetry in electron distribution which is the source of polarity-induced effects on excited state dynamics.20–23 Instead, adding two symmetric phenylhydrazone groups extends the electron distribution along the conjugated chain, making the effective conjugation of BPH-Asx longer than of Asx.
N groups but both located at the same side of the molecule, generating large asymmetry in the electron distribution along the conjugated chain. BPH-Asx in contrast, has two CN groups positioned symmetrically at ends of the conjugated chain, preventing asymmetry in electron distribution which is the source of polarity-induced effects on excited state dynamics.20–23 Instead, adding two symmetric phenylhydrazone groups extends the electron distribution along the conjugated chain, making the effective conjugation of BPH-Asx longer than of Asx.
In conclusion, we have demonstrated that the carotenoid astaxanthin can be successfully modified with organic compounds via a new and simple synthetic pathway, adding non-native groups to the astaxanthin conjugated system, which may provide a basis for synthesis of further non-natural carotenoids. The modification of Asx presented here results in significant changes of photophysical properties, opening a way to study effects that are not present in natural carotenoids. This approach may help to shed more light on complicated structure of carotenoid excited states as well as on intricate relaxation pathways involving dark excited states, eventually contributing to a deeper understanding of complex photophysical properties of carotenoids.
Author contributions
Emrah Özcan: conceptualization, investigation, data curation, formal analysis, methodology, writing – original draft, writing – review & editing. Pavel Chábera: methodology, investigation. Gürkan Keşan: methodology, investigation. Radek Litvín: conceptualization, methodology, supervision, writing – original draft, writing – review & editing. Tomáš Polívka: conceptualization, funding acquisition, writing – original draft, writing – review & editing, supervision.Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research leading to these results has received funding from the Czech Science Foundation (grant 19-28323X), and LASERLAB-EUROPE (grant agreement no. 871124, European Union's Horizon 2020 research and innovation programme). EO further thanks Gebze Technical University for technical support in enabling mass spectroscopy, NMR, and FTIR measurements, and Profs Bünyemin Çoşut and İbrahim Fazıl Şengül for their help and support. The authors thank to Petr Štěpnička and Martin Štícha from Charles University for their help with ESI-MS measurements.References
- K. K. Namitha and P. S. Negi, Crit. Rev. Food Sci. Nutr., 2010, 50, 728–760 CrossRef CAS PubMed.
- T. Polívka and H. A. Frank, Acc. Chem. Res., 2010, 43, 1125–1134 CrossRef PubMed.
- R. Croce and H. van Amerongen, Science, 2020, 369, 2058 CrossRef PubMed.
- D. Zigmantas, T. Polívka, P. Persson and V. Sundström, Chem. Phys. Rev., 2022, 3, 041303 CrossRef CAS.
- A. V. Ruban, R. Berera, C. Ilioaia, I. H. van Stokkum, J. T. Kennis, A. A. Pascal, H. van Amerongen, B. Robert, P. Horton and R. van Grondelle, Nature, 2007, 450, 575–578 CrossRef CAS PubMed.
- N. E. Holt, D. Zigmantas, L. Valkunas, X. P. Li, K. K. Niyogi and G. R. Fleming, Science, 2005, 307, 433–436 CrossRef CAS PubMed.
- H. Staleva, J. Komenda, M. K. Shukla, V. Slouf, R. Kana, T. Polivka and R. Sobotka, Nat. Chem. Biol., 2015, 11, 287–291 CrossRef CAS PubMed.
- C. D. P. Duffy and A. V. Ruban, J. Photochem. Photobiol., B, 2015, 152, 215–226 CrossRef CAS PubMed.
- T. G. Monger, R. J. Cogdell and W. W. Parson, Biochim. Biophys. Acta, 1976, 449, 136–153 CrossRef CAS PubMed.
- Z. Kvíčalová, J. Alster, E. Hofmann, P. Khoroshyy, R. Litvín, D. Bína, T. Polívka and J. Pšenčík, Biochim. Biophys. Acta, Bioenerg., 1857, 2016, 341–349 Search PubMed.
- H. A. Frank and R. J. Cogdell, Photochem. Photobiol., 1996, 63, 257–264 CrossRef CAS PubMed.
- C. S. Foote, Y. C. Chang and R. W. Denny, J. Am. Chem. Soc., 1970, 92, 5216–5218 CrossRef CAS PubMed.
- T. W. M. Boileau, A. C. Moore and J. W. Erdman, J. Antioxid. Act., 1999, 133–158 CAS.
- S. A. R. Paiva and R. M. Russell, J. Am. Coll. Nutr., 1999, 18, 426–433 CrossRef CAS PubMed.
- N. I. Krinsky and K.-J. Yeum, Biochem. Biophys. Res. Commun., 2003, 305, 754–760 CrossRef CAS PubMed.
- M. Kobayashi and Y. Sakamoto, Biotechnol. Lett., 1999, 21, 265–269 CrossRef CAS.
- T. Polívka and V. Sundström, Chem. Rev., 2004, 104, 2021–2072 CrossRef PubMed.
- T. Polívka and V. Sundström, Chem. Phys. Lett., 2009, 477, 1–11 CrossRef.
- M. R. Wasielewski and L. D. Kispert, Chem. Phys. Lett., 1986, 128, 238–243 CrossRef CAS.
- H. A. Frank, J. A. Bautista, J. Josue, Z. Pendon, R. G. Hiller, F. P. Sharples, D. Gosztola and M. R. Wasielewski, J. Phys. Chem. B, 2000, 104, 4569–4577 CrossRef CAS.
- D. Zigmantas, R. G. Hiller, F. P. Sharples, H. A. Frank, V. Sundstrom and T. Polivka, Phys. Chem. Chem. Phys., 2004, 6, 3009–3016 RSC.
- M. M. Enriquez, M. Fuciman, A. M. LaFountain, N. L. Wagner, R. R. Birge and H. A. Frank, J. Phys. Chem. B, 2010, 114, 12416–12426 CrossRef CAS PubMed.
- P. Chábera, M. Fuciman, P. Hříbek and T. Polívka, Phys. Chem. Chem. Phys., 2009, 11, 8795–8803 RSC.
- E. Özcan, V. Kuznetsova, G. Keşan, M. Fuciman, R. Litvín and T. Polívka, J. Photochem. Photobiol., A, 2023, 441, 114737 CrossRef.
- R. P. Ilagan, R. L. Christensen, T. W. Chapp, G. N. Gibson, T. Pascher, T. Polivka and H. A. Frank, J. Phys. Chem. A, 2005, 109, 3120–3127 CrossRef CAS PubMed.
- N. Christensson, T. Polivka, A. Yartsev and T. Pullerits, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 245118 CrossRef.
- C. C. Gradinaru, J. T. M. Kennis, E. Papagiannakis, I. H. M. van Stokkum, R. J. Cogdell, G. R. Fleming, R. A. Niederman and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2364 CrossRef CAS PubMed.
- P. O. Andersson and T. Gillbro, J. Chem. Phys., 1995, 103, 2509 CrossRef CAS.
- W. Wohlleben, T. Buckup, H. Hashimoto, R. J. Cogdell, J. L. Herek and M. Motzkus, J. Phys. Chem. B, 2004, 108, 3320 CrossRef CAS.
- T. Lenzer, F. Ehlers, M. Scholz, R. Oswald and K. Oum, Phys. Chem. Chem. Phys., 2010, 12, 8832 RSC.
- E. Papagiannakis, I. H. M. van Stokkum, M. Vengris, R. J. Cogdell, R. van Grondelle and D. S. Larsen, J. Phys. Chem. B, 2006, 110, 5727 CrossRef CAS PubMed.
- D. M. Niedzwiedzki, J. O. Sullivan, T. Polivka, R. R. Birge and H. A. Frank, J. Phys. Chem. B, 2006, 110, 22872–22885 CrossRef CAS PubMed.
- V. Balevičius, D. Abramavicius, T. Polívka, A. Galestian Pour and J. Hauer, J. Phys. Chem. Lett., 2016, 7, 3347–3352 CrossRef PubMed.
- D. Kosumi, M. Fujiwara, R. Fujii, R. J. Cogdell, H. Hashimoto and M. Yoshizawa, J. Chem. Phys., 2009, 130, 214506 CrossRef PubMed.
- H. Staleva, M. Zeeshan, P. Chábera, V. Partali, H. R. Sliwka and T. Polívka, J. Phys. Chem. A, 2015, 119, 11304 CrossRef CAS PubMed.
- D. M. Niedzwiedzki, T. Kajikawa, K. Aoki, S. Katsumura and H. A. Frank, J. Chem. Phys. B, 2013, 117, 6874–6887 CrossRef CAS PubMed.
- D. Kosumi, T. Kajikawa, S. Okumura, M. Sugisaki, K. Sakaguchi, S. Katsumura and H. Hashimoto, J. Phys. Chem. Lett., 2014, 5, 792–797 CrossRef CAS PubMed.
- T. Kajikawa, K. Aoki, R. S. Singh, T. Iwashita, T. Kusumoto, H. A. Frank, H. Hashimoto and S. Katsumura, Org. Biomol. Chem., 2009, 7, 3723–3733 RSC.
- D. Kosumi, T. Kajikawa, K. Yano, S. Okumura, M. Sugisaki, K. Sakaguchi, S. Katsumura and H. Hashimoto, Chem. Phys. Lett., 2014, 602, 75–79 CrossRef CAS.
- M. M. Enriquez, S. Hananoki, S. Hasegawa, T. Kajikawa, S. Katsumura, N. L. Wagner, R. R. Birge and H. A. Frank, J. Phys. Chem. B, 2012, 116, 10748–10756 CrossRef CAS PubMed.
- H.-R. Sliwka and V. Partali, Molecules, 2012, 17, 2877–2928 CrossRef CAS PubMed.
- M. P. O’Neil, M. R. Wasielewski, M. M. Khaled and L. D. Kispert, J. Chem. Phys., 1991, 95, 7212–7218 CrossRef.
- P. Chábera, M. Fuciman, K. Razi Naqvi and T. Polívka, Chem. Phys., 2010, 373, 56–64 CrossRef.
- H. Fukami, K. Namikawa, N. Sugiura-Tomimori, M. Sumida, K. Katano and M. Nakao, J. Oleo Sci., 2006, 55, 653–656 CrossRef CAS.
- H. L. Jackson, A. J. Cardounel, J. L. Zweier and S. F. Lockwood, Bioorg. Med. Chem. Lett., 2004, 14, 3985–3991 CrossRef CAS PubMed.
- J. Willibald, S. Rennebaum, S. Breukers, S. H. Abdel Hafez, A. Patel, C. L. Øpstad, R. Schmid, S. N. Naess, H.-R. Sliwka and V. Partali, Chem. Phys. Lipids, 2009, 161, 32–37 CrossRef CAS PubMed.
- R. Litvín, D. Bína, M. Herbstová and Z. Gardian, Photosynth. Res., 2016, 130, 137–150 CrossRef PubMed.
- I. F. Sengul, K. Wood, N. Kumar and D. S. Black, Tetrahedron, 2012, 68, 9050–9055 CrossRef CAS.
- K. Holtin, M. Kuehnle, J. Rehbein, P. Schuler, G. Nicholson and K. Albert, Anal. Bioanal. Chem., 2009, 395, 1613–1622 CrossRef CAS PubMed.
- J. Kevin, H. Stapelfeldt and L. H. Skibsted, Chem. Phys. Lett., 1992, 190, 514–519 CrossRef.
- T. Khan, R. Litvín, V. Šebelík and T. Polívka, ChemPhysChem, 2021, 22, 471–480 CrossRef CAS PubMed.
- M. Fuciman, M. Durchan, V. Slouf, G. Kesan and T. Polivka, Chem. Phys. Lett., 2013, 568, 21–25 CrossRef.
- M. Fuciman, G. Kesan, A. M. LaFountain, H. A. Frank and T. Polivka, J. Phys. Chem. B, 2015, 119, 1457–1467 CrossRef CAS PubMed.
- H. Hashimoto, T. Yoda, T. Kobayashi and A. J. Young, J. Mol. Struct., 2002, 604, 125–146 CrossRef CAS.
- H. Staleva-Musto, V. Kuznetsova, D. Bína, R. Litvín and T. Polívka, Photosynth. Res., 2020, 144, 127–135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02282c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |