
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Opportunities and challenges of RiPP-based therapeutics
Isabel P.-M.
Pfeiffer†
 ,
Maria-Paula
Schröder†
and
Silja
Mordhorst
*
,
Maria-Paula
Schröder†
and
Silja
Mordhorst
*
University of Tübingen, Pharmaceutical Institute, Department of Pharmaceutical Biology, Auf der Morgenstelle 8, 72076 Tübingen, Germany. E-mail: silja.mordhorst@uni-tuebingen.de
First published on 27th February 2024
Abstract
Covering: up to 2024
Ribosomally synthesised and post-translationally modified peptides (RiPPs) comprise a substantial group of peptide natural products exhibiting noteworthy bioactivities ranging from antiinfective to anticancer and analgesic effects. Furthermore, RiPP biosynthetic pathways represent promising production routes for complex peptide drugs, and the RiPP technology is well-suited for peptide engineering to produce derivatives with specific functions. Thus, RiPP natural products possess features that render them potentially ideal candidates for drug discovery and development. Nonetheless, only a small number of RiPP-derived compounds have successfully reached the market thus far. This review initially outlines the therapeutic opportunities that RiPP-based compounds can offer, whilst subsequently discussing the limitations that require resolution in order to fully exploit the potential of RiPPs towards the development of innovative drugs.
 Isabel P.-M. Pfeiffer | Isabel Pfeiffer studied Biology (BSc) and Microbiology (MSc) at the University of Tübingen, Germany. Since 2022, she has been a PhD student in the lab of Silja Mordhorst at the department of Pharmaceutical Biology, University of Tübingen. Her research focuses on the investigation of novel bacterial RiPP natural product biosynthetic pathways. |
 Maria-Paula Schröder | Currently Maria-Paula Schröder is pursuing her PhD at the University of Tübingen in the group of Silja Mordhorst. She has a Bachelor of Science in Chemistry and a Master of Science in Biochemistry from TU Dresden (Germany). Her research focus lies in elucidating new RiPP natural products, exploring their biosynthetic pathways, uncovering the final products, and investigating their potential applications. |
 Silja Mordhorst | Silja Mordhorst studied Pharmaceutical Sciences and earned her doctoral degree at the University of Freiburg, Germany. In 2019, she started her postdoc as a Feodor-Lynen fellow of the Alexander-von-Humboldt foundation in the natural product lab of Professor Jörn Piel at ETH Zurich, Switzerland, where she investigated novel peptide-modifying enzymes from RiPP biosynthetic gene clusters. In 2022, she was appointed assistant professor for Pharmaceutical Biology at the University of Tübingen, Germany. Her research includes studies on the discovery and biosynthesis of (mainly RiPP) natural products and the biochemical characterisation of enzymes with a special focus on their potential for peptide engineering. |
1 Introduction
Ribosomally synthesised and post-translationally modified peptides (RiPPs) constitute a large superfamily of peptide natural products and possess an extensive structural diversity.1 The different structural elements lead to various biological functions, including a range of therapeutic effects. The biological effects of these compounds rely on chemical motifs, which are introduced by post-translational modifications in precursor peptides that are produced by ribosomes. Most precursor peptides comprise an N-terminal leader peptide and a C-terminal core peptide, while some precursors bear an additional follower peptide at the C-terminus. RiPP biosynthetic gene clusters encode the precursor peptide(s), post-translationally acting maturases, transporter proteins, and often proteases to cleave the modified core sequences from the leader peptide releasing the mature natural product.2The family of RiPP natural products is continuously expanding and its members encompass redox cofactors,3–5 chalkophores,6 siderophores,7–9 and antimicrobials that participate in natural competition within microbial communities. Although most researchers categorise “classical” RiPPs as bacterial, fungal, and plant specialised metabolites, we also classify certain human, amphibian, and mollusk hormones or toxins as RiPPs in this review, since they also fall under the category of ribosomally synthesised and post-translationally modified peptides. In contrast to “classical” RiPPs, these molecules only contain disulphide bridges as post-translational modifications. Peptides facilitate an array of biological processes in all domains of life,9 and are believed to be the pharmaceuticals of tomorrow. Their large chemical diversity, typically low toxicity/immunogenicity and their notable target specificity make them a prime subject for drug discovery.10,11 The molecular weight of peptide drugs falls between small molecules and large biologicals, such as antibodies. Therefore, peptide drugs are deemed to combine the high specificity of biologicals that are capable of inhibiting protein–protein interactions, with the favourable physicochemical properties of classical small molecule drugs, that are essential for bioavailability. While peptides tend to have low metabolic stability, and linear peptides can display lower binding affinity to their target structure owing to their structural flexibility, modifications can be introduced that incorporate structural characteristics to enhance peptide stability and binding affinity.12,13
Numerous review articles have been published on RiPP research, including general overviews of RiPPs,1,2 their physiological and ecological roles,9 their modes of action,14,15 their antiviral activities,16 their engineering potential,17–19 and reviews on certain RiPP families.20–25 This review will concentrate on the potential therapeutic applications of RiPPs and will describe the promising opportunities RiPPs can offer in drug discovery, as well as the limitations currently preventing their application.
2 Opportunities
RiPP natural products exhibit significant bioactivities in laboratory-based in vitro and in vivo experiments. Numerous research groups have directed their focus on antibiotic RiPPs, due to the prevalent issue of antibiotic resistance, commonly referred to as “the silent pandemic”. Nonetheless, RiPP natural products have demonstrated additional pharmaceutically relevant effects including antifungal, antiviral, antiparasitic, antitumour, and analgesic activities. In the upcoming sections we will present sample compounds for each of the bioactivities mentioned, underscoring the immense potential of this natural product class for pharmaceutical purposes.2.1 Antibiotic RiPPs
Over 80 antibiotically active RiPPs with distinct targets and modes of action are described to date. Most of them have already been reviewed elsewhere;15 therefore, only a few examples shall be highlighted here. Along with members of popular RiPP classes, peptides with remarkable activity against drug-resistant and Gram-negative bacteria, and RiPPs with unique targets or modes of action have been chosen for this chapter. First, members of the RiPP class of lanthipeptides, including lanthidins and lipolanthins, are described, followed by linear azol(in)e containing peptides (LAPs), glycocins, thiopeptides, and lasso peptides.Cinnamycin (also known as Ro 09-0198 or lanthiopeptin) and duramycin (syn. PA48009 or leucopeptin, Fig. 1) are two closely related class II lanthipeptides, which are produced by Streptomyces cinnamoneus.26 They differ solely in one amino acid and display antibiotic activity against Gram-positive bacteria,27–29 by binding to the cell membrane component phosphatidylethanolamine (PE) in a highly specific manner. While cinnamycin causes membrane disruption, duramycin induces membrane permeabilisation. The precise mechanism of action for both peptides, however, remains unclear.15,30
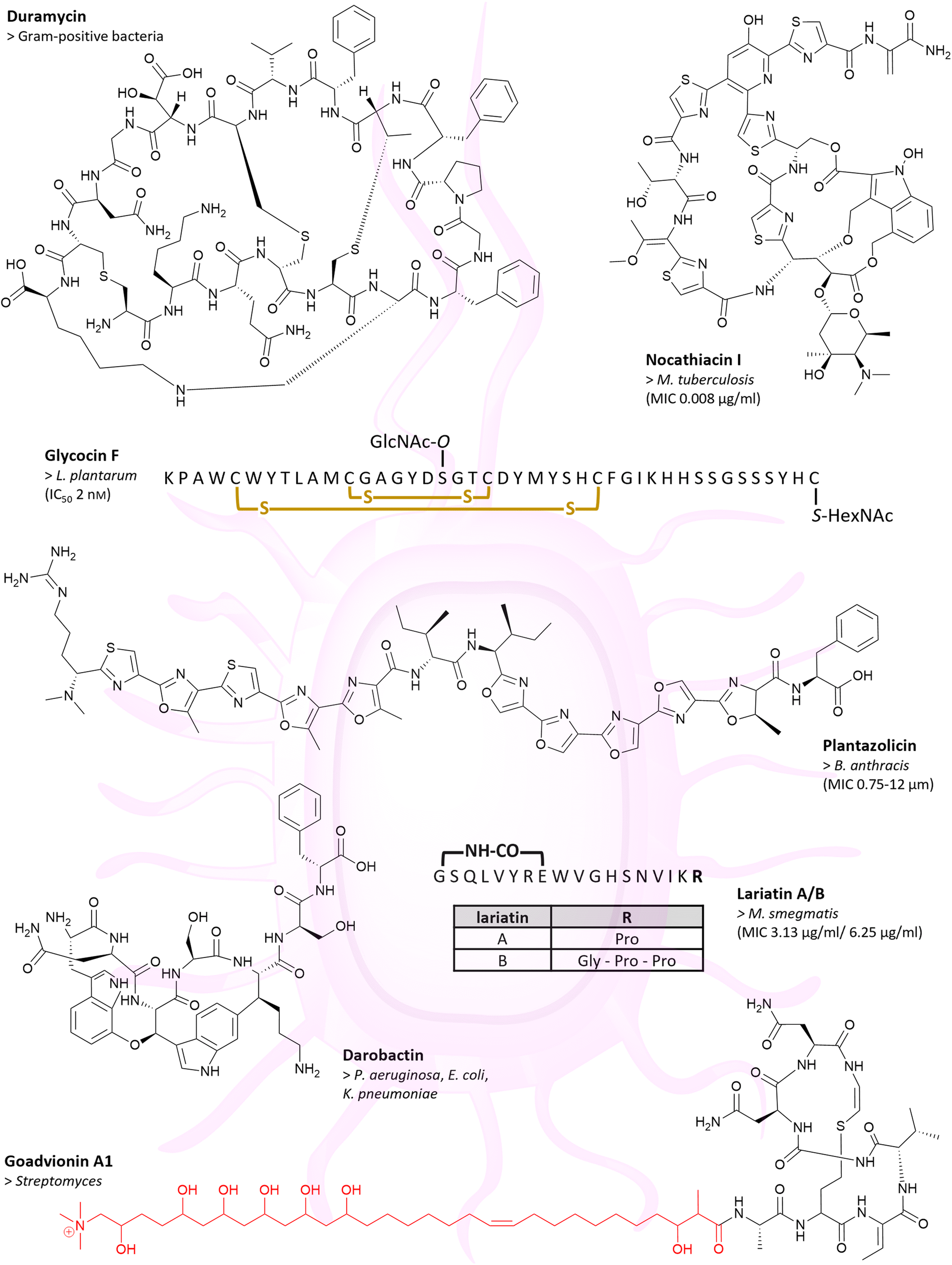 | ||
| Fig. 1 Many RiPPs show antibiotic activities. Chemical structures of duramycin, nocathiacin I, glycocin F, plantazolicin, darobactin, lariatin A/B and goadvionin A1 are depicted. Example(s) of affected bacteria with MIC or IC50 value are listed below the compound name. For glycocin F, amino acids are shown in the single-letter code and yellow indicated crosslinks are disulphide bridges. The acyl group of goadvionin A1 is indicated in red. | ||
Lexapeptide and cacaoidin represent the first members of the class V lanthipeptides, which are characterised by an atypical three-component lanthionine synthetase. The two peptides exhibit an N,N-dimethylation at their N-termini.31 Lexapeptide, derived from Streptomyces rochei Sal35, displays antibiotic activity against various Gram-positive bacteria in a low micromolar range, encompassing Mycobacterium smegmatis (MIC‡ 0.26 μM, Table 1),§Enterococcus faecalis (MIC 0.52 μM), methicillin-resistant Staphylococcus aureus (MRSA; MIC 0.52 μM) and Staphylococcus epidermidis (MRSE; MIC 1.03 μM). Neither the target nor the mode of action have yet been elucidated.32 Cacaoidin is produced by Streptomyces cacaoi CA-170360 and shows antimicrobial activity against MRSA (MIC 0.22 μM) and Clostridioides (formerly Clostridium) difficile (MIC 1.7 μM). It is believed that cacaoidin targets cell wall biosynthesis, through the inhibition of transglycosylases in the cell wall and through the binding of the cell wall precursor lipid II.31,33
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined. | |||
| Achromonodins | Gram-negative bacteria | RNA polymerase | A. pulmonis: MIC 1.3 μM (ref. 55) |
| Acinetodin | Not observed | RNA polymerase63 | n.d. |
| Cacaoidin | Gram-positive bacteria | Cell wall | MRSA: MIC 0.22 μM |
| C. difficile: MIC 1.7 μM (ref. 33) | |||
| Capistruin | Gram-negative bacteria | DNA-dependent RNA polymerase | P. aeruginosa: MIC 50 μM |
| B. caledonica: MIC 12 μM (ref. 57) | |||
| Cinnamycin | Gram-positive bacteria | Cell membrane (PE)30 | n.d. |
| Citrocin | Gram-negative bacteria | RNA polymerase | E. coli BW25113: MIC 31 μM (ref. 64) |
| Cloacaenodin | Gram-negative bacteria | RNA polymerase | E. cloacae ATCC 13047: MIC 0.94 μM (ref. 65) |
| Darobactin | Gram-negative bacteria | Outer membrane protein BamA59 | n.d. |
| Duramycin | Gram-positive bacteria | Cell membrane (PE)30 | n.d. |
| Glycocin F | Gram-positive bacteria | n.d. | L. plantarum: IC50 2 nM (ref. 44) |
| Goadsporin | Gram-positive bacteria41 | n.d. | n.d. |
| Goadvionins | Gram-positive bacteria35 | n.d. | n.d. |
| Klebsidin | Gram-negative bacteria | RNA polymerase | K. pneumoniae: MIC 256 μM (ref. 63) |
| Lariatin A & B | Mycobacteria | Mycobacterial cell wall | Lariatin A: M. smegmatis: MIC 3.13 μg ml−1 |
| M tuberculosis: MIC 0.39 μg ml−1 | |||
| Lariatin B: M. smegmatis: MIC 6.25 μg ml−1 (ref. 52) | |||
| Lassomycin | Mycobacteria | ClpC1 ATPase51 | n.d. |
| Lexapeptide | Gram-positive bacteria | n.d. | M. smegmatis: MIC 0.26 μM |
| E. faecalis: MIC 0.52 μM | |||
| MRSA: MIC 0.52 μM | |||
| MRSE: MIC 1.03 μM (ref. 32) | |||
| Microcin B17 | Gram-negative bacteria66 | Bacterial DNA gyrase, subunit B38 | n.d. |
| Microcin J25 | Gram-negative bacteria | DNA-dependent RNA polymerase | S. newport: MIC 0.01 μg ml−1 (ref. 56) |
| Microcin Y | Gram-positive and Gram-negative bacteria | RNA polymerase | B. subtilis ATCC 6633: MIC 4 μM |
| S. infantis: MIC 0.04 μM (ref. 67 and 68) | |||
| Microvionin | Gram-positive bacteria | n.d. | MRSA: MIC 0.46 μg ml−1 |
| S. pneumoniae: MIC 0.15 μg ml−1 (ref. 34) | |||
| Nocathiacin | Gram-positive bacteria | Bacterial protein biosynthesis | MRSA: MIC 0.007 μg ml−1 |
| VRE: MIC 0.03 μg ml−1 (ref. 48) | |||
| M. tuberculosis: MIC 0.008 μg ml−1 (ref. 49) | |||
| PRSP: MIC <0.002 μg ml−1 (ref. 48) | |||
| Nosiheptide | Gram-positive bacteria | Bacterial protein biosynthesis | MRSA: MIC 0.03 μg ml−1 |
| VRE: MIC 0.125 μg ml−1 | |||
| C. difficile: MIC 0.008 μg ml−1 (ref. 50) | |||
| Plantazolicin | Gram-positive bacteria | Cell membrane | B. anthracis: MIC 0.75–12 μM (ref. 36) |
| Siamycins | Gram-positive bacteria | Cell wall/lipid II54 | n.d. |
| Thiazomycin | Gram-positive bacteria | Bacterial protein biosynthesis | MRSA: MIC 0.032 μg ml−1 |
| VRE: MIC 0.064 μg ml−1 (ref. 48) | |||
| Thiostrepton | Gram-positive bacteria | Bacterial protein biosynthesis47 | n.d. |
| Ubonodin | Gram-negative bacteria | RNA polymerase | B. cepacia ATCC 25416: MIC 4 μM (ref. 69) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Marketed (non-RiPP) antibiotics | |||
| Ceftazidime | Gram-positive and Gram-negative bacteria | Cell wall | E. coli: MIC 0.06–0.25 μg ml−1 (ref. 70) |
| Vancomycin | Gram-positive bacteria | Cell wall | Streptococcus pneumoniae: MIC 0.25–0.5 μg ml−1 (ref. 71) |
Lipidated lanthipeptides are found in the class of lipolanthines. The first lipolanthine, microvionin, was identified through a bioactivity-guided screening of culture extracts from Microbacterium arborescens 5913. This lipolanthine shows potent antibacterial activity against Gram-positive bacteria, notably against MRSA and Streptococcus pneumoniae with low minimal inhibitory concentrations of 0.46 μg ml−1 and 0.15 μg ml−1, respectively.34 Another family of lipolanthines are the goadvionins, which comprise currently eight polyketide/RiPP hybrid lipopeptides, named goadvionin A1–A4 and B1–B4 (Fig. 2). Extracts from a strain that produces goadvionin, namely Streptomyces sp. TP-A0584, displayed antibacterial effects against Streptomyces and other Gram-positive bacteria, but no effect was observed on Gram-negative bacteria.35
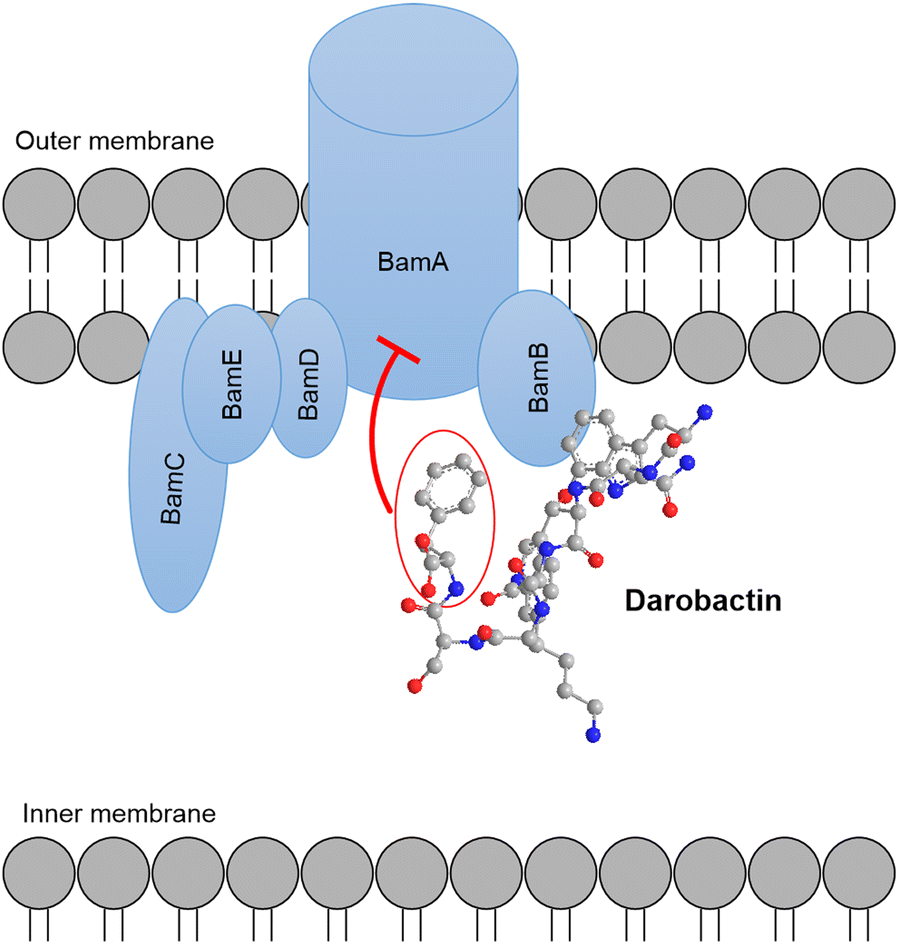 | ||
| Fig. 2 Darobactin targets the outer membrane protein complex Bam of Gram-negative bacteria. Binding to the subunit BamA leads to inhibition of activity. | ||
Plantazolicin (Fig. 2), microcin B17, and goadsporin are classified as LAPs. Plantazolicin, a highly selective, narrow-spectrum antibiotic was isolated from Bacillus amyloliquefaciens FZB42. It demonstrated noteworthy efficacy against Bacillus anthracis (MIC 0.75–12 μM). Following localisation to the cell envelop of B. anthracis, depolarisation and lysis of the cellular membrane was observed as a result of plantazolicin treatment. Plantazolicin utilises a distinct mechanism of action in contrast to other antibiotics targeting the cell envelope. It is suggested that it takes advantage of a transient weakened cell membrane, caused by increased membrane fluidity or changes in the lipid composition, such as an aberrant cardiolipin portion. Cell death ultimately occurs through cell lysis.36 The prototypical example of class B microcins is microcin B17 (MccB17),37 which was isolated from Escherichia coli and demonstrated to block DNA replication. In contrast to other DNA gyrase inhibitors, like the quinolones, MccB17 does not bind the A subunit but the B subunit of bacterial DNA gyrase.38 This causes a cascade of reactions. Double-stranded DNA breaks initiate the SOS response, resulting in DNA degradation and, finally, cell death. A single molecule of MccB17 is enough to kill a bacterium.39,40 Antibiotic activity of goadsporin, produced by Streptomyces sp. TP-A0584, was found against actinomycetes, but not against other bacteria and fungi. Goadsporin was demonstrated to bind the intracellular signal recognition particle (SRP) and interfering with the correct cellular localisation of nascent proteins. But the target of its antibiotic activity remains unknown.41,42
Further compounds with antibiotic activity can be found in the RiPP family of glycosylated bacteriocins, the glycocins. Bacteriocins are synthesised by bacteria and belong to the class of antimicrobial peptides, which demonstrate antibiotic activity only against highly similar strains.43 Glycocin F (or plantaricin KW30, Fig. 1), produced by Lactobacillus plantarum KW30, exhibits uncommon structural features. These include an N-acetylglucosamine (GlcNAc), which is β-O-linked to serine 18 of the peptide, and an N-acetylhexosamine (HexNAc), which is S-linked to the C-terminal cysteine 43. Inhibitory activity of glycocin F was observed in Lactobacillus strains, particularly L. plantarum strains were strongly inhibited (IC50‡ 2 nM). It was demonstrated that GlcNAc is required for bacteriostasis and HexNAc crucial for a maximal efficacy. To our knowledge, the exact mode of action has not yet been reported.44,45
Several thiopeptide antibiotics with diverse structures have been characterised so far. They are unified by the presence of a six-membered heterocycle and a common mode of action, the inhibition of bacterial protein biosynthesis.46 Thiostrepton was already isolated in the 1950s from Streptomyces sp. and inhibits different strains of Streptococcus and Staphylococcus.47 Potent antibacterial activity against MRSA and vancomycin-resistant Enterococcus (VRE) was observed by the thiopeptides thiazomycin (MIC MRSA: 0.032 μg ml−1; VRE: 0.064 μg ml−1) produced by Amycolatopsis fastidiosa,48 nocathiacin (Fig. 1, MIC MRSA: 0.007 μg ml−1; VRE: 0.03 μg ml−1)48,49 and nosiheptide (MIC MRSA: 0.03 μg ml−1; VRE: 0.125 μg ml−1).50 Nocathiacins, isolated from Nocardia sp. ATCC202099, were additionally effective against Mycobacterium tuberculosis (MIC 0.008 μg ml−1), Mycobacterium avium (MIC 0.06 μg ml−1) and like thiazomycin against penicillin-resistant Streptococcus pneumoniae (PRSP) with a minimal inhibitory concentration <0.002 μg ml−1.48,49 Nosiheptide, which is produced by Streptomyces sp. CNT-373, was further shown to inhibit a hypervirulent B1 strain of Clostridioides difficile (MIC 0.008 μg ml−1).50
Antimycobacterial activity was detected for the lasso peptides lassomycin and lariatin A and B (Fig. 1). Lassomycin is a unique bactericidal compound produced by Lentzea kentuckyensis, targeting the ClpC1 ATPase. It triggers the ATPase activity and in parallel abolishes the ClpC1's proteolytic activity. In addition, lassomycin exhibits a high specificity for mycobacteria, but is inactive against bacteria of the human microbiota.51 Lariatin A is an 18 amino acid peptide, and lariatin B contains two additional amino acids in the tail region, glycine and proline. Both peptides were isolated from Rhodococcus jostii K01B0171 and specifically inhibited growth of M. smegmatis (lariatin A: MIC 3.13 μg ml−1; lariatin B: MIC 6.25 μg ml−1); M. tuberculosis was only inhibited by lariatin A with a MIC of 0.39 μg ml−1. They displayed no activity against other bacterial and fungal test organisms. Mycobacteria possess an uncommon cell wall structure, differentiating them from other bacteria. Since the lariatins only inhibited mycobacteria, it is assumed they are targeting a step specific for mycobacterial cell wall biosynthesis.52Streptomyces sp. AA6532 produces siamycin I (Fig. 5), which also belongs to the class of lasso peptides. Strong antibiotic activity was observed against Gram-positive bacteria, including MRSA and VRE. Siamycin I binds to the pyrophosphate-sugar motif of lipid II at the outside of Gram-positive cell walls. Incorporation of the N-acetyl glucosamine–N-acetylmuramic acid disaccharide into the cell wall is thought to be hindered by that.53,54 The lasso peptides achromonodin-1 and achromonodin-2 identified from Achromobacter sp. show narrow-spectrum antibiotic activity against other pathogenic Achromobacter sp., which often infect cystic fibrosis patients.55
Compounds targeting Gram-negative bacteria are scarce and novel anti-Gram-negative compounds are rarely discovered. The lasso peptides microcin J25 (MccJ25) and capistruin are both highly active against Gram-negative bacteria. MccJ25, isolated from E. coli, was found to inhibit the DNA-dependent RNA polymerase (RNAP) and antibiotic activity with a MIC of 0.01 μg ml−1 against Salmonella newport was demonstrated.56 Capistruin, produced by Burkholderia thailandensis E264, displays antibiotic activity against Pseudomonas and Burkholderia strains that are closely related to the producer. For Pseudomonas aeruginosa AT27853 a MIC of 50 μM and for Burkholderia caledonica a MIC of 12 μM were determined.57 Like MccJ25, capsitruin was found to bind and inhibit RNAP.58 A bactericidal antibiotic with negligible anti-Gram-positive, but strong anti-Gram-negative activity is darobactin (Fig. 1). It is produced by Photorhabdus khanii HGB1456. Inhibition of clinically relevant drug resistant pathogens, including P. aeruginosa, E. coli and Klebsiella pneumoniae strains, was demonstrated. The effect was explained by darobactin binding the outer membrane protein BamA, thereby preventing proper assembly of the Gram-negatives outer membrane (Fig. 2).59 The binding site of darobactin at BamA is not addressed by other commercially available antibiotics making darobactin a valuable compound to treat infections with multidrug resistant strains. Genetically engineered derivatives of darobactin show an increased antibacterial activity (128-fold increase compared to darobactin) and a promising ADMET (absorption, distribution, metabolism, excretion, and toxicity) profile.60–62
2.2 Antifungal RiPPs
Fungal diseases pose a serious threat to human health. Mortality rates above 50% of invasive fungal infections underline the need for novel antifungal drugs.72 Antifungal RiPPs are found in several RiPP families, including lanthipeptides, cyanobactins, thiopeptides, and lasso peptides.Pinensin A and B (Fig. 3) are two new lanthipeptides obtained from Chitinophaga pinensis DSM 28390, representing the first antifungal lantibiotics produced by a Gram-negative bacterium. The pinensins A and B both contain two methyllanthionine rings, and differ by an additional alanine at the C-terminus in pinensin A. Both peptides were found to exhibit a weak antibacterial but in combination a broad antifungal activity against filamentous fungi and yeast (MIC 2.1–4.2 μg ml−1, Table 2). Their target and the mechanism behind the activity remain elusive.73
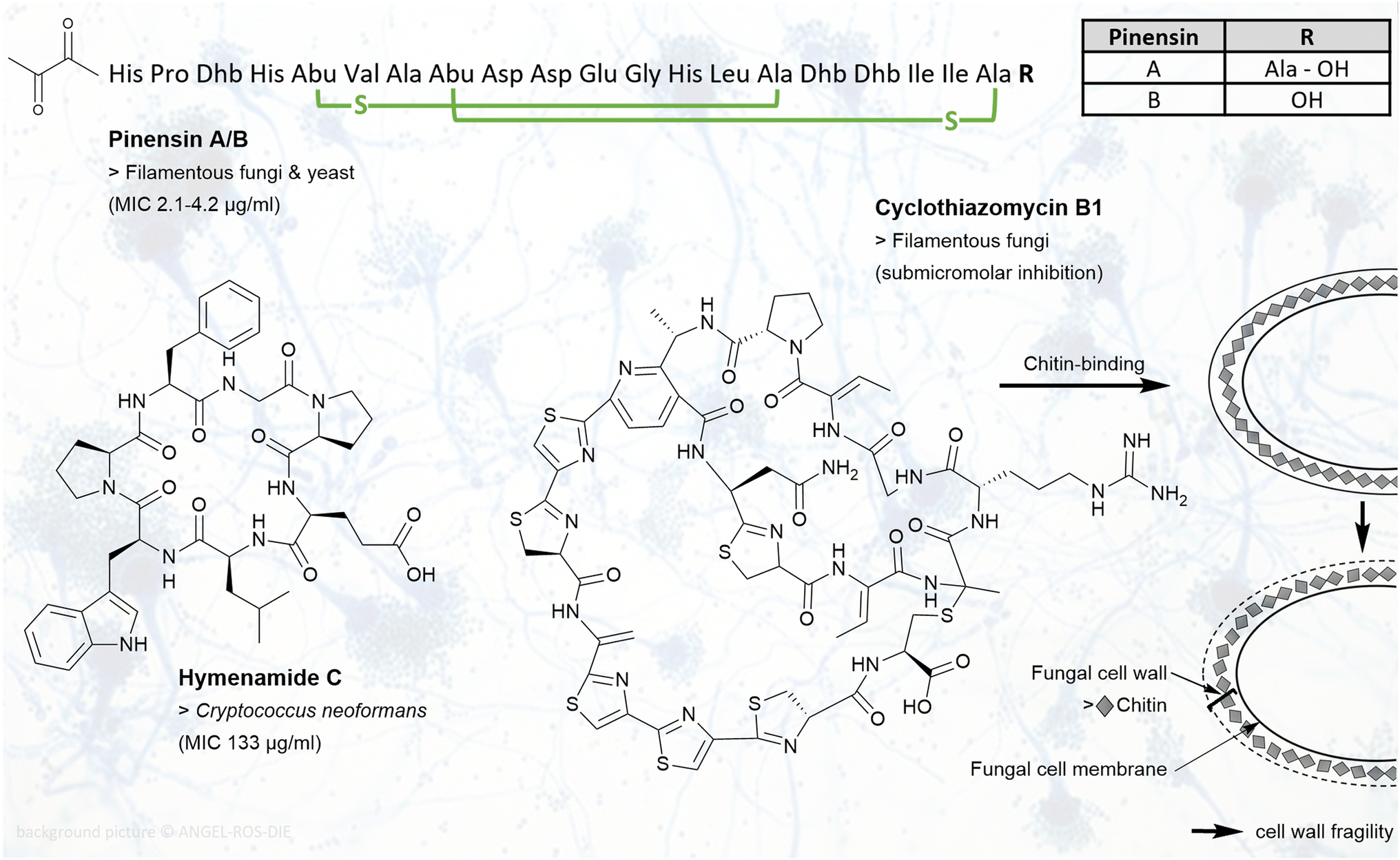 | ||
| Fig. 3 RiPPs with antifungal activities: chemical structures of pinensin A/B, hymenamide C, and cyclothiazomycin B1 are shown. Example(s) of affected fungi with MIC values are listed below the compound name. For pinensin A/B, the three-letter code is used. Dhb, dehydrobutyrine; Abu, 2-aminobutyric acid. Green indicated crosslinks are lanthionine bridges. For cyclothiazomycin B1, the target chitin in the fungal cell wall is shown. Binding of cyclothiamycin B1 to chitin leads to cell wall fragility. | ||
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined. | |||
| Cyclothiazomycin B1 | Filamentous fungi | Fungal cell wall | Submicromolar inhibition76 |
| Humidimycin | Antifungal enhancer | HOG pathway | +Caspofungin IC50 0.007 μg ml−1 (ref. 77) |
| Hymenamides | Non-filamentous fungi | n.d. | C. neoformans: MIC 133 μg ml−1 (hymenamides A, C, and E),74,75 |
| 33 μg ml−1 (hymenamide B) | |||
| C. albicans: MIC 33 μg ml−1 (hymenamide A) | |||
| 66 μg ml−1 (hymenamide B)75 | |||
| Pinensins | Filamentous fungi and yeast | n.d. | Yeast: MIC 2.1–4.2 μg ml−1 (ref. 73) |
|
|||
| Marketed (non-RiPP) antifungal drug | |||
| Caspofungin | Filamentous fungi and yeast | Fungal cell wall78 | Candida sp.: MIC 0.015–2 μg ml−1 (ref. 79) |
| Aspergillus sp.: MIC 0.25–16 μg ml−1 (ref. 80) | |||
Four cyclic heptapeptides of the cyanobactin family, hymenamide A–C (Fig. 3) and hymenamide E, were isolated from the marine sponge Hymeniacidon sp. Antifungal activity of these proline-rich peptides was observed against Cryptococcus neoformans with a MIC of 133 μg ml−1 for congeners A, C, E and 33 μg ml−1 for B. Hymenamide A and B showed additionally activity against Candida albicans with a MIC of 33 μg ml−1 and 66 μg ml−1, respectively.74,75
Cyclothiazomycin B1 (Fig. 3) is a cyclic peptide that belongs to the class of thiopeptides. It was isolated from a Streptomyces sp. strain and found to be fungistatic. By binding chitin in the fungal cell wall, it inhibits growth of filamentous fungi in a submicromolar range. This leads putatively to cell wall fragility. Activity against bacteria, yeast, or cytotoxic effects on mammalian cells were not observed.76
A lasso peptide with high structural similarities to the siamycins, is humidimycin (or MDN-0010, Fig. 5). It was isolated from Streptomyces humidus F-100.629. The peptide is not antifungal itself but exhibits a synergistic effect with approved antifungal drugs, like caspofungin (CAS). This makes humidimycin an antifungal enhancer. The antifungal effect of CAS in combination with humidimycin was shown to be enhanced by 4.5-fold, resulting in a remarkable low IC50 of 0.007 μg ml−1. Its target and the mechanism behind this effect are not yet fully understood. The high osmolarity glycerol (HOG) pathway is assumed to be involved in the response to humidimycin. When treated only with CAS, the HOG pathway may be employed to bypass the CAS-blocked pathway. Thus, tackling a different target than CAS could explain the observed synergistic effect. This could present a possible starting point to enhance CAS activity.77
2.3 Antiviral RiPPs
Epidemics and pandemics of viral diseases recurred frequently throughout human history. The recent emergence of the severe acute respiratory syndrome coronavirus (SARS-CoV2) and the coronavirus disease 2019 (COVID-19) represents just one example of the keen interest in new antiviral therapeutics.81 More than 40 peptides from different RiPP families are known to have antiviral activity, including members of proteusins, lasso peptides, and lanthipeptides.16Landornamide A (Fig. 3) is a member of the proteusin family and produced by Kamptonema sp. PCC 6506. It exhibits rare antiarenaviral activity, inhibiting the infection with lymphocytic choriomeningitis virus (LCMV) with an IC50 of 1.4–2.9 μM (Table 3).82 Investigations aiming to determine the target and mode of action of landornamide A are ongoing.
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined. | |||
| Cinnamycin | HSV-1 | Viral proliferation15,88 | n.d. |
| Divamide A | HIV | Cell entry or exit91 | n.d. |
| Duramycin | HSV-1, Ebola, West Nile virus, Dengue virus89 | Cell entry15,88 | n.d. |
| Labyrinthopeptins | HIV & HSV | Cell entry | LabyA1: HIV: EC502 0.79–3.3 μM |
| HSV: EC50 0.29–2.7 μM | |||
| LabyA2: HIV: EC50 of 26 μM (ref. 92) | |||
| Landornamide A | Arenavirus | n.d. | LCMV: IC50 1.4–2.9 μM (ref. 82) |
| Siamycins I-III | HIV & HSV | Viral cell fusion | (I) HIV: ID502 7 μg ml−1 HSV: ID50 48 μg ml−1 (ref. 93) |
| (II) HIV: ID50 9 μg ml−1 | |||
| HSV: ID50 27 μg ml−1 (ref. 93) | |||
| (III) HIV-1 reverse transcriptase: IC50 4 μg ml−1, HIV aspartyl protease: IC50 48 μg ml−1 (ref. 84) | |||
|
|||
| Marketed (non-RiPP) antiviral drugs | |||
| Docosanol | HSV | Cell entry | EC50 2.5 mg ml−1 (ref. 94) |
| Maraviroc | HIV | Cell entry | IC50 43 nM (ref. 95) |
| Saquinavir | HIV | HIV protease | IC50 1–10 nM (ref. 96) |
Several RiPPs with anti-HIV activity are known to date. The lasso peptides siamycin I and II differ in position 4: the valine residue is replaced by isoleucine in siamycin II. An ID50‡ 7 μg ml−1 for HIV and ID50 48 μg ml−1 for HSV (herpes simplex virus) was measured for siamycin I. Siamycin II, produced by Streptomyces sp. AA3891, displayed and ID50 of 9 μg ml−1 for HIV and an ID50 of 27 μg ml−1 for HSV. Their anti-HIV and anti-HSV activity is hypothesised to be related to blocking viral cell fusion.53 Structural similarities to gp41 transmembrane protein functional domains of HIV-1 were observed in siamycin III (or RP 71955 or arborycin), implying an analogous mode of action of all siamycins. Siamycin III is produced by Streptomyces sp. SP9440 and contains an isoleucine at position 4 like siamycin II, and a valine instead of isoleucine at position 17.83 Furthermore, siamycin III was found to affect HIV-1 by inhibiting the production of the reverse transcriptase (IC50 4 μg ml−1) and the aspartyl protease (IC50 48 μg ml−1) of HIV. Knowledge on the exact mechanism of action of the siamycins is not available yet.15,84,85 Based on its structural relatedness to the siamycins, humidimycin (mentioned above) is thought to have anti-HIV properties as well.86
Activity against HIV and HSV can be attributed to labyrinthopeptin A1 (LabyA1, Fig. 5) and A2 (LabyA2), which belong to a new class of carbacyclic lantibiotics, isolated from Actinomadura namibiensis DSM 6313. These compounds contain labionin, an αC quaternary substituted amino acid. This was the first time such a carbocyclic side chain linkage was described. Two serine and one cysteine residue(s) are linked by LabKC to form this unique structural feature. The cell entry of HIV and HSV is inhibited, in case of HIV by interaction of LabyA1 with the envelope protein gp120. Depending on the cell line tested, an EC50‡ of 0.79–3.3 μM for HIV and an EC50 of 0.29–2.7 μM for HSV was observed. LabyA1 further acts as anti-HIV enhancer. It showed an additive to synergistic effect together with anti(retro)viral drugs such as aciclovir, tenofovir and saquinavir. In contrast, LabyA2 was found to be about 10-fold less potent against HSV than LabyA1 and displayed only an EC50 of 26 μM against HIV. This notable difference in potency may be explained by the structural differences of the labyrinthopeptins. LabyA1 has two additional amino acids and LabyA1 and LabyA2 vary at five amino acid positions.87
In addition to their antimicrobial activity, cinnamycin and duramycin exhibit antiviral activity against HSV-1. By targeting phosphatidylethanolamine (PE), the viral proliferation is inhibited.15,88 Duramycin effects furthermore Ebola, West Nile and Dengue virus specifically by preventing virus attachment, thereby stopping cellular entry (Fig. 4).89
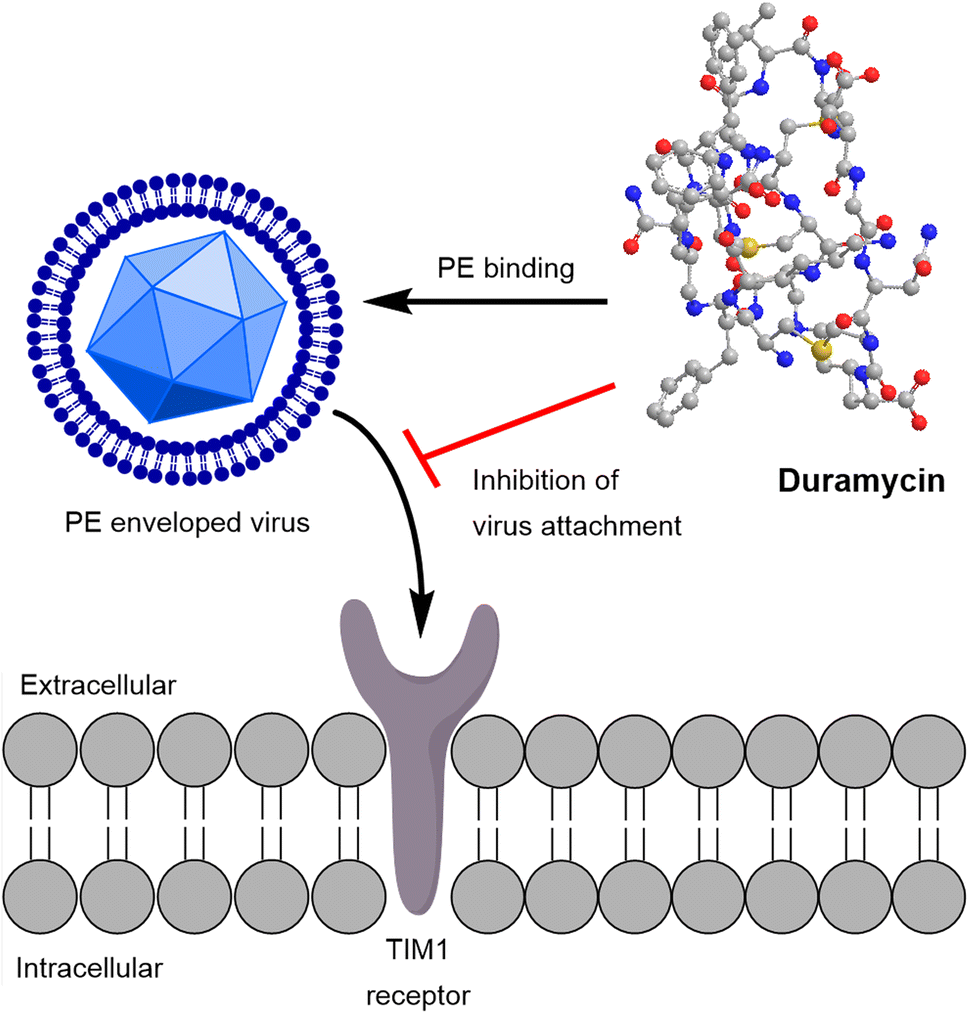 | ||
| Fig. 4 Duramycin targets enveloped viruses by binding PE (phosphatidylethanolamine). This leads to inhibition of virus attachment to the TIM1 membrane receptor in human cells and blocks cell entry. | ||
The divamides are cinnamycin-like anti-HIV peptides that were originally isolated from a marine tunicate. Later, it was found that divamide A–C were produced by a symbiont of the tunicate, the cyanobacterium Prochloron didemnid. Employing bioassay-guided fractionation, the anti-HIV activity could be assigned only to divamide A (Fig. 5). By structure activity relationship studies, the lysinoalanine residue was shown to be crucial for its antiviral activity. The exact underlying mode of action could not be clarified yet. Interaction of divamide A with T-cell membranes was observed, thus it was concluded it may block the viral cell entry or exit. Another bioactivity related discovery was the splitting of divamide A's cytotoxic and antiviral effect by minor changes in the sequence of amino acids.90
 | ||
| Fig. 5 RiPPs with antiviral activities: chemical structures of divamide A, siamycins and humidimycin, landornamide A, labyrinthopeptin A1, and XY3-3 are depicted. Example(s) of affected viruses with ID50, IC50 or EC50 values are listed below the compound name. For labyrinthopeptin A1, the three-letter code is used. Lab, labionin; Dhb, dehydrobutyrine. Yellow indicated crosslinks are disulphide bridges, blue indicated crosslinks are labionin bridges. | ||
2.4 Antiparasitic RiPPs
Parasitic diseases affect 3.5 billion people worldwide according to WHO.97 Economical and public health effects disproportionally burden the poorest nations and the drugs currently in use to treat these infections often show sub-optimal efficacy or serious toxicity. They were mostly discovered over 50 years ago, and today many drug-resistant strains exist. To combat these infections and mitigate the long lasting effects, it is imperative to find new drug leads.97,98 First, we will discuss antimicrobial RiPPs that have additional antiparasitic properties. Subsequently, we will provide examples from the defensin, cyanobactin, and cyclotide families. Defensins are antimicrobial peptides found in eukaryotes and prokaryotes; they are ribosomally synthesised and post-translationally modified, and can therefore be attributed to the natural product class of RiPPs.2,99,100Besides its antimicrobial activity (see Section 2.1), thiostrepton (Fig. 8) has potential as an antimalarial drug lead. It was shown to target the proteasome of the human malaria parasite Plasmodium falciparum. The natural compound itself shows modest antimalarial activity with an IC50 of 8.9 μM (Table 4), while optimised thiostrepton derivatives achieved IC50 values as low as 1.0 μM.101 Two independent targets were identified, the parasitic 20S proteasome and the large ribosomal subunit of the prokaryotic apicoplast, an essential cell organelle found in parasites belonging to the phylum Apicomplexa. Addressing two targets presents a substantial advantage, as it lessens the risk of resistance development. Additionally thiostrepton-based derivatives show considerable selectivity for the parasite proteasome over the human proteasome and no toxicity to human cell lines, making these compounds attractive antimalarial drug leads.101
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined. | |||
| Aeshna defensin | Antimalaria | n.d. | Dosed at 1 μg (ref. 107) |
| Balgacyclamides A-C | Antimalaria | n.d. | IC50 8.2–9 μM (ref. 110) |
| Balgacyclamides A-C | Anti-trypanosomatid | n.d. | IC50 51–59 μM (ref. 110) |
| Cycloviolacin O14 | Antihelmintic | n.d. | IC50 1.40 μM (ref. 116) |
| Kalata B1 | Antihelmintic | n.d. | IC50 3.36 μM (ref. 116) |
| Mollamide B | Antimalaria | n.d. | IC50 2 μg ml−1 (ref. 111) |
| Mollamide B | Leishmanicidal | n.d. | IC50 18 μg ml−1 (ref. 111) |
| Phlebotumus defensin | Leishmanicidal | n.d. | IC5068-85 μM (ref. 108) |
| Phormicin | Antimalaria | n.d. | Dosed at 1 μg (ref. 107) |
| Thiostrepton | Antimalaria | Proteasome | IC50 8.9 μM (ref. 101) |
| Venturamides | Antimalaria | n.d. | IC50 5.2–8.2 μM (ref. 112) |
| Venturamides | Anti-trypanosomatid | n.d. | IC50 14.6–15.8 μM (ref. 112) |
|
|||
| Marketed (non-RiPP) antiparasitic drugs | |||
| Atovaquone/Proguanil | Antimalaria | Mitochondrial cytb gene | IC50 3.4 nM and 36.5 μM (ref. 117) |
| Nifurtimox | Anti-trypanosomatid | Inhibition trypanozhione reductase, oxidative stress118 | IC50 0.9–3.4 μM (ref. 119) |
| Meglumine antimoniate | Leishmanicidal | n.d. | EC50 29.1–60.1 μg ml−1 (depending on strain)120 |
The defensins found in the dragonfly Aeshna cyanea, known as Aeshna defensin, and in the flesh fly Protophormia terraenovae, referred to as phormicin, exhibit antiparasitic activity against P. gallinaceum.102–104P. gallinaceum is a malarial parasite to birds of the genus Gallus and it is used as a model system in malaria research.105,106 Injecting Aeshna defensin or phormicin into the haemolymph of Aedes aegypti mosquitos 3–4 days after parasite ingestion significantly reduced the oocyte density, without showing toxicity against the mosquito hosts.107 A defensin from the sandfly Phlebotomus duboscqi active against Leishmania major was identified in a Leishmania infected sand fly. Leishmania spp. are parasites that cause leishmaniasis, a tropical disease with potentially fatal consequences. Recombinant Phlebotomus defensin was then tested against L. major, i.e. strain MHOM/YE/84, in vitro and IC50 values ranged from 68–85 μM, demonstrating antiparasitic activity for the Phlebotomus defensin.108,109
Balgacyclamides A–C (Fig. 8) belong to the family of cyanobactins and were isolated from Microcystis aeruginosa EAWAG 251. They were tested against a range of parasites, such as P. falciparum, Trypanosoma brucei rhodesiense, Trypanosoma cruzi, and Leishmania donovani. T.b. rhodesiense is the cause of African trypanosomiasis, which can lead to coma and death if left untreated. The best antiparasitic activity of balgacyclamides was detected against P. falciparum K1 with an IC50 of 8.2–9 μM, depending on the derivative. Besides, moderate activity against T.b. rhodesiense with IC50 values between 51–59 μM, and no activity against other parasites was reported.110 The cyanobactin mollamide B (Fig. 6), isolated from the tunicate Didemnum molle, was tested for its antiparasitic activity against the chloroquine-sensitive P. falciparum strain D6, the chloroquine-resistant P. falciparum strain W2, and L. donovani. It showed moderate activity with IC50 values of 2 μg ml−1, 2.1 μg ml−1, and 18 μg ml−1, respectively.111 Other peptides isolated from cyanobacteria, venturamide A and B (Fig. 6), were also tested against tropical parasites. They were isolated from Oscillatoria sp., a marine cyanobacterium. In bioassays both compounds showed moderate activity against P. falciparum and T. cruzi. Venturamide A and B showed IC50 values of 8.2 μM and 5.2 μM for P. falciparum, and 14.6 μM and 15.8 μM for T. cruzi, respectively.112
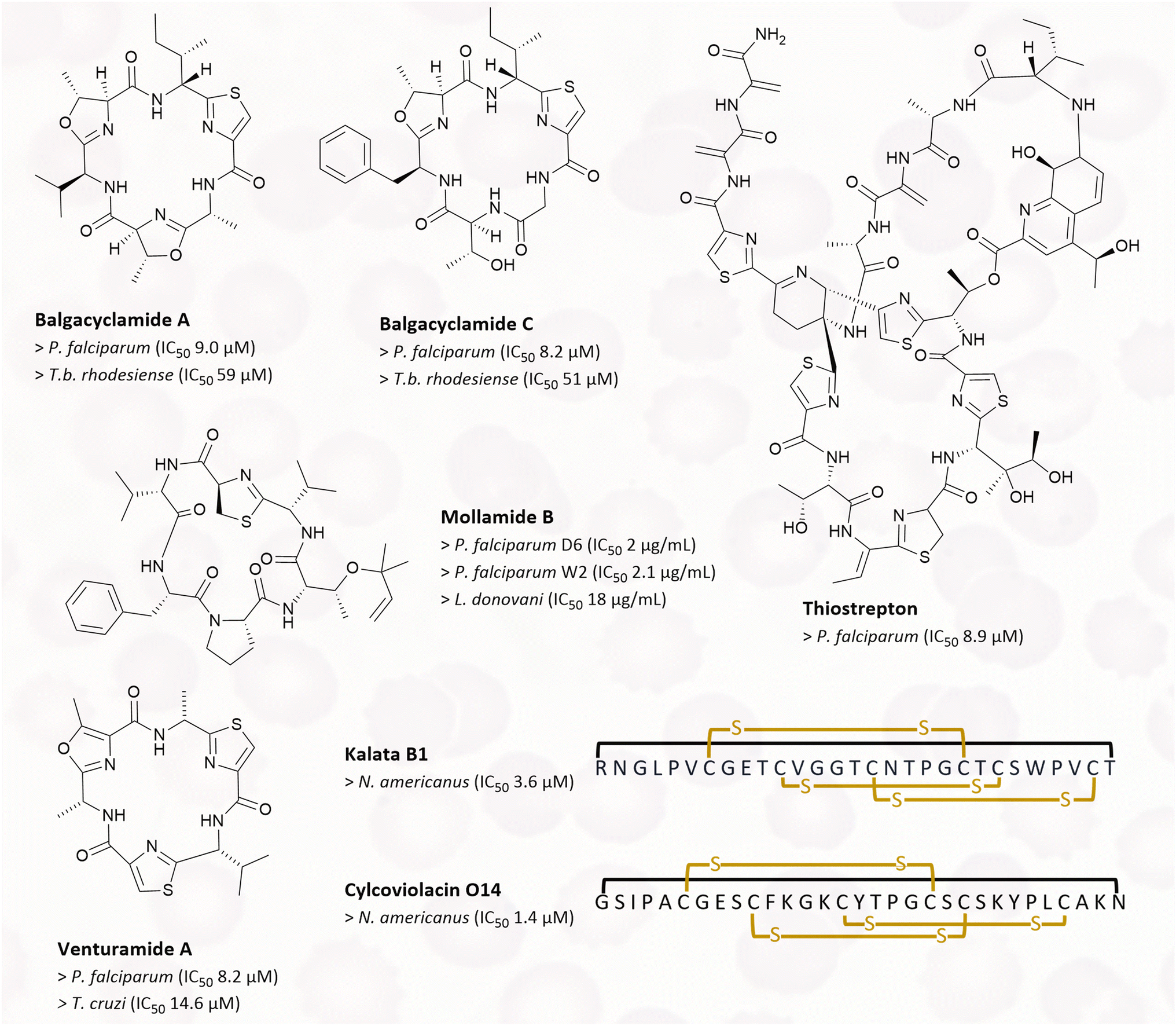 | ||
| Fig. 6 RiPPs with antiparasitic properties: chemical structures of balgacyclamide A and C, thiostrepton, mollamide B, venturamide A and B, kalata B1, and cycloviolacin O14 are shown. For kalata B1 and cycloviolacin O14, the one-letter code is used. Yellow indicated crosslinks are disulphide bridges, the black link indicates a macrocyclisation. IC50 values of affected parasites are listed below the respective compound names. | ||
Cyclotides are plant derived RiPPs, and some representatives of this family show anthelmintic activities. Kalata B1 was isolated from Oldenlandia affinis, and cycloviolacin O14 was isolated from Viola odorata (Fig. 6).113,114 Both peptides were screened for their anthelmintic properties against the human hookworm Necator americanus. Hookworm infections are a human health issue in the tropics and subtropics, and they are the leading cause of maternal and child morbidity. Infections in children can result in intellectual, cognitive and growth retardation.115In vitro tests conducted on N. americanus larvae showed that kalata B1 has an IC50 of 3.63 μM, while cycloviolacin O14 has an IC50 of 1.40 μM.116
2.5 Anticancer/antitumour RiPPs
Cancer is among the foremost causes of death around the globe, resulting in almost 10 million deaths in 2020.121 Consequently, there is a pressing need for improved clinical treatment options. Peptides and proteins are of particular interest, as they possess a significant advantage over small molecule drug candidates: they show a reduced probability of off-target interactions.122 In this section, we will first discuss thiostrepton and its analogues, followed by cyanobactins, bacteriocins, ammosamides and related pyrroloquinoline RiPPs, defensins, and RiPPs of an unknown class. The final section will discuss RiPP natural products that have no cytotoxic properties but inhibit lung cancer cell migration and RiPPs that have synergistic properties with chemotherapy drugs.Siomycin A was originally described as an antibiotic similar in structure to thiostrepton isolated from Streptomyces sp. H-690.123 In a study by Radhakrishnan et al. it was identified as a potential inhibitor of the transcription factor forkhead box M1 (FoxM1).124,125 Dysregulation of FoxM1 has been associated with lung cancer and basal cell breast carcinoma.126 Siomycin A appears to be a negative regulator of FoxM1, which at least partly contributes to its anticancer and proapoptotic activities. It was demonstrated that siomycin A treatment induces apoptosis only in transformed lung fibroblasts in culture, leaving non-cancerous cells nearly unaffected.124 Unsurprisingly, thiostrepton also has anticancer properties. It directly interacts with FoxM1 as shown in MCF7 breast cancer cell lines by Hegde et al.125
Cyanobactins are better known for their antimicrobial activities, but some also display cytotoxicity against human cancer cell lines. For instance, wewakazole B (Fig. 8), isolated from Moorea producens has an IC50 of 1.0 μM (Table 5) against human H460 lung and an IC50 of 0.58 μM against human MCF7 breast cancer cells.127 Lissoclinamides are a group of cyanobactins isolated from the ascidian Lissoclinum patella. Initial cytotoxicity assays revealed lissoclinamide 7 to be the most potent compound, with an IC50 of 0.06 μg ml−1 for urinary bladder carcinoma cells.128 Another cyanobactin from L. patella is patellin 6. In cytotoxicity assays, an IC50 of 2 μg ml−1 for P388 (leukaemia), A549 (lung carcinoma), HT29 (colon cancer) and CV1 (non-cancerous cells) cells was measured.129 Further expanding the group of cytotoxic cyanobactins, hymenamide B from Hymeniacidon sp. exhibited an IC50 of 3.2 and 6.0 μg ml−1 against murine lymphoma L1210 cells and human epidermoid carcinoma KB cells, respectively.75 Hymenamide B also has antifungal properties (see Section 2.2 above). Similarly, several peptides from Didemnum molle symbionts, comoramides A and B, and mayotamides A and B (Fig. 5) show mild cytotoxicity against A549 lung cancer cells, HT29 colorectal cancer cells, and MEL-28 malignant melanoma cells.130
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined, cancer cell type in brackets (if applicable).135 | |||
| 6OTD | Proapoptotic, anti-proliferative | Telomerase | GI50 21 nM (U251), 180 nM (SNB-78), 0.30 μM (average of 39 human cancer cell lines)158 |
| Ammosamides | Cytotoxic | Myosin | IC50 320 μM (HCT-116 cells)140 |
| Ammosester A | Cytotoxic | n.d. | EC50 56 μM (MDA-MB-231), 21 μM (SK-MEL-5), 140 μM (SF-295), 15 μM (NCI-H226), 17 μM (OVCAR-3)146 |
| Chaxapeptin | Inhibition of cell motility | 48% reduced motility speed at 50 μM (ref. 162) | |
| Comoramides and mayotamides | Cytotoxic | n.d. | IC50 5–10 μg ml−1 (A549, HT29, MEL-28)130 |
| Defb14 | Anticancer | n.d. | 50 μg per day (Lewis lung carcinoma)151 |
| Dendroamide A | Chemo-sensitisation | P-glycoprotein | 0.6 μM (MCF7-MDR)164 |
| Felipeptin A1 and A2 | Chemo-sensitisation | Downregulation of tumour suppressor Rb | Synergistic effects at 6.25 and 12.5 μM166 |
| Gymnopeptide A | Anticancer | n.d. | 88.4 nM (HeLa), 66.4 nM (A431), 26.5 nM (MCF7) |
| 37.4 nM (MDA-MB-231), 18.0 nM (T47D)153 | |||
| Gymnopeptide B | Anticancer | n.d. | 42.5 nM (HeLa), 44.3 nM (A431), 18.5 nM (MCF7) |
| 30.7 nM (MDA-MB-231), 14.0 nM (T47D)153 | |||
| Hymenamide B | Cytotoxic | n.d. | IC50 3.5 μg ml−1 (L1210) |
| 6.0 μg ml−1 (epidermoid carcinoma KB)75 | |||
| Kintamdin | Cytotoxic | n.d. | IC50 0.6 (HTB-22), 2.4 μM (CRL-11147), 12.0 μM (HTB-38)152 |
| Lissoclinamide 7 | Cytotoxic | Metal chelating167 | IC50 0.06 μg ml−1 (T24), 0.04 μg ml−1 (MRC5CV1)128 |
| Lymphostin | Antitumour | mTOR inhibition | IC50 1.7 nM (ref. 148) |
| Microcin E492 | Antitumour | n.d. | Induces apoptosis at ∼20 μg ml−1 in HeLa cells133 |
| Nisin A | Cytotoxic, proapoptotic | Induction of CHAC1 expression135 | IC50 600 μM (SWA480)134 |
| In vivo dosed at 200 mg kg−1 (UM-SCC-17B)135 | |||
| Nisin ZP | Antitumour | Calpain | 800 mg kg−1 (UM-SCC-17B)136 |
| Patellamide D | Chemo-sensitisation | n.d. | 3.3 μM (CEM/VLB100)165 |
| Patellin 6 | Cytotoxic | n.d. | 2 μg ml−1 (P388, A549, HT29 and CV1)129 |
| P. vulgaris defensins | Anticancer | n.d. | 4.1 μM (HepG2), 8.3 μM (MCF7)150 |
| Siomycin A | Anticancer, proapoptotic | Forkhead box M1 (ref. 168) | Inhibition in vivo shown at 20 μM (ref. 124) |
| Sungsanpin | Inhibition of cell motility | Induces expression of metalloproteinase 1 and 2 (ref. 160) | 53% reduced motility speed at 50 μM (ref. 162) |
| Telomestatin | Proapoptotic, anti-proliferative | Telomerase | GI50 6.5 μM (average of 39 human cancer cell lines)158 |
| Thiostrepton | Anticancer, proapoptotic | Forkhead box M1 | IC50 42.6 μM (ref. 125) |
| Ulleungdin | Inhibition of cell motility | 56% reduced motility speed at 50 μM (ref. 162) | |
| Wewakazole B | Anticancer | n.d. | IC50 1.0 μM (H460), IC50 0.58 μM (MCF7)127 |
|
|||
| Marketed (non-RiPP) anticancer drug | |||
| 5-Fluorouracil (5-FU) | Anticancer | Inhibition of thymidylate synthase169 | IC50 45–5063 ng ml−1 (range across 19 cancer cell lines)170 |
Class I bacteriocins represent a group of RiPPs that were initially reported as antimicrobials, and were subsequently discovered to possess a range of properties, including anticancer effects.131 One example is microcin E492 (MccE492), which showed activity against SW620 human colorectal cancer. Cancer cell viability decreased in a dose-dependent manner when treated with 0, 30 and 60 μg ml−1 MccE492 suspension in an in vivo model using zebrafish larvae. Additionally, MccE492 induces apoptosis in HeLa cells at about 20 μg ml−1 (Table 5). It is produced by Klebsiella pneumoniae RYC492 and it is mainly active against Enterobacteriaceae.132,133 Similarly, nisin A, a bacteriocin well known for its antibacterial application in food preservation, also displays cytotoxic properties. It induces apoptosis in cells of colon cancer, breast cancer, and hepatic cancer, with an IC50 of around 600 μM.134 Furthermore nisin A was tested in a floor-of-mouth oral cancer xenograft mouse model. Treatment with 200 mg kg−1 per day of nisin A led to significant tumour volume decrease with no observed adverse effects.135 Additionally Kamarajan et al.136 tested nisin ZP, a natural occurring variant of nisin A. Both nisin A and nisin ZP are produced by Lactococcus lactis. Nisin ZP was tested in a similar oral cancer floor-of-mouth mouse model. It was found to decelerate tumour growth, inhibit cancer cell proliferation, prevent angiogenic processes, suppress orasphere formation and tumorigenesis in vivo at dosages up to 800 mg kg−1 per day. Converting this dose to human administration would entail giving 66.7 mg kg−1 to humans, which falls well within the no observed adverse effect level (NOAEL) range of 225 mg nisin A kg−1 bodyweight identified by the European Food and Safety Administration. It is theorized that nisin ZP activates calpain which then leads to apoptosis in cancerous cells (Fig. 7).136–138
 | ||
| Fig. 7 Nisin ZP activates calpain in cancerous cells; this activation leads to the hydrolysis of different cytosolic and membrane proteins and induces apoptosis. Schematic is based on ref. 136 and 138. | ||
Ammosamides belong to the natural product class of pyrroloquinoline alkaloids, and their biosynthetic pathway contains cryptic RiPP genes.139 Ammosamides were initially isolated from the Streptomyces strain CNR-698 and are compounds with intriguing structures and bright colours, but with low solubility.140 Though they were originally thought to constitute a 16-member family, it has been shown that most ammosamides are artefacts of ammosamide C, forming derivatives when exposed to nucleophiles, air, and light.140,141 Ammosamides A and B (Fig. 8) display in vitro cytotoxicity towards the human colorectal cancer cell line HCT-116, with an IC50 of 320 nM. Myosin appears to be the intracellular target.142 Ammosamide B derivatives demonstrate potent quinone reductase 2 (QR2) inhibition. QR2 has been identified as a potential target for the development of chemotherapeutic drugs.143 Research by Li et al. indicates that ammosamides can be a highly effective scaffold for the development of targeted therapies.144 Several derivatives were synthesised using the pyrroloquinoline base to optimise inhibition of BRD4. BRD4 is a transcriptional and epigenetic regulator, well-established as a target in cancer research.145 Derivative 49 showed the highest inhibition against BRD4, when screened against MV4-11 and 22RV1, leukaemia and prostate cancer cell lines. Additionally, it also showed good anti-proliferative effects. Consequently, this compound displays potential as a new drug lead for chemotherapeutics.144 The ammosester subfamily of ammosamides was discovered in the genome of Streptomyces unicalis DCA2648 and exhibits modest cytotoxicity against several human cancer cell lines, including melanoma (SK-MEL-5), breast (MDA-MB-231), central nervous system (SF-295), non-small cell lung (NCI-H226), and ovarian (OVCAR-3) cancer.146
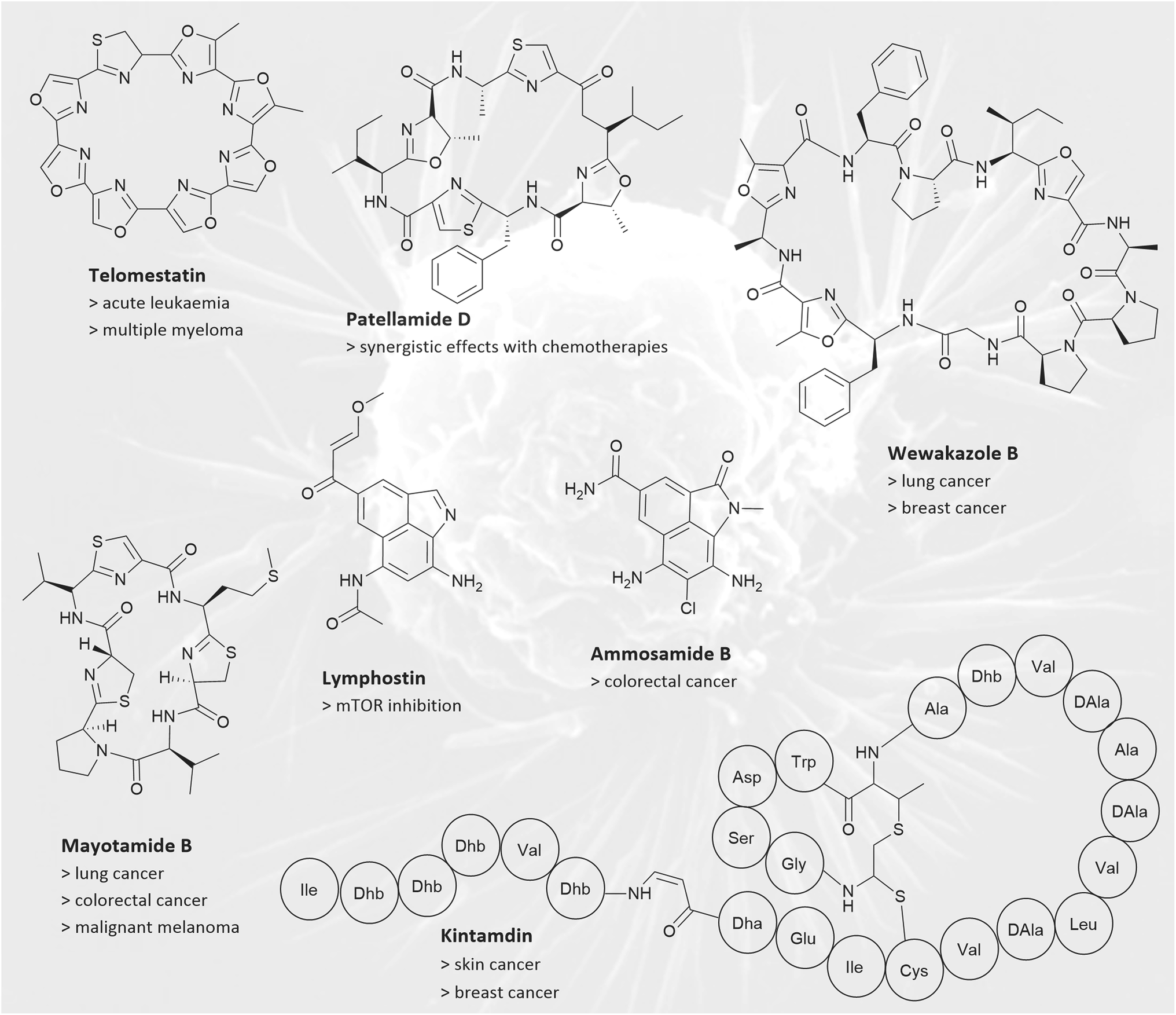 | ||
| Fig. 8 RiPPs with anticancer activities: chemical structures of ammosamide B, kintamidin, lymphostin, wewakazole B, telomestatin, patellamide D, and mayolamide B are shown. Examples for affected cancer cell lines are given below the compound names. For kintamdin, the three-letter code is used. | ||
Additional members of the pyrroloquinoline natural product family are the lymphostins (Fig. 8). Like ammosamides, they possess a non-canonical biosynthesis, involving genes associated with RiPP biosynthesis, the exact function of which require further investigation.139 Originally isolated from Streptomyces sp. KY11783, lymphostins and their derivatives are potent inhibitors of mTOR (mechanistic Target of Rapamycin), exhibiting IC50 values ranging between 0.8 and 1.8 nM.147,148 The serine/threonine kinase mTOR is involved in regulating cell survival, cell growth, cell metabolism, protein synthesis, and autophagy. As shown in animal models and clinical cancer patients, dysfunction of mTOR contributes to tumorigenesis.149
Some defensins (see Section 2.4) show anticancer activity both in vitro and in vivo. For instance, a defensin from Phaseolus vulgaris cv. “extra-long purple pole bean” displays potent antiproliferative properties against tumour cells. The IC50 values for hepatoma (HepG2) cells and breast cancer (MCF7) cells are 4.1 μM and 8.3 μM, respectively.150 Additionally, Defb14, a mouse defensin, was shown to restrain the growth of Lewis lung carcinoma in a mouse model without causing any observable damage to the surrounding tissue.151
Kintamdin is a novel, macrocyclic RiPP that does not fall into any known RiPP category (Fig. 8). Kintamdin was isolated from Streptomyces sp. RK44 and shows cytotoxic activity against skin and breast cancer cell lines, with IC50 values of 2.4 μM and 0.6 μM respectively.152 Other macrocyclic peptides of an unknown class were obtained from the mushroom Gymnopus fusipes, the cyclic gymnopeptides A and B. They show antiproliferative properties against cervical, epidermoid, and breast cancer cell lines with IC50 values ranging from10–90 nM.153 Telomestatin (Fig. 8), a macrocyclic RiPP isolated from Streptomyces anulatus, has demonstrated great potential as a chemotherapeutic agent.154,155 It interacts specifically with telomerase, resulting in proapoptotic and antiproliferative effects in acute leukaemia and multiple myeloma.156,157 Telomestatin has more than one mode of action, making it an ideal candidate for further development of anticancer agents. It causes telomere dysfunction, downregulates the proto-oncogene c-Myb, and induces a higher level of replication stress response in cancer cells compared to non-cancerous cells. The telomestatin analogue 6OTD shows activity in lower concentrations compared to the native peptide when exerting its anticancer effects on a human cancer cell line panel. On average 6OTD had an GI50‡ value of 0.30 μM for 39 cell lines, while telomestatin had an average GI50 of 6.5 μM.158 Additionally, 6OTD showed potent antitumour activity against human glioblastoma U251 cells, with treated tumours only reaching 33% of the non-treated tumour sizes in an in vivo mouse model. Other analogues are under investigation.158,159
Another approach to cancer treatment involves the use of drugs that inhibit cancer cell motility. Although not cytotoxic in nature, these compounds can help reduce the risk of cancer metastasis in patients. One group of compounds that can achieve this are certain lasso peptides, which have closely related sequences and structures. This cluster of lasso peptides is made up of sungsanpin, chaxapeptin, and ulleungdin. These compounds were isolated from Streptomyces sp. SNJ013, Streptomyces leeuwenhoekii C58 and Streptomyces sp. KCB13f003, respectively. Sungsanpin was first discovered in a deep-sea sediment sample, while chaxapeptin was found in the Atacama desert, and ulleungdin on a volcanic island.160–162 Despite their different origins, these natural products exhibit similar bioactivity. At a concentration of 50 μM, all three compounds inhibit cell migration of A549 lung cancer cells by approximately 50% compared to untreated cells.160–162 These three RiPPs have the potential to inspire a derivative compound for clinical applications. Digal et al. have already demonstrated that the knot motif is not necessary for retaining function. Macrocyclic and head-to-side chain derivatives of sungsanpin and ulleungdin have retained comparable bioactivity.163
In addition to classical cytotoxic antitumour medications, pharmaceuticals that work in synergy with chemotherapy drugs offer a promising approach, particularly for targeting multidrug-resistant tumours. One such compound is dendroamide A, which was isolated from Stigonema dendroideum and has been shown to increase the accumulation of vinblastine, a chemotherapy drug, in P-glycoprotein-overexpressing breast carcinoma cells (MCF7/ADR).164 Similarly, patellamide D (Fig. 8) from the marine tunicate Lissaclinum patella, reduced IC50 values for the cytotoxins vinblastine, colchicine, and adriamycin in CEM/VLB100 human leukaemic cells, reversing the cell line's multi drug resistance.165 The findings suggest potential for the use of patellamide D as a therapeutic agent for drug-resistant cancers. Felipeptin A1 and A2 are lasso peptides isolated from Amycolatopsis sp. YIM10. They have been shown to promote cell proliferation in cancer cell lines MCF7, HCT-116 and A375 (malignant melanoma). When applied individually, only marginal effects were observed. However, when both felipeptin A1 and A2 were combined, a synergistic pro-proliferation effect was observed in cell viability assays. Pre-treating A375 cell lines with 6.25 or 12.5 μM of each felipeptin increased the sensitivity to the cytotoxic chemotherapy drug doxorubicin. Pre-treating doxorubicin-resistant MCF7 cancer cells with felipeptins A1 and A2 re-sensitised them to the chemotherapeutic drug. This study demonstrates that felipeptin A1 and A2 have potential in future chemotherapy applications to reduce dosage and overcome resistance in clinical settings.166
2.6 Analgesic RiPPs
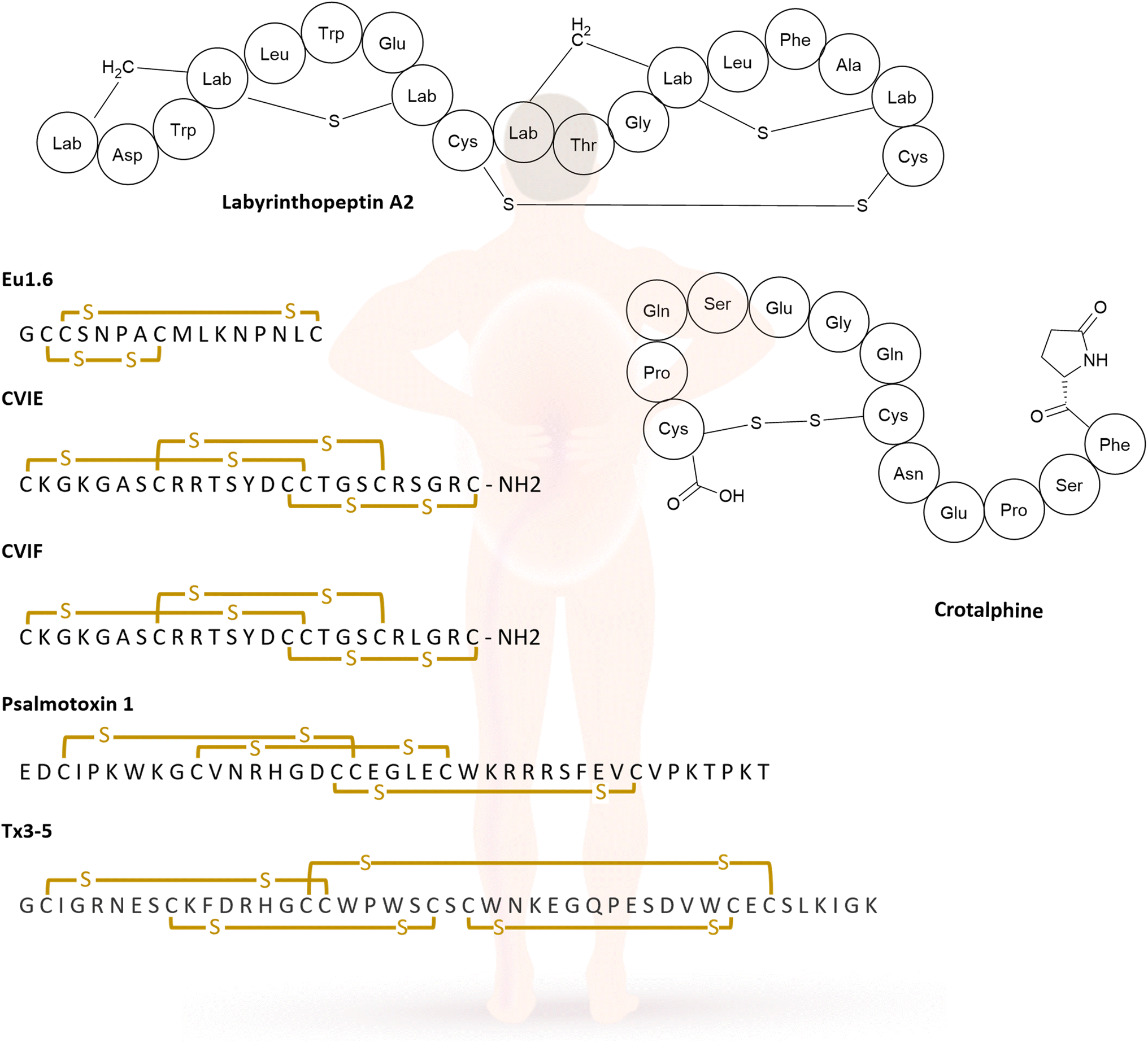
Chronic pain is an immense burden on people and public health providers worldwide; according to the US Center for Disease Control and Prevention point prevalence is at 20.9% for chronic pain in the United States.171 In contrast to acute pain, chronic pain has little evolutionary benefit and becomes a disease in its own right. Pain management is therefore of the utmost importance for patients to mitigate not only the pain itself but other biological, social, and psychological factors that occur alongside this condition.172 There a several RiPPs with analgesic properties. This section will begin by exploring two lanthipeptides, followed by an examination of conotoxins and finally, other venom peptides that possess analgesic properties. Conotoxins are peptides isolated from the cone snails venom, and are used by the snails for immobilising prey, which can later be swallowed whole.173The lanthipeptide labyrinthopeptin A2 (LabyA2, see Section 2.3 above) was first isolated because of its antiviral activity in culture extracts of Actinomadura namibiensis DSM 6313, further investigation was prompted by its unusual post-translational modifications. LabyA2 (Fig. 6) contains labionin, an unprecedented non-canonical amino acid. LabyA2 showed in vivo efficacy in a mouse model of neuropathic pain. In a spared nerve injury mouse model, intravenous administration of LabyA2 led to significant attenuation of tactile allodynia over a 6 h observation period (Table 6).174 Iorio et al. identified another lanthipeptide that contains labionin in Actinoplanes sp. DSM14059, known as NAI-112. In addition to labionin, NAI-112 has another unusual modification: the indole nitrogen of Trp13 carries a 6-deoxyhexose moiety. This is the first example of an N-glycosylated lanthipeptide and the first natural product in which a tryptophan residue is N-glycosylated. After observing similarities with LabyA2, the researchers tested for antinociceptive activity. In these assays, NAI-112 proved to be effective in alleviating acute pain induced by formalin injections, by reducing hyperalgesia and allodynia in a dose-dependent manner. It also demonstrated efficacy on established chronic pains in a mouse model with chronic constriction injury, with full effect at a dose of 30 mg kg−1 of NAI-112. Furthermore, there was no indication of NAI-112 affecting motor coordination up to 60 minutes after administration and no signs of toxicity were observed.175
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined. | |||
| Crotalphine | Antinociceptive | κ and δ opioid receptors | Dosed at 1 μg kg−1 (ref. 183) |
| CVIE and CVIF | Antiallodynic | Neuronal-type Ca2+ channels | Dosed at 1 nM (ref. 184) |
| Eu1.6 | Analgesic | Neuronal-type Ca2+ channels | IC50 1.1 nM (ref. 103) |
| Labyrinthopeptin A2 | Antiallodynic | n.d. | ED50 50 μg kg−1 (ref. 174) |
| NAI-112 | Antinociceptive | n.d. | Dosed at 30 mg kg−1 (ref. 175) |
| Psalmotoxin 1 | Analgesic | ASIC1a | Dosed at 0.1 nmol per mouse181 |
| Tx3–5 | Antinociceptive | n.d. | ID50 16.6 fmol per site, max inhibition dose of 30 fmol per site182 |
|
|||
| Marketed (RiPP) analgesic drug | |||
| Ziconotide | Antiallodynic | Neuronal-type Ca2+ channels | IC50 2–29 pmol (binding to Ca2+ channel), in vivo models: ID50 3–30 pmol (acute pain), ID50 30–1000 ng (chronic pain)185 |
The most prominent group of analgesic peptide drug leads comprise conotoxins. One example is the α-conopeptide Eu1.6 from Conus eburneus (Fig. 10), which shows significant analgesic activity in neuropathic pain models at low doses, surpassing the positive control of morphine and gabapentin. Eu1.6 represents the first conopeptide showing this effect with intravenous administration in contrast to intrathecal injections, which are injected into the spinal canal. Additionally, no significant impact on the motor, cardiac or respiratory function of mice was observed even at a dose that was 100 times higher than the effective dose. This result renders Eu1.6 a promising lead structure in treating neuropathic pain.176
The ω-conotoxins CVIE and CVIF (Fig. 10) were isolated from Conus catus venom, and they are selective inhibitors of neuronal-type Ca2+ channel (Fig. 9). In a rat model of persistent pain, intrathecal administration of 1 nM led to a significant reversal of mechanical allodynia to the pre-injury baseline levels. Concurrently side effects of shakes, tail twitching, and serpentine tail movements were recorded.177 Those side effects are typical, and they do not necessarily represent a hurdle in developing these compounds into pharmaceuticals. Ziconotide, a synthetic derivative of the conotoxin MVIIA also belongs to the ω-conotoxin family and is the first RiPP drug to be approved by the FDA (see Section 3.2 below).178
 | ||
| Fig. 9 The conotoxin CVIE selectively blocks N-type Cav2.2 calcium channels, thereby inhibiting nociception. | ||
The group of venom ribosomal peptide natural product is vast, with many valuable peptides found not only in cone snail venom but also spider venoms. Some examples are described below; for a more in-depth review see Wu et al.179 Psalmotoxin 1 (PcTx1, Fig. 10) was isolated from the South American tarantula Psalmopoeus cambridgei. It is non-lethal and blocks ASIC1a neurons that are associated with a variety of pain sensations.180 In different rodent models, PcTx1 showed similar pain relief as morphine, for both acute and neuropathic pain. In cases of chronic constriction injury (CCI) of the sciatic nerve in rats, thermal hyperalgesia and tactile allodynia were reversed by PcTx1. The peptide needs to be injected intrathecally or intracerebroventricularly, as intraperitoneal or subcutaneous injections had no effect.181 Another example is the peptide Tx3–5 (Fig. 10) from Phoneutria nigriventer venom. It has been proven that intrathecal injection of Tx3–5 in different mouse models can prevent or reverse postoperative nociception, show partial antinociceptive effects in a neuropathic pain model, and can nearly abolish cancer related nociception. However, it did not change noxious heat sensitivity or mechanical threshold, implying no effects on physiological pain. Additionally, no visible adverse effects were observed at the administered dose, with a TD50 about 50-fold higher than maximum inhibition dose.182
 | ||
| Fig. 10 Examples for RiPPs with analgesic activities. Structures of labyrinthopeptin A2, crotalphine, Eu1.6, CVIE, CVIF, psalmotoxin 1, and Tx3–5 are depicted. For Eu1.6, CVIE, CVIF, psalmotoxin 1, and Tx3–5, the one-letter code is used. Yellow indicated crosslinks are disulphide bridges. | ||
Crotalphine (Fig. 10), a venom-derived peptide obtained from the South American rattlesnake Crotalus durissus terrificus, also exhibited antinociceptive properties. The oral administration of crotalphine effectively blocked hyperalgesia induced by PGE2, an inflammatory agent. The peptide demonstrated a dose-dependent, antinociceptive effect over a course of 5 days. Injecting crotalphine intravenously and intraplantarly (into the sole of the foot) also showed long lasting antinociceptive effects, both systematically and locally, for PGE2-induced hyperalgesia. The effects are most likely mediated by activation of κ-opioid receptors. Oral availability and long lasting effects indicate the therapeutic potential of crotalphine and its derivatives for treating chronic pain.183
2.7 Further bioactivities
Newly discovered natural products are usually tested for the most common or most urgently needed bioactivities, such as antibiotic, antiviral, antifungal, and anticancer activities. However, there are some other interesting bioactivities such as antiinflammatory, antidiabetic, antihypertensive, and antiparkinsonian effects. Examples of RiPPs showing these bioactivities are described for different RiPP families, i.e., lanthipeptides, lasso peptides, and cyanobactins.The recently discovered lanthipeptides myxococin A and B from Myxococcus fulvus show antiinflammatory effects in lipopolysaccharide-induced mouse macrophages without any detectable cytotoxicity, which makes them interesting drug candidates (Table 7).186 Ancovenin is a cinnamycin derivative isolated from Streptomyces sp. No. A647P-2 and belongs to the lanthipeptide subfamily as well. It shows antihypertensive activity by inhibiting the angiotensin 1-converting enzyme (ACE) with an IC50 of 0.87 μM and was named after this activity (angiotensin converting enzyme inhibitor). ACE inhibition is clinically used for the treatment of high blood pressure.187–189
| Compound name | Bioactivity | Target (if known) | Activity metrics |
|---|---|---|---|
| a n.d. = not determined. | |||
| Acalitide | Neuroprotective/anti-parkinsonian | Dopamine neurons204 | n.d. |
| Agardhipeptin A | Regulation of fibrinolysis | Plasmin | IC50 65 μg ml−1 (ref. 202) |
| Anantin | Antihypertensive | Atrial natriuretic peptide receptor | IC50 1.0 μM (ref. 190) |
| Ancovenin | Antihypertensive | Angiotensin converting enzyme | IC50 0.87 μM (ref. 188) |
| BI-32169 | Antidiabetic | Glucagon receptor | IC50 440 nM (ref. 199) |
| BI-32169–CH3 | Antidiabetic | Glucagon receptor | IC50 320 nM (ref. 199) |
| Myxococin A + B | Antiinflammatory186 | n.d. | n.d. |
| RES-701-1 | Antihypertensive | Type B endothelin receptor | IC50 10 nM (ref. 192) |
In the class II of lasso peptides, two receptor antagonists are found: Anantin and RES-701-1. Anantin, a peptide isolated from Streptomyces coerulescens, binds to the atrial natriuretic peptide (ANP) receptor. ANP is the natural agonist of this membrane receptor, and it is involved in blood pressure homeostasis; Anantin is the first microbially produced antagonist of ANP.190,191 RES-701-1 was isolated from Streptomyces sp. RE-701 and binds strongly to the type B endothelin receptor with an IC50 of 10 nM. Endothelins are a group of potent vasoactive peptides and high endothelin levels are found in several pathophysiological conditions such as systemic hypertension, cardiac ischemia, and asthmatic attacks.192 The endothelin type B receptor mediates vasoconstriction and vasodilatation, hence it is involved in blood pressure regulation. Several RES-701-1-related compounds have been isolated: the derivative RES-701-2 from the RES-701-1 producer and RES-701-3 and RES-701-4 from Streptomyces sp. RE-896. The derivatives differ in two positions; 1 and 2 contain an alanine in position 7, while 3 and 4 contain a serine in this position, and 1 and 3 contain a tryptophan in position 16, whereas 2 and 4 contain 7-hydroxy-tryptophan.193 The biosynthetic gene cluster of RES-701-3 and -4 has recently been identified in the marine bacterium Streptomyces caniferus CA-271066.194 RES-701-1 and its derivatives are selective endothelin antagonists with RES-701-3 > -1 > -2 > -4 (order of potency).193 Moreover, synthetic derivatives of RES-701-1 have been developed: a hybrid peptide chemically synthesised from the RES-701-1 N-terminus and the endothelin-1 C-terminus exhibited an IC50 of 0.24 nM.195 Further derivatives also showed antagonistic activity for the endothelin type A receptor.196,197 These results nicely exemplify that natural products are promising lead structures for drug development.
The compound BI-32169 belongs to class III of the lasso peptide subfamily.198 BI-32169 is produced by Streptomyces sp. (DSM 14996) and inhibits the human glucagon receptor with an IC50 of 440 nM. Its methyl ester derivative exhibits slightly more potent inhibitory activity with an IC50 of 320 nM.199,200 Glucagon antagonists are considered promising candidates for new antidiabetic therapies.201
The macrocyclic peptide agardhipeptin A was isolated from the cyanobacterium Oscillatoria agardhii (NIES-204) and belongs to the RiPP family of cyanobactins. Agardhipeptin A is a weak plasmin inhibitor (IC50 of 65 μg ml−1); the protease plasmin is involved in the regulation of fibrinolysis and plasmin inhibitors are clinically used to treat hyperfibrinolysis-associated bleeding events.202,203
The recently discovered fungal RiPP natural product acalitide is a macrotricyclised compound with two disulphide bridges; disulphide bridges are a common motif in marketed peptide drugs. Acalitide was isolated from the ascomycete Acaulium album and shows neuroprotective effects. It is a promising drug candidate to treat the Parkinson's disease.204
2.8 Imaging and diagnostic agents
Therapeutic applications not only encompass medication but also diagnostics such as radiocontrast agents, medical probes, or microscopic dyes. A few RiPPs, mainly analogues of peptide hormones, are used for such applications.Phalloidin is a macrocyclic RiPP of the phallotoxin group produced by the fungus Amanita phalloides.205 Functionalised with a fluorophore, phalloidin derivatives are commonly used as a stain in immunofluorescence microscopy because of their high affinity for actin filaments in cells and tissues.206
Bombesin is a peptide hormone isolated from the skin of the toad Bombina bombina.207 It binds with high affinity to receptors of certain cancer cells, i.e. gastrin-releasing peptide receptor (GRPR)-positive tumours such as human prostate and breast tumours, as well as small-cell lung, ovarian, and endometrial cancer. Fluorescently labelled bombesin derivatives have been investigated for the application in peptide receptor imaging.208–210 Later on, bombesin analogues for targeted tumour therapy approaches were developed, since they might be used as drug shuttles for the intracellular delivery of different cytotoxic compounds.211
Several other compounds derived from peptide hormones can be used in radiopharmaceutical applications. Examples include somatostatin analogues for the diagnosis of neuroendocrine tumours,212,213 glucagon-like peptide-1 analogues for imaging of beta cell function in diabetes patients,214,215 and cholecystokinin analogues for the visualisation of medullary thyroid carcinomas, small-cell lung cancers, and stromal ovarian cancers.216,217
2.9 Synthetic, engineered RiPP natural products
The previous sections have shown that RiPPs exhibit a broad range of bioactivities and are interesting drug candidates for treating a variety of diseases. In addition, the RiPP biosynthesis represents a possible biotechnological production route, and it is particularly well suited for engineering approaches to produce customised peptide products for pharmaceutical applications. For more comprehensive reviews on RiPP engineering, the reader is referred to ref. 17–19.Several bioengineered RiPPs with new activities have been identified in different screening approaches. The protein–protein interaction (PPI) of the HIV p6 protein with the human TSG101 protein at its UEV domain is crucial for viral cell exit. Aiming to find an inhibitor of this PPI, a lanthipeptide library with the prochlorosin leader peptide and diversified core peptides was constructed. To generate bicyclic peptides, the promiscuous synthetase ProcM from Prochlorococcus was employed. One selective inhibitor, the peptide variant XY3-3 (Fig. 3), was obtained. Binding to UEV at a distinct site to p6 protein was proven. Interaction of p6-UEV was shown to be disrupted with an IC50 of 3.6 μM. Moreover, the requirement of both thioether rings for binding and thus for the activity of XY3-3 was demonstrated.218 XY3-3 is an interesting antiviral drug candidate. Further examples for bioengineered RiPPs are compounds containing the ‘RGD’ binding epitope. The human integrin αVβ3 is a potential drug target that recognises the ‘RGD’ motif. Compounds targeting the αVβ3 integrin receptor may be used as antiangiogenics in cancer diagnostics and therapy. By precursor engineering, this ‘RGD’ motif has been introduced into many different RiPPs, including lanthipeptides,219,220 lasso peptides,221–223 knottins,224,225 and cyclotides (θ-defensins).226
In general, RiPP pathways are relatively short and streamlined; the biosynthetic gene clusters consist of separate precursor peptide(s) and post-translationally acting enzymes that are often promiscuous. This modularity facilitates the gene cluster manipulation and allows to mix and match enzymes from different pathways to generate tailored designer peptides. The promiscuity of the maturases is founded on the separation of the substrate recognition site and the modification site; this spatial segregation allows the modification of various core sequences. Furthermore, the gene-encoded precursor peptides can be modified easily by simple mutagenesis and enable the rapid generation of novel peptide variants.19
Even though combinatorial biosynthesis of RiPP pathways is theoretically easy, the combination of enzymes from different pathways can be challenging. Many RiPP maturases contain so-called RiPP recognition elements (RRE) to recognise and bind the leader part of their cognate precursor peptides.227 Consequently, they do not accept precursor peptides with different or without leader peptides. For such naturally incompatible maturase-precursor combinations, where maturases are inactive towards designed core peptides, various engineering strategies have been described, see ref. 18 for a comprehensive overview. Here, we will only briefly mention the most common strategies. The leader peptide part can be engineered to promote correct protein–protein interactions between the leader peptide and the maturase, e.g. rationally designed chimeric leader peptides harbouring RREs for different maturases can be employed.228 However, this approach is quite time-consuming, since the leader parts required for maturase activity need to be experimentally determined for every single maturase. In a second step, the chimeric leader needs to be produced and tested. Leader peptide complementation could be a simpler strategy. The required leader peptide can be supplied either in cis, meaning it is covalently linked to the maturase, or in trans, meaning it is expressed as a separate peptide molecule.229–232 However, this strategy is not generally applicable to all types of RiPP systems, but it works for specific RiPPs. Another option would be a sortase-based leader-peptide exchange approach,233 in which a sortase A recognition site is introduced between the leader peptide and the core peptide. The sortase exchanges the leader peptide of one maturase with the leader peptide of a second or third maturase. To incorporate unnatural amino acids that cannot be introduced by post-translational modifications of proteinogenic amino acids, the flexizyme technology can be applied.234,235 This technology enables the reprogramming of codons to accommodate unnatural amino acids using aptamers to charge tRNAs in vitro.
In summary, several options to circumvent compatibility issues have been described. The bioengineering of RiPPs is still in its early stages, and several obstacles must be overcome before RiPP processing enzymes from various subfamilies can be effectively combined for targeted peptide engineering. The RiPP technology represents a great chance for drug development by offering opportunities to generate a broad range of drug candidate derivatives in peptide libraries. Moreover, these engineered pathways could at the same time be suitable to produce peptide drugs at a large scale, superseding multi-step total synthesis of these compounds.
3 Advanced RiPP (candidate) drugs
3.1 RiPPs in clinical trials
Although many RiPPs have been discovered exhibiting pharmaceutically interesting bioactivities, only few entered clinical trials so far. Data about clinical trials is hard to access since most of it is not published in the peer-reviewed literature. Therefore, the following enumeration is not comprehensive, but contains the candidates we were able to identify with our means.Cystic fibrosis is caused by a defective or missing cystic fibrosis transmembrane conductance regulator (CFTR) anion channel. Lancovutide (or Moli1901 or duramycin) was demonstrated to activate chloride channels, thereby having the potential to compensate for the dysfunctional CFTR. It entered phase IIb clinical trial for treatment of cystic fibrosis in 2007. Since the administration of lancovutide compared to placebo showed no significant positive effect on cystic fibrosis patients, no further clinical investigations were carried out to our knowledge.236,237
Two semisynthetic derivatives of thiopeptide GE2270A, LFF571 and CB-06-01 (or NAI-003), completed both phase II clinical trials. Novartis Pharmaceuticals developed and tested LFF571 successfully against moderate C. difficile infections. Knowledge about the target of LFF571 is not published. By terminating the antibacterial research in 2018, the LFF571 project was stopped by Novartis.238
The company Cassiopea SpA made a proof-of-concept investigation of CB-06-01 for treatment of moderate-to-severe acne. CB-06-01 assumably binds the elongation factor Tu in Propionibacterium acnes, thereby abolishing protein biosynthesis. Derivatisation of GE2270A to CB-06-01 reduced the spectrum of activity but led to a lower minimal inhibitory concentration against P. acnes. Phase II dose-ranging studies of CB-06-01 were planned to be performed in the future according to Cassiopea SpA.239–241
There are multiple conotoxins known that possess clinically relevant properties. Conotoxin Vc1.1 was isolated from Conus victoriae and discovered to act as a nicotinic acetylcholine receptor antagonist (nAChR). The synthetic version of Vc1.1, named ACV1, was found to be a potential therapeutic for the treatment of diabetic neuropathy, post herpetic neuralgia, and sciatic neuropathic pain.242 Metabolic Pharmaceuticals Ltd tested ACV1 for treatment of neuropathic pain in a phase I clinical trial, aiming to assess the safety, tolerability, pharmacokinetics and -dynamics.243 Furthermore, they examined the application of ACV1 as medication for diabetic peripheral neuropathy and post-herpetic neuralgia in a phase II study.244 Due to remarkably lower efficiency of ACV1 in humans compared to rats and thus immensely higher doses necessary in human treatment, Metabolic Pharmaceuticals Ltd eventually terminated its ACV1 clinical research project.245
MrIA and MrIB, two closely related conopeptides produced by Conus marmoreus, founded the class of χ-conopeptides. They allosterically target the neuronal norepinephrine transporter (NET).246 To encounter the instability of natural MrIA peptide, Xenome Ltd synthesised a derivate of MrIA, Xen2174. Considering the enhanced stability, duration of analgesia and an equivalent efficacy vs. side effects window of Xen2174, clinical studies have been conducted.247–249 Two different clinical trials with healthy subjects were carried out under Xenome Ltd, investigating its therapeutic potential in pain management. Besides evaluating safety, tolerability, pharmacokinetics and -dynamics, pain modalities affected by Xen2174 and dose-dependent effects were investigated. The outcome was positively evaluated in both studies.250,251 However, a third trial comparing administration of Xen2174 alone and in interplay of Xen2174 with the well-established local anaesthetic bupivacaine, was rated negative after completion.252,253 Several other conopeptides are described as currently being in or having been in (pre-)clinical trials.254
3.2 RiPPs on the market
In the previous chapters, we presented an overview of potential drug candidates in the clinical trial pipeline of RiPPs. In this chapter, we will review those RiPPs that have been approved for a variety of indications.Ziconotide (Fig. 11), known as Prialt®, is perhaps the most prominent example of a RiPP drug. It is a synthetic form of the ω-conotoxin MVIIA, which was originally discovered in Conus magus, and subsequently reported on by Olivera et al. in 1987.255 Ziconotide binds to N-type calcium channels in the spinal cord, and it is the only non-opioid analgesic approved for the treatment of chronic pain in Europe and the USA. It is administered intrathecally and recommended as first line treatment for neuropathic and nociceptive pain. Although adverse side effects continue to pose a concern, ziconotide has significant benefits, including the absence of bone marrow toxicity and respiratory depression, as well as displaying no signs of withdrawal symptoms post-treatment.256 Presently, investigation is underway to optimise treatment plans and mitigate adverse side effects (http://ClinicalTrials.gov Identifier NCT04321408), which underlines the importance of this drug.
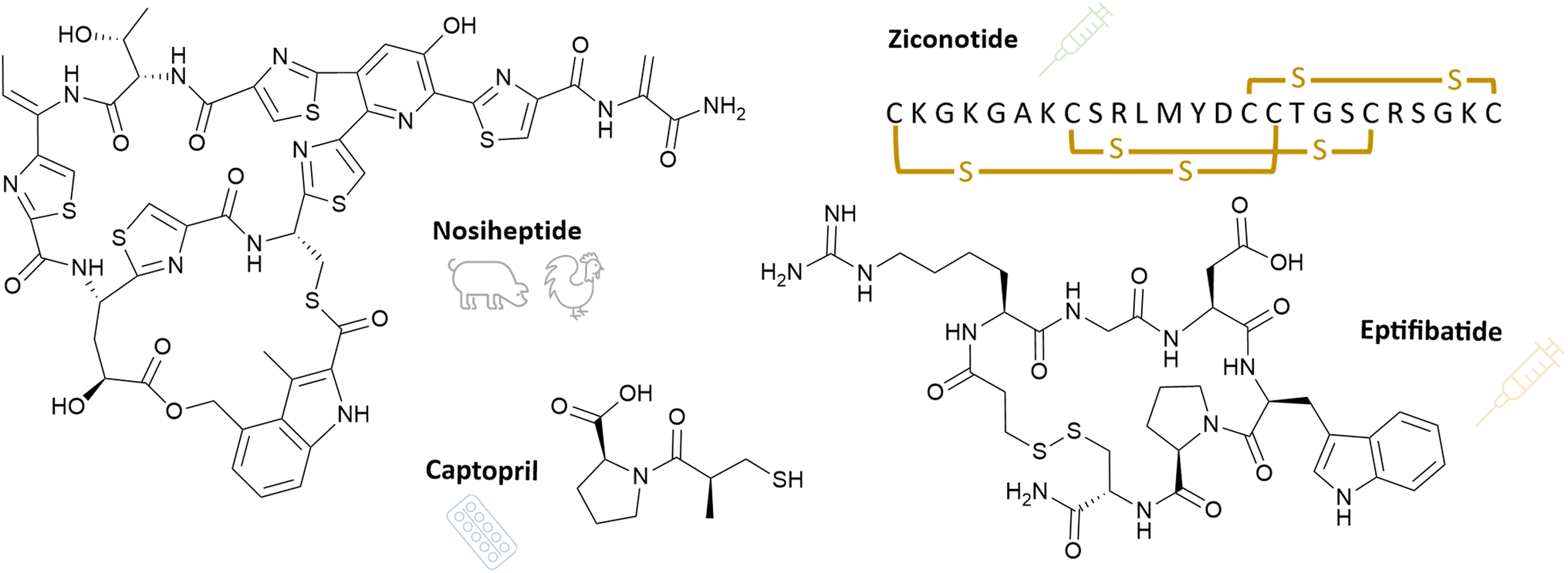 | ||
| Fig. 11 RiPP(-based) compounds, that are currently in clinical use. Chemical structures of nosiheptide, captopril, eptifibatide, and ziconotide are depicted. For ziconotide, the one-letter code is used. Yellow indicated crosslinks are disulphide bridges. | ||
Eptifibatide (Fig. 11) has been approved by both by the FDA and EMA for treating acute coronary syndrome since the late 1990s. It is derived from barbourin, a venom peptide found in the southeastern pygmy rattlesnake, Sistrurus milliarius barbourin.257 Eptifibatide is a cyclic heptapeptide and contains a modified version of the common ‘KGD motif’. The ‘RGD motif’ allows it to bind to glycoprotein IIb/IIIa, the target protein. Additionally, its tertiary structure is essential to the specificity. Glycoprotein IIb/IIIa constitutes the final common pathway accountable for platelet aggregation. Inhibiting this pathway confers eptifibatide with its antithrombotic effect. More recently other possible indications have been investigated, i.e., the treatment of ischemic stroke, carotid stenting, stenting of intracranial aneurysms, and septic shock. While initial studies have shown promising results more extensive investigations are required to confirm its therapeutic value.258,259
Captopril (Fig. 11) is the first angiotensin-converting enzyme (ACE) inhibitor, a drug class used to treat high blood pressure. It is derived from a venom peptide, called bradykinin-potentiating factor, isolated from Bothrops jararaca venom.260,261 While intravenous applications showed antihypertensive properties, derivatising this peptide into an orally available drug while retaining antihypertensive properties paved the way for drug approval in the early eighties.262,263 Captopril is the prototype of all ACE inhibitor drugs, a class still used as first line treatment in cardiovascular diseases, and new indications are emerging. These include but are not limited to atherosclerosis, heart attacks, and diabetic nephropathy.264
Thiostrepton is prominently featured throughout this article. Although it lacks approval for human use as of yet, thiostrepton finds veterinary application. It is an approved antibiotic for skin and eye infections, such as in Animax Ointment (NDC 17033-122-75).265 Like thiostrepton, nosiheptide (Fig. 11) belongs to the thiopeptide class of antibiotics. It was isolated from Streptomyces actuous 4003 and exhibits potent activity against Gram-positive bacteria.266 Additionally, nosiheptide possesses growth promoting properties, making it a beneficial feed additive for enhancing feed efficiency and weight gain in swine and chicken.267–269
4 Challenges and limitations
Compared to the significant number of RiPP natural products that exhibit intriguing bioactivities, as discussed in the first chapter ‘Opportunities’, only a small number of RiPPs are currently in clinical trials or on the market. The causes of this gap are diverse. The study of RiPPs and their biosynthetic machinery is much more recent compared to other natural product superfamilies, such as non-ribosomal peptides and polyketide natural products. Additional reasons include problems of stability and permeability, rapid renal elimination, and harmful side effects, among other factors. Traditionally, peptides have been considered as weak drug candidates due to their tendency to exhibit suboptimal ADME (Absorption, Distribution, Metabolism and Excretion) properties.2704.1 Oral bioavailability/gastrointestinal stability
Oral drug administration is generally preferred for reasons of compliance and convenience. However, canonical peptides, which are composed of proteinogenic amino acids, are often labile compounds. The acidic pH in the stomach, as well as pancreatic and intestinal proteases, cause rapid degradation. Therefore, it is a significant challenge to deliver peptides orally, and research in this area has been ongoing for over 100 years. Since the discovery of insulin, attempts have been made by researchers to design an oral form, but without success to date.271 Two prime examples of marketed, orally available peptides are cyclosporine A and desmopressin. The macrocyclic structure of both peptides enhances their protease and acid stability. Although RiPPs are not solely linear canonical peptides and certain post-translational modifications including epimerisations, backbone N-methylations, β-amino acids, and macrocyclisations can aid in improving their stability,19 acidic hydrolysis or proteases can result in partial degradation of the peptide compounds.4.2 Absorption
Another challenge posed by RiPP candidate drugs relates to their ability to enter cells. Their large molecular weight (>500 g mol−1) means that in most cases they do not satisfy the “Lipinski rule of five” criteria, and therefore have limited cell permeability, particularly when administered orally where there are additional restrictions due to intestinal epithelial permeability. This is because peptides, which are often hydrophilic, are not able to diffuse passively through physiological barriers and cell membranes efficiently.272One example of limited uptake is ziconotide (Prialt®), which is a drug available on the market (see Section 3.2). It cannot penetrate the blood–brain barrier and is therefore administered via intrathecal application. However, this approach carries the risk of severe infections, impeding the widespread use of ziconotide in clinical practice.185
It should be noted that a small number of macrocyclic peptides have been approved as orally available peptide drugs. Backbone N-methylations can enhance their cell penetration: cyclosporine A, a nonribosomal peptide that is used as an immunosuppressant in clinical settings, serves as an example.273
4.3 Metabolic stability
Therapeutic peptides undergo rapid elimination. In particular, oral administration leads to a significant first pass effect that reduces the amount of active ingredient systemically available after absorption in the gastrointestinal tract. The peptides have a short half-life in blood plasma, and the liver and kidneys clear them from circulation in minutes.272Glycosylation of ribosomal peptides can enhance the metabolic stability by reducing the clearance rate and/or preventing side-chain oxidation.274,275 Furthermore, the conjugation with lipids or synthetic polyethylene glycol (PEG) units has been shown to increase the serum half-life of peptides.276,277 Similar post-translational modifications, specifically glycosylation and lipidation, are naturally occurring in the families of glycocins,1 lipolanthines,34 and selidamides278 within the RiPP natural product class, among others. These modifications can improve the circulation of peptides in the bloodstream, which is a critical factor in their therapeutic application. Thus, these glyco- and lipopeptides are likely to exhibit enhanced metabolic stability, rendering them favourable candidates for therapeutic peptides.
4.4 Toxicity
Canonical peptides generally possess high conformational flexibility, which can contribute to a lack of selectivity. This may cause activation of multiple target receptors, leading to undesired side effects.272 In contrast, RiPPs exhibit greater specificity due to the imposition of conformational constraints via post-translational modifications.Peptides have a predictable metabolism, and their metabolites are seldom toxic. Thus, ribosomal peptides have a lower incidence of undesired side effects compared to small molecules.276 However, like every small molecule or large biological drug, ribosomal peptides can also have toxic side effects. For instance, ziconotide shows systemic and central nervous system side effects in higher doses, including dizziness, blurred vision, nystagmus, and sedation, which ultimately subside after discontinuing the drug.185
Although post-translational modifications can improve the physicochemical properties of ribosomal peptides, including oral bioavailability (via macrocyclisation), cell permeability (using N-methylamides) and metabolic stability (through glycosylation or lipidation), these structural characteristics must be combined and cannot be universally applied to every drug candidate. This leads to biosynthetic challenges and may decrease binding affinity at the target, thus diminishing the bioactivity.
5 Concluding remarks
The previous chapter highlights that RiPP(-based) therapies still have a considerable way to progress. The currently available strategies necessitate improvement, and new strategies must be developed to conquer the encountered obstacles. One of the concerns surrounding peptide medications available via oral administration is their stability, which can be enhanced via numerous post-translational modifications, such as macrocyclisation or conjugation with fatty acids. Some RiPPs exhibit inherent stability due to their specific structural features. One prominent example is the family of lasso peptides which are robust against high temperatures, acidic conditions, and proteases.279,280 Although oral administration may not be possible, beneficial bioactivities can result in therapeutic medicines, as different methods of treatment are attainable (for example, topical or parenteral, e.g., IM, SC, IV, IT,¶etc.). In particular, for serious illnesses, patients frequently stay in the hospital, which enables drug administration beyond the oral route. Besides, a new form of drug administration addressing the buccal mucosa is currently under development: a suction patch inspired by octopus suckers that is loaded with a peptide drug and a permeation enhancer, and may serve as a viable alternative for parenteral drugs.281In addition to diverse methods for extending the half-life of peptide drugs, including PEGylation282 and fusion proteins,283 various approaches for regulating drug release at specific body sites and over a certain period of time have been described. These encompass the use of biodegradable polymer matrices,284 engineered living materials,285 pulmonary and intranasal drug delivery systems,286,287 vector-based methods and nanoparticles.288,289 Nanoparticle systems have already been employed in RiPPs: Nisin A, commonly used as a food preservative, is a narrow-spectrum lanthibiotic that is effective against Gram-positive bacteria. However, when nisin A is combined with gold-coated nanoparticles forming so called nanocomposites, it can exhibit activity against Gram-negative bacteria.290 This demonstrates how new formulations can enhance the activity spectrum of antibiotics. In general, the application of new formulations represents a promising measure for unstable drug candidates. This has already been demonstrated by mRNA vaccines that utilise lipid-based vectors. A similar liposome encapsulation technique using liposomes has recently been applied to the anti-parkinsonian drug candidate acalitide. To enhance the affinity to the transferrin receptor and enable brain targeting, the liposome carrying acalitide was equipped with surface-bound transferrin. The experimental treatment was effectively administered to a mouse model, ultimately reaching the mouse brain and culminating six hours after being administered.204 This achievement underscores the significance of novel strategies for drug delivery.
RiPPs show potential as drug candidates and can also facilitate the construction of drug delivery systems. A stable conjugation method for protein therapeutics is the tetrazine ligation approach, named “TyrEx cycloaddition”, which relies on a post-translational modification installed by a RiPP maturase. This method facilitates the bonding of a drug molecule with an antibody that specifically targets tumour cells. The process of conjugation is based on an aminopyruvate unit that is introduced at a short tag through a post-translational splicing reaction catalysed by a splicease. This short tag can be genetically incorporated into a target protein or an antibody, which enables the visualisation and localisation of tumour cells as well as attacking them by releasing the drug after entering the tumour cell. Such a methodology is believed to be a significant advance towards more effective cancer therapies.291
In the future, it is necessary to apply or adapt the drug delivery strategies mentioned earlier to promising RiPP candidate drugs. This step will enable the full realisation of RiPPs' or RiPP-based compounds' potential and widen their availability for various medical treatments.
6 Author contributions
All authors worked together on the manuscript, in detail: conceptualisation: S. M.; supervision: S. M.; visualisation: I. P., M.-P. S.; writing – original draft: I. P., M.-P. S., S. M.; writing – review and editing: S. M.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgements
The authors thank Harald Gross (Pharmaceutical Institute, University of Tübingen) for proofreading and critical discussions. Research in the Mordhorst lab is funded by the Baden-Württemberg Stiftung through the Postdoctoral fellowship for leading early career researchers and by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments.9 Notes and references
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS PubMed.
- R. Ayikpoe, V. Govindarajan and J. A. Latham, Appl. Microbiol. Biotechnol., 2019, 103, 2903–2912 CrossRef CAS.
- R. S. Ayikpoe and J. A. Latham, J. Am. Chem. Soc., 2019, 141, 13582–13591 CrossRef CAS PubMed.
- G. E. Kenney and A. C. Rosenzweig, J. Biol. Chem., 2018, 293, 4606–4615 CrossRef CAS PubMed.
- X. Thomas, D. Destoumieux-Garzón, J. Peduzzi, C. Afonso, A. Blond, N. Birlirakis, C. Goulard, L. Dubost, R. Thai, J.-C. Tabet and S. Rebuffat, J. Biol. Chem., 2004, 279, 28233–28242 CrossRef CAS PubMed.
- G. Vassiliadis, J. Peduzzi, S. Zirah, X. Thomas, S. Rebuffat and D. Destoumieux-Garzón, Antimicrob. Agents Chemother., 2007, 51, 3546–3553 CrossRef CAS PubMed.
- Y. Li and S. Rebuffat, J. Biol. Chem., 2020, 295, 34–54 CrossRef CAS PubMed.
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS.
- X. Jing and K. Jin, Med. Res. Rev., 2020, 40, 753–810 CrossRef CAS PubMed.
- A. Tapeinou, M.-T. Matsoukas, C. Simal and T. Tselios, Pept. Sci., 2015, 104, 453–461 CrossRef CAS PubMed.
- L. Cao, T. Do and A. J. Link, J. Ind. Microbiol. Biotechnol., 2021, 48, kuab005 CrossRef CAS PubMed.
- C. Ongpipattanakul, E. K. Desormeaux, A. DiCaprio, W. A. van der Donk, D. A. Mitchell and S. K. Nair, Chem. Rev., 2022, 122, 14722–14814 CrossRef CAS PubMed.
- Y. Fu, A. H. Jaarsma and O. P. Kuipers, Cell. Mol. Life Sci., 2021, 78, 3921–3940 CrossRef CAS PubMed.
- G. A. Hudson and D. A. Mitchell, Curr. Opin. Microbiol., 2018, 45, 61–69 CrossRef CAS PubMed.
- A. L. Vagstad, Curr. Opin. Biotechnol., 2023, 80, 102891 CrossRef CAS PubMed.
- S. Mordhorst, F. Ruijne, A. L. Vagstad, O. P. Kuipers and J. Piel, RSC Chem. Biol., 2023, 4, 7–36 RSC.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457–5520 CrossRef CAS PubMed.
- W. Gu, S.-H. Dong, S. Sarkar, S. K. Nair and E. W. Schmidt, in Methods in Enzymology, ed. B. S. Moore, Academic Press, 2018, vol. 604, pp. 113–163 Search PubMed.
- W. L. Cheung-Lee and A. J. Link, J. Ind. Microbiol. Biotechnol., 2019, 46, 1371–1379 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed.
- J. Martins and V. Vasconcelos, Mar. Drugs, 2015, 13, 6910–6946 CrossRef CAS PubMed.
- F. Hubrich, A. Lotti, T. A. Scott and J. Piel, Chimia, 2021, 75, 543 CrossRef CAS PubMed.
- C. Kaletta, K.-D. Entian and G. Jung, Eur. J. Biochem., 1991, 199, 411–415 CrossRef CAS PubMed.
- L. A. Lindenfelser, T. G. Pridham and C. E. Kemp, Antibiot. Chemother., 1959, 9, 690–695 CAS.
- A. Ökesli, L. E. Cooper, E. J. Fogle and W. A. Van Der Donk, J. Am. Chem. Soc., 2011, 133, 13753–13760 CrossRef PubMed.
- L. Huo, A. Ökesli, M. Zhao and W. A. Van Der Donk, Appl. Environ. Microbiol., 2017, 83, e02698 Search PubMed.
- F. Märki, E. Hänni, A. Fredenhagen and J. Van Oostrum, Biochem. Pharmacol., 1991, 42, 2027–2035 CrossRef PubMed.
- J. P. Deisinger, M. Arts, I. Kotsogianni, J.-S. Puls, F. Grein, F. J. Ortiz-López, N. I. Martin, A. Müller, O. Genilloud and T. Schneider, iScience, 2023, 26, 106394 CrossRef CAS PubMed.
- M. Xu, F. Zhang, Z. Cheng, G. Bashiri, J. Wang, J. Hong, Y. Wang, L. Xu, X. Chen, S. Huang, S. Lin, Z. Deng and M. Tao, Angew. Chem., Int. Ed., 2020, 59, 18029–18035 CrossRef CAS PubMed.
- F. J. Ortiz-López, D. Carretero-Molina, M. Sánchez-Hidalgo, J. Martín, I. González, F. Román-Hurtado, M. Cruz, S. García-Fernández, F. Reyes, J. P. Deisinger, A. Müller, T. Schneider and O. Genilloud, Angew. Chem., Int. Ed., 2020, 59, 12654–12658 CrossRef PubMed.
- V. Wiebach, A. Mainz, M.-A. J. Siegert, N. A. Jungmann, G. Lesquame, S. Tirat, A. Dreux-Zigha, J. Aszodi, D. Le Beller and R. D. Süssmuth, Nat. Chem. Biol., 2018, 14, 652–654 CrossRef CAS PubMed.
- R. Kozakai, T. Ono, S. Hoshino, H. Takahashi, Y. Katsuyama, Y. Sugai, T. Ozaki, K. Teramoto, K. Teramoto, K. Tanaka, I. Abe, S. Asamizu and H. Onaka, Nat. Chem., 2020, 12, 869–877 CrossRef CAS PubMed.
- K. J. Molohon, J. O. Melby, J. Lee, B. S. Evans, K. L. Dunbar, S. B. Bumpus, N. L. Kelleher and D. A. Mitchell, ACS Chem. Biol., 2011, 6, 1307–1313 CrossRef CAS PubMed.
- F. Baquero and F. Moreno, FEMS Microbiol. Lett., 1984, 23, 117–124 CrossRef CAS.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, Med. Chem. Commun., 2019, 10, 1719–1739 RSC.
- J. Davagnino, M. Herrero, D. Furlong, F. Moreno and R. Kolter, Proteins, 1986, 1, 230–238 CrossRef CAS PubMed.
- M. Herrero and F. Moreno, Microbiology, 1986, 132, 393–402 CAS.
- H. Onaka, H. Tabata, Y. Igarashi, Y. Sato and T. Furumai, J. Antibiot., 2001, 54, 1036–1044 CrossRef CAS PubMed.
- H. Onaka, M. Nakaho, K. Hayashi, Y. Igarashi and T. Furumai, Microbiology, 2005, 151, 3923–3933 CrossRef CAS PubMed.
- H. Mathur, D. Field, M. Upton and P. D. Cotter, Front. Microbiol., 2021, 12, 695081 CrossRef PubMed.
- J. Stepper, S. Shastri, T. S. Loo, J. C. Preston, P. Novak, P. Man, C. H. Moore, V. Havlíček, M. L. Patchett and G. E. Norris, FEBS Lett., 2011, 585, 645–650 CrossRef CAS PubMed.
- W. J. Kelly, R. V. Asmundson and C. M. Huang, J. Appl. Bacteriol., 1996, 81, 657–662 CrossRef CAS.
- A. A. Vinogradov and H. Suga, Cell Chem. Biol., 2020, 27, 1032–1051 CrossRef CAS PubMed.
- A. H. Kutscher, L. Seguin, E. V. Zegarelli and J. D. Piro, J. Am. Dent. Assoc., 1959, 59, 715–720 CrossRef CAS PubMed.
- S. B. Singh, J. Occi, H. Jayasuriya, K. Herath, M. Motyl, K. Dorso, C. Gill, E. Hickey, K. M. Overbye, J. F. Barrett and P. Masurekar, J. Antibiot., 2007, 60, 565–571 CrossRef CAS PubMed.
- M. J. Pucci, J. J. Bronson, J. F. Barrett, K. L. DenBleyker, L. F. Discotto, J. C. Fung-Tomc and Y. Ueda, Antimicrob. Agents Chemother., 2004, 48, 3697–3701 CrossRef CAS PubMed.
- N. M. Haste, W. Thienphrapa, D. N. Tran, S. Loesgen, P. Sun, S.-J. Nam, P. R. Jensen, W. Fenical, G. Sakoulas, V. Nizet and M. E. Hensler, J. Antibiot., 2012, 65, 593–598 CrossRef CAS PubMed.
- E. Gavrish, C. S. Sit, S. Cao, O. Kandror, A. Spoering, A. Peoples, L. Ling, A. Fetterman, D. Hughes, A. Bissell, H. Torrey, T. Akopian, A. Mueller, S. Epstein, A. Goldberg, J. Clardy and K. Lewis, Chem. Biol., 2014, 21, 509–518 CrossRef CAS PubMed.
- M. Iwatsuki, R. Uchida, Y. Takakusagi, A. Matsumoto, C.-L. Jiang, Y. Takahashi, M. Arai, S. Kobayashi, M. Matsumoto, J. Inokoshi, H. Tomoda and S. Ōmura, J. Antibiot., 2007, 60, 357–363 CrossRef CAS PubMed.
- M. Tsunakawa, S.-L. Hu, Y. Hoshino, D. J. Detlefson, S. E. Hill, T. Furumai, R. J. White, M. Nishio, K. Kawano, S. Yamamoto, Y. Fukagawa and T. Oki, J. Antibiot., 1995, 48, 433–434 CrossRef CAS PubMed.
- S. Tan, K. C. Ludwig, A. Müller, T. Schneider and J. R. Nodwell, ACS Chem. Biol., 2019, 14, 966–974 CrossRef CAS PubMed.
- D. V. Carson, Y. Zhang, L. So, W. L. Cheung-Lee, A. J. Cartagena, S. A. Darst and A. J. Link, bioRxiv, 2023, preprint, DOI:10.1101/2023.06.21.545946.
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1992, 174, 7428–7435 CrossRef PubMed.
- T. A. Knappe, U. Linne, S. Zirah, S. Rebuffat, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2008, 130, 11446–11454 CrossRef CAS PubMed.
- K. Kuznedelov, E. Semenova, T. A. Knappe, D. Mukhamedyarov, A. Srivastava, S. Chatterjee, R. H. Ebright, M. A. Marahiel and K. Severinov, J. Mol. Biol., 2011, 412, 842–848 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS PubMed.
- S. Groß, F. Panter, D. Pogorevc, C. E. Seyfert, S. Deckarm, C. D. Bader, J. Herrmann and R. Müller, Chem. Sci., 2021, 12, 11882–11893 RSC.
- C. E. Seyfert, C. Porten, B. Yuan, S. Deckarm, F. Panter, C. D. Bader, J. Coetzee, F. Deschner, K. H. M. E. Tehrani, P. G. Higgins, H. Seifert, T. C. Marlovits, J. Herrmann and R. Müller, Angew. Chem., Int. Ed., 2023, 62, e202214094 CrossRef CAS PubMed.
- C. E. Seyfert, A. V. Müller, D. J. Walsh, J. Birkelbach, A. M. Kany, C. Porten, B. Yuan, D. Krug, J. Herrmann, T. C. Marlovits, A. K. H. Hirsch and R. Müller, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-24tmj.
- M. Metelev, A. Arseniev, L. B. Bushin, K. Kuznedelov, T. O. Artamonova, R. Kondratenko, M. Khodorkovskii, M. R. Seyedsayamdost and K. Severinov, ACS Chem. Biol., 2017, 12, 814–824 CrossRef CAS PubMed.
- W. L. Cheung-Lee, M. E. Parry, A. Jaramillo Cartagena, S. A. Darst and A. J. Link, J. Biol. Chem., 2019, 294, 6822–6830 CrossRef CAS PubMed.
- D. V. Carson, M. Patiño, H. E. Elashal, A. J. Cartagena, Y. Zhang, M. E. Whitley, L. So, A. K. Kayser-Browne, A. M. Earl, R. P. Bhattacharyya and A. J. Link, ACS Infect. Dis., 2023, 9, 111–121 CrossRef CAS PubMed.
- C. Asensio, J. C. Pérez-Díaz, M. C. Martínez and F. Baquero, Biochem. Biophys. Res. Commun., 1976, 69, 7–14 CrossRef CAS PubMed.
- Y. Li, Y. Han, Z. Zeng, W. Li, S. Feng and W. Cao, J. Agric. Food Chem., 2021, 69, 8758–8767 CrossRef CAS PubMed.
- Y. Han, Y. Li, Z. Zeng, W. Li, S. Feng and W. Cao, Microbiol. Spectrum, 2022, 10, e01859 Search PubMed.
- W. L. Cheung-Lee, M. E. Parry, C. Zong, A. J. Cartagena, S. A. Darst, N. D. Connell, R. Russo and A. J. Link, ChemBioChem, 2020, 21, 1335–1340 CrossRef CAS PubMed.
- H. Knothe and G. A. Dette, J. Antimicrob. Chemother., 1981, 8, 33–41 CrossRef CAS PubMed.
- G. A. Pankuch, M. R. Jacobs and P. C. Appelbaum, Antimicrob. Agents Chemother., 1994, 38, 2065–2072 CrossRef CAS PubMed.
- G. D. Brown, D. W. Denning, N. A. R. Gow, S. M. Levitz, M. G. Netea and T. C. White, Sci. Transl. Med., 2012, 4, 165rv13 Search PubMed.
- K. I. Mohr, C. Volz, R. Jansen, V. Wray, J. Hoffmann, S. Bernecker, J. Wink, K. Gerth, M. Stadler and R. Müller, Angew. Chem., Int. Ed., 2015, 54, 11254–11258 CrossRef CAS PubMed.
- M. Tsuda, H. Shigemori, Y. Mikami and J. Kobayashi, Tetrahedron, 1993, 49, 6785–6796 CrossRef CAS.
- J. Kobayashi, M. Tsuda, T. Nakamura, Y. Mikami and H. Shigemori, Tetrahedron, 1993, 49, 2391–2402 CrossRef CAS.
- N. Mizuhara, M. Kuroda, A. Ogita, T. Tanaka, Y. Usuki and K. Fujita, Bioorg. Med. Chem., 2011, 19, 5300–5310 CrossRef CAS PubMed.
- V. Valiante, M. C. Monteiro, J. Martín, R. Altwasser, N. El Aouad, I. González, O. Kniemeyer, E. Mellado, S. Palomo, N. de Pedro, I. Pérez-Victoria, J. R. Tormo, F. Vicente, F. Reyes, O. Genilloud and A. A. Brakhage, Antimicrob. Agents Chemother., 2015, 59, 5145–5153 CrossRef CAS PubMed.
- A. J. Sucher, E. B. Chahine and H. E. Balcer, Ann. Pharmacother., 2009, 43, 1647–1657 CrossRef CAS PubMed.
- F. Marco, M. A. Pfaller, S. A. Messer and R. N. Jones, Diagn. Microbiol. Infect. Dis., 1998, 32, 33–37 CrossRef CAS PubMed.
- S. Arikan, M. Lozano-Chiu, V. Paetznick and J. H. Rex, Antimicrob. Agents Chemother., 2001, 45, 327–330 CrossRef CAS PubMed.
- J. Piret and G. Boivin, Front. Microbiol., 2021, 11, 631736 CrossRef PubMed.
- N. M. Bösch, M. Borsa, U. Greczmiel, B. I. Morinaka, M. Gugger, A. Oxenius, A. L. Vagstad and J. Piel, Angew. Chem., Int. Ed., 2020, 59, 11763–11768 CrossRef PubMed.
- M. Shao, J. Ma, Q. Li and J. Ju, Mar. Drugs, 2019, 17, 127 CrossRef CAS PubMed.
- G. Helynck, C. Dubertret, J.-F. Mayaux and J. Leboul, J. Antibiot., 1993, 46, 1756–1757 CrossRef CAS PubMed.
- D. Fréchet, J. D. Guitton, F. Herman, D. Faucher, G. Helynck, B. Monegier du Sorbier, J. P. Ridoux, E. James-Surcouf and M. Vuilhorgne, Biochemistry, 1994, 33, 42–50 CrossRef PubMed.
- M. Sánchez-Hidalgo, J. Martín and O. Genilloud, Antibiotics, 2020, 9, 67 CrossRef PubMed.
- G. Férir, M. I. Petrova, G. Andrei, D. Huskens, B. Hoorelbeke, R. Snoeck, J. Vanderleyden, J. Balzarini, S. Bartoschek, M. Brönstrup, R. D. Süssmuth and D. Schols, PLoS One, 2013, 8, e64010 CrossRef PubMed.
- N. Naruse, O. Tenmyo, K. Tomita, M. Konishi, T. Miyaki, H. Kawaguchi, K. Fukase, T. Wakamiya and T. Shiba, J. Antibiot., 1989, 42, 837–845 CrossRef CAS PubMed.
- A. S. Richard, A. Zhang, S.-J. Park, M. Farzan, M. Zong and H. Choe, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 14682–14687 CrossRef CAS PubMed.
- T. E. Smith, C. D. Pond, E. Pierce, Z. P. Harmer, J. Kwan, M. M. Zachariah, M. K. Harper, T. P. Wyche, T. K. Matainaho, T. S. Bugni, L. R. Barrows, C. M. Ireland and E. W. Schmidt, Nat. Chem. Biol., 2018, 14, 179–185 CrossRef CAS PubMed.
- T. E. Smith, C. D. Pond, E. Pierce, Z. P. Harmer, J. Kwan, M. M. Zachariah, M. K. Harper, T. P. Wyche, T. K. Matainaho, T. S. Bugni, L. R. Barrows, C. M. Ireland and E. W. Schmidt, Nat. Chem. Biol., 2018, 14, 179–185 CrossRef CAS PubMed.
- G. Férir, M. I. Petrova, G. Andrei, D. Huskens, B. Hoorelbeke, R. Snoeck, J. Vanderleyden, J. Balzarini, S. Bartoschek, M. Brönstrup, R. D. Süssmuth and D. Schols, PLoS One, 2013, 8, e64010 CrossRef PubMed.
- M. Tsunakawa, S.-L. Hu, Y. Hoshino, D. J. Detlefson, S. E. Hill, T. Furumai, R. J. White, M. Nishio, K. Kawano, S. Yamamoto, Y. Fukagawa and T. Oki, J. Antibiot., 1995, 48, 433–434 CrossRef CAS PubMed.
- D. H. Katz, J. F. Marcelletti, M. H. Khalil, L. E. Pope and L. R. Katz, Proc. Natl. Acad. Sci. U.S.A., 1991, 88, 10825–10829 CrossRef CAS PubMed.
- J. Barretina Ginesta, J. Castaner, J. Bozzo and M. Bayes, Drugs Future, 2005, 30, 0469 CrossRef.
- Product Monograph ‘Invirase’, Hoffmann-La Roche Limited, 2020 Search PubMed.
- B. M. Watkins, Trends Parasitol., 2003, 19, 477–478 CrossRef CAS PubMed.
- Neglected tropical diseases, https://www.who.int/news/item/16-07-2020-neglected-tropical-diseases-treating-more-than-one-billion-people-for-the-fifth-consecutive-year, accessed 23 August 2023.
- M. Papagianni, Biotechnol. Adv., 2003, 21, 465–499 CrossRef CAS PubMed.
- C. M. Scheidler, L. M. Kick and S. Schneider, ChemBioChem, 2019, 20, 1479–1486 CrossRef CAS PubMed.
- M. N. Aminake, S. Schoof, L. Sologub, M. Leubner, M. Kirschner, H.-D. Arndt and G. Pradel, Antimicrob. Agents Chemother., 2011, 55, 1338–1348 CrossRef CAS PubMed.
- J. Lambert, E. Keppi, J. L. Dimarcq, C. Wicker, J. M. Reichhart, B. Dunbar, P. Lepage, A. Van Dorsselaer, J. Hoffmann and J. Fothergill, Proc. Natl. Acad. Sci. U.S.A., 1989, 86, 262–266 CrossRef CAS PubMed.
- S.-H. Liu, H.-F. Li, Y. Yang, D. Wei, H.-B. Jiang, W. Dou, G.-R. Yuan and J.-J. Wang, AMB Express, 2018, 8, 5 CrossRef PubMed.
- P. Bulet, S. Cociancich, M. Reuland, F. Sauber, R. Bischoff, G. Hegy, A. Van DORSSELAER, C. Hetru and J. A. Hoffmann, Eur. J. Biochem., 1992, 209, 977–984 CrossRef CAS PubMed.
- R. B. Williams, Avian Pathol., 2005, 34, 29–47 CrossRef CAS PubMed.
- R. Pigeault, J. Vézilier, S. Cornet, F. Zélé, A. Nicot, P. Perret, S. Gandon and A. Rivero, Philos. Trans. R. Soc., B, 2015, 370, 20140300 CrossRef PubMed.
- M. Shahabuddin, I. Fields, P. Bulet, J. A. Hoffmann and L. H. Miller, Exp. Parasitol., 1998, 89, 103–112 CrossRef CAS PubMed.
- N. Boulanger, C. Lowenberger, P. Volf, R. Ursic, L. Sigutova, L. Sabatier, M. Svobodova, S. M. Beverley, G. Späth, R. Brun, B. Pesson and P. Bulet, Infect. Immun., 2004, 72, 7140–7146 CrossRef CAS PubMed.
- E. Torres-Guerrero, M. R. Quintanilla-Cedillo, J. Ruiz-Esmenjaud and R. Arenas, F1000Research, 2017, 6, 750 Search PubMed.
- C. Portmann, S. Sieber, S. Wirthensohn, J. F. Blom, L. Da Silva, E. Baudat, M. Kaiser, R. Brun and K. Gademann, J. Nat. Prod., 2014, 77, 557–562 CrossRef CAS PubMed.
- M. S. Donia, B. Wang, D. C. Dunbar, P. V. Desai, A. Patny, M. Avery and M. T. Hamann, J. Nat. Prod., 2008, 71, 941–945 CrossRef CAS PubMed.
- R. G. Linington, J. González, L.-D. Ureña, L. I. Romero, E. Ortega-Barría and W. H. Gerwick, J. Nat. Prod., 2007, 70, 397–401 CrossRef CAS PubMed.
- O. Saether, D. J. Craik, I. D. Campbell, K. Sletten, J. Juul and D. G. Norman, Biochemistry, 1995, 34, 4147–4158 CrossRef CAS PubMed.
- D. C. Ireland, M. L. Colgrave and D. J. Craik, Biochem. J., 2006, 400, 1–12 CrossRef CAS PubMed.
- D. J. Diemert, J. M. Bethony and P. J. Hotez, Clin. Infect. Dis., 2008, 46, 282–288 CrossRef PubMed.
- M. L. Colgrave, A. C. Kotze, S. Kopp, J. S. McCarthy, G. T. Coleman and D. J. Craik, Acta Trop., 2009, 109, 163–166 CrossRef CAS PubMed.
- R. Khositnithikul, P. Tan-ariya and M. Mungthin, Malar. J., 2008, 7, 23 CrossRef PubMed.
- S. W. Page, in Small Animal Clinical Pharmacology, ed. J. E. Maddison, S. W. Page and D. B. Church, W. B. Saunders, Edinburgh, Second Edition, 2008, pp. 198–260 Search PubMed.
- M. V. Díaz, M. R. Miranda, C. Campos-Estrada, C. Reigada, J. D. Maya, C. A. Pereira and R. López-Muñoz, Acta Trop., 2014, 134, 1–9 CrossRef PubMed.
- R. C. Zauli-Nascimento, D. C. Miguel, J. K. U. Yokoyama-Yasunaka, L. I. A. Pereira, M. A. Pelli de Oliveira, F. Ribeiro-Dias, M. L. Dorta and S. R. B. Uliana, Trop. Med. Int. Health, 2010, 15, 68–76 CAS.
- Cancer, https://www.who.int/news-room/fact-sheets/detail/cancer, accessed 29 August 2023.
- A. Kamb, S. Wee and C. Lengauer, Nat. Rev. Drug Discovery, 2007, 6, 115–120 CrossRef CAS PubMed.
- H. Nishimura, S. Okamoto, M. Mayama, H. Ohtsuka, K. Nakajima, K. Tawara, M. Shimohira and N. Shimaoka, J. Antibiot., 1961, 14, 255–263 CAS.
- S. K. Radhakrishnan, U. G. Bhat, D. E. Hughes, I.-C. Wang, R. H. Costa and A. L. Gartel, Cancer Res., 2006, 66, 9731–9735 CrossRef CAS PubMed.
- N. S. Hegde, D. A. Sanders, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2011, 3, 725–731 CrossRef CAS PubMed.
- S. S. Myatt and E. W.-F. Lam, Nat. Rev. Cancer, 2007, 7, 847–859 CrossRef CAS PubMed.
- J. A. V. Lopez, S. S. Al-Lihaibi, W. M. Alarif, A. Abdel-Lateff, Y. Nogata, K. Washio, M. Morikawa and T. Okino, J. Nat. Prod., 2016, 79, 1213–1218 CrossRef CAS PubMed.
- C. J. Hawkins, M. F. Lavin, K. A. Marshall, A. L. Van den Brenk and D. J. Watters, J. Med. Chem., 1990, 33, 1634–1638 CrossRef CAS PubMed.
- A. R. Carroll, J. C. Coll, D. J. Bourne, J. K. Macleod, T. M. Zabriskie, C. M. Ireland and B. F. Bowden, Aust. J. Chem., 1996, 49, 659–667 CrossRef CAS.
- A. Rudi, M. Aknin, E. M. Gaydou and Y. Kashman, Tetrahedron, 1998, 54, 13203–13210 CrossRef CAS.
- S. Soltani, R. Hammami, P. D. Cotter, S. Rebuffat, L. B. Said, H. Gaudreau, F. Bédard, E. Biron, D. Drider and I. Fliss, FEMS Microbiol. Rev., 2021, 45, fuaa039 CrossRef PubMed.
- M. A. Varas, C. Muñoz-Montecinos, V. Kallens, V. Simon, M. L. Allende, A. E. Marcoleta and R. Lagos, Front. Microbiol., 2020, 11, 405 CrossRef PubMed.
- C. Hetz, M. R. Bono, L. F. Barros and R. Lagos, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 2696–2701 CrossRef CAS PubMed.
- S. Ahmadi, M. Ghollasi and H. M. Hosseini, Microb. Pathog., 2017, 111, 193–197 CrossRef CAS PubMed.
- N. E. Joo, K. Ritchie, P. Kamarajan, D. Miao and Y. L. Kapila, Cancer Med., 2012, 1, 295–305 CrossRef CAS PubMed.
- P. Kamarajan, T. Hayami, B. Matte, Y. Liu, T. Danciu, A. Ramamoorthy, F. Worden, S. Kapila and Y. Kapila, PLoS One, 2015, 10, e0131008 CrossRef PubMed.
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS), M. Younes, P. Aggett, F. Aguilar, R. Crebelli, B. Dusemund, M. Filipič, M. J. Frutos, P. Galtier, U. Gundert-Remy, G. G. Kuhnle, C. Lambré, J.-C. Leblanc, I. T. Lillegaard, P. Moldeus, A. Mortensen, A. Oskarsson, I. Stankovic, I. Waalkens-Berendsen, R. A. Woutersen, M. Wright, L. Herman, P. Tobback, F. Pizzo, C. Smeraldi, A. Tard, A. Papaioannou and D. Gott, EFSA J., 2017, 15, e05063 Search PubMed.
- H. R. Momeni, Cell J., 2011, 13, 65 CAS.
- P. A. Jordan and B. S. Moore, Cell Chem. Biol., 2016, 23, 1504–1514 CrossRef CAS PubMed.
- C. C. Hughes, J. B. MacMillan, S. P. Gaudêncio, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2009, 48, 725–727 CrossRef CAS PubMed.
- D. Reimer and C. C. Hughes, J. Nat. Prod., 2017, 80, 126–133 CrossRef CAS PubMed.
- C. C. Hughes, J. B. MacMillan, S. P. Gaudêncio, W. Fenical and J. J. La Clair, Angew. Chem., Int. Ed., 2008, 121, 742–746 CrossRef.
- C. M. Celli, N. Tran, R. Knox and A. K. Jaiswal, Biochem. Pharmacol., 2006, 72, 366–376 CrossRef CAS PubMed.
- W. Li, C. Zhang, H. Zhang, R. Dong, J. Liu, C. Wang, M. Wang, Y. Wang, C. Wang, Y. Zhang, L. Shi, Y. Xu and L.-P. Sun, Bioorg. Chem., 2022, 127, 105917 CrossRef CAS PubMed.
- B. Donati, E. Lorenzini and A. Ciarrocchi, Mol. Cancer, 2018, 17, 164 CrossRef CAS PubMed.
- J. Luo, D. Yang, Hindra, A. Adhikari, L.-B. Dong, F. Ye, X. Yan, C. Rader and B. Shen, J. Ind. Microbiol. Biotechnol., 2021, 48, kuab027 CrossRef CAS PubMed.
- H. Nagata, K. Ochiai, Y. Aotani, K. Ando, M. Yoshida, I. Takahashi and T. Tamaoki, J. Antibiot., 1997, 50, 537–542 CrossRef CAS PubMed.
- A. Miyanaga, J. E. Janso, L. McDonald, M. He, H. Liu, L. Barbieri, A. S. Eustáquio, E. N. Fielding, G. T. Carter, P. R. Jensen, X. Feng, M. Leighton, F. E. Koehn and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 13311–13313 CrossRef CAS PubMed.
- T. Tian, X. Li and J. Zhang, Int. J. Mol. Sci., 2019, 20, 755 CrossRef CAS PubMed.
- P. Lin, J. H. Wong and T. B. Ng, Biosci. Rep., 2009, 30, 101–109 CrossRef PubMed.
- Y. Hanaoka, Y. Yamaguchi, H. Yamamoto, M. Ishii, T. Nagase, H. Kurihara, M. Akishita and Y. Ouchi, Anticancer Res., 2016, 36, 5999–6004 CrossRef CAS PubMed.
- S. Wang, S. Lin, Q. Fang, R. Gyampoh, Z. Lu, Y. Gao, D. J. Clarke, K. Wu, L. Trembleau, Y. Yu, K. Kyeremeh, B. F. Milne, J. Tabudravu and H. Deng, Nat. Commun., 2022, 13, 5044 CrossRef CAS PubMed.
- A. Ványolós, M. Dékány, B. Kovács, B. Krámos, P. Bérdi, I. Zupkó, J. Hohmann and Z. Béni, Org. Lett., 2016, 18, 2688–2691 CrossRef PubMed.
- K. Shin-ya, K. Wierzba, K. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Seto, J. Am. Chem. Soc., 2001, 123, 1262–1263 CrossRef CAS PubMed.
- K. Amagai, H. Ikeda, J. Hashimoto, I. Kozone, M. Izumikawa, F. Kudo, T. Eguchi, T. Nakamura, H. Osada, S. Takahashi and K. Shin-ya, Sci. Rep., 2017, 7, 3382 CrossRef PubMed.
- A. Nakajima, T. Tauchi, G. Sashida, M. Sumi, K. Abe, K. Yamamoto, J. H. Ohyashiki and K. Ohyashiki, Leukemia, 2003, 17, 560–567 CrossRef CAS PubMed.
- M. A. Shammas, R. J. S. Reis, C. Li, H. Koley, L. H. Hurley, K. C. Anderson and N. C. Munshi, Clin. Cancer Res., 2004, 10, 770–776 CrossRef CAS PubMed.
- T. Nakamura, S. Okabe, H. Yoshida, K. Iida, Y. Ma, S. Sasaki, T. Yamori, K. Shin-ya, I. Nakano, K. Nagasawa and H. Seimiya, Sci. Rep., 2017, 7, 3605 CrossRef PubMed.
- M. Yasuda, Y. Ma, S. Okabe, Y. Wakabayashi, D. Su, Y.-T. Chang, H. Seimiya, M. Tera and K. Nagasawa, Chem. Commun., 2020, 56, 12905–12908 RSC.
- S. Um, Y.-J. Kim, H. Kwon, H. Wen, S.-H. Kim, H. C. Kwon, S. Park, J. Shin and D.-C. Oh, J. Nat. Prod., 2013, 76, 873–879 CrossRef CAS PubMed.
- S. S. Elsayed, F. Trusch, H. Deng, A. Raab, I. Prokes, K. Busarakam, J. A. Asenjo, B. A. Andrews, P. van West, A. T. Bull, M. Goodfellow, Y. Yi, R. Ebel, M. Jaspars and M. E. Rateb, J. Org. Chem., 2015, 80, 10252–10260 CrossRef CAS PubMed.
- S. Son, M. Jang, B. Lee, Y.-S. Hong, S.-K. Ko, J.-H. Jang and J. S. Ahn, J. Nat. Prod., 2018, 81, 2205–2211 CrossRef CAS PubMed.
- L. Digal, S. C. Samson, M. A. Stevens, A. Ghorai, H. Kim, M. C. Mifflin, K. R. Carney, D. L. Williamson, S. Um, G. Nagy, D.-C. Oh, M. C. Mendoza and A. G. Roberts, ACS Chem. Biol., 2024, 19, 81–88 CrossRef CAS PubMed.
- J. Ogino, R. E. Moore, G. M. L. Patterson and C. D. Smith, J. Nat. Prod., 1996, 59, 581–586 CrossRef CAS PubMed.
- A. B. Williams and R. S. Jacobs, Cancer Lett., 1993, 71, 97–102 CrossRef CAS PubMed.
- J. F. Guerrero-Garzón, E. Madland, M. Zehl, M. Singh, S. Rezaei, F. L. Aachmann, G. Courtade, E. Urban, C. Rückert, T. Busche, J. Kalinowski, Y.-R. Cao, Y. Jiang, C. Jiang, G. Selivanova and S. B. Zotchev, iScience, 2020, 23, 101785 CrossRef PubMed.
- L. A. Salvador-Reyes and H. Luesch, Nat. Prod. Rep., 2015, 32, 478–503 RSC.
- I. Nakano, K. Joshi, K. Visnyei, B. Hu, M. Watanabe, D. Lam, E. Wexler, K. Saigusa, Y. Nakamura, D. R. Laks, P. S. Mischel, M. Viapiano and H. I. Kornblum, J. Neuro-Oncol., 2011, 13, 622–634 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- A. Beck, M. C. Etienne, S. Chéradame, J. L. Fischel, P. Formento, N. Renée and G. Milano, Eur. J. Cancer, 1994, 30, 1517–1522 CrossRef PubMed.
- S. M. Rikard, MMWR Morb. Mortal. Wkly. Rep., 2023, 72, 379–385 CrossRef PubMed.
- S. P. Cohen, L. Vase and W. M. Hooten, Lancet, 2021, 397, 2082–2097 CrossRef PubMed.
- A. J. Kohn, P. R. Saunders and S. Wiener, Ann. N. Y. Acad. Sci., 1960, 90, 706–725 CrossRef CAS PubMed.
- K. Meindl, T. Schmiederer, K. Schneider, A. Reicke, D. Butz, S. Keller, H. Gühring, L. Vértesy, J. Wink, H. Hoffmann, M. Brönstrup, G. M. Sheldrick and R. D. Süssmuth, Angew. Chem., Int. Ed., 2010, 49, 1151–1154 CrossRef CAS PubMed.
- M. Iorio, O. Sasso, S. I. Maffioli, R. Bertorelli, P. Monciardini, M. Sosio, F. Bonezzi, M. Summa, C. Brunati, R. Bordoni, G. Corti, G. Tarozzo, D. Piomelli, A. Reggiani and S. Donadio, ACS Chem. Biol., 2014, 9, 398–404 CrossRef CAS PubMed.
- Z. Liu, P. Bartels, M. Sadeghi, T. Du, Q. Dai, C. Zhu, S. Yu, S. Wang, M. Dong, T. Sun, J. Guo, S. Peng, L. Jiang, D. J. Adams and Q. Dai, Sci. Rep., 2018, 8, 1004 CrossRef PubMed.
- G. Berecki, L. Motin, A. Haythornthwaite, S. Vink, P. Bansal, R. Drinkwater, C. I. Wang, M. Moretta, R. J. Lewis, P. F. Alewood, M. J. Christie and D. J. Adams, Mol. Pharmacol., 2010, 77, 139–148 CrossRef CAS PubMed.
- C. I. Schroeder, C. J. Doering, G. W. Zamponi and R. J. Lewis, Med. Chem., 2006, 2, 535–543 CrossRef CAS PubMed.
- T. Wu, M. Wang, W. Wu, Q. Luo, L. Jiang, H. Tao and M. Deng, J. Venomous Anim. Toxins Incl. Trop. Dis., 2019, 25, e146318 CrossRef CAS PubMed.
- P. Escoubas, J. R. De Weille, A. Lecoq, S. Diochot, R. Waldmann, G. Champigny, D. Moinier, A. Ménez and M. Lazdunski, J. Biol. Chem., 2000, 275, 25116–25121 CrossRef CAS PubMed.
- M. Mazzuca, C. Heurteaux, A. Alloui, S. Diochot, A. Baron, N. Voilley, N. Blondeau, P. Escoubas, A. Gélot, A. Cupo, A. Zimmer, A. M. Zimmer, A. Eschalier and M. Lazdunski, Nat. Neurosci., 2007, 10, 943–945 CrossRef CAS PubMed.
- S. M. Oliveira, C. R. Silva, G. Trevisan, J. G. Villarinho, M. N. Cordeiro, M. Richardson, M. H. Borges, C. J. Castro, M. V. Gomez and J. Ferreira, Pflug. Arch. Eur. J. Physiol., 2016, 468, 881–894 CrossRef CAS PubMed.
- K. Konno, G. Picolo, V. P. Gutierrez, P. Brigatte, V. O. Zambelli, A. C. M. Camargo and Y. Cury, Peptides, 2008, 29, 1293–1304 CrossRef CAS PubMed.
- G. Berecki, L. Motin, A. Haythornthwaite, S. Vink, P. Bansal, R. Drinkwater, C. I. Wang, M. Moretta, R. J. Lewis, P. F. Alewood, M. J. Christie and D. J. Adams, Mol. Pharmacol., 2010, 77, 139–148 CrossRef CAS PubMed.
- J. G. McGivern, Neuropsychiatr. Dis. Treat., 2007, 3, 69–85 CrossRef CAS PubMed.
- X. Wang, X. Chen, Z.-J. Wang, M. Zhuang, L. Zhong, C. Fu, R. Garcia, R. Müller, Y. Zhang, J. Yan, D. Wu and L. Huo, J. Am. Chem. Soc., 2023, 145, 16924–16937 CrossRef CAS PubMed.
- Y. Kido, T. Hamakado, T. Yoshida, M. Anno, Y. Motoki, T. Wakamiya and T. Shiba, J. Antibiot., 1983, 36, 1295–1299 CrossRef CAS PubMed.
- T. Wakamiya, Y. Ueki, T. Shiba, Y. Kido and Y. Motoki, Tetrahedron Lett., 1985, 26, 665–668 CrossRef CAS.
- T. Wakamiya, Y. Ueki, T. Shiba, Y. Kido and Y. Motoki, Bull. Chem. Soc. Jpn., 1990, 63, 1032–1038 CrossRef CAS.
- W. Weber, W. Fischli, E. Hochuli, E. Kupfer and E. K. Weibel, J. Antibiot., 1991, 44, 164–171 CrossRef CAS PubMed.
- D. F. Wyss, H.-W. Lahm, M. Manneberg and A. M. Labhardt, J. Antibiot., 1991, 44, 172–180 CrossRef CAS PubMed.
- Y. Morishita, S. Chiba, E. Tsukuda, T. Tanaka, T. Ogawa, M. Yamasaki, M. Yoshida, I. Kawamoto and Y. Matsuda, J. Antibiot., 1994, 47, 269–275 CrossRef CAS PubMed.
- T. Ogawa, K. Ochiai, T. Tanaka, E. Tsukuda, S. Chiba, K. Yano, M. Yamasaki, M. Yoshida and Y. Matsuda, J. Antibiot., 1995, 48, 1213–1220 CrossRef CAS PubMed.
- D. Oves-Costales, M. Sánchez-Hidalgo, J. Martín and O. Genilloud, Mar. Drugs, 2020, 18, 238 CrossRef CAS PubMed.
- T. Suzawa, K. Shibata, T. Tanaka, Y. Matsuda and M. Yamasaki, Bioorg. Med. Chem. Lett., 1997, 7, 1715–1720 CrossRef CAS.
- K. Shibata, T. Suzawa, T. Ohno, K. Yamada, T. Tanaka, E. Tsukuda, Y. Matsuda and M. Yamasaki, Bioorg. Med. Chem., 1998, 6, 2459–2467 CrossRef CAS PubMed.
- K. Shibata, K. Yano, T. Tanaka, Y. Matsuda and M. Yamasaki, Lett. Pept. Sci., 1997, 4, 167–170 CAS.
- T. A. Knappe, U. Linne, X. Xie and M. A. Marahiel, FEBS Lett., 2010, 584, 785–789 CrossRef CAS PubMed.
- O. Potterat, K. Wagner, G. Gemmecker, J. Mack, C. Puder, R. Vettermann and R. Streicher, J. Nat. Prod., 2004, 67, 1528–1531 CrossRef CAS PubMed.
- M. Lotfy, H. Kalasz, G. Szalai, J. Singh and E. Adeghate, Open Med. Chem. J., 2014, 8, 28–35 CrossRef PubMed.
- J. G. McCormack, N. Westergaard, M. Kristiansen, C. L. Brand and J. Lau, Curr. Pharm. Des., 2001, 7, 1451–1474 CrossRef CAS PubMed.
- H. J. Shin, H. Matsuda, M. Murakami and K. Yamaguchi, Tetrahedron, 1996, 52, 13129–13136 CrossRef CAS.
- R. A. Al-Horani and U. R. Desai, Med. Res. Rev., 2014, 34, 1168–1216 CrossRef CAS PubMed.
- Z. Tong, X. Xie, H. Ge, R. Jiao, T. Wang, X. Wang, W. Zhuang, G. Hu and R. Tan, Acta Pharm. Sin. B., 2024, 14, 881–892 CrossRef CAS PubMed.
- J. D. Walton, H. E. Hallen-Adams and H. Luo, Pept. Sci., 2010, 94, 659–664 CrossRef CAS PubMed.
- M. Romani and J. Auwerx, Bio-Protoc., 2021, 11, e4183 CAS.
- A. Anastasi, V. Erspamer and M. Bucci, Experientia, 1971, 27, 166–167 CrossRef CAS PubMed.
- C. Schweinsberg, V. Maes, L. Brans, P. Bläuenstein, D. A. Tourwé, P. A. Schubiger, R. Schibli and E. G. Garayoa, Bioconjugate Chem., 2008, 19, 2432–2439 CrossRef CAS PubMed.
- S. M. Okarvi and I. A. Jammaz, Nucl. Med. Biol., 2012, 39, 795–804 CrossRef CAS PubMed.
- N. Sadeghzadeh, M. Ahmadzadeh and M. Erfani, J. Radioanal. Nucl. Chem., 2013, 298, 287–293 CrossRef CAS PubMed.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, J. Pept. Sci., 2019, 25, e3224 CrossRef CAS PubMed.
- R. Eychenne, C. Bouvry, M. Bourgeois, P. Loyer, E. Benoist and N. Lepareur, Molecules, 2020, 25, 4012 CrossRef CAS PubMed.
- V. Ambrosini, L. Zanoni, A. Filice, G. Lamberti, G. Argalia, E. Fortunati, D. Campana, A. Versari and S. Fanti, Cancers, 2022, 14, 1055 CrossRef CAS PubMed.
- L. Joosten, M. Brom, H. Peeters, S. Heskamp, M. Béhé, O. Boerman and M. Gotthardt, Mol. Pharm., 2018, 15, 486–494 CrossRef CAS PubMed.
- I. Velikyan and O. Eriksson, Theranostics, 2020, 10, 437–461 CrossRef CAS PubMed.
- L. Aloj, M. R. Panico, C. Caracó, A. Zannetti, S. Del Vecchio, C. Di Nuzzo, C. Arra, G. Morelli, D. Tesauro, S. De Luca, C. Pedone and M. Salvatore, Biopolymers, 2002, 66, 370–380 CrossRef CAS PubMed.
- S. Roosenburg, P. Laverman, F. L. van Delft and O. C. Boerman, Amino Acids, 2011, 41, 1049–1058 CrossRef CAS PubMed.
- X. Yang, K. R. Lennard, C. He, M. C. Walker, A. T. Ball, C. Doigneaux, A. Tavassoli and W. A. Van Der Donk, Nat. Chem. Biol., 2018, 14, 375–380 CrossRef CAS PubMed.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, ACS Cent. Sci., 2018, 4, 458–467 CrossRef CAS PubMed.
- J. D. Hegemann, S. C. Bobeica, M. C. Walker, I. R. Bothwell and W. A. van der Donk, ACS Synth. Biol., 2019, 8, 1204–1214 CrossRef CAS PubMed.
- T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2011, 50, 8714–8717 CrossRef CAS PubMed.
- J. D. Hegemann, M. De Simone, M. Zimmermann, T. A. Knappe, X. Xie, F. S. Di Leva, L. Marinelli, E. Novellino, S. Zahler, H. Kessler and M. A. Marahiel, J. Med. Chem., 2014, 57, 5829–5834 CrossRef CAS PubMed.
- K. Mohri, K. P. H. Nhat, M. Zouda, S. Warashina, Y. Wada, Y. Watanabe, S. Tagami and H. Mukai, Eur. J. Pharm. Sci., 2023, 180, 106339 CrossRef CAS PubMed.
- R. H. Kimura, A. M. Levin, F. V. Cochran and J. R. Cochran, Proteins: Struct., Funct., Bioinf., 2009, 77, 359–369 CrossRef CAS PubMed.
- A. P. Silverman, A. M. Levin, J. L. Lahti and J. R. Cochran, J. Mol. Biol., 2009, 385, 1064–1075 CrossRef CAS PubMed.
- A. C. Conibear, A. Bochen, K. J. Rosengren, P. Stupar, C. Wang, H. Kessler and D. J. Craik, ChemBioChem, 2014, 15, 451–459 CrossRef CAS PubMed.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS PubMed.
- B. J. Burkhart, N. Kakkar, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, ACS Cent. Sci., 2017, 3, 629–638 CrossRef CAS PubMed.
- E. Reyna-González, B. Schmid, D. Petras, R. D. Süssmuth and E. Dittmann, Angew. Chem., Int. Ed., 2016, 55, 9398–9401 CrossRef PubMed.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Velásquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952–6955 CrossRef CAS PubMed.
- Y. Goto, Y. Ito, Y. Kato, S. Tsunoda and H. Suga, Chem. Biol., 2014, 21, 766–774 CrossRef CAS PubMed.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. E. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS PubMed.
- L. Franz and J. Koehnke, Chem. Commun., 2021, 57, 6372–6375 RSC.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- S. R. Fleming, T. E. Bartges, A. A. Vinogradov, C. L. Kirkpatrick, Y. Goto, H. Suga, L. M. Hicks and A. A. Bowers, J. Am. Chem. Soc., 2019, 141, 758–762 CrossRef CAS PubMed.
- E. Eber, M. Trawinska-Bartnicka, D. Sands, G. Bellon, U. Mellies, K. Bolbás, S. Quattrucci, H. Mazurek, R. Widmann, C. Schoergenhofer, B. Jilma and F. Ratjen, J. Cystic Fibrosis, 2021, 20, 61–67 CrossRef CAS PubMed.
- E. Eber, Lancovutide (Moli1901) Inhalation Solution Study in Adolescents and Adults With Cystic Fibrosis, ClinicalTrials.gov, Identifier: NCT00671736, 2007 Search PubMed.
- Safety and Efficacy of Multiple Daily Dosing of Oral LFF571 in Patients With Moderate Clostridium Difficile Infections, ClinicalTrials.gov, Identifier: NCT01232595, 2010 Search PubMed.
- D. C. K. Chan and L. L. Burrows, J. Antibiot., 2021, 74, 161–175 CrossRef CAS PubMed.
- A. Fabbretti, C.-G. He, E. Gaspari, S. Maffioli, L. Brandi, R. Spurio, M. Sosio, D. Jabes and S. Donadio, Antimicrob. Agents Chemother., 2015, 59, 4560–4568 CrossRef CAS PubMed.
- Clinical efficacy and safety of NAI-Acne gel 3% applied twice-a-day to patients with facial acne vulgaris, European Union Clinical Trials Register, Identifier: 2014-001491-62, 2014 Search PubMed.
- B. G. Livett, D. W. Sandall, D. Keays, J. Down, K. R. Gayler, N. Satkunanathan and Z. Khalil, Toxicon, 2006, 48, 810–829 CrossRef CAS PubMed.
- C. Herd, A randomised, placebo-controlled, double-blind, single and multiple ascending dose study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of subcutaneous doses of ACV1 in healthy adult male subjects, Australian New Zealand Clinical Trials Registry, Identifier: ACTRN12605000408684, 2005 Search PubMed.
- C. Herd, A randomised, double-blind, placebo-controlled study to assess the safety, tolerability, pharmacodynamics, and pharmacokinetics of subcutaneous doses of ACV1 in patients with diabetic peripheral neuropathic pain or post-herpetic neuralgia, Australian New Zealand Clinical Trials Registry, Identifier: ACTRN12607000201471, 2007 Search PubMed.
- Metabolic discontinues clinical trial programme for neuropathic pain drug, ACV1, Metabolic Pharmaceuticals Ltd, 2007 Search PubMed.
- I. A. Sharpe, J. Gehrmann, M. L. Loughnan, L. Thomas, D. A. Adams, A. Atkins, E. Palant, D. J. Craik, D. J. Adams, P. F. Alewood and R. J. Lewis, Nat. Neurosci., 2001, 4, 902–907 CrossRef CAS PubMed.
- A. Brust, E. Palant, D. E. Croker, B. Colless, R. Drinkwater, B. Patterson, C. I. Schroeder, D. Wilson, C. K. Nielsen, M. T. Smith, D. Alewood, P. F. Alewood and R. J. Lewis, J. Med. Chem., 2009, 52, 6991–7002 CrossRef CAS PubMed.
- A. Brust, Conopeptide to drug: the development, structure and activity correlation of Xen2174, 29th European Peptide Symposium, September 3 – 8, 2006, Gdansk, Poland, 2006 Search PubMed.
- P. Okkerse, J. L. Hay, E. Sitsen, A. Dahan, E. Klaassen, W. Houghton and G. J. Groeneveld, Br. J. Clin. Pharmacol., 2017, 83, 751–763 CrossRef CAS PubMed.
- A randomized, double-blind, placebo-controlled, serial-cohort, single ascending dose of Xen2174 PK/PD study administered intrathecally in healthy volunteers, Centrale Commissie Mensgebonden Onderzoek, Identifier: NL38941.056.11, 2011 Search PubMed.
- A randomized, double-blind, placebo-controlled Phase I study to evaluate the safety and tolerability of intrathecally administered single ascending bolus doses of Xen2174 in healthy subjects, Centrale Commissie Mensgebonden Onderzoek, Identifier: NL29372.040.09, 2009 Search PubMed.
- A randomized, double-blind, placebo-controlled, serial-cohort, single ascending dose of Xen2174 with bupivacaine interaction study administered intrathecally in healthy volunteers, Centrale Commissie Mensgebonden Onderzoek, Identifier: NL37832.058.11, 2011 Search PubMed.
- F. T. Shafiei, R. K. McAllister and J. Lopez, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2023 Search PubMed.
- C. Allerton and Royal Society of Chemistry, Pain therapeutics: current and future treatment paradigms, RSC Publishing, Cambridge, 2014 Search PubMed.
- B. M. Olivera, L. J. Cruz, V. De Santos, G. LeCheminant, D. Griffin, R. Zeikus, J. M. McIntosh, R. Galyean and J. Varga, Biochemistry, 1987, 26, 2086–2090 CrossRef CAS PubMed.
- J. E. Pope and T. R. Deer, Expert Opin. Pharmacother., 2013, 14, 957–966 CrossRef CAS PubMed.
- R. M. Scarborough, J. W. Rose, M. A. Hsu, D. R. Phillips, V. A. Fried, A. M. Campbell, L. Nannizzi and I. F. Charo, J. Biol. Chem., 1991, 266, 9359–9362 CrossRef CAS PubMed.
- R. M. Scarborough, Am. Heart J., 1999, 138, 1093–1104 CrossRef CAS PubMed.
- G. Tonin and J. Klen, Int. J. Mol. Sci., 2023, 24, 5446 CrossRef CAS PubMed.
- S. H. Ferreira, Br. J. Pharmacol., 1965, 24, 163–169 CrossRef CAS PubMed.
- Y. S. Bakhle, Nature, 1968, 220, 919–921 CrossRef CAS PubMed.
- H. Gavras, H. R. Brunner, G. A. Turini, G. R. Kershaw, C. P. Tifft, S. Cuttelod, I. Gavras, R. A. Vukovich and D. N. McKinstry, N. Engl. J. Med., 1978, 298, 991–995 CrossRef CAS PubMed.
- A. B. Atkinson and J. I. S. Robertson, Lancet, 1979, 314, 836–839 CrossRef PubMed.
- K. Hanif, H. K. Bid and R. Konwar, Hypertens. Res., 2010, 33, 11–21 CrossRef CAS PubMed.
- C. Bailly, Eur. J. Pharmacol., 2022, 914, 174661 CrossRef CAS PubMed.
- F. Benazet, M. Cartier, J. Florent, C. Godard, G. Jung, J. Lunel, D. Mancy, C. Pascal, J. Renaut, P. Tarridec, J. Theilleux, R. Tissier, M. Dubost and L. Ninet, Experientia, 1980, 36, 414–416 CrossRef CAS PubMed.
- C. H. McGinnis, C. A. Johnson and J. E. Fox, Poult. Sci., 1978, 57, 1641–1645 CrossRef.
- F. Benazet and J. R. Cartier, Poult. Sci., 1980, 59, 1405–1415 CrossRef CAS PubMed.
- G. L. Cromwell, T. S. Stahly, V. C. Speer and R. O'Kelly, J. Anim. Sci., 1984, 59, 1125–1128 CrossRef CAS PubMed.
- M. R. Naylor, A. T. Bockus, M.-J. Blanco and R. S. Lokey, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS PubMed.
- D. J. Brayden and M.-J. Alonso, Adv. Drug Delivery Rev., 2016, 106, 193–195 CrossRef CAS PubMed.
- Y. Haggag, S. El-Gizawy and M. Osman, Biomed. J. Sci. Tech. Res., 2018, 8, 6659–6662 Search PubMed.
- M. Dreyfuss, E. Härri, H. Hofmann, H. Kobel, W. Pache and H. Tscherter, Eur. J. Appl. Microbiol. Biotechnol., 1976, 3, 125–133 CrossRef CAS.
- E. Uchida, K. Morimoto, N. Kawasaki, Y. Ahmed, A. Said and T. Hayakawa, Free Radical Res., 1997, 27, 311–323 CrossRef CAS PubMed.
- A. R. Costa, M. E. Rodrigues, M. Henriques, R. Oliveira and J. Azeredo, Crit. Rev. Biotechnol., 2014, 34, 281–299 CrossRef CAS PubMed.
- M. Erak, K. Bellmann-Sickert, S. Els-Heindl and A. G. Beck-Sickinger, Bioorg. Med. Chem., 2018, 26, 2759–2765 CrossRef CAS PubMed.
- E. M. Bech, S. L. Pedersen and K. J. Jensen, ACS Med. Chem. Lett., 2018, 9, 577–580 CrossRef CAS PubMed.
- F. Hubrich, N. M. Bösch, C. Chepkirui, B. I. Morinaka, M. Rust, M. Gugger, S. L. Robinson, A. L. Vagstad and J. Piel, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2113120119 CrossRef CAS PubMed.
- S. Soltani, S. Zirah, S. Rebuffat, F. Couture, Y. Boutin, E. Biron, M. Subirade and I. Fliss, Front. Microbiol., 2022, 12, 780355 CrossRef PubMed.
- J. D. Hegemann, ChemBioChem, 2020, 21, 7–18 CrossRef CAS PubMed.
- Z. Luo, D. Klein Cerrejon, S. Römer, N. Zoratto and J.-C. Leroux, Sci. Transl. Med., 2023, 15, eabq1887 CrossRef CAS PubMed.
- S. Jevsevar, M. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef CAS PubMed.
- W. R. Strohl, BioDrugs, 2015, 29, 215–239 CrossRef CAS PubMed.
- W. R. Gombotz and D. K. Pettit, Bioconjugate Chem., 1995, 6, 332–351 CrossRef CAS PubMed.
- P. Dhakane, V. S. Tadimarri and S. Sankaran, Adv. Funct. Mater., 2023, 33, 2212695 CrossRef CAS.
- D. K. Chellappan, P. Prasher, V. Saravanan, V. S. Vern Yee, W. C. Wen Chi, J. W. Wong, J. K. Wong, J. T. Wong, W. Wan, J. Chellian, N. Molugulu, S. L. Prabu, R. Ibrahim, T. Darmarajan, M. Candasamy, P. K. Singh, V. Mishra, M. D. Shastri, F. C. Zacconi, A. Chakraborty, M. Mehta, P. K. Gupta, H. Dureja, M. Gulati, S. K. Singh, G. Gupta, N. K. Jha, B. G. George Oliver and K. Dua, Chem.-Biol. Interact., 2022, 351, 109706 CrossRef CAS PubMed.
- H. R. Costantino, L. Illum, G. Brandt, P. H. Johnson and S. C. Quay, Int. J. Pharm., 2007, 337, 1–24 CrossRef CAS PubMed.
- O. Afzal, A. S. A. Altamimi, M. S. Nadeem, S. I. Alzarea, W. H. Almalki, A. Tariq, B. Mubeen, B. N. Murtaza, S. Iftikhar, N. Riaz and I. Kazmi, Nanomaterials, 2022, 12, 4494 CrossRef CAS PubMed.
- L. Chen, W. Hong, W. Ren, T. Xu, Z. Qian and Z. He, Signal Transduction Targeted Ther., 2021, 6, 1–25 CrossRef PubMed.
- M. Vukomanović, V. Žunič, Š. Kunej, B. Jančar, S. Jeverica, R. Podlipec and D. Suvorov, Sci. Rep., 2017, 7, 4324 CrossRef PubMed.
- D. Richter, E. Lakis and J. Piel, Nat. Chem., 2023, 15, 1422–1430 CrossRef CAS PubMed.
Footnotes |
| † These authors have contributed equally. |
| ‡ The following different ratings are employed to characterise the activity/strength of the different compounds. Please find their respective definitions below. EC50 = half maximal effective concentration; GI50 = concentration required for 50% growth inhibition; IC50 = half maximal inhibitory concentration; ID50 = median infectious dose; LC100 = lethal concentration (kills 100% of a test sample); LD50 = median lethal dose; LD90 = 90% lethal dose (kills 90% of a test sample); MIC = minimal inhibitory concentration; TD50 = median toxic dose. The lower the value, the more potent (or toxic) the compound is. |
| § To help the reader classify the values mentioned, we summarise below typical values of drugs that are in clinical use: MIC values of potent antibiotics and antifungals are in the low μM range. IC50 values of potent antifungals and antivirals are in the submicromolar to low μM range. First line treatments for antiparasitic drugs usually have IC50 values in the nM to low μM range. Chemotherapy drugs that have made it through clinical trial show IC50 values in the nM range for their target. |
| ¶ IM = intramuscular; IT = intrathecal; IV = intravenous; SC = subcutaneous |
| This journal is © The Royal Society of Chemistry 2024 |