
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of acyclic analogues of adenosine sulfonamides and their activity against RNA cap guanine N7-methyltransferase of SARS-CoV-2†
Rostom
Ahmed-Belkacem‡
a,
Adrien
Delpal‡
b,
Bruno
Canard
b,
Jean-Jacques
Vasseur
 a,
Etienne
Decroly
*b and
Françoise
Debart
*a
a,
Etienne
Decroly
*b and
Françoise
Debart
*a
aInstitute of Biomolecules Max Mousseron (IBMM), University of Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: francoise.debart@umontpellier.fr
bLaboratoire Architecture et Fonction des Macomolécules Biologiques (AFMB), University of Aix-Marseille, CNRS, Marseille, France. E-mail: Etienne.decroly@univ-amu.fr
First published on 1st October 2024
Abstract
N-Arylsulfonamide-based adenosine analogues were previously shown to be potent inhibitors of SARS-CoV-2 RNA cap guanine N7-methyltransferase nsp14. Here, we synthesized three series of N-arylsulfonamide acyclic analogues of adenosine as bisubstrates of nsp14. Most of these acyclic compounds were barely active at 50 μM against this cap N7-methyltransferase.
The RNA cap guanine N7-methyltransferase (MTase) of SARS-CoV-2 has been validated as an antiviral target as mutations in the N7-MTase catalytic site have been shown to impair SARS-CoV replication.1 Indeed, reverse genetics experiments identified two amino acid substitutions abrogating virus viability and a mutation producing a crippled phenotype in several betacoronaviruses. N7-MTase is thus a critical enzyme for betacoronavirus replication. The CoV N7-MTase nsp14 is the first enzyme involved in the enzymatic cascade of viral RNA cap methylations and is responsible for N7-methylation of the cap guanosine, which is essential for viral RNA translation into proteins. The N7-methylation of the cap structure is followed by its 2′-O-methylation of the first RNA transcribed nucleotide by the nsp10/nsp16 complex.2,3
In recent years, we and other groups have followed a rational drug design approach to synthesize competitive inhibitors and have identified a number of potent nanomolar N7-MTase nsp14 inhibitors that mimic SAM/SAH derivatives.4–11 Some SAR data supported by molecular docking led to the elucidation of a bisubstrate-based mechanism of action and was used to optimize the design of bisubstrate inhibitors.7,9,10 These inhibitors were designed as compounds that occupy both the methyl donor (S-adenosylmethionine, SAM) binding pocket and the RNA cap binding pocket of the SARS-CoV-2 N7-MTase. In our previous work and that of Chen and Nencka's groups,7,9,10 the most efficient bisubstrate inhibitors were composed of an N-arylsulfonamide core, a pharmacophore found in many biologically active compounds, and occupying the RNA cap binding site, attached to the 5′ position of an adenosine or a derivative, mimicking that of SAM (Fig. 1). The N-arylsulfonamide core has been shown to account for much of the compound's high affinity for SARS-CoV-2 nsp14, resulting in potent inhibition.
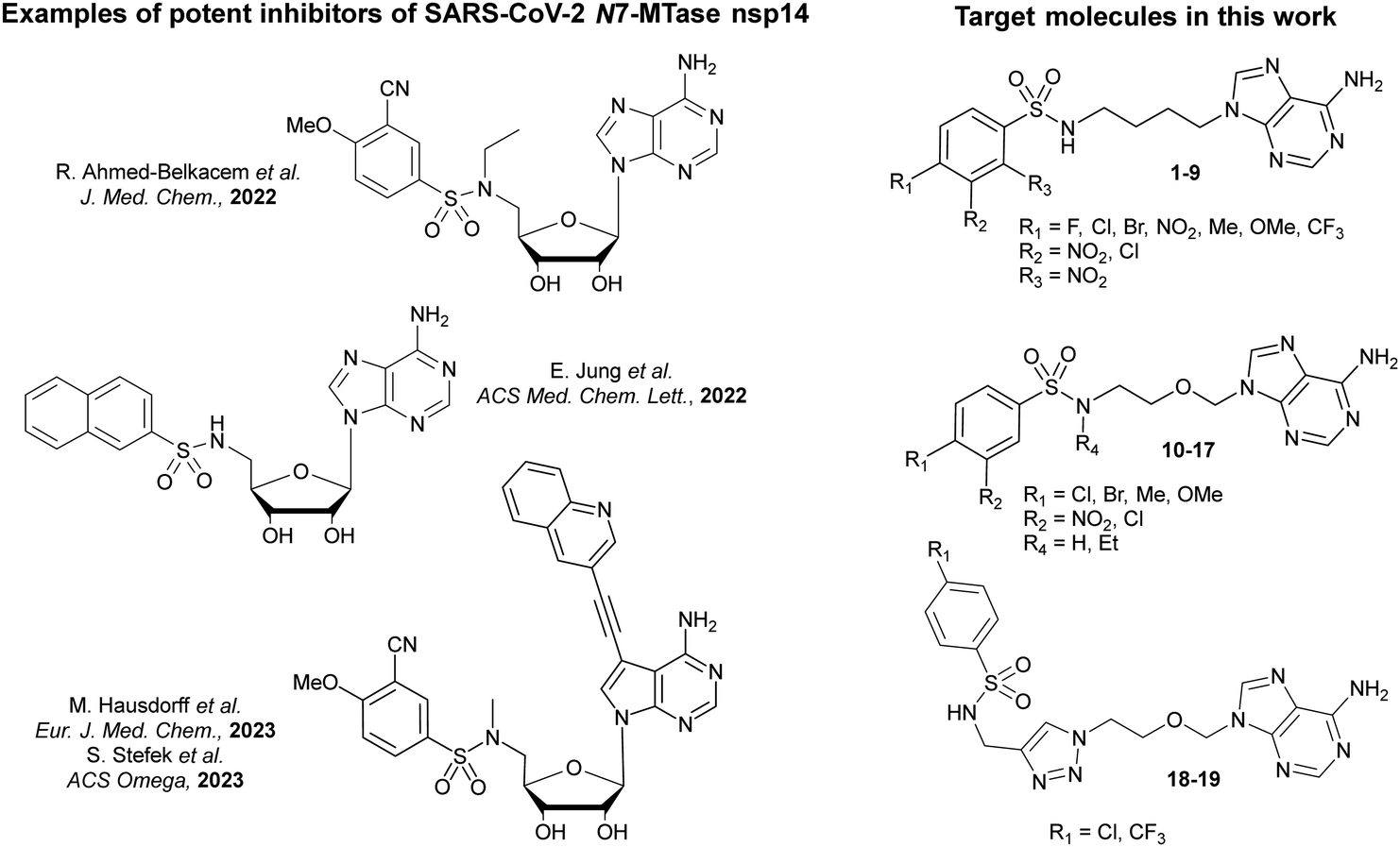 | ||
| Fig. 1 Examples of previous N-arylsulfonamide-based adenosine analogues showing low nanomolar inhibitory activity against SARS-CoV-2 N7-methyltransferase nsp14 (left panel).6,7,9,10 Chemical structures of the target molecules as acyclic analogues of adenosine sulfonamides 1–19 (right panel). | ||
In this study, we designed molecules with the essential N-arylsulfonamide core, and the sugar scaffold has been removed, leading to acyclic nucleosides. The glycosidic linkage (cleavable by purine nucleoside phosphorylases) of our previous adenosine-derived inhibitors is replaced by an acyclic alkyl chain that could stabilize the compounds against metabolic degradation (Fig. 1). Moreover, the abstraction of 2′- and 3′-hydroxyls could improve the lipophilicity of these analogues. Similar replacement of the ribose moiety has already been used many years ago in antiviral research.12–14 One of the most medically important acyclic nucleosides is acyclovir, an acyclic guanosine mimic with high activity and specificity against HSV-1 and HSV-2.15 It has been shown that the flexibility of the acyclic alkyl chain can optimize interactions with the target enzyme binding sites. Adefovir and tenofovir are two other well-known phosphonate carrying acyclic nucleosides. These two adenosine mimetics have been reported as potent antiviral drugs against HIV and HBV.16,17 In addition, numerous examples on the synthesis and activity of nucleoside analogues with acyclic sugar mimic as antiviral or anticancer agents have been widely reported.18–26 In this work, we propose the possibility of preventing the N7 methylation of SARS-CoV-2 RNA cap structure thereby limiting the viral RNA translation into protein through the inhibitory action of acyclic nucleosides on the N7- MTase nsp14. Thus, we synthesized three series of novel acyclic analogues of adenosine sulfonamides containing a butyl chain (1–9), an N-ethyloxymethyl chain (10–17) or a 1,2,3-triazolylethyloxymethyl linker between the N-arylsulfonamide core and adenine (18–19).
Molecular docking of compound 1 showed a suitable overlay with SAH in SARS-CoV-2 nsp14 (PDB ID: 7R2V, resolution 2.53 Å, data not shown) (Scheme 1). As we had shown the relevance of the p-OMe substituent in combination with m-NO2 on the N-ethyl-N-arylsulfonamide ring,6 compound 15 was docked into nsp14 and the overlay of its adenosine moiety with the SAH present in the SARS-CoV-2 nsp14 structure was confirmed (ESI†). It is noteworthy that even in the absence of the ribose, the acyclic nucleoside adopts a conformation that preserves hydrogen interactions with Arg310 and Asn386. In addition, the π–π stacking interaction previously reported between the phenylsulfonamide core and the Phe426 is retained. However, this occurred as a “parallel displaced” rather than a “face-to-face” interaction observed with the previous nucleoside inhibitors.6,9 Finally, the p-substituents (OMe, Me, F, Cl, Br) always fit correctly into the hydrophobic pocket formed by Phe506, Phe401 and Tyr420. Overall, these findings supported the synthesis and evaluation of these novel acyclic nucleosides.
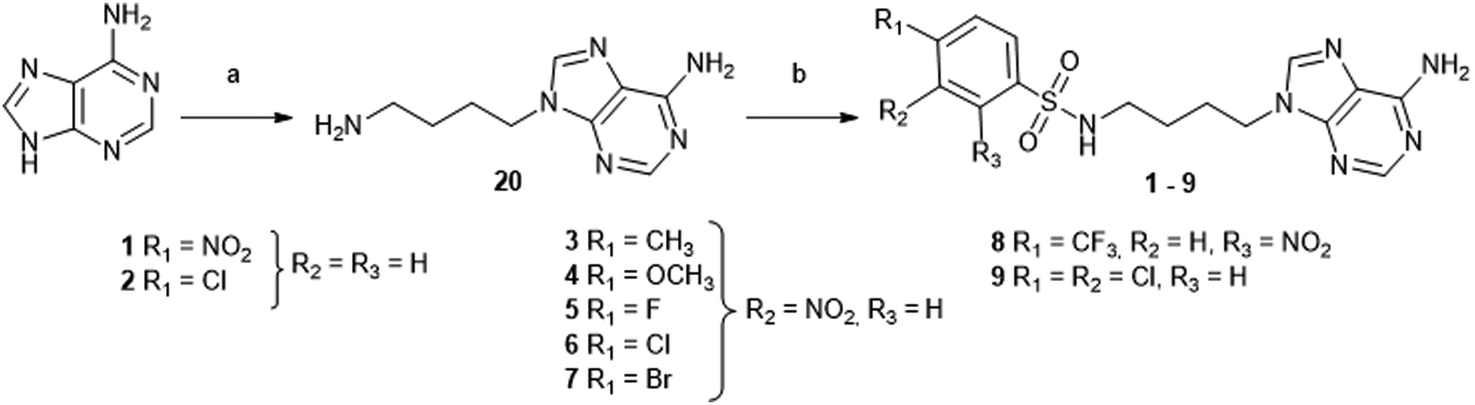 | ||
| Scheme 1 Synthesis of acyclic adenosine analogues 1–9. Reagents and conditions: (a) (i) N-(4-bromobutyl)phthalimide, K2CO3, DMF, 70 °C, 18 h; (ii) hydrazine hydrate (50–60%), EtOH, 65 °C, 2 h, 60%.27 (b) Arylsulfonyl chloride, Et3N, DMF, 3 h, 34–81%. | ||
The first target molecules were acyclic nucleosides 1–9, which contain a 4-carbon chain between the N9-adenine and the sulfonamide function, instead of ribose. The synthesis started with an alkylation step on adenine to introduce the butyl chain. Adenine was deprotonated by potassium carbonate in DMF and alkylated with N-(4-bromobutyl)phthalimide to give an intermediate N-protected acyclic nucleoside (Scheme 1). A further hydrazine treatment afforded compound 20 with 60% yield27 and the resulting primary amine was reacted with various commercially available arylsulfonyl chlorides to give the N-arylsulfonamide-containing acyclic nucleosides 1–9.28
Next, we designed acyclic adenosine analogues 10–17, in which the sugar was replaced by an aliphatic ethyloxymethyl linker decorated with various arylsulfonamides that mimic the cap guanosine. These compounds were prepared in 5 steps from adenine (Scheme 2). After protection of the N6-amino function of adenine with a benzoyl group,29 the alkylation of N6-benzoyl adenine 21 was performed using 1,3-dioxolane and acetic anhydride, affording intermediate 22 in 55% yield.24 The acetyl group at the extremity of the ethyloxymethyl chain and the N-benzoyl group were then cleaved in basic medium and the resulting hydroxyl in 23 was converted to primary amine using Mitsunobu conditions, giving 24 in quantitative yield.30 The free amine of 24 was finally reacted with various arylsulfonyl chlorides to provide eight N-arylsulfonamide-containing acyclic nucleosides 10–17.
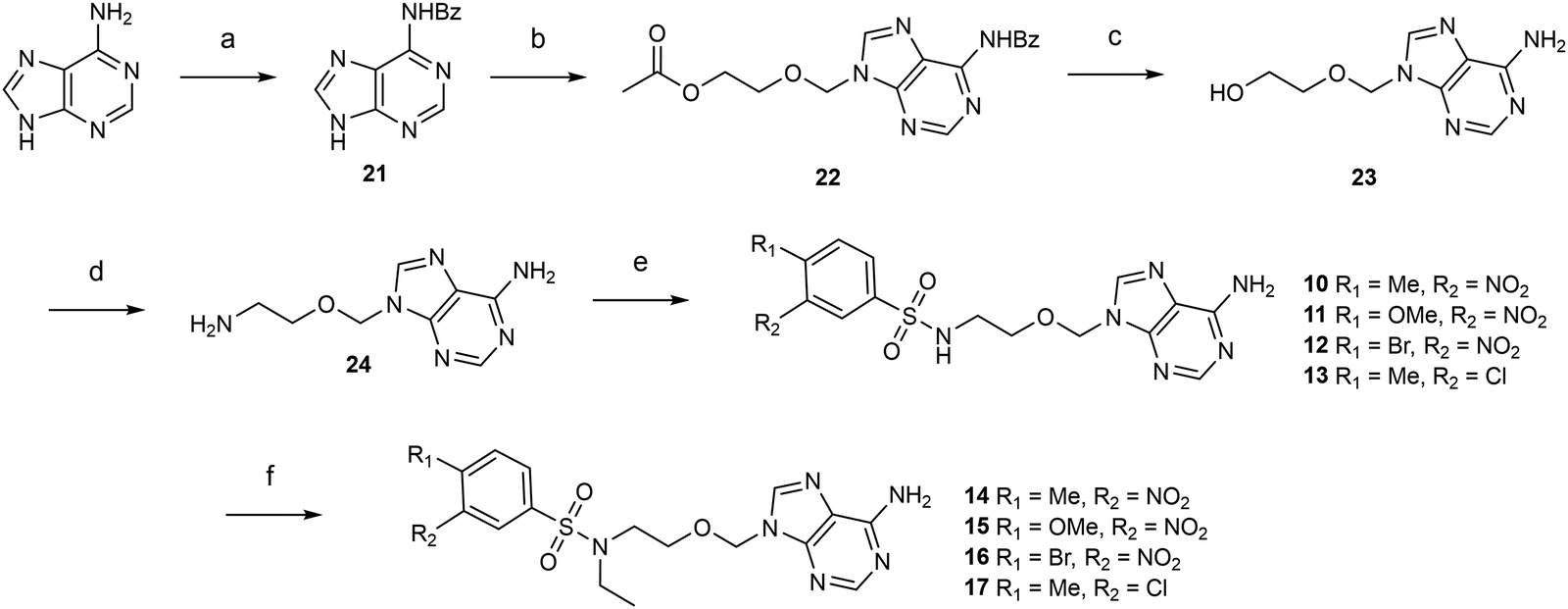 | ||
| Scheme 2 Synthesis of acyclic adenosine analogues 10–17. Reagents and conditions: (a) BzCl, pyridine, 100 °C, 3 h then 25 °C, 18 h, 62%. (b) 1,3-dioxolane, acetic anhydride, TMSOTf, MeCN, 25 °C, 15 min, 55%. (c) NH4OH/MeOH 3/1 (v:v), 25 °C, 18 h, quant. (d) (i) Phtalimide, DIAD, PPh3, THF, 25 °C, 2.5 h; (ii) hydrazine hydrate (50–60%), EtOH, 65 °C, 2 h, quant. (e) Arylsulfonyl chloride, Et3N, DMF, 3 h, 61–74%. (f) Ethyl p-toluenesulfonate, KI, K2CO3, DMF, 50 °C, 16 h, 47–54%. | ||
Acyclic nucleosides 18 and 19 containing a 1,2,3-triazole ring were the last target compounds to be synthesized from 23 and N-propargyl-N-arylsulfonamide reagents previously prepared in-house (Scheme 3).11 First, an azide group was introduced using diphenyl phosphoryl azide (DPPA) in 1,4-dioxane reacting with 23 to give 26 with 74% yield. Then, a copper-catalyzed azide–alkyne cycloaddition in the presence of CuSO4 and sodium ascorbate afforded acyclic nucleoside analogues 18 and 19 in 74% yield and 69% yield, respectiveley.31
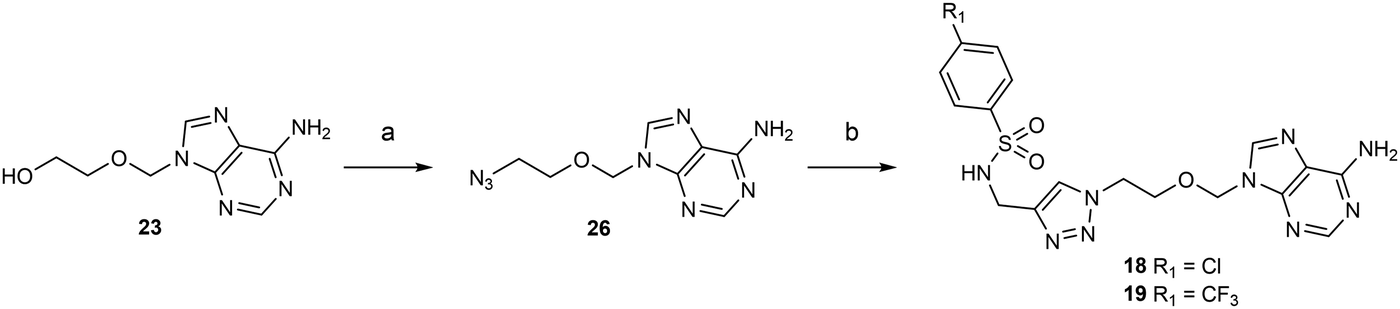 | ||
| Scheme 3 Synthesis of acyclic adenosine analogues 18–19. Reagents and conditions: (a) (i) DPPA, DBU, 1,4-dioxane, 25 °C, 16 h; (ii) NaN3, 15-C-5, 100 °C, 4 h, 74%. (b) N-arylsulfonamide-N-propargyl reagents,11 CuSO4, sodium ascorbate, H2O/dioxane, 0 °C, 3 h, 74% (for p-Cl), 69% (for p-CF3). | ||
All synthesized compounds 1–19 were screened for inhibitory activity of SARS-CoV-2 N7-MTase nsp14 using a radioactive MTase assay. Briefly, the nsp14 protein was incubated together with 1–19 at 5 μM and 50 μM concentrations and the [3H]-radiolabeled methyl transferred from the SAM methyl donor onto the cap structure of a short synthetic RNA substrate (GpppAC4) was measured by a filter binding assay.32 All acyclic nucleoside analogues show no inhibitory activity at 5 μM concentration, and most of them are barely active at 50 μM against the N7-MTase nsp14, as enzyme activity remains above 50% at this concentration. Replacement of the ribose between the adenine nucleobase and the N-arylsulfonamide moieties is clearly detrimental for the inhibitory activity of these acyclic analogues, mimics of our previous nsp14 inhibitors (0.146 μM < IC50 < 14.1 μM).6 However, two acyclic compounds 1 and 4 with a butyl chain display a modest inhibitory effect at 50 μM on N7- MTase (N7-MTase activity < 35%). Overall, the butyl chain seems to be less detrimental than ethyloxymethyl or triazole-containing linkers for the inhibitory activity of these acyclic adenosine sulfonamides against SARS-CoV-2 N7-MTase nsp14. The lack of activity could be explained by the high flexibility of the alkyl chain, which might prevent the simultaneous binding of adenine in the SAM binding pocket and the arylsulfonamide ring in the cap binding pocket observed when ribose is present in the nucleoside inhibitors.6,9
Conclusions
We have successfully prepared three series of acyclic nucleoside sulfonamides as analogues of N-arylsulfonamide-based adenosines, which had been found to be potent inhibitors of SARS-CoV-2 RNA cap N7-methyltransferase nsp14. Replacement of the ribose moiety by a butyl, ethyloxymethyl or triazolylethyloxymethyl linker impaired the inhibitory activity of these acyclic compounds. However, these results are valuable for SAR and will be useful for further design of nsp14 inhibitor bisubstrates.Author contributions
R. A.-B.: conceptualization, investigation, methodology, validation, writing – original draft. A. D.: investigation, methodology, validation. B. C.: resources, writing – review & editing. J.-J. V.: resources, supervision, writing – review & editing. E. D.: data curation, supervision, validation, funding acquisition, project administration, writing – review & editing. F. D.: funding acquisition, project administration, supervision, validation, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors (R. A.-B., J.-J. V., F. D.) thank the French “Agence Nationale de la Recherche (ANR)” (Project MetInCoV: ANR-21-CO14-0004-01) for financial support and Amina Tahir for HPLC, RMN et HRMS analyses of the final compounds. This project has received funding from the European Union's Horizon 2020 Research and Innovation program under grants no. 101003627 (the SCORE project) and no. 101005077 (the CARE project) (E. D., B. C., A. D.).Notes and references
- N. S. Ogando, P. El Kazzi, J. C. Zevenhoven-Dobbe, B. W. Bontes, A. Decombe, C. C. Posthuma, V. Thiel, B. Canard, F. Ferron, E. Decroly and E. J. Snijder, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108709118 CrossRef CAS.
- M. Bouvet, C. Debarnot, I. Imbert, B. Selisko, E. J. Snijder, B. Canard and E. Decroly, PLoS Pathog., 2010, 6, e1000863 CrossRef.
- M. Romano, A. Ruggiero, F. Squeglia, G. Maga and R. Berisio, Cells, 2020, 9, 1267 CrossRef CAS.
- O. Bobileva, R. Bobrovs, I. Kanepe, L. Patetko, G. Kalnins, M. Sisovs, A. L. Bula, S. Gri Nberga, M. R. Boroduskis, A. Ramata-Stunda, N. Rostoks, A. Jirgensons, K. Ta Rs and K. Jaudzems, ACS Med. Chem. Lett., 2021, 12, 1102–1107 CrossRef CAS.
- T. Otava, M. Šála, F. Li, J. Fanfrlík, K. Devkota, S. Perveen, I. Chau, P. Pakarian, P. Hobza, M. Vedadi, E. Boura and R. Nencka, ACS Infect. Dis., 2021, 7, 2214–2220 CrossRef CAS PubMed.
- R. Ahmed-Belkacem, M. Hausdorff, A. Delpal, P. Sutto-Ortiz, A. M. G. Colmant, F. Touret, N. S. Ogando, E. J. Snijder, B. Canard, B. Coutard, J. J. Vasseur, E. Decroly and F. Debart, J. Med. Chem., 2022, 65, 6231–6249 CrossRef CAS.
- E. Jung, R. Soto-Acosta, J. Xie, D. J. Wilson, C. D. Dreis, R. Majima, T. C. Edwards, R. J. Geraghty and L. Chen, ACS Med. Chem. Lett., 2022, 13, 1477–1484 CrossRef CAS.
- O. Bobileva, R. Bobrovs, E. E. Sirma, I. Kanepe, A. L. Bula, L. Patetko, A. Ramata-Stunda, S. Grinberga, A. Jirgensons and K. Jaudzems, Molecules, 2023, 28, 768 CrossRef CAS PubMed.
- M. Hausdorff, A. Delpal, S. Barelier, L. Nicollet, B. Canard, F. Touret, A. Colmant, B. Coutard, J. J. Vasseur, E. Decroly and F. Debart, Eur. J. Med. Chem., 2023, 256, 115474 CrossRef CAS.
- M. Štefek, D. Chalupská, K. Chalupský, M. Zgarbová, A. Dvořáková, P. Krafčíková, A. S. M. Li, M. Šála, M. Dejmek, T. Otava, E. Chaloupecká, J. Kozák, J. Kozic, M. Vedadi, J. Weber, H. Mertlíková-Kaiserová and R. Nencka, ACS Omega, 2023, 8, 27410–27418 CrossRef PubMed.
- R. Ahmed-Belkacem, J. Troussier, A. Delpal, B. Canard, J.-J. Vasseur, E. Decroly and F. Debart, RSC Med. Chem., 2024, 15, 839–847 RSC.
- S. Freeman and J. M. Gardiner, Mol. Biotechnol., 1996, 5, 125–137 CrossRef CAS.
- E. D. Clercq, Biochem. Pharmacol., 2011, 82, 99–109 CrossRef PubMed.
- K. L. Seley-Radtke and M. K. Yates, Antiviral Res., 2018, 154, 66–86 CrossRef CAS PubMed.
- D. H. King, J. Am. Acad. Dermatol., 1988, 18, 176–179 CrossRef CAS.
- E. De Clercq and A. Holý, Nat. Rev. Drug Discovery, 2005, 4, 928–940 CrossRef CAS PubMed.
- A. Holý, Antiviral Res., 2006, 71, 248–253 CrossRef PubMed.
- R. Gawin, E. De Clercq, L. Naesens and M. Koszytkowska-Stawińska, Bioorg. Med. Chem., 2008, 16, 8379–8389 CrossRef CAS PubMed.
- H. Elayadi, M. Smietana, C. Pannecouque, P. Leyssen, J. Neyts, J.-J. Vasseur and H. B. Lazrek, Bioorg. Med. Chem. Lett., 2010, 20, 7365–7368 CrossRef CAS.
- I. A. Khalymbadzha, T. S. Shestakova, J. O. Subbotina, O. S. Eltsov, A. A. Musikhina, V. L. Rusinov, O. N. Chupakhin, I. L. Karpenko, M. V. Jasko, M. K. Kukhanova and S. L. Deev, Tetrahedron, 2014, 70, 1298–1305 CrossRef CAS.
- H. L. Peters, D. Jochmans, A. H. de Wilde, C. C. Posthuma, E. J. Snijder, J. Neyts and K. L. Seley-Radtke, Bioorg. Med. Chem. Lett., 2015, 25, 2923–2926 CrossRef CAS PubMed.
- M. Gładysz, P. Ruszkowski and J. Milecki, Nucleosides, Nucleotides Nucleic Acids, 2018, 37, 53–66 CrossRef.
- V. Malnuit, S. Smoleń, M. Tichý, L. Poštová Slavětínská and M. Hocek, Eur. J. Org. Chem., 2019, 5409–5423 CrossRef CAS.
- J. Frydrych, L. Poštová Slavětínská, M. Dračínský and Z. Janeba, Molecules, 2020, 25, 4307 CrossRef CAS.
- J. E. Thames, C. D. Waters, C. Valle, M. Bassetto, W. Aouadi, B. Martin, B. Selisko, A. Falat, B. Coutard, A. Brancale, B. Canard, E. Decroly and K. L. Seley-Radtke, Bioorg. Med. Chem., 2020, 28, 115713 CrossRef CAS.
- E.-J. Hao, G.-X. Li, Y.-R. Liang, M.-S. Xie, D.-C. Wang, X.-H. Jiang, J.-Y. Cheng, Z.-X. Shi, Y. Wang and H.-M. Guo, J. Med. Chem., 2021, 64, 2077–2109 CrossRef CAS.
- K. J. Lee, W. Tieu, B. Blanco-Rodriguez, A. S. Paparella, J. Yu, A. Hayes, J. Feng, A. C. Marshall, B. Noll, R. Milne, D. Cini, M. C. J. Wilce, G. W. Booker, J. B. Bruning, S. W. Polyak and A. D. Abell, ACS Chem. Biol., 2019, 14, 1990–1997 CrossRef CAS.
- G. Zhen, G. Zeng, K. Jiang, F. Wang, X. Cao and B. Yin, Chem. – Eur. J., 2023, 29, e202203217 CrossRef CAS.
- C. F. Liu, Y. Zeng and X. Lu, Singapore Pat., WO2010027326A1, 2008 Search PubMed.
- A. Granzhan, E. Largy, N. Saettel and M. P. Teulade-Fichou, Chem. – Eur. J., 2010, 16, 878–889 CrossRef CAS.
- D. Coelho, L. Le Corre, K. Bartosik, L. Iannazzo, E. Braud and M. Ethève-Quelquejeu, Chem. Eur. J., 2023, 29, e202301134 CrossRef CAS.
- F. Peyrane, B. Selisko, E. Decroly, J. J. Vasseur, D. Benarroch, B. Canard and K. Alvarez, Nucleic Acids Res., 2007, 35, e26 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular docking; general and detailed synthetic procedures, and spectral characterization data for synthesized compounds. See DOI: https://doi.org/10.1039/d4nj02481h |
| ‡ These two first authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |