
Architecture of full-length type I modular polyketide synthases revealed by X-ray crystallography, cryo-electron microscopy, and AlphaFold2
Saket R.
Bagde
 ab and
Chu-Young
Kim
*c
ab and
Chu-Young
Kim
*c
aProgram in Cellular and Molecular Medicine, Boston Children's Hospital, Boston, MA 02115, USA
bDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA. E-mail: sbagde@crystal.harvard.edu
cDepartment of Biochemistry, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA. E-mail: chuyoung@illinois.edu
First published on 19th March 2024
Abstract
Covering: up to the end of 2023
Type I modular polyketide synthases construct polyketide natural products in an assembly line-like fashion, where the growing polyketide chain attached to an acyl carrier protein is passed from catalytic domain to catalytic domain. These enzymes have immense potential in drug development since they can be engineered to produce non-natural polyketides by strategically adding, exchanging, and deleting individual catalytic domains. In practice, however, this approach frequently results in complete failures or dramatically reduced product yields. A comprehensive understanding of modular polyketide synthase architecture is expected to resolve these issues. We summarize the three-dimensional structures and the proposed mechanisms of three full-length modular polyketide synthases, Lsd14, DEBS module 1, and PikAIII. We also describe the advantages and limitations of using X-ray crystallography, cryo-electron microscopy, and AlphaFold2 to study intact type I polyketide synthases.
 Saket R. Bagde | Saket Bagde completed his PhD at Cornell University under Professor Chris Fromme's mentorship. He conducted his Master's research at the University of Texas at El Paso in Professor Chu-Young Kim's lab and received his Master's Degree in Biology from the Indian Institute of Science Education and Research Pune. He is interested in studying large macromolecular complexes and has determined structures of a modular polyketide synthase, Lsd14, and a guanine nucleotide exchange factor, TRAPPII complex. Saket is currently studying molecular assemblies involved in cell adhesion as a Post-Doctoral Researcher at Boston Children's Hospital and Harvard Medical School in Professor Timothy Springer's lab. |
 Chu-Young Kim | Chu-Young Kim was born in Seoul, South Korea. He received a BA degree in Chemistry from Cornell University, and MSE degree in Bioengineering and PhD degree in Chemistry from University of Pennsylvania (David W. Christianson). He then carried out postdoctoral research at Stanford University (Chaitan Khosla). He was a faculty member at the National University of Singapore and the University of Texas at El Paso. He is currently a Professor of Biochemistry at University of Illinois Urbana-Champaign. His research involves structural and biochemical characterization of enzymes involved in natural product biosynthesis and DNA repair. |
1 Introduction
Polyketide natural products have fascinated synthetic chemists, medicinal chemists, enzymologists, structural biologists, protein engineers, and drug developers due to their complex chemical structure and useful biological activities. Polyketides contain multiple ketone groups which are reduced to different levels and a myriad of stereocenters, making them challenging to synthesize using chemical methods. In nature, polyketides are produced by various organisms including bacteria, fungi, and algae. Natural polyketides often exhibit potent therapeutic properties, and many have been developed into clinical drugs, including antibiotics, immunosuppressants, and anticancer agents.1 This early success has led scientists to search for an efficient method for preparing novel polyketides for drug development. One attractive approach is to genetically engineer polyketide-producing organisms to produce non-natural polyketides. Polyketide-producing organisms possess polyketide synthases (PKSs), multifunctional enzymes that synthesize complex polyketides from simple carboxylic acid building blocks. These biosynthetic enzymes can be categorized into type I, type II, and type III PKSs, which differ in how the required catalytic activities are distributed and organized across different polypeptides. Here, we focus exclusively on the type I cis-acyltransferase PKSs.Type I PKSs are an attractive engineering target due to the direct relationship between their domain composition and the structure of the polyketide product.2 This allows for the creation of artificial polyketide biosynthesis pathways for the production of novel polyketides. The first step toward achieving reliable and routine PKS engineering is to elucidate their three-dimensional structure and understand their mechanism of action. While high-resolution structures of modular PKS fragments have revealed important information about how the individual domains catalyze reactions, structural analysis of intact modular PKSs at high resolution has been lacking. To this end, the recently determined three-dimensional structures of Lsd14 from Streptomyces lasalocidi and 6-deoxyerythronolide B synthase module 1 (DEBS M1) from Saccharopolyspora erythraea have provided much-needed new structural details of intact modular PKSs which we summarize in the current article.3,4
2 Structural methods for studying modular polyketide synthases
X-ray crystallography and single-particle cryo-electron microscopy (cryo-EM) were employed in the recent PKS structural studies. The structure of Lsd14 in which the acyl carrier protein is docked to the acyltransferase (AT) domain was determined using X-ray crystallography, while the structure of Lsd14 in which the acyl carrier protein is docked to the ketosynthase (KS) domain was solved using cryo-EM.3 The structure of DEBS M1 was elucidated using cryo-EM.4 These achievements illustrate that X-ray crystallography and cryo-EM can both be used for studying intact modular PKSs. In the Lsd14 study, the two structural techniques were complementary. We were able to crystallize Lsd14 in one specific conformation only, despite extensive effort to crystallize the protein in other possible conformations. Ultimately, we solved the structure of Lsd14 in an alternative conformation using cryo-EM. This example shows the benefits of using multiple structural techniques to study the same PKS.Crystallographic study of modular PKSs is challenging at multiple levels. Due to their sheer size, recombinant protein expression and purification steps require much optimization. The large size and flexibility of PKSs prevent their crystallization. Even after obtaining the first Lsd14 crystals, we spent over a year optimizing the crystallization condition to obtain well-diffracting crystals. X-ray crystallography typically provides higher-resolution structural information than cryo-EM, and this held true for Lsd14 and DEBS M1. X-ray crystallography yielded the highest resolution structure, at 2.4 Å resolution, while cryo-EM produced structures having a resolution ranging from 3.1 to 4.3 Å. Such difference in resolution is significant because protein side-chain atoms and water molecules are well defined at 2.4 Å but not at 3.1 Å.
Sample preparation for cryo-EM is typically more straightforward than X-ray crystallography because the protein need not be crystallized. However, for Lsd14 and DEBS M1, sample preparation for single-particle cryo-EM proved to be highly involved. Initial electron micrographs of Lsd14 on the cryo-EM grid indicated that the PKS homodimer had fallen apart and the individual protomers lacked a well-defined tertiary structure. Such a phenomenon is frequently encountered in single-particle cryo-EM and is often attributed to the unfavorable condition created at the air–water interface during grid preparation.5,6 Khosla group solved this problem by incubating DEBS M1 with a Fab antibody fragment that binds to the N-terminal docking domain (DD), a coiled-coil structure that mediates interaction with the upstream PKS in the biosynthesis pathway.4 Because this region of the PKS is part of the dimer interface, Fab binding directly enhances dimer stability. In addition to the Fab, the Khosla group included citrate as an additive in the buffer used for cryo-EM analysis. Citrate improved thermostability and catalytic activity of DEBS M1 and M3 chimeras fused to the DEBS TE domain. Further, citrate also promoted dimerization of the KS-AT di-domain of DEBS M3.4
To solve the cryo-EM structure of Lsd14, we used the same Fab that was used for DEBS M1 structure determination. To utilize this Fab, we created a chimeric Lsd14 protein where its native N-terminal DD was genetically replaced with the corresponding domain in DEBS M1. As expected, incubating the engineered Lsd14 protein with the Fab had the same favorable effect as for DEBS M1, preserving dimeric Lsd14 particles on cryo-EM grids, and this enabled us to solve the Lsd14 cryo-EM structure. Using citrate buffer along with the addition of substrates and substrate analogs further stabilized domain–domain interactions.3 Encouraged by this success, we tried the Fab strategy on several other type I PKSs but they were unsuccessful. In some cases, the PKS homodimer disintegrated even when the Fab was added, and in one case Fab caused protein aggregation. In other cases, Fab stabilized the PKS dimer, and we were able to observe the ketosynthase and acyltransferase domains, but the remaining domains were unresolved, presumably due to their flexibility. At the moment, structural study of type I PKSs using X-ray crystallography and cryo-EM are equally challenging.
Recent advances in protein structural prediction methods have shown great promise in accurately predicting the three-dimensional structures of many proteins.7 AlphaFold2 (AF2) employs a deep-learning-based approach to predict protein structures based on amino acid sequence coevolution information.8 An extension of AlphaFold2, AlphaFold2-Multimer can be used to predict structures of protein complexes.9 Despite these advances, accurate structure prediction of large and flexible protein complexes remains a challenge.10 In a later section, we exemplify this for modular PKSs by comparing AlphaFold2-multimer predicted structures of modular PKSs to their experimentally determined models. Notably, AlphaFold2 was unable to predict the asymmetric architecture that was observed in the experimental structures of Lsd14 and DEBS M1.
3 Structure and proposed mechanism of the Lsd14 modular polyketide synthase
3.1 Polyether polyketides
Polyether polyketides are characterized by the presence of two or more ether and acetal rings.11 Polyether ionophores function as antibiotics presumably by binding monovalent and divalent metal cations and transporting them across the cell membrane which leads to disruption of the physiological ion gradient.12 Examples of polyether antibiotics include lasalocid A, monensin, nanchangamycin, narasin, nigericin, salinomycin, and tetronomycin. The biosynthetic pathway of polyether polyketides involves modular PKSs that build the polyketide backbone and at least two additional enzymes – a monooxygenase and an epoxide hydrolase – that form the characteristic cyclic ether groups.11 Lasalocid A is produced by the actinomycete bacterium Streptomyces lasalocidi. The lasalocid A biosynthesis pathway consists of seven modular PKS proteins, Lsd11 to Lsd17 (Fig. 1A), a flavin-dependent monooxygenase that catalyzes the epoxidation reaction, Lsd18, and an epoxide hydrolase, Lsd19 (ref. 13–16).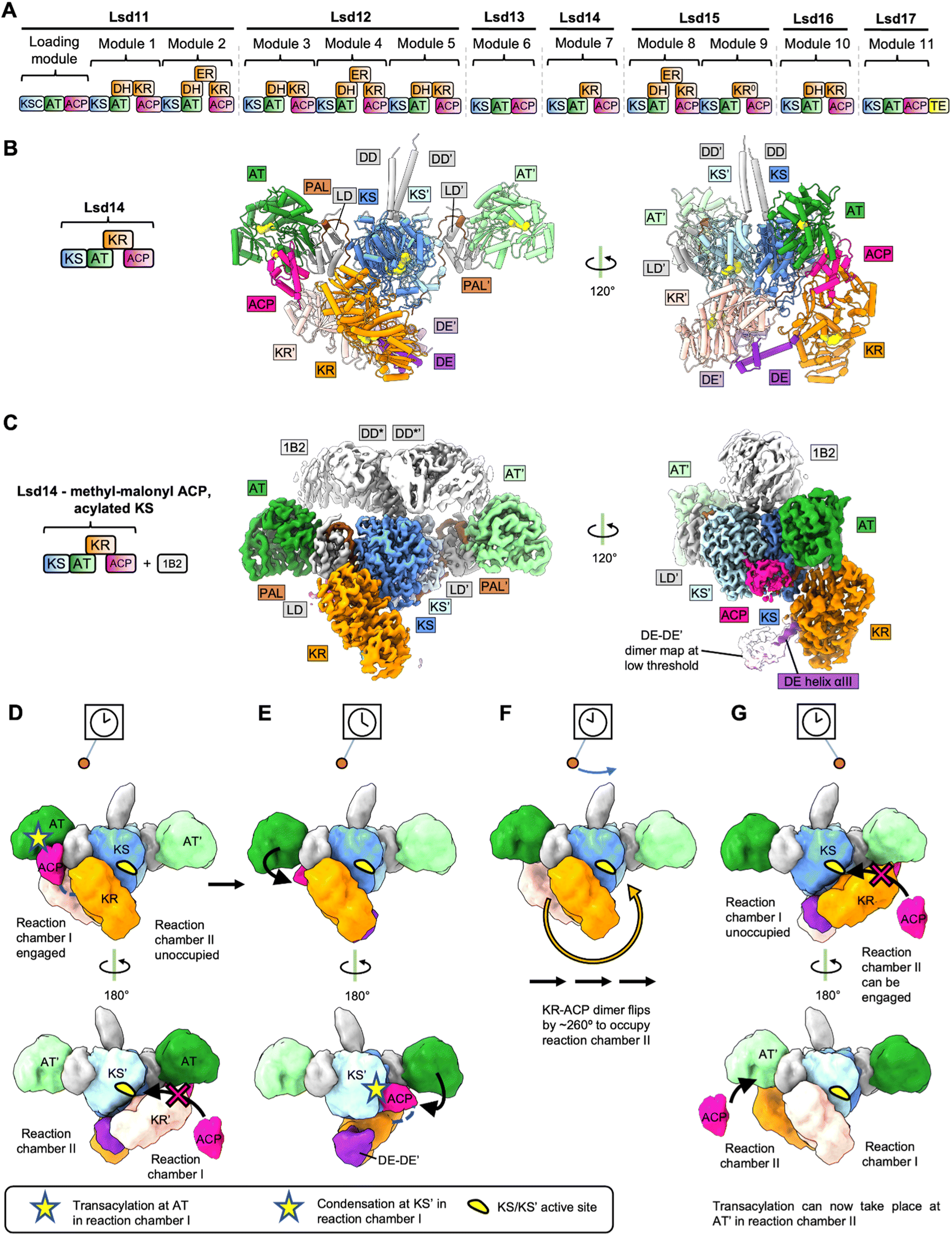 | ||
| Fig. 1 Structures and model for Lsd14. (A) Modular PKS involved in Lasalocid biosynthesis. (B) Crystal structure of Lsd14 in the transacylation state. (C) Cryo-EM map of Lsd14 stalled at the condensation state. (D)-(G) Pendulum clock model for Lsd14. (D) Both KRs and the ACPs tethered to the KR domains are present in reaction chamber I. ACPs tethered to the respective KRs cannot reach reaction chamber II due to limited linker length. Hence, reaction chamber II is unoccupied. Access to the KS′ entrance is blocked by the KR′ domain, so one ACP docks at the AT, and the transacylation reaction takes place in reaction chamber I. (E) KR′ undocks from KS, opening access to KS′ active site. ACP loaded with extender unit docks onto KS′ and the condensation reaction takes place in reaction chamber I. (F) Reduction reaction at the KR domain and the transfer of the growing polyketide chain to the next module take place (domain organization details unknown). The KR–KR′ dimer flips by ∼260° along with the tethered ACPs. (G) Reaction chamber I is now unoccupied whereas reaction chamber II is occupied. The PKS reaction cycle can now start in reaction chamber II. | ||
3.2 Structures of intact Lsd14
The X-ray crystal and cryo-EM structures of the intact Lsd14 PKS were reported recently.3 Lsd14 is a single module PKS and contains a ketosynthase domain (KS), an acyltransferase domain (AT), a ketoreductase domain (KR), and an acyl carrier protein (ACP).13 Overall, three different conformations of Lsd14 were reported in this study. First, a 2.4 Å resolution crystal structure of apo-Lsd14 stalled at the transacylation step. Second, a 3.1 Å resolution cryo-EM structure of holo-Lsd14 stalled at the condensation step. Third, an additional 3.1 Å resolution cryo-EM structure of Lsd14 in which the ACP is not docked at any of the catalytic domains. Notably, cryo-EM structures were determined for a chimeric Lsd14 containing the DD from DEBS module 3 in complex with the 1B2 antibody fragment.17 The X-ray and cryo-EM structures revealed that the domains in the Lsd14 dimer attain an elaborate asymmetric architecture.3.3 Crystal structure of apo-Lsd14 stalled at the transacylation step
Seven out of the eight catalytic domains, including two copies of each of the KS, AT, and KR domains and one copy of the ACP domain were resolved in the Lsd14 crystal structure. One ACP domain was not detected, likely because it is not locked into a single position. The Lsd14 homodimer adopts an extended architecture where the KS-AT dimer forms an extended platform, and the KR domains are found below this platform. Importantly, the locations of active site entrances in the Lsd14 structure give rise to two separate reaction chambers where two copies of polyketide chains can be synthesized independently. The most surprising aspect of the Lsd14 structure is its overall asymmetric architecture leading to the formation of two unequal reaction chambers. While the KS-AT didomains form a symmetric dimer spanning the two reaction chambers, the two KR domains and the resolved ACP are located in the same reaction chamber in all the three Lsd14 structures reported in this study. In this configuration, only reaction chamber I (Fig. 1B and C) has the full complement of the KS, AT, KR, and ACP domains whereas reaction chamber II is incomplete.The KS domain forms a compact dimeric structure, similar to the structures of the KS domains from other modular PKSs reported previously.18 The KS and the AT domains are connected via the KS-AT linker domain (LD). The AT and the KS-AT linker domains are located on the sides of the KS dimer, extending out in opposite directions, giving rise to an elongated platform. This extended architecture of the KS-LD-AT dimer is similar to that reported for the excised KS-AT-LD fragments from DEBS module 3, DEBS module 5, and curacin synthase module CurL.18–20 An important consequence of the extended architecture of the KS-LD-AT dimer is that the active site of the AT domain is located ∼80 Å away from the KS active site. Based on this observation, it was suggested that substantial domain reorganization is needed for the ACP to interact with KS after picking up the extender unit from the AT active site.18 The structures of Lsd14 in the transacylation state and the condensation state discussed later in this section suggest the rearrangements that may enable access of ACP to the AT and KS active sites sequentially.
In the Lsd14 crystal structure both copies of the KR domains, KR and KR′, are docked to the KS dimer. KR/KR′ domains form hydrogen bonding and electrostatic interactions at the interface with the KS dimer. While the complementary nature of charged surfaces at the KR/KS interface is generally conserved in other modular PKS sequences, the functional importance of these interfaces awaits biochemical characterization. Because of the cyclic arrangements of the KR domains in the KR-DE dimer, the active site entrance of one KR domain faces the KS dimer, and the other faces away from the reaction chamber. Importantly, (DE-KR)2 is docked on the KS/KS′ dimer in such a way that it largely occupies reaction chamber I. This docking mode of the KR dimer on the KS dimer is responsible for the overall asymmetry of the Lsd14 architecture and has important mechanistic implications. The ACP domains are tethered to the KR domains via linkers of limited length that are not long enough for the ACPs to reach reaction chamber II. As a result, docking of the KRs in reaction chamber I is also predicted to restrict the ACPs to reaction chamber I.
3.4 Cryo-EM structure of holo-Lsd14 stalled at the condensation step
Two strategies were used to stabilize Lsd14 for cryo-EM analysis. First, an antibody Fab fragment, 1B2, was used to stabilize the Lsd14 dimer on cryo-EM grids. 1B2 was developed in a previously reported study to bind to DEBS module 3 and was reported to form extensive contacts with the docking domain of DEBS module 3 (DD*).17 Substitution of the docking domain of Lsd14 with DD* (to obtain chimeric Lsd14-DD*) was required to form a complex of Lsd14 with 1B2. Second, stabilization of the KR domain was achieved by the addition of the native AT substrate, methyl malonyl-CoA. Cryo-EM analysis of holo-Lsd14-DD*-1B2 complex incubated with a KS substrate analog, 2-acetaminoethyl-thio-3-oxobutanoate, yielded a 3.1 Å resolution map of holo-Lsd14-DD*-1B2 stalled at the condensation step. In this structure, six of the eight domains were resolved, including both copies of the KS and the AT domains and one copy each of the KR and the ACP domains. One KR and one ACP domain were not resolved.In this structure, the ACP was docked on the KS domain at its dedicated side entrance. The phosphopantetheine group on the ACP was clearly resolved and it extended into the KS active site tunnel. Docking of ACP on the KS domain appears to depend on the acylation state of the KS domain as the ACP did not dock to the KS domain in the cryo-EM structure of Lsd14-DD*-1B2 determined in the absence of KS substrate analog. The ACP interacts with the KS domain via loop I and helix II. This binding mode is consistent with previously reported structural and biochemical characterization of other type I PKSs including the CTB1-iterative PKS fragment and DEBS.30–32
A comparison of the structures of Lsd14 in the transacylation and the condensation steps revealed an overlap between the docking sites of the ACP and the KR′ domains on the KS/KS′ dimer. This suggests that the KR′ domain needs to undock from its docking site on the KS/KS dimer after the transacylation step to enable docking of ACP to the KS/KS′ dimer for condensation reaction to occur. Furthermore, a helix-loop motif within the KS (residues 175–184) undergoes a conformational change upon ACP binding. Importantly, the cryo-EM structure of Lsd14-DD*-1B2 stalled at the condensation step also attains the overall asymmetric architecture discussed above for Lsd14 stalled at the transacylation step. The lone resolved ACP and the KR domains occupy reaction chamber I which leaves reaction chamber II incomplete. This suggests that only one reaction chamber is used at a time in both transacylation and condensation states.
3.5 Pendulum clock model for PKS reaction cycle
Based on the structural analysis of Lsd14, we have proposed a pendulum clock model to explain the non-synchronous use of the two reaction chambers in Lsd14 (Fig. 1D–G).3 In this model, the KS-AT platform remains static whereas the KR-DE dimer and the tethered ACP domains swing back and forth like a pendulum, occupying only one reaction chamber at a time. In this case, reaction chamber I is hypothesized to perform extender group transacylation, condensation, and β-keto group reduction reactions while the ACP from the upstream PKS transfers the growing polyketide chain intermediate to the KS active site of reaction chamber II. Next, the KR-DE dimer swings to reaction chamber II so that extender group transacylation, condensation, and the β-keto group reduction reactions can take place in reaction chamber II.While the pendulum mechanism provides a plausible explanation for the observed asymmetric architecture of Lsd14, further functional experiments are required to test the validity of this model. Furthermore, the architecture of Lsd14 in the ketoreduction step has not been reported. Based on the current data, it is unclear what drives the movement of the pendulum. The pendulum movement may be triggered by the free energy released during the KS-catalyzed decarboxylative condensation reaction.
One advantage of utilizing such an elaborate mechanism, as opposed to synchronous catalysis across the two reaction chambers, is that it prevents iterative chain elongation by the same module, thus serving the same purpose as the turnstile mechanism proposed for the DEBS PKS discussed below. In the pendulum clock model, the KS active site opens for translocation when (DE-KR-ACP)2 swings over to the opposite reaction chamber. Thus, ACP is prevented from transacylating the product back to the same KS because it is moved out of the reaction chamber by the pendulum movement.
4 Structure and proposed mechanism of the DEBS modular polyketide synthase
4.1 Macrolide polyketides
Macrolide polyketides are characterized by the presence of a large macrocyclic lactone ring with one or more deoxy sugars attached. Macrolides are an important class of antibiotics that function by binding to the 50S subunit of 70S bacterial ribosomes and inhibiting or interfering with protein synthesis. Examples of macrolide antibiotics include erythromycin, clarithromycin, and tylosin. The polyketide portion of erythromycin A, 6-deoxyerythronolide B (6-dEB), that forms the 14-membered macrolactone ring, is synthesized by DEBS in Saccharopolyspora erythraea (Fig. 2A). The 6-deoxyerythronolide B biosynthetic pathway consists of three type I modular PKSs, DEBS1, DEBS2, and DEBS3, which are encoded by the genes eryAI, eryAII and eryAIII respectively. The loading module and 6 extension modules of DEBS form the 14-membered macrolactone ring.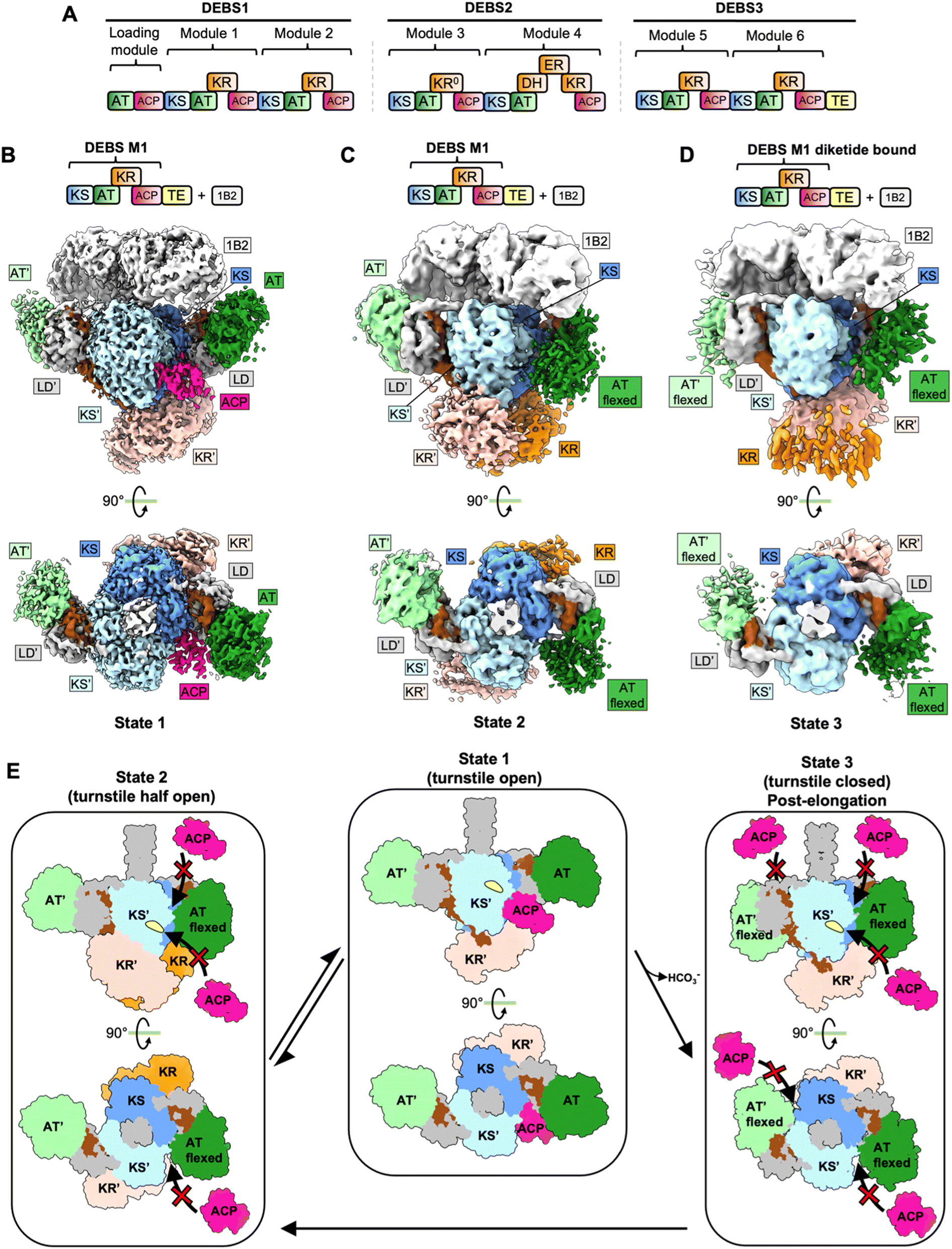 | ||
| Fig. 2 Structures and model for DEBS M1. (A) DEBS modular PKS. (B) Cryo-EM maps of DEBS M1 stalled at the condensation state. This map represents the turnstile open state (State 1). (C) Cryo-EM map of DEBS M1 in the turnstile half-open state (State 2). (D) Cryo-EM map of DEBS M1 in the turnstile closed state (State 2). (E) Turnstile model for DEBS M1. In the turnstile open state, the AT domains are not flexed, and the ACP can dock on either of the KS side entrances. Notably, only one ACP is docked on the KS dimer. In the turnstile half open, one of the AT domains attains a flexed conformation and prevents the docking of ACP on the KS′ side entrance. Upon completion of the elongation reaction cycle, DEBS M1 adopts a turnstile closed state in which both the ATs attain a flexed conformation, blocking access to both KS side entrances. | ||
4.2 Structures of DEBS fragments and the turnstile model
DEBS is by far the most extensively studied modular PKS. Structures of multiple DEBS module fragments have been reported.18,33–38 The crystal structures and small-angle X-ray scattering (SAXS) analysis of DEBS KS-AT didomains with and without a conformation stabilizing antibody support an extended KS-AT dimer architecture, like that described for Lsd14 above.17,38 A “turnstile” mechanism has been proposed to explain substrate channeling across multiple modules of an assembly line PKS.39 In this model, the initiation of chain elongation cycles is coupled over multiple modules via the translocation and decarboxylative condensation reactions within each module. In the “turnstile closed” state, translocation of the polyketide intermediate from the previous module is prevented, and the polyketide intermediate elongated within the same module is attached to the ACP. The turnstile is opened upon translocation of the elongated polyketide intermediate to the next module and the KS domain of the module in the “turnstile open” state can now be acylated with the polyketide intermediate from the previous module. However, in the absence of the structure of an intact PKS module in the “turnstile open” or “turnstile closed” states, the mechanistic details of this model remained unclear.4.3 Cryo-EM analysis of intact DEBS module 1
Recently reported cryo-EM analysis of intact DEBS module chimeras suggests that structural rearrangements at the KS/AT interface can explain the opening and closing of the turnstile by modulating access to the KS active site entrance.4 In this study, a chimeric module in which DEBS module 1 (DEBS M1) fused to the DEBS thioesterase domain was subjected to cryo-EM analysis. DEBS M1 contains KS, AT, KR, and ACP domains. Like the strategy used for stabilizing the Lsd14 dimer on cryo-EM grids described above, the docking domain of DEBS M1 was substituted with that of DEBS M3. The resulting holo-DEBS M1-TE construct bound to Fab 1B2 was used for cryo-EM analysis. Three main conformations of DEBS M1 were reported in this study, each attaining an architecture forming two reaction chambers. Two different conformations were observed from the same cryo-EM dataset for holo-DEBS M1-TE. A “turnstile open” conformation, denoted State 1, and a “turnstile half-open” conformation, denoted State 2, were resolved to overall resolutions of 3.2 Å and 4.1 Å, respectively (Fig. 2B, C and E). A holo-DEBS M1 construct without the addition of a TE domain, and upon incubation with native substrate, extender unit, NADPH, and TCEP in the presence of Fab 1B2, attained a third “turnstile closed” conformation.In State 1, holo-DEBS M1 has an asymmetric conformation like that observed for Lsd14 structures3 (Fig. 1B, C and 2C). While the KS-AT domains of holo-DEBS M1 in State 1 form a symmetric extended architecture similar to that observed for structures of excised KS-AT fragments reported before,18–20 the overall domain organization is asymmetric. Only one of the two KR domains is observed, and it is docked at the KS dimer forming electrostatic interactions at the KS/KR interface. Tracing the connectivity of the KS-KR linker revealed that the observed KR domain (KR′) is attached to the KS domain of the other subunit. Furthermore, only one of the two ACPs is observed, and it is docked at the KS domain in the cleft formed by the KS domain, the KS-AT linker, and the AT domain in a configuration that agrees with previous functional studies.30,40 The ACP-KS docking mode in the DEBS-M1 cryo-EM map is similar to that observed for Lsd14, described above. Interestingly, the crystal structure of the ketosynthase-like decarboxylase (KSQ)-AT fragment from the loading module of Gfs PKS in complex with ACP revealed that the GfsA ACP-KSQ docking mode is also similar to that observed for Lsd14 and DEBS M1 KS-ACP.3,4,54 The DEBS-M1 ACP interacts with the KS domain via loop I. The KS-docked ACP also contacts the LD in addition to the KS dimer. While only one ACP is docked at the KS dimer, both of the KS active site entrances are accessible to ACPs for the translocation reactions in State 1, which thus represents a “turnstile open” conformation (Fig. 2E). Furthermore, 3D classification of particles in State 1 aimed at resolving the linker between the sole observed ACP docked at the KS′ active site entrance and the KR domain revealed that this ACP belongs to the partner subunit, in agreement with the model that condensation reaction occurs intermolecularly.3
In State 2, both KR domains are docked symmetrically on the KS dimer forming interfaces similar to that observed in State 1. Interestingly, the KS-KR linker connectivity in this map revealed that the KR domains interact with the KS domain belonging to the same subunit, as opposed to the intermolecular KS:KR′ docking observed in State 1. This supports a model in which the reductive domains can undergo a swiveling rotational motion with respect to the condensing domains, similar to that proposed for the metazoan fatty acid synthase.3 A notable observation is that while the overall architecture in State 2 is also asymmetric, as observed for State 1, the asymmetry in State 2 originates from the architecture of the KS-AT dimer. This is because in State 2, one of the AT domains is flexed down compared to the conformation observed in the KSAT fragment structures, Lsd14 structures, and that observed in State 1. The flexing of AT in holo-DEBS M1 is different from the arched conformation described for PikAIII module structures described below, as the corresponding LD in holo-DEBS M1 still attains an extended conformation whereas the LD is also flexed down in the PikAIII cryo-EM structures.3 While the local resolution of the map for the flexed AT is low, the position of the flexed AT, as inferred from unrestrained fitting in the map density, revealed a KS-AT-LD configuration with a constricted KS-AT cleft. This constriction blocks access of the ACP to the KS active site tunnel entrance and may prevent translocation and condensation reactions. The flexed AT conformation may provide a structural explanation for the turnstile closing. On the other hand, the second AT in State 2 is not flexed, and ACP can access the KS active site tunnel. State 2 thus represents a “turnstile half-open” conformation (Fig. 2E).
In State 3, both AT domains attain a flexed-down conformation leading to constriction of the KS-LD-AT cleft in both reaction chambers. In this conformation, the access of ACP to the KS active site entrances in both reaction chambers is blocked and hence it depicts a “turnstile closed” conformation. While the identity of the acyl group on the ACP was not directly confirmed, this conformation likely represents a post-elongation diketide product-bound state as the sample was incubated with native substrates prior to vitrification.
5 Structure and proposed mechanism of PikAIII modular polyketide synthase
5.1 Ketolide polyketides
Pikromycin is a ketolide polyketide which are distinguished from macrolides based on the substitution of the L-cladinose at position C3 with a ketone group.41 Ketolides have the potential to be developed into novel macrolide antibiotics. Pikromycin is naturally produced by Streptomyces venezuelae.42 The pikromycin biosynthetic gene cluster contains four type I PKS genes (PIKAI-PIKAIV) which harbor one loading module and six extension modules that produce the 14-membered ring macrolactone narbonolide which is the polyketide core of pikromycin43 (Fig. 3A).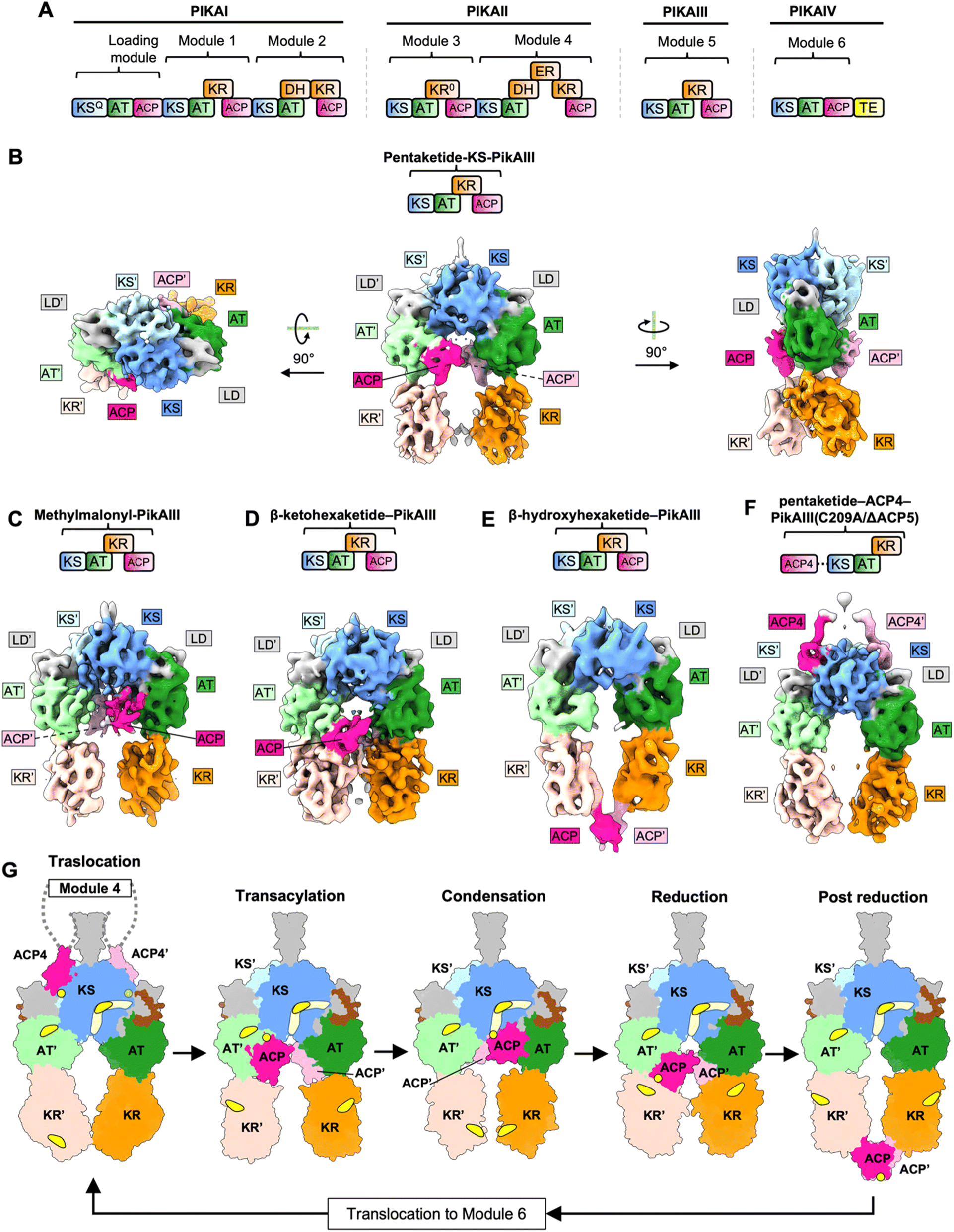 | ||
| Fig. 3 Structures and model for PikAIII. (A) Pikromycin modular PKS. (B) Cryo-EM maps of DEBS M1 stalled at the condensation state. (B) Cryo-EM map of pentaketide loaded PikAIII stalled at the transacylation state. (C) Cryo-EM map of methyl-malonyl loaded PikAIII stalled at the condensation state. (D) Cryo-EM map of β-ketohexaketide loaded PikAIII stalled at the reduction state. (E) Cryo-EM map of β-hydroxyhexaketide loaded PikAIII stalled at the post-reduction state. (F) Cryo-EM map of pentaketide–ACP4–PikAIII(C209A/ΔACP5) depicting PikAIII stalled at the translocation state. (G) Model of PikAIII reaction cycle. In the translocation state, ACPs from module 4 access side entrances of the KS domains in PikAIII and acylate the active site cysteine of the KS domain with the pentaketide intermediate. ACPs then dock to the AT domain of pentaketide-PikAIII for the transacylation reaction. The difference in orientation of the KR domains between the transacylation and the translocation steps is likely not physiological and appears to be due to lack of ACP in the pentaketide–ACP4–PikAIII(C209A/ΔACP5) construct. In the condensation state, the methylmalonyl-loaded ACPs access the bottom entrance of the KS domains. This is accompanied by a rearrangement of the orientation of the KR domain. In the reduction state, the KRs rearrange back to the orientation in which the KR active site is closer to the AT domain. Upon reduction, the ACPs are ejected out of the reaction chamber in the post-reduction state, and the elongated β-hydroxyhexaketide intermediate can be translocated to module 6. | ||
5.2 Cryo-EM analysis of intact PikAIII
Pikromycin PKS module 5 (PikAIII) contains a pair of each of the KS, AT, KR, and ACP domains. Analysis of PikAIII using cryo-EM yielded medium-resolution maps of the intact module in different catalytic states at overall resolution values of 7.3–9.5 Å44,45 (Fig. 3B–F). Homologous structures of the KS, AT, KR, and ACP domains from DEBS were fitted into the PikAIII maps using rigid body fitting, to yield pseudo-atomic models. To identify the architecture of the PikAIII module in different biochemical states, holo-PikAIII was loaded with chemically synthesized substrate intermediates. The identity of the acyl group on the phosphopantetheine arm of ACP and the catalytic cysteine of the KS domains was experimentally confirmed using bottom-up liquid chromatography/Fourier transform ion cyclotron resonance mass spectrometry (LC/FT-ICR MS). Using this approach, architectures of PikAIII in each of the steps of a KS-AT-KR-ACP containing PKS module were resolved (Fig. 3B–F). A key feature of all these structures is that the PikAIII module attains an arch-shaped architecture harboring a single reaction chamber, as opposed to an extended architecture harboring two reaction chambers that was observed for DEBS modules, Lsd14 and the related FAS.3,4,44–46 Furthermore, all the models reported in the PikAIII cryo-EM analysis display a 2-fold symmetry as opposed to asymmetric architectures observed for DEBS modules, Lsd14, and the related FAS.3,4,44,45,47Cryo-EM analysis of holo-PikAIII incubated with MM–CoA, thiophenol-activated pentaketide and NADPH yielded ∼11 Å maps of β-hydroxyhexaketide-PikAIII revealing the architecture of PikAIII in the post-reduction step (Fig. 3E). Three maps were resolved for this sample, all having an overall configuration of KS-AT-KR domains similar to that observed in the reduction step. Strikingly, the ACPs are located outside of the reaction chamber, docked below the KR domains in the post-reduction state. Based on this observation, ejection of the ACPs upon completion of the PKS elongation and modification cycle was proposed as a mechanism to maintain directed transfer of the PKS intermediates to the next module. In both reduction and post-reduction steps, the AT domain was shifted towards the KS, thereby occluding the KS domain side entrances. This was proposed to prevent intermediate transfer from the previous module during the reduction and post-reduction steps, further contributing to directionality of the PKS reaction cycle.
Overall, the maps of PikAIII in different biochemical states of the PKS reaction cycle reveal how an arch-shaped architecture of PikAIII allows for access of the intramodular ACP to the catalytic sites of the KS, AT, and KR domains within the same reaction chamber (Fig. 3G). On the other hand, ACP from the previous module uses a different KS active site entrance located outside the reaction chamber leading to spatial separation of the intermodular and intramodular intermediate transfer docking sites. Furthermore, the location of the ACP, KR, and AT domains appears to be dependent on the identity of the acyl group loaded on the ACP and at the KS active site revealing the roles of interdomain crosstalk and the influence of substrate identity on the architecture of PKS modules. While the post-ACP dimerization elements were not resolved in the reported maps, the ACPs dock at equivalent sites symmetrically and are within the range of distances that would allow dimerization, suggesting that the ACPs move in concert throughout the PikAIII reaction cycle.
6 Comparison of Lsd14, DEBS, and PikAIII architectures and mechanisms
6.1 Overall architectures of Lsd14, DEBS and PikAIII
The overall extended architecture observed for Lsd14 and DEBS M1 is highly divergent from the arch-shaped architecture observed for PikAIII. This architectural difference has important mechanistic implications as the extended architecture gives rise to two reaction chambers, whereas a single reaction chamber is formed in the arch-shaped architecture. Additionally, both Lsd14 and DEBS M1 adopt an overall asymmetric architecture such that the two reaction chambers in Lsd14 and DEBS are not equivalent. In Lsd14, the KR-DE-ACP domains are the symmetry-breaking elements. Importantly, the presence of the KR-DE dimer in either of the reaction chambers appears to dictate which reaction chamber will contain the ACP domains, as observed for both the transacylation and the condensation step structures of Lsd14. For DEBS M1, the AT and KR domains are the symmetry-breaking elements. In the turnstile open state, only one reaction chamber contains the full complement of KS, ACP, AT, and KR domains, whereas the other reaction chamber lacks the KR and the ACP domains. In the turnstile half-open state, while the KR domains are docked symmetrically in both the reaction chambers, only one of the AT domains is flexed down, resulting in an overall asymmetric architecture in which access to the KS domain active site entrance is blocked in one reaction chamber but not in the other. Finally, in the turnstile closed state, while both the AT domains attain a flexed-down conformation, only one of the KR domains is stably docked to the KS dimer, resulting in an overall asymmetric architecture. Interestingly, the flexed AT conformation was not observed in any of the Lsd14 structures.6.2 Conformational rearrangements for sequential use of two reaction chambers in Lsd14 and DEBS
As the docking of the KR domains in either reaction chambers of DEBS and Lsd14 appears to dictate which reaction chamber is used, large-scale conformational change is required to move the KR domains from one reaction chamber to the next to enable sequential use of reaction chambers. This can be achieved by a 180° swiveling or a pendulum-like motion of the KR domains with respect to the KS-AT dimer.3 The conformational flexibility observed for the metazoan fatty acid synthase (FAS) suggests a similar swiveling and pendulum-like motion of the reducing half of the FAS with respect to the condensing half.47 In the case of Lsd14, since the KR domains dimerize via the DE resulting in a cyclic dimer where the active site of only one KR domain faces the reaction chamber, the DE-KR dimer likely needs to be flipped in a pendulum-like motion when going from one reaction chamber to another so that both KR active sites can be used. DEBS M1 does not have a DE, in which case the orientation of the KR domains is not dictated by dimerization. Therefore, a swiveling motion of the KR domains should suffice for sequential use of the two reaction chambers and the KR active sites.46.3 Determinants of ACP localization
Mechanistic details of how the substrate identity of the ACP may dictate docking to specific domains remain unclear. The identity of the substrate attached to the ACP and the acylation at the KS domains appears to dictate what conformation PikAIII adopts, indicating an important role for the tethered substrate on the localization of ACP. In the case of Lsd14, the ACP was docked to the KS only when the KS active site contained a substrate analog. On the other hand, loading of an acyl group on KS catalytic cysteine was not required for ACP/KS docking in PikAIII. Importantly, even though acylation of the KS active site cysteine was observed in both KS active sites of Lsd14 in the presence of KS substrate analog, the ACP domain was docked only in the reaction chamber where the KR domain was docked, suggesting both substrate identity and protein–protein interactions likely dictate the localization of ACP in Lsd14.7 AlphaFold2 predictions of Lsd14, DEBS, and PikAIII
To test the applicability of Alphafold2 (AF2) for structural analysis of intact PKS modules, we predicted homodimer structures of intact Lsd14, PikAIII, and DEBS module 1 using Alphafold2-Multimer-v3 via colabfold 1.5.0 (Fig. 4). We used the following parameters – num_recycles = 10, recycle_early_stop_tolerance = 0.5, no relaxation, msa_mode = mmseqs2_uniref_env, num_models = 5, stop_at_score = 100, recompile_padding = 10, use_templates = true, num_seeds = 1, and left the rest of the parameters at default values. We analyzed the top three predicted models based on the predicted interface TM (ipTM) scores. Structures of the individual domains of all three PKSs were predicted accurately by AF2. However, AF2 predicted models failed to capture the asymmetric architecture observed experimentally for Lsd14 and DEBS module 1 and the flexed AT conformations observed for DEBS module 1.3,4,8,9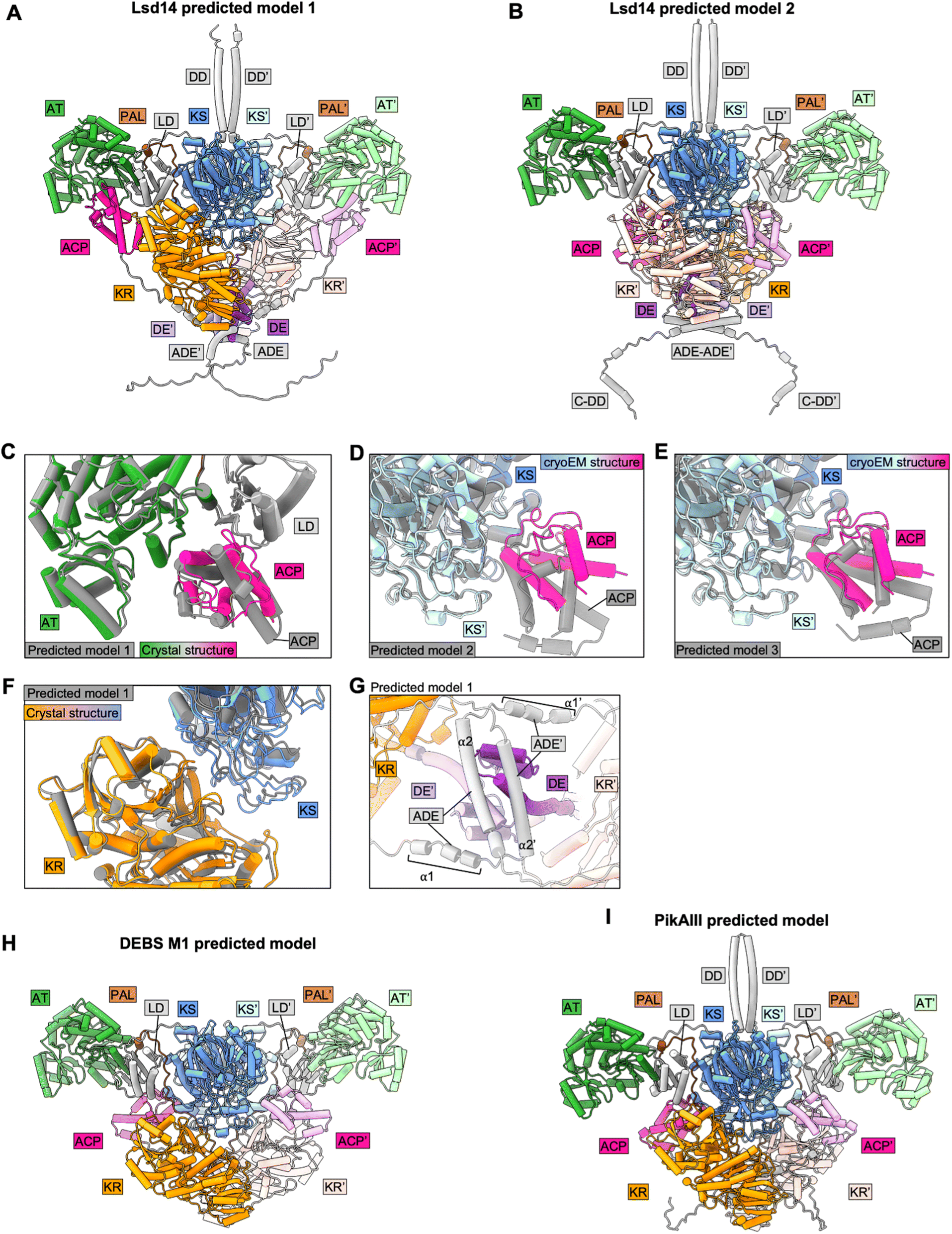 | ||
| Fig. 4 Analysis of AF2-predicted structures of intact PKS modules. (A) and (B) Predicted dimeric models of Lsd14. (C) and (F) Crystal structure of Lsd14 superposed on the predicted model. (D) and (E) Cryo-EM structure of Lsd14 superposed on the predicted models. (G) Zoomed view showing DE and ADE in the predicted model of Lsd14. (H) Predicted dimeric model of DEBS M1. (I) Predicted dimeric model of PikAIII. | ||
7.1 Predicted models of Lsd14
In one of the top three predicted models of Lsd14 (predicted model 1 with an iPTM score of 0.7), the ACPs are docked to the AT domains. While the predicted ACP/AT docking mode is similar to that observed experimentally in the crystal structure of Lsd14 (RMSD = 0.72 Å), these two docking modes are not identical (Fig. 4C). The ACP in the crystal structure appears to dock closer to the LD domain, than that in the predicted structure. Further, both copies of ACP are docked on the two AT domains in the AF2 predicted structure whereas only one ACP is docked to the AT domain in the crystal structure.In the other two top predicted models (predicted models 2 and 3 with iPTM scores of 0.68 and 0.68, respectively) of Lsd14, the ACPs are docked to the KS dimer (Fig. 4A and B). The ACP/KS docking mode observed in predicted model 2 is different from that observed experimentally in the cryo-EM structure of Lsd14 (Fig. 4D). The ACP/KS docking mode observed in predicted model 3 appears to be similar to that observed experimentally (Fig. 4E). Again, both copies of ACP in the predicted models 2 and 3 are docked at the side entrances of the KS active site tunnel whereas only one ACP was docked on the KS dimer in the cryo-EM structures.
In all three models, both copies of the KR domain are docked on the KS dimer making an interface similar to that observed for the KR domain in the experimental structures of Lsd14 (Fig. 4A, B, and F).3 Interestingly, none of the predicted models captured the second KS-KR docking mode observed between the KR′ domain and the KS dimer in the Lsd14 crystal structure (Fig. 2B, 4A and B). The predicted model of the post-ACP dimerization element (ADE) is different from that of the experimentally determined ADE structure of a related PKS, DEBS2.3 The ADE consists of two helices α1 and α2 which are expected to form an antiparallel four-helix bundle. In the AF2 predicted Lsd14 model, α2 and α2′ dimerize, whereas α1 and α1′ do not, possibly to accommodate the binding of the ACPs to the distantly located AT docking sites. If the ADEs were to form the expected four-helix bundle, the ACPs would not be able to dock to both of the AT domains due to limited post-ACP linker length. Furthermore, the ADE helices α2 and α2′ seem to be placed at an incorrect location in the predicted model as they undergo steric clash with the pre-KR dimerization element (DE) (Fig. 4G).
7.2 Predicted models of DEBS module 1
In all three predicted models of DEBS M1 (ipTM scores 0.74, 0.73, and 0.73), the ACPs are docked to the KS domains via a docking mode that is similar to that observed experimentally in the DEBS M1 cryo-EM structures (Fig. 4H). As observed for Lsd14, both copies of the ACPs and the KRs are docked symmetrically to the KS dimer in the two reaction chambers giving rise to a symmetric architecture that is different from that observed experimentally, in the cryo-EM structure of DEBS module 1 with a KS docked ACP.4 Furthermore, the AF2-predicted models failed to capture the flexed AT conformations observed in the cryo-EM structures.47.3 Predicted models of PikAIII
Interestingly, the architecture of AF2 predicted models of PikAIII (ipTM scores 0.74, 0.73, and 0.69) is vastly different from that observed in the PikAIII cryo-EM structures44,45 (Fig. 4I). In all of the top three predicted structures of PikAIII, the KS-AT dimer attains an extended architecture containing two reaction chambers, similar to that observed experimentally for DEBS and Lsd14, unlike the experimentally observed arch-shaped architecture of PikAIII, harboring a single reaction chamber.Overall, while AF2 performed well in predicting the structures of the individual domains and some of the interdomain interfaces in the PKS modules, a number of the expected inter-domain interfaces based on experimentally derived structures were not captured in the predicted models. Importantly, all the predicted models of Lsd14 and DEBS M1 attained symmetric architectures that are different from the asymmetric architectures observed experimentally. Given that the asymmetry in the architecture of PKSs has been proposed to have important mechanistic implications, our analysis of predicted structures of Lsd14 and DEBS M1 suggests predicted models of PKSs may fail to capture important aspects of PKS function.
8 Outlook
Three-dimensional structures of three full-length type I PKS modules are now available, that of Lsd14, DEBS M1, and PikAIII. Serendipitously, they all share the same domain composition, KS-AT-KR-ACP, which allowed direct comparison of the three structures. Lsd14 and DEBS M1 have an extended quaternary structure that is reminiscent of mammalian fatty acid synthases. This architecture harbors two separate active site chambers. Interestingly, the ACP is observed in only one of the two chambers, which suggests that the PKS module utilizes only one chamber at a time. We predict that this feature enables the growing polyketide chain to be passed to the downstream module while preventing a second cycle of chain extension from taking place on the already chain-extended substrate. However, this mechanism requires further experimental investigation. Notably, SAXS analysis and cross-linking mass spectrometry with coarse-grained structural modeling of DEBS module 2 derived constructs also suggested an extended conformation for DEBS module 2.48 In contrast, PikAIII has an arch-shaped architecture that harbors a fused, oversized active site chamber. Here, the active sites belonging to either protomer can potentially be utilized to construct the polyketide chain, and furthermore, two polyketide chains may be synthesized simultaneously. The pros and cons of each architecture and in what context one form is employed over the other is not understood. But for future PKS engineering, it will be important to know whether a given PKS module has an extended or an arch-shaped architecture as this information will likely impact how the domain-added, substituted, or deleted PKS modules are designed.All currently available full-length PKS module structures are of the KS-AT-KR-ACP type that catalyzes partial reduction of the polyketide chain. Cryo-EM analysis of an iterative lovastatin PKS LovB containing a KS-MAT-DH-CMeT-ψER-KR-ACP-CON configuration in complex with LovC comprising a standalone ER domain has been reported.49 Most of the domains were resolved in the LovB-LovC cryo-EM map revealing an X-shaped architecture harboring two L-shaped reaction chambers. Interestingly, the ACP and the C-terminal CON domains were not resolved in the cryo-EM maps. On the other hand, the structure of a full-length non-iterative modular PKS harboring fully-reducing domain configuration has not been reported. The structure of a fragment of module 5 of the modular PKS involved in juvenimicin biosynthesis containing a fully-reducing region including the DH, KR, and ER domains revealed a divergence in the architecture of the reducing regions in fully-reducing modular PKS with respect to the iterative systems.50
To achieve a comprehensive understanding of PKS structure and function, and to realize routine engineering of PKSs, structural information of full-length PKS modules with different combinations of KS, AT, KR, ER, DH, MT, and ACP domains is needed. We also need to structurally characterize modular PKSs that catalyze non-canonical reactions as this information will be helpful for engineering novel catalytic capabilities in the PKS modules. The amidation reaction-based chain-release catalyzed by a modular PKS involved in calcimycin biosynthesis, CalA3, is an interesting example.51 CalA3 comprises KS-AT-DH-KR domains and lacks an ACP domain. Cryo-EM and mutational analysis of CalA3 revealed how the KS domain that typically catalyzes C–C bond formation is repurposed to catalyze C–N bond formation in CalA3.51 Interestingly, CalA3 adopted a compact architecture in which the likely-inactive reducing region (DH-KR) forms extensive contacts with the KS-AT domains which may prevent conformational dynamics.
We also need to determine the structure of two-module, three-module, and higher-order modules to obtain a general understanding of type I PKS architecture. Recently, cryo-EM analysis of a tri-modular chimeric PKS from the pikromycin assembly line, Pik127, made by combining the loading module, module 2, and module 7 in a single polypeptide chain was reported. Only the loading module, comprising the KSQ and AT domains was resolved, likely due to inter-modular flexibility.52 Cryo-EM analysis of mycobacterial Pks13 module containing an ACP1-KS-AT-ACP2-ACP3-TE configuration also revealed intermodular flexibility as only the ACP1-KS-AT domains were well resolved.53 Interestingly, only one of the two copies of ACP1 in dimeric Pks13 was resolved in the reported maps, suggesting an asynchronous mechanism for trans-thioesterification to the KS.
The advancement of cryo-EM methodology was expected to rapidly increase the number of high-resolution full-length modular PKS structures. Unfortunately, Lsd14 and DEBS M1 structural studies have shown that PKS modules are not particularly cryo-EM grid-friendly. Furthermore, the examples noted above suggest that intra-modular and inter-modular flexibility remains a major challenge in the structural characterization of PKS containing different combinations of catalytic domains and consisting of more than just one module. Thus, the elucidation of additional full-length modular PKS structures will require significant effort. We must also pursue creative biochemical studies and daring PKS engineering trials using the currently available structural information.
9 Conflicts of interest
The authors declare no competing interests.10 Acknowledgements
This work was supported by the National Institutes of Health grant R01GM138990 to C.-Y.K.11 Notes and references
- D. J. Newman and G. M. Cragg, Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, Modular organization of genes required for complex polyketide biosynthesis, Science, 1991, 252, 675–679 CrossRef CAS PubMed.
- S. R. Bagde, I. I. Mathews, J. C. Fromme and C.-Y. Kim, Modular polyketide synthase contains two reaction chambers that operate asynchronously, Science, 2021, 374, 723–729 CrossRef CAS PubMed.
- D. P. Cogan, et al., Mapping the catalytic conformations of an assembly-line polyketide synthase module, Science, 2021, 374, 729–734 CrossRef CAS PubMed.
- A. J. Noble, et al., Routine single particle CryoEM sample and grid characterization by tomography, eLife, 2018, 7, e34257 CrossRef PubMed.
- R. M. Glaeser and B.-G. O. Han, hazards faced by macromolecules when confined to thin aqueous films, Biophys. Rep., 2017, 3, 1–7 CrossRef CAS PubMed.
- B. Kuhlman and P. Bradley, Advances in protein structure prediction and design, Nat. Rev. Mol. Cell Biol., 2019, 20, 681–697 CrossRef CAS PubMed.
- J. Jumper, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- R. Evans and et al., Protein complex prediction with AlphaFold-Multimer, bioRxiv, 2022, Preprint, 10.04.463034, DOI:10.1101/2021.10.04.463034.
- M. F. Lensink, et al., Impact of AlphaFold on structure prediction of protein complexes: The CASP15-CAPRI experiment, Proteins: Struct., Funct., Bioinf., 2023, 91(12), 1658–1683 CrossRef CAS PubMed.
- T. Liu, D. E. Cane and Z. Deng, The enzymology of polyether biosynthesis, Methods Enzymol., 2009, 459, 187–214 CAS.
- D. A. Kevin, D. A. Meujo and M. T. Hamann, Polyether ionophores: broad-spectrum and promising biologically active molecules for the control of drug-resistant bacteria and parasites, Expert Opin. Drug Discovery, 2009, 4, 109–146 CrossRef CAS PubMed.
- A. Migita, et al., Identification of a gene cluster of polyether antibiotic lasalocid from Streptomyces lasaliensis, Biosci., Biotechnol., Biochem., 2009, 73, 169–176 CrossRef CAS PubMed.
- K. Hotta, et al., Enzymatic catalysis of anti-Baldwin ring closure in polyether biosynthesis, Nature, 2012, 483, 355–358 CrossRef CAS PubMed.
- F. T. Wong, et al., Epoxide hydrolase-lasalocid a structure provides mechanistic insight into polyether natural product biosynthesis, J. Am. Chem. Soc., 2015, 137, 86–89 CrossRef CAS PubMed.
- A. Minami, et al., Sequential Enzymatic Epoxidation Involved in Polyether Lasalocid Biosynthesis, J. Am. Chem. Soc., 2012, 134, 7246–7249 CrossRef CAS PubMed.
- X. Li, et al., Structure–Function Analysis of the Extended Conformation of a Polyketide Synthase Module, J. Am. Chem. Soc., 2018, 140, 6518–6521 CrossRef CAS PubMed.
- Y. Tang, C.-Y. Kim, I. I. Mathews, D. E. Cane and C. Khosla, The 2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 11124–11129 CrossRef CAS PubMed.
- Y. Tang, A. Y. Chen, C.-Y. Kim, D. E. Cane and C. Khosla, Structural and Mechanistic Analysis of Protein Interactions in Module 3 of the 6-Deoxyerythronolide B Synthase, Chem. Biol., 2007, 14, 931–943 CrossRef CAS PubMed.
- J. R. Whicher, et al., Cyanobacterial Polyketide Synthase Docking Domains: A Tool for Engineering Natural Product Biosynthesis, Chem. Biol., 2013, 20, 1340–1351 CrossRef CAS PubMed.
- A. Miyanaga, et al., Structural Basis of Protein-Protein Interactions between a trans-Acting Acyltransferase and Acyl Carrier Protein in Polyketide Disorazole Biosynthesis, J. Am. Chem. Soc., 2018, 140, 7970–7978 CrossRef CAS PubMed.
- A. Miyanaga, S. Iwasawa, Y. Shinohara, F. Kudo and T. Eguchi, Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1802–1807 CrossRef CAS PubMed.
- Y. Feng, et al., Structural visualization of transient interactions between the cis-acting acyltransferase and acyl carrier protein of the salinomycin modular polyketide synthase, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2022, 78, 779–791 CrossRef CAS PubMed.
- A. Miyanaga, K. Kawada, T. Chisuga, F. Kudo and T. Eguchi, Structural Basis of Transient Interactions of Acyltransferase VinK with the Loading Acyl Carrier Protein of the Vicenistatin Modular Polyketide Synthase, Biochemistry, 2023, 62, 17–21 CrossRef CAS PubMed.
- M. Hans, A. Hornung, A. Dziarnowski, D. E. Cane and C. Khosla, Mechanistic Analysis of Acyl Transferase Domain Exchange in Polyketide Synthase Modules, J. Am. Chem. Soc., 2003, 125, 5366–5374 CrossRef CAS PubMed.
- K. Patel, et al., Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition, Chem. Biol., 2004, 11, 1625–1633 CrossRef CAS PubMed.
- R. McDaniel, et al., Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1846–1851 CrossRef CAS PubMed.
- J. Zheng, S. K. Piasecki and A. T. Keatinge-Clay, Structural Studies of an A2-Type Modular Polyketide Synthase Ketoreductase Reveal Features Controlling α-Substituent Stereochemistry, ACS Chem. Biol., 2013, 8, 1964–1971 CrossRef CAS PubMed.
- J. Zheng, C. D. Fage, B. Demeler, D. W. Hoffman and A. T. Keatinge-Clay, The Missing Linker: A Dimerization Motif Located within Polyketide Synthase Modules, ACS Chem. Biol., 2013, 8, 1263–1270 CrossRef CAS PubMed.
- S. Kapur, A. Y. Chen, D. E. Cane and C. Khosla, Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide B synthase, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22066–22071 CrossRef CAS PubMed.
- T. Robbins, J. Kapilivsky, D. E. Cane and C. Khosla, Roles of Conserved Active Site Residues in the Ketosynthase Domain of an Assembly Line Polyketide Synthase, Biochemistry, 2016, 55, 4476–4484 CrossRef CAS PubMed.
- D. A. Herbst, et al., The structural organization of substrate loading in iterative polyketide synthases, Nat. Chem. Biol., 2018, 14, 474–479 CrossRef CAS PubMed.
- R. W. Broadhurst, D. Nietlispach, M. P. Wheatcroft, P. F. Leadlay and K. J. Weissman, The Structure of Docking Domains in Modular Polyketide Synthases, Chem. Biol., 2003, 10, 723–731 CrossRef CAS PubMed.
- P. Argyropoulos, et al., Towards a characterization of the structural determinants of specificity in the macrocyclizing thioesterase for deoxyerythronolide B biosynthesis, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 486–497 CrossRef CAS PubMed.
- S.-C. Tsai, H. Lu, D. E. Cane, C. Khosla and R. M. Stroud, Insights into Channel Architecture and Substrate Specificity from Crystal Structures of Two Macrocycle-Forming Thioesterases of Modular Polyketide Synthases, Biochemistry, 2002, 41, 12598–12606 CrossRef CAS PubMed.
- V. Y. Alekseyev, C. W. Liu, D. E. Cane, J. D. Puglisi and C. Khosla, Solution structure and proposed domain–domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase, Protein Sci., 2007, 16, 2093–2107 CrossRef CAS PubMed.
- S.-C. Tsai, et al., Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: Versatility from a unique substrate channel, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14808–14813 CrossRef CAS PubMed.
- D. P. Cogan, et al., Antibody Probes of Module 1 of the 6-Deoxyerythronolide B Synthase Reveal an Extended Conformation During Ketoreduction, J. Am. Chem. Soc., 2020, 142, 14933–14939 CrossRef CAS PubMed.
- B. Lowry, X. Li, T. Robbins, D. E. Cane and C. Khosla, A Turnstile Mechanism for the Controlled Growth of Biosynthetic Intermediates on Assembly Line Polyketide Synthases, ACS Cent. Sci., 2016, 2, 14–20 CrossRef CAS PubMed.
- S. Kapur, et al., Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 4110–4115 CrossRef CAS PubMed.
- G. G. Zhanel, et al., The Ketolides, Drugs, 2002, 62, 1771–1804 CrossRef CAS PubMed.
- H. Brockmann and W. P. Henkel, ein bitter schmeckendes Antibioticum aus Actinomyceten (Antibiotica aus Actinomyceten, VI. Mitteil, Chem. Ber., 1951, 84, 284–288 CrossRef CAS.
- Y. Xue, L. Zhao, H. Liu and D. H. Sherman, A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12111–12116 CrossRef CAS PubMed.
- S. Dutta, et al., Structure of a modular polyketide synthase, Nature, 2014, 510, 512–517 CrossRef CAS PubMed.
- J. R. Whicher, et al., Structural rearrangements of a polyketide synthase module during its catalytic cycle, Nature, 2014, 510, 560–564 CrossRef CAS PubMed.
- F. J. Asturias, et al., Structure and molecular organization of mammalian fatty acid synthase, Nat. Struct. Mol. Biol., 2005, 12, 225–232 CrossRef CAS PubMed.
- E. J. Brignole, S. Smith and F. J. Asturias, Conformational flexibility of metazoan fatty acid synthase enables catalysis, Nat. Struct. Mol. Biol., 2009, 16, 190–197 CrossRef CAS PubMed.
- M. Klaus, et al., Solution Structure and Conformational Flexibility of a Polyketide Synthase Module, JACS Au, 2021, 1, 2162–2171 CrossRef CAS PubMed.
- J. Wang, et al., Structural basis for the biosynthesis of lovastatin, Nat. Commun., 2021, 12, 867 CrossRef CAS PubMed.
- T. M. McCullough, et al., Structure of a modular polyketide synthase reducing region, Structure, 2023, 31, 1109–1120.e3 CrossRef CAS PubMed.
- J. Wang, et al., C–N bond formation by a polyketide synthase, Nat. Commun., 2023, 14, 1319 CrossRef CAS PubMed.
- M. S. Dickinson, T. Miyazawa, R. S. McCool and A. T. Keatinge-Clay, Priming enzymes from the pikromycin synthase reveal how assembly-line ketosynthases catalyze carbon-carbon chemistry, Structure, 2022, 30, 1331–1339.e3 CrossRef CAS PubMed.
- S. K. Kim, et al., Structure and dynamics of the essential endogenous mycobacterial polyketide synthase Pks13, Nat. Struct. Mol. Biol., 2023, 30, 296–308 CrossRef CAS PubMed.
- T. Chisuga, S. Murakami, A. Miyanaga, F. Kudo and T. Eguchi, Structure-Based Analysis of Transient Interactions between Ketosynthase-like Decarboxylase and Acyl Carrier Protein in a Loading Module of Modular Polyketide Synthase, ACS Chem. Biol., 2023, 18(6), 1398–1404 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |