
Chemical transformation mechanism for blue-to-green emitting CsPbBr3 nanocrystals†
Yuling
Liu‡
abc,
Rui
Yun‡
abc,
Yue
Li
abc,
Wenda
Sun
abc,
Tiancheng
Zheng
abc,
Qian
Huang
abc,
Libing
Zhang
 d and
Xiyan
Li
*abc
d and
Xiyan
Li
*abc
aInstitute of Photoelectronic Thin Film Devices and Technology, Solar Energy Conversion Center, Nankai University, Tianjin 300350, P. R. China
bKey Laboratory of Efficient Utilization of solar energy of Tianjin, Tianjin 300071, P. R. China
cEngineering Research Center of Thin Film Photoelectronic Technology of Ministry of Education, Tianjin 300350, P. R. China. E-mail: xiyan.li@nankai.edu.cn
dTianjin Key Laboratory of Molecular Optoelectronic, Department of Chemistry, Tianjin University, Tianjin, 300072, P. R. China
First published on 6th March 2024
Abstract
Recently, metal–halide perovskites have rapidly emerged as efficient light emitters with near-unity quantum yield and size-dependent optical and electronic properties, which have attracted considerable attention from researchers. However, the ultrafast nucleation rate of ionic perovskite counterparts severely limits the in-depth exploration of the growth mechanism of colloidal nanocrystals (NCs). Herein, we used an inorganic ligand nitrosonium tetrafluoroborate (NOBF4) to trigger a slow post-synthesis transformation process, converting non-luminescent Cs4PbBr6 NCs into bright green luminescent CsPbBr3 NCs to elucidate the concrete transformation mechanism via four stages: (i) the dissociation of pristine NCs, (ii) the formation of Pb–Br intermediates, (iii) low-dimensional nanoplatelets (NPLs) and (iv) cubic CsPbBr3 NCs, corresponding to the blue-to-green emission process. The desorption and reorganization of organic ligands induced by NO+ and the involvement of BF4− in the ligand exchange process played pivotal roles in this dissolution–recrystallization of NCs. Moreover, controlled shape evolution from anisotropic NPLs to NCs was investigated through variations in the amount of NOBF4. This further validates that additives exert a decisive role in the symmetry and growth of nanostructured perovskite crystals during phase transition based on the ligand-exchange mechanism. This finding serves as a source of inspiration for the synthesis of highly luminescent CsPbBr3 NCs, providing valuable insights into the chemical mechanism in post-synthesis transformation.
 Xiyan Li | Xiyan Li is currently a full professor in the College of Electronic Information and Optical Engineering at Nankai University of China. She received her Ph.D. degree from Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 2013. She then worked as a post-doctoral fellow at National University of Singapore from 2013 to 2015, and at the University of Toronto from 2015 to 2019, respectively. Her current research focuses on the controlled synthesis, novel properties, and optoelectronic applications of semiconductor nanostructures, especially perovskite-based optoelectronic devices. |
Introduction
Metal–halide perovskites (MHPs), which are ideal for next-generation light emitters, have drawn substantial interest in a number of optoelectrical applications such as high-definition displays, field-effect transistors and wearable electronics.1–7 Especially, major advances have been made in multicolor perovskite light-emitting diodes in terms of desired emission wavelengths, solution processing characteristics and the exceptional balance of cost and performance. Different from conventional nanocrystals (NCs) with covalent bonds and rigid crystal structures, MHP-NCs with soft lattices are primarily bound by ionic chemical bonds. The difference in binding strength results in MHP-NCs having low lattice formation energy, high lattice ionicity, and sub-second formation kinetics. These properties make it challenging to stabilize MHP-NCs at small sizes and understand the mechanism of their dynamic growth via direct synthesis methods, which is critical for further optimizing the performance of MHP NCs. Therefore, it is necessary to explore some tactics to retard the nucleation and growth rate of nanocrystals to investigate the specific crystal growth kinetics. Tian's group devised a stepwise synthesis method triggered by a polar alcohol to control the reaction rate and analyze the formation process of CsPbBr3 NCs.8 Li et al. developed a microwave-irradiation-assisted method to obtain intermediate products during the growth of CsPbBr3 NCs.9 These studies provide useful insights into the formation mechanism of CsPbBr3 NCs. However, the improvement of emission properties in the above-mentioned studies was not satisfactory and PLQYs were not high enough owing to the polar solvent and surface defects.In addition, the noncovalent and high dynamic binding of surface capping ligands result in their easy desorption from the surface of NCs and further destabilize the morphology and crystal structure of pristine NCs.10–14 Nonetheless, this unique characteristic also presents an opportunity to achieve novel morphologies or different crystal phases through post-treatment. Thus, chemical transformation from zero-dimensional (0D) Cs4PbBr6 NCs to three-dimensional (3D) CsPbBr3 NCs with desired optical properties has been proven to be a feasible and interesting method.15–17 Recent studies suggest that phase transition in a solution is easily generated by the stripping of CsBr or the insertion of excess PbBr2, while the 0D and 3D phases maintain a similar size distribution.18–20 Instead, Baranov et al. proposed a different post-synthetic strategy of converting oleylamine (OAm)/oleate (OA)-capped Cs4PbBr6 NCs into strongly emissive polymer-capped CsPbBr3 NCs using poly(maleic anhydride-alt-1-octadecene) (PMAO). Due to the mild reactivity of the organic polymer, the intermediate Cs4PbBr6–CsPbBr3 heterostructures were monitored during a slow transformation process. The partially converted particles were obtained by the reaction of the succinic anhydride units of PMAO and OAm ligands bound to the surface of Cs4PbBr6 NCs.21 Such nanoscale transformation proves that this reaction is driven by the reactivity of the regents, opening up strategies for designing and precisely controlling the crystal growth at the nanoscale.
In this regard, the adsorption and desorption of ligands on the surfaces of NCs are crucial for the post-synthesis transformation of their morphology and the crystallographic phase. Introducing new additives to as-prepared MHP-NCs can not only alter their surface properties but also trigger the post-synthesis transformation, leading to morphology evolution and crystallographic phase transition of NCs.22–25 Inorganic ligand (nitrosonium tetrafluoroborate, NOBF4) is applicable for surface modification with nanoparticles (e.g., NaYF4, Fe3O4, TiO2, FePt and Bi2S3).26–30 Sequential surface functionalization of colloidal nanomaterials can be achieved without altering their size and shape. This process replaces the hydrophobic ligands on the NCs’ surface with short-chain ligands, making the nanoparticles accessible for various applications in biology, photovoltaics and other fields. Very recently, we have reported an inorganic ligand (NOBF4) mediated transformation strategy from non-luminescent Cs4PbBr6 NCs to the precisely controllable blue-emitting CsPbBr3 NPLs with a two-monolayer (2 ML) unit cell.31 This treatment revealed a dynamic dissolution–recrystallization process along with the appearance of deep-blue light (444 nm), where the nitrite ion (NO+) rendered the intrinsic Cs4PbBr6 NCs unstable by stripping the organic species from their surface. Meanwhile, the NO+-induced acidic environment can protonate OAm into OAm+ cations, thus enabling the slow precipitation of the precisely controllable 2 ML CsPbBr3 NPLs in the non-polar solvent (several hours).
Herein, we will extend our investigation by studying the influence of the content of dissociating agent NOBF4 on the Cs4PbBr6 NCs. Benefiting from the non-polar environment, the reaction process with different amounts of NOBF4 was monitored by time-dependent photoluminescence (PL) and absorption spectra due to a slow reaction rate. We found that the shape of the products was controllable from the anisotropic NPLs to bulk CsPbBr3 NCs by increasing the amount of NOBF4, accompanied by the slow transformation of blue into green light in the solution, corresponding to several specified emissions (Fig. 1, and S1†). More details on the conversion mechanism and surface capping properties were explored carefully by detecting the intermediate products during the phase transition process from non-luminescent Cs4PbBr6 NCs into brightly green luminescent CsPbBr3 NCs. It was noted that the ligand desorption depended on the amount of NO+, while BF4− also participated in the ligand exchange process and was adsorbed on the surface of the reconstructed crystal. In addition, the fully-transformed CsPbBr3 NCs exhibited a narrow emission line width of 18 nm and high photoluminescence quantum yield in the solution (up to 80%). Thus, this post-processing provides valuable insight into the mechanism of crystal growth and has potential application in the synthesis of highly luminescent NCs.
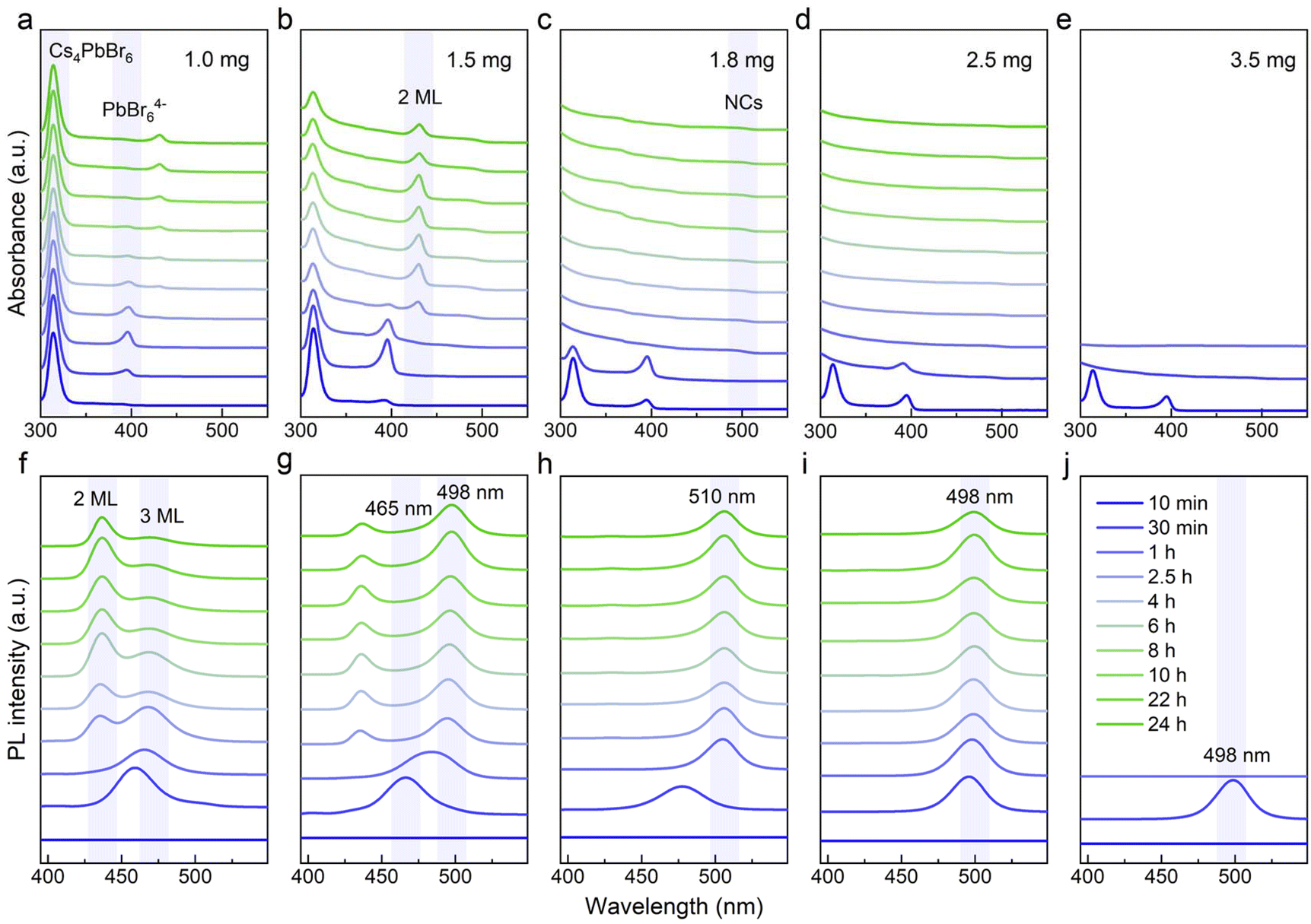 | ||
| Fig. 1 (a–e) Absorption and (f–j) PL spectra of luminescent products obtained with different amounts of NOBF4 (1.0 mg, 1.5 mg, 1.8 mg, 2.5 mg and 3.5 mg) at different reaction times. | ||
Results and discussion
Initially, the typical Cs4PbBr6 samples were synthesized through our previous approach, and then simply separated by centrifugation. The resulting white precipitate was redispersed in cyclohexane for further use, as detailed in the ESI.† The transmission electron microscopy (TEM) images are shown in Fig. S2a, we observed regular and spherical nanoparticles with an average diameter of approximately 11.2 nm (Fig. S2b†). From the X-ray diffraction (XRD) pattern and distinct absorption peak in Fig. S2c and d,† the formation of 0D perovskite counterparts was further confirmed. Next, the crude solution (100 μL) was injected into 2 mL cyclohexane containing a trace of NOBF4 under vigorous stirring, and the entire phase transformation process triggered by inorganic additives was completed within 24 hours.As the additive increases (1.0, 1.5, 1.8, 2.5 and 3.5 mg), we noticed that a series of sequential spectral changes spanning the region from 300 nm to 520 nm, as depicted in Fig. 1, corresponding to the obvious change in the luminescent color under the ambient light (left) as well as 365 nm UV irradiation (right) (Fig. S1†). It was worth noting that the multiple strong absorption and emission bands observed in the evolutionary spectra were similar to those from the previous reports, which were identified as the characteristic peaks of the 0D Cs4PbBr6, Pb–Br intermediates, low-dimensional CsPbBr3 NPLs (2 ML and 3 ML) and 3D CsPbBr3 NCs.31,32 Hence, it is reasonable to speculate that these intermediate processes were closely related to the final precipitation of the brightly green-emitting MHP-NCs. In order to clarify the chemical transformation mechanism for this blue-to-green emitting perovskite analogous, we recorded the time-dependent absorption and photoluminescence (PL) spectra in detail over a long period of time. In Fig. 1a and b, it was found that the excitonic absorption band at 314 nm of Cs4PbBr6 NCs coexisted with Pb–Br intermediates (396 nm) or 2 ML anisotropic NPLs (430 nm) because the intrinsic Cs4PbBr6 NCs in the crude solution could not be completely consumed when the additive content was below 1.5 mg. As shown in Fig. 1f and g, we also monitored the 2 ML (436 nm) to 3 ML (468 nm) NPLs and 3 ML NPLs (465 nm) to 3D CsPbBr3 NCs (498 nm) transformation process in PL evolution along with the residual intermediates (436-to-468 nm and 465-to-498 nm). Especially, the characteristic peak of 3 ML NPLs within 30 minutes always appears first, that is, the long-wavelength products in the dual-emission prefer to precipitate, reflecting that the conversion of intermediates was easier than its formation.
When the additive was sufficient, the characteristic absorption peak of the original Cs4PbBr6 NCs at 314 nm and PbBr64− octahedra at 396 nm gradually disappeared accompanied by the emergence of a new peak at approximately 497 nm after 30 minutes, assigned to CsPbBr3 NCs (Fig. 1c and d). The corresponding emission shape was a single green-emitting band around 500 nm (Fig. 1h and i). It was seen that the additive amount of 1.8 to 2.5 mg was just in the range of 0D-to-3D complete conversion (Fig. 1c, d, and h, i). Notably, the minor differences in the additive content led to the final emission position shifting to a shorter wavelength (from 510 nm to 498 nm, Fig. 1h and i), which may be due to the etching effect of the nitrite ion.26,31 From Fig. 3b and d, it was found that the quantum yield (QY) of the conversion products increases dramatically within four hours and was gradually stabilized at approximately 80%. In Fig. 3c and e, the time-resolved PL measurements reflected the average lifetimes of these CsPbBr3 NCs ranging from 4.10 to 5.50 ns (Tables S1, and S2†).33,34
As the additive increases to 3.5 mg, the initially observed green light appearing within 30 minutes, rapidly diminished due to an intensified attack by the nitrite ion on the perovskite counterparts in the non-polar solvent. Meanwhile, it was noted that the non-luminescent impurities of CsBF4 and (CsF)Br2 phases were formed (Fig. S3†). Through the above analysis, we conclude that the intrinsic Cs4PbBr6 NCs underwent a dissociation–recrystallization process, where the free ions first aggregate to form the Pb–Br octahedra, and then grow from 2 ML to 3 ML and eventually into CsPbBr3 NCs, accompanied with the color evolution from deep blue (∼436 nm) to pure blue (∼468 nm) and finally green emitting (∼500 nm). The NOBF4 concentration and time-dependent chemical transformation process are summarized in Fig. 2.
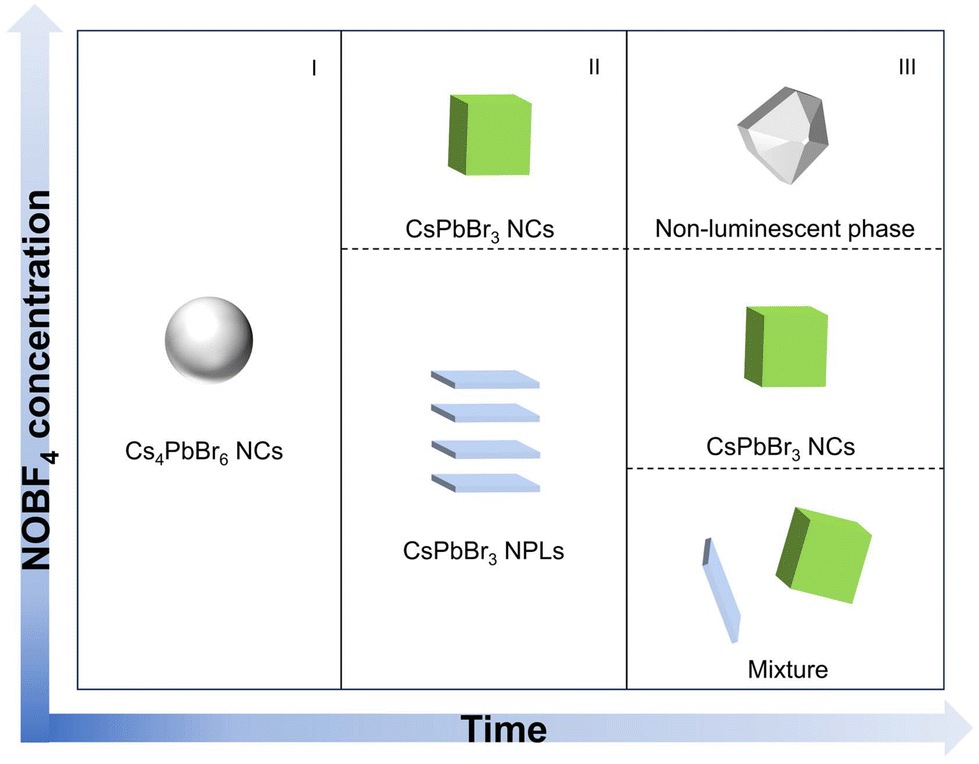 | ||
| Fig. 2 Schematic presentation of the nanostructures in the reaction medium with the reaction time and NOBF4 concentration. | ||
The XRD patterns of the perovskite counterparts obtained at 15 min, 30 min, 1 h, 4 h, 8 h and 24 h are depicted in Fig. 3a, which agreed with the hexagonal Cs4PbBr6 (blue) and orthorhombic CsPbBr3 (green) reference. As the reaction proceeded, it was noted that the 0D phase initially coexisted with the 3D phase, followed by a gradual decrease of the diffraction peak of Cs4PbBr6. The periodic diffraction peak within 30 min further confirmed the existence of 2 ML NPLs in the early stage of the reaction, along with a stacking distance of 4.6 nm and a uniform thickness of the inorganic layer at 1.1 nm (Fig. S4†). As shown in Fig. 4, transmission electron microscopy (TEM) was employed to monitor the chemical transformation process. The small particles within 30 min were proven to be completely dissociated Cs4PbBr6 nanoparticles, as described in previous reports, gradually growing into white square particles, corresponding to the increased average size from 10.9 nm to 14.9 nm (Fig. S5, and 6†). Meanwhile, the size of partially dissolved Cs4PbBr6 nanoparticles slightly decreased (from 11.2 nm to 7.9 nm), indicating that the dissolution and recrystallization processes occurred simultaneously (Fig. S7†). The phase structure of white square particles was further identified by their lattice fringe in the HRTEM image, and the spacing of 0.29 nm corresponded to the (002) plane in CsPbBr3 NCs (Fig. 4h).35 Taken together, the sequential step growth was monitored optically and microscopically, evidenced by the possible growth mechanism of bulk CsPbBr3 NCs along with their shape and phase transformation.
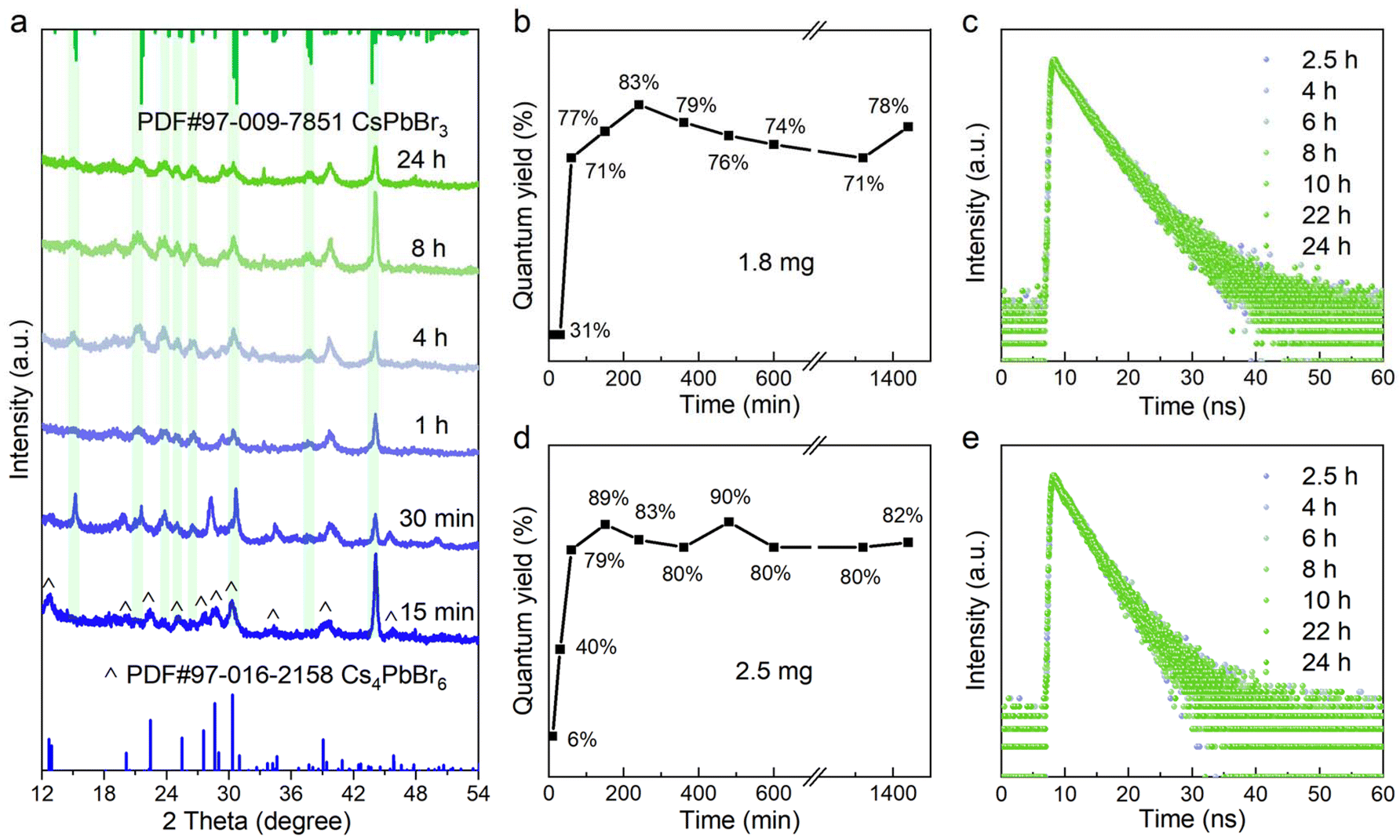 | ||
| Fig. 3 (a) XRD patterns of the luminescent products synthesized with 1.8 mg NOBF4 at different reaction times. (b and d) Quantum yield and (c and e) PL decay spectra of the luminescent products synthesized with 1.8 mg and 2.5 mg NOBF4 at different reaction times. | ||
 | ||
| Fig. 4 (a–f) TEM images of the luminescent products obtained with 1.8 mg NOBF4 at 15 min, 30 min, 1 h, 4 h, 8 h and 24 h. (g) TEM and (h) HRTEM images of the purified luminescent products obtained with 1.8 mg NOBF4. | ||
Currently, it is widely accepted that reaction temperature is crucial for the formation of anisotropic MHP-NPLs: by lowering the temperature it is possible to have an atomic-scale control over the thickness down to the few monolayers that are thin enough to exhibit the characteristics of quantum confinement.36 Therefore, the question arises, how does this additive precisely control the lateral dimension of perovskite counterparts at the atomic-scale thickness? To address this, we conducted the Fourier transform infrared (FTIR) measurements on the pristine Cs4PbBr6 treated with/without NOBF4 (Fig. 5a). After the NOBF4 treatment, it was observed that a significant reduction occurred in the characteristic C–H stretching vibrations at 2800–3000 cm−1 and 1470 cm−1, as well as the vibration of N–H bending at 1570 cm−1. The band around 1710 cm−1 was assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of OA.31 These results indicated that a substantial number of ligands bound to the Cs4PbBr6 NCs surface were effectively eliminated since the nitrite ion in the additive was reactive towards amine species. Unlike the untreated sample, a new emerging peak at 1084 cm−1, was identified to the BF4− anion.26,37 This phenomenon of BF4− anions adsorption on the surface is common in surface modification of various colloidal nanoparticles, but it was not observed in our previous studies on 2 ML blue-emitting NPLs.31 Moreover, no signal attributable to NO+ was detectable in the region from 2100 to 2200 cm−1.26,38 Therefore, we hypothesize that the surface capping properties of the sample treated with more additives were distinct from those reported previously for 2 ML NPLs.31
O stretching vibration of OA.31 These results indicated that a substantial number of ligands bound to the Cs4PbBr6 NCs surface were effectively eliminated since the nitrite ion in the additive was reactive towards amine species. Unlike the untreated sample, a new emerging peak at 1084 cm−1, was identified to the BF4− anion.26,37 This phenomenon of BF4− anions adsorption on the surface is common in surface modification of various colloidal nanoparticles, but it was not observed in our previous studies on 2 ML blue-emitting NPLs.31 Moreover, no signal attributable to NO+ was detectable in the region from 2100 to 2200 cm−1.26,38 Therefore, we hypothesize that the surface capping properties of the sample treated with more additives were distinct from those reported previously for 2 ML NPLs.31
 | ||
| Fig. 5 . (a) FTIR and (b) full XPS spectra of pristine Cs4PbBr6 NCs (0 h) and the newly formed CsPbBr3 NCs after 24 h with 1.8 mg NOBF4. High-solution XPS spectra of (c) Pb 4f, (d) Br 3d and (e) N 1s. | ||
X-ray photoelectron spectroscopy (XPS) measurement was performed to analyze the surface binding ligands. The full XPS survey spectra of Cs4PbBr6 NCs (0 h) and CsPbBr3 NCs (24 h) revealed the existence of Cs, Pb, Br, N, B and F (Fig. 5b), and the bonding information was investigated in detail by the high-resolution XPS. As shown in Fig. 5c, d and S8,† CsPbBr3 NCs gradually shifted towards higher binding energies for Cs 3d, Pb 4f and Br 3d compared with the pristine Cs4PbBr6 NCs, demonstrating the stronger Pb–Br interactions in the [PbBr6]4− octahedra. The signal of B 1s and F 1s were detected in CsPbBr3 NCs (Fig. S8†); this observation aligned with the FTIR results, implying that a ligand-exchange process occurred between the organic ligands and inorganic BF4− anions. Moreover, for the treated samples, we observed that a dominant peak at 401.6 eV coexisted with the relatively weak peak at 399.8 eV in N 1s spectra (Fig. 5e), which were associated with the protonated amines (–NH3+) and –NH2 groups of OAm, respectively.39,40 Compared to the reported organic species on the NPLs’ surface, it was noted that partial –NH3+ was reduced to –NH2 groups of OAm in the green-emitting nanocubes since the abundant nitrite ions in the system would cause a stronger surface etching for the Cs4PbBr6 nanoparticles and accelerate the fusion of NPLs through the bare-surface contact.41 Thus, we concluded that the nanoplatelet–nanocube ripening was driven by the additive-induced ligand desorption.
In previous studies, NOBF4 was employed for surface modification based on a generalized ligand-exchange strategy in semiconductor and metal NC systems, allowing surface functionalization of nanoparticles and reversible phase transfer between hydrophobic and hydrophilic media without altering their size and shape. In principle, NO+ was found to remove the native capping molecules adhered to the NC surface, and readily reacted with water molecules in the solvent to make the conditions acidic, while the weakly coordinated BF4− imparted a higher colloidal solubility for NCs in polar solvents, especially for large-sized NCs.26 For ionic MHP-NCs, it is extremely sensitive to tiny changes in the ligand species due to the significant influence of surface chemistry on the growth of nanoscale perovskite materials. Thus, the removal of amine ligands from the NC surface after adding NOBF4 causes the pristine NCs to dissociate into free ions, and the surface ligand species are likely to undergo reassignment because the acidic protons turn OAm into OAm cations, as evidenced by our previous work. Benefiting from the non-polar solvent, the intrinsic growth and highly anisotropic crystal shape of the recrystallized perovskite counterparts can be engineered precisely at the atomic-level thickness. Such surface modification mechanism is also applicable to blue-green emitting MHP-NCs having a variety of sizes and shapes in this work.
The entire transformation process can be divided into four stages, as illustrated in Fig. 6, involving (i) the pristine 0D Cs4PbBr6 NCs dissociated into free ions after adding the inorganic ligand into the non-polar solvent; (ii) a series of Pb–Br intermediates formed initially when these ions are encountered; (iii) the presence of Cs+ in the system along with the emerging protonated OAm ions were scrambling to fill the bromoplumbate ionic scaffold, accompanied by the appearance of deep-blue light with respect to the low-dimensional NPLs; (iv) as time goes on, a larger nanostructure was generated due to the NPLs losing strong quantum confinement following exposure to more additives, corresponding to slight ripening of nanoplatelets–nanocubes, that is the blue-emitting NPLs were consumed during the growth of larger and more stable cube-like particles. It is well-established that the oriented attachment often appears in a wide variety of nanomaterials, in which these NPLs with identical crystallographic facets were stacked together and subsequently assembled into larger nanostructures with lower surface energy.36,41–43 Such an attachment process is driven by the ligand desorption with respect to the poor binding organic ligands. Accordingly, it was anticipated that some factors were able to accelerate the ligand desorption and thereby ripened NPLs to bulk nanostructures, such as temperature, polar solvents and light.44–46
 | ||
| Fig. 6 Schematic illustration of the proposed transformation mechanism. | ||
Based on the above analysis, we found that the amounts of the additives are essential for the emission color of final perovskite products, as depicted in Fig. 1. Following the increased NOBF4, it was noted that the number of layers of NPLs gradually increased in a sequence and the blue-green transformation process was promoted. The multiple emission peaks indicated that small amounts of additives are likely to capture the intermediate state of partially converted products as a full coalescence of 2D NPLs difficult to access for minor NO+. Moreover, this monitored slow transition process from low-dimensional NPLs revealed the dynamic growth kinetics of the bulk nanostructured CsPbBr3 crystal. In addition, the PL properties of the green-emitting CsPbBr3 NCs dispersed in a non-polar cyclohexane system were investigated to evaluate their environmental stability. As shown in Fig. S9a,† it always maintains good monochromaticity and its luminescence position has no obvious fluctuation after 12 days of storage in ambient atmosphere, and the FWHM remained constant at 18 nm (Fig. S9b†). The luminescence intensity decreased by 20% within two weeks (Fig. S9b†), which is related to the highly dynamic binding character of surface organic species. Overall, this approach not only synthesizes highly luminescent CsPbBr3 NCs but also provides an effective pathway to understand the dynamic growth mechanism of MHP-NCs and precisely control their optical behavior.
Conclusion
In summary, we utilized the stripping effect of NO+ as a dissociating agent to induce the chemical transformation from non-luminescent Cs4PbBr6 to bright green-emitting CsPbBr3. It was found that more additives accelerated the ligand desorption of the weakly binding capping molecules and facilitated the coalescence of NPLs into cube-like nanoparticles. This phenomenon corresponds to the noticeable characteristic red-shifts in the absorption and PL spectra, portraying a comprehensive evolution in morphology and structure changes of the non-luminescent nanoparticles over 24 h. Interestingly, BF4− is also adsorbed onto the surface of the reconstructed nanocrystal to enrich its surface properties. This surface modification approach proves to be applicable to MHP-NCs, endowing them with various surface functionalizations to precisely engineer their surface properties.Author contributions
Yuling Liu and Rui Yun: conceptualization, data curation, writing – original draft and writing – review. Yue Li: investigation. Wenda Sun and Tiancheng Zheng: formal analysis. Qian Huang and Libing Zhang: supervision and funding acquisition. Xiyan Li: supervision, funding acquisition, and final writing – review. All authors approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22275101).References
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X. G. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C. J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Sero, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Perez-Prieto, L. Li;, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Muller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, State of the Art and Prospects for Halide Perovskite Nanocrystals, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed.
- R. Yun, H. Yang, W. Sun, L. Zhang, X. Liu, X. Zhang and X. Li, Recent Advances on Mn2+-Doping in Diverse Metal Halide Perovskites, Laser Photonics Rev., 2022, 17, 2200524 CrossRef.
- Q. Van Le, H. W. Jang and S. Y. Kim, Recent Advances toward High-Efficiency Halide Perovskite Light-Emitting Diodes: Review and Perspective, Small Methods, 2018, 2, 1700419 CrossRef.
- J. Chen, W. Du, J. Shi, M. Li, Y. Wang, Q. Zhang and X. Liu, Perovskite quantum dot lasers, InfoMat, 2019, 2, 170–183 CrossRef.
- P. Ramasamy, D. H. Lim, B. Kim, S. H. Lee, M. S. Lee and J. S. Lee, All-inorganic cesium lead halide perovskite nanocrystals for photodetector applications, Chem. Commun., 2016, 52, 2067 RSC.
- X. Liu, D. Yu, X. Song and H. Zeng, Metal Halide Perovskites: Synthesis, Ion Migration, and Application in Field-Effect Transistors, Small, 2018, 14, 1801460 CrossRef PubMed.
- J. Zhao, L.-W. Lo, Z. Yu and C. Wang, Handwriting of perovskite optoelectronic devices on diverse substrates, Nat. Photonics, 2023, 17, 964–971 CrossRef CAS.
- A. Liu, C. Bi, X. Qu and J. Tian, Demystifying the Formation of Colloidal Perovskite Nanocrystals via Controlling Stepwise Synthesis, J. Phys. Chem. C, 2021, 125, 14204–14211 CrossRef CAS.
- Y. Li, H. Huang, Y. Xiong, S. V. Kershaw and A. L. Rogach, Revealing the Formation Mechanism of CsPbBr3 Perovskite Nanocrystals Produced via a Slowed-Down Microwave-Assisted Synthesis, Angew. Chem., Int. Ed., 2018, 57, 5833–5837 CrossRef CAS PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, Role of Acid-Base Equilibria in the Size, Shape, and Phase Control of Cesium Lead Bromide Nanocrystals, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS PubMed.
- J. De Roo, M. Ibanez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, Highly Dynamic Ligand Binding and Light Absorption Coefficient of Cesium Lead Bromide Perovskite Nanocrystals, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Surface Chemistry Engineering of Perovskite Quantum Dots: Strategies, Applications, and Perspectives, Adv. Mater., 2022, 34, 2105958 CrossRef CAS PubMed.
- F. Haydous, J. M. Gardner and U. B. Cappel, The impact of ligands on the synthesis and application of metal halide perovskite nanocrystals, J. Mater. Chem. A, 2021, 9, 23419–23443 RSC.
- S. R. Smock, T. J. Williams and R. L. Brutchey, Quantifying the Thermodynamics of Ligand Binding to CsPbBr3 Quantum Dots, Angew. Chem., Int. Ed., 2018, 57, 11711–11715 CrossRef CAS PubMed.
- H. Yang, T. Cai, L. Dube, K. Hills-Kimball and O. Chen, Synthesis of Ultrathin Perovskite Nanowires via a Postsynthetic Transformation Reaction of Zero-Dimensional Perovskite Nanocrystals, Cryst. Growth Des., 2021, 21, 1924–1930 CrossRef CAS.
- A. Pramanik, S. Patibandla, Y. Gao, K. Gates and P. C. Ray, Water Triggered Synthesis of Highly Stable and Biocompatible 1D Nanowire, 2D Nanoplatelet, and 3D Nanocube CsPbBr3 Perovskites for Multicolor Two-Photon Cell Imaging, JACS Au, 2021, 1, 53–65 CrossRef CAS PubMed.
- S. Bera, R. K. Behera, S. D. Adhikari, A. K. Guria and N. Pradhan, Equilibriums in Formation of Lead Halide Perovskite Nanocrystals, J. Phys. Chem. Lett., 2021, 12, 11824–11833 CrossRef CAS PubMed.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, From Nonluminescent Cs4PbX6 (X = Cl, Br, I) Nanocrystals to Highly Luminescent CsPbX3 Nanocrystals: Water-Triggered Transformation through a CsX-Stripping Mechanism, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nearly Monodisperse Insulator Cs4PbX6 (X = Cl, Br, I) Nanocrystals, Their Mixed Halide Compositions, and Their Transformation into CsPbX3 Nanocrystals, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- K. Xu, Q. Wei, H. Wang, B. Yao, W. Zhou, R. Gao, H. Chen, H. Li, J. Wang and Z. Ning, The 3D-structure-mediated growth of zero-dimensional Cs4SnX6 nanocrystals, Nanoscale, 2022, 14, 2248–2255 RSC.
- D. Baranov, G. Caputo, L. Goldoni, Z. Dang, R. Scarfiello, L. De Trizio, A. Portone, F. Fabbri, A. Camposeo, D. Pisignano and L. Manna, Transforming colloidal Cs4PbBr6 nanocrystals with poly(maleic anhydride-alt-1-octadecene) into stable CsPbBr3 perovskite emitters through intermediate heterostructures, Chem. Sci., 2020, 11, 3986–3995 RSC.
- S. Paul and S. Acharya, Postsynthesis Transformation of Halide Perovskite Nanocrystals, ACS Energy Lett., 2022, 7, 2136–2155 CrossRef CAS.
- N. Pradhan, Alkylammonium Halides for Facet Reconstruction and Shape Modulation in Lead Halide Perovskite Nanocrystals, Acc. Chem. Res., 2021, 54, 1200–1208 CrossRef CAS PubMed.
- L. Ruan, W. Shen, A. Wang, A. Xiang and Z. Deng, Alkyl-Thiol Ligand-Induced Shape- and Crystalline Phase-Controlled Synthesis of Stable Perovskite-Related CsPb2Br5 Nanocrystals at Room Temperature, J. Phys. Chem. Lett., 2017, 8, 3853–3860 CrossRef CAS PubMed.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. Kazes, A Mechanistic Study of Phase Transformation in Perovskite Nanocrystals Driven by Ligand Passivation, Chem. Mater., 2017, 30, 84–93 CrossRef.
- A. Dong, X. Ye, J. Chen, Y. Kang, T. Gordon, J. M. Kikkawa and C. B. Murray, A generalized ligand-exchange strategy enabling sequential surface functionalization of colloidal nanocrystals, J. Am. Chem. Soc., 2011, 133, 998–1006 CrossRef CAS PubMed.
- X. Lai, X. Li, X. Lv, Y.-Z. Zheng, F. Meng and X. Tao, Broadband dye-sensitized upconverting nanocrystals enabled near-infrared planar perovskite solar cells, J. Power Sources, 2017, 372, 125–133 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Ultra-large-scale syntheses of monodisperse nanocrystals, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- R. Malakooti, L. Cademartiri, Y. Akçakir, S. Petrov, A. Migliori and G. A. Ozin, Shape-Controlled Bi2S3 Nanocrystals and Their Plasma Polymerization into Flexible Films, Adv. Mater., 2006, 18, 2189–2194 CrossRef CAS.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Monodisperse FePt Nanoparticles and Ferromagnetic FePt Nanocrystal Superlattices, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed.
- R. Yun, H. Yang, Y. Li, Y. Liu, Y. Chu, S. Wu, X. Liu, X. Zhang, L. Zhang and X. Li, Inorganic Ligand Triggered Transformation from Cs4PbBr6 Nanocrystals to Blue-Emitting CsPbBr3 Nanoplatelets, Chem. Mater., 2023, 35, 424–431 CrossRef CAS.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, Highly Luminescent Colloidal Nanoplates of Perovskite Cesium Lead Halide and Their Oriented Assemblies, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- H. Huang, W. Zhao, H. Yang, X. Zhang, J. Su, K. Hu, Z. Nie, Y. Li and J. Zhong, In situ synthesis of blue-emitting bromide-based perovskite nanoplatelets towards unity quantum efficiency and ultrahigh stability, J. Mater. Chem. C, 2021, 9, 5535–5543 RSC.
- J. Shamsi, D. Kubicki, M. Anaya, Y. Liu, K. Ji, K. Frohna, C. P. Grey, R. H. Friend and S. D. Stranks, Stable Hexylphosphonate-Capped Blue-Emitting Quantum-Confined CsPbBr3 Nanoplatelets, ACS Energy Lett., 2020, 5, 1900–1907 CrossRef CAS PubMed.
- X. Xiao, Y. Li and R. J. Xie, Blue-emitting and self-assembled thinner perovskite CsPbBr3 nanoplates: synthesis and formation mechanism, Nanoscale, 2020, 12, 9231–9239 RSC.
- N. T. Thanh, N. Maclean and S. Mahiddine, Mechanisms of nucleation and growth of nanoparticles in solution, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- H. D. Lutz, J. Himmrich and M. Schmidt, Lattice vibration spectra. Part LXXXVI. Infrared and Raman spectra of baryte-type TlClO4, TlBF4, and NH4BF4 single crystals and of11 B-enriched NH4BF4, J. Alloys Compd., 1996, 241, 1–9 CrossRef CAS.
- T. Gerlach, F.-W. Schütze and M. Baerns, An FTIR Study on the Mechanism of the Reaction between Nitrogen Dioxide and Propene over Acidic Mordenites, J. Catal., 1999, 185, 131–137 CrossRef CAS.
- C. Bi, Z. Yao, X. Sun, X. Wei, J. Wang and J. Tian, Perovskite Quantum Dots with Ultralow Trap Density by Acid Etching-Driven Ligand Exchange for High Luminance and Stable Pure-Blue Light-Emitting Diodes, Adv. Mater., 2021, 33, 2006722 CrossRef CAS PubMed.
- Q. Zhong, M. Cao, Y. Xu, P. Li, Y. Zhang, H. Hu, D. Yang, Y. Xu, L. Wang, Y. Li, X. Zhang and Q. Zhang, L-Type Ligand-Assisted Acid-Free Synthesis of CsPbBr3 Nanocrystals with Near-Unity Photoluminescence Quantum Yield and High Stability, Nano Lett., 2019, 19, 4151–4157 CrossRef CAS PubMed.
- H. Zhang, J. J. De Yoreo and J. F. Banfield, A Unified Description of Attachment-Based Crystal Growth, ACS Nano, 2014, 8, 6526–6530 CrossRef CAS PubMed.
- D. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Direction-Specific Interactions Control Crystal Growth by Oriented Attachment, Science, 2012, 336, 1014–1018 CrossRef CAS PubMed.
- S. Bhaumik, Oriented Attachment of Perovskite Cesium Lead Bromide Nanocrystals, ChemistrySelect, 2019, 4, 4538–4543 CrossRef CAS.
- L. Peng, A. Dutta, R. Xie, W. Yang and N. Pradhan, Dot–Wire–Platelet–Cube: Step Growth and Structural Transformations in CsPbBr3 Perovskite Nanocrystals, ACS Energy Lett., 2018, 3, 2014–2020 CrossRef CAS.
- J. Liu, K. Song, Y. Shin, X. Liu, J. Chen, K. X. Yao, J. Pan, C. Yang, J. Yin, L.-J. Xu, H. Yang, A. M. El-Zohry, B. Xin, S. Mitra, M. N. Hedhili, I. S. Roqan, O. F. Mohammed, Y. Han and O. M. Bakr, Light-Induced Self-Assembly of Cubic CsPbBr3 Perovskite Nanocrystals into Nanowires, Chem. Mater., 2019, 31, 6642–6649 CrossRef CAS.
- J. T. DuBose, A. Christy, J. Chakkamalayath and P. V. Kamat, Transformation of Perovskite Nanoplatelets to Large Nanostructures Driven by Solvent Polarity, ACS Mater. Lett., 2021, 4, 93–101 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05215j |
| ‡ Equally contributed author. |
| This journal is © The Royal Society of Chemistry 2024 |