
Engineered nanomicelles targeting proliferation and angiogenesis inhibit tumour progression by impairing the synthesis of ceramide-1-phosphate†
Poonam
Yadav
 a,
Kajal
Rana
a,
Ruchira
Chakraborty
a,
Ali
Khan
b,
Devashish
Mehta
b,
Dolly
Jain
a,
Bharti
Aggarwal
a,
Somesh K.
Jha
a,
Ujjaini
Dasgupta
b and
Avinash
Bajaj
*a
a,
Kajal
Rana
a,
Ruchira
Chakraborty
a,
Ali
Khan
b,
Devashish
Mehta
b,
Dolly
Jain
a,
Bharti
Aggarwal
a,
Somesh K.
Jha
a,
Ujjaini
Dasgupta
b and
Avinash
Bajaj
*a
aLaboratory of Nanotechnology and Chemical Biology, Regional Centre for Biotechnology, NCR Biotech Science Cluster, 3rd Milestone Faridabad-Gurgaon Expressway, Faridabad-121001, Haryana, India. E-mail: bajaj@rcb.res.in
bAmity Institute of Integrative Sciences and Health, Amity University Haryana, Manesar, Gurgaon-122413, Haryana, India
First published on 1st May 2024
Abstract
Tumour cells secrete various proangiogenic factors like VEGF, PDGF, and EGF that result in the formation of highly vascularized tumours with an immunosuppressive tumour microenvironment. As tumour growth and metastasis are highly dependent on angiogenesis, targeting tumour vasculature along with rapidly dividing tumour cells is a potential approach for cancer treatment. Here, we specifically engineered sub-100 sized nanomicelles (DTX-CA4 NMs) targeting proliferation and angiogenesis using an esterase-sensitive phosphocholine-tethered docetaxel conjugate of lithocholic acid (LCA) (PC-LCA-DTX) and a poly(ethylene glycol) (PEG) derivative of an LCA–combretastatin A4 conjugate (PEG-LCA-CA4). DTX-CA4 NMs effectively inhibit the tumour growth in syngeneic (CT26) and xenograft (HCT116) colorectal cancer models, inhibit tumour recurrence, and enhance the percentage survival in comparison with individual drug-loaded NMs. DTX-CA4 NMs enhance the T cell-mediated anti-tumour immune response and DTX-CA4 NMs in combination with an immune checkpoint inhibitor, anti-PDL1 antibody, enhance the anti-tumour response. We additionally showed that DTX-CA4 NMs effectively attenuate the production of ceramide-1-phosphate, a key metabolite of the sphingolipid pathway, by downregulating the expression of ceramide kinase at both transcriptional and translational levels. Therefore, this study presents the engineering of effective DTX-CA4 NMs for targeting the tumour microenvironment that can be explored further for clinical applications.
Introduction
The tumour microenvironment (TME) encompasses malignant cells along with stromal cells that include different immune cell types, cancer-associated fibroblasts, and endothelial cells.1 Interactions between tumour and stromal cells mediated by soluble factors like cytokines, chemokines, growth factors, enzymes, and metabolites create a proangiogenic niche in the TME and restrict the antigen presentation, inhibit dendritic cell maturation, and recruit immunosuppressive immune cells.2–5 In addition, abnormal tumour vasculature characterized by leaky blood vessels and ineffective lymphatic drainage hampers T cell infiltration, impedes their effector functions and severely interferes with anti-cancer immunity.6 The complex TME is also responsible for high interstitial fluid pressure, impaired blood supply, hypoxia, and acidosis that ultimately constrain the delivery of therapeutic agents, leading to poor efficacy of chemotherapy.7 Therefore, inhibition of tumour-associated angiogenesis along with proliferative tumour cells has been a potential approach for developing new therapeutic strategies.8Several FDA-approved antiangiogenic agents like monoclonal antibodies and tyrosine kinase inhibitors showed modest positive outcomes with limited impact on overall survival.9,10 Chemotherapeutic drugs like docetaxel (DTX) are also known to inhibit angiogenesis,11 but the presence of proangiogenic factors, including Vascular Endothelial Growth Factor (VEGF) in the TME, protects the endothelial cells and limits the antiangiogenic effect of DTX.12 Preclinical studies have shown that a combination of DTX with therapeutic agents targeting VEGF can reverse the protective effect and enhance the anti-tumour efficacy.13 Combretastatin A4 (CA4), an antiangiogenic agent, effectively inhibits the VEGF expression and secretion, thus hindering the activation of the VEGF receptor in endothelial cells.14 Although combination therapies targeting proliferation and angiogenesis have shown synergistic effects in tumour regression, poor drug accumulation at the tumour site, non-specific distribution, and short half-life of chemotherapeutics result in severe toxicities along with the emergence of drug resistance, poor survival rates, and chances of tumour relapse.15–17 Therefore, new approaches need to be developed that can effectively co-deliver chemotherapeutic agents like DTX and antiangiogenic agents like CA4 at the tumour site.
Sphingolipids are key lipid metabolites that form the structural modules of cell membranes and also act as bioactive signalling metabolites.18 Ceramides are the central component of the sphingolipid metabolic pathway and are synthesized from serine and palmitic acid through a multistep reaction pathway.19 Ceramides can further get converted to different metabolites like ceramide-1-phosphates, sphingomyelins, glucosylceramides, or sphingosine-1-phosphate with diverse physiological functions in proliferation, inflammation, angiogenesis, and drug resistance.20,21 Synthesis of ceramide-1-phosphates (C1Ps) involves tethering the phosphate group at the hydroxyl group of ceramides by ceramide kinase (CERK).22 C1Ps can activate cellular signalling cascades responsible for cancer cell survival, migration, inflammation, and angiogenesis.23,24 Earlier studies demonstrated that different chemotherapeutic drugs or their combinations alter the sphingolipid metabolites to execute their therapeutic functions, for example, a combination of doxorubicin and celastrol causes an increase in ceramide levels responsible for apoptotic cell death.25 A combination of doxorubicin, CA4 and dexamethasone decreases glucosylceramide levels and inhibits drug resistance.26 However, the impact of chemotherapeutic drugs, especially antiproliferative and antiangiogenic drugs, on the sphingolipid metabolic pathway, especially C1Ps, is not known.
Among different biomaterials used for the delivery of chemotherapeutics, nanomicelles (NMs) are supramolecular assemblies of lipid or polymer-derived amphiphilic molecules comprising a hydrophilic head and a hydrophobic tail.27 Bile acids are amphiphilic steroid molecules synthesized from cholesterol catabolism and their self-assembling properties can be utilized to create supramolecular nanomaterials.28–30 In this study, we demonstrate the development of DTX-CA4 NMs by combining a phospholipid derivative of the lithocholic acid (LCA)–docetaxel conjugate (PC-LCA-DTX) with a PEGylated-derivative of the LCA–CA4 conjugate (PEG-LCA-CA4) that show improved anticancer efficacy in both syngeneic and xenograft murine colon cancer models. We further demonstrated the higher accumulation of CD4+, CD8+, and granzyme B+ effector cytotoxic T cells upon treatment with DTX-CA4 NMs, altering the immunosuppressive TME. Moreover, these NMs effectively inhibit the activity of CERK and restrict the production of C1P.
Results and discussion
Rationale and design for the engineering of DTX-CA4 NMs
Tumour tissues have high expression of several enzymes, including esterases, matrix metalloproteinases (MMPs), and phospholipases, in comparison with normal tissues.31 Therefore, prodrugs are being developed utilizing enzyme- or pH-sensitive linkages that allow the specific cleavage of the chemical bonds in the tumour tissues for release of drugs. Prodrug strategies for the delivery of anticancer drugs are preferred over physical encapsulation as these strategies prevent rapid drug release in the bloodstream and enhance drug accumulation in tumour tissues.32 In our earlier study, we showed the synthesis of PC-LCA-DTX where DTX was conjugated at the 24′-COOH terminal of LCA through the ester bond, and phosphocholine was attached at the 3′-OH end of LCA. We also synthesized a PEGylated LCA (PEG-LCA), a helper lipid, where PEG was conjugated at the 3′-OH end of LCA.29 Our study demonstrated that a mixture of PC-LCA-DTX and PEG-LCA at a defined ratio self-assembles to form DTX NMs, and these DTX NMs possess superior anticancer efficacy than clinically used Taxotere® formulation in a murine breast cancer model with improved pharmacokinetic and biodistribution profiles.29 Our studies also demonstrated that these NMs do not allow the release of any free drug in the systemic circulation, and the presence of esterase-sensitive linkages allowed the release of DTX in the tumour tissues.As a combination of chemotherapeutic agents and monoclonal antibodies used against VEGF in clinics for cancer treatment, we propose to develop DTX-CA4 NMs that can effectively co-deliver an anticancer drug (DTX) and an antiangiogenic drug (CA4). As both DTX and CA4 are poorly soluble BCS Class IV drugs, we hypothesize that an amalgamation of PC-LCA-DTX with a PEGylated lipid–drug conjugate of CA4 (LCA-CA4-PEG) can undergo self-assembly, resulting in the formation of DTX-CA4 NMs (Fig. 1A and B). We conjugated CA4 and DTX with LCA using an ester linkage, as these molecules can utilize the high expression of esterases in the TME for drug release (Fig. 1A). Conjugation of PEG and phosphatidylcholine will improve the physical stability, solubility and internalization of NMs, thereby preventing their clearance by the reticuloendothelial system.32
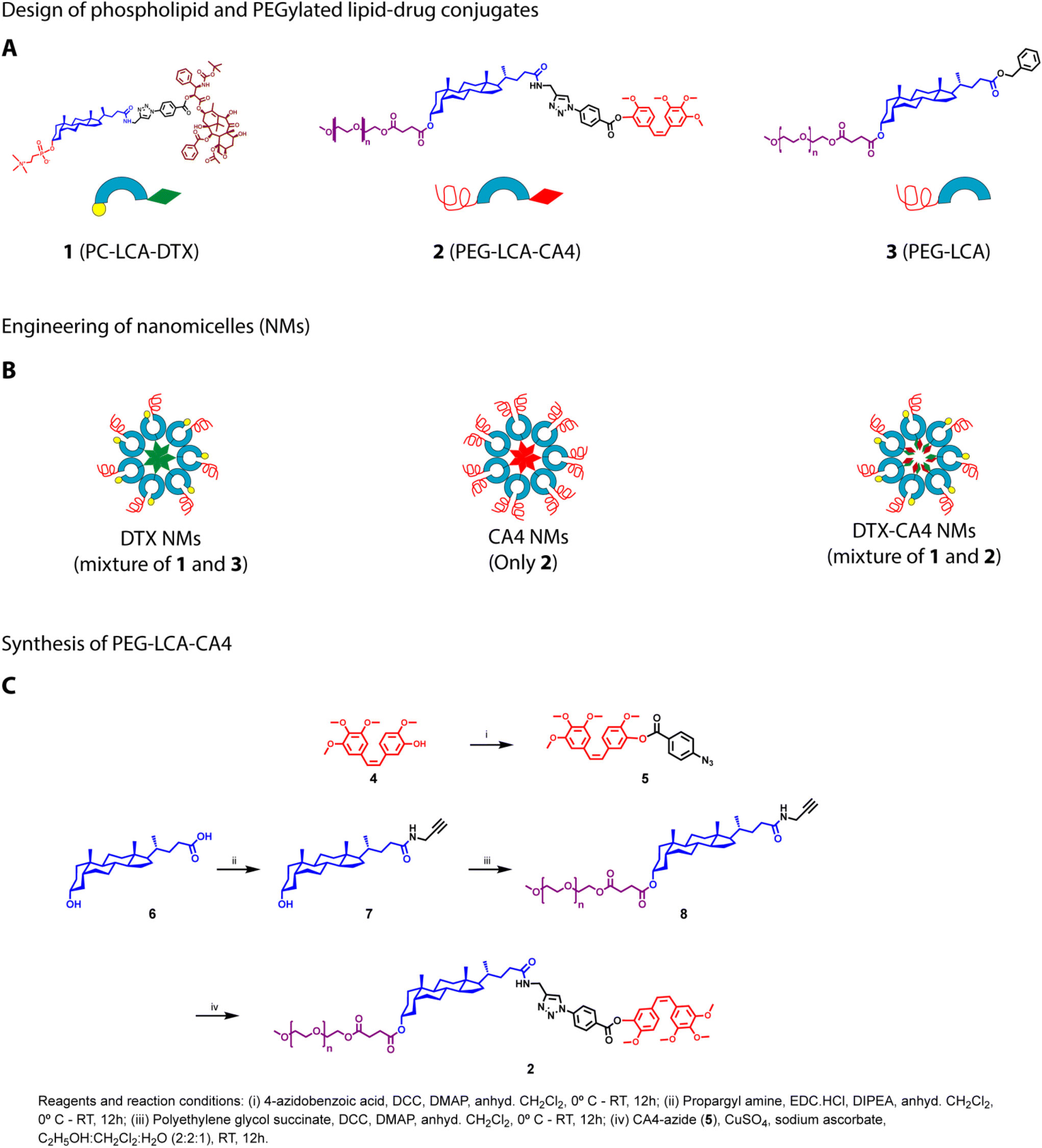 | ||
| Fig. 1 Design and engineering of lipid–drug conjugates and their NMs. (A) Chemical structures and representation of lipid–drug conjugates: PC-LCA-DTX (1), PEG-LCA-CA4 (2), and PEG-LCA (3). (B) Design of engineered DTX NMs, CA4 NMs and DTX-CA4 NMs. (C) Synthesis of PEG-LCA-CA4. | ||
Synthesis of PC-LCA-DTX, PEG-LCA-CA4, and PEG-LCA
We synthesized PC-LCA-DTX (1), as mentioned earlier.29 To synthesize PEG-LCA-CA4 (2), CA4 (4) was reacted with 4-azido benzoic acid under DCC–DMAP coupling conditions to get CA4 azide (5) (Fig. 1C). For the synthesis of propargyl amine-derived LCA,30 propargyl amine was added at the carboxy-terminal of LCA (6) by employing EDC·HCl, resulting in the propargylamide derivative of LCA (compound 7). Compound 7 was further conjugated with polyethylene glycol succinate employing DCC/DMAP coupling to get PEG-LCA-PA (compound 8). Lastly, CA4 azide (5) was conjugated with PEG-LCA-PA (8) using a click reaction to get the PEGylated LCA–CA4 conjugate (PEG-LCA-CA4) (2) with a yield of ∼88% (Fig. 1C). Intermediates and final compounds were purified using flash column chromatography and characterized by 1H-NMR, HRMS, and HPLC. PEG-LCA (3) was synthesized and characterized as mentioned previously.30Preparation and characterization of NMs
Three distinct types of NMs (DTX NMs, CA4 NMs, DTX-CA4 NMs) were prepared using the solvent displacement method.29 We dissolved PC-LCA-DTX and LCA-PEG (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w)) in tetrahydrofuran (THF) and the solution was slowly added to saline. THF was then removed using a rotary evaporator to get the DTX NMs (Fig. 1B). Similarly, we prepared CA4 NMs where a THF solution of PEG-LCA-CA4 was injected dropwise into saline, and THF was removed to get CA4 NMs (Fig. 1B). For the preparation of DTX-CA4 NMs, we added a THF solution of PC-LCA-DTX and PEG-LCA-CA4 (1:4 w/w with respect to drugs) in saline dropwise and then removed THF to get DTX-CA4 NMs (Fig. 1B).
:1 (w/w)) in tetrahydrofuran (THF) and the solution was slowly added to saline. THF was then removed using a rotary evaporator to get the DTX NMs (Fig. 1B). Similarly, we prepared CA4 NMs where a THF solution of PEG-LCA-CA4 was injected dropwise into saline, and THF was removed to get CA4 NMs (Fig. 1B). For the preparation of DTX-CA4 NMs, we added a THF solution of PC-LCA-DTX and PEG-LCA-CA4 (1:4 w/w with respect to drugs) in saline dropwise and then removed THF to get DTX-CA4 NMs (Fig. 1B).
Characterization of these NMs by dynamic light scattering (DLS) demonstrated that DTX NMs were 71.10 ± 4.1 nm in size with a polydispersity index (PDI) of 0.18 ± 0.01 as reported earlier,29 whereas CA4 NMs have a hydrodynamic diameter of 13.62 ± 1.5 nm with PDI of 0.04 ± 0.1 (Fig. 2A), and DTX-CA4 NMs were of 13.0 ± 0.45 nm in size with a PDI of 0.2 ± 0.02 (Fig. 2B). NMs were negatively stained (1% phosphotungstic acid) for transmission electron microscopy (TEM), and TEM images confirmed the spherical shape of CA4 NMs and DTX-CA4 NMs with similar dimensions (Fig. 2C and D). Stability studies confirmed the storage stability of DTX-CA4 NMs at room temperature and at 4 °C (Fig. 2E) and serum stability under 10% and 50% FBS conditions for 24 h at 37 °C (Fig. 2F). As the TME is enriched in esterases and is acidic in nature, we measured the release of DTX and CA4 from DTX-CA4 NMs in the presence and absence of esterase at pH 7.4 and 4.5. Drug release studies confirmed an esterase-responsive release of DTX and CA4 from DTX-CA4 NMs at physiological pH (Fig. 2G and H). The acidic conditions induced an early release of DTX and CA4 from DTX-CA4 NMs compared to physiological pH conditions (Fig. 2I and J), demonstrating the enzyme- and pH-responsive release of chemotherapeutics from these NMs.
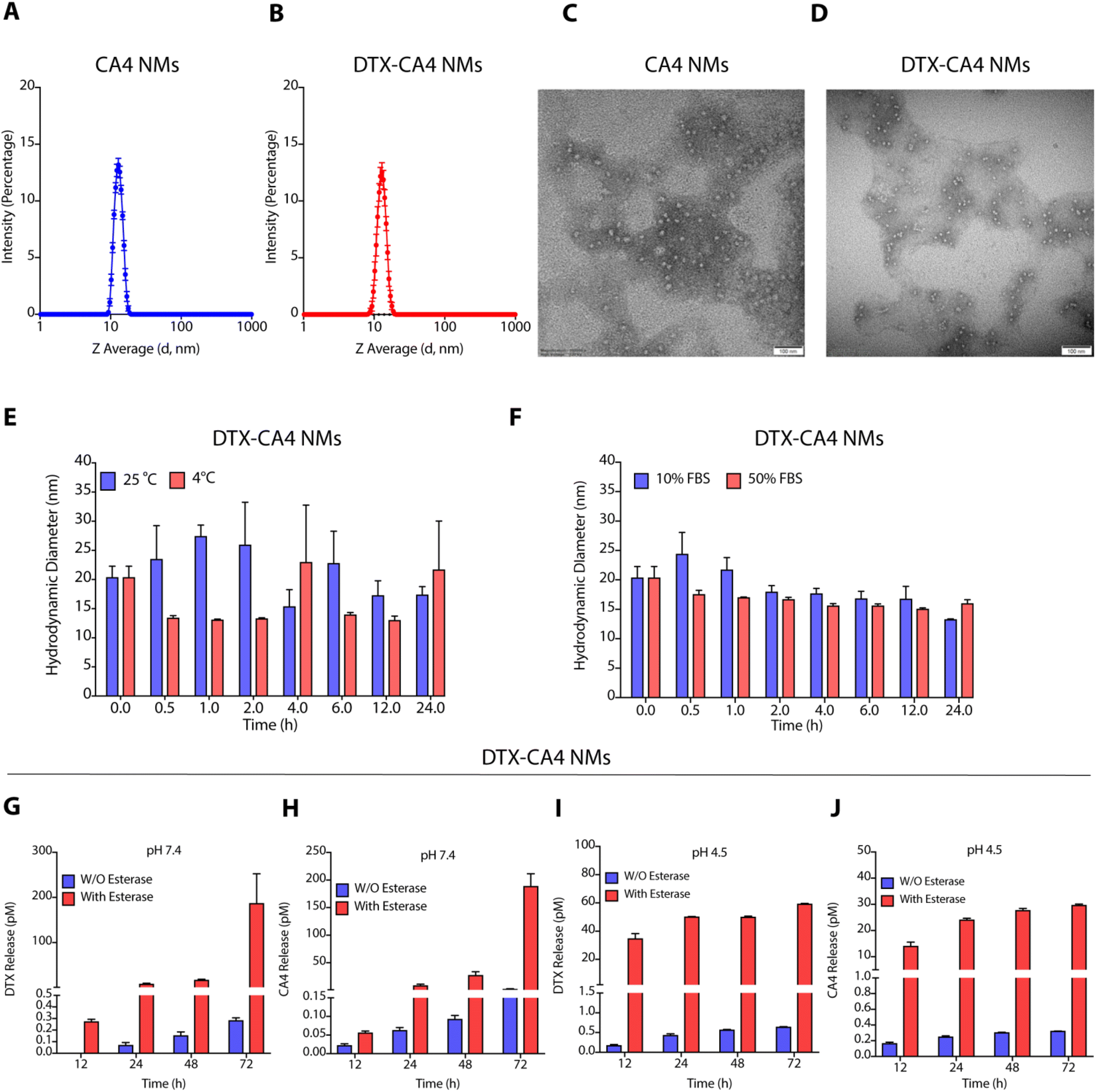 | ||
| Fig. 2 Characterization of engineered NMs. Hydrodynamic diameters of CA4 NMs (A) and DTX-CA4 NMs (B) using dynamic light scattering studies. Transmission electron microscopic images of CA4 NMs (C) and DTX-CA4 NMs (D). Stability studies (n = 3) of DTX-CA4 NMs at 25 and 4 °C (E) and under 10 and 50% FBS conditions at 37 °C (F). Release of DTX (G and I) and CA4 (H and J) from DTX-CA4 NMs at physiological pH 7.4 (G and H) and acidic pH (4.5) (I and J) in the absence and presence of esterase. | ||
Maximum tolerated dose (MTD) studies
As nanoformulations are known to avoid the severe toxicity associated with free chemotherapeutic agents, we tested the effect of DTX-CA4 NMs and a solution of CA4 and DTX in combination (DTX-CA4 Sol) with the body weight and survival of BALB/c mice. We administered five doses (5 mg kg−1 of DTX and 20 mg kg−1 of CA4 or 10 mg kg−1 of DTX with 40 mg kg−1 of CA4) of DTX-CA4 NMs and DTX-CA4 Sol for five days intravenously in BALB/c mice and the body weight and mouse survivability were recorded during the experiment. The mice did not show any reduction in body weight with DTX-CA4 Sol and DTX-CA4 NMs at the 5 mg kg−1 dose of DTX with the 20 mg kg−1 dose of CA4 (Fig. 3A). Similarly, there was no change in the percentage survival of the mice with any of the treatments (Fig. 3B). In contrast, there was a ∼10% reduction in the body weight of mice with DTX-CA4 Sol upon administration of 10 mg kg−1 of DTX with 40 mg kg−1 of CA4 compared to DTX-CA4 NMs, and 25% of mice showed mortality after day 10 of the treatment with DTX-CA4 Sol (Fig. 3C and D). Interestingly, there was no effect on the body weight and mouse survival upon administration of DTX-CA4 NMs at 10 mg kg−1 of DTX with 40 mg kg−1 of CA4, thereby confirming the safety profile of DTX-CA4 NMs.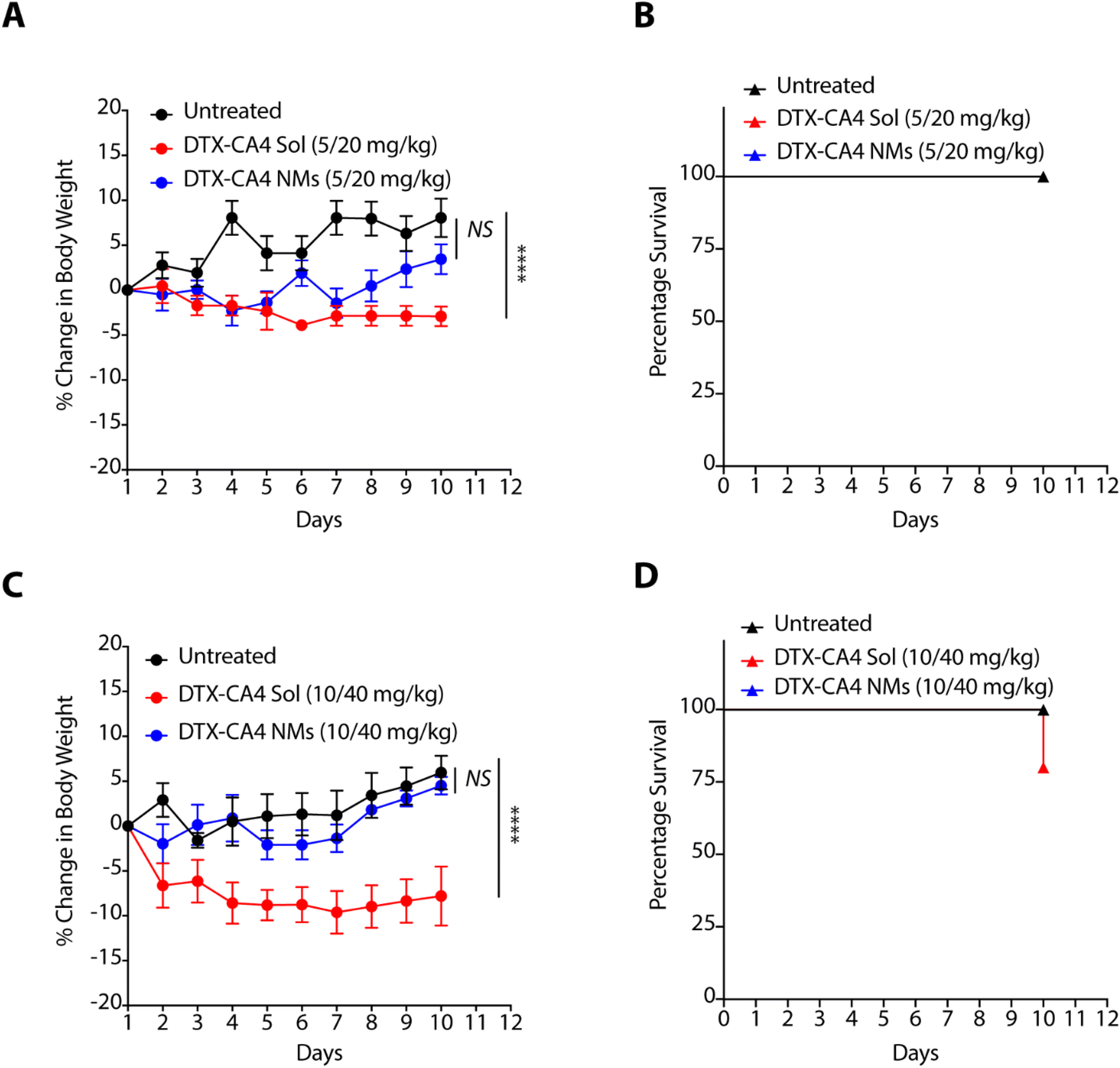 | ||
| Fig. 3 Percentage change in the body weight (A and C) and mice survival (B and D) on administration of five doses of the combination of DTX and CA4 in solution (DTX-CA4 Sol) or as nanomicelles (DTX-CA4 NMs) at different doses where a combination of DTX and CA4 was either used as 5 and 20 mg kg−1 or 10 and 40 mg kg−1, respectively (n = 5 per group). | ||
DTX-CA4 NMs are effective against murine colon cancer models
We used a xenograft colon cancer model to examine the anticancer efficacy of DTX-CA4 NMs, where we injected human colon cancer cells (HCT116) subcutaneously in immunocompromised NOD-SCID male mice. Tumour-bearing mice (∼50 mm3) were randomly categorized into four sets where set 1 was left untreated, set 2 was treated with DTX NMs, set 3 was treated with CA4 NMs and set 4 was treated with DTX-CA4 NMs. We have administered a dose of 5 mg kg−1 of DTX and a dose of 20 mg kg−1 for CA4 intravenously every third day. The tumour volume and body weights were recorded during the experiment on every alternate day. Administration of DTX-CA4 NMs significantly inhibited the tumour growth of HCT116 tumours in comparison with untreated mice (Fig. 4A). There was a ∼3-fold reduction in the final day tumour volume on treatment with DTX-CA4 NMs compared to untreated mice and a >1.5-fold alleviation of tumour growth over treatment with DTX NMs and CA4 NMs alone (Fig. 4A). Body weight measurements showed a ∼5% decrease in the body weight after day 17 of treatment with DTX-CA4 NMs, but mice recovered after that (Fig. 4B). DTX-CA4 NMs-treated mice showed a median survival of 43 days in comparison with only 35 days in untreated mice (Fig. 4C), and an effective reduction in tumour growth was observed from excised tumours as well (Fig. 4D).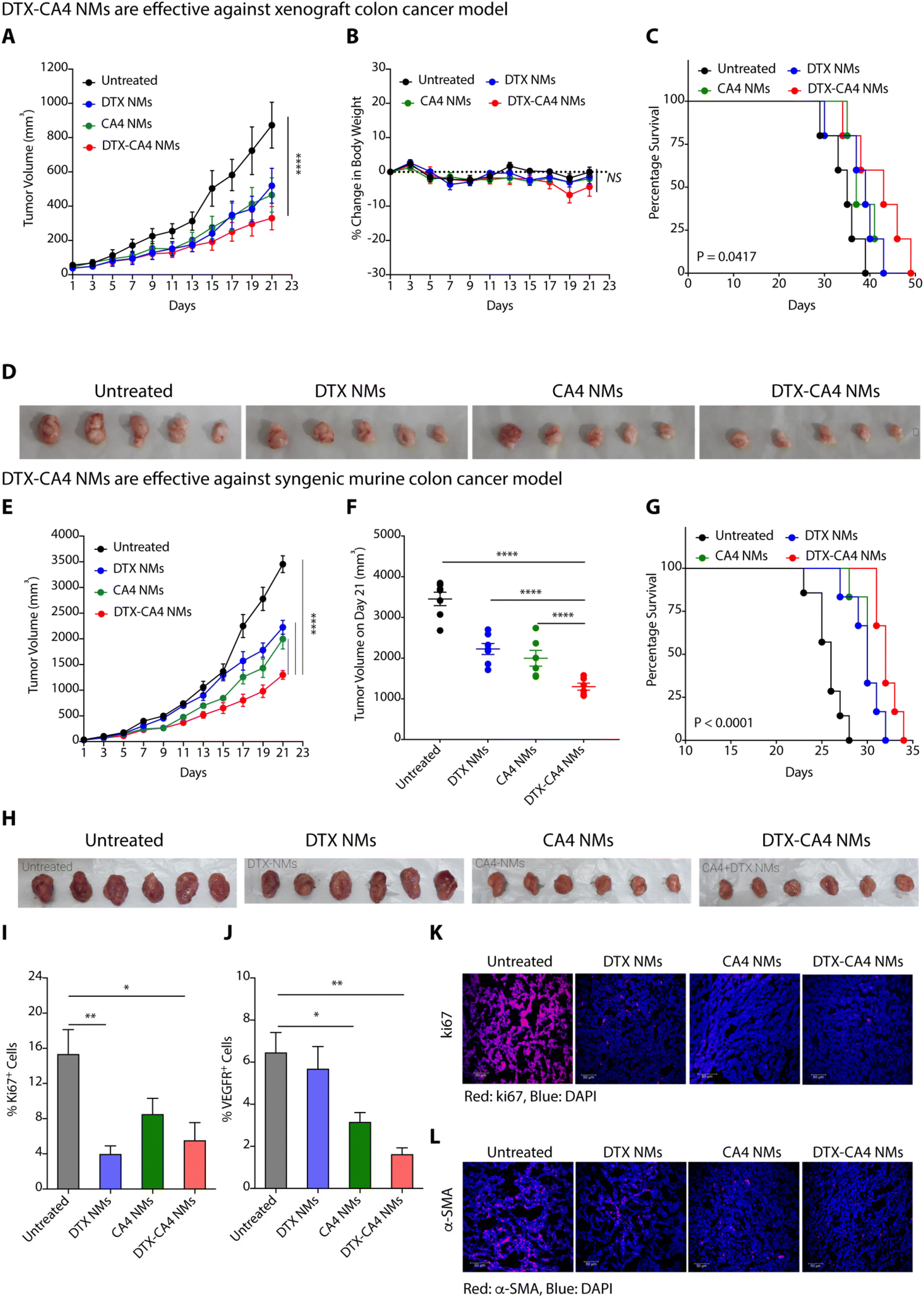 | ||
| Fig. 4 DTX-CA4 NMs restrict tumour growth and enhance mouse survival. Effect of DTX-CA4 NMs on tumour growth kinetics (mean ± SEM) (A), body weight change (mean ± SEM) (B), and percentage survival (C) in HCT116 tumour-bearing NOD-SCID mice (n = 6 per group). (D) Tumour images of excised HCT116 tumours after different treatments. Effect of DTX-CA4 NMs on tumour growth kinetics (mean ± SEM) (E), last day tumour volume (mean ± SEM) (F), and percentage survival (G) in CT26 tumour-bearing BALB/c mice (n = 6 per group). (H) Representative images of excised CT26 tumours after different treatments. Percentage of Ki67+ (I) and VEGFR+ cells (J) (mean ± SD, n = 4–6) in untreated and different NM-treated CT26 tumours quantified by flow cytometry. Immunofluorescence images showing reduced expression of Ki67 (K) and α-SMA (L) in DTX-CA4 NMs-treated tumour tissues in comparison with other groups. | ||
Next, we investigated the anticancer efficacy of different NMs against a murine colon (CT26) cancer model where 1.5 × 106 CT26 cells were implanted subcutaneously in the left flank of immunocompetent BALB/c mice. Mice with ∼50 mm3 of tumour volume were randomly segregated into four groups, and treatment was given in a similar manner as mentioned above. We observed a reduction in the kinetics of tumour growth on treatment with DTX-CA4 NMs in contrast to other treatments (Fig. 4E). From the final day tumour volume data, we observed that DTX-CA4 NMs caused a >2.5-fold reduction in comparison with untreated mice and a 1.5-fold alleviation compared to treatment with other NMs (Fig. 4F). Survival studies demonstrated a median survival of 34 days with the treatment of DTX-CA4 NMs, whereas untreated mice survived only for 28 days (Fig. 4G) with no changes in the body weight in any treatment groups. Tumour images clearly showed that DTX-CA4 NMs effectively inhibit tumour growth (Fig. 4H).
As DTX-CA4 NMs target the proliferating cancer cells and tumour-associated angiogenesis, we performed an in-depth immunostaining and flow cytometric analysis to evaluate the effect of NMs on the TME. At the end of the animal experiment, tumour tissues were dissected and excised tumours were processed to get a single-cell suspension. The cells were then stained with anti-Ki67 and anti-VEGFR antibodies and analysed by flow cytometry. We observed a ∼3-fold decrease in the percentage of Ki67+ cells and a >3.5-fold decrease in % VEGFR+ cells upon treatment with DTX-CA4 NMs as compared to untreated tumours, suggesting the reduced proliferation and angiogenesis (Fig. 4I and J). The flow cytometry data were further validated by immunofluorescence staining, where tumour tissue sections were fixed, sectioned and stained with Ki67, TUNEL, α-FAP, and α-SMA. Immunofluorescence staining of tumour sections exhibited a significant decrease in cell proliferation marker Ki67 in DTX-CA4-treated tumour sections compared to other treatments and untreated tumour tissues (Fig. 4K). Immunofluorescence staining also confirmed a significant decrease in vascular smooth muscle cell marker α-SMA levels that predominate in connective tissues on treatment with DTX-CA4 NMs (Fig. 4L). In addition to this, the TUNEL assay confirmed the enhanced apoptosis in DTX-CA4 NM-treated tissues (Fig. S1A†) and staining with α-FAP showed a decrease in cancer-associated fibroblasts (Fig. S1B†). Therefore, DTX-CA4 NMs enhance apoptotic cell death, reduce cell proliferation, and mitigate angiogenesis in the TME.
DTX-CA4 NMs activate T-cell immunity in the TME
Abnormal tumour vasculature is known to recruit immunosuppressive immune cells, delay dendritic cell maturation, and hamper the infiltration of cytotoxic T cells.33 The tumour-supportive immune microenvironment inhibits the effector functions of T cells responsible for cancer cell death and develops a tumour-friendly milieu for cancer progression and metastasis.34 Along with the proapoptotic effect, DTX also modifies the immunosuppressive TME in breast and prostate cancer models by improving anti-tumour CD4+ and CD8+ T cells through the production of IFN-γ and by restricting the infiltration of pro-tumoural myeloid-derived suppressive cells (MDSCs).35,36 Therefore, there is a need to understand the impact of DTX-CA4 NMs on the TME.We investigated the changes in the infiltration of total immune cells, T cells, and effector T cells by flow cytometry in CT26 tumours on treatment with DTX-CA4 NMs and compared to untreated and DTX NMs- and CA4 NMs-treated tumours. CT26 tumour tissues were chopped, homogenized, and processed further to prepare a single-cell suspension. The samples were then incubated and stained with a mixture of antibodies, and different immune cell populations were analyzed using a flow cytometer (Fig. S2†). We found a >3-fold increase in the infiltration of CD45+ immune cells in CT26 tumours treated with DTX-CA4 NMs than in untreated tumours (Fig. 5A). Further quantification revealed a ∼20-fold increase of CD3+ T cell population in DTX-CA4 NMs-treated CT26 tumours compared to untreated tumours (Fig. 5B), and we observed a ∼1.25-fold increase in CD4+ T cells as well (Fig. 5C). Among different CD8 T cell populations, DTX-CA4 NMs treatment caused an increase in CD8+ T cells and a >10-fold increase in the effector population of granzyme B+ CD8+ T cells than untreated mice, thereby confirming the alteration of the immunosuppressive TME (Fig. 5D and E). Therefore, these results confirmed that DTX-CA4 NMs treatment augmented the infiltration of CD3+ and CD4+ T cells and enhanced the activation of CD8+ T cells. We also observed an increase in PD-1-expressing CD8+ cytotoxic T cells on treatment with DTX-CA4 NMs, as PD1 expression is correlated with the activation of CD8+ T cells (Fig. 5F).
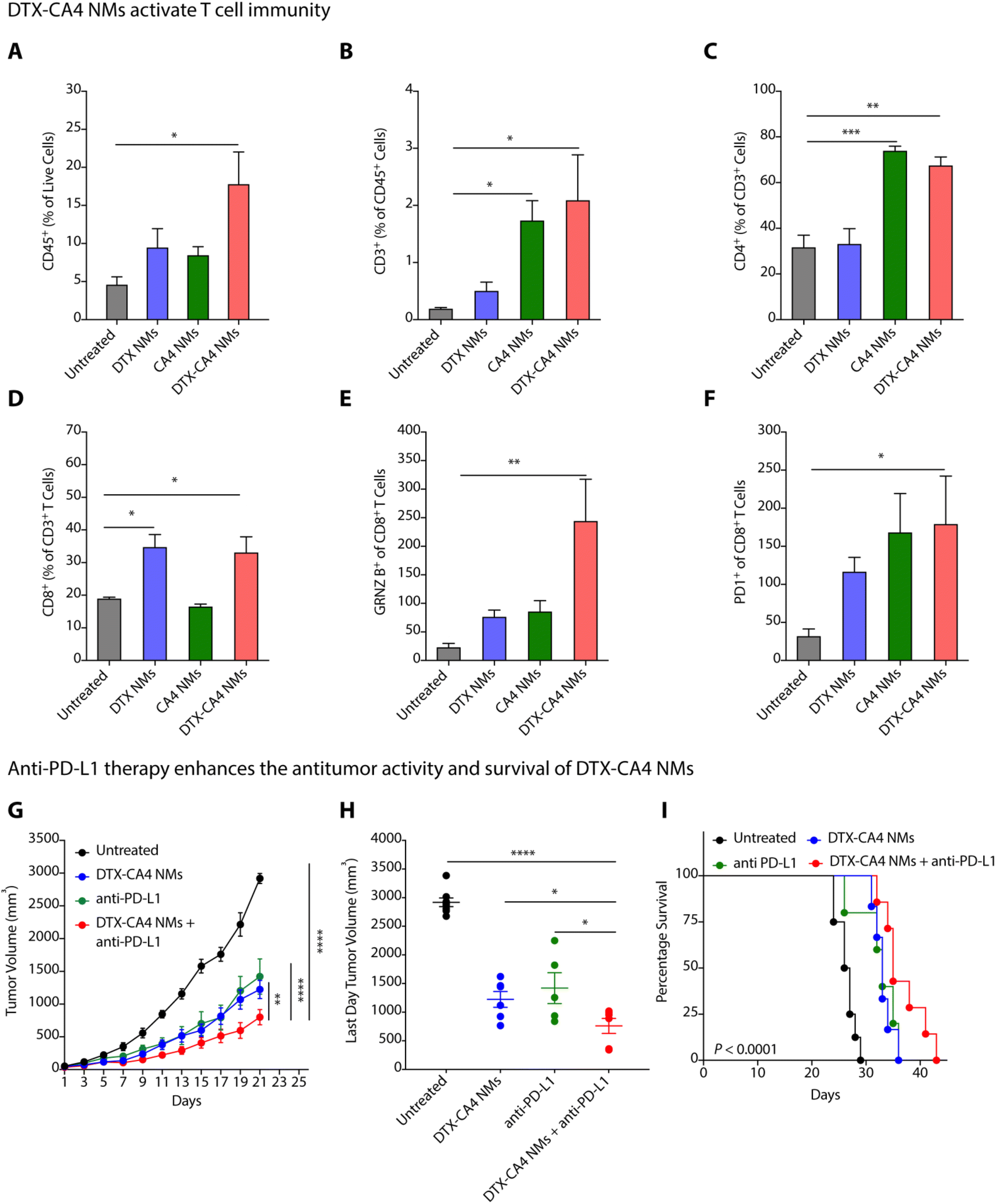 | ||
| Fig. 5 DTX-CA4 NMs alter the TME and activate T cell immunity. Changes (mean ± SEM) in CD45+ immune cells (A), CD3+ (B), CD4+ (C), CD8+ (D), GRNZ B+CD8+ (E), and PD1+CD8+ T cells (F) in DTX-CA4 NMs-treated CT26 tumour tissues compared to tumour tissues from untreated, DTX NMs- and CA4 NMs-treated mice (n = 5–6). Effect of DTX-CA4 NMs in combination with anti-PD-L1 antibody on tumour growth kinetics (mean ± SEM) (G), tumour volume at the last day (mean ± SEM) (H), and mouse percentage survival (I) in CT26 tumour-bearing BALB/c mice (n = 6 per group). | ||
Anti-PD-L1 enhances the anti-tumour efficacy of DTX-CA4 NMs
PD-1, an immune checkpoint expressed by T cells, acts as a regulator of immune response.37 Cancer cells express its ligand PD-L1, and PD1–PDL1 interactions inhibit T cell activation.37,38 Therefore, blocking PD-1/PDL1 interactions using anti-PD-1 or anti-PD-L1 antibodies can lead to T cell activation and stimulate immune responses against tumour cells. To test the combination of DTX-CA4 NMs with anti-PD-L1 therapy, we segregated CT26-tumour bearing mice into different batches where batch 1 remained untreated, and anti-PD-L1, DTX-CA4 NMs and a combination of DTX-CA4 NMs and anti-PD-L1 were administered in the mice of batch 2, 3, and 4, respectively. Anti-PD-L1 (100 μg) was given intraperitoneally on every 4th day and DTX-CA4 NMs intravenously on every third day. DTX-CA4 NMs, in combination with anti-PD-L1, caused a >1.5-fold reduction in tumour growth in comparison with other treatments (Fig. 5G and H) and also improved the mouse survival as well (Fig. 5I). Consequently, these results reveal that engineered DTX-CA4 NMs target cell proliferation and angiogenesis activates T cell immunity by enhancing the granzyme B+ CD8+ T cells.DTX-CA4 NMs inhibit tumour recurrence from residual tumour cells
To examine the impact of DTX-CA4 NMs as an adjuvant therapy following surgical intervention, we performed surgical removal of tumours from CT26 tumour-bearing mice when the tumour volume reached 50–80 mm3. We randomly divided the mice into four groups (Fig. 6A): untreated (group 1), DTX NMs-treated (group 2), CA4 NMs-treated (group 3) and DTX-CA4 NMs-treated (group 4) and treatment was started from the very next day of surgery. Doses were injected intravenously on every third day, and a total of 7 doses were given. Tumour recurrence was observed in untreated mice as a result of residual tumour cells, while DTX-CA4 NMs treatment did not allow any noticeable tumour growth in mice. (Fig. 6B and C). We observed a clear response (CR) in 6 out of 10 mice in the DTX-CA4 NMs-treated group (Fig. 6B), and all the mice survived until 50 days (Fig. 6D). We rechallenged the mice with cancer cells that showed a CR from the DTX-CA4 NMs-treated group, and new mice were used for the untreated group (Fig. 6A). Interestingly, we observed no tumour growth in the DTX-CA4 NMs-treated group after rechallenge (Fig. 6E). Therefore, these results suggest that DTX-CA4 NMs inhibit tumour recurrence after surgery and can be a potential candidate to be used as an adjuvant therapy.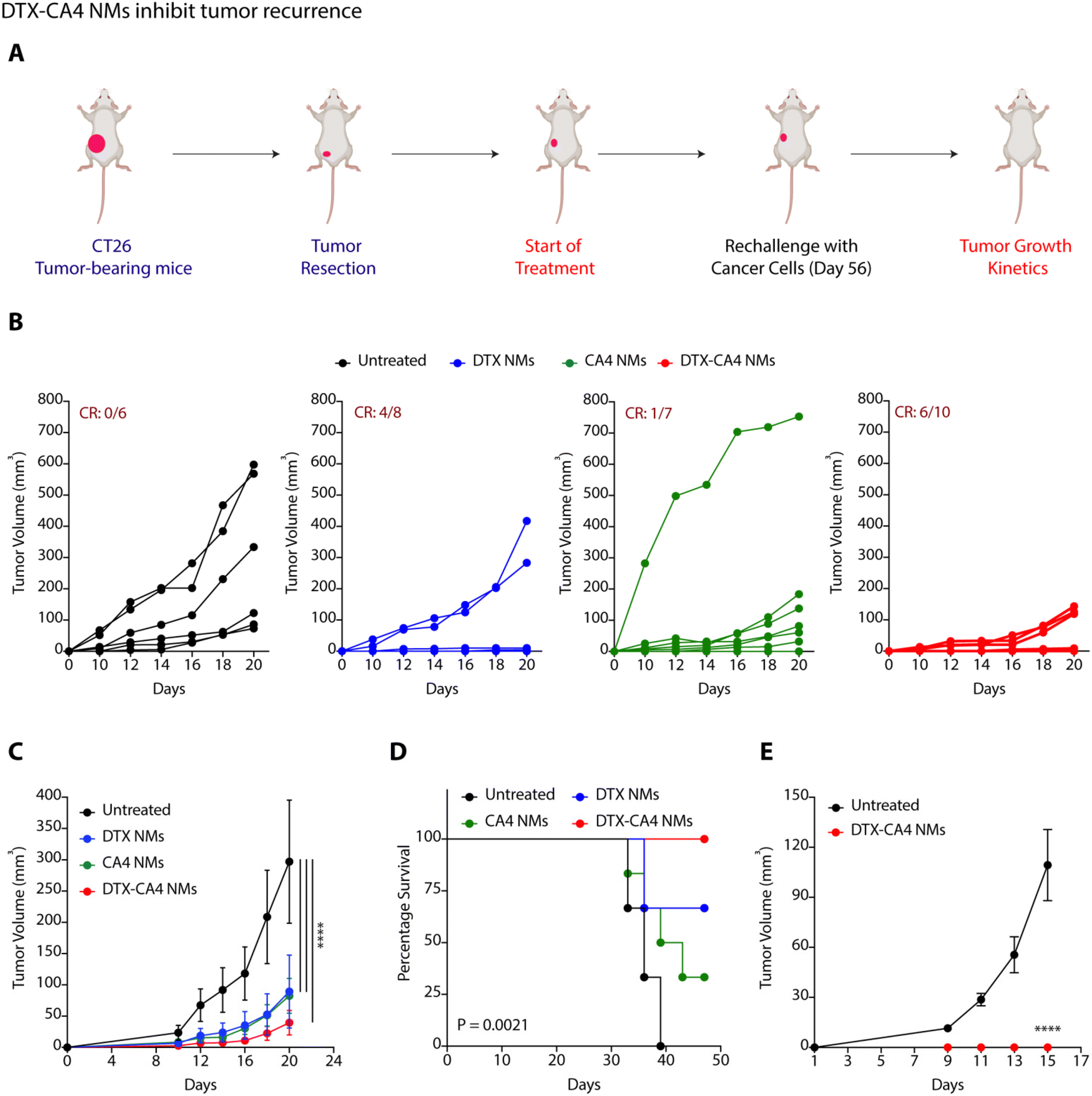 | ||
| Fig. 6 DTX-CA4 NMs inhibit tumour recurrence. (A) Schematic illustration of the experiment to test the efficacy of DTX-CA4 NMs on tumour recurrence. (B) Effect of treatment of different NMs on tumour growth kinetics of individual mice of each group after surgical removal of CT26 tumours (n = 6–10 per group). (C) Change in the tumour volume on treatment with different NMs after surgical removal of tumours. (D) Change in the percentage survival of mice after different treatments. (E) Effect on the tumour growth kinetics after rechallenge with CT26 cells. | ||
DTX-CA4 NMs inhibit C1P synthesis
C1Ps are one of the key metabolites of the sphingolipid pathway and are synthesized from ceramide using CERK (Fig. 7A).39 Earlier studies have shown that C1Ps can modulate cell proliferation, migration, cell survival, apoptotic cell death, and inflammation.40 CERK-mediated generation of C1Ps acts as a key mediator in cPLA2 activation and the release of arachidonic acid, and plays a key role in wound healing.41 C1P is also a potent pro-inflammatory agent that stimulates macrophage chemotaxis.42 Therefore, CERK and C1Ps are potential targets for anti-cancer therapies, and their inhibition can be explored further. As we observed significant alterations in the TME with an increase in the level of cytotoxic T cells in DTX-CA4 NMs-treated tumours, we hypothesized that DTX-CA4 NMs might alter the lipid metabolites like pro-inflammatory C1Ps. Therefore, we quantified the levels of C1Ps in DTX-CA4 NMs-treated and untreated CT26 tumour tissues using LC-MS/MS. We noticed a ∼1.5-fold decrease in the level of C1Ps in DTX-CA4 NMs-treated tumour tissues compared to untreated tissues (Fig. 7B). As CERK is the sole enzyme responsible for the synthesis of C1Ps, we quantified the changes in gene expression of CERK in the CT26 tumour model using qRT-PCR, which revealed a >2.5-fold reduction in CERK expression following DTX-CA4 NMs treatment (Fig. 7C). Next, we validated the changes in CERK expression by immunoblotting (Fig. 7D and Fig. S3†) and observed a >5-fold decrease in CERK protein expression in DTX-CA4 NMs-treated tumours compared to untreated tumours (Fig. 7E). Therefore, qRT-PCR and immunoblot quantification confirmed the reduction in CERK levels on treatment with DTX-CA4 NMs, suggesting CERK as a potential therapeutic target for this combination therapy.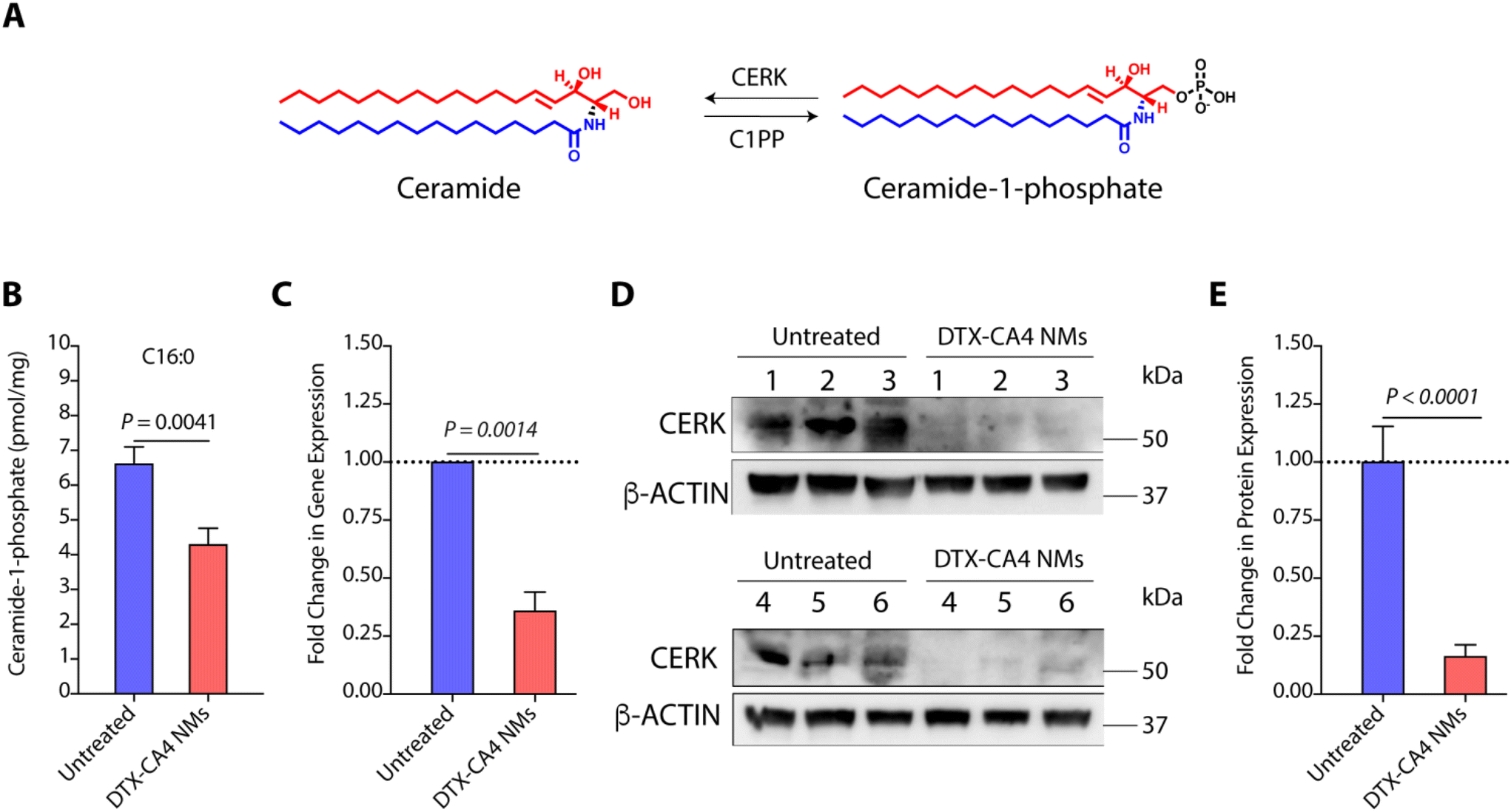 | ||
| Fig. 7 DTX-CA4 NMs inhibit ceramide-1-phosphate, a key mediator, for tumour progression. (A) An illustration depicting the conversion of ceramide into C1P facilitated by CERK. (B) LC-MS/MS-based estimation of C1P levels in untreated and DTX-CA4 NMs-treated CT26 tumours (n = 4–5). (C) qRT-PCR data showing the fold change in the gene expression of CERK in untreated and DTX-CA4 NMs-treated CT26 tumours. Immunoblots for CERK expression (D) and its quantification (n = 6) (E) in untreated and DTX-CA4 NMs-treated tumours. | ||
Conclusions
In summary, we presented the development of DTX-CA4 NMs, which are designed to target the proliferating cancer cells and angiogenesis. We used a combination of a phosphocholine derivative of DTX with a PEGylated derivative of CA4 that self-assembles and forms DTX-CA4 NMs. Engineered DTX-CA4 NMs cause a reduction in tumour growth kinetics more effectively than individual drug-treated tumours, along with a reduction in cell proliferation, angiogenesis, and fibroblasts with an increase in apoptosis. Engineered DTX-CA4 NMs also enhance the infiltration and activation of CD8+ T cells in the TME, along with an increase in the expression of PD-1, and administration of anti-PD-L1 therapy enhances the efficacy of DTX-CA4 NMs. Additionally, we have shown that the DTX-CA4 NMs also impede the synthesis of C1Ps by inhibiting the expression of CERK at the transcriptional level. These results collectively indicate that DTX-CA4 NMs enable the co-delivery of antiproliferative and antiangiogenic agents that reduce the level of proinflammatory molecules like C1Ps and enhance the anti-tumour immunity by recruiting T cells in the TME to promote tumour regression. Hence, the engineering of DTX-CA4 NMs has paved the way for new possibilities, which can be further investigated for the delivery of therapeutic agents for cancer therapy (Fig. 8).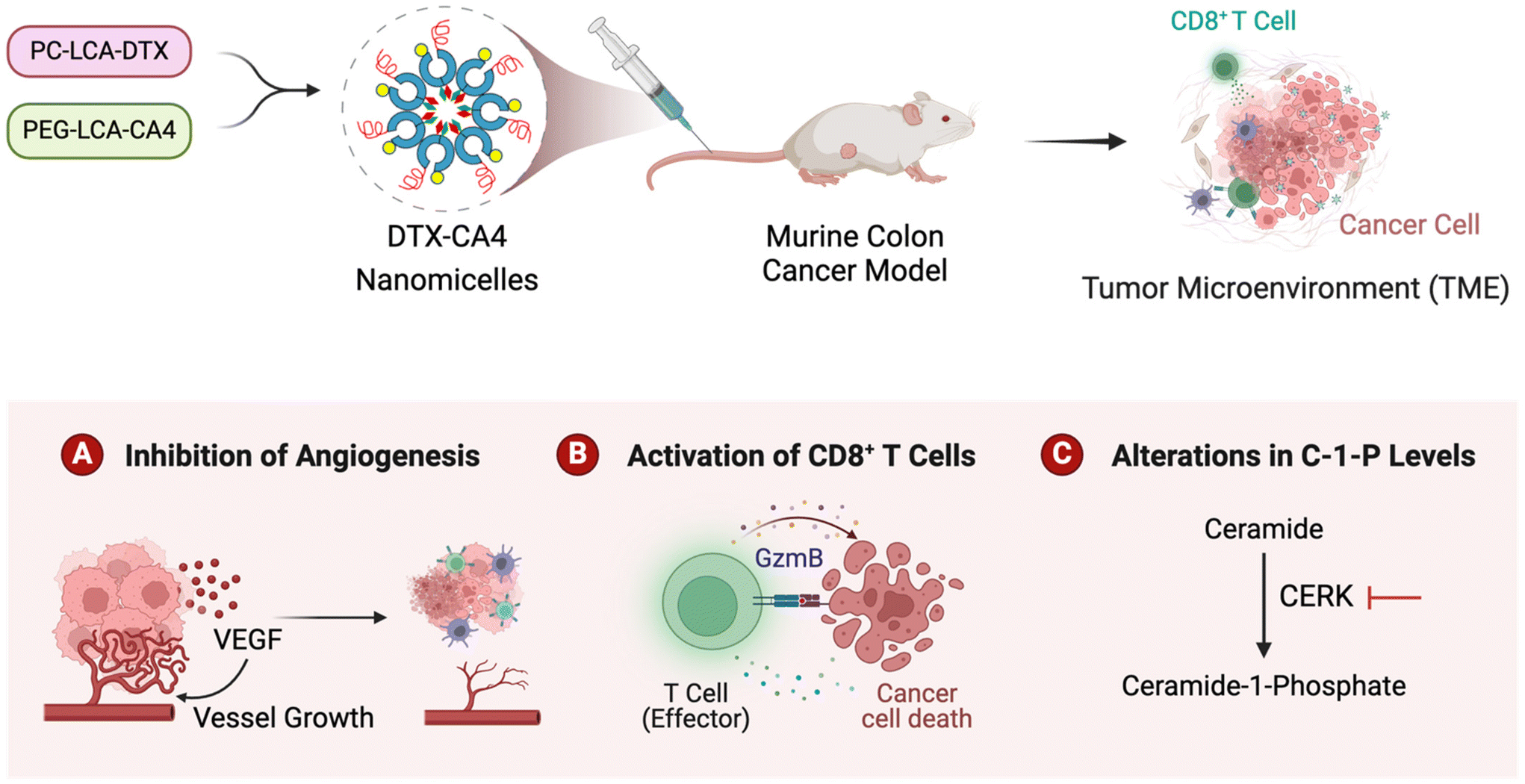 | ||
| Fig. 8 An illustration depicting the engineering of DTX-CA4 NMs aimed at inhibiting cell proliferation and angiogenesis to induce tumour regression, activate T cell immunity and inhibit the synthesis of proinflammatory C1Ps. | ||
Experimental section
Methods
Compound 5. Anhydrous dichloromethane (DCM) (Merck, 270997) (5 mL) was utilized to dissolve 4-azidobenzoic acid (1 g) and N,N′-dicyclohexylcarbodiimide (DCC) (Merck, D80002) (1.3 g), followed by the addition of 4-dimethyl aminopyridine (DMAP) (Spectrochem, 1004194) (0.772 g). The solution was stirred for 10 min, followed by the addition of DCM (5 mL) solution of combretastatin A4 (compound 4) (Labex cooperation, 117048-59-6) (1 g). The reaction was continued overnight at room temperature and quenched using water. The crude product was extracted from the reaction mixture using ethyl acetate (Merck, 319902). Flash column chromatography was used to purify compound 5 (CA4-azide) using hexane/ethyl acetate (80
:20). Yield: ∼88%. 1H-NMR (400 MHz, CDCl3): δ 3.725 (s, 6H), 3.792–3.813 (m, 6H), δ 6.440–6.527 (m, 4H), δ 6.883–6.904 (d, 1H, J = 8.4 Hz), δ 7.101–7.180 (m, 4H, m), δ 8.140–8.160 (d, 2H, J = 8.0 Hz). 13C-NMR (75 MHz, CDCl3) δ: 29.7176, 55.9631, 55.9979, 60.9077, 76.6399, 77.0634, 77.2647, 77.4867, 105.8804, 112.0997, 118.9762, 123.3167, 125.7529, 128.5801, 129.5981, 130.1758, 132.1255, 132.4592, 137.1825, 139.5488, 145.3577, 150.4253, 152.9923, 163.7938. HRMS (ESI) m/z: calculated for (C25H23N3O6)+ 461.1586, observed: (M + H)+ 462.1659.
Compound 7. The propargylamine derivative of LCA was synthesized as per the published procedure.30 Briefly, N,N-ethylcarbodiimide hydrochloride (EDC HCl) (Spectrochem, 0104244) (2 g) and LCA (6) (Labex cooperation, 434-13-9) (2 g) were solubilized in anhydrous DCM, followed by the addition of N,N-diisopropylethylamine (DIPEA) (Spectrochem, 0104261) (5.74 mL). After stirring for 20 minutes, propargyl amine (0.73 mL) (Tokyo Chemical Industry Corporation, P0911) was introduced, and the solution was left stirring for 12 hours. The crude product was subjected to extraction and purification through silica column chromatography with hexane/ethyl acetate (60
:40) as the elution solvent. Yield: 95%. 1H-NMR (300 MHz, CDCl3): δ 0.626 (s, 3H), δ 0.802–2.14 (m, steroid), δ 2.21–2.226 (m, 2H), δ 3.59–3.66 (m, 1H), δ 4.04–4.06 (m, 2H), δ 5.69 (bs, 1H). 13C-NMR (75 MHz, CDCl3) δ: 12.06, 18.39, 20.83, 23.38, 24.21, 26.42, 27.2, 28.26, 29.18, 30.54, 31.57, 33.30, 34.58, 35.35, 35.45, 35.85, 36.46, 40.19, 40.44, 42.10, 42.75, 55.98, 55.51, 71.54, 71.87, 173.18. HRMS (ESI) m/z: calculated for (C27H43NO2)+ 413.3294, observed: (M + H)+ 414.3375.
Compound 8. Polyethylene glycol succinate was synthesized as per the published procedure.30 The propargyl amine derivative of LCA (7) and polyethylene glycol succinate (1.4 g) were dissolved in dry DCM (5 mL), followed by the addition of DCC (0.678 g) and DMAP (0.401 g). The mixture was stirred overnight at room temperature, and the resulting product was extracted in DCM and purified with DCM/methanol (95
:05) as the eluent through flash chromatography. Yield: 85.36%. 1H-NMR (400 MHz, CDCl3): δ 0.630 (s, 3H), δ 0.901–1.963 (m, steroid), δ 2.052–2.097 (m, 1H), δ 2.223–2.246 (m, 2H), δ 2.567–2.658 (m, 4H), δ 3.369 (s, 3H), δ 3.472 (s, 1H), δ 3.526–3.549 (m, 3H), δ 3.672–3.696 (m, 62H), δ 4.035–4.041 (d, 2H, J = 2.4 Hz), δ 4.223–4.247 (t, 2H, J = 4.8 Hz), δ 4.652–4.780 (m, 1H), δ 5.677 (s, 1H). 13C-NMR (300 MHz, CDCl3) δ: 29.04, 29.10, 29.43, 29.56, 29.65, 30.70, 31.55, 32.16, 32.49, 33.19, 34.55, 34.98, 35.44, 35.75, 40.11, 40.38, 41.86, 42.71, 49.94, 51.83, 55.19, 56.01, 56.44, 59.00, 63.78, 63.84, 64.01, 64.08, 68.96, 69.02, 70.47, 71.41, 71.89, 74.74, 76.71, 79.84, 153.92, 171.73, 172.26, 172.37, 172.69, 173.19, 173.48. HRMS (ESI) m/z: calculated for (C64H113NO21)+ 1231.7805; observed: (M + Na)+ 1254.7697.
Compound 2 (PEG-LCA-CA4). PEG-LCA-PA (8) (1 g) and CA4-azide (5) (0.562 g) were dissolved in a solution of DCM
:MeOH:H2O (2:2:1), followed by the addition of sodium ascorbate (Acros Organics, 352685000) (80 mg) and copper(II) sulphate (SRL, 61298) (64 mg). The mixture was stirred for 12 hours, after which the crude product was extracted using DCM and purified in DCM/methanol (90:10) using flash column chromatography. Yield: 88%. 1H-NMR (400 MHz, CDCl3): δ 0.605 (s, 3H), 0.904–2.32 (m, 45H), 2.597–2.659 (m, 5H), 3.370 (s, 4H), 3.459–3.809 (m, 102H), 4.225–4.235 (d, 2H, J = 4 Hz), 4.580–4.594 (d, 2H, J = 5.6 Hz), 4.710–4.811 (m, 1H), 6.250 (s, 1H), 6.432–6.551 (m, 4H), 6.890–6.945 (m, 1H), 7.152 (s, 1H), 7.152–7.199 (m, 1H), 7.854–7.894 (m, 2H), δ 8.145 (s, 1H), δ 8.310–8.332 (d, 2H, J = 8.8 Hz). HRMS (ESI) m/z: calculated for (C89H136N4O27)+ 1692.9392, observed: (M + K)+ 1731.9009.
Engineering and characterization of nanomicelles
Nanomicelles were engineered and characterized as per the previously reported protocol.30 Tetrahydrofuran (THF) solution of PC-LCA-DTX (1) (0.95 mg) and PEG-LCA-CA4 (2) (10.7 mg) was combined and added slowly in ice cold saline (2 mL) under continuous stirring at the maximum speed. The complete evaporation of THF (Merck, 186562) with the help of a rota evaporator resulted in DTX-CA4 NMs. In the same manner, CA4 NMs were formulated exclusively with PEG-LCA-CA4, while DTX NMs were formulated with PC-LCA-DTX along with LCA-PEG using a 1:1 ratio. A Zetasizer nano ZS90 (Malvern, UK) was utilized for dynamic light scattering studies to assess the hydrodynamic size and polydispersity index (PDI) of all nanomicelles. NMs were negatively stained with 1% phosphotungstic acid (PTA) to determine the morphology using a JEM-1400Flash transmission electron microscope. The hydrodynamic size of DTX-CA4 NMs was monitored after storage at room temperature (RT) and 4 °C and under 10% and 50% FBS conditions at 37 °C to assess the storage and serum stability.
Drug release studies
DTX-CA4 NMs, containing 10.7 mg mL−1 of PEG-LCA-CA4 (2) and 0.95 mg mL−1 of PC-LCA-DTX (1), were added to the dialysis tubes with a 100 kDa cutoff and subjected to dialysis in release buffer (1× PBS). The drug release was quantified in the presence and absence of esterase enzyme (100 UI) in the release buffer of physiological (pH 7.4) and acidic pH (pH 4.5) at 37 °C. At different time points, 1 mL of aqueous sample was withdrawn and replenished with fresh release buffer. The samples were dried and then extracted using 100 μL of methanol. Following reconstitution, the samples were vortexed for 5 minutes and then sonicated for an additional 5 minutes. Next, the samples were spun at 5000 rpm for 5 min, and the resulting supernatant was collected in fresh tubes. This process was repeated two times more, and the extracted samples were dried under high vacuum. The dried extracted samples were finally reconstituted in 100 μL of methanol and subjected to HPLC analysis. We employed a reverse-phase Waters Alliance HPLC system to assess the concentrations of free DTX and CA4 released from DTX-CA4 NMs using a C18 silica column (250 × 4.6 mm) with a pore size of 5 μm. The mobile phase comprised of solvent A ACN and solvent B water (0.1% NH4OH) in an 8:2 (A:B) ratio, flowing at a rate of 0.5 mL min−1. The system was equipped with a PDA detector, and quantification was performed at 254 nm.
Cell culture
RPMI 1640 media (Merck, R6504) supplemented with 10% (v/v) fetal bovine serum (FBS) (Merck, RM1112), 1% penicillin and streptomycin were used to culture human colon cancer cells (HCT116) and murine colon cancer cells (CT26). The cells were maintained in an incubator at 37 °C with 5% carbon dioxide and harvested for further experiments using 1× trypsin.Maximum tolerated dose studies
Normal BALB/c mice were randomized into two batches and each batch had three groups: untreated, DTX-CA4 solution and DTX-CA4 NMs. In the first batch, DTX-CA4 solution and DTX-CA4 NMs were injected at a dosage of 5 mg kg−1 and 20 mg kg−1 for DTX and CA4, respectively. On the other hand, the second batch received a dose of 10 mg kg−1 for DTX and 40 mg kg−1 for CA4. Doses were injected intravenously for consecutive five days, and body weight and mouse mortality were recorded during the experiment.Animal studies
Flow cytometry
Tumour tissues were dissected and chopped into smaller pieces, followed by their homogenization using Gentle MACS dissociator. The homogenates were centrifuged at 400g for 5 minutes, and then the resulting pellets were resuspended in an RPMI-based enzymatic mixture containing collagenase IV (0.75 mg mL−1) and DNase I (50 U mL−1) for digestion. The samples were then placed in an incubator at constant shaking at 37 °C for 30–45 min. Digested samples were stained with a 70 μm cell strainer to make single cell suspension, and the mixture was centrifuged at 400g for 5 min. The pellets were dissolved in FACS buffer containing 2 mM EDTA and 4% goat serum in 1× PBS and incubated for 5–10 minutes to prevent non-specific binding. Approximately 5 × 106 cells in 100 μL of FACS buffer were used for staining following counting with a LUNAII™ Automated cell counter from Logos Biosystems, USA. Cell viability was determined by staining live/dead cells using the Zombie Aqua Fixable Viability Kit following the manufacturer's instructions. The cells were incubated with a mixture of antibodies for staining the surface markers at 4 °C in the dark for 20 min. For T cell analysis, a combination of anti-CD45-PerCP-Cy5.5 (BioLegend, 103132), anti-CD3-BV421 (BioLegend, 100336), anti-CD4-PE-Cy7 (BioLegend, 100422), anti-CD8-FITC (BioLegend, 100706), and anti-PD-1-APC-Cy7 (BioLegend, 135224) was used. After staining, the samples were washed with FACS buffer (400 μL in each sample) to remove the excess antibody and finally resuspended with FACS buffer.For intracellular granzyme B staining, the cells were fixed and permeabilized in Cyto-fast fix/perm buffer (150 μL), followed by incubation with anti-granzyme B-PE antibody (BioLegend, 372207) in the dark for 25–30 min. Furthermore, after being washed with Cyto-fast perm wash buffer, the cells were suspended in FACS buffer. Next, a BD FACS Verse flow cytometer from BD Biosciences, USA, was used to acquire the samples. The acquired data were subsequently analyzed using Flowjo v10 software (USA).
Furthermore, angiogenesis and cell proliferation were quantified employing the previously published protocol.30
Immunofluorescence studies, TUNEL staining, and immunoblot studies
Immunofluorescence studies, TUNEL staining, and immunoblot studies (CERK, Abcam, 155061) were performed as per the published protocol.30C1P quantification using LC-MS/MS
C1P levels were measured in both untreated and treated tumour tissues using an established method our laboratory.40qRT-PCR studies
Tissues were homogenized in liquid nitrogen and the homogenate was dissolved in trizol (Qiagen, 79306), and RNA was isolated using the chloroform (Qualigen, Q22465)–isopropanol (Qualigen, Q13825) method. Subsequently, 1 μg of total RNA was subjected to reverse transcription using the Biorad cDNA kit according to the manufacturer's standard protocol. The RNA was incubated at 46 °C for 20 min in the reverse transcription process, followed by terminating the reaction through heating at 95 °C for 1 min. The SYBR kit from Biorad was utilized to assess the level of CERK mRNA following the manufacturer's instructions. The master mix provided in the kit includes all the necessary reagents for qRT-PCR, such as a hot start DNA polymerase, reaction buffer, dNTPs, a salt, and other components. To conduct the assay, only template cDNA and specific primers were combined with the master mix. For the mouse CERK analysis, the primers employed are 5′-GAGC′TGACTT′AAGACCGA-3′ and 5′-CGTAG′GATGGAAGCCAAGG-3′. These primers were carefully designed to amplify ceramide kinase while ensuring they span at least one exon/intron boundary, effectively preventing the amplification of genomic DNA.Statistical analysis
The data are reported as the mean ± standard error of the mean (SEM) or the mean ± standard deviation (SD) of three or more samples. Statistical analysis involves the use of unpaired student t-test, one-way ANOVA, or two-way analyses of variance (ANOVA). A p value of less than 0.05 (*p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001) was deemed to indicate statistical significance.Author contributions
PY synthesized the compounds, prepared nanomicelles, and performed all in vitro and in vivo experiments. KR and DJ performed all immune profiling analyses. RC helped in nanomicelle preparation, animal experiments and drug release studies. AK helped in the synthesis of compounds, standardization of nanomicelles and performed mass spectrometry to quantify the lipids. DM performed HPLC for the drug release study and helped in the characterization of compounds. BA checked gene expression using qRT-PCR. DJ performed immunoblot studies. UD supervised the mass spectrometry analyses. AB envisioned the idea and superintended the project.Ethical statement
The Institutional Animal Ethics Committee of the Regional Centre for Biotechnology (RCB/IAEC/2016/010) reviewed and approved the animal experimental protocols. The experiments were conducted in accordance with the guidelines provided by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.Conflicts of interest
The authors have proclaimed that there are no contending financial interests.Acknowledgements
We express our gratitude to RCB for providing intramural funding, as well as to the Department of Biotechnology (DBT), Science and Engineering Research Board (SERB) and the Department of Science and Technology, Government of India for their support towards the research conducted in AB's laboratory. The students extend their thanks to funding agencies such as CSIR, DBT, UGC, and ICMR for their research fellowship. The animal work carried out in the small animal facility at the Regional Centre for Biotechnology was made possible through the support of BT/PR5480/INF/22/158/2012 (DBT). Furthermore, we would like to acknowledge the assistance of the DBT e-Library Consortium (DeLCON) for granting access to e-resources. The Amity Lipidomics Research Facility at Amity University Haryana receives support from the DST-FIST grant, SR/FST/LSI-664/2016.References
- T. L. Whiteside, The tumour microenvironment and its role in promoting tumour growth, Oncogene, 2008, 27, 5904–5912 CrossRef CAS PubMed.
- M. D. Palma, D. Biziato and T. V. Petrova, Microenvironmental regulation of tumour angiogenesis, Nat. Rev. Cancer, 2017, 17, 457–474 CrossRef PubMed.
- J. Fang, Y. Lu, J. Zheng, X. Jiang, H. Shen, X. Shang, Y. Lu and P. Fu, Exploring the crosstalk between endothelial cells, immune cells, and immune checkpoints in the tumour microenvironment: new insights and therapeutic implications, Cell Death Dis., 2023, 14, 586 CrossRef PubMed.
- I. Zuazo-Gaztelu and O. Casanovas, Unravelling the role of angiogenesis in cancer ecosystems, Front. Oncol., 2018, 8, 248 CrossRef PubMed.
- D. Fukumura, J. Kloepper, Z. Amoozgar, D. G. Duda and R. K. Jain, Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges, Nat. Rev. Clin. Oncol., 2018, 15, 325–340 CrossRef CAS PubMed.
- L. L. Munn and R. K. Jain, Vascular regulation of antitumour immunity, Science, 2019, 365, 544–545 CrossRef CAS PubMed.
- Y. Zhou, X. Chen, J. Cao and H. Gao, Overcoming the biological barriers in the tumour microenvironment for improving drug delivery and efficacy, J. Mater. Chem. B, 2020, 8, 6765–6781 RSC.
- R. I. Teleanu, C. Chircov, A. M. Grumezescu and D. M. Teleanu, Tumour angiogenesis and anti-angiogenic strategies for cancer treatment, J. Clin. Med., 2019, 9, 84 CrossRef PubMed.
- J. Garcia, H. I. Hurwitz, A. B. Sandler, D. Miles, R. L. Coleman, R. Deurloo and O. L. Chinot, Bevacizumab in cancer treatment: a review of 15 years of clinical experience and future outlook, Cancer Treat. Rev., 2020, 86, 102017 CrossRef CAS PubMed.
- Z. Hao and I. Sadek, Sunitinib: The antiangiogenic effect and beyond, OncoTargets Ther., 2016, 9, 5495–5505 CrossRef CAS PubMed.
- M. Imran, S. Saleem, A. Chaudhuri, J. Ali and S. Baboota, Docetaxel: An update on its molecular mechanisms, therapeutic trajectory and nanotechnology in the treatment of breast, lung and prostate cancer, J. Drug Delivery Sci. Technol., 2020, 60, 101959 CrossRef CAS.
- C. J. Sweeney, K. D. Miller, S. E. Sissons, S. Nozaki, D. K. Heilman, J. Shen and G. W. Sledge, The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors, Cancer Res., 2001, 61, 3369–3372 CAS.
- X. L. Guo, G. J. Lin, H. Zhao, Y. Gao, L. P. Qian, S. R. Xu, L. N. Fu, Q. Xu and J. J. Wang, Inhibitory effects of docetaxel on expression of VEGF, bFGF and MMPs of LS174T cell, World J. Gastroenterol., 2003, 9, 1995–1998 CrossRef CAS PubMed.
- M. Su, J. Huang, S. Liu, Y. Xiao, X. Qin, J. Liu, C. Pi, T. Luo, J. Li, X. Chen and Z. Luo, The anti-angiogenic effect and novel mechanisms of action of Combretastatin A-4, Sci. Rep., 2016, 6, 28139 CrossRef CAS PubMed.
- S. W. Morton, M. J. Lee, Z. J. Deng, E. C. Dreaden, E. Siouve, K. E. Shopsowitz, N. J. Shah, M. B. Yaffe and P. T. Hammond, A nanoparticle-based combination chemotherapy delivery system for enhanced tumour killing by dynamic rewiring of signaling pathways, Sci. Signaling, 2014, 7, ra44 CrossRef PubMed.
- S. Sengupta, D. Eavarone, I. Capila, G. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system, Nature, 2005, 436, 568–572 CrossRef CAS PubMed.
- Y. Wang, H. Chen, Y. Liu, J. Wu, P. Zhou, Y. Wang, R. Li, X. Yang and N. Zhang, pH-sensitive pullulan-based nanoparticle carrier of methotrexate and combretastatin A4 for the combination therapy against hepatocellular carcinoma, Biomaterials, 2013, 34, 7181–7190 CrossRef CAS PubMed.
- Y. A. Hannun and L. M. Obeid, Sphingolipid and their metabolism in physiology and disease, Nat. Rev. Mol. Cell Biol., 2018, 19, 175–191 CrossRef CAS PubMed.
- B. Ogretmen and Y. A. Hannun, Biologically active sphingolipids in cancer pathogenesis and treatment, Nat. Rev. Cancer, 2004, 4, 604–616 CrossRef CAS PubMed.
- B. Ogretmen, Sphingolipid metabolism in cancer signalling and therapy, Nat. Rev. Cancer, 2018, 18, 33–50 CrossRef CAS PubMed.
- S. A. F. Morad and M. C. Cabot, Ceramide-orchestrated signalling in cancer cells, Nat. Rev. Cancer, 2013, 13, 51–65 CrossRef CAS PubMed.
- A. Gomez-Muñoz, The role of ceramide-1-phosphate in tumour cell survival and dissemination, Adv. Cancer Res., 2018, 140, 217–234 CrossRef PubMed.
- H. A. Mena, P. R. Zubiry, B. Dizier, V. Mignon, F. Parborell, M. Schattner, C. Boisson-Vidal and S. Negrotto, Ceramide 1-phosphate protects endothelial colony–forming cells from apoptosis and increases vasculogenesis in vitro and in vivo, Arterioscler. Thromb. Vasc. Biol., 2019, 39, E219–e232 CrossRef CAS PubMed.
- A. Gomez-Munoz, N. Presa, A. Gomez-Larrauri, I. Rivera, M. Trueba and M. Ordonez, Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate, Prog. Lipid Res., 2016, 61, 51–62 CrossRef CAS PubMed.
- N. Medatwal, M. N. Ansari, S. Kumar, S. Pal, S. K. Jha, P. Verma, K. Rana, U. Dasgupta and A. Bajaj, Hydrogel-mediated delivery of celastrol and doxorubicin induces a synergistic effect on tumour regression via upregulation of ceramides, Nanoscale, 2020, 12, 18463–18475 RSC.
- S. Pal, N. Medatwal, S. Kumar, A. Kar, V. Komalla, P. S. Yavvari, D. Mishra, Z. R. Rizvi, S. Nandan, D. Malakar, M. Pillai, A. Awasthi, P. Das, R. D. Sharma, A. Srivastava, S. Sengupta, U. Dasgupta and A. Bajaj, A localized chimeric hydrogel therapy combats tumour progression through alteration of sphingolipid metabolism, ACS Cent. Sci., 2019, 5, 1648–1662 CrossRef CAS PubMed.
- S. M. Tawfik, S. Azizov, M. R. Elmasry, M. Sharipov and Y. Lee, Recent advances in nanomicelles delivery systems, Nanomaterials, 2021, 11, 70 CrossRef CAS PubMed.
- N. Pavlovic, S. Golocorbin-Kon, M. Danic, B. Stanimirov, H. A. Salami, K. Stankov and M. Mikov, Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles, Front. Pharmacol., 2018, 9, 1283 CrossRef PubMed.
- V. Sreekanth, A. Kar, S. Kumar, S. Pal, P. Yadav, Y. Sharma, V. Komalla, S. Sharma, R. Shyam, R. D. Sharma, A. Mukhopadhyay, S. Sengupta, U. Dasgupta and A. Bajaj, Bile-acid-tethered docetaxel-based nanomicelles mitigates the tumour progression via epigenetic changes, Angew. Chem., Int. Ed., 2021, 60, 5394–5399 CrossRef CAS PubMed.
- P. Yadav, K. Rana, V. Nardini, A. Khan, T. Pani, A. Kar, D. Jain, R. Chakraborty, R. Singh, S. K. Jha, D. Mehta, H. Sharma, R. D. Sharma, S. V. S. Deo, S. Sengupta, V. S. Patil, L. H. Faccioli, U. Dasgupta and A. Bajaj, Engineered nanomicelles inhibit the tumour progression via abrogating the prostaglandin-mediated immunosuppression, J. Controlled Release, 2024, 368, 548–565 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Prodrugs: design and clinical applications, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- H. Dong, L. Pang, H. Cong, Y. Shen and B. Yu, Application and design of esterase-responsive nanoparticles for cancer therapy, Drug Delivery, 2019, 26, 416–432 CrossRef CAS PubMed.
- O. E. Rahma and F. S. Hodi, The intersection between tumour angiogenesis and immune suppression, Clin. Cancer Res., 2019, 25, 5449–5457 CrossRef CAS PubMed.
- Y. Huang, B. Y. S. Kim, C. K. Chan, S. M. Hahn, I. L. Weissman and W. Jiang, Improving immune-vascular crosstalk for cancer immunotherapy, Nat. Rev. Immunol., 2018, 18, 195–203 CrossRef CAS PubMed.
- K. N. Kodumudi, K. Woan, D. L. Gilvary, E. Sahakian, S. Wei and J. Y. Djeu, A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumour bearers, Clin. Cancer Res., 2010, 16, 4583–4594 CrossRef CAS PubMed.
- Z. Ma, W. Zhang, B. Dong, Z. Xin, Y. Ji, R. Su, K. Shen, J. Pan, Q. Wang and W. Xue, Docetaxel remodels prostate cancer immune microenvironment and enhances checkpoint inhibitor-based immunotherapy, Theranostics, 2022, 12, 4965–4979 CrossRef CAS PubMed.
- A. Filippone, M. Lanza, D. Mannino, G. Raciti, C. Colarossi, D. Sciassa, S. Cuzzocrea and I. Paternity, PD1/PD-L1 immune checkpoint as a potential target for preventing brain tumour progression, Cancer Immunol. Immunother., 2022, 71, 2067–2075 CrossRef CAS PubMed.
- B. Zhao, Z. Dong, W. Liu, F. Lou, Q. Wang, H. Hong and Y. Wang, Co-administration of combretastatin A4 nanoparticles and anti-PD-L1 for synergistic therapy of hepatocellular carcinoma, J. Nanobiotechnol., 2021, 19, 124 CrossRef CAS PubMed.
- A. Gomez-Larrauri, A. Ouro, M. Trueba and A. Gomez-Muñoz, Regulation of cell growth, survival and migration by ceramide 1-phosphate - implications in lung cancer progression and inflammation, Cell Signaling, 2021, 83, 109980 CrossRef CAS PubMed.
- K. Rajput, M. N. Ansari, S. J. Jha, T. Pani, N. Medatwal, S. Chattopadhyay, A. Bajaj and U. Dasgupta, Ceramide Kinase (CERK) emerges as a common therapeutic target for triple positive and triple negative breast cancer cells, Cancer, 2022, 14, 4496 CrossRef CAS PubMed.
- K. D. Maus, D. J. Stephenson, A. N. Ali, H. P. MacKnight, H. Huang, J. Serrats, M. Kim, R. F. Diegelmann and C. E. Chalfant, Ceramide kinase regulates acute wound healing by supressing 5-oxo-ETE biosynthesis and signaling via its receptor OXER1, J. Lipid Res., 2022, 63, 100187 CrossRef CAS PubMed.
- L. Arana, P. Gangoiti, A. Ouro, M. Trueba and A. Gomez-Munoz, Ceramide and ceramide 1-phosphate in health and disease, Lipids Health Dis., 2010, 9, 15 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1and S2, original immunoblot (Fig. S3) and spectra. See DOI: https://doi.org/10.1039/d3nr04806c |
| This journal is © The Royal Society of Chemistry 2024 |