
MnO2 nanoparticles supported on graphitic carbon nitride as an electrocatalyst for oxygen reduction and evolution
Aušrinė
Zabielaitė
 *,
Virginija
Kepenienė
*,
Dijana
Šimkūnaitė
,
Raminta
Stagniūnaitė
,
Vitalija
Jasulaitienė
,
Giedrius
Stalnionis
,
Jūratė
Vaičiūnienė
,
Loreta
Tamašauskaitė-Tamašiūnaitė
and
Eugenijus
Norkus
*,
Virginija
Kepenienė
*,
Dijana
Šimkūnaitė
,
Raminta
Stagniūnaitė
,
Vitalija
Jasulaitienė
,
Giedrius
Stalnionis
,
Jūratė
Vaičiūnienė
,
Loreta
Tamašauskaitė-Tamašiūnaitė
and
Eugenijus
Norkus
Department of Catalysis, Center for Physical Sciences and Technology, Saulėtekio Ave. 3, Vilnius, LT-10257, Lithuania. E-mail: virginija.kepeniene@ftmc.lt; ausrine.zabielaite@ftmc.lt; Fax: +370 52649774; Tel: +370 52648845
First published on 6th November 2024
Abstract
The aim of this study is to present a straightforward methodology for the preparation of non-precious metal catalysts comprising MnO2 and carbonaceous materials, namely graphite powder (C), graphitic carbon nitride (gCN), and graphitic carbon nitride/graphite powder (gCN/C) substrates. The morphology and composition of the prepared MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts have been investigated using X-ray diffraction (XRD), field-emission scanning electron microscopy (FE-SEM), X-ray photoelectron spectroscopy (XPS), and inductively coupled plasma optical emission spectroscopy (ICP-OES). The electrochemical performance of the prepared MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts has been investigated for the oxygen reduction reaction (ORR) and oxygen evolution (OER) reaction using cyclic and linear voltammetry. All of the investigated catalysts exhibited enhanced electrocatalytic activity with regard to the ORR and OER processes when compared with the bare substrates. The MnO2–gCN/C catalyst was found to be the most efficient catalyst for both investigated reactions when compared with MnO2/C and MnO2–gCN. The MnO2–gCN/C catalyst demonstrated the most positive ORR onset potential of 0.9 V and the most negative OER onset potential of 1.53 V. Furthermore, it demonstrated remarkable stability, retaining approximately 85% of its initial signal after a continuous test of 24 hours in both long-term ORR and OER processes.
Introduction
Energy is essential in our technology-driven age. However, the majority of energy production is currently based on fossil fuels, which emit carbon dioxide (CO2), the main greenhouse gas linked with global warming and climate change. Finding alternative renewable, affordable and sustainable energy sources is therefore the greatest challenge of this century, leading to the rapid development of advanced sustainable energy technologies.1–3 Among these, the photo-/electrochemical energy, which is generated and stored in an environmentally friendly manner using devices such as fuel cells, metal–air batteries and photo-electrochemical cells, has attracted attention due to its cost-effectiveness, reliability, availability and safety. All of these systems involve electrocatalytic reactions such as the hydrogen evolution reaction (HER), the oxygen evolution reaction (OER) or the oxygen reduction reaction (ORR), and/or photo-catalytic water splitting.4–9 All these reactions are in need of efficient HER/OER electrocatalysts to reduce the activation energy and overpotentials.4,9,10 Noble metals, including Pt or Pt-group materials (such as Pt, Pd, Ru, Ir, and Rh), have been the most widely studied and are currently considered to be the state-of-the-art catalysts for water splitting.11–16 Given the impressively high cost of precious metals and their scarcity in nature, significant progress has been made in the search for alternative, cost-effective and innovative substitutes. Evidence of this is seen in a rapidly increasing number of publications on earth-abundant transition/noble metal-free (TMs, M = Co, Ni, Fe, Mn, Mo) and TM-based alloys or non-metallic (TMXs, X = N, O, S, C, P, etc.) compounds, aimed to function as active HER/OER electrocatalysts.17–21 TMs have received a lot of attention due to their distinctive structural features, abundant active sites, tunable electronic properties, compositions and ease of application for large-scale production. Co, Ni and Fe are typically characterized as the most powerful materials for water splitting.22–25 Meanwhile, MnO2, due to its structural flexibility, excellent photostability, non-toxicity, wider solar irradiation utilization capacity and, most of all, narrower band gap, has attracted enormous attention as a visible-light active oxygen evolution photocatalyst.26 MnO2, with its high theoretical capacitance, low cost, good redox behaviour and environmentally friendly nature, has proved successful as an electrode material for pseudo capacitors as well as batteries.27–29 Its rich crystal structure in terms of δ, α, γ and β phases, morphological diversity and multiple surface valence states are of great importance for the efficient catalysis of oxygen-related reactions.30–33 However, intrinsically poor electrical conductivity of MnO2 is a limitation to its widespread commercial applications.34 In addition, MnO2 often suffers from particle agglomeration during electrode preparation. It can also undergo volume expansion during the charge/discharge process. This reduces its electrocatalytic activity for oxygen-related responses. Therefore, the formation of binary or ternary composites by integrating MnO2 with highly conductive materials and selecting suitable supports is crucial for obtaining stable and catalytically active catalysts. So far, many materials, including metal oxides, organic polymers,35 porous materials,36 carbon-based materials,37–39etc., have been broadly investigated to stabilize metal nanoparticles and to boost oxygen-related reactions. It is a proven strategy that increasing the surface area and creating porous structures with more active sites optimize the catalyst structure for more efficient interfacial reactions, mass transfer and adsorption of oxygen-based species, all of which dictate faster oxygen reduction and evolution.39 In addition, the use of inherently conductive carbon-based structures facilitates the electron transfer steps and significantly enhances the sluggish kinetics of the ORR and OER. Moreover, heteroatom dopants (N, P, S or B) coupled with the transition metal in the carbon/carbon composite can help uniformly disperse active sites in the carbon frameworks and accelerate charge transfer rates. For example, coupling α-MnO2 with N-doped graphite nanofibers (N-GNFs) resulted in improved conductivity and increased surface areas of α-MnO2/N-GNFs.40 Due to enhanced ORR and OER activities, this combination showed better rechargeability. S-doped graphitized carbon (S-GC) with MnOx (MnOx/S-GC) exhibited much higher ORR and OER activities than S-GC owing to their synergistic interactions, demonstrating the active coupling effect between MnOx and S-GC.41 However, their catalytic performance and stability over a wide range of conditions do not yet fully meet the requirements for practical use.Recently, g-C3N4 has been widely used as a base carrier for the deposition of nanoparticles of various metals (Ni, Co, Mn, Cu, Fe, etc.) and their oxides. g-C3N4 has excellent properties such as high bulk modulus, good thermal conductivity, small mechanical friction coefficient, high elasticity, and chemical inertness. Moreover, g-C3N4 is also a very promising material that can replace the commonly used carbon for the production of catalysts due to its high nitrogen (N) content. Among the many carbon-based materials, g-C3N4 has great potential in various photocatalytic and electrocatalytic applications due to its tunable physicochemical and electrical properties, as well as its non-toxicity, metal-free nature, visible light optical response, environmentally friendly nature, biocompatibility, etc.42–56 Moreover, the production of g-C3N4 is cheap and does not involve high cost. Polymeric g-C3N4, as a non-metallic semiconductor, fully satisfies the thermodynamic requirements for photocatalytic water splitting and has become a major focus of research in recent years.57 However, efficient photocatalytic total water splitting via g-C3N4 still faces serious limitations due to the sluggish transfer of the photogenerated carriers and fast recombination rate.58 Therefore, to accelerate the kinetics of the photogenerated carrier transfer, new materials and structures have been developed by incorporating g-C3N4 with other semiconductors, in particular by constructing a Z-scheme system.26,59,60 Z. Mo et al.59 proposed an efficient Z-scheme system consisting of 2D MnO2/monolayer g-C3N4 with defective Mn3+ active sites for overall water splitting. These defective Mn3+ active sites were able to enhance H2O adsorption and optimize the interfacial charge separation/transfer during the photocatalytic process by introducing the Mn3+/Mn4+ redox couple for excellent overall water splitting performance. In ref. 26, the oxygen vacancy rich α-MnO2@B/O-g-C3N4 photocatalyst was prepared by embedding 1D α-MnO2in situ over the 2D BCN matrix for photocatalytic O2 and H2 evolution. The as-synthesized photocatalysts exhibited better photocatalytic performance than their counterparts of pristine materials. Among the synthesized photocatalysts, MBOCN-20 exhibited O2 and H2 evolution rates of 295.1 and 560.1 μmol h−1, respectively.
However, for electrochemical applications, the optimal use of g-C3N4 also requires the improvement of its poor conductivity and low catalytic activity, which can be increased in several ways: physically mixing g-C3N4 with conductive carbon materials,61 immobilizing g-C3N4 on carbon bases (carriers)62,63 or depositing metal nanoparticles using microwave-assisted processes, hydrothermal and solvothermal syntheses routes, sol–gel processes, chemical reduction, etc.51,56,64 Although MnO2 based g-C3N4 materials have great potential for oxygen-related photocatalytic reactions, they have received significantly less attention for their use in electrochemical reactions. Recently, a novel nanocomposite, namely one-dimensional (1D) MnO2 nanowires grown in situ within 2D mesoporous carbon nitride (MnO2@mpg-C3N4), has been shown to be a highly efficient electrocatalyst for the OER.65 The outstanding OER performance of MnO2@mpg-C3N4 was attributed to the effective stabilization of Mn3+ species (Mn2O3) in the electrocatalyst via the nitrogen functional groups of mpg-C3N4 and the formation of a 3D heterostructure, which resulted in improved aerophobicity due to orientation modifications of the growing 1D MnO2 nanowires, an open structure facilitating the rapid detachment of gas bubbles from the electrode surface, and a large number of transport channels for the penetration of the electrolyte, ions and electrons. However, there is a lack of research on MnO2-based g-C3N4 materials supported on or mixed with graphitic carbon, operating as ternary electrocatalysts in electrochemical reactions for green energy production. Therefore, it is important to investigate their potential in this area, especially given that the incorporation of graphitic carbon components is essential in improving the transport efficiency of photogenerated charge carriers in g-C3N4-based heterostructures.66 Moreover, carbon is notable for its good conductivity, substantial surface area with numerous active sites, and stability in various environments. It prevents catalyst aggregation and provides synergistic effects when combined with g-C3N4, leading to improved performance.67–69
This paper presents a straightforward, one-step approach for preparing an efficient ternary electrocatalyst, MnO2 supported on graphitic carbon nitride (gCN), and its mixture with graphite powder (C), exhibiting enhanced characteristics for both the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). The advantages of combining gCN with C and MnO2 are highlighted in the paper.
Experimental
Chemicals
Melamine (99%), KMnO4 (99%), graphite powder (99.9995%), ethanol (C2H5OH, 96%), and 5 wt% Nafion were purchased from Sigma-Aldrich and used as received without further purification. Ultra-pure water with a resistivity of 18.2 MΩ cm−1 was used to prepare all the solutions. All the reagents used were of analytical grade.Fabrication of catalysts
Melamine was subjected to a 4-hour annealing process at 520 °C in order to obtain gCN. The precursor was placed in a closed high-alumina crucible and heated to temperature with a rate of 5 °C min−1. After the synthesis, it was ground into fine powder.The pure MnO2 powder was prepared using a microwave-assisted method. In a typical experiment, 0.063 g of KMnO4 and 0.1 g of MnSO4·H2O were dispersed in 20 ml of ultra-pure water and sonicated for 30 minutes. Then, the reaction mixture was put into a microwave reactor Monowave 300 (Anton Paar). The synthesis was conducted at a temperature of 150 °C for a period of five minutes. The precipitate obtained was filtered out, washed with ultra-pure water, and subsequently dried in a vacuum oven at 80 °C for a period of two hours. The preparation process of pure MnO2 is illustrated using eqn (1):70
| 2KMnO4 + 3MnSO4 + 2H2O → 5MnO2 + 2H2SO4 + K2SO4 | (1) |
Then, the obtained MnO2 powder was mixed with graphite powder, gCN, and gCN/C substrates in a ratio of 80 wt% MnO2 and 20 wt% of carbonaceous substrates, as illustrated in Table 1.
| Catalyst | Material, wt% | ||
|---|---|---|---|
| MnO2 | gCN | C | |
| MnO2/C | 80 | — | 20 |
| MnO2–gCN | 80 | 20 | — |
| MnO2–gCN/C | 80 | 10 | 10 |
Characterization of catalysts
The surface morphology of the prepared catalysts was characterized using a SEM/FIB workstation Helios Nanolab 650 with an energy dispersive X-ray (EDX) spectrometer INCA Energy 350 X-Max 20.X-ray diffraction (XRD) patterns of the studied powders were obtained using an X-ray diffractometer (D2 PHASER, Bruker, Karlsruhe, Germany) equipped with an LYNXEYE XE-T detector with an axial Soller slit of 2.3° and air-scatter-screen. The energy resolution of the instrument was 380 eV or less. The instrument is equipped with a Cu anode X-ray source. X-ray generation: 30 kV/10 mA; accuracy: ±0.02° 2θ over the whole angular range. The measurements were conducted in the 2θ range of 10–90° with a step size of 0.05° 2θ.
The surface elemental composition of the synthesized MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts was determined employing X-ray photoelectron spectroscopy (XPS) using a Kratos AXIS Supra+ spectrometer (Thermo Scientific, East Grinstead, UK) with monochromatic Al Kα (1486.6 eV) operated at a fixed pass energy of 20 eV.
Electrochemical measurements
The electrochemical experiments were conducted using a PGSTAT100 potentiostat/galvanostat (Metrohm Autolab B.V., Utrecht, The Netherlands) and a three-electrode standard cell, wherein the working electrode was a glassy carbon (GC) electrode that had been modified with the synthesized catalyst's ink. The geometric surface area of the GC electrode was 0.196 cm2. An Ag/AgCl (3 M KCl) electrode and GC rod were employed as the reference and counter electrodes, respectively. The catalyst ink was prepared by combining 20 mg of the MnO2/C, MnO2–gCN, or MnO2–gCN/C catalysts with 3.96 ml of C2H5OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1 vol) and 40 μl of 5 wt% Nafion. The obtained mixtures of different catalysts were subjected to sonication for a period of 24 hours. Subsequently, 10 μl of each catalyst ink was pipetted onto the GC electrode and allowed to dry for a period of two hours at room temperature. The loading of the prepared catalysts was 254.7 μg cm−2.
:H2O (1:1 vol) and 40 μl of 5 wt% Nafion. The obtained mixtures of different catalysts were subjected to sonication for a period of 24 hours. Subsequently, 10 μl of each catalyst ink was pipetted onto the GC electrode and allowed to dry for a period of two hours at room temperature. The loading of the prepared catalysts was 254.7 μg cm−2.
Linear sweep voltammograms (LSVs) were recorded in an O2-saturated 0.1 M KOH solution for the ORR at a scan rate of 10 mV s−1 and N2-saturated 1 M KOH for the OER at a scan rate of 2 mV s−1. Chronoamperometric analysis (CA) was performed in an O2-saturated 0.1 M KOH solution at 0.55 V for the ORR and in N2-saturated 1 M KOH solution at an overpotential of 1.62 V for the OER for a period of 24 hours. All reported potentials were referenced with respect to the reversible hydrogen electrode (RHE) in accordance with eqn (2):
| ERHE = Emeasured + 0.059·pH + EAg/AgCl(3MKCl) | (2) |
KCl) = 0.210 V. The measured current densities were normalized to the geometric area of the GC electrode.
The number of electrons transferred per O2 molecule (n) was calculated using the Koutecky–Levich (K–L) eqn (3)–(5):71
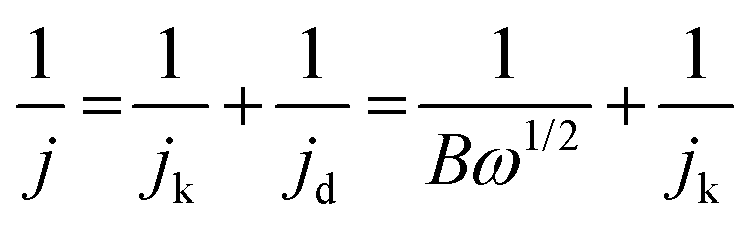 | (3) |
| B = 0.62nFC0D02/3v−1/6 | (4) |
| jk = nFkC0 | (5) |
485 C mol−1), C0 is the concentration of oxygen in bulk (1.2 × 10−6 mol cm−3), D0 is the diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), ν is the kinematic viscosity of the solution (0.01 cm2 s−1), and ω (rad s−1) is the rotation rate of the electrode.72
Results and discussion
Herein, we present a straightforward method for the synthesis of an efficient non-precious ternary electrocatalyst, MnO2 supported on gCN, and its mixture with C, which exhibits enhanced characteristics for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). The synthesis of MnO2 was achieved through the application of microwave-assisted heating to a mixture of KMnO4 and MnSO4. Fig. 1 depicts the SEM images of the MnO2 deposits on the C (a), gCN (b), and gCN/C (c) substrates. These images illustrate that the MnO2 deposits on the C, gCN and gCN/C substrates assume the form of needles, which coalesce to form a network-like pattern (Fig. 1a–c). The images also reveal that MnO2/C exhibits a distinct morphology of needles across the entire surface, with no discernible presence of empty spaces on the surface (Fig. 1a). In the case of MnO2–gCN, a distinct surface layout is observed but with the same needles network-like pattern (Fig. 1b). In contrast, in the case of MnO2–gCN/C, a large porous three-dimensional structure with developed empty voids inside is formed (Fig. 1c). Concurrently, the generation of a three-dimensional structure with the deepest volumetric cavities is observed when MnO2 is deposited on the mixed gCN/C substrate, resulting in a markedly increased surface area. This is particularly favourable for enhancing active site availability and accessibility, guiding charge mobility and transport direction through the creation of transfer channels for electro-active species. It is clearly evident that the deposition of MnO2 on the mixed gCN/C substrate results in the formation of a larger surface area than that observed in the deposition of MnO2 on the individual C and gCN components. | ||
| Fig. 1 SEM views of MnO2 supported on C (a), gCN (b), and gCN/C (c) substrates. | ||
The XRD pattern of the as-prepared gCN exhibited a typical pattern, with two pronounced peaks centered approximately at 12.7° and 27.3°, which may be assigned to the (100) and (002) planes of the trigonal N bond of tri-s-triazine and the layered packing of conjugated aromatic units in g-C3N4, respectively (Fig. 2a).73–75 The XRD pattern for the as-synthesized MnO2 sample is shown in Fig. 2b. The symbols indicated positions of the XRD peaks of α-MnO2 (COD 9016667), indicating a tetragonal unit cell with lattice parameters of a = b = 9.815 Å and c = 2.847 Å. The presence of broad peaks implied that the synthesized α-MnO2 is essentially a mixture of amorphous and nanocrystalline phases. The peaks at 2θ° values of 12.74°, 18.06°, 28.74°, 37.63°, 42.04°, 49.89°, 56.19°, 60.24°, and 65.52° correspond to the crystal planes of (110), (200), (310), (211), (301), (411), (600), (521), and (002), respectively of the α-MnO2. In addition, the synthesized powder is composed of small crystallites with an average size of approximately 13–20 nm.
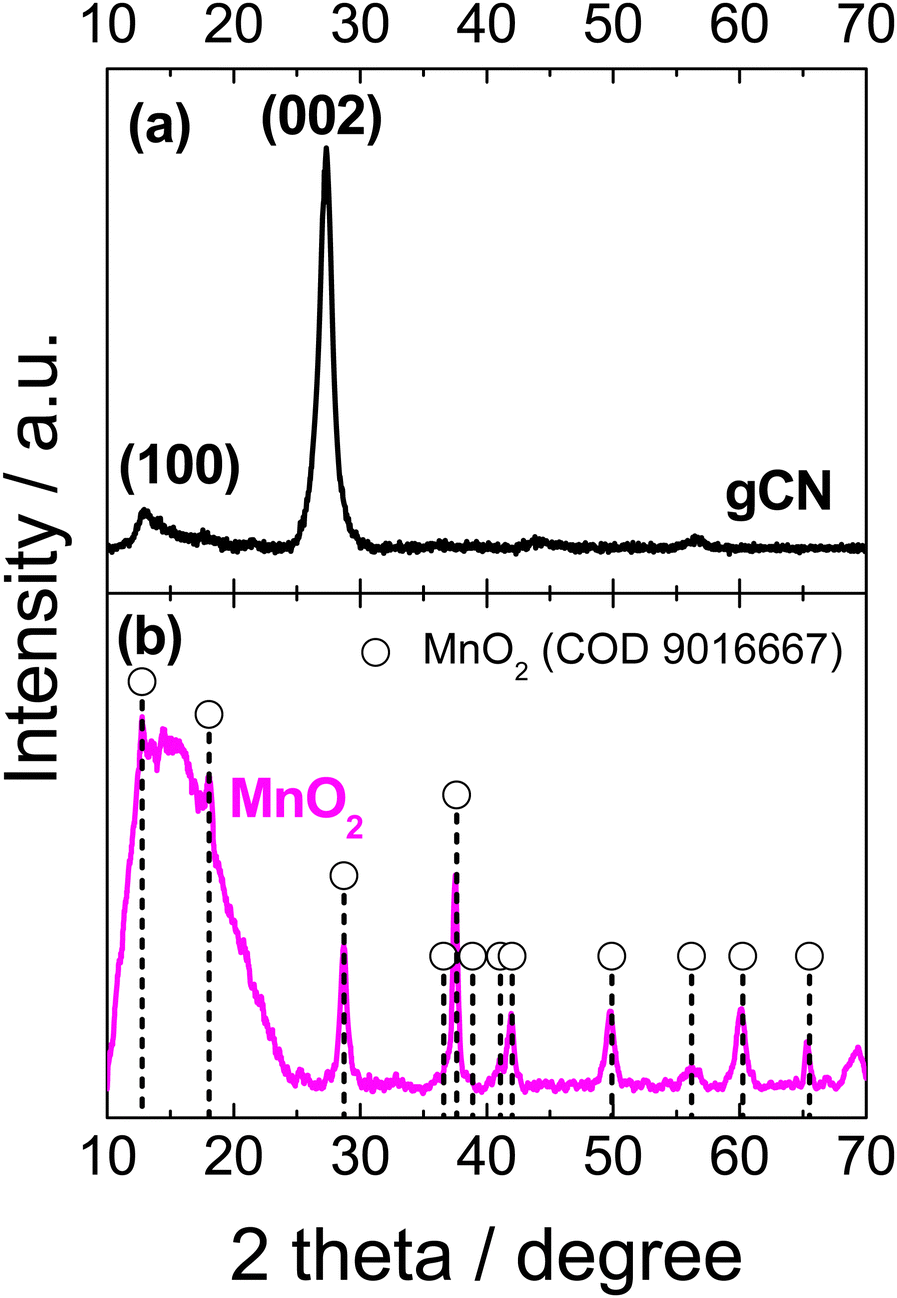 | ||
| Fig. 2 XRD patterns of the prepared gCN (a) and MnO2 (b) catalysts. Peak positions are indicated according to the Crystallography Open Database COD data cards: MnO2 – 9016667 (b). | ||
The XPS study yielded results that were consistent with those of the XRD analysis. The C 1s, Mn 2p, and O 1s peaks are clearly observed in the XPS survey spectra obtained for all MnO2/C, MnO2–gCN, and MnO2–gCN/C samples as well as N 1s signals for MnO2–gCN and MnO2–gCN/C samples. This indicates the successful synthesis of MnO2 on substrates. Furthermore, the XPS survey spectra reveal the presence of two peaks at binding energies (Eb) of 292 and 377 eV, which can be attributed to K 2p and K 2s, respectively (Fig. 3).
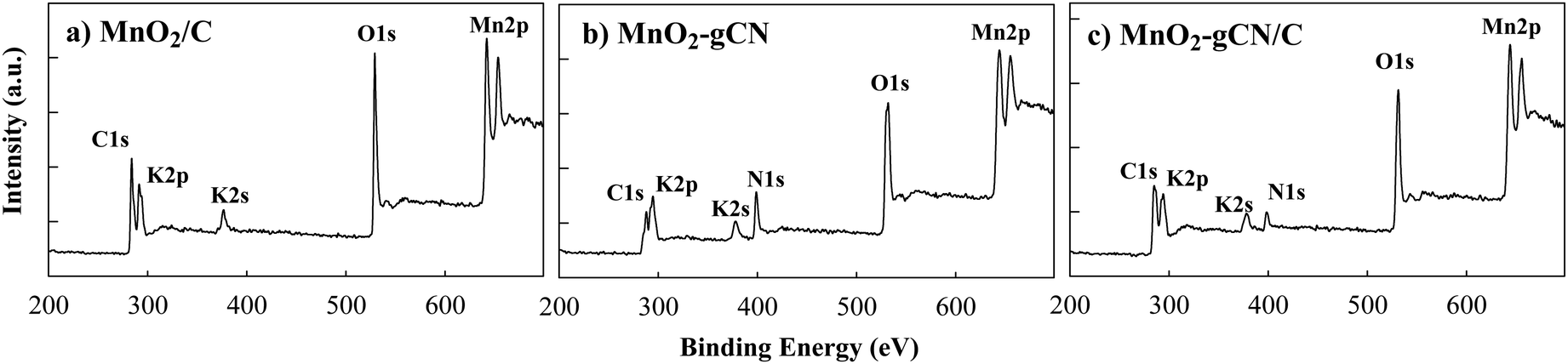 | ||
| Fig. 3 XPS survey spectra of MnO2/C (a), MnO2–gCN (b), and MnO2–gCN/C (c) catalysts. | ||
As illustrated in Fig. 4, the Mn 2p core-level spectra exhibit a spin–orbit doublet comprising the main Mn 2p3/2 and Mn 2p1/2 peaks. These are located at 641.8–645.3 eV and 653.0–657.2 eV for all investigated samples, respectively. The spin-energy separation is 11.5 ± 0.2 eV, confirming the presence of MnO2.
 | ||
| Fig. 4 High-resolution XPS spectra of Mn 2p (a)–(c), O 1s (d)–(f), C 1s (g)–(i), and N 1s (j) and (k) for MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts. | ||
The deconvoluted Mn 2p3/2 peaks at 641.8–644.0 eV, obtained from the splitting of the XPS Mn 2p spectra, correspond to the Mn(IV) oxidation state and indicate the successful formation of MnO2 during the synthesis as evidenced by the presence of the characteristic peaks in the spectra. These findings are in accordance with previous studies.76–79 The XPS Mn 2p spectra exhibited peaks at approximately 645.6 eV for all samples, indicating the presence of Mn(VII) species in the form of permanganate.80 This also provides an explanation for the presence of K 2p and K 2s peaks in the survey of the investigated samples (Fig. 3). It is conceivable that not all of the initial KMnO4 was oxidized to MnO2; further investigation may be required to ascertain this. The O 1s spectra were deconvoluted into four peaks centered at 529.2, 530.1, 531.0, and 532.4 eV for MnO2/C (Fig. 4d), 529.5, 531.3, 532.3, and 533.4 eV for MnO2–gCN (Fig. 4e), and 530.0, 531.3, 532.2, and 533.3 eV for MnO2–gCN/C (Fig. 4f). Typically, the peaks with a lower binding energy of 529–531 eV indicate the presence of lattice oxygen species (Oα), which correspond to metal–O bonds. These data corroborate the formation of MnO2 as well. Peaks with a higher Eb of 532–533 eV are indicative of oxygen chemisorbed by the surface (Oβ).80–82
In the case of the MnO2/C catalyst, the high-resolution C 1s spectrum was deconvoluted into three peaks centered at Eb values of 284.0, 284.8 and 286.0 eV (Fig. 4g). The peak at 284.0 eV can be assigned to carbon atoms with a C–C bond, while other peaks at higher binding energies can be assigned to oxygen-functionalized carbon atoms, such as C–O or C–OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.83–85 Upon combining the MnO2 with gCN and gCN/C substrates, the obtained C 1s spectra resolved into several peaks centered at around 284.8, 286.3, 288.2, and 289.3 eV for MnO2–gCN and 284.8, 285.9, 287.0 and 287.8 eV for MnO2–gCN/C, indicating that the existence of the g–C3N4 phase corresponded to adventitious sp2 C–C carbon species (284.42 eV), sp2 hybridized carbon atoms in N–C–N (286.38 eV), and sp2 CN bond (287.36 eV) in the s-triazine ring, respectively.86–89 In turn, the N 1s signals for these two catalysts are located in the binding energy region of 397–401 eV and can be fitted into three peaks located at 398.2 eV, 399.5 eV, and 400.7 eV for sp2-hybridized nitrogen in triazine rings (C–NC), tertiary nitrogen N–(C)3 groups, and amino functions with hydrogen (C–N–H), respectively.86,88,89
O.83–85 Upon combining the MnO2 with gCN and gCN/C substrates, the obtained C 1s spectra resolved into several peaks centered at around 284.8, 286.3, 288.2, and 289.3 eV for MnO2–gCN and 284.8, 285.9, 287.0 and 287.8 eV for MnO2–gCN/C, indicating that the existence of the g–C3N4 phase corresponded to adventitious sp2 C–C carbon species (284.42 eV), sp2 hybridized carbon atoms in N–C–N (286.38 eV), and sp2 CN bond (287.36 eV) in the s-triazine ring, respectively.86–89 In turn, the N 1s signals for these two catalysts are located in the binding energy region of 397–401 eV and can be fitted into three peaks located at 398.2 eV, 399.5 eV, and 400.7 eV for sp2-hybridized nitrogen in triazine rings (C–NC), tertiary nitrogen N–(C)3 groups, and amino functions with hydrogen (C–N–H), respectively.86,88,89
Investigation of oxygen reduction
The catalytic activity of MnO2/C, MnO2–gCN, and MnO2–gCN/C towards the ORR was initially evaluated through cyclic voltammetry (CV) in an Ar- or O2-saturated 0.1 M KOH solution. All the samples showed definite reduction peaks in the O2-saturated solution, while no such peaks were observed in the Ar-saturated electrolyte (Fig. 5a–c). In the case of the O2-saturated solution, reduction peaks were observed with an onset potential of approximately 0.9 V vs. RHE for all the investigated catalysts.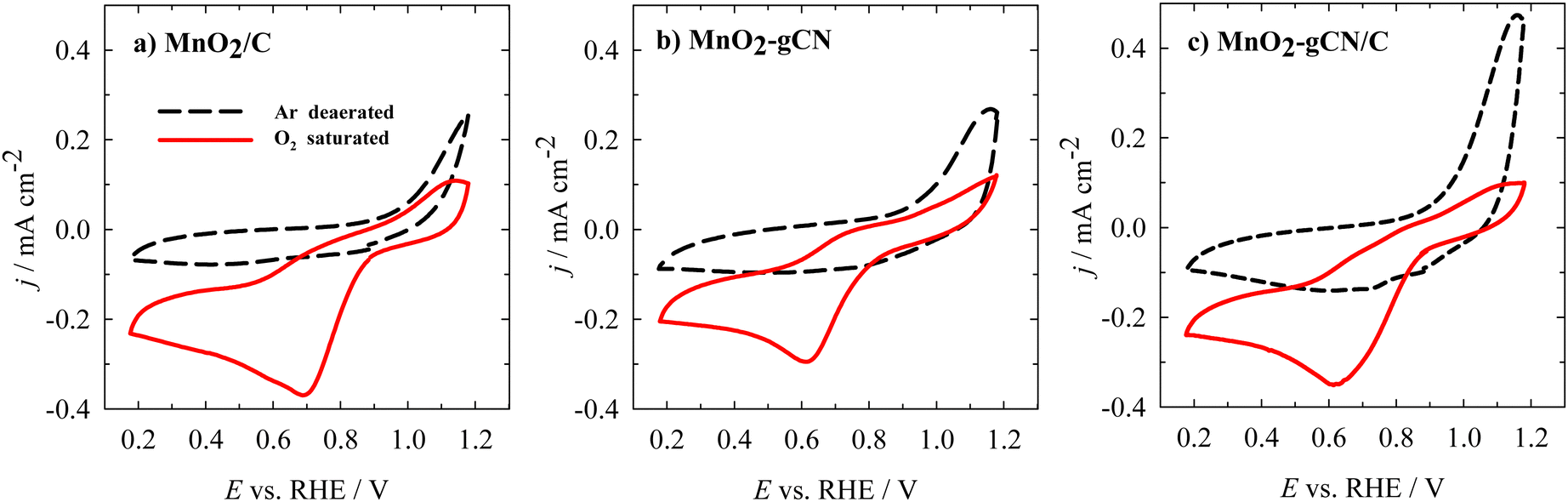 | ||
| Fig. 5 CVs of the investigated catalysts recorded in an Ar- or O2-saturated 0.1 M KOH solution with a scan rate of 10 mV s−1 at 0 rpm. | ||
Therefore, to further reveal the ORR kinetics of the MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts, the reduction of oxygen was investigated by linear sweep voltammetry (LSV) in an O2–saturated 0.1 M KOH solution at different rotation speeds. The electrode potential was scanned from 1.2 V in the cathodic direction to 0.2 V vs. RHE at a scan rate of 10 mV s−1. The rotation speed was varied between 400 and 2400 rpm. The theory states that at potential values between 0.7 and 0.9 V vs. RHE, the ORR response should be dominated by the kinetics of the electrocatalysts.90 The electrochemical LSV measurements demonstrated that all the investigated catalysts exhibited typical ORR curves, but with different onset potentials. Fig. 4 depicts the corresponding LSV curves of O2 reduction on C, gCN, and gCN/C substrates (Fig. 6a) and MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts (Fig. 6b) recorded in an O2-saturated 0.1 M KOH solution at a constant rotation rate of 1600 rpm. As could be expected, the substrates do not exhibit typical LSV curves for O2 reduction. Only the gCN/C substrate exhibits the most analogous O2 reduction LSV curve; however, the onset potential of 0.67 V is insufficient to substantiate the occurrence of O2 reduction on this substrate. A similar tendency is observed for the gCN and C substrates with an onset potential of 0.67 and 0.72 V, respectively (marked as the potential at a current density of −0.1 mA cm−2) (Fig. 6a).
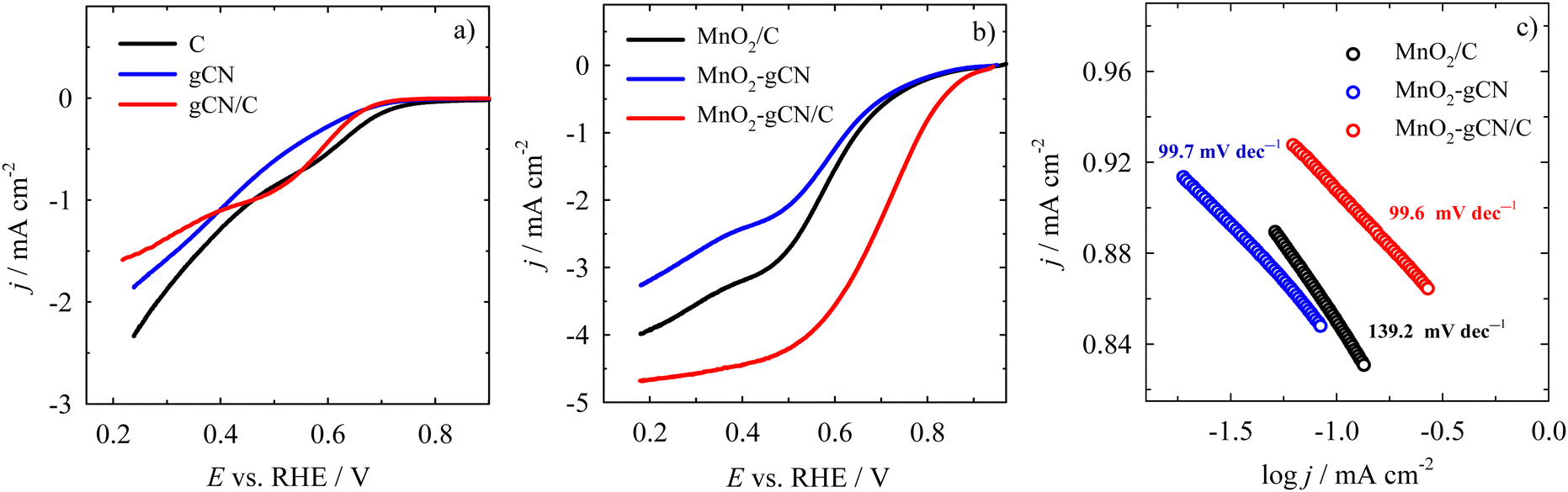 | ||
| Fig. 6 Comparison of LSV curves recorded in O2-saturated 0.1 M KOH solution at 1600 rpm at 10 mV s−1 for the substrates (a) and for the MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts (b). (c) The corresponding Tafel slopes for the investigated catalysts. | ||
Meanwhile, the onset potential for each catalyst, estimated from the measurement data at the same current density (−0.1 mA cm−2), was found to be equal to 0.85, 0.84, and 0.90 V for the MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts, respectively (Fig. 6b). The half-wave potential (E1/2) was found to be approximately 0.6 V for the MnO2/C and MnO2–gCN catalysts and 0.72 V for the MnO2–gCN/C catalyst. Additionally, the MnO2–gCN/C catalyst exhibited a current density at a potential of 0.4 V approximately twofold higher than that of the MnO2/C and MnO2–gCN catalysts. Furthermore, the calculated Tafel slopes for the MnO2/C, MnO2–gCN, and MnO2–gCN/C electrocatalysts demonstrate that the MnO2–gCN/C (99.6 mV dec−1) and MnO2–gCN (99.7 mV dec−1) catalysts exhibit the fastest ORR kinetics in comparison to the MnO2/C (139.2 mV dec−1) catalyst (Fig. 6c). As illustrated, the ORR on the MnO2–gCN/C catalyst commences at more positive electrode potential values than on the MnO2-supported separate C and gCN substrates. This suggests that the catalyst exhibits enhanced electrocatalytic activity for the ORR. This heightened activity may be ascribed to the generation of more expansive nanosphere structures with augmented exposed surfaces on a gCN/C substrate, which serves to enhance the catalytic efficacy of the catalyst.
The detailed voltammograms at varying rotation rates for the MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts are presented in Fig. 7a, c and e. It is evident that the measured current density (j) increased with an increase in the rotation rate (ω). Fig. 7b, d, and f present K–L plots of the investigated catalysts in an O2-saturated 0.1 M NaOH solution. In general, the plotted K–L curves exhibited a good linearity between j−1 and ω−1/2, indicating that the ORR kinetics at different potentials (0.2–0.5 V) exhibited first-order dependence as shown in Fig. 7b, d and f. The non-zero intercepts of the extrapolated K–L lines demonstrate that the process of O2 reduction is not wholly diffusion-controlled, even at more negative potentials. It has been demonstrated that the oxygen reduction reaction occurs via a four-electron pathway. In accordance with the K–L eqn (1)–(3), the calculated averaged number of electrons transferred (n) was equal to 3.8 (Fig. 7b, d and f insets).
 | ||
| Fig. 7 LSVs of the (a) MnO2/C, (c) MnO2–gCN, and (e) MnO2–gCN/C catalysts recorded in an O2–saturated 0.1 M KOH solution at 10 mV s−1 and varying the rotation speed from 400 to 2400 rpm; (b), (d) and (f) Koutecky–Levich plots of the ORR for each catalyst. The insets show the calculated electron transferred for each catalyst. | ||
The ORR mechanism typically follows one of the two pathways: the direct four-electron pathway and the successive two-electron pathway.91,92 In an alkaline medium, the direct four-electron transfer pathway can be expressed in a concise manner as follows:30,91–93
| O2 + 2H2O + 4e− → 4OH− E0 = +0.401 V | (6) |
The successive two-electron pathway involves the formation of peroxide (e.g., HO2− as shown in eqn (7)) with the subsequent reduction of peroxide to OH− (eqn (8)) or the disproportionation of peroxide (eqn (9)):
| O2 + 2H2O + 2e− → HO2− + OH− E0 = −0.065 V | (7) |
| HO2− + H2O + 2e− → 3OH− E0 = +0.87 V | (8) |
| 2HO2− → 3OH− + O2 | (9) |
The calculated number of electrons transferred (n) in this study was approximately 4 for the examined MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts, which is in line with the reported values in ref. 91 and 94–96 and close to that observed for the commercial Pt/C,91 which is beneficial to improving the ORR catalytic activity. The aforementioned results are primarily attributable to the quasi-4e reduction pathway of MnO2, which encompasses a negligible yield of hydrogen peroxide. This phenomenon has been ascribed to the disproportionation of H2O2, namely the disproportionation of HO2− into OH− and O2viaeqn (7) and (9). MnO2 is involved in the reduction of oxygen through a series of typical reactions, which are summarized in eqn (10)–(13) and outlined in ref. 94 and 97:
| MnO2 + xH2O + xe− ↔ MnOOHx + xOH− | (10) |
| 2MnOOH + O2 ↔ 2(MnOOH⋯O,ads) | (11) |
| (MnOOH)2⋯O2,ads → MnOOH⋯Oads + OH− + MnO2,rds | (12) |
| MnOOH⋯Oads + e− ↔ MnO2 + OH− | (13) |
In the case of the MnO2 catalyst, the initial step of the oxygen reduction reaction involves the reaction between MnO2 and H2O molecules, resulting in the formation of MnIIIOOH and OH−. Subsequently, the MnIIIOOH functions as an active site for the adsorption and reduction of oxygen.95,97 The catalytic activity of carbon-based MnO2 catalysts is enhanced due to the ability of manganese dioxide to facilitate the decomposition of HO2− into OH−,98 which in turn facilitates the reduction of oxygen (eqn (7) and (8)). As for the catalytic activity of porous g-C3N4, the graphitic N (N bonded to three carbon atoms) provides electrons to the π-conjugated system, which increases the nucleophilic strength of adjacent carbon rings [C(δ-)], and enhances O2 adsorption and thus accelerates the ORR.98 Therefore, the examined MnO2–gCN and MnO2–gCN/C catalysts showed enhanced catalytic activity towards the ORR.
Investigation of oxygen evolution
The activity of the prepared MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts was also evaluated for the oxygen evolution reaction (OER). The transfer of four electrons per oxygen molecule is required by the OER. This transfer can occur in multiple steps. It is therefore generally accepted that the OER is a complex multistep four-electron oxidation process. The theoretical thermodynamic voltage for the OER is 1.23 V, as reported in ref. 99 and 100. Various OER mechanisms have been proposed due to the difficulty in detecting intermediates. Here, we present the most classical and widely accepted mechanism for alkaline or neutral media, which is divided into four steps and is applicable in a variety of contexts:99,101,102| step 1: OH− → *OH + e− |
| step 2: *OH + OH− → *O + H2O + e− |
| step 3: *O + OH− → *OOH + e− |
| step 4: *OOH + OH− → O2 + H2O + e− |
It is proposed that the active sites of the electrocatalyst are indicated by the symbol *. The four-step process involves three reaction intermediates (*OH, *O, and *OOH), which represent the adsorbed oxygen-related species on the catalyst surface. The oxidation of water/hydroxyl ions is typically initiated under electrochemical oxidation conditions, resulting in the formation of *OH. This is followed by deprotonation and further oxidation, leading to the generation of the *O and *OOH intermediates, which ultimately produce O2 products. In acidic electrolytes, water molecules serve as the source of oxygen, whereas in neutral or alkaline solutions, oxygen is derived from hydroxyl ions.
The data obtained from the OER experiment indicate that catalysts containing MnO2 exhibit enhanced electrocatalytic activity in the OER when compared to the bare substrates (Fig. 8a and b). Fig. 8 illustrates that the highest current densities and the lowest overpotential were observed for MnO2–gCN/C among the investigated catalysts, indicating a notable enhancement in catalytic activity towards the OER. The Eonset values (marked as the potential at an OER current density of 1 mA cm−2) were observed to decrease in a gradual order, as follows: MnO2/C (1.58 V) < MnO2–gCN (1.55 V) < MnO2–gCN/C (1.53 V) with overpotential values of 350, 320, and 300 mV, respectively (Fig. 8b). Meanwhile the Eonset values of substrates were more positive for C (1.78 V) < gCN (1.73 V) < gCN/C (1.70 V), with overpotential values of 550, 500 and 470 mV, respectively (Fig. 8a). The MnO2–gCN/C catalyst exhibited an overpotential of only 390 mV, enabling the achievement of a current density of 10 mA cm−2. This was in contrast to the overpotentials of 410 mV for the MnO2–gCN catalyst and 430 mV for the MnO2/C catalyst. Furthermore, the substrates did not attain the 10 mA cm−2 current density value. The Tafel slope of MnO2–gCN/C (85.2 mV dec−1) is lower than those of MnO2–gCN (88.5 mV dec−1), and MnO2/C (86.4 mV dec−1) (Fig. 8c), indicating superior catalytic activity for the OER.
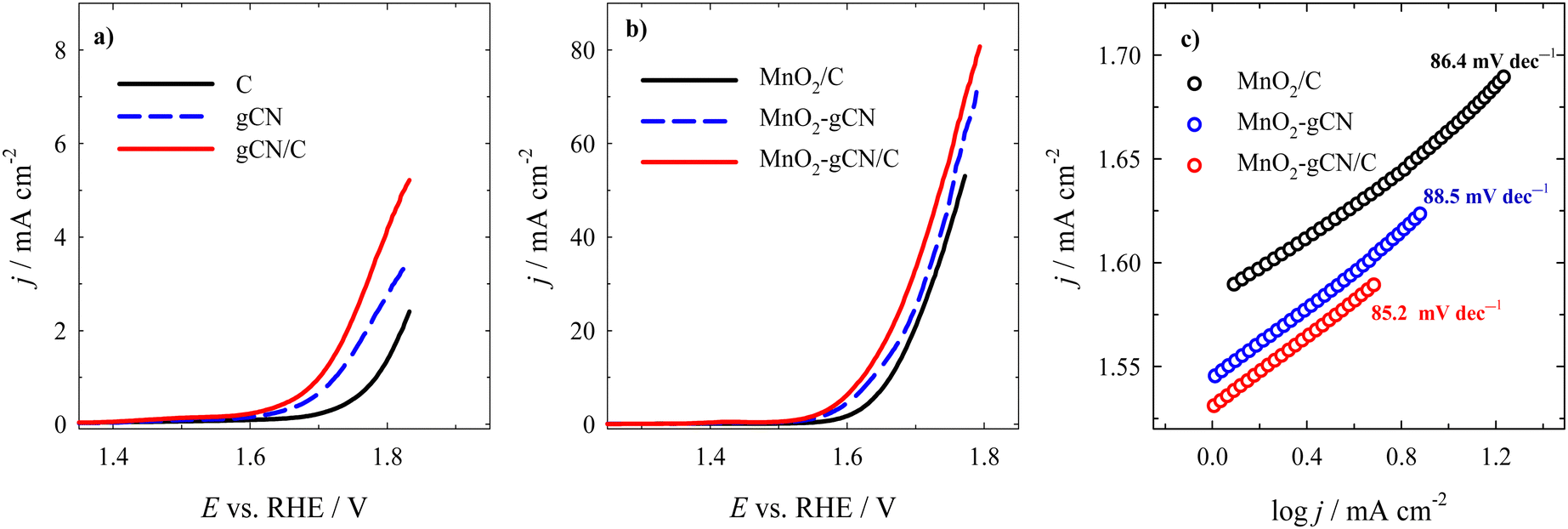 | ||
| Fig. 8 OER polarization curves recorded in a N2-saturated 1 M KOH solution at a potential scan rate of 2 mV s−1 on C, gCN, and gCN/C substrates (a) and MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts (b). (c) The corresponding Tafel slopes for the MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts. | ||
Given that g-CN has a distinctive structural configuration, the integration of additional highly conductive materials can further enhance its electrochemical performance. Furthermore, in the context of integration, it has been observed that the carbon (C) atoms are able to penetrate the lattice structure of g-CN by replacing some of the bridging N atoms, resulting in the formation of delocalised π bonds between the exchanged carbon atoms and the hexaatomic skeletal frame. The delocalization of π bonds is responsible for increasing the electrical conductivity of the hybridized g-CN, due to the high electron transfer kinetics of the delocalized π bonds.103 Furthermore, carbon nanomaterials (CNMs) can be seamlessly integrated within g-CN, preserving its intrinsic properties while offering additional benefits. This is crucial because defects can act as trapping sites for electrons, hindering their mobility and reducing the overall conductivity.103 Moreover, the integration of carbon atoms into g-CN by replacing a two-coordinated N atom (N2C) has been shown to facilitate the decomposition of the O–H bond in water, enabling the capture of released OH, and thereby improving the catalytic performance for the OER.104 This evidence substantiates the synergistic effect of the C dopant, which is further amplified in the presence of MnO2. The high surface area of CNMs provides numerous sites for electrolyte adsorption, thereby promoting ion diffusion and enhancing electrochemical performance. Furthermore, the formation of surface interactions between materials via interfacial contact is of great significance, as it increases active site availability and accessibility, and guides charge mobility/transport direction by creating transfer channels of electro-active species.
Moreover, the overall oxygen overpotential ΔE (ΔE = Ej10 − E1/2, where Ej10 is the operating potential of the OER at 10 mA cm−2, E1/2 is the half-wave potential for the ORR), reflecting the energy efficiency during change/discharge cycles, is commonly applied to evaluate the performance of bifunctional catalysts. The calculated oxygen overpotential for our investigated catalysts was equal 0.91, 1.04 and 1.06 for MnO2–gCN/C, MnO2–gCN and MnO2/C, respectively. It is seen that MnO2–gCN/C demonstrates the lowest ΔE among all the reported values. This confirms that the use of the hybrid gCN/C substrate significantly enhances the bifunctional performance of MnO2. Furthermore, the results obtained are competitive with those previously presented in the literature (Table 2).
| Catalyst | E 1/2 vs. RHE (V) | E j10 vs. RHE (V) | ΔE = Ej10 − E1/2vs. RHE (V) | Ref. |
|---|---|---|---|---|
| a Values are presented for j = 3 mA cm−2. | ||||
| MnO2/g-CN/C | 0.72 | 1.62 | 0.91 | This work |
| MnO2/g-CN | 0.61 | 1.64 | 1.03 | This work |
| MnO2/C | 0.60 | 1.66 | 1.06 | This work |
| Ni/α-MnO2 | 0.82 | 1.61 | 0.79 | 105 |
| RuO2 | 0.74 | 1.52 | 0.78 | 105 |
| α-MnO2 pristine | 0.78 | 1.78 | 1.00 | 105 |
| Pt/C | 0.85 | 1.82 | 0.97 | 105 |
| MnO@Cu–N–C | 0.81 | 1.64 | 0.83 | 106 |
| MnO/rGo | 0.70 | 1.71 | 1.01 | 107 |
| Ni–MnO/rGO | 0.78 | 1.60 | 0.82 | 107 |
| α-MnO2/XC-72 | 0.78 | 1.81 | 1.03 | 108 |
| MnO2/CNTsa | 0.83 | 1.68 | 0.85 | 109 |
| MnO2/CNTs | 0.67 | 1.72 | 1.05 | 96 |
| MnO2/CNTs−400 | 0.74 | 1.65 | 0.91 | 96 |
| MnOx/CNTs−400 | 0.77 | 1.69 | 0.92 | 96 |
| MnO2-NWRs/CNTs350 | 0.78 | 1.76 | 0.98 | 110 |
| MnO2-150-0.5/N-KB | 0.76 | 1.83 | 1.07 | 111 |
| MnO@Co–N/C | 0.83 | 1.76 | 0.93 | 112 |
| α-MnO2 nanowires | 0.83 | 1.62 | 0.79 | 113 |
| Co–MnO2/CNTs | 0.87 | 1.68 | 0.80 | 114 |
Given the superior performance of the MnO2–gCN/C catalyst in the ORR and OER, a comprehensive assessment of its stability and catalytic activity was conducted through chronoamperometric measurements in the alkaline electrolyte (Fig. 9) at a fixed potential of 0.55 V in an O2-saturated 0.1 M KOH solution for the ORR (Fig. 9a) and at a fixed potential of 1.62 V in N2-saturated 1.0 M KOH solution for the OER (Fig. 9b), with further analysis of the percentage of current density retained over time. As illustrated in Fig. 9, there was no discernible change in current density observed in the polarization curves during the chronoamperometric measurements of both the ORR and OER on MnO2–gCN/C. Following a continuous test period of 24 hours, the MnO2–gCN/C exhibited a residual signal of approximately 85.7 and 84.5% of its initial value during the long-term ORR and OER processes, respectively, thereby demonstrating a stable electrocatalytic performance. This result is comparable to that previously observed for the commercial Pt/C during the ORR process in our own research.93 Moreover, we conducted a comparative analysis of the electrocatalytic activity of our prepared catalysts towards the ORR and OER in comparison to the other Mn-, Ru-, Ir-, and Pt-supported catalysts that have been recently reported in the literature. Tables 2 and 3 present the data collected. A lot of research has been conducted on commercial Pt- and Ru-supported catalysts. There is no doubt that commercial Pt/C or Pt-supported catalysts exhibit high activity for the ORR as does commercial RuO2 for the OER.30,93,105,115 Nevertheless, recently developed materials demonstrate the potential to replace Pt and Ru during the ORR and OER processes (Tables 2 and 3). It can be observed that the electrochemical data of the ORR and OER as Eonset, E1/2, Ej10, ΔE and Tafel slopes for the as-prepared MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts exhibited comparable or even enhanced electrocatalytic activity towards these two reactions in alkaline media, when compared to the data presented in the literature (Tables 2 and 3).
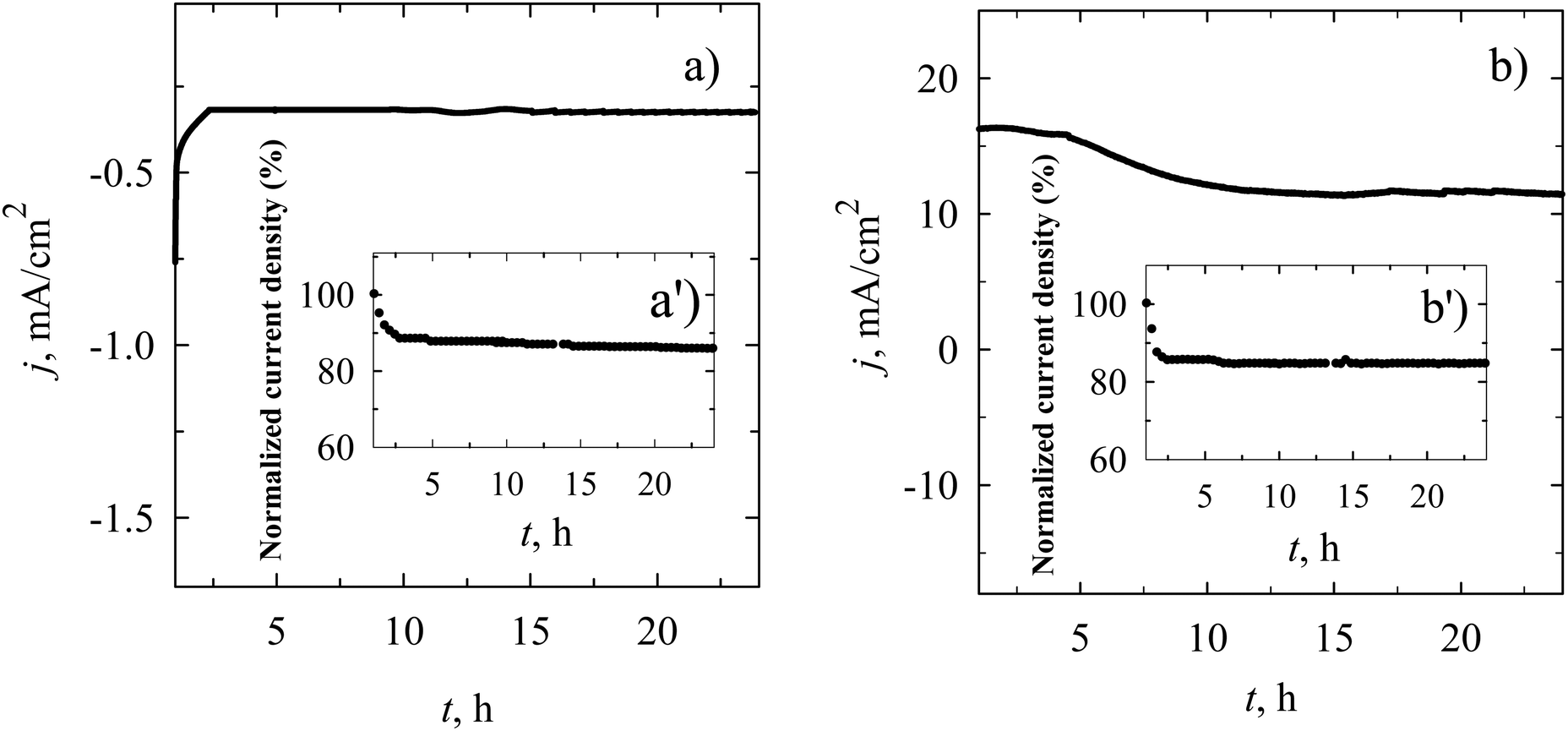 | ||
| Fig. 9 (a) Chronoamperometric curves of the MnO2–gCN/C catalyst recorded at a potential of 0.55 V in an O2-saturated 0.1 M KOH solution for 24 hours; (b) chronoamperometric curves of the MnO2–gCN/C catalyst recorded at a potential of 1.62 V in a N2-saturated 1 M KOH solution for 24 hours; (a′ and b′ insets) chronoamperometric responses (percentage of current density retained vs. operation time) of the MnO2–gCN/C catalyst. | ||
| Material | Electrolyte | ORR | OER | Ref. | |||
|---|---|---|---|---|---|---|---|
| E onset (V) | Tafel slope (mV dec−1) | E onset (V) | E (V) at 10 mA cm−2 | Tafel slope (mV dec−1) | |||
| Where “—” means not given. | |||||||
| MnO2/C | 0.1/1 M KOH | 0.850 | 139.2 | 1.58 | 0.430 | 86.4 | This work |
| MnO2/g-CN | 0.1/1 M KOH | 0.840 | 99.7 | 1.55 | 0.410 | 88.5 | This work |
| MnO2/g-CN/C | 0.1/1 M KOH | 0.900 | 99.6 | 1.530 | 0.390 | 85.2 | This work |
| α-MnO2 | 0.1 M KOH | 0.742 | 58.0 | 1.569 | 0.521 | 62.0 | 30 |
| β-MnO2 | 0.1 M KOH | 0.587 | 176.0 | 1.865 | — | 135.0 | 30 |
| γ-MnO2 | 0.1 M KOH | 0.524 | 195.0 | 1.706 | — | 144.0 | 30 |
| δ-MnO2 | 0.1 M KOH | 0.629 | 96.0 | 1.565 | 0.542 | 69.0 | 30 |
| λ-MnO2 | 0.1 M KOH | 0.590 | 168.0 | 1.719 | — | 97.0 | 30 |
| Pt/C | 0.1 M KOH | 0.862 | — | 1.520 | 0.582 | — | 30 |
| Pt/C | 0.1 M NaOH | 0.95 | — | — | — | — | 93 |
| RuO2 | 0.1 M KOH | 0.741 | — | 1.392 | 0.284 | — | 30 |
| MnO@Cu–N–C | 0.1 M KOH | — | 102.0 | 1.530 | 0.410 | — | 106 |
| g-C3N4/α-MnO2 | 0.1 M KOH | 0.920 | — | 1.678 | — | — | 116 |
| RuO2 comm | 1 M KOH | — | — | 1.470 | 0.350 | 106.0 | 115 |
| RuO2 comm | 1 M NaOH | — | — | — | 0.380 | 122.0 | 115 |
| IrO2 comm | 1 M KOH | — | — | 1.480 | 0.380 | 83.0 | 115 |
It can therefore be concluded that the MnO2–gCN/C catalyst, which can be prepared easily and without much cost, exhibits promising activity towards the ORR and OER processes. It may therefore be offered as a potential replacement for Pt or other noble metals in the generation of new energy.
Conclusions
This work presents the straightforward synthesis of non-precious catalysts, namely MnO2/C, MnO2–gCN, and MnO2–gCN/C. In comparison to the metal-free C, gCN, and gCN/C substrates that were used, the immobilization of MnO2 nanoparticles on the surface resulted in an enhancement of the electrocatalytic activity and the selectivity of the catalyst towards the four-electron reduction reaction of O2 to H2O. It has been demonstrated that the MnO2–gCN/C catalyst exhibits the most positive onset potential for the ORR, equal to 0.9 V vs. RHE, half-wave potential equal to 0.72 V vs. RHE, and a current density that is twofold that of the MnO2/C and MnO2–gCN catalysts. Furthermore, the immobilization of MnO2 nanoparticles on carbonaceous substrates has been observed to increase the activity of the OER, enhancing the onset potential to 0.15 V for all investigated catalysts. However, the most effective catalyst for the OER was the MnO2–gCN/C catalyst, which exhibited an onset potential of 1.53 V with an overpotential of 300 mV and the lowest overpotential of 390 mV at a current density of 10 mA cm−2. Additionally, it displayed a Tafel slope of 85.2 mV dec−1. Moreover, the calculated overall oxygen overpotential (ΔE) value was 0.91 V, representing the lowest value among the investigated MnO2/C and MnO2–gCN catalysts. This resulted in a notable enhancement in the bifunctional performance of the ORR and OER. Furthermore, the MnO2–gCN/C catalyst exhibited significant stability and catalytic activity, retaining approximately 85% of its initial signal in both long-term ORR and OER processes following continuous testing during chronoamperometric measurements for 24 hours. These results demonstrate that the MnO2/C, MnO2–gCN, and MnO2–gCN/C catalysts, which have been prepared in this study, have the potential to be employed as promising catalysts for low-cost and efficient non-precious metal electrocatalysts for the ORR and OER.Author contributions
This study was conducted through contributions from all authors. Conceptualization: L. T.-T., A. Z. and E. N.; methodology: V. J., G. S. and J. V.; formal analysis: A. Z., R. S. and D. Š.; investigation: A. Z. and R. S.; data curation: D. Š., V. K. and L. T.-T.; visualization: V. K. and A. Z.; writing—original draft preparation: L. T.-T. and V. K.; writing—review and editing: E. N.; all authors have read and agreed to the prepared version of the manuscript.Data availability
There are no data available to provide.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Social Fund (project No. 09.3.3-LMT-K-712-23-0188) under a grant agreement with the Research Council of Lithuania (LMTLT).References
- A. Hussain, S. M. Arif and M. Aslam, Renewable Sustainable Energy Rev., 2017, 71, 12, DOI:10.1016/j.rser.2016.12.033.
- W. Zhang, A. Valencia, G. Lixing, Q. P. Zheng and N. B. Chang, Appl. Energy, 2020, 279, 115716, DOI:10.1016/j.apenergy.2020.115716.
- S. P. S. Badwal, S. S. Giddey, C. Munnings, A. I. Bhattand and A. F. Hollenkamp, Front. Chem., 2014, 2, 1, DOI:10.3389/fchem.2014.00079.
- Y. Gong, J. Yao, P. Wang, Z. Li, H. Zhouand and C. Xu, Chin. J. Chem. Eng., 2022, 43, 282, DOI:10.1016/j.cjche.2022.02.010.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100, DOI:10.1002/adma.202007100.
- Y. Yao, X. Gao and X. Meng, Int. J. Hydrogen Energy, 2021, 46, 9087, DOI:10.1016/j.ijhydene.2020.12.212.
- L. G. Li, P. T. Wang, Q. Shao and X. Q. Huang, Chem. Soc. Rev., 2020, 49, 3072, 10.1039/D0CS00013B.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571, DOI:10.1021/acs.accounts.8b00002.
- B. Xiong, L. Chen and J. Shi, ACS Catal., 2018, 8, 3688, DOI:10.1021/acscatal.7b04286.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337, 10.1039/C6CS00328A.
- S. Wang, A. Lu and C. J. Zhong, Nano Convergence, 2021, 8, 4, DOI:10.1186/s40580-021-00254-x.
- M. V. Kaneva, L. B. Gulina and V. P. Tolstoy, J. Alloys Compd., 2022, 901, 163640, DOI:10.1016/j.jallcom.2022.163640.
- F. Nichols, J. E. Lu, R. Mercado, R. Dudschus, F. Bridges and S. W. Chen, Chem. – Eur. J., 2020, 26, 4136, DOI:10.1002/chem.201904559.
- D. Pan, Q. Liu, F. Nichols, R. Mercado, H. L. Kuo, J. Q. Lu and F. Bridges, J. Colloid Interface Sci., 2023, 629, 591, DOI:10.1016/j.jcis.2022.09.080.
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Appl. Catal., B, 2022, 310, 121318, DOI:10.1016/j.apcatb.2022.121318.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347, DOI:10.1021/ja510442p.
- Y. Li, L. Zhou and S. Guo, EnergyChem, 2021, 3, 100053, DOI:10.1016/j.enchem.2021.100053.
- S. Li, E. Li, X. An, X. Hao, Z. Jiange and G. Guan, Nanoscale, 2021, 13, 12788, 10.1039/D1NR02592A.
- H. Wu, C. Feng, L. Zhang, J. Zhang and D. P. Wilkinson, Electrochem. Energy Rev., 2021, 4, 473, DOI:10.1007/s41918-020-00086-z.
- A. H. Al-Naggar, N. M. Shinde, J. S. Kim and R. S. Mane, Coord. Chem. Rev., 2023, 474, 214864, DOI:10.1016/j.ccr.2022.214864.
- Z. Chen, W. Wei and B. J. Ni, Curr. Opin. Green Sustainable Chem., 2021, 27, 100398, DOI:10.1016/j.cogsc.2020.100398.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012, 10.1039/C2EE03250C.
- X. Cao, T. Wang and L. Jiao, Adv. Fiber Mater., 2021, 3, 210, DOI:10.1007/s42765-021-00065-z.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266, DOI:10.1002/adma.201602270.
- D. Liu, H. Ai, M. Chen, P. Zhou and B. Li, Small, 2021, 17, 2007557, DOI:10.1002/smll.202007557.
- B. P. Mishra, L. Acharya, S. Subudhi and K. Parida, Int. J. Hydrogen Energy, 2022, 47, 32107, DOI:10.1016/j.ijhydene.2022.07.125.
- H. M. Abuzeid, S. A. Elsherif, N. A. AbdelGhany and A. M. Hashem, J. Energy Storage, 2019, 21, 156, DOI:10.1016/j.est.2018.11.021.
- A. Elsherif, H. M. Abuzeid, A. M. Hashem and N. A. A. Ghany, J. Energy Storage, 2023, 74, 109341, DOI:10.1016/j.est.2023.109341.
- C. Cui, G. Du, K. Zhang, T. An, B. Li, X. Liu and Z. Liu, J. Alloys Compd., 2020, 814, 152239, DOI:10.1016/j.jallcom.2019.152239.
- T. Subramaniam, M. B. Idris, G. Harshini Sai and S. Devaraj, Mater. Chem. Phys., 2023, 303, 127845, DOI:10.1016/j.matchemphys.2023.127845.
- M. He, Z. Turup, X. Jin and F. Chen, Int. J. Hydrogen Energy, 2023, 48, 25770, DOI:10.1016/j.ijhydene.2023.03.283.
- B. Xu, H. Lu, W. Cai, Y. Cao, Y. Deng and W. Yang, Electrochim. Acta, 2019, 305, 360, DOI:10.1016/j.electacta.2019.03.062.
- G. Cheng, P. Liu, S. Chen, Y. Wu, L. Huang, M. Chen, C. Hu, B. Lan, X. Su, M. Sun and L. Yu, Colloids Surf., A, 2022, 637, 128228, DOI:10.1016/j.colsurfa.2021.128228.
- X. Bai, X. Tong, Y. Gao, W. Zhu, C. Fu, J. Ma, T. Tan, C. Wang, Y. Luo and H. Sun, Electrochim. Acta, 2018, 281, 525, DOI:10.1016/j.electacta.2018.06.003.
- W. He, C. Wang, F. Zhuge, X. Deng, X. Xu and T. Zhai, Nano Energy, 2017, 35, 242, DOI:10.1016/j.nanoen.2017.03.045.
- X. Hu, F. Cheng, X. Han, T. Zhang and J. Chen, Small, 2015, 11, 809–813, DOI:10.1002/smll.201401790.
- L. Peng, X. Peng, B. Liu, C. Wu, Y. Xie and G. Yu, Nano Lett., 2013, 13, 2151, DOI:10.1021/nl400600x.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2017, 90, 75, DOI:10.1016/j.pmatsci.2017.07.004.
- Y. J. Wang, H. Fan, A. Ignaszak, L. Zhang, S. Shao, D. P. Wilkinson and J. Zhang, J. Chem. Eng., 2018, 348, 416, DOI:10.1016/j.cej.2018.04.208.
- A. Zahoor, M. Christy, H. Jang, K. S. Nahm and Y. S. Lee, Electrochim. Acta, 2015, 157, 299, DOI:10.1016/j.electacta.2015.01.058.
- Y. Gao, H. Zhao, D. Chen, C. Chen and F. Ciucci, Carbon, 2015, 94, 1028, DOI:10.1016/j.carbon.2015.07.084.
- G. Dong, Y. Zhang, Q. Pan and J. Qiu, J. Photochem. Photobiol., C, 2014, 20, 33, DOI:10.1016/j.jphotochemrev.2014.04.002.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15, 10.1039/C4NR03008G.
- P. Suja, J. John, T. P. D. Rajan, G. M. Anilkumar, T. Yamaguchi, S. C. Pillai and U. S. Hareesh, J. Mater. Chem. A, 2023, 11, 8599, 10.1039/d2ta09776a.
- A. Alaghmandfard and K. Ghandi, Nanomaterials, 2022, 12, 294, DOI:10.3390/nano12020294.
- A. K. Mrinalini Kalyani, R. Rajeev, L. Benny, A. R. Cherian and A. Varghese, Mater. Today Chem., 2023, 30, 101523, DOI:10.1016/j.mtchem.2023.101523.
- M. Ismael, J. Alloys Compd., 2020, 846, 156446, DOI:10.1016/j.jallcom.2020.156446.
- M. J. Molaei, Int. J. Hydrogen Energy, 2023, 48, 32708, DOI:10.1016/j.ijhydene.2023.05.066.
- O. Iqbal, H. Ali, N. Li, A. I. Al-Sulami, K. F. Alshammari, H. S. M. Abd-Rabboh, Y. Al-Hadeethi, I. Ud Din, A. I. Alharthi, R. Altamimi, A. Zada, Z. Wang, A. Hayat and M. Z. Ansari, Mater. Today Phys., 2023, 34, 101080, DOI:10.1016/j.mtphys.2023.101080.
- M. Talukdar and P. Deb, Carbon, 2022, 192, 308, DOI:10.1016/j.carbon.2022.02.060.
- V. R. Govindaraju, K. Sureshkumar, T. Ramakrishnappa, S. Muralikrishna, D. Samrat, R. K. Pai, V. Kumar, K. Vikrant and K. H. Kim, J. Cleaner Prod., 2021, 320, 128693, DOI:10.1016/j.jclepro.2021.128693.
- T. O. Ajiboye, A. T. Kuvarega and D. C. Onwudiwe, Nano-Struct. Nano-Objects, 2020, 24, 100577, DOI:10.1016/j.nanoso.2020.100577.
- L. Kong, J. Wang, F. Ma, M. Sun and J. Quan, Appl. Mater. Today, 2019, 16, 388, DOI:10.1016/j.apmt.2019.06.003.
- F. Besharat, F. Ahmadpoor, Z. Nezafat, M. Nasrollahzadeh, N. R. Manwar, P. Fornasiero and M. B. Gawande, ACS Catal., 2022, 12, 5605, DOI:10.1021/acscatal.1c05728.
- W. K. Darkwah and Y. Ao, Nanoscale Res. Lett., 2018, 13, 388, DOI:10.1186/s11671-018-2702-3.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338, DOI:10.1016/j.ccr.2021.214338.
- D. Qu, J. Liu, X. Miao, M. M. Han, H. C. Zhang, Z. Cui, S. R. Sun, Z. H. Kang, H. Y. Fan and Z. C. Sun, Appl. Catal., B, 2018, 227, 418, DOI:10.1016/j.apcatb.2018.01.030.
- W. Che, W. Cheng and T. Yao, J. Am. Chem. Soc., 2017, 139, 3021, DOI:10.1021/jacs.6b11878.
- Z. Mo, H. Xu, Z. Chen, X. She, Y. Song, J. Lian, X. Zhu, P. Yan, Y. Lei, S. Yuan and H. Li, Appl. Catal., B, 2019, 241, 452, DOI:10.1016/j.apcatb.2018.08.073.
- X. Liu, L. Yan, X. Hu, H. Feng, B. Guo, X. Ha, H. Xu, R. Wang, X. Tan and H. Huang, Int. J. Hydrogen Energy, 2022, 47, 36110, DOI:10.1016/j.ijhydene.2022.08.174.
- J. Tian, Q. Liu, A. M. Asiri, K. A. Alamry and X. Sun, ChemSusChem, 2014, 7, 2125, DOI:10.1002/cssc.201402118.
- X. Wang, L. Wang, F. Zhao, C. Hu, Y. Zhao and Z. Zhang, Nanoscale, 2015, 7, 3035, 10.1039/C4NR05343E.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281, DOI:10.1002/anie.201403946.
- J. Wang, H. Kong, J. Zhang, Y. Hao, Z. Shao and F. Ciucci, Prog. Mater. Sci., 2021, 116, 100717, DOI:10.1016/j.pmatsci.2020.100717.
- G. Elmacı, A. S. Ertürk, M. Sevim and Ö. Metin, Int. J. Hydrogen Energy, 2019, 44, 17995, DOI:10.1016/j.ijhydene.2019.05.089.
- X. Zhang and P. Yang, Carbon, 2024, 216, 118584, DOI:10.1016/j.carbon.2023.118584.
- H. E. Zilli-Tomita, J. O. Saucedo-Lucero, V. A. Suárez-Toriello, J. R. Rangel-Mendez, M. Avalos-Borja and J. A. Arcibar-Orozco, Sustainable Chem. Environ., 2024, 6, 100084, DOI:10.1016/j.scenv.2024.100084.
- W. Zhang, J. Li and Z. Wei, Chin. J. Catal., 2023, 48, 15, DOI:10.1016/S1872-2067(23)64427-4.
- B. Jaleh, M. Nasrollahzadeh, M. Eslamipanah, A. Nasri, E. Shabanlou, N. R. Manwar, R. Zboril, P. Fornasiero and M. B. Gawande, Carbon, 2022, 198, 301, DOI:10.1016/j.carbon.2022.07.023.
- V. S. Bagotsky, A. M. Skundin and Y. M. Volfkovich, Electrochemical power sources – batteries, fuel cells, and supercapacitors, John Wiley & Sons, 2015 DOI:10.1002/9781118942857.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Ration, 82nd edn, 2001 Search PubMed.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134, DOI:10.1016/S0022-0728(00)00407-1.
- Q. Gu, Z. Gao, H. Zhao, Z. Lou, Y. Liao and C. Xue, RSC Adv., 2015, 5, 49317, 10.1039/C5RA07284K.
- C. Hu, Y. C. Chu, M. S. Wang and W. Xiao-Han, J. Photochem. Photobiol., A, 2017, 348, 8, DOI:10.1016/j.jphotochem.2017.08.006.
- H. Chen, Y. Fan, H. Xu, D. Cui, Ch Xue and W. Zhang, J. Alloys Compd., 2021, 863, 158448, DOI:10.1016/j.jallcom.2020.158448.
- A. Ramirez, P. Hillebrand and D. Stellmach, J. Phys. Chem. C, 2014, 118, 14073, DOI:10.1021/jp500939d.
- A. Moses Ezhil Raj, S. Grace Victoria and V. Bena Jothy, Appl. Surf. Sci., 2010, 256, 2920, DOI:10.1016/j.apsusc.2009.11.051.
- J. Dong, G. Lu, J. Yue, Z. Cheng and X. Kang, Appl. Surf. Sci., 2019, 480, 1116, DOI:10.1016/j.apsusc.2019.02.245.
- Y. Wang, W. Lai and N. Wang, Energy Environ. Sci., 2017, 10, 941, 10.1039/C6EE03773A.
- M. C. Biesinger, B. P. Payne and A. P. Grosvenor, Appl. Surf. Sci., 2011, 257, 2717, DOI:10.1016/j.apsusc.2010.10.051.
- W. Yang, Y. Zhua and F. You, Appl. Catal., B, 2018, 233, 184, DOI:10.1016/j.apcatb.2018.03.107.
- X-ray Photoelectron Spectroscopy (XPS) Reference Pages [https://www.xpsfitting.com].
- D. Zhou, H. Lin and F. Zhang, Electrochim. Acta, 2015, 161, 427, DOI:10.1016/j.electacta.2015.02.085.
- B. S. Singu and R. Yoon, J. Alloys Compd., 2019, 770, 1189, DOI:10.1016/j.jallcom.2018.08.145.
- H. Z. Chi, Y. Q. Wu and Y. K. Shen, Electrochim. Acta, 2018, 290, 487, DOI:10.1016/j.electacta.2018.09.029.
- Q. Gu, Z. Gao, H. Zhao, Z. Lou, Y. Liao and C. Xue, RSC Adv., 2015, 5, 49317, 10.1039/C5RA07284K.
- C. Hu, Y. C. Chu, M. S. Wang and W. Xiao-Han, J. Photochem. Photobiol., A, 2017, 348, 8, DOI:10.1016/j.jphotochem.2017.08.006.
- Q. Yang, J. An, Z. Xu, S. Liang and H. Wang, Colloids Surf., A, 2021, 614, 126161, DOI:10.1016/j.colsurfa.2021.126161.
- A. Zabielaite, A. Balciunaite, D. Upskuviene, D. Simkunaite, R. Levinas, G. Niaura, J. Vaiciuniene, V. Jasulaitiene, L. Tamasauskaite-Tamasiunaite and E. Norkus, Materials, 2023, 16, 5923, DOI:10.3390/ma16175923.
- K. M. Shi, X. Cheng, Z. Y. Jia, J. W. Guo, C. Wang and J. Wang, Int. J. Hydrogen Energy, 2016, 41, 16903, DOI:10.1016/j.ijhydene.2016.06.244.
- P. C. Li, C. C. Hu, T. H. You and P. Y. Chen, Carbon, 2017, 111, 813, DOI:10.1016/j.carbon.2016.10.057.
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon, 2019, 151, 160, DOI:10.1016/j.carbon.2019.05.075.
- V. Kepeniene, R. Stagniunaitė, L. Tamasauskaite-Tamasiunaite, V. Pakstas, V. Jasulaitiene, B. Léger, J. Rousseau, A. Ponchel, E. Monflier and E. Norkus, Mater. Chem. Phys., 2020, 241, 122332, DOI:10.1016/j.matchemphys.2019.122332.
- A. Ullah, B. Hussain, M. Waqar, M. R. Khan, J. Jehanzeb, K. Alam, P. Rosaiah, M. Ali, Annu, B. Akkinepally and I. Hussain, Diamond Relat. Mater., 2024, 149, 111611, DOI:10.1016/j.diamond.2024.111611.
- H. Zheng, X. Shi, S. Jiang, X. Han and L. Wen, J. Phys. Chem. Solids, 2024, 185, 111753, DOI:10.1016/j.jpcs.2023.111753.
- M. He, X. Jin, F. Chen, J. Chen, J. Min, H. Duan, X. Kuang, J. Li, Z. Wu and J. Li, J. Alloys Compd., 2023, 933, 167728, DOI:10.1016/j.jallcom.2022.167728.
- A. Ke Worku, De. W. Ayele, N. G. Habtu, M. A. Teshager and Z. G. Workineh, Mater. Today Sustainability, 2021, 13, 100072, DOI:10.1016/j.mtsust.2021.100072.
- Y. Hanga, C. Zhanga, X. Luo, Y. Xie, S. Xin, Y. Li, D. Zhang and J. B. Goodenough, J. Power Sources, 2018, 392, 15, DOI:10.1016/j.jpowsour.2018.04.078.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang and Y. J. Xu, Chem. Soc. Rev., 2017, 46, 337, 10.1039/C6CS00328A.
- Z. F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329, DOI:10.1038/s41560-019-0355-9.
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544, DOI:10.1002/aenm.201700544.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134, DOI:10.1002/adma.201807134.
- A. Haruna, K. B. Dönmez, S. Hooshmand, E. Avcı, M. Qamar, S. A. Zaidi, F. Shahzad, T. S. Miller, B. K. Chakrabarti, C. A. Howard and M. K. Bayazıt, Carbon, 2024, 226, 119177, DOI:10.1016/j.carbon.2024.119177.
- Y. Yang, S. Chen, J. Zhou and R. Zhang, Int. J. Hydrogen Energy, 2024, 71, 709, DOI:10.1016/j.ijhydene.2024.05.277.
- J. Xie, Y. Chen, Z. He, S. Liu, Y. Liu, B. Li, T. Xu, X. Ning, S. Chen, T. Zeng and H. He, ACS Appl. Nano Mater., 2024, 7, 18027, DOI:10.1021/acsanm.4c03521.
- Y. Ruan, H. Lei, W. Xue, T. Wang, S. Song and H. Xu, J. Alloys Compd., 2023, 934, 167781, DOI:10.1016/j.jallcom.2022.167781.
- G. Fu, X. Yan, Y. Chen, L. Xu, D. Sun, J. M. Lee and Y. Tang, Adv. Mater., 2018, 301, 704609, DOI:10.1002/adma.201704609.
- P. C. Li, C. C. Hu, T. H. You and P. Y. Chen, Carbon, 2017, 111, 813, DOI:10.1016/j.carbon.2016.10.057.
- N. Xu, Q. Nie, L. Luo, C. Yao, Q. Gong, Y. Liu, X. D. Zhou and J. Qiao, ACS Appl. Mater. Interfaces, 2019, 11, 578, DOI:10.1021/acsami.8b15047.
- N. N. Xu, J. W. Liu, J. L. Qiao, H. T. Huang and X. D. Zhou, J. Power Sources, 2020, 455, 227992, DOI:10.1016/j.jpowsour.2020.227992.
- M. Wang, K. Chen, J. Liu, Q. G. He, G. L. Li and F. Z. Li, Catalysts, 2018, 8, 138, DOI:10.3390/catal8040138.
- W. H. Hu, Q. Wang, S. S. Wu and Y. M. Huang, J. Mater. Chem. A, 2016, 4, 16920, 10.1039/C6TA08103G.
- Y. Gu, G. Yan, Y. Lian, P. Qi, Q. Mu, C. Zhang, Z. Deng and Y. Peng, Energy Storage Mater., 2019, 23, 252, DOI:10.1016/j.ensm.2019.05.006.
- N. Xu, Y. Zhang, Y. Wang, M. Wang, T. Su, C. Coco, J. Qiao and X.-D. Zhou, Appl. Energy, 2020, 279, 115876, DOI:10.1016/j.apenergy.2020.115876.
- R. Samanta, P. Panda, R. Mishra and S. Barman, Energy Fuels, 2022, 36, 1015, DOI:10.1021/acs.energyfuels.1c04082.
- Y. Hang, C. Zhang, X. Luo, Y. Xie, S. Xin, Y. Li, D. Zhang and J. B. Goodenough, J. Power Sources, 2018, 392, 15, DOI:10.1016/j.jpowsour.2018.04.078.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |