
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of intramolecular Fe oxidation with distance and driving force in Ru–Fe photocatalysts†
Christian
Herrero
 a,
Frédéric
Banse
*a,
Winfried
Leibl
b and
Annamaria
Quaranta
*b
a,
Frédéric
Banse
*a,
Winfried
Leibl
b and
Annamaria
Quaranta
*b
aUniversité Paris-Saclay, CNRS, Institut de Chimie Moléculaire et des Matériaux d’Orsay, 91400 Orsay, France. E-mail: frederic.banse@universite-paris-saclay.fr
bUniversité Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France. E-mail: annamaria.quaranta@cea.fr
First published on 16th September 2024
Abstract
In this paper we have investigated the efficiency of the intramolecular electron transfer process in a family of RuII–FeII dyads, previously shown to perform the light-driven activation of an iron-bound water molecule. The Ru chromophore and Fe catalytic units are connected at different lengths through a triazole group attached to an alkyl chain containing either three or five carbon atoms. The driving force for Fe oxidation is modified by adding ethyl ester substituents on the bipyridines completing the coordination sphere of a [Ru(bpy)3]2+-like chromophore. Transient absorption measurements and simulations were used to obtain the rate constants of internal charge transfer for the different complexes. The parameters governing the electron transfer process, reorganisation energy λ and electronic coupling HAB were obtained, and the observed variations are discussed within the framework of Marcus theory, which can help in understanding how to optimize the design of molecular photocatalysts.
Introduction
The past decades have witnessed a tremendous development of efficient and selective light-driven catalytic processes, notably in the fields of synthetic photochemistry1 and solar fuels.2–5 When compared to electro- or thermo-catalytic procedures, a challenge inherent to photocatalysis is the synchronisation of the fast physical processes of photon absorption and generation of excited states (fs to ns timescale) with the slow chemical reactions involving multi-electron, multi-proton transfers and structural rearrangements necessary to perform catalysis. These two functions, light absorption and catalysis, can either be assumed by the same molecule, a ‘photocatalyst’, or by two distinct units: one capable of performing photo-induced redox processes (photoredox unit) activating a second catalytic unit for chemical transformations. Only a few catalysts behave as photocatalysts,6–10 as the requirements associated with the absorption of visible light and generation of excited states well-suited to undergo redox reactions limit their scope.11 Moreover, sustaining efficient light absorption throughout its different redox states may interfere with the multi-electron catalytic processes. These limits can be overcome with the addition of an appropriate chromophore. In photocatalytic systems where the functions of light absorption and catalysis are assumed by different units, the initial step consists in generating efficiently, that is in high yield, a long-lived charge-separated (CS) state between the two moieties via photo-induced electron transfer (ET).A family of complexes that well serves as a photoredox component is that of ruthenium polypyridines, widely used for their stability and tuneable properties.12 These complexes are employed either in bimolecular configurations,13,14 or in covalently linked chromophore–catalyst assemblies.12,15 The latter approach offers the possibility to go beyond the diffusion-limited processes which characterise the bimolecular approach and, at the same time, allows optimising the structural elements that influence the formation of a CS state which, within the framework of Marcus theory,16 is controlled by the driving force (−ΔG0), the reorganisation energy (λ), and the electronic coupling (HAB) between the two redox partners. In linked chromophore–catalyst systems, the electronic coupling depends on the nature of the donor and acceptor units as well as the bridge that connects those two moieties. Indeed, the bridge not only sets the distance between the donor and acceptor, but it also determines the electron transfer mechanism that operates between them, that is via tunnelling or hopping through the bridge.17–19 The molecular orbitals of the bridge, if significantly higher in energy than those of the donor and acceptor, do not participate directly in the process, but these represent virtual states determining the electronic coupling, HAB, between the two units: in this case, the electron transfer reaction proceeds via a super-exchange mechanism, and the ET rates exhibit an exponential dependence on the distance. On the other hand, if the molecular orbitals of the bridge involved in the electron transfer lie at similar energies as the donor and/or acceptor, the bridge participates directly in the electron transfer process which occurs via a hopping mechanism, less sensitive to the distance between the donor and the acceptor.20,21
In this work, we examine the impact of distance and driving force on the intramolecular electron transfer process in a family of covalently linked ruthenium chromophore–catalyst complexes. The aim is to evaluate the role of the intervening triazole-alkyl chain bridge, as well as the influence of the driving force, on the efficiency of the intramolecular ET, in an effort to go toward a mechanistically driven approach to photocatalysis. The complexes investigated, designed to perform the visible light activation of the catalytic unit,22 are shown in Fig. 1. The chromophore units are bis-heteroleptic ruthenium complexes bearing either bipyridine ligands (1 and 2), or modified bipyridines featuring four electron-withdrawing ethyl ester units on their periphery (3 and 4) having different lengths between their components. The catalyst, [FeII(L52)Cl]+, is an FeII ion wrapped by a pentadentate amine/pyridine ligand (L52) reported to perform the catalytic oxidation of organic substrates in the presence of oxygen-containing oxidants, and for which the spectroscopic signature of the active FeIV(O) intermediate has been reported.23,24 In a previous study involving 1-OH2, where the chloride ligand is substituted by a water molecule, we have shown that upon the absorption of visible light, the complex undergoes two sequential one-electron photo-oxidations of the iron catalyst leading to the formation of an active FeIV(O) species, able to perform oxygen atom transfer onto a substrate.22
 | ||
| Fig. 1 Structure of the compounds studied. | ||
As mentioned above, the bridge connecting the Ru-chromophore and the Fe-based catalyst is a triazole unit, resulting from “click” synthetic procedures,25 connected to a saturated alkyl chain containing either three (3C) or five (5C) –CH2– units. Ruthenium chromophores are well-suited for click procedures, with numerous examples being reported.26–34 The role of the triazole bridge in photo-induced and thermal electron transfer processes has been investigated in the literature.30,35 In the donor–acceptor systems examined, distinctive photo-induced electron transfer processes have been identified: hopping pathways resulting in fast charge separation;36–38 tunnelling pathways in Zn-porphyrin–fullerene dyads;39–41 but also cases where the photo-induced CS process was slow42 or blocked43,44 although thermodynamically feasible. An asymmetric role of the bridge with respect to photo-induced oxidation or reduction within the same system has also been reported.36
A successful method, to increase the driving force for the intramolecular electron transfer and stabilise it over time, is the flash-quench method which consists in quenching the excited state of the chromophore either by adding in excess amounts, or by grafting on it, an electron donor or acceptor, in order to generate either the reduced or oxidised state of the chromophore, respectively. This species is a better reductant (oxidant) than the excited state and is longer-lived because its disappearance relies on second-order charge recombination with the oxidised (reduced) quencher. Under such conditions (i.e. thermal electron transfer), electron transfer through the triazole bridge could be observed even when hindered in the excited state.26,27,34,41,45–47
Results and discussion
Electrochemical properties
Cyclic voltammetry studies have been performed on the reference catalyst compound [FeII(L52)Cl]+ and on dyads to obtain the driving force for possible electron transfer processes occurring between the chromophore and the catalyst. For [FeII(L52)Cl]+, the FeIII/FeII redox event is observed at E1/2 = 0.60 V vs. SCE in acetonitrile.22 Compounds 1 and 2 containing unmodified bipyridines on the chromophores present the same electrochemical profile (Fig. S1, ESI†): oxidation of FeII to FeIII appears at 0.58 V, while the RuIII/RuII redox process is observed at E1/2 = 1.25 V (1.24 V for 2), allowing for a driving force −ΔG0 = 0.66 (0.67) eV for intramolecular FeII oxidation by RuIII. For both compounds bearing ester bipyridine groups, 3 and 4, FeII oxidation occurs at E1/2 = 0.59 V, while the RuIII/RuII wave appears at E1/2 = 1.45 V, exhibiting a +0.20 V anodic shift as compared to the unmodified bipyridine analogues. This can be explained by the incorporation of four electron-withdrawing groups which induce a decrease in the electronic density at the metal centre. This results in an increased driving force (−ΔG0 = 0.86 eV) to perform FeII oxidation. Thus, we anticipate that the ester-containing dyads should be more efficient in generating the FeIII species.On the cathodic side, reduction waves are observed at −1.32 V vs. SCE for 1 and 2 and at −0.92 V for 3 and 4, respectively. These processes, assigned to the reduction of a bipyridine of the ruthenium chromophore ([RuII(bpy)2(bpy)˙−]+), will be short-handed as RuI in the following text. The reduction potential of the ruthenium excited state (Ru*) for the process Ru* → RuI, given by
| E1/2(Ru*/RuI) = E1/2(RuII/RuI) − E00(Ru*) |
| Complex | E 1/2(FeIII/FeII)/V | E 1/2(RuIII/RuII)/V | ΔG0/eV | E 1/2(RuII/RuI)/V |
|---|---|---|---|---|
| 1 | 0.58 | 1.25 | −0.67 | −1.32 |
| 2 | 0.58 | 1.24 | −0.66 | −1.32 |
| 3 | 0.59 | 1.45 | −0.86 | −0.92 |
| 4 | 0.59 | 1.45 | −0.86 | −0.92 |
Photophysical properties
The absorption spectrum of the FeII redox state of the [FeII(L52)Cl]+ catalyst exhibits a maximum at 400 nm (ε = 5000 M−1 cm−1), originating from an MLCT FeII → pyridine transition.22 Upon excitation at 355 nm, an emission spectrum (Fig. 2) with a maximum at 475 nm is observed at early times (10 ns) which broadens at longer times (100 ns), hinting at the presence of a shoulder at ∼530 nm. This suggests the presence of heterogeneity in the excited states, some possibly close in energy to the chromophore's ones. No transient absorption was observed, even at short times (Fig. S2, ESI†). The decay kinetics of the observed emission could not be resolved as it disappears within the time resolution of our apparatus; however, from the spectra evolution, it could be estimated to be ∼30 ns. Such lifetime, uncommonly long for FeII polypyridine complexes, has previously been observed for similar iron complexes for which high-spin (HS) FeII excited states, lower in energy than 1MLCT, are populated and deactivated via a transition to a low-spin (LS) state.49,50 Equilibration between different HS states has been observed to cause biphasic decays.50 | ||
Fig. 2 Evolution of the emission spectrum of the catalyst moiety in the H2O/CH3CN (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. Emission-gated spectra (gate: 10 ns) were obtained upon excitation with 355 nm laser flash (energy: 10 mJ) of an argon purged solution. :1) mixture. Emission-gated spectra (gate: 10 ns) were obtained upon excitation with 355 nm laser flash (energy: 10 mJ) of an argon purged solution. | ||
In the case of the chromophore–catalyst complexes, the absorption spectra (Fig. 3) exhibit a band at 290 nm (310 nm for the ester-bearing bipyridines), originating from the chromophore π–π* transition, and broad MLCT bands in the 400–500 nm region. In line with previously published results,51,52 absorption bands for the ester-bipyridines complexes are red-shifted by ∼20 nm. Such a shift is reflected in the emission spectra (Fig. S3, ESI†) where maxima are observed at 610 nm for complexes 1 and 2, and at 640 nm for 3 and 4. This evidences, in agreement with the electrochemical results, the presence of a low-lying LUMO in the latter group. No emission bands associated with the catalyst were observed because excitation light at 460 nm is only absorbed by the ruthenium moiety (ε[FeII(L52)Cl]+ < 1000 M−1 cm−1).
 | ||
| Fig. 3 Ground state absorption spectra for the complexes investigated in H2O/CH3CN (4:1). | ||
All the complexes studied exhibit biexponential emission decay kinetics (Fig. 4 and Table 2), sharing a long-living component (τ = 1100 ns), slightly longer than that of the reference [Ru(bpy)3]2+.48 This has previously been observed for ruthenium chromophores functionalised with the triazole attached via its carbon atom.27,47 Biexponential decay of emission, often observed for heterobimetallic complexes,51,53,54 may have several causes. It can be due to the presence of conformers with different efficiencies for quenching of the excited state by energy/electron transfer, or from the equilibration between close-lying excited states. It is worth noting that in the compounds studied, the bridge might present some flexibility due to the alkyl chain,55 allowing for different conformations, some possibly more prone to quenching. Regarding the possibility of having quenching by energy transfer, we notice that the relative amplitude of the fast emission phase is significantly lower for the ester-containing complexes, a fact that is tentatively attributed to the preferential localization of the electron on the ester-substituted bipyridine groups in the MLCT excited state (*Ru).52,53 Such fast phase is largest for complex 1 where the shortest distance between the Fe atom and the excited state on the Ru chromophore is expected, in particular if the latter is localised on the bipyridine with the attached triazole linker. These observations are consistent with a possible energy transfer occurring in the conformers presenting a short-lived emission. However, the energy levels of the catalysts should lie higher than, or close to, those of the chromophore, a fact implying a slow energy transfer rate and inefficient quenching. Another possible explanation for the observed short-lived component could be a reductive quenching of *RuII state by FeII, which, as previously discussed, is an exergonic reaction. This process, which should be mediated by a hole-transfer superexchange path between the acceptor LUMO and the HOMO bridge unit,21 has often been observed in dyads bearing a triazole bridge.35 Transient absorption spectra (Fig. 4) do not provide evidence for stable intermediates resulting from a photoinduced intramolecular reductive quenching of *RuII by FeII. The absence of a detectable fraction of a RuI state absorbing at 510 nm56 could, however, be explained with the recombination rate, for which ΔG0 = −0.7 eV, being faster than the CS rate, a scenario that prevents the observation of the transient CS state. With regard to driving force, the photoinduced reductive quenching of the ruthenium excited state should be favoured in ester-complexes 3 and 4 (ΔG0 ∼ −0.52 eV), which, however, exhibit longer lifetimes. In consequence, the occurrence of reductive quenching as an explanation for the biphasic emission decay is considered as unlikely and we privilege the alternative explanation for the observed biexponential decay that is the equilibration between excited states close in energy. The ordering of the multiple MLCT excited states might be altered in dyads.57 In particular, in agreement with the emission spectrum of [FeII(L52)Cl]+, the lower energy levels of the catalyst might be close to the higher MLCT levels of the chromophores, the more so for the unmodified-bipyridine.
 | ||
| Fig. 4 Emission kinetics at the emission maximum (left) and transient absorption spectra at 100 ns (right) after the laser pulse, for the different compounds in H2O/CH3CN (4:1). Optically matched absorption at 460 nm. Decays have been fitted with a biexponential curve (red traces). On the right, the amplitude of the absorption spectra, taken at 100 ns after the laser pulse, is consistent with the excited state lifetimes. | ||
| Complex | λ EM/nm | τ 1/ns | τ 2/ns |
|---|---|---|---|
| [FeII(L52)Cl]+ | 470, 530 | ∼30 | >30 |
| 1 | 610 | 1100 (39%) | 70 (61%) |
| 2 | 610 | 1100 (74%) | 50 (26%) |
| 3 | 650 | 1100 (81%) | 120 (19%) |
| 4 | 650 | 1150 (98%) | 100 (2%) |
To summarise, the photophysical studies have evidenced an heterogeneity of the excited states in these dyads with an excited state population having a short lifetime (∼100 ns). This population is smaller in the presence of a long alkyl linker (5C) and for the ester-substituted groups on the ruthenium chromophore, both modifications resulting in locating the MLCT state away from the catalyst, which possibly reduces the interaction between the energy levels of the chromophore and the catalyst. Moreover, the collected results show that although the formation of a RuI–FeIII CS state is thermodynamically accessible, it is not formed. Thus, we investigated the possibility to generate a long-lived FeIII species using an external electron acceptor.
Photoinduced electron transfer in the presence of an external electron acceptor
We have previously observed22 the intramolecular generation of FeIII in complex 1, using [Ru(NH3)5]2+, which is an optically transparent reversible electron acceptor.58 We employ here methyl viologen (MV2+) which is also able to oxidatively quench the chromophore (E1/2(MV2+/MV˙+) = −0.45 vs. SCE)59 in a bimolecular process creating a MV˙+ radical and a RuIII species which is then able to oxidise FeII. Reduction of MV2+ results in characteristic absorption bands at 400 nm (ε = 41800 M−1 cm−1) and 605 nm (ε = 13900 M−1 cm−1),60 whereas conversion of RuII into RuIII produces a depletion of absorption at 450 nm (Δε = −11300 M−1 cm−1).61 The optical features of the MV˙+ radical can thus be used to probe the efficiency of the initial bimolecular CS, to infer, using the kinetic evolution of the system, the subsequent steps, and to quantify the yields.
In the presence of 20 mM MV2+, the emission of the excited state was quenched in all the compounds examined, although with different rates (kET) and yields. Interestingly, the quenching rate was smaller than diffusion-limited, varying from kET ∼ 107 M−1 s−1 for the ester-containing complexes, to an order of magnitude higher with kET ∼ 3 × 108 M−1 s−1 for unmodified-bipyridine ones. This can be rationalised by the driving force values, which amounts to −ΔG0 ∼ 0.12 eV and 0.42 eV, respectively. With ΔG0 being certainly smaller than the reorganisation energy λ, the ET process is placed in the Marcus normal region. As such, the rate increases with −ΔG0 but it is smaller than the maximum rate (occurring at −ΔG0 = λ), which is, in the case of bimolecular reactions, limited by diffusion.
After the decay of the excited state, the presence of MV˙+ is attested by the appearance of peaks at 400 and 605 nm (Fig. S5, ESI†), while the bleaching at 450 (470, 480 for the esters) nm confirms the presence of the RuIII species.
To investigate the light-induced internal ET events, we measured and analysed absorption transients at the wavelengths characteristic for RuIII (450–480 nm) and the MV˙+ radical (605 nm) (Fig. 5). In all cases, we found the decay of the RuIII state to be faster than the decay of the reduced electron acceptor, evidencing that the reduction of RuIII by FeII is competitive with back ET. To obtain the rate constants describing the dynamics of the system, we fitted the experimental data shown in Fig. 5, using the target model given below, which includes the initial bimolecular quenching by MV2+ (kET), both intramolecular (kIET) and the final charge recombination between MV˙+ and either RuIII (kBET1) or FeIII (kBET2).
| Ru(II)–Fe(II) + MV2+ → Ru(III)–Fe(II) + MV˙+ kET |
| Ru(III)–Fe(II) → Ru(II)–Fe(III) kIET |
| Ru(III)–Fe(II) + MV˙+ → Ru(II)–Fe(II) + MV2+ kBET1 |
| Ru(II)–Fe(III) + MV˙+ → Ru(II)–Fe(II) + MV2+ kBET2 |
000 s−1) is obtained, for intramolecular FeIII generation in complex 3. Finally, the longer metal-to-metal distance in 4 qualitatively explains the smaller rate constant compared to 3. In an effort to rationalise the effect of driving force and distance on kIET, the rate of the intramolecular electron transfer was measured at different temperatures for compounds 1, 3 and 4 and the temperature dependence was analysed to obtain an estimation of the reorganisation energy λ and electron coupling HAB for the different compounds (cf. ESI†). The values obtained (Fig. S14, ESI†) are reported in Table 3.
 | ||
| Fig. 5 Differential transient absorption kinetics at selected wavelengths in the presence of 20 mM MV2+ for the compounds studied in the H2O/CH3CN (4:1) argon-purged solution at 293 K. The solutions excited at 460 nm using a laser energy of ∼10 mJ and absorption at the excitation wavelength of 0.40. | ||
| Complex | ΔG0/eV | λ/eV | H AB/meV | k IET/s−1 |
|---|---|---|---|---|
| 1 | −0.66 | 2.03 | 0.12 | 3100 |
| 2 | −0.67 | nd | nd | 620 |
| 3 | −0.86 | 2.39 | 0.25 | 11000 |
| 4 | −0.86 | 2.12 | 0.07 | 8300 |
Among the complexes investigated, the fastest and most efficient intramolecular electron transfer was observed for complex 3, which features a high oxidation potential of the Ru chromophore and the shorter Ru–Fe distance. The complex least efficient in generating FeIII appears to be 2, as the RuIII/RuII redox potential is lower and the Ru–Fe distance is longer. These results could be rationalised within the framework of Marcus theory for ET. The reorganisation energy λ was found to be high (>2 eV), explaining the rather slow intramolecular ET observed for all the dyads. Since for the prototypal chromophore [RuII(bpy)3]2+ no changes in the bonds occur upon reduction and λ is given only by the solvent contribution (λ ∼ 0.7 eV),62,63 we attribute the high reorganisation energy to significant changes in the bond lengths occurring on the Fe catalyst upon oxidation. This assignment is consistent with the spin state switch from high spin (S = 2) for [FeII(L52)Cl]+ and [FeII(L52)(OH2)]2+ (ref. 64) to low spin (S = 1/2) for [FeIII(L52)(OH)]+.22 Such a spin state change in this family of complexes is accompanied by a Fe–N bond decrease of ca. 0.2 Å.65 Although high, λ is quite similar among the different complexes, thus, it does not seem to be the reason for the differences observed among them. An increase of driving force, −ΔG0, by about 200 meV, due to the ester substitutions on the periphery of the Ru-chromophore, impacts instead the activation energy EA (cf. ESI†) and leads, as expected, to an increased rate kIET, by a factor of ca. 3.5 and 13 for the short and long variants, respectively. Finally, the main parameter describing the distance dependence of the ET rate, the electronic coupling HAB, was found to be strongly reduced (by at least a factor of 10, from ca. 0.20 meV to 0.07) by the introduction of two additional CH2 groups in the linker.
Conclusions
From the results obtained, some conclusions can be drawn on the role of the bridge, as well as of the driving force, on both photoinduced energy and electron transfer processes in the chromophore–catalyst complexes. The observation of two excited state populations can be explained either by the presence of two conformers, due to some flexibility of the linker, or with the equilibration of the excited state between two close-lying energy levels. A photo-induced reductive quenching of the chromophore is thermodynamically favourable and it could explain the short-lived component for one of the conformers. However, this explanation seems unlikely considering that the ester-modified bipyridine complexes 3 and 4, for which the driving force is considerably higher, exhibit the longest excited state lifetimes. Also, an energy transfer process seems improbable because the excited states of the catalyst lie higher in energy than that of the chromophore. We favour instead the presence of close-lying excited states that can equilibrate and this process is significantly reduced in the dyads with ester-modified bipyridines on the Ru-chromophore. Since the transient oxidation of FeII could not be observed via photo-induced intramolecular electron transfer, in order to generate the FeIII species required to advance toward the active catalyst species, an external electron acceptor was added in the solution. In this case, the intramolecular oxidation of FeII to FeIII was found to occur at different rates and with different yields depending on the driving force and the distance between the two metals. The triazole was not observed as an intermediate in the thermal intramolecular electron transfer process, consistent with the absence of a redox wave in the electrochemical measurements, confirming that the reaction takes place via tunnelling through the bridge. Among the parameters governing the electron transfer process, it was found that the reorganisation energy was very similar, while the electronic coupling HAB was an order of magnitude smaller for the bigger metal–metal distances, making the intramolecular electron transfer considerably slower. Another parameter having a strong influence on the rate was found to be the driving force −ΔG0.These results highlight the importance of the bridge and of ΔG0 when designing complexes for photocatalysis. Optimising the efficiency of intramolecular electron transfer as described in this work is necessary for an efficient photodriven catalysis. However, for multi-electronic transformations, a further challenge consists also in optimising efficient charge accumulation on the catalyst. This requires similar characterisation of photo-induced intramolecular ET to intermediate redox states of the catalyst, which might not only present different driving force and excited state quenching properties. More importantly, the bridge connecting the photosensitizer and the catalyst must be designed in a way to minimize deleterious charge annihilation due to fast ET from the photosensitizer excited state to the previously created oxidized catalyst species.66,67 Extension of the mechanistic studies presented here using double-flash excitation procedures can help disentangle the reaction sequences operating for different redox states of the catalyst and these studies are currently ongoing in our lab.
Experimental
Chemicals were purchased from Acros and used without further purification. All solvents except for dichloromethane, acetonitrile and methanol were purchased from Aldrich or VWR and were used as received. Dichloromethane and acetonitrile were distilled over CaH2, whereas methanol was distilled over Mg. NMR spectra were taken either on a Bruker AV 300 MHz or a Bruker 600 MHz spectrometer using the residual protonated solvent as an internal standard. Chemical shifts (δ) are given in parts per million (ppm) and coupling constants (J) are reported in hertz (Hz). Splitting patterns are designated as singlet (s), doublet (d), triplet (t), doublet of doublet (dd), and doublet of doublet of doublet (ddd). Splitting patterns that could not be interpreted or easily visualized are designated as multiplet (m). Electrospray mass spectra were recorded on a Thermo Scientific TSQ, or on a Bruker micrOTOFq mass spectrometer in the positive mode of detection (ESI+).Synthetic procedure
The synthetic procedure is shown in Fig. 6.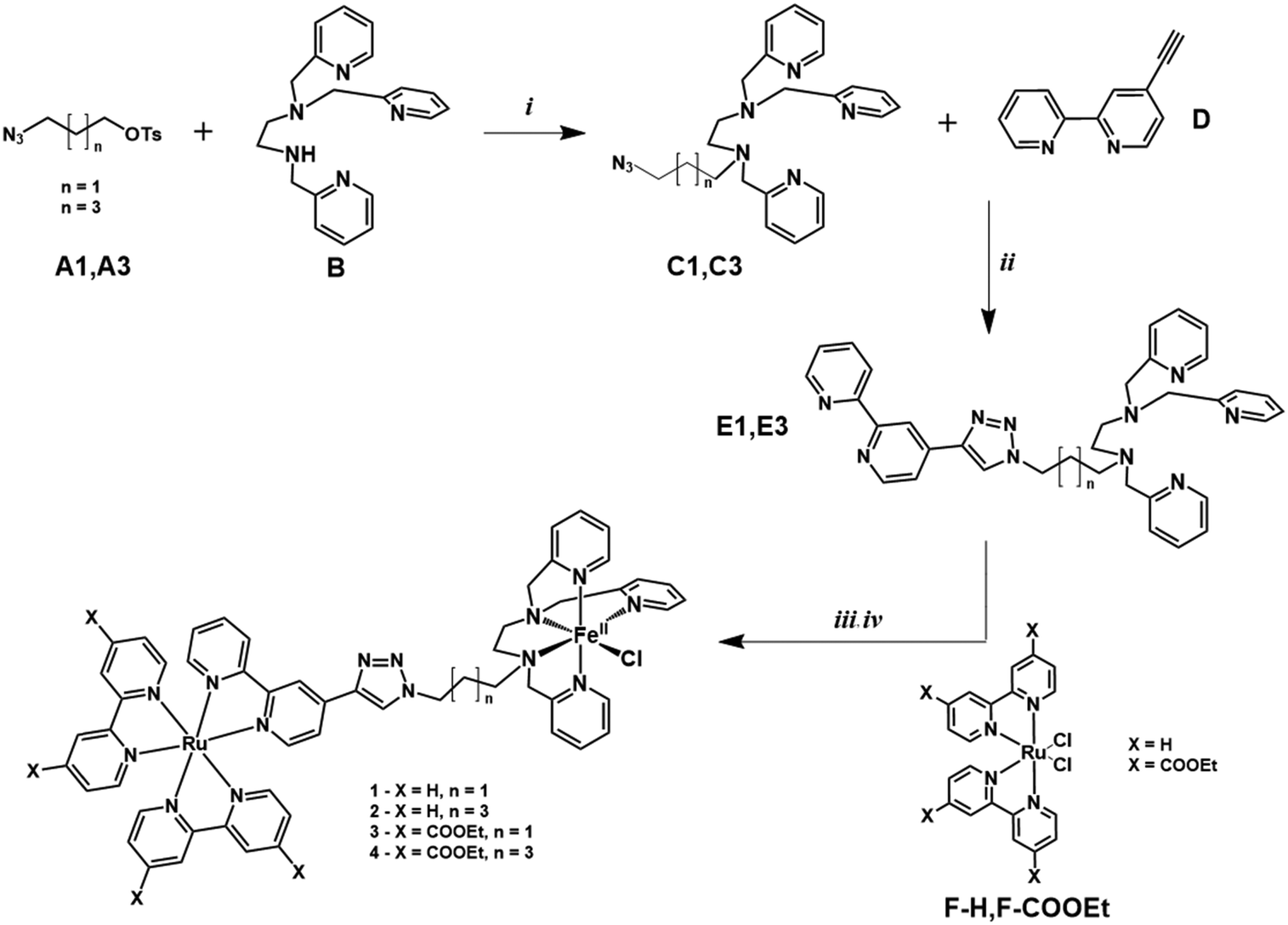 | ||
| Fig. 6 Synthetic procedure for complex Ru–FeCl (1-Cl). (i) CH3CN, K2CO3, 90 °C, 72 h (ii) CuSO4·5H2O, Na ascorbate, CH2Cl2:H2O (iii) Ru(bpy)2Cl2, AgNO3, MeOH, NaPF6 (iv) FeCl2 CH3CN:MeOH. C1,22D,27E1, F–H,51F-COOEt51 and 1 have been previously reported. | ||
:1 CH2Cl2/H2O). The solution was purged and degassed 3 times with argon and vacuum. To this solution were added CuSO4·5H2O (326 mg, 1.30 mmol, 1.50 eq.) and sodium ascorbate (522 mg, 2.61 mmol, 3 eq.). The mixture was allowed to react overnight under an argon atmosphere. At this time, the reaction mixture was transferred to a round bottom flask and 20 mL of a mixture containing 1.3 g of HEDTA and 0.6 g of NaOH was added, followed by mixing for one hour. The solution was extracted using CH2Cl2 and the product was purified via column chromatography (alumina. CH2Cl2 to CH2Cl2/2.5% MeOH) (227 mg, 41% yield). 1H NMR (300 MHz, CDCl3): δ 8.74 (d, 1H, J = 5.0 Hz, bpy-H); 8.68 (m, 2H, bpy-H); 8.49 (m, 4H, 3 Py-H, 1 bpy-H); 8.05 (s, 1H, tria-H); 7.95 (dd, 1H, J = 1.5, 5.0 Hz, bpy-H); 7.84 (td, 1H, J = 1.5, 7.8 Hz, bpy-H); 7.57 (m, 3H, pyr-H); 7.46 (d, J = 7.7 Hz, 2H, pyr-H); 7.34 (m, 2H, bpy-H); 7.30 (d, 1H, J = 5.5 Hz, Pyr-H); 7.11 (m, 3H, Py-H); 4.39 (t, 2H, J = 7.2 Hz, CH2-Trz); 3.80 (s, 4H, CH2-Pyr); 3.66 (s, 2H, CH2-Pyr), 2.70 (s, 4H, CH2-CH2); 2.43 (t, 2H, J = 7.2 Hz, N-CH2-CH2-CH2-CH2-CH2-Trz), 1.88 (q, 2H, J = 7.2 Hz, CH2-CH2-Trz); 1.49 (q, 2H, J = 7.2 Hz, CH2-CH2-CH2-CH2-Trz), 1.29 (q, 2H, J = 7.2 Hz, CH2-CH2-CH2-Trz); HRMS(ESI+) [M + H]+ = 625.3520. Calculated for C37H41N10 = 625.3510.
1H NMR (300 MHz, CD3OCD3): δ 8.99 (s broad, 2H, bpy-H); 8.95 (d, 1H, J = 8.1 Hz, bpy-H); 8.84 (d, 4H, J = 5.0 Hz, bpy-H); 8.55 (m, 3H, J = 8.0 Hz, Ru-Tz, Ru-bpy-H); 8.23 (m broad, 6H, bpy-H); 8.11 (q, 5H, J = 9 Hz, Trz-bpy-H, Ru-bpy-H); 7.91 (m, 4H Py-H); Ru-bpy, 7.59 (d, 5H, J = 8.0 Hz, bpy-H); 7.49 (broad, 4H, J = 7.6 Hz, bpy-H); 7.35 (7, 2H, Py-H); 4.35 (t, 2H, J = 7 Hz, CH2-Trz); 4.03 (broad, 2H, CH2-N); 3.79 (broad, 2H, CH2-CH2-N); 3.60 (s, 2H, N-CH2-CH2-N); 3.00 (broad, 2H, N-CH2-CH2-N); 2.91–2.84 (broad, 6H, CH2-Pyr); 2.32 (dd, 2H, CH2-CH2-CH2-Trz); 2.11 (m, 2H, CH2-CH2-Trz). HRMS (ESI+) [M + H]2+ = 645.1268. Calculated for C57H56F6N14PRu = 645.1276.
1H NMR (300 MHz, CD3OCD3) δ: 9.05 (s, 1H, bpy-H); 8.95 (dd, 2H, J = 5 Hz, 1 Hz, Ester-bpy-H); 8.83 (d, 5H, J = 7 Hz); 8.71 (s, 1H, Trz-H); 8.62 (d, 1H, J = 5 Hz, bpy-H); 8.20 (m, 6H-Pyr-H); 8.08 (m, 5H, bpy-H); 7.94 (m, 2H, bpy-H); 7.70 (m, 3H, pyr-H); 7.61 (m, 5H, Pyr-H); 7.48 (dd, 1H, J = 5 Hz, 1 Hz, bpy-H); 4.59 (t, 2H, J = 7.0 Hz, Trz-CH2-CH2-CH2-N); 4.48 (m, 10 H, J = 7 Hz, O-CH2CH3 and CH2); 3.82 (t, 2H, J = 5 Hz, CH2-Py); 3.62 (t, 2H, J = 5 Hz, CH2-Py); 3.42 (q broad, 2 H, J = 8 Hz); 2.55 (q, 2H, J = 7 Hz, Trz-CH2-CH2-CH2-N); 1.44 (t, 12H, J = 7 Hz, O-CH2CH3). HRMS(ESI+) [M + PF6]3+ = 775.1537. Calculated for C67H68 AgF6N14O8PRu = 775.1544.
1H NMR (300 MHz, CD3OCD3): δ 8.99 (m, 2H, Pyr-H); 8.83 (d, 5H, J = 8 Hz, Ester-bpy-H); 8.59 (s, 1H, Trz-H); 8.54 (s broad, 1H, bpy-H); 8.22 (t, 6H, H = 8 Hz, Ester bpy-H); 8.08 (q, 5H, J = 5 Hz, Ester-bpy-H); 7.87 (m, 3H, Pyr-H); 7.60 (broad, 4H, bpy-H); 7.48 (q, 4H, J = 8 Hz); 7.35 (q, 1H, J = 8 Hz, bpy-H); 4.48 (q, 8H, J = 7 Hz, O-CH2CH3); 4.34 (m, 2H); 4.06 (broad, 2H); 3.82 (broad, 2H); 3.61 (m, 2H); 2.83 (m, 8H, J = 7 Hz, 2 Hz, CH2-Py); 2.33 (broad, 2 H); 1.44 (t, 12H, J = 8 Hz, O-CH2-CH3); HRMS(ESI+) [M–CH3CH2 + PF6]3+ = 762.1382. Calculated for C65H64 AgF6N14O6PRu = 762.1361.
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Labex CHARMMMAT for a postdoctoral fellowship to CH as well as the ANR for financial support (project ANR Blanc Cathymetoxy). This work was also supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01, EU-COST Actions CM1003 and Perspect-H2O.References
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70 RSC.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- J. Grodkowski, T. Dhanasekaran, P. Neta, P. Hambright, B. S. Brunschwig, K. Shinozaki and E. Fujita, J. Phys. Chem. A, 2000, 104, 11332–11339 CrossRef CAS.
- S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734–5740 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- F. Kuttassery, S. Mathew, S. Sagawa, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemSusChem, 2017, 10, 1909–1915 CrossRef CAS PubMed.
- A. Y. Chan, A. Ghosh, J. T. Yarranton, J. Twilton, J. Jin, D. M. Arias-Rotondo, H. A. Sakai, J. K. McCusker and D. W. C. MacMillan, Science, 2023, 382, 191–197 CrossRef CAS PubMed.
- V. Balzani, P. Ceroni, A. Credi and M. Venturi, Coord. Chem. Rev., 2021, 433, 213758 CrossRef CAS.
- K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS.
- O. S. Wenger, Acc. Chem. Res., 2011, 44, 25–35 CrossRef CAS PubMed.
- M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051–5066 CrossRef CAS PubMed.
- A. Arrigo, A. Santoro, M. T. Indelli, M. Natali, F. Scandola and S. Campagna, Phys. Chem. Chem. Phys., 2014, 16, 818–826 RSC.
- M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC.
- C. Herrero, A. Quaranta, M. Sircoglou, K. Sénéchal-David, A. Baron, I. M. Marín, C. Buron, J.-P. Baltaze, W. Leibl, A. Aukauloo and F. Banse, Chem. Sci., 2015, 6, 2323–2327 RSC.
- V. Balland, M.-F. Charlot, F. Banse, J.-J. Girerd, T. A. Mattioli, E. Bill, J.-F. Bartoli, P. Battioni and D. Mansuy, Eur. J. Inorg. Chem., 2004, 301–308 CrossRef CAS.
- A. Bohn, C. Chinaux-Chaix, K. Cheaib, R. Guillot, C. Herrero, K. Sénéchal-David, J.-N. Rebilly and F. Banse, Dalton Trans., 2019, 48, 17045–17051 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- A. Baron, C. Herrero, A. Quaranta, M. F. Charlot, W. Leibl, B. Vauzeilles and A. Aukauloo, Chem. Commun., 2011, 47, 11011–11013 RSC.
- A. Baron, C. Herrero, A. Quaranta, M.-F. Charlot, W. Leibl, B. Vauzeilles and A. Aukauloo, Inorg. Chem., 2012, 51, 5985–5987 CrossRef CAS PubMed.
- K. P. Chitre, E. Guillén, A. S. Yoon and E. Galoppini, Eur. J. Inorg. Chem., 2012, 5461–5464 CrossRef CAS.
- C. Herrero, A. Quaranta, S. E. Ghachtouli, B. Vauzeilles, W. Leibl and A. Aukauloo, Phys. Chem. Chem. Phys., 2014, 16, 12067–12072 RSC.
- N. Zabarska, A. Stumper and S. Rau, Dalton Trans., 2016, 45, 2338–2351 RSC.
- S. Hohloch, D. Schweinfurth, M. G. Sommer, F. Weisser, N. Deibel, F. Ehret and B. Sarkar, Dalton Trans., 2014, 43, 4437–4450 RSC.
- M. Braumüller, M. Staniszewska, J. Guthmuller and S. Rau, Eur. J. Inorg. Chem., 2016, 4958–4963 CrossRef.
- M. Ramachandran, S. Anandan and M. Ashokkumar, New J. Chem., 2019, 43, 9832–9842 RSC.
- A. Charisiadis, E. Giannoudis, Z. Pournara, A. Kosma, V. Nikolaou, G. Charalambidis, V. Artero, M. Chavarot-Kerlidou and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2021, 1122–1129 CrossRef CAS.
- C. Müller, S. Bold, M. Chavarot-Kerlidou and B. Dietzek-Ivanšić, Coord. Chem. Rev., 2022, 472, 214764 CrossRef.
- M. Natali, M. Ravaglia, F. Scandola, J. Boixel, Y. Pellegrin, E. Blart and F. Odobel, J. Phys. Chem. C, 2013, 117, 19334–19345 CrossRef CAS.
- F. Zieschang, M. H. Schreck, A. Schmiedel, M. Holzapfel, J. H. Klein, C. Walter, B. Engels and C. Lambert, J. Phys. Chem. C, 2014, 118, 27698–27714 CrossRef CAS.
- K. Yoosaf, J. Iehl, I. Nierengarten, M. Hmadeh, A.-M. Albrecht-Gary, J.-F. Nierengarten and N. Armaroli, Chem. – Eur. J., 2014, 20, 223–231 CrossRef CAS PubMed.
- G. de Miguel, M. Wielopolski, D. I. Schuster, M. A. Fazio, O. P. Lee, C. K. Haley, A. L. Ortiz, L. Echegoyen, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2011, 133, 13036–13054 CrossRef CAS PubMed.
- H. Imahori, K. Tamaki, D. M. Guldi, C. P. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS PubMed.
- V. Nikolaou, F. Plass, A. Planchat, A. Charisiadis, G. Charalambidis, P. A. Angaridis, A. Kahnt, F. Odobel and A. G. Coutsolelos, Phys. Chem. Chem. Phys., 2018, 20, 24477–24489 RSC.
- A. Harriman, K. J. Elliott, M. A. H. Alamiry, L. Le Pleux, M. Severac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer and F. Odobel, J. Phys. Chem. C, 2009, 113, 5834–5842 CrossRef CAS.
- L. Zedler, P. Wintergerst, A. K. Mengele, C. Müller, C. Li, B. Dietzek-Ivanšić and S. Rau, Nat. Commun., 2022, 13, 2538 CrossRef CAS PubMed.
- S. Bold, T. Straistari, A. B. Muñoz-García, M. Pavone, V. Artero, M. Chavarot-Kerlidou and B. Dietzek, Catalysts, 2020, 10, 1340 CrossRef CAS.
- P. Farras, S. Maji, J. Benet-Buchholz and A. Llobet, Chem. – Eur. J., 2013, 19, 7162–7172 CrossRef CAS PubMed.
- S. Sheth, A. Baron, C. Herrero, B. Vauzeilles, A. Aukauloo and W. Leibl, Photochem. Photobiol. Sci., 2013, 12, 1074–1078 CrossRef CAS PubMed.
- P. Gotico, T.-T. Tran, A. Baron, B. Vauzeilles, C. Lefumeux, M.-H. Ha-Thi, T. Pino, Z. Halime, A. Quaranta, W. Leibl and A. Aukauloo, ChemPhotoChem, 2021, 5, 654–664 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- S. Schenker, P. C. Stein, J. A. Wolny, C. Brady, J. J. McGarvey, H. Toftlund and A. Hauser, Inorg. Chem., 2001, 40, 134–139 CrossRef CAS PubMed.
- A. Draksharapu, Q. Li, H. Logtenberg, T. A. van den Berg, A. Meetsma, J. S. Killeen, B. L. Feringa, R. Hage, G. Roelfes and W. R. Browne, Inorg. Chem., 2012, 51, 900–913 CrossRef CAS PubMed.
- C. Herrero, A. Quaranta, R.-A. Fallahpour, W. Leibl and A. Aukauloo, J. Phys. Chem. C, 2013, 117, 9605–9612 CrossRef CAS.
- A. G. Tebo, S. Das, R. Farran, C. Herrero, A. Quaranta, R. Fallahpour, S. Protti, M.-F. Charlot, A. Aukauloo and W. Leibl, C. R. Chim, 2017, 20, 323–332 CrossRef CAS.
- M. L. A. Abrahamsson, H. B. Baudin, A. Tran, C. Philouze, K. E. Berg, M. K. Raymond-Johansson, L. Sun, B. Åkermark, S. Styring and L. Hammarström, Inorg. Chem., 2002, 41, 1534–1544 CrossRef CAS PubMed.
- A. Johansson, M. Abrahamsson, A. Magnuson, P. Huang, J. Mårtensson, S. Styring, L. Hammarström, L. Sun and B. Åkermark, Inorg. Chem., 2003, 42, 7502–7511 CrossRef CAS PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–834 CrossRef CAS PubMed.
- C. P. Anderson, D. J. Salmon, T. J. Meyer and R. C. Young, J. Am. Chem. Soc., 1977, 99, 1980–1982 CrossRef CAS.
- L. Flamigni, S. Encinas, F. Barigelletti, F. M. MacDonnell, K.-J. Kim, F. Puntoriero and S. Campagna, Chem. Commun., 2000, 1185–1186 RSC.
- J. R. Pladziewicz, J. A. Broomhead and H. Taube, Inorg. Chem., 1973, 12, 639–643 CrossRef CAS.
- J. C. Luong, L. Nadjo and M. S. Wrighton, J. Am. Chem. Soc., 1978, 100, 5790–5795 CrossRef CAS.
- T. Watanabe and K. Honda, J. Phys. Chem., 1982, 86, 2617–2619 CrossRef CAS.
- P. Mueller and K. Brettel, Photochem. Photobiol. Sci., 2012, 11, 632–636 CrossRef CAS PubMed.
- P. Siders and R. A. Marcus, J. Am. Chem. Soc., 1981, 103, 741–747 CrossRef CAS.
- E. J. Piechota and G. J. Meyer, J. Chem. Educ., 2019, 96, 2450–2466 CrossRef CAS.
- P. Mialane, A. Nivorojkine, G. Pratviel, L. Azéma, M. Slany, F. Godde, A. Simaan, F. Banse, T. Kargar-Grisel, G. Bouchoux, J. Sainton, O. Horner, J. Guilhem, L. Tchertanova, B. Meunier and J.-J. Girerd, Inorg. Chem., 1999, 38, 1085–1092 CrossRef CAS PubMed.
- N. Ségaud, J.-N. Rebilly, K. Sénéchal-David, R. Guillot, L. Billon, J.-P. Baltaze, J. Farjon, O. Reinaud and F. Banse, Inorg. Chem., 2013, 52, 691–700 CrossRef PubMed.
- M.-H. Ha-Thi, V.-T. Pham, T. Pino, V. Maslova, A. Quaranta, C. Lefumeux, W. Leibl and A. Aukauloo, Photochem. Photobiol. Sci., 2018, 17, 903–909 CrossRef CAS PubMed.
- P. Gotico, A. DelVecchio, D. Audisio, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, ChemPhotoChem, 2018, 2, 715–719 CrossRef CAS.
- V. Fourmond, Anal. Chem., 2016, 88, 5050–5052 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02755h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |