
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable skeletal reorganizations in natural product synthesis
Zeliang
Zhang†
 a,
Xiao
Qian†
a,
Yucheng
Gu
b and
Jinghan
Gui
*a
a,
Xiao
Qian†
a,
Yucheng
Gu
b and
Jinghan
Gui
*a
aState Key Laboratory of Chemical Biology, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: guijh@sioc.ac.cn
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 31st January 2024
Abstract
Covering: 2016 to 2023
The synthetic chemistry community is always in pursuit of efficient routes to natural products. Among the many available general strategies, skeletal reorganization, which involves the formation, cleavage, and migration of C–C and C–heteroatom bonds, stands out as a particularly useful approach for the efficient assembly of molecular skeletons. In addition, it allows for late-stage modification of natural products for quick access to other family members or unnatural derivatives. This review summarizes efficient syntheses of steroid, terpenoid, and alkaloid natural products that have been achieved by means of this strategy in the past eight years. Our goal is to illustrate the strategy's potency and reveal the spectacular human ingenuity demonstrated in its use and development.
 Zeliang Zhang | Zeliang Zhang was born in Shandong, China and obtained his BSc degree in Applied Chemistry from Shandong University in 2018. Then he joined the group of Prof. Jinghan Gui at Shanghai Institute of Organic Chemistry (SIOC) to pursue his PhD degree, working on the semisynthesis of heavily rearranged steroids from inexpensive, commercially available steroid materials. |
 Xiao Qian | Xiao Qian was born in Yangzhou, Jiangsu Province, China, in 1999. He received his BSc degree in chemistry in 2021 from Fudan University. In 2022, he joined Prof. Jinghan Gui's group at SIOC to pursue his PhD research in the synthesis of natural products, especially those with heavily rearranged steroidal skeletons. |
 Yucheng Gu | Yucheng Gu received his BSc degree from Hebei Medical University in 1984. He then received his MSc degree at China Academy of Traditional Chinese Medicine (Prof. Youyou Tu, 1989). He joined the China Japan Friendship Hospital as a research associate. In 1994, he went to the UK and received his PhD at Edinburgh Napier University in Scotland (1998), followed by a postdoctoral fellowship at Huddersfield University in England for three years. In 2002, he joined Syngenta as a natural products chemist and is now a principal technical specialist. His research interests are natural products, biological active compounds, and their applications. |
 Jinghan Gui | Jinghan Gui received his BSc degree from Anhui Normal University in 2007 and completed his PhD at SIOC with Prof. Weisheng Tian in 2012. He went on to pursue his postdoctoral research with Prof. Phil S. Baran at Scripps Research in February 2013. In March 2016, he moved back to SIOC to begin his independent research career. His current research interests focus on the efficient synthesis of complex, biologically active natural products and the development of novel synthetic methods with practical applications in organic synthesis. |
1. Introduction
Organic synthesis has evolved considerably since Friedrich Wöhler synthesized urea in 1828. Over the centuries, remarkable developments in synthetic methodologies and strategies have enriched the chemist's repertoire of techniques for taming the structural complexity of natural products.1–9 The resulting impressive achievements in total synthesis have convinced the chemistry community that almost any natural product can be synthesized in the laboratory.10,11 As a result, the major focus of modern synthetic studies has switched to improving synthetic efficiency,12–14 which is typically represented by shortening synthetic routes,15 increasing overall yields, and improving scalability.16Towards this end, there is a continuous need for the development of novel synthetic strategies and methods that make the synthesis of complex molecules more direct. Generally speaking, synthesis of natural products can be divided into two main tasks: assembly of the molecular skeleton and modulation of oxidation states. The former task has received more attention than the latter because rapid construction of the skeleton, which often features formidable fused and bridged polycyclic ring systems, is the key to an efficient synthetic route. Over the years, the art and science of total synthesis have advanced together, and many ingenious strategies have been developed. For example, cyclization reactions can swiftly convert simple linear molecules to involuted cyclic ones;17–19 and convergent fragment-coupling approaches can shorten the longest linear sequence of steps, thereby improving the overall yield and decreasing synthetic cost.20–23 Alternatively, reorganization of the skeletons of readily available substrates is also a feasible strategy for quickly and efficiently generating scaffold diversity and complexity.24–29 In fact, skeletal reorganization is one of the most important tools used by nature to generate the huge variety of natural products.30–33
Skeletal reorganization can utilize either a natural product or a human-made synthetic intermediate as a substrate. In the former case, generation of target molecules by skeletal reorganization of inexpensive commercially available compounds derived from natural sources is often inspired by proposed biosynthetic pathways. In the latter case, the latent relationship between the target molecule and readily accessible intermediates may become apparent only after careful structural analysis. However, once realized, this tactic can enable really efficient construction of the target skeleton. In addition, it can also be utilized in the late stages of syntheses, thereby allowing the divergent syntheses of a collection of natural products with distinct but closely related scaffolds.34–36
Despite the advantages of the skeletal reorganization strategy, it poses some challenges. A major one is controlling oxidation states. Because skeletal reorganization usually relies on controlled rearrangements, appropriate oxidation patterns are necessary to effect the desired reactions. Therefore, careful selection of starting materials and design of the substrates is vital to the successful application of this strategy.
In this review, impressive synthesis examples are selected to showcase the power of the skeletal reorganization strategy in natural product synthesis. The review is divided into sections based on three classes of target natural products: steroids, terpenoids, and alkaloids. In each section, five sets of examples will be introduced to showcase the achievements that have been made with this fantastic strategy.
2. Steroids
Steroids, which feature a 6/6/6/5-tetracyclic skeleton, are ubiquitous natural products found in a wide variety of plants, fungi, and animals (including marine organisms). In recent years, rearranged steroids37 (i.e., abeo-steroids and secosteroids) have attracted significant attention because of their interesting structures and biological activities. Given the biosynthetic relationships between classical steroids and rearranged steroids, it is not surprising that the complex core framework of the latter can be efficiently constructed by means of skeletal reorganizations of abundant steroid starting materials; and semisyntheses of rearranged steroids from inexpensive commercially available steroids have been actively pursued by synthetic chemists.Biosynthetically, site-specific enzymatic C–H oxidation of classical steroids, followed by selective bond migration and cleavage, leads to rapid preparation of molecules with a diverse array of rearranged steroid skeletons (Scheme 1). For example, enzymatic hydroxylation at C14 of the classical steroid skeleton can facilitate migration of C13 from C14 to C8, delivering 13(14→8)abeo-steroids such as the natural product dankasterone B (1). Alternatively, oxidation at C19, C7 or C9, in combination with subsequent C5, C11, and C1 migration, can afford 5(10→19)abeo-, 11(9→7)abeo-, and 1(10→6)abeo-steroids, respectively. Representative members of these three families include cyclocitrinol (2), pleurocin A (3), and asperfloketal A (4). In this section, five examples will be presented to demonstrate the advantages of skeletal reorganization for efficient synthesis of rearranged steroids.
 | ||
| Scheme 1 Proposed biosynthesis of rearranged steroids and structures of representative examples. | ||
2.1 Gui's syntheses of cyclocitrinols
Cyclocitrinol (2), a C25 5(10→19)abeo-steroid with a unique bicyclo[4.4.1]undecane A/B ring system, was first isolated from a terrestrial Penicillium citrinum by Gräfe and co-workers in 2000.38 In 2003, Crews and co-workers revised the structure of 2 on the basis of X-ray crystallographic analysis.39 To date, more than 30 C25 steroids sharing a bicyclo[4.4.1]undecane A/B ring system have been isolated,38–46 mostly from Penicillium species. Members of this family display diverse bioactivities, including antibacterial, cAMP-inducing, and anti-osteoporosis activities, as well as moderate cytotoxicity against KB and MCF-7 cancer cell lines.39,41,46,47To date, five groups had reported construction of the cyclocitrinol skeleton: the Schmalz group,48 the Leighton group,49 the Li group,50 the Ding group51 and the Tanino group.52 In 2018, Li and co-workers reported the first total synthesis of 2, which was accomplished in 18 steps and featured an intramolecular type II [5 + 2] cycloaddition.53,54 Soon after, the Gui group disclosed unified syntheses of ten cyclocitrinols from pregnenolone (5), an inexpensive commercially available steroid, by means of a bioinspired skeletal reorganization approach.55
The key challenge in the semisynthetic approach to 2 lies in the transformation of the decalin motif of classical steroids to the bicyclo[4.4.1]undecane ring system. Inspired by the proposed biosynthetic pathway, Gui and co-workers surmised that the desired skeletal reorganization could be realized through cyclopropane formation and subsequent fragmentation (Scheme 2), that is, formal C5–C19 bond formation and C5–C10 bond cleavage. Their synthetic endeavors commenced with the preparation of a diastereomeric mixture of sulfoxides 6 from 5 in seven steps. Upon treatment with O-methylquinine at 130 °C, sulfoxides 6 underwent syn elimination followed by cyclopropane formation and ring expansion to deliver trienes 9a (54%) and 9b (11%). In this transformation, O-methylquinine first acted as a nucleophile to enable the formation of cyclopropane 8 and then as a base and a leaving group to facilitate the cyclopropane fragmentation to afford the trienes. Subsequently, triene 9a was transformed to ketone 10via a hydroboration/oxidation sequence. Notably, this biomimetic skeletal reorganization strategy enabled the gram-scale preparation of key intermediate 10 in only nine steps from 5. With ketone 10 in hand, various side chains were introduced to complete divergent syntheses of ten cyclocitrinols in an additional one to three steps (i.e., 10–12 steps overall from 5).56
 | ||
| Scheme 2 Gui's syntheses of cyclocitrinols. | ||
2.2 Heretsch's and Ma's syntheses of dankasterones
Dankasterones A (23) and B (1), two 13(14→8)abeo-steroids with a 6/6/5/6-tetracyclic skeleton, were isolated from Gymnascella dankaliensis by the Numata group and found to exhibit significant cytotoxicity against murine P388 leukemia cells and human cancer cell lines.57,58 Later, 13(14→8),14(8→7)di-abeo-steroids swinhoeisterols A–F were isolated from the Xisha sponge Theonella swinhoei by the W. Zhang group.59,60 Two of these compounds, swinhoeisterols A (13) and C, show cytotoxicity against A549 and MG-63 cancer cells and inhibit the histone acetyltransferase (h)p300.60 In 2019, the Y. Zhang group isolated periconiastone A (12), which has a 13(14→8)abeo-4,14-cyclo-system skeleton, from Periconia sp. TJ403-rc01 and demonstrated that it has antibacterial activities.61 Periconiastone A is proposed to be biosynthesized from dankasterone B by means of an intramolecular aldol reaction that forms the C4–C14 bond, and the swinhoeisterol skeleton might be formed from the dankasterone skeleton via C14 migration from C8 to C7 (Scheme 3). | ||
| Scheme 3 Proposed biosyntheses of periconiastone and swinhoeisterol from dankasterone. | ||
Till now, three groups have finished syntheses of dankasterones including total syntheses by the Ma group,62,63 the Snyder group64 and semisynthesis by the Heretsch group.65,66 From a semisynthetic perspective, three pivotal transformations are necessary to obtain the complex core framework of these rearranged steroids from classical steroids: (1) migration of C13 from C14 to C8 to produce the dankasterone skeleton, (2) formation of a C4–C14 bond in dankasterone skeleton to generate the periconiastone skeleton, and (3) migration of C14 from C8 to C7 in dankasterone skeleton to furnish the swinhoeisterol skeleton. In 2020, Heretsch and co-workers completed elegant bioinspired syntheses of swinhoeisterol A (13), dankasterones A (23) and B (1), and periconiastone A (12), which were achieved by means of radical-mediated rearrangements (Scheme 4A).65,66 The syntheses started with 14-hydroxy steroid 15, which was prepared from ergosterol (14) in four steps. Treatment of 15 with either PhI(OAc)2/I2 or HgO/I2 generated 14-alkoxy radical 16. Subsequent migration of C13 from C14 to C8 via an alkoxy radical fragmentation/radical addition sequence afforded 18, which either was trapped with iodine to afford 22 in 76% yield under the PhI(OAc)2/I2 conditions or underwent a Dowd–Beckwith rearrangement to produce 21 in 68% yield under the HgO/I2 conditions. Iodide 22 was converted to 1 in four steps. Dehydrogenation of 1via a Saegusa oxidation produced 23, and an intramolecular aldol reaction of 1 yielded 12. Compound 21 was transformed to 13 in 16 steps.
 | ||
| Scheme 4 Heretsch's and Ma's syntheses of dankasterones A and B and periconiastone A. | ||
In 2021, Ma and co-workers reported total syntheses of dankasterones A (23) and B (1) and periconiastone A (12) (Scheme 4B).62,63 Notably, this synthesis also involved the use of an alkoxy radical rearrangement to realize the skeletal reorganization. To begin their syntheses, Ma et al. prepared 26 through a seven-step fragment-coupling sequence. Subsequently, the C8-hydroperoxyl group of 27 was introduced by reaction of 26 with HClO4/O2. Treatment of 27 with FeSO4·7H2O initiated the alkoxy radical rearrangement/radical addition sequence that resulted in migration of C13 from C14 to C8 to generate 30 in 55% yield from 26. Then 30 was transformed to 1 in two steps or 23 in three steps. Finally, as was the case with the Heretsch group's synthesis,651 was converted to 12 through an aldol reaction.
2.3 Heretsch's synthesis of asperfloketal A
Anthrasteroids, which possess an aromatic B ring flanked by A and C rings in a linear geometry, can be traced back to 1963, when the first member of this class of compounds was obtained by chemical synthesis.67,68 The anthrasteroid asperfloketal A (4), which was isolated from Aspergillus flocculosus 16D-1 by Lin et al. in 2020, shows anti-inflammatory activity in CuSO4-induced transgenic zebrafish.69 Two years later, the Heretsch group finished the first synthesis of 4, which was accomplished in a bioinspired manner.70These investigators devised a double Wagner–Meerwein rearrangement strategy as a stepwise version of the rearrangement that reorganizes the classical steroid skeleton into the anthrasteroid skeleton (Scheme 5), that is, migration of C1 from C10 to C5 and migration of C4 from C5 to C6. As in their semisynthesis of strophasterol A,71 vinyl chloride 31 was prepared from ergosterol (14) in nine steps. Treatment of 31 with KOH resulted in C14–C15 bond cleavage, which was followed by keto–enol isomerization, E1cB elimination, and alkene isomerization to produce 35 in quantitative yield. Subsequently, 35 was converted to 36 in three steps; 36 was reduced by NaBH4; and the resulting diol (not shown) underwent cyclopropane opening/C9–OH dehydration, which triggered the double Wagner–Meerwein rearrangement cascade. Notably, the second Wagner–Meerwein rearrangement delivered a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomeric acetates 39b and 39a, which were derived from migration of C1 from C5 to C6 and migration of C4 from C5 to C6, respectively. From 39b, 4 was synthesized in an additional four steps.
:1 mixture of regioisomeric acetates 39b and 39a, which were derived from migration of C1 from C5 to C6 and migration of C4 from C5 to C6, respectively. From 39b, 4 was synthesized in an additional four steps.
 | ||
| Scheme 5 Heretsch's synthesis of asperfloketal A. | ||
2.4 Heretsch's synthesis of pleurocin A
Pleurocin A (3) (also known as matsutakone), which has a unique 11(9→7)abeo-steroid skeleton, was first isolated from Tricholoma matsutake by the Liu group in 2017.72 Soon after, Tanaka and co-workers isolated 3 from Pleurotus eryngii and found that it inhibits NO production.73The pivotal task in the semisynthesis of pleurocin A (3) lies in reorganization of the classical steroid skeleton to the pleurocin skeleton through C9–C11 bond cleavage and C7–C11 bond formation. Inspired by the proposed polar biosynthesis,72,73 Heretsch and co-workers devised a biomimetic approach involving polar cleavage of the C9–C11 bond and subsequent radical formation of the C11–C7 bond (Scheme 6).74 Specifically, starting from ergosterol (14), they obtained triol 40 in eight steps. Upon treatment with PhI(OAc)2, 40 was smoothly converted to 41 in 98% yield through C9–C11 bond cleavage and a formal dioxa-[4 + 2]-cycloaddition. Then iodide 42, which was synthesized in six steps from 41, underwent a diastereoselective 6-endo-trig radical cyclization (Giese addition) initiated by AIBN/n-Bu3SnCl/NaBH3CN; and trapping of the resulting C8-alkyl radical (not shown) by oxygen delivered 44 in 74% yield. During this process, two contiguous stereogenic centers were established stereospecifically. Finally, deacetylation of 44 afforded 3.
 | ||
| Scheme 6 Heretsch's synthesis of pleurocin A. | ||
2.5 Gui's syntheses of pinnigorgiols
9,11-Secosteroids, most of which are isolated from marine organisms, are a subfamily of secosteroids. Pinnigorgiols are a group of heavily rearranged 9,11-secosteroids that possess a unique tricyclo[5.2.2.04,8]decane skeleton. Pinnigorgiols B (60) and E (59), which were isolated by Sung and co-workers from a gorgonian coral Pinnigorgia sp., induce apoptosis of hepatic stellate cells.75,76 Aplysiasecosterol A, which shares its core skeleton with the pinnigorgiols, was convergently synthesized by the Li group77 and the Kigoshi group.78 In 2021, the Gui group reported the first synthesis of pinnigorgiols, which they accomplished by means of a bioinspired skeletal reorganization for converting the classical steroid skeleton to the complex cage skeleton.79 In 2016, the Kigoshi group proposed that the structurally similar natural product aplysiasecosterol A (48) might be derived from aplysiasecosterol B (45) through an α-ketol rearrangement, followed by a vinylogous α-ketol rearrangement and hemiketal formation (Scheme 7A).80,81 Inspired by this proposed biosynthesis (Scheme 7A), Gui and co-workers devised a stepwise bond migration, bond cleavage, and bond formation sequence to access the core tricyclo[5.2.2.04,8]decane framework; this sequence involved C10 migration from C5 to C6, cleavage of the C5–C6 bond, and C5–C7 bond formation/hemiketalization (Scheme 7B). | ||
| Scheme 7 Kigoshi's proposed biosynthesis of aplysiasecosterol A and Gui's bioinspired strategy for installing the tricyclic cage skeleton. | ||
Specifically, ergosterol (14) was first converted to diol 53 in five steps (Scheme 8), and 53 was transformed into ketone 54 by means of a semi-pinacol rearrangement. Ozonolysis of the double bond of 54, followed by acetylation, afforded diketone 55, which was converted to the corresponding enol silyl ether and subjected to another ozonolysis to cleave the C5–C6 bond, affording diseco intermediate 56. Then thioester 57 was obtained in three steps from 56. Upon treatment with AIBN/n-Bu3SnH, 57 underwent an acyl radical cyclization/hemiketalization cascade, thereby completing the semisynthesis of 59 in 15 steps from 14. Notably, two rings and three contiguous stereogenic centers were established during this process. Hydrolysis of 59 afforded 60 in 60% yield.
 | ||
| Scheme 8 Gui's syntheses of pinnigorgiols. | ||
3. Terpenoids
Terpenes and terpenoids are the largest classes of natural products. Although the term “terpene” is often used to refer only to compounds with an empirical formula of C5H8, “terpenoid” refers to terpenes in which methyl groups have been moved or removed or oxygen atoms have been added. Although all terpenes and terpenoids are conceptually derived from isoprenes, they can have different numbers of isoprene units, complicated linkage patterns, diverse redox states, and rearranged skeletons, all of which result in the existence of many dramatically distinct structural classes. Therefore, the skeletal reorganization strategy is a flexible option for their chemical synthesis. Although proposed biosynthesis pathways or co-isolation of structurally related species can inspire biomimetic rearrangements for straightforward access to the target molecules, other innovative reorganization approaches have been devised to achieve interconversions of seemingly unrelated skeletons. Furthermore, those approaches have emerged from both semisynthesis and total synthesis studies, which have greatly expanded the scope of the skeletal reorganization strategy. In this section, remarkable recent accomplishments in this area will be demonstrated by five examples.3.1 Heretsch's and Deng's synthesis of spirochensilide A
Triterpenoids are often structurally very similar to steroidal compounds because the triterpene squalene is the biosynthetic precursor of lanosterol (62) and cycloartenol, the key intermediates in steroid biosynthesis. Thus, it is not surprising that rapid skeletal reorganization can be used for the synthesis of complex triterpenoids from inexpensive triterpenoid starting materials.Good examples were reported in 2022, when both the Heretsch group and the Deng group reported synthesis of the triterpenoid spirochensilide A (61).82,83 This compound was isolated from Abies chensiensis by Gao and co-workers in 2015 and moderately inhibits NO production.84 The first total synthesis of 61, which was reported by Yang and co-workers in 2020, featured a Meinwald rearrangement and a Pauson–Khand reaction.85–87 In contrast, inspired by the biosynthesis proposed by the investigators who isolated 61, the Heretsch and Deng groups utilized a semisynthetic strategy to access it from 62via a skeletal reorganization approach (Scheme 9).82,83
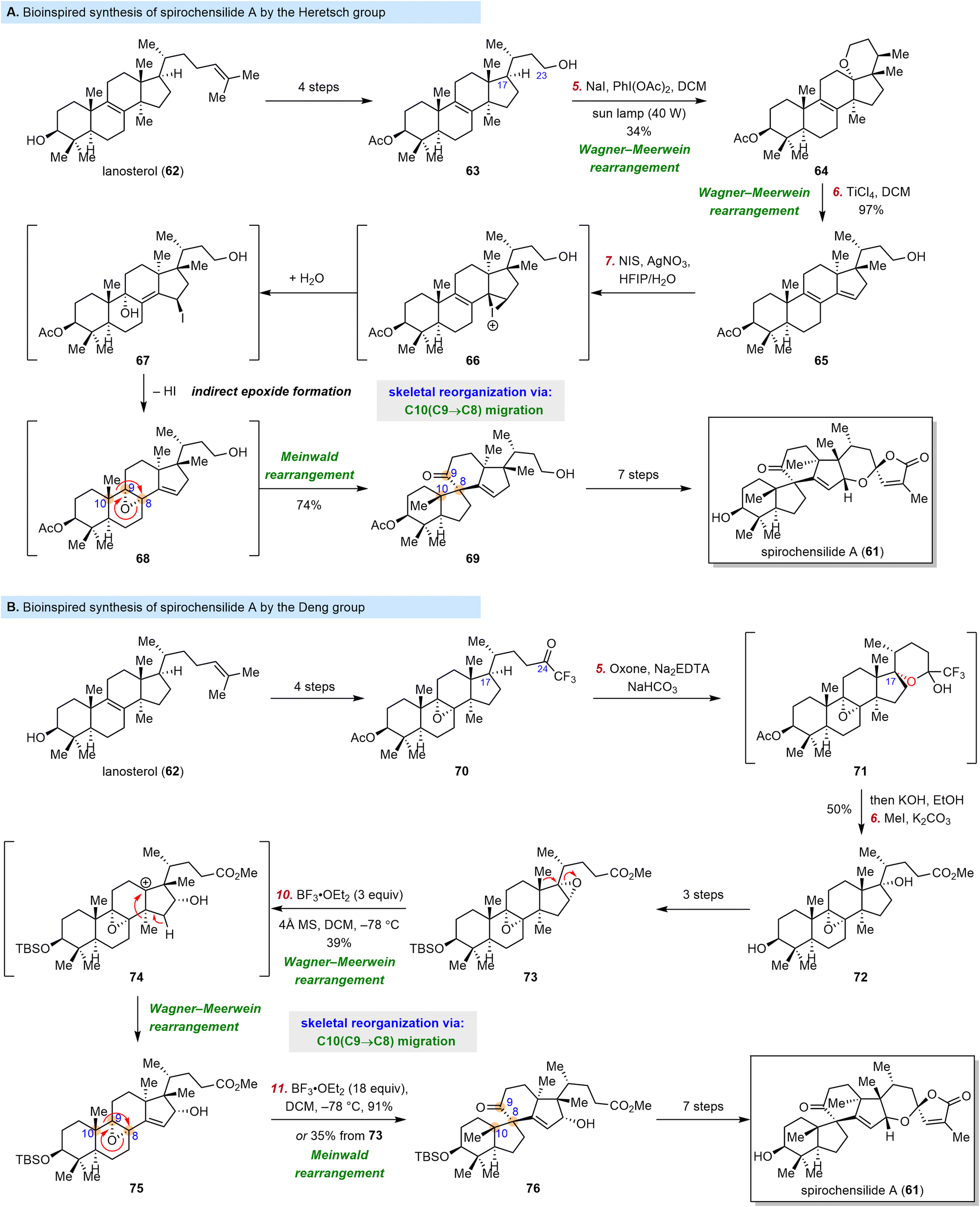 | ||
| Scheme 9 Heretsch's and Deng's bioinspired synthesis of spirochensilide A. | ||
This approach posed two main challenges: (1) a site- and stereoselective C–H oxidation at C17 to drive the migration of two methyl groups and (2) a Meinwald rearrangement to install the spiro[4.5]decane motif. As shown in Scheme 9A, the Heretsch group surmounted the first challenge by carrying out the C–H oxidation at C17 of 63 (which was prepared in four steps from 62) through activation of the C23 hydroxyl group, 1,5-hydrogen atom transfer, and radical-polar crossover. Specifically, upon photoirradiation of 63 in the presence of NaI/PhI(OAc)2, the first methyl migration took place spontaneously to generate 64, and the second migration was initiated by treatment of 64 with TiCl4 to give 65. Then Heretsch et al. investigated the formation of an α-8,9-epoxide, which was the substrate for the desired Meinwald rearrangement. They eventually discovered an indirect epoxidation method involving reaction with N-iodosuccinimide/AgNO3via an iodination-double SN2′ mechanism to afford epoxide 68, which was not isolable and could not be obtained by direct epoxidation of 65. Interestingly, under the above-described epoxidation conditions, 68 spontaneously underwent a Meinwald rearrangement to give 69; in this way, the key 10(9→8) migration was achieved. With the target reorganized skeleton in hand, Heretsch et al. were able to synthesize spirochensilide A (61) in another seven steps.
In contrast, the Deng group sought a reorganization substrate with the required oxidation pattern in place (Scheme 9B), which allowed them to develop a rearrangement cascade. Specifically, they managed to install the α-8,9-epoxide at an early stage, and then they oxidized the C17 position of trifluoromethyl ketone 70. Dioxirane-mediated intramolecular hydroxylation of 70 delivered hemiketal 71, which was smoothly elaborated to diepoxide 73. Upon treatment with BF3·OEt2 to activate the less hindered 16,17-epoxide, 73 underwent two consecutive Wagner–Meerwein rearrangements to accomplish the migration of two angular methyl groups, furnishing epoxide 75, which was then transformed into desired spiro[4.5]decane compound 76via a Meinwald rearrangement. Notably, the three rearrangement steps could be achieved in one pot to access 76 directly from 73 in 35% yield. With 73 in hand, Deng et al. synthesized spirochensilide A (61) in another seven steps.
3.2 Lei's synthesis of jungermannenone C
The jungermannenones are ent-kaurane-type diterpenoids isolated from liverwort Jungermannia species and are considered to be promising candidates for cancer chemotherapy on the basis of preliminary biological studies.88–90 In 2016, Lei and co-workers reported the first scalable total syntheses of rac-jungermannenones B and C (77), which involved a regioselective 1,6-dienyne reductive cyclization reaction as the key assembly step.91 In 2019, these investigators reported a second-generation route, leading to the total synthesis of 77 and featuring a photoinduced reorganization from the ent-kaurane-type skeleton to the jungermannenone-type skeleton.92 Of note, unlike the skeletal reorganization precursor used in the synthesis of spirochensilide A (61), the precursor for the ent-kaurane-type skeleton was prepared by total synthesis.Specifically, as shown in Scheme 10, intermediate 78, which was prepared in three steps from simple building blocks, underwent an enantioselective bicyclization via a radical cation generated by means of organo-SOMO (singly occupied molecular orbital) catalysis according to MacMillan's protocol93 to give aldehyde 80. This aldehyde was converted to dienyne 81 in five steps, and 81 underwent a reductive radical cyclization of the dienyne mediated by n-Bu3SnH to afford 82, which was converted to 83 in three simple steps. Both 82 and 83 have the ent-kaurane-type skeleton.
 | ||
| Scheme 10 Lei's synthesis of jungermannenone C. | ||
The skeletal reorganization step was accomplished by irradiation of 83 at 254 nm, which afforded 77 in 58% isolated yield, along with a 28% yield of recovered 83. The photochemical 1,3-acyl migration was an equilibrium reaction, and Lei et al. found that 83 could be regenerated from 77 by irradiation at 365 nm in 21% yield, along with a 71% yield of recovered 77. These results revealed that a photoinduced radical rearrangement is another way to access jungermannenones, in addition to carbocationic rearrangement pathways.
3.3 Sarpong's syntheses of phomactins, xishacorene B, and longiborneol
Naturally occurring “chiral pool terpenes” are widely used as starting materials for total synthesis because they are inexpensive. However, they are typically used in situations where their structures can easily be superimposed on some portion of the target structures. When this is not the case, their use becomes less straightforward and more challenging to implement, but they nevertheless offer access to many possible scaffolds.In 2006, Bermejo and co-workers discovered that epoxidation of the isopropenyl moiety of (S)-carvone (84) and subsequent Ti(III)-mediated radical epoxide opening/cyclization could furnish pinene cyclobutanols 85 (as the major diastereomer) and 86 (as the minor diastereomer) (Scheme 11A).94 This cascade reorganizes the carvone skeleton to the α-pinene skeleton and, more importantly, introduces two hydroxyl groups, one at C1 and one at C9. The more intuitive route to 85 and 86, through selective C–H oxidation of α-pinene (87), is clearly much more synthetically challenging.
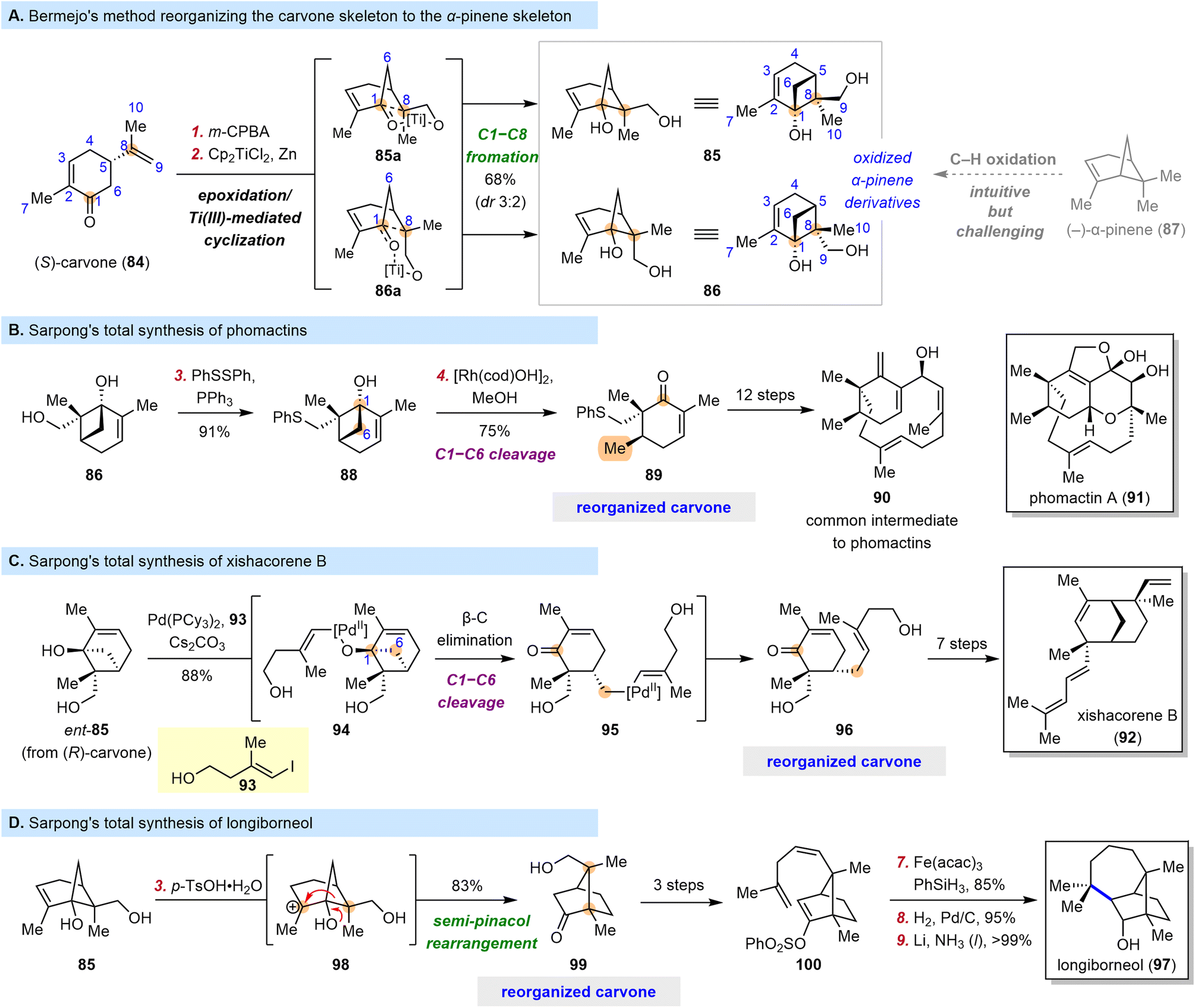 | ||
| Scheme 11 Sarpong's syntheses of phomactins, xishacorene B and longiborneol. | ||
In 2009, Bermejo et al. showcased the power of this tactic in a concise synthesis of paeonisuffrone, which contains a bicyclo[3.1.1] motif;95 and the Sarpong group recognized that cyclobutanols such as 85 and 86 could provide a platform for further elaboration. In fact, in 2018, the latter group published efficient syntheses of phomactin diterpenoids from diol 86 (Scheme 11B).96 They prepared compound 88 from 86 by a Mitsunobu-type reaction and subjected it to rhodium-catalyzed C1–C6 bond cleavage via a β-carbon elimination to obtain cyclohexenone 89, which appears similar to carvone but actually bears a completely reorganized skeleton and a six-membered ring with an unusual substitution pattern. Intermediates with analogous cyclohexenyl cores are common in previously reported synthetic investigations of the phomactin family,97–104 but the approach reported by Sarpong et al.96 arguably stands out as a creative, simplified solution to the synthetic challenge posed by these compounds. From compound 89, 12 straightforward transformations led to 90, which served as a common intermediate in the syntheses of five phomactins.
The C1–C6 bond cleavage described above resulted in a methyl group at C6, and the Sarpong group proposed that a C–C coupling reaction could be integrated into the bond cleavage process to extend its application. In 2018, they reported the total synthesis of xishacorene B (92) from (R)-carvone (Scheme 11C).105 This time, palladium was used as the transition-metal catalyst for the coupling reaction. The synthesis began with treatment of ent-85 (obtained from (R)-carvone) and 93 with Pd(PCy3)2, generating Pd(II)-alkoxide 94 through oxidative addition followed by ligand exchange. Cleavage of the C1–C6 bond through β-carbon elimination afforded alkyl-Pd(II) intermediate 95, which, upon reductive elimination, furnished enone 96, another compound with a reorganized carvone skeleton. Subsequently, 96 was converted to 92 in seven steps.
In addition to the cyclobutanol cleavage reactions leveraged above, polar rearrangements of the bicyclo[3.1.1] carboskeleton of 85 and 86 can obviously be used to produce other bicyclic compounds, and such rearrangements have long been recognized as reliable synthetic tools. Indeed, in 2022, the Sarpong group reported a nine-step synthesis of longiborneol (97) by means of such a strategy (Scheme 11D).106 They found that treatment of 85 with p-TsOH led to reorganized carvone 99, which has a bicyclo[2.2.1] skeleton, through a semi-pinacol rearrangement pathway. Recognizing a carvone in 99 is difficult even though only three reactions were involved in its preparation from (S)-carvone (84), but the resemblance of 99 to 97 is comparatively obvious. Compound 99 was converted to 100 in three steps, and a subsequent cyclization induced by metal hydride hydrogen atom transfer furnished the seven-membered ring of the target molecule; vinyl phenylsulfonate was used as a novel radical acceptor for this reaction. Overall, 97 was synthesized in nine steps, and this route was much shorter than previously reported routes,107–109 owing to the efficiency of the skeletal reorganization process at the outset.
3.4 Maimone's syntheses of berkeleyone A, andrastin D, and terretonin L
In the examples described above, all the substrates for the skeletal reorganizations were related to natural products. However, if this limitation could be removed, the possibilities would be much broader. In nature, the union of 3,5-dimethylorsellinic acid (DMOA) with farnesyl pyrophosphate leads to the assembly of over 100 meroterpenoid natural products.110 Although these DMOA-derived meroterpenoids have been the subject of a number of biosynthetic studies, few chemical synthesis have been disclosed.111,112 In 2016, the Maimone group described the first total synthesis of a complex DMOA-derived meroterpenoid, berkeleyone A (101).113 The berkeleyones were isolated in 2011 from the fungus Penicillium rubrum, which is found in deep water acid mine waste.114 Berkeleyones were found to inhibit caspase-1 and interleukin-1β production, and 101 was the most potent of the tested compounds.114As depicted in Scheme 12A, the Maimone group's synthesis commenced with farnesyl bromide 102, which was converted to 103via a polyene cyclization. A ring-opening/annulation process was effected by treatment of 103 with lithium tetramethylpiperidide and diketene, and this formal [4 + 2] reaction afforded 104. At this stage, Maimone et al. explored the conversion of the 5,6-fused ring system of 104 to the hallmark bicyclo[3.3.1]nonane skeleton of 101.
 | ||
| Scheme 12 Maimone's synthesis of berkeleyone A, and Maimone and Newhouse's syntheses of andrastin D and terretonin L. | ||
Following O-methylation of the 1,3-diketone moiety, they tested their previously developed I(III)-mediated oxidative ring expansion method (with PhI(OAc)2 and KOH in MeOH)115–117 in this polycyclic setting and isolated 106 in 84% yield. This skeletal reorganization probably proceeded through intermediate 105. Subsequently, six steps transformed 106 into 101via intermediates 107 and 108.
A year later, the Maimone group, together with the Newhouse group, reported syntheses of the meroterpenoids andrastin D (115) and terretonin L (116) (Scheme 12B).118 These molecules are biogenetically related to 101 but have 6,5-fused C/D rings instead of a bicyclo[3.3.1] ring system. Inspired by Shigehisa's work,119 the authors reasoned that with an oxidative variant of the classic Mukaiyama hydration process, the bicyclo[3.3.1] motif could be reorganized into the 6,5-fused C/D rings. Their endeavors started from 109, which was prepared easily from 108. Tertiary radical 110, which was formed via Co–H addition to 109, underwent a 1,4-addition to give cyclopropylcarbinyl radical 111; and a subsequent radical fragmentation gave radical 112. This process can be thought of as a vinylogous Dowd–Beckwith rearrangement. Depending on the reaction conditions, intermediate 112 could undergo either a carbocation-based process to give 113 (90% yield) or a radical-based process to give 114 (43% yield, 85% based on recovered starting material). This concise process accomplished the reorganization of the berkeleyone skeleton to the andrastin/terrenoid skeleton. Compound 113 was then converted to 115 by a simple Krapcho-type demethylation, and 114 was converted to 116 in three steps.
3.5 Ding's syntheses of crinipellins and retigeranic acid A
Tetracyclic diterpenoids, especially those with bridged ring systems, have received considerable attention from the synthetic community. In 2017, the Ding group disclosed a novel oxidative-dearomatization-induced (ODI) [5 + 2] cycloaddition/pinacol rearrangement cascade reaction,120 and have since synthesized the diterpenoid natural products pharicin A,120 stemarin,121 rhodomolleins XX and XXII,122 and rhodomollanol A.123 The general pattern of reactions is shown in Scheme 13A.124 Vinylphenols 117 were subjected to oxidative dearomatization conditions, and the resulting oxidized phenol moiety (not shown) underwent [5 + 2] cycloaddition with the pendant olefin to give cationic intermediates 118. A subsequent pinacol-type 1,2-acyl migration gave products 119, which had a cyclopentane- or cyclohexane-fused bicyclo[3.2.1] ring system with a sufficient number of oxidized sites for further elaboration. In addition to developing this ingenious skeletal reorganization process, the Ding group also exploited the synthetic potential of the skeleton of 119 by reorganizing it into angular triquinanes 121. | ||
| Scheme 13 Ding's syntheses of crinipellins and retigeranic acid A. | ||
Crinipellins are a class of natural products that have a linear cis,anti,cis-triquinane (ABC rings) and an angular triquinane (BCD rings) system and were isolated from a basidiomycete Crinipellis species.125–127 Piers and Renaud in 1993,128 Piers in 1998,129 Lee in 2014,130 and Yang in 2018131 accomplished total syntheses of crinipellin A (122) or B in racemic or enantiopure form. In 2022, the Ding group reported collective, divergent syntheses of crinipellins A–F and dihydrocrinipellins A and B.132 As shown in Scheme 13B, Ding et al. started by subjecting vinylphenol 123 to bromination followed by the above-mentioned ODI-[5 + 2]/pinacol rearrangement cascade, which afforded 124. Notably, the bromine atom installed prior to the cascade was used to control the diastereoselectivity of the cycloaddition reaction. Compound 124 was converted to vinylketone 125 in four steps, which set the stage for the key hydrogen-atom-transfer-initiated Dowd–Beckwith rearrangement. The tertiary radical of compound 126, which formed under metal hydride hydrogen atom transfer conditions, added to the ketone to form a cyclopropane ring, fragmentation of which gave rise to 129 in a remarkable 95% yield via radical 128. An additional four to eight transformations afforded eight members of the crinipellin family.
Notably, this type of skeletal reorganization usually led to products with quaternary stereocenters. However, in the vast majority of angular triquinanes—including retigeranic acid A (130), silphinenes, and bipolarolides—at least one of these stereocenters is tertiary or oxa-quaternary. Therefore, to synthesize 130,133–138 the Ding group developed a new rearrangement in 2023, when they reported a reductive skeletal reorganization strategy (Scheme 13C).139 First, vinylphenol 131, which was prepared in eight steps from commercially available compounds, was exposed to the ODI-[5 + 2]/pinacol rearrangement cascade conditions, which furnished diketone 132. This time, there was no ketone olefination step; instead, 132 was directly treated with SmI2 to effect a reductive coupling of the two ketone groups, giving intermediate 134. Cyclopropane fragmentation completed a Dowd–Beckwith rearrangement process, and resulting intermediate 135 underwent an acyloin rearrangement to afford 136, which was then doubly reduced to diol 137. In this new skeletal reorganization, one tertiary bridgehead stereocenter (C6) was retained. After another five steps—including a Wolff ring contraction—130 was obtained in a total of 15 steps.
4. Alkaloids
Alkaloid natural products contain at least one nitrogen atom and are usually basic. They are characterized by a greater structural diversity than most other classes of natural products because they generally do not share the same biosynthetic precursors. However, biogenetic relationships between different molecular scaffolds are common, and this renders skeletal reorganization an important strategy for the chemical synthesis of alkaloids. Moreover, the nitrogen atoms not only can act as polar handles to facilitate C–C bond reorganization but also can be directly involved in C–N bond reorganization. In this section, the power and diversity of skeletal reorganizations in alkaloid synthesis will be demonstrated with five examples.4.1 Li's synthesis of arcutinidine
Diterpenoid alkaloids, which originate from the amination of tetracyclic diterpenes, are a large group of natural products with complex structures and various biological activities. In 2000, a new arcutine-type C20 diterpenoid alkaloid, which features a unique skeleton with a C5–C20 bond rather than the usual C10–C20 bond, was isolated from Aconitum arcuatum and Aconitum carmichaelii.140,141In 2015, Sarpong and co-workers proposed that the arcutine skeleton is biosynthesized from the hetidine skeleton (Scheme 14A).142,143 In their proposal, protonation of the C5–OH group of 138 leads to 139, which has a C5 carbocation that induces a 1,2-alkyl shift of C20 from C10 to C5, generating a compound 140 with a carbocation at C10. Trapping 140 with H2O then completes the skeletal reorganization to give arcutine skeleton 141. From 2019 to 2021, three elegant total syntheses of arcutine alkaloids were published by the Qin group,144–146 the Sarpong group147,148 and the Li group.149 Unlike Qin's and Sarpong's syntheses, Li's synthesis employed an intriguing bioinspired strategy based on consideration of the interrelationship of diterpenoid alkaloids150 and the biosyntheses postulated by Liang and Wang151 and by the Sarpong group.142,143
 | ||
| Scheme 14 Sarpong's proposed biosynthesis of the arcutine skeleton and Li's bioinspired total synthesis of arcutinidine. | ||
Li's synthesis utilized a Prins cyclization/Wagner–Meerwein rearrangement cascade to convert the hetidine skeleton to the arcutine skeleton (Scheme 14B). The synthesis began with the preparation of α,β-unsaturated enone 145 through the convergent coupling of 142, 143, and 144. Upon deprotonation with lithium hexamethyldisilazane, 145 underwent an intramolecular, anionic Diels–Alder reaction to afford an 84% yield of 146, which has the key hetidine skeleton. Dehydration of 146 with SOCl2/pyridine afforded 147 regioselectively. Notably, the methoxymethyl group was designed for use as a precursor of the oxonium ion of 148, the formation of which triggered a skeletal reorganization involving C20 migration from C10 to C5. Treatment of 147 with the Lewis acid SnCl4 smoothly produced oxonium ion 148, initiating the Prins cyclization; and the resultant C5 tertiary carbocation of 149 induced the desired Wagner–Meerwein rearrangement to afford a 63% yield of 150, which has the arcutine skeleton. Arcutinidine (151), arcutinine, and arcutine (not shown) were synthesized from 150.
4.2 Boger's synthesis of strempeliopine
Strempeliopine (164), the parent schizozygane alkaloid, was first isolated by Laguna and co-workers from the roots of Strempeliopsis strempelioides K. Schum.152 Later, its absolute configuration was established by Hájíček and Trojánek through stereoselective synthesis.153–155To date, six groups have reported syntheses of strempeliopine (164).153–160 In 2021, Boger and co-workers disclosed a total synthesis of 164 enabled by a Grob-type fragmentation of the C12–C19 bond and a reductive radical cyclization mediated by BF3·Et2O/SmI2 to form the C2–C19 bond (Scheme 15).159 Hájíček and Trojánek reported a similar reductive rearrangement strategy, but it suffered from poor reproducibility.153–155 To address this issue, Boger and co-workers developed a stepwise version of the skeletal reorganization. This version commenced with the preparation of pentacyclic intermediate 153, which has the core skeleton of the aspidoperma alkaloids, from 1,3,4-oxadiazole 152 in six steps by means of a [4 + 2]/[3 + 2]cycloaddition cascade.161–165 Removal of the protecting groups from 153 afforded acid 154, which was unstable and was therefore immediately subjected to thermal decarboxylation to generate 155. Grob-type fragmentation of 155 cleaved the C12–C19 bond to afford 156, the iminium ion of which was trapped by the pendent primary alcohol to furnish 157 in 95% yield from 153. Protection of the indole moiety of 157 as a benzyl carbamate delivered 158, which was the precursor for the reductive coupling. When subjected to Huang's conditions,166–168158 underwent a radical cyclization to afford 163 in 63% yield. Boger et al. proposed that the transformation proceeded by the following mechanism: (1) 158 was activated by BF3·OEt2 to yield iminium ion 159, which was reduced by SmI2 to α-aminoalkyl radical 160; (2) radical 160 underwent a radical 6-endo-trig cyclization that formed the C2–C19 bond; and (3) the resulting radical at C12 was reduced by SmI2 to afford anion 162, protonation of which generated 163. With 163 in hand, these investigators completed the synthesis of 164 in three additional steps.
 | ||
| Scheme 15 Boger's synthesis of strempeliopine. | ||
4.3 Fan's synthesis of lycojaponicumin D
Lycopodium alkaloids have been known for centuries and have drawn significant attention from both synthetic and medicinal chemists. In 2012, the structurally unprecedented Lycopodium alkaloid lycojaponicumin D (165), which features a unique 5/7/6/6 tetracyclic skeleton and an unusual C3–C13 linkage, was isolated from Lycopodium japonicum Thunb. ex Murray by Yu et al.169 These investigators proposed a plausible biosynthetic pathway from fawcettimine to 165,169 but Fan et al. instead proposed a possible biogenetic relationship170 between lycojaponicumin D (165), lycoposerramine F (166) (also known as miyoshianine A),171 and lycodoline (167), a proposal that provided valuable inspiration for the chemical synthesis of 165.Fan et al. set out to develop a new, efficient synthetic route to lycodoline (167) and to realize its biomimetic conversion to lycojaponicumin D (165) (Scheme 16).170 To this end, they began by developing a novel bridgehead C–H heterofunctionalization reaction for the formation of the challenging bridgehead C–heteroatom bond. Specifically, hemiacetal ketone 168, which was prepared in six steps from 4-methylcyclohexanone, underwent a Saegusa–Ito oxidation to produce a highly strained bicyclo[3.3.1] bridgehead enone (not shown), which was immediately attacked by acetate anion via an oxa-Michael addition to give β-functionalized product 169. Lycodoline (167) was then obtained in three steps from 169, which set the stage for biomimetic conversion of 167 to 166 and 165. The synthesis of 166 was smoothly achieved via double oxidation. After extensive investigation of reaction conditions, Fan et al. used triphosgene as an activating reagent to facilitate cleavage of the N–O bond of 166, which triggered the C4–C13 bond fragmentation to give intermediate 171. Subsequent keto–enol tautomerization and a Mannich reaction effected the C3–C13 bond linkage, thus completing the synthesis of 165 in three steps from 167 by a bioinspired skeletal reorganization strategy.
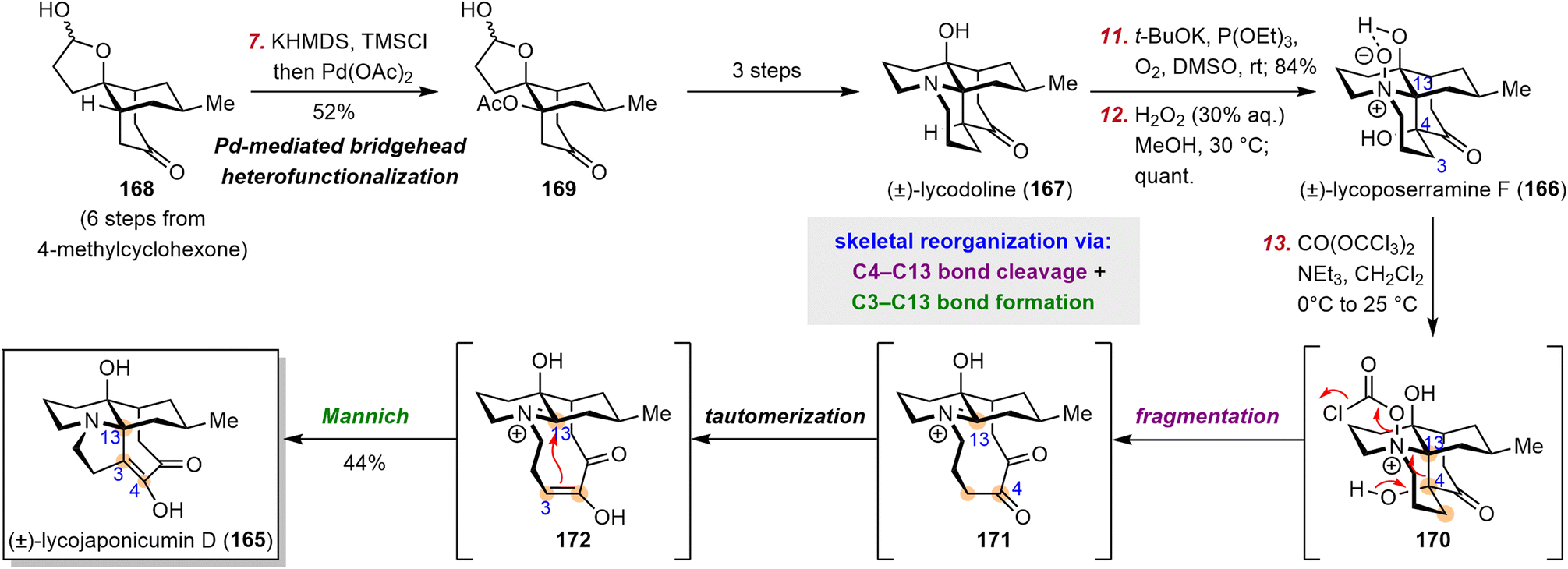 | ||
| Scheme 16 Fan's synthesis of lycojaponicumin D. | ||
4.4 Zhu's syntheses of condyfoline and tubifoline
The aspidospermatan-type alkaloid condyfoline (173) was first synthesized in 1963, by Schumann and Schmid, who subjected condylocarpine (174) to hydrogenation and acidic decarboxylation (Scheme 17A).172 Subsequently, condyfoline (173) was found to be readily converted to tubifoline (175), whose configuration was established through derivatization of akuammicine (176). In this way, the absolute configuration of 174 was also established. Since then, five groups have reported synthetic studies of 173, which culminated in racemic syntheses by the Harley-Mason group173 and the Ban group,174 enantioselective syntheses by the Amat group175,176 and the Zhu group,177 and epimer synthesis by the Andrade group.178 The elegant enantioselective synthesis of 173 by Zhu et al.177 is notable in featuring a TiCl3-mediated intramolecular reductive cyclization, leading to C2–N1 bond formation and C21 migration from C2 to C3 in one pot. | ||
| Scheme 17 Determination of the structures of condyfoline and tubifoline and Zhu's syntheses of the two molecules. | ||
Starting from 177, Zhu and co-workers prepared 2-nitrophenyl alkene 178 in six steps (Scheme 17B). Subsequently, 178 was treated with N-iodosuccinimide to afford putative intermediate 179. Treatment of this aziridinium salt with t-BuOK resulted in an elimination reaction to produce desired alkene 180. To complete the synthesis of 173, Zhu et al. developed an efficient cascade reaction to install the indolenine moiety. Upon treatment with aqueous TiCl3, 180 underwent 6π electrocyclization and a [1,5]-carbon shift, accomplishing C2–N1 bond formation and C21 migration from C2 to C3, to furnish 173 in 86% yield. Schumann and Schmid had reported the conversion of 173 to 175 in a low yield,172 but Zhu and co-workers reinvestigated this transformation and found that upon storage at −20 °C for 20 days, 173 was cleanly converted to 175 in 93% yield; they proposed that this transformation occurred through a retro-Mannich/1,3-prototropy/Mannich cascade reaction.
4.5 Jiang's synthesis of calycanthine
Dimeric cyclotryptamine alkaloids have had a rich history since the isolation of calycanthine (187) in 1888.179 Five basic dimeric scaffolds have been identified from various isolated natural products.180–184 Numerous pioneering studies suggest that tryptamine, tryptophan, and oxindole derivatives are the biosynthetic precursors for cyclotryptamine dimerization.185 Despite these precedents, the Jiang group developed a different dimerization tactic to access the relatively underexplored isocalycanthine scaffold.185They selected isocalycanthine (188) as their first synthetic target (Scheme 18). They designed chiral substrate 189, a constitutional isomer of tryptophan, as their dimerization substrate. When 189 was exposed to a combination of Fe(OTf)3 as the catalyst and dilauroyl peroxide as the terminal oxidant, dimeric product 190 was obtained in 48% yield. Notably, this reaction constructed two vicinal all-carbon quaternary centers with superb stereocontrol. Compound 190 was then converted smoothly to 191 in five steps, and at this point, it seemed that 188 was only one reduction step away. Several promising reductants were tested, and Jiang et al. were surprised to find that only Red-Al afforded a major product, which was identified as 187. They proposed that the driving force for this unexpected skeletal reorganization, which involved sequential ring opening and ring closing, was release of strain in the congested skeleton. Intriguingly, this transformation is as an example wherein the nitrogen atoms themselves were directly involved in the reorganization process.
 | ||
| Scheme 18 Jiang's synthesis of calycanthine. | ||
5. Conclusions
In this review, we have highlighted recent examples of efficient syntheses of steroid, terpenoid, and alkaloid natural products by means of a skeletal reorganization strategy, which offers an expedient approach for quick, easy access to structurally complex molecular frameworks. This strategy can be expected to continue to inspire synthetic chemists to realize additional creative syntheses of complex molecules.Admittedly, many of the skeletal reorganizations described above may not have been obvious upon first inspection of the molecule, but clues to the emergence of their application can be discerned. The foremost source of reorganization strategies for the construction of complex core frameworks may be based on the proposed biosynthetic pathways, which account for the majority of the steroid and alkaloid syntheses discussed in this review. Similarly, structural comparison between co-isolated or structurally related molecules can provide insights into late-stage interconversions between natural products, processes that frequently involve skeletal reorganizations. Apart from these biogenesis-guided strategies, in-depth exploration of rearrangement reactions opens up a much larger space for the development of skeletal reorganizations. Detailed investigation of a given rearrangement reaction, elaborative design of substrates, and creative derivatization of rearrangement products can bring about impressive reorganizations that can facilitate access to architecturally distinct natural products. Related inspirations may come from careful study of the literature, from the chemist's imagination, and from openness to serendipitous discoveries.
Although significant progress has been made in the development of the skeletal reorganization strategy, inefficient preparation of reorganization precursors somewhat hampers its application. Among the various difficulties encountered, precise installation of appropriate oxidation patterns has proven to be exceedingly challenging. Chemoenzymatic approaches that combine chemical transformations with enzymatic oxidation appear to be an attractive solution to this challenge, owing to the boom in research on enzyme engineering.186–192 Moreover, integration of functionalization and rearrangement processes into a single transformation can result in synthetic routes that are much more concise and direct. The thriving field of transition-metal catalysis is sure to facilitate such integration because transition-metal catalysts can activate C–H and C–C bonds that are traditionally considered to be inert and can also mediate various C–C bond forming reactions. Such activities have been exhibited by many transition metals, including (but not limited to) Cu,193,194 Rh,195–197 Pd,198,199 Pt,200 and Au;200–202 and some of these metals have already found applications in natural product synthesis. Enzymes are another potential tool. Various enzymatic transformations involving oxidation or functionalization followed by rearrangements have been reported over the years.203–211 To date, such transformations have made only limited contributions to natural product synthesis, but they will undoubtedly receive increasing attention from synthetic chemists. Overall, if the reorganization precursors could be more accessible, skeletal reorganizations will play a more important role in organic synthesis.
6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
Financial support was provided by the National Key Research and Development Program of China (2022YFC2303100, 2021YFF0502400), the National Natural Science Foundation of China (22122112), the Talent Plan of Shanghai Branch, Chinese Academy of Sciences (CASSHB-QNPD-2023-009), the CAS Project for Young Scientists in Basic Research (YSBR-095), the Science and Technology Commission of Shanghai Municipality (21ZR1476400), the Shanghai Sailing Program (21YF1456400) and the Syngenta PhD Fellowship (Z. Z.). This work has been supported by the New Cornerstone Science Foundation through the XPLORER PRIZE (J. G.).8. Notes and references
- E. J. Corey, W. J. Howe, H. W. Orf, D. A. Pensak and G. Petersson, J. Am. Chem. Soc., 1975, 97, 6116–6124 CrossRef CAS.
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS PubMed.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS PubMed.
- R. W. Hoffmann, Angew. Chem., Int. Ed., 2013, 52, 123–130 CrossRef CAS PubMed.
- P. S. Baran, J. Am. Chem. Soc., 2018, 140, 4751–4755 CrossRef CAS PubMed.
- C. Hui, F. Chen, F. Pu and J. Xu, Nat. Rev. Chem, 2019, 3, 85–107 CrossRef.
- A. J. E. Novak and D. Trauner, Trends Chem., 2020, 2, 1052–1065 CrossRef CAS.
- I. Bakanas, R. F. Lusi, S. Wiesler, J. H. Cooke and R. Sarpong, Nat. Rev. Chem, 2023, 7, 783–799 CrossRef CAS PubMed.
- S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183–188 CrossRef CAS PubMed.
- K. C. Nicolaou, S. Rigol and R. Yu, CCS Chem., 2019, 1, 3–37 CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- P.-Q. Huang, Z.-J. Yao and R. P. Hsung, Efficiency in Natural Product Total Synthesis, John Wiley & Sons, Inc., Hoboken, 2018 Search PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- C. A. Kuttruff, M. D. Eastgate and P. S. Baran, Nat. Prod. Rep., 2014, 31, 419–432 RSC.
- D. J. Edmonds, D. Johnston and D. J. Procter, Chem. Rev., 2004, 104, 3371–3403 CrossRef CAS PubMed.
- T. Nakata, Chem. Soc. Rev., 2010, 39, 1955–1972 RSC.
- X. Han, G. R. Peh and P. E. Floreancig, Eur. J. Org Chem., 2013, 2013, 1193–1208 CrossRef CAS.
- D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed.
- M. Inoue, Acc. Chem. Res., 2017, 50, 460–464 CrossRef CAS PubMed.
- Y. Gao and D. Ma, Acc. Chem. Res., 2021, 54, 569–582 CrossRef CAS PubMed.
- J. K. Kerkovius, A. R. Wong, V. W. Mak and S. E. Reisman, Chem. Sci., 2023, 14, 4397–4400 RSC.
- X. Xie and L. Zu, Synlett, 2018, 29, 1008–1013 CrossRef CAS.
- S. Yokoshima, Synlett, 2020, 31, 1967–1975 CrossRef CAS.
- B. Delayre, Q. Wang and J. Zhu, ACS Cent. Sci., 2021, 7, 559–569 CrossRef CAS PubMed.
- A. W. Schuppe, Y. Liu and T. R. Newhouse, Nat. Prod. Rep., 2021, 38, 510–527 RSC.
- L. Chen, G. Li and L. Zu, Org. Chem. Front., 2022, 9, 5383–5394 RSC.
- R. F. Lusi, M. A. Perea and R. Sarpong, Acc. Chem. Res., 2022, 55, 746–758 CrossRef CAS PubMed.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- Y. Qu, M. E. A. M. Easson, R. Simionescu, J. Hajicek, A. M. K. Thamm, V. Salim and V. De Luca, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 3180–3185 CrossRef CAS PubMed.
- H.-W. Liu and T. P. Begley, Comprehensive Natural Products III : chemistry and biology, Elsevier, California, 2020 Search PubMed.
- X. Pan, J. D. Rudolf and L.-B. Dong, Nat. Prod. Rep., 2024 10.1039/d3np00033h.
- X. Zhang, B. N. Kakde, R. Guo, S. Yadav, Y. Gu and A. Li, Angew. Chem., Int. Ed., 2019, 58, 6053–6058 CrossRef CAS PubMed.
- P. Fu, T. Liu, Y. Shen, X. Lei, T. Xiao, P. Chen, D. Qiu, Z. Wang and Y. Zhang, J. Am. Chem. Soc., 2023, 145, 18642–18648 CrossRef CAS PubMed.
- W. Zhang, M. Lu, L. Ren, X. Zhang, S. Liu, M. Ba, P. Yang and A. Li, J. Am. Chem. Soc., 2023, 145, 26569–26579 CrossRef CAS PubMed.
- F. L. Duecker, F. Reuß and P. Heretsch, Org. Biomol. Chem., 2019, 17, 1624–1633 RSC.
- A. G. Kozlovsky, V. P. Zhelifonova, S. M. Ozerskaya, N. G. Vinokurova, V. M. Adanin and U. Gräfe, Pharmazie, 2000, 55, 470–471 CAS.
- T. Amagata, A. Amagata, K. Tenney, F. A. Valeriote, E. Lobkovsky, J. Clardy and P. Crews, Org. Lett., 2003, 5, 4393–4396 CrossRef CAS PubMed.
- A. M. R. Marinho, E. Rodrigues-Filho, A. G. Ferreira and L. S. Santos, J. Braz. Chem. Soc., 2005, 16, 1342–1346 CrossRef.
- L. Du, T. Zhu, Y. Fang, Q. Gu and W. Zhu, J. Nat. Prod., 2008, 71, 1343–1351 CrossRef CAS PubMed.
- M.-W. Xia, C.-B. Cui, C.-W. Li and C.-J. Wu, Mar. Drugs, 2014, 12, 1545–1568 CrossRef PubMed.
- Y.-M. Ying, Z.-Z. Zheng, L.-W. Zhang, W.-G. Shan, J.-W. Wang and Z.-J. Zhan, Helv. Chim. Acta, 2014, 97, 95–101 CrossRef CAS.
- S. Lin, K.-Y. Chen, P. Fu, J. Ye, Y.-Q. Su, X.-W. Yang, Z.-X. Zhang, L. Shan, H.-L. Li, Y.-H. Shen, R.-H. Liu, X.-K. Xu and W.-D. Zhang, Magn. Reson. Chem., 2015, 53, 223–226 CrossRef CAS PubMed.
- F.-X. Yu, Z. Li, Y. Chen, Y.-H. Yang, G.-H. Li and P.-J. Zhao, Fitoterapia, 2017, 117, 41–46 CrossRef CAS PubMed.
- T. P. T. Hoang, C. Roullier, G. Genta-Jouve, M.-C. Boumard, T. R. du Pont, H. Nazih, Y.-F. Pouchus and O. Grovel, Phytochem. Lett., 2019, 34, 18–24 CrossRef CAS.
- Z.-H. He, C.-L. Xie, T. Wu, Y.-T. Yue, C.-F. Wang, L. Xu, M.-M. Xie, Y. Zhang, Y.-J. Hao, R. Xu and X.-W. Yang, J. Nat. Prod., 2023, 86, 157–165 CrossRef CAS PubMed.
- S. El Sheikh, A. Meier zu Greffen, J. Lex, J.-M. Neudörfl and H.-G. Schmalz, Synlett, 2007, 2007, 1881–1884 CrossRef.
- C. W. Plummer, C. S. Wei, C. E. Yozwiak, A. Soheili, S. O. Smithback and J. L. Leighton, J. Am. Chem. Soc., 2014, 136, 9878–9881 CrossRef CAS PubMed.
- G. Mei, X. Liu, C. Qiao, W. Chen and C.-C. Li, Angew. Chem., Int. Ed., 2014, 54, 1754–1758 CrossRef PubMed.
- T. Liu, Y. Yan, H. Ma and K. Ding, Chin. J. Org. Chem., 2014, 34, 1793–1799 CrossRef CAS.
- K. Sato and K. Tanino, Synlett, 2021, 32, 674–678 CrossRef CAS.
- J. Liu, J. Wu, J.-H. Fan, X. Yan, G. Mei and C.-C. Li, J. Am. Chem. Soc., 2018, 140, 5365–5369 CrossRef CAS PubMed.
- J. Wu, J. Liu, J.-H. Fan, Z.-D. Xie, H. Qin and C.-C. Li, Chin. J. Chem., 2021, 39, 1247–1254 CrossRef CAS.
- Y. Wang, W. Ju, H. Tian, W. Tian and J. Gui, J. Am. Chem. Soc., 2018, 140, 9413–9416 CrossRef CAS PubMed.
- Y. Wang, W. Ju, H. Tian, S. Sun, X. Li, W. Tian and J. Gui, J. Am. Chem. Soc., 2019, 141, 5021–5033 CrossRef CAS PubMed.
- T. Amagata, M. Doi, M. Tohgo, K. Minoura and A. Numata, Chem. Commun., 1999, 1321–1322 RSC.
- T. Amagata, M. Tanaka, T. Yamada, M. Doi, K. Minoura, H. Ohishi, T. Yamori and A. Numata, J. Nat. Prod., 2007, 70, 1731–1740 CrossRef CAS PubMed.
- J. Gong, P. Sun, N. Jiang, R. Riccio, G. Lauro, G. Bifulco, T.-J. Li, W. H. Gerwick and W. Zhang, Org. Lett., 2014, 16, 2224–2227 CrossRef CAS PubMed.
- J. Li, H. Tang, T. Kurtán, A. Mándi, C.-L. Zhuang, L. Su, G.-L. Zheng and W. Zhang, J. Nat. Prod., 2018, 81, 1645–1650 CrossRef CAS PubMed.
- W. Gao, C. Chai, Y. He, F. Li, X. Hao, F. Cao, L. Gu, J. Liu, Z. Hu and Y. Zhang, Org. Lett., 2019, 21, 8469–8472 CrossRef CAS PubMed.
- P. Chen, C. Wang, R. Yang, H. Xu, J. Wu, H. Jiang, K. Chen and Z. Ma, Angew. Chem., Int. Ed., 2021, 60, 5512–5518 CrossRef CAS PubMed.
- S. Deng, H. Xu, H. Jiang and Z. Ma, Org. Chem. Front., 2022, 9, 3961–3965 RSC.
- V. G. Lisnyak, PhD thesis, The University of Chicago, 2021 Search PubMed.
- F. L. Duecker, R. C. Heinze and P. Heretsch, J. Am. Chem. Soc., 2019, 142, 104–108 CrossRef PubMed.
- F. L. Duecker, R. C. Heinze, S. Steinhauer and P. Heretsch, Chem.–Eur. J., 2020, 26, 9971–9981 CrossRef CAS PubMed.
- O. Tanaka and E. Mosettig, J. Am. Chem. Soc., 1963, 85, 1131–1133 CrossRef CAS.
- K. Tsuda, R. Hayatsu, J. A. Steele, O. Tanaka and E. Mosettig, J. Am. Chem. Soc., 1963, 85, 1126–1131 CrossRef CAS.
- F.-R. Jiao, B.-B. Gu, H.-R. Zhu, Y. Zhang, K.-C. Liu, W. Zhang, H. Han, S.-H. Xu and H.-W. Lin, J. Org. Chem., 2021, 86, 10954–10961 CrossRef CAS PubMed.
- M. Alekseychuk and P. Heretsch, J. Am. Chem. Soc., 2022, 144, 21867–21871 CrossRef CAS PubMed.
- R. C. Heinze, D. Lentz and P. Heretsch, Angew. Chem., Int. Ed., 2016, 55, 11656–11659 CrossRef CAS PubMed.
- Z.-Z. Zhao, H.-P. Chen, B. Wu, L. Zhang, Z.-H. Li, T. Feng and J.-K. Liu, J. Org. Chem., 2017, 82, 7974–7979 CrossRef CAS PubMed.
- T. Kikuchi, Y. Horii, Y. Maekawa, Y. Masumoto, Y. In, K. Tomoo, H. Sato, A. Yamano, T. Yamada and R. Tanaka, J. Org. Chem., 2017, 82, 10611–10616 CrossRef CAS PubMed.
- R. C. Heinze and P. Heretsch, J. Am. Chem. Soc., 2019, 141, 1222–1226 CrossRef CAS PubMed.
- Y.-C. Chang, T.-L. Hwang, J.-H. Sheu, Y.-C. Wu and P.-J. Sung, Mar. Drugs, 2016, 14, 218 CrossRef PubMed.
- Y.-C. Chang, L.-M. Kuo, J.-H. Su, T.-L. Hwang, Y.-H. Kuo, C.-S. Lin, Y.-C. Wu, J.-H. Sheu and P.-J. Sung, Tetrahedron, 2016, 72, 999–1004 CrossRef CAS.
- Z. Lu, X. Zhang, Z. Guo, Y. Chen, T. Mu and A. Li, J. Am. Chem. Soc., 2018, 140, 9211–9218 CrossRef CAS PubMed.
- T. Ohyoshi, H. Iizumi, S. Hosono, H. Tano and H. Kigoshi, Org. Lett., 2023, 25, 4725–4729 CrossRef CAS PubMed.
- X. Li, Z. Zhang, H. Fan, Y. Miao, H. Tian, Y. Gu and J. Gui, J. Am. Chem. Soc., 2021, 143, 4886–4890 CrossRef CAS PubMed.
- A. Kawamura, M. Kita and H. Kigoshi, Angew. Chem., Int. Ed., 2015, 54, 7073–7076 CrossRef CAS PubMed.
- M. Kita, A. Kawamura and H. Kigoshi, Tetrahedron Lett., 2016, 57, 858–860 CrossRef CAS.
- M. Alekseychuk, S. Adrian, R. C. Heinze and P. Heretsch, J. Am. Chem. Soc., 2022, 144, 11574–11579 CrossRef CAS PubMed.
- X. Long, J. Li, F. Gao, H. Wu and J. Deng, J. Am. Chem. Soc., 2022, 144, 16292–16297 CrossRef CAS PubMed.
- Q.-Q. Zhao, Q.-Y. Song, K. Jiang, G.-D. Li, W.-J. Wei, Y. Li and K. Gao, Org. Lett., 2015, 17, 2760–2763 CrossRef CAS PubMed.
- X.-T. Liang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2020, 142, 8116–8121 CrossRef CAS PubMed.
- X.-T. Liang, B.-C. Sun, C. Liu, Y.-H. Li, N. Zhang, Q.-Q. Xu, Z.-C. Zhang, Y.-X. Han, J.-H. Chen and Z. Yang, J. Org. Chem., 2021, 86, 2135–2157 CrossRef CAS PubMed.
- X.-T. Liang, B.-C. Sun, N. Zhang, Z.-C. Zhang, Y.-H. Li, Q.-Q. Xu, C. Liu, J.-H. Chen and Z. Yang, J. Org. Chem., 2021, 86, 2158–2172 CrossRef CAS PubMed.
- F. Nagashima, W. Kasai, M. Kondoh, M. Fujii, Y. Watanabe, J. E. Braggins and Y. Asakawa, Chem. Pharm. Bull., 2003, 51, 1189–1192 CrossRef CAS PubMed.
- F. Nagashima, M. Kondoh, M. Fujii, S. Takaoka, Y. Watanabe and Y. Asakawa, Tetrahedron, 2005, 61, 4531–4544 CrossRef CAS.
- J.-B. Qu, R.-L. Zhu, Y.-L. Zhang, H.-F. Guo, X.-N. Wang, C.-F. Xie, W.-T. Yu, M. Ji and H.-X. Lou, J. Nat. Prod., 2008, 71, 1418–1422 CrossRef CAS PubMed.
- W. Liu, H. Li, P.-J. Cai, Z. Wang, Z.-X. Yu and X. Lei, Angew. Chem., Int. Ed., 2016, 55, 3112–3116 CrossRef CAS PubMed.
- B. Hong, W. Liu, J. Wang, J. Wu, Y. Kadonaga, P.-J. Cai, H.-X. Lou, Z.-X. Yu, H. Li and X. Lei, Chem, 2019, 5, 1671–1681 CAS.
- S. Rendler and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 5027–5029 CrossRef CAS PubMed.
- F. A. Bermejo, A. Fernández Mateos, A. Marcos Escribano, R. Martín Lago, L. Mateos Burón, M. Rodríguez López and R. Rubio González, Tetrahedron, 2006, 62, 8933–8942 CrossRef CAS.
- M. Martín-Rodríguez, R. Galán-Fernández, A. Marcos-Escribano and F. A. Bermejo, J. Org. Chem., 2009, 74, 1798–1801 CrossRef PubMed.
- Y. Kuroda, K. J. Nicacio, I. Alves da Silva Jr, P. R. Leger, S. Chang, J. R. Gubiani, V. M. Deflon, N. Nagashima, A. Rode, K. Blackford, A. G. Ferreira, L. D. Sette, D. E. Williams, R. J. Andersen, S. Jancar, R. G. S. Berlinck and R. Sarpong, Nat. Chem., 2018, 10, 938–945 CrossRef CAS PubMed.
- H. Miyaoka, Y. Saka, S. Miura and Y. Yamada, Tetrahedron Lett., 1996, 37, 7107–7110 CrossRef CAS.
- W. P. D. Goldring and G. Pattenden, Chem. Commun., 2002, 2002, 1736–1737 RSC.
- P. J. Mohr and R. L. Halcomb, J. Am. Chem. Soc., 2003, 125, 1712–1713 CrossRef CAS PubMed.
- W. P. D. Goldring and G. Pattenden, Org. Biomol. Chem., 2004, 2, 466–473 RSC.
- J. Huang, C. Wu and W. D. Wulff, J. Am. Chem. Soc., 2007, 129, 13366–13367 CrossRef CAS PubMed.
- Y. Tang, K. P. Cole, G. S. Buchanan, G. Li and R. P. Hsung, Org. Lett., 2009, 11, 1591–1594 CrossRef CAS PubMed.
- J. Huang, W. Bao, S. Huang, W. Yang, L. Zhu, G. Du and C.-S. Lee, Org. Lett., 2018, 20, 7466–7469 CrossRef CAS PubMed.
- L. Sun, J. Huang, B. Wang, G. Du and C.-S. Lee, ChemistrySelect, 2023, 8, e202304204 CrossRef CAS.
- I. Kerschgens, A. R. Rovira and R. Sarpong, J. Am. Chem. Soc., 2018, 140, 9810–9813 CrossRef CAS PubMed.
- R. F. Lusi, G. Sennari and R. Sarpong, Nat. Chem., 2022, 14, 450–456 CrossRef CAS PubMed.
- S. C. Welch and R. L. Walters, J. Org. Chem., 1974, 39, 2665–2673 CrossRef CAS.
- D. L. Kuo and T. Money, J. Chem. Soc., Chem. Commun., 1986, 1986, 1691–1692 RSC.
- K. Takasu, S. Mizutani, M. Noguchi, K. Makita and M. Ihara, J. Org. Chem., 2000, 65, 4112–4119 CrossRef CAS PubMed.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- M. Elkin, S. M. Szewczyk, A. C. Scruse and T. R. Newhouse, J. Am. Chem. Soc., 2017, 139, 1790–1793 CrossRef CAS PubMed.
- Y. Zhang, Y. Ji, I. Franzoni, C. Guo, H. Jia, B. Hong and H. Li, Angew. Chem., Int. Ed., 2021, 60, 14869–14874 CrossRef CAS PubMed.
- C. P. Ting, G. Xu, X. Zeng and T. J. Maimone, J. Am. Chem. Soc., 2016, 138, 14868–14871 CrossRef CAS PubMed.
- D. B. Stierle, A. A. Stierle, B. Patacini, K. McIntyre, T. Girtsman and E. Bolstad, J. Nat. Prod., 2011, 74, 2273–2277 CrossRef CAS PubMed.
- C. P. Ting and T. J. Maimone, J. Am. Chem. Soc., 2015, 137, 10516–10519 CrossRef CAS PubMed.
- C. P. Ting and T. J. Maimone, Synlett, 2016, 27, 1443–1449 CrossRef CAS.
- X. Shen, C. P. Ting, G. Xu and T. J. Maimone, Nat. Commun., 2020, 11, 508 CrossRef CAS PubMed.
- G. Xu, M. Elkin, D. J. Tantillo, T. R. Newhouse and T. J. Maimone, Angew. Chem., Int. Ed., 2017, 56, 12498–12502 CrossRef CAS PubMed.
- H. Shigehisa, T. Aoki, S. Yamaguchi, N. Shimizu and K. Hiroya, J. Am. Chem. Soc., 2013, 135, 10306–10309 CrossRef CAS PubMed.
- C. He, J. Hu, Y. Wu and H. Ding, J. Am. Chem. Soc., 2017, 139, 6098–6101 CrossRef CAS PubMed.
- J. Hu, Z. Jia, K. Xu and H. Ding, Org. Lett., 2020, 22, 1426–1430 CrossRef CAS PubMed.
- K. Yu, Z.-N. Yang, C.-H. Liu, S.-Q. Wu, X. Hong, X.-L. Zhao and H. Ding, Angew. Chem., Int. Ed., 2019, 58, 8556–8560 CrossRef CAS PubMed.
- J. Gao, P. Rao, K. Xu, S. Wang, Y. Wu, C. He and H. Ding, J. Am. Chem. Soc., 2020, 142, 4592–4597 CrossRef CAS PubMed.
- K. Gao, J. Hu and H. Ding, Acc. Chem. Res., 2021, 54, 875–889 CrossRef CAS PubMed.
- T. Anke, J. Heim, F. Knoch, U. Mocek, B. Steffan and W. Steglich, Angew. Chem., Int. Ed., 1985, 24, 709–711 CrossRef.
- Y.-Y. Li and Y.-M. Shen, Helv. Chim. Acta, 2010, 93, 2151–2157 CrossRef CAS.
- M. Rohr, K. Oleinikov, M. Jung, L. P. Sandjo, T. Opatz and G. Erkel, Bioorg. Med. Chem., 2017, 25, 514–522 CrossRef CAS PubMed.
- E. Piers and J. Renaud, J. Org. Chem., 1993, 58, 11–13 CrossRef CAS.
- E. Piers, J. Renaud and S. J. Rettig, Synthesis, 1998, 1998, 590–602 CrossRef.
- T. Kang, S. B. Song, W.-Y. Kim, B. G. Kim and H.-Y. Lee, J. Am. Chem. Soc., 2014, 136, 10274–10276 CrossRef CAS PubMed.
- Z. Huang, J. Huang, Y. Qu, W. Zhang, J. Gong and Z. Yang, Angew. Chem., Int. Ed., 2018, 57, 8744–8748 CrossRef CAS PubMed.
- Y. Zhao, J. Hu, R. Chen, F. Xiong, H. Xie and H. Ding, J. Am. Chem. Soc., 2022, 144, 2495–2500 CrossRef CAS PubMed.
- E. J. Corey, M. C. Desai and T. A. Engler, J. Am. Chem. Soc., 1985, 107, 4339–4341 CrossRef CAS.
- L. A. Paquette, J. Wright, G. J. Drtina and R. A. Roberts, J. Org. Chem., 1987, 52, 2960–2962 CrossRef.
- J. Wright, G. J. Drtina, R. A. Roberts and L. A. Paquette, J. Am. Chem. Soc., 1988, 110, 5806–5817 CrossRef CAS.
- T. Hudlicky, A. Fleming and L. Radesca, J. Am. Chem. Soc., 1989, 111, 6691–6707 CrossRef CAS.
- P. A. Wender and S. K. Singh, Tetrahedron Lett., 1990, 31, 2517–2520 CrossRef CAS.
- X. Chen, W. Yao, H. Zheng, H. Wang, P.-P. Zhou, D.-Y. Zhu and S.-H. Wang, J. Am. Chem. Soc., 2023, 145, 13549–13555 CrossRef CAS PubMed.
- D. Sun, R. Chen, D. Tang, Q. Xia, Y. Zhao, C.-H. Liu and H. Ding, J. Am. Chem. Soc., 2023, 145, 11927–11932 CrossRef CAS.
- B. Tashkhodzhaev, S. A. Saidkhodzhaeva, I. A. Bessonova and M. Y. Antipin, Chem. Nat. Compd., 2000, 36, 79–83 CrossRef CAS.
- S. A. Saidkhodzhaeva, I. A. Bessonova and N. D. Abdullaev, Chem. Nat. Compd., 2001, 37, 466–469 CrossRef.
- C. J. Marth, G. M. Gallego, J. C. Lee, T. P. Lebold, S. Kulyk, K. G. M. Kou, J. Qin, R. Lilien and R. Sarpong, Nature, 2015, 528, 493–498 CrossRef CAS PubMed.
- M. Weber, K. Owens and R. Sarpong, Tetrahedron Lett., 2015, 56, 3600–3603 CrossRef CAS PubMed.
- W. Nie, J. Gong, Z. Chen, J. Liu, D. Tian, H. Song, X.-Y. Liu and Y. Qin, J. Am. Chem. Soc., 2019, 141, 9712–9718 CrossRef CAS PubMed.
- X.-Y. Liu, F.-P. Wang and Y. Qin, Acc. Chem. Res., 2021, 54, 22–34 CrossRef CAS PubMed.
- X. Wu, W. Nie, M. Fu, X.-Y. Liu, F. Xue and Y. Qin, Tetrahedron, 2021, 86, 132092 CrossRef CAS.
- K. R. Owens, S. V. McCowen, K. A. Blackford, S. Ueno, Y. Hirooka, M. Weber and R. Sarpong, J. Am. Chem. Soc., 2019, 141, 13713–13717 CrossRef CAS PubMed.
- S. V. McCowen, N. A. Doering and R. Sarpong, Chem. Sci., 2020, 11, 7538–7552 RSC.
- S. Zhou, K. Xia, X. Leng and A. Li, J. Am. Chem. Soc., 2019, 141, 13718–13723 CrossRef CAS.
- S. Zhou, R. Guo, P. Yang and A. Li, J. Am. Chem. Soc., 2018, 140, 9025–9029 CrossRef CAS PubMed.
- F.-P. Wang and X.-T. Liang, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Elsevier Science, New York, 2002, ch. 1, vol. 59, pp. 1–280 Search PubMed.
- A. Laguna, L. Novotný, L. Dolejš and M. Buděšínský, Planta Med., 1984, 50, 285–288 CrossRef CAS PubMed.
- J. Hájíček and J. Trojánek, Tetrahedron Lett., 1981, 22, 2927–2928 CrossRef.
- J. Hájíček and J. Trojánek, Tetrahedron Lett., 1982, 23, 365–368 CrossRef.
- J. Hájíček and J. Trojánek, Collect. Czech. Chem. Commun., 1986, 51, 1731–1742 CrossRef.
- D. R. Bobeck, H. I. Lee, A. C. Flick and A. Padwa, J. Org. Chem., 2009, 74, 7389–7402 CrossRef CAS PubMed.
- Q. Zhou, X. Dai, H. Song, H. He, X. Wang, X.-Y. Liu and Y. Qin, Chem. Commun., 2018, 54, 9510–9512 RSC.
- X. Zhang and J. C. Anderson, Angew. Chem., Int. Ed., 2019, 58, 18040–18045 CrossRef CAS PubMed.
- X. Zeng and D. L. Boger, J. Am. Chem. Soc., 2021, 143, 12412–12417 CrossRef CAS PubMed.
- Y. An, M. Wu, W. Li, Y. Li, Z. Wang, Y. Xue, P. Tang and F. Chen, Chem. Commun., 2022, 58, 1402–1405 RSC.
- E. L. Campbell, A. M. Zuhl, C. M. Liu and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 3009–3012 CrossRef CAS PubMed.
- K. Lee and D. L. Boger, J. Am. Chem. Soc., 2014, 136, 3312–3317 CrossRef CAS PubMed.
- K. Lee and D. L. Boger, Tetrahedron, 2015, 71, 3741–3746 CrossRef CAS PubMed.
- X. Zeng, V. Shukla and D. L. Boger, J. Org. Chem., 2020, 85, 14817–14826 CrossRef CAS PubMed.
- J. Zhang, V. Shukla and D. L. Boger, J. Org. Chem., 2019, 84, 9397–9445 CrossRef CAS PubMed.
- Y.-G. Xiang, X.-W. Wang, X. Zheng, Y.-P. Ruan and P.-Q. Huang, Chem. Commun., 2009, 7045–7047 RSC.
- K.-Z. Hu, J. Ma, S. Qiu, X. Zheng and P.-Q. Huang, J. Org. Chem., 2013, 78, 1790–1801 CrossRef CAS PubMed.
- X.-K. Liu, X. Zheng, Y.-P. Ruan, J. Ma and P.-Q. Huang, Org. Biomol. Chem., 2012, 10, 1275–1284 RSC.
- X.-J. Wang, Y.-B. Liu, L. Li, S.-S. Yu, H.-N. Lv, S.-G. Ma, X.-Q. Bao, D. Zhang, J. Qu and Y. Li, Org. Lett., 2012, 14, 5688–5691 CrossRef CAS PubMed.
- X.-H. Zhao, Q. Zhang, J.-Y. Du, X.-Y. Lu, Y.-X. Cao, Y.-H. Deng and C.-A. Fan, J. Am. Chem. Soc., 2017, 139, 7095–7103 CrossRef CAS PubMed.
- H. Takayama, K. Katakawa, M. Kitajima, K. Yamaguchi and N. Aimi, Chem. Pharm. Bull., 2003, 51, 1163–1169 CrossRef CAS PubMed.
- D. Schumann and H. Schmid, Helv. Chim. Acta, 1963, 46, 1996–2003 CrossRef CAS.
- B. A. Dadson, J. Harley-Mason and G. H. Foster, Chem. Commun., 1968, 1233–1233 RSC.
- Y. Ban, K. Yoshida, J. Goto and T. Oishi, J. Am. Chem. Soc., 1981, 103, 6990–6992 CrossRef CAS.
- M. Amat, M.-D. Coll, D. Passarella and J. Bosch, Tetrahedron: Asymmetry, 1996, 7, 2775–2778 CrossRef CAS.
- M. Amat, M. D. Coll, J. Bosch, E. Espinosa and E. Molins, Tetrahedron: Asymmetry, 1997, 8, 935–948 CrossRef CAS.
- B. Delayre, C. Piemontesi, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 13990–13997 CrossRef CAS PubMed.
- P. Kokkonda and R. B. Andrade, Org. Lett., 2019, 21, 9594–9597 CrossRef CAS PubMed.
- R. G. Eccles, Drug. Circ. Chem. Gaz., 1888, 32, 65 Search PubMed.
- H. M. Gordin, J. Am. Chem. Soc., 1909, 31, 1305–1312 CrossRef.
- H. F. Hodson, B. Robinson and G. F. Smith, Proc. Chem. Soc., 1961, 465–466 CAS.
- S. M. Verbitski, C. L. Mayne, R. A. Davis, G. P. Concepcion and C. M. Ireland, J. Org. Chem., 2002, 67, 7124–7126 CrossRef CAS PubMed.
- J. J. Dotson, J. L. Bachman, M. A. Garcia-Garibay and N. K. Garg, J. Am. Chem. Soc., 2020, 142, 11685–11690 CrossRef CAS PubMed.
- J. J. Dotson, I. Liepuoniute, J. L. Bachman, V. M. Hipwell, S. I. Khan, K. N. Houk, N. K. Garg and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2021, 143, 4043–4054 CrossRef CAS PubMed.
- L. Bai, Y. Ma and X. Jiang, J. Am. Chem. Soc., 2021, 143, 20609–20615 CrossRef CAS PubMed ; and the references therein..
- J. Li, A. Amatuni and H. Renata, Curr. Opin. Chem. Biol., 2020, 55, 111–118 CrossRef CAS PubMed.
- X. Zhang, E. King-Smith, L.-B. Dong, L.-Y. Yang, J. D. Rudolf, B. Shen and H. Renata, Science, 2020, 369, 799–806 CrossRef CAS PubMed.
- H. Renata, Synlett, 2021, 32, 775–784 CrossRef CAS PubMed.
- C. N. Stout and H. Renata, Acc. Chem. Res., 2021, 54, 1143–1156 CrossRef CAS PubMed.
- E. P. Vanable, L. G. Habgood and J. D. Patrone, Molecules, 2022, 27, 6373 CrossRef CAS PubMed.
- D. Wu and X. Lei, Tetrahedron, 2022, 127, 133099 CrossRef CAS.
- C. N. Stout, N. M. Wasfy, F. Chen and H. Renata, J. Am. Chem. Soc., 2023, 145, 18161–18181 CrossRef CAS PubMed.
- I. Nakamura and M. Terada, Chem. Rec., 2015, 15, 429–444 CrossRef CAS PubMed.
- Q. Tan, Z. Yang, D. Jiang, Y. Cheng, J. Yang, S. Xi and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6420–6424 CrossRef CAS PubMed.
- X. Wang, A. Lerchen, T. Gensch, T. Knecht, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 1381–1384 CrossRef CAS PubMed.
- X. Wang, Y. Li, T. Knecht, C. G. Daniliuc, K. N. Houk and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 5520–5524 CrossRef CAS PubMed.
- Y. Xue and G. Dong, Acc. Chem. Res., 2022, 55, 2341–2354 CrossRef CAS PubMed.
- Y. Ano, D. Takahashi, Y. Yamada and N. Chatani, ACS Catal., 2023, 13, 2234–2239 CrossRef CAS.
- R. Li, X. Shi and D. Zhao, Chin. J. Chem., 2023, 41, 1679–1683 CrossRef CAS.
- A. Fürstner, Acc. Chem. Res., 2014, 47, 925–938 CrossRef PubMed.
- L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano and S. D. Nelson, Arch. Biochem. Biophys., 2011, 507, 95–110 CrossRef CAS PubMed.
- S. Niwayama, S. Kobayashi and M. Ohno, J. Am. Chem. Soc., 1994, 116, 3290–3295 CrossRef CAS.
- D. Lee and M.-J. Kim, Org. Lett., 1999, 1, 925–927 CrossRef CAS PubMed.
- D. Thiel, D. Doknić and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS PubMed.
- N. Thevenet, V. de la Sovera, M. A. Vila, N. Veiga, D. Gonzalez, G. Seoane and I. Carrera, Org. Lett., 2015, 17, 684–687 CrossRef CAS PubMed.
- E. Brenna, M. Crotti, M. De Pieri, F. G. Gatti, G. Manenti and D. Monti, Adv. Synth. Catal., 2018, 360, 3677–3686 CrossRef CAS.
- A. Tazawa, Y. Ye, T. Ozaki, C. Liu, Y. Ogasawara, T. Dairi, Y. Higuchi, N. Kato, K. Gomi, A. Minami and H. Oikawa, Org. Lett., 2018, 20, 6178–6182 CrossRef CAS PubMed.
- R. Xin, W. W. L. See, H. Yun, X. Li and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202204889 CrossRef CAS PubMed.
- A. Schneider, J. Ruppert, T. B. Lystbæk, S. Bastian and B. Hauer, ACS Catal., 2023, 13, 1946–1951 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |