DOI:
10.1039/D3NR05570A
(Paper)
Nanoscale, 2024,
16, 4678-4690
Nanoparticle-mediated co-delivery of inflammasome inhibitors provides protection against sepsis†
Received
2nd November 2023
, Accepted 8th January 2024
First published on 10th January 2024
Abstract
The NLRP3 inflammasome, a multiprotein complex responsible for triggering the release of pro-inflammatory cytokines, plays a crucial role in inducing the inflammatory response associated with sepsis. While small molecule inhibitors of the NLRP3 inflammasome have been investigated for sepsis management, delivering NLRP3 inhibitors has been accompanied by several challenges, primarily related to the drug formulation, delivery route, stability, and toxicity. Many existing inflammasome inhibitors either show higher liver toxicity or require a high dosage to efficiently impede the inflammasome complex assembly. Moreover, the potential synergistic effects of combining multiple inflammasome inhibitors in sepsis therapy remain largely unexplored. Therefore, a rational approach is essential for presenting the potential administration of NLRP3 small molecule inhibitors to inhibit NLRP3 inflammasome activation effectively. In this context, we present a lipid nanoparticle-based dual-drug delivery system loaded with MCC 950 and disulfiram, demonstrating markedly higher efficiency compared to an equivalent amount of free-drug combinations and individual drug nanoparticles in vitro. This combination therapy substantially improved the in vivo survival rate of mice for LPS-induced septic peritonitis. Additionally, the synergistic approach illustrated a significant reduction in the expression of active caspase-1 as well as IL-1β inhibition integral components in the NLRP3 pathway. This study underscores the importance of integrating combination therapies facilitated by nanoparticle delivery to address the limitations of small molecule inflammasome inhibitors.
Introduction
Sepsis is a complex and life-threatening medical condition resulting from an imbalance in the host immune response following an infection. It could lead to several complications such as systemic inflammation, hypotension, organ dysfunction, and, in severe cases, death.1,2 Despite the significant advancements in critical care and therapies, sepsis still poses a substantial mortality threat of over 20%, making it the foremost cause of death among ICU (intensive care unit) patients. The management of sepsis poses a formidable challenge due to its multifaceted etiology, rapid pathogenic progression, and the intricacies of achieving an optimal initial therapeutic dose as well as regimen.3–5 Recent studies and discussions within the sepsis research domain have predominantly focused on elucidating the principal determinant of sepsis survival. Central to this discourse is the ongoing debate concerning whether the greater determinant lies in innate and adaptive immune dysfunction or in the delicate equilibrium between inflammatory and anti-inflammatory processes.6,7 As inflammatory mediators, proinflammatory cytokines like tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6) have shown to cause amplified immune responses in sepsis; mitigating the excessive inflammatory response has emerged as a pivotal focal point in sepsis treatment research, as reported by numerous studies.8–10 In line with this, the NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome has been studied as a potential therapeutic target for sepsis due to its role in regulating the inflammatory response.11–13 NLRP3 inflammasome activation is a two-step process that mediates caspase-1 activation and the secretion of proinflammatory cytokines IL-1β/IL-18 in response to microbial infection and cellular damage.14–18 The literature lists several inflammasome inhibiting drugs that could help in alleviating inflammation and be utilized for treating several inflammatory diseases.19–21 However, concerns of toxicity, in vivo stability, bioavailability, and delivery represent a significant challenge while developing and using anti-inflammatory drugs, especially in treating conditions like sepsis.22–24 Therefore, given the complexity and diverse manifestations of sepsis, a multifaceted approach addressing multiple aspects of the condition is needed, in order to improve treatment outcomes.
Trials targeting single components of the inflammatory cascade have often failed to significantly reduce multiple organ injury, dysfunction or mortality rates associated with sepsis.10,12,25–27 In this context, combination therapies can offer a promising approach for targeting multiple components of the NLRP3 inflammasome pathway to treat sepsis.28–30 Also, nanoparticle-based drug delivery holds great potential for mitigating issues31–33 related to liver toxicity and high dosage34 associated with small molecule inhibitors, including those targeting the inflammasome. Nanoparticles can serve as a platform to safely package a rational combination of drugs and ensure their efficient delivery.35,36 In this manner, the combination drugs can exhibit synergistic effects by inhibiting inflammasome-associated components either simultaneously or sequentially by following horizontal or vertical inhibition. In addition, liposomes will also ensure an increased retention time of drugs and their sustained release over time, reducing the need for a repeated high dosage of free drugs and their several off-target effects.37,38 Thus, we hypothesized that co-delivery of rationally combined inflammasome-inhibiting drugs via a nanoparticle platform offers a synergistic therapy due to the enhanced retention of drugs acting simultaneously on several targets participating in the inflammasome signaling pathway. Here, we proposed to simultaneously deliver a combination of the NLRP3 complex inhibitor MCC-95039–42 and the gasdermin D inhibitor disulfiram43–46 drug using a 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipid nanoparticle37 system, which will ensure their increased solubility, enhanced bioavailability, and increased retention time in circulation.
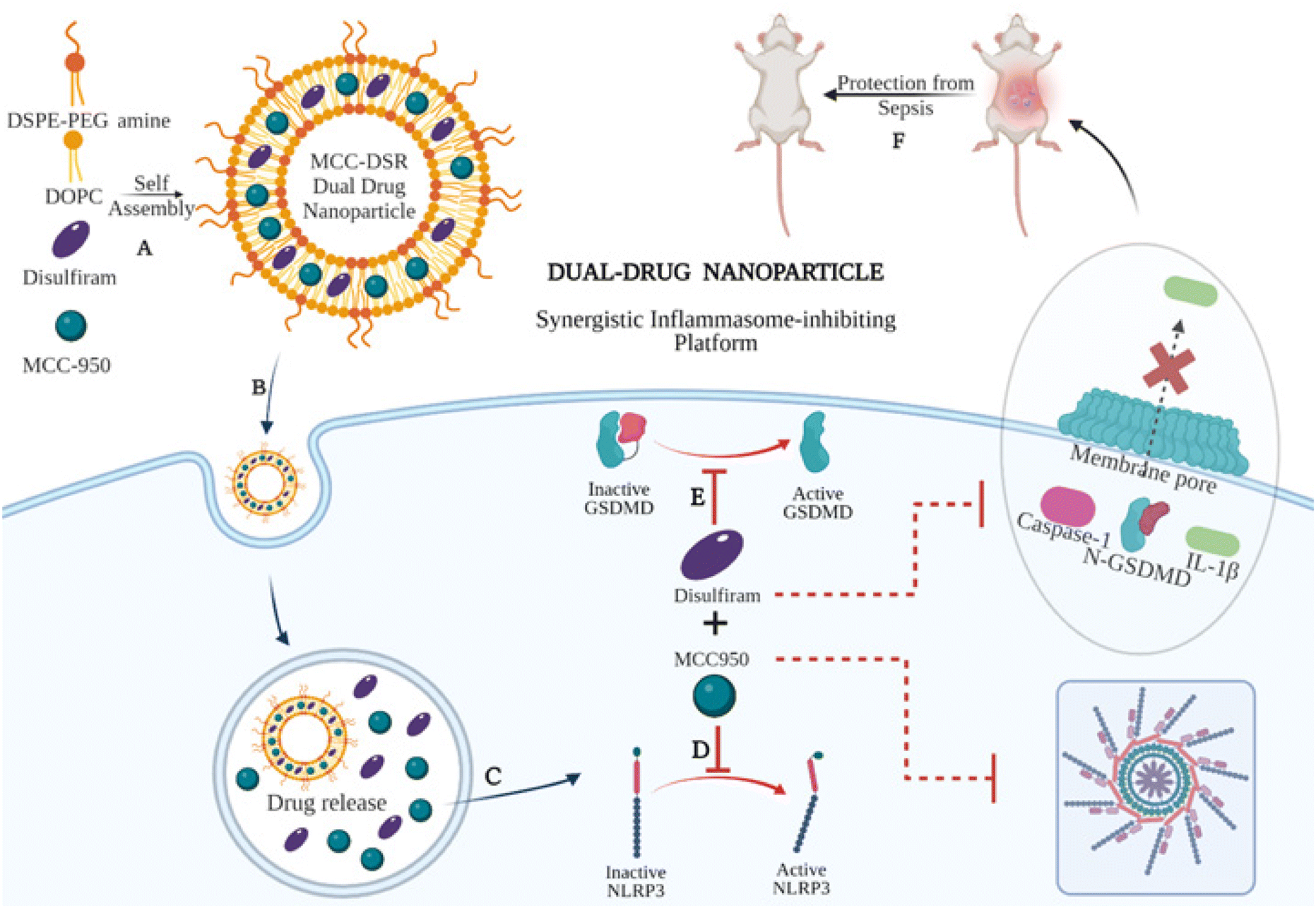
In this study, we first synthesized and characterized single-drug nanoparticle systems, MCC Nps and DSR Nps, and tested their efficiency to inhibit IL-1β along with their respective free drugs. We found single-drug nanoparticles to be as efficient as that of the respective equivalent amount of free drugs at later time points, especially after 10 h, even at a lower dose of 100 nM of drug. After identifying the optimum time points and concentrations, we next synthesized dual-drug loaded nanoparticles, MCC–DSR Nps, (Fig. 1A) and analyzed their kinetics for IL-1β inhibition in comparison to that of combination of equivalent free drugs and single-drug nanoparticles. The dual-drug nanoparticles could internalize efficiently in macrophages (Fig. 1B) and cause sustained release of both the drugs (Fig. 1C), thus making MCC–DSR Nps the most efficient platform among all other platforms after 24 h of treatment. MCC-950 prevents the NLRP3 oligomerization (Fig. 1D) and disulfiram acts on gasdermin D to prevent pore formation on the cell surface, thus inhibiting IL-1β release (Fig. 1E). These dual effects were confirmed by studying the expression of active caspase-1 and IL-1β cytokine release anticipated to exhibit a direct influence due to the activity of individual drugs. Additionally, we tested these nanoparticles in an LPS-induced peritonitis animal model to identify their in vivo efficacy and found that MCC–DSR co-encapsulated nanoparticles were able to provide complete protection against LPS-mediated sepsis through their synergistic response (Fig. 1F).
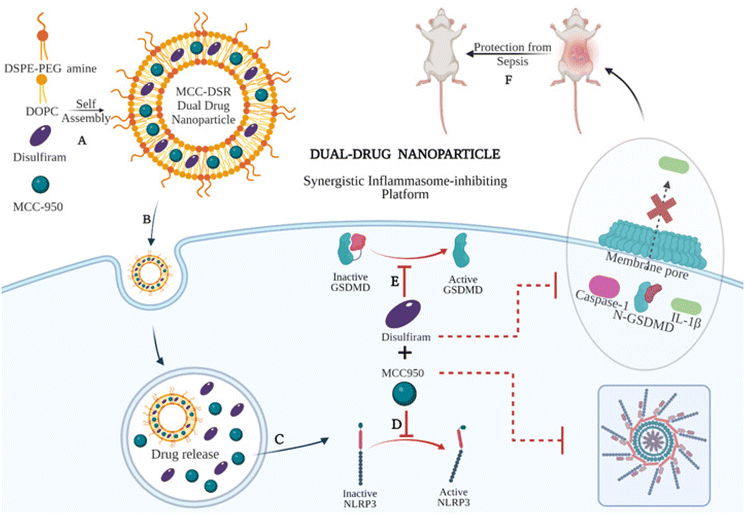 |
| | Fig. 1 Schematic showing the synergistic therapeutic platform for dual-drug nanoparticles. (A) Self assembly of DSPE-PEG amine, DOPC lipids along with MCC-950 (NLRP3 inhibitor) and disulfiram (gasdermin D inhibitor) for the preparation of the dual-drug lipid nanoparticle system. (B) Internalization of MCC–DSR Nps in macrophages. (C) Release of the drugs, MCC-950 and disulfiram, inside the endosome. (D) MCC-950 binds to NLRP3 and prevents its oligomerization and thus inhibits the activation of the NLRP3 inflammasome complex. (E) Disulfiram blocks the activation of gasdermin D, which prevents the pore formation and the subsequent release of cytokines outside the cell. (F) The dual-drug nanoparticle system provides protection against LPS-induced sepsis in mice. | |
Results and discussion
Synthesis and characterization of single- and dual-inflammasome-inhibiting drug nanoparticles
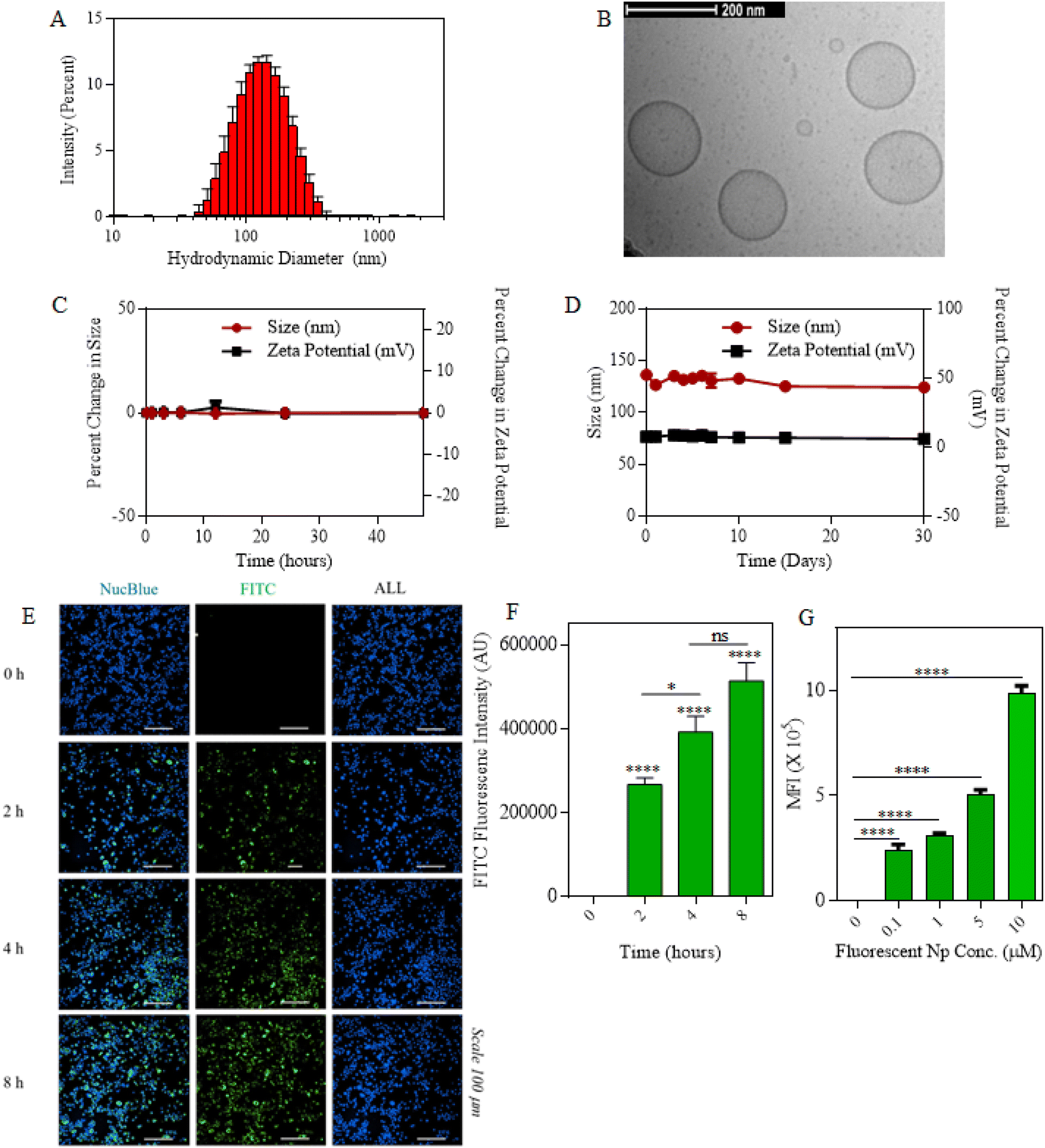
We first synthesized single- and dual-drug nanoparticles by utilizing the co-lipid nanoparticle backbone obtained from 60 mole percentage of DOPC lipid (1,2-dioleoyl-sn-glycero-3-phosphocholine) and 30 mole percentage of DSPE-PEG (2000) amine (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol)-2000]). The co-lipid bilayer was then physically encapsulated with either MCC-950 or/and disulfiram (DSR) inflammasome inhibiting drugs, accounting for the remaining 10 mole percentage either individually or in combination in order to form single- or dual-drug nanoparticles. Fig. S1† presents the characterization of the size and stability of MCC950/DSR single-drug Nps. For MCC–DSR Nps, both drugs were encapsulated in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with each contributing towards 5 mole percentage. All the nanoparticles were synthesized by the thin lipid film hydration method,47 where the thin lipid film obtained via rotavap was hydrated in PBS for 1.5 h at 60 °C so as to allow their self-assembling into a lipid bilayer along with entrapment of the drugs.48 The encapsulation efficiency of disulfiram accounted for around 60%, whereas that of MCC was around 20%.49 All the drug nanoparticles; DSR Nps, MCC Nps and MCC–DSR Nps were stably formed and had a hydrodynamic diameter less than 200 nm. Fig. 2A shows the hydrodynamic diameter of MCC–DSR Nps to be around 150 nm, obtained via dynamic light scattering using a Malvern Nano Zetasizer. The size and morphology of these liposomes were also confirmed using cryo-TEM as shown in Fig. 2B. These dual-drug nanoparticles were found to be stable in 10% human serum for about 48 h with almost no change in size and zeta potential (Fig. 2C). They were also stable under the storage conditions of PBS at 4 °C for about a month with their size maintained at almost 150 nm and neutral surface charge (Fig. 2D). Next, we determined the macrophage internalization of these particles by encapsulating FITC at 5 mole percentage to obtain fluorescent nanoparticles detectable by microscopy and flow cytometry. Fig. 2E presents the microscopic images of the time-dependent cellular uptake of FITC Nps from 0 h to 8 h. Fig. 2F displays the quantification of FITC nanoparticle internalization at different time points using microscopy and it shows that the internalization increased with higher time points, with 4–8 hours showing sufficient internalization for later studies. Fig. 2G plots the concentration-dependent internalization of these fluorescent particles, showing increased uptake with higher concentration. Overall, these results show that the drug-encapsulating liposomes were stably synthesized and successfully internalized by immortalized macrophages in a concentration- and time-dependent manner.
:1 with each contributing towards 5 mole percentage. All the nanoparticles were synthesized by the thin lipid film hydration method,47 where the thin lipid film obtained via rotavap was hydrated in PBS for 1.5 h at 60 °C so as to allow their self-assembling into a lipid bilayer along with entrapment of the drugs.48 The encapsulation efficiency of disulfiram accounted for around 60%, whereas that of MCC was around 20%.49 All the drug nanoparticles; DSR Nps, MCC Nps and MCC–DSR Nps were stably formed and had a hydrodynamic diameter less than 200 nm. Fig. 2A shows the hydrodynamic diameter of MCC–DSR Nps to be around 150 nm, obtained via dynamic light scattering using a Malvern Nano Zetasizer. The size and morphology of these liposomes were also confirmed using cryo-TEM as shown in Fig. 2B. These dual-drug nanoparticles were found to be stable in 10% human serum for about 48 h with almost no change in size and zeta potential (Fig. 2C). They were also stable under the storage conditions of PBS at 4 °C for about a month with their size maintained at almost 150 nm and neutral surface charge (Fig. 2D). Next, we determined the macrophage internalization of these particles by encapsulating FITC at 5 mole percentage to obtain fluorescent nanoparticles detectable by microscopy and flow cytometry. Fig. 2E presents the microscopic images of the time-dependent cellular uptake of FITC Nps from 0 h to 8 h. Fig. 2F displays the quantification of FITC nanoparticle internalization at different time points using microscopy and it shows that the internalization increased with higher time points, with 4–8 hours showing sufficient internalization for later studies. Fig. 2G plots the concentration-dependent internalization of these fluorescent particles, showing increased uptake with higher concentration. Overall, these results show that the drug-encapsulating liposomes were stably synthesized and successfully internalized by immortalized macrophages in a concentration- and time-dependent manner.
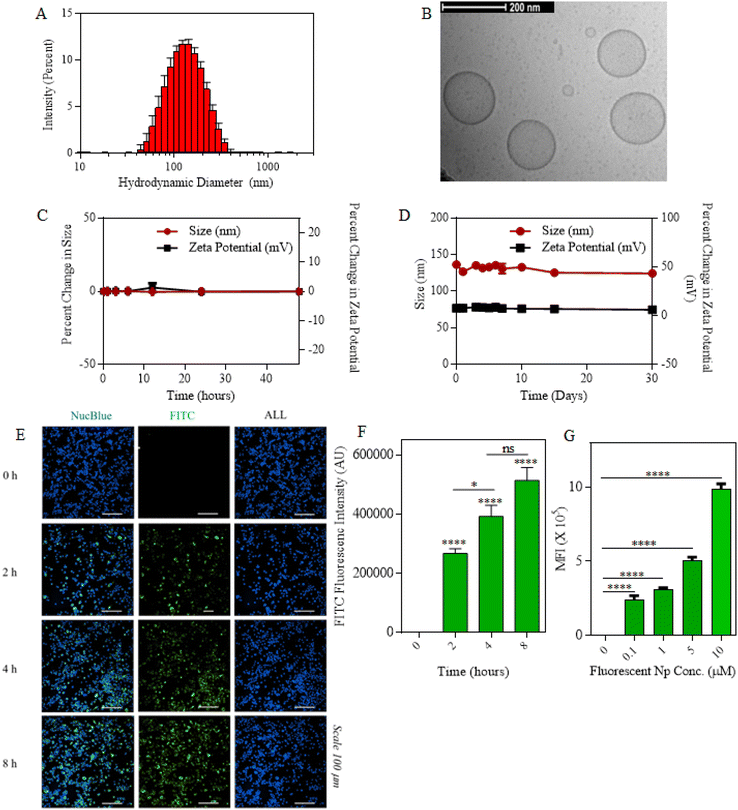 |
| | Fig. 2 Characterization and internalization of MCC–DSR Nps. (A) Graph plots of the hydrodynamic diameters of MCC–DSR dual-drug nanoparticles. Data are shown as the mean ± S.D. (n = 3). (B) Cryo-TEM image of dual-drug nanoparticles. Scale bar: 200 nm. (C) Graph displays the percentage of change in the size and zeta potential of MCC–DSR dual-Nps incubated in human serum for a total duration of 48hours. (D) PBS stability plot for the size and zeta potential of MCC–DSR Nps in PBS over a period of 30 days. Data shown in c and d are the mean ± S.E.M. (n = 3). (E) Representative microscopy images of iBMDMs internalized with FITC-encapsulated fluorescent particles in a time-dependent manner from 0 h to 8 h. Nuclei were stained with NucBlue. Scale bar: 100 μm. (F) Quantitative analysis for the cellular uptake of fluorescent nanoparticles imaged via confocal microscopy. (G) Flow cytometry analysis of the cellular uptake of different concentrations of FAM particles using iBMDMs. Data are shown as the mean ± S.E.M. (n = 3). Statistical analysis was performed by one-way ANOVA and Dunnett's multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001. | |
Single-drug nanoparticles display efficient IL-1β inhibition as compared to free drugs overtime at different concentrations
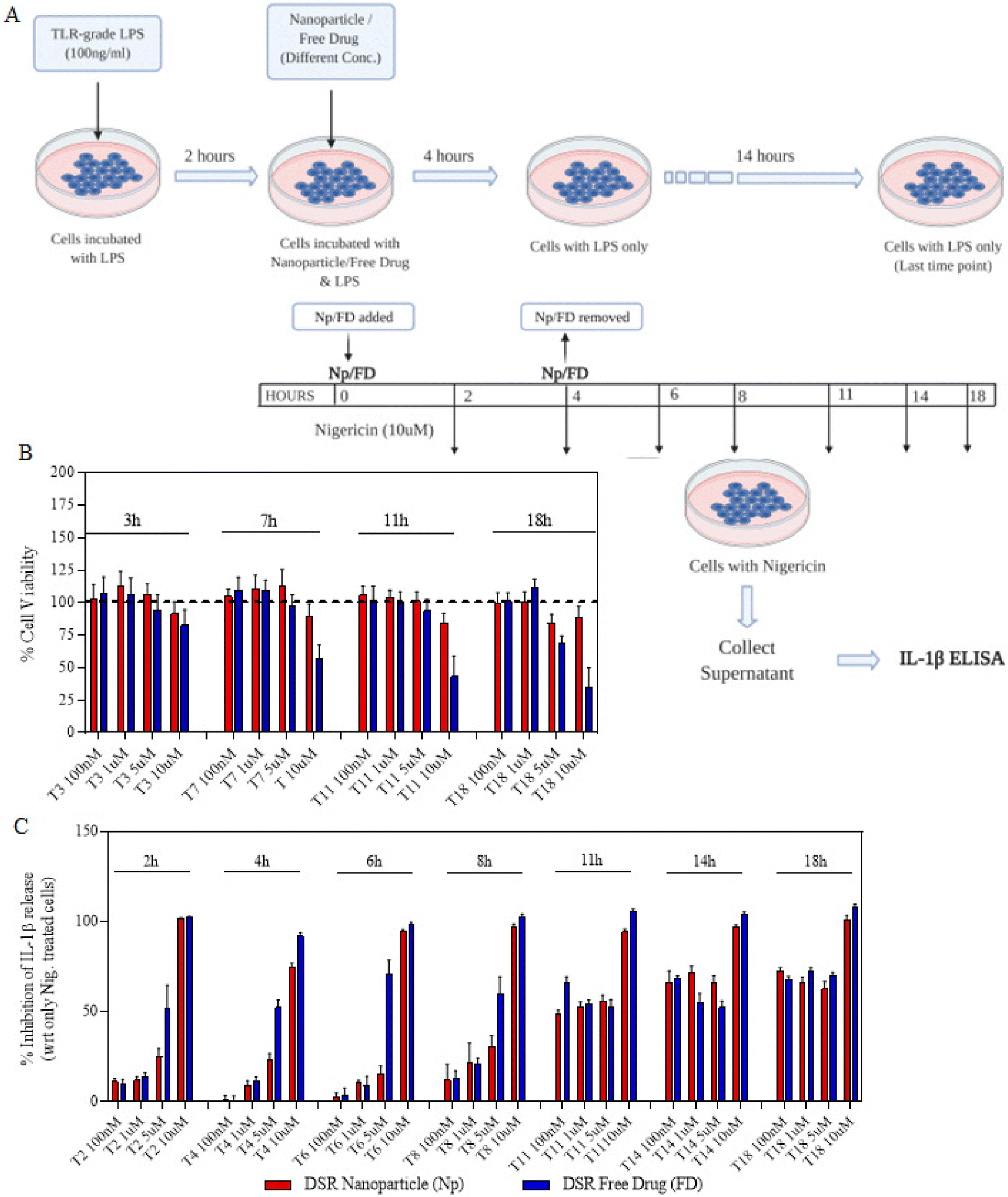
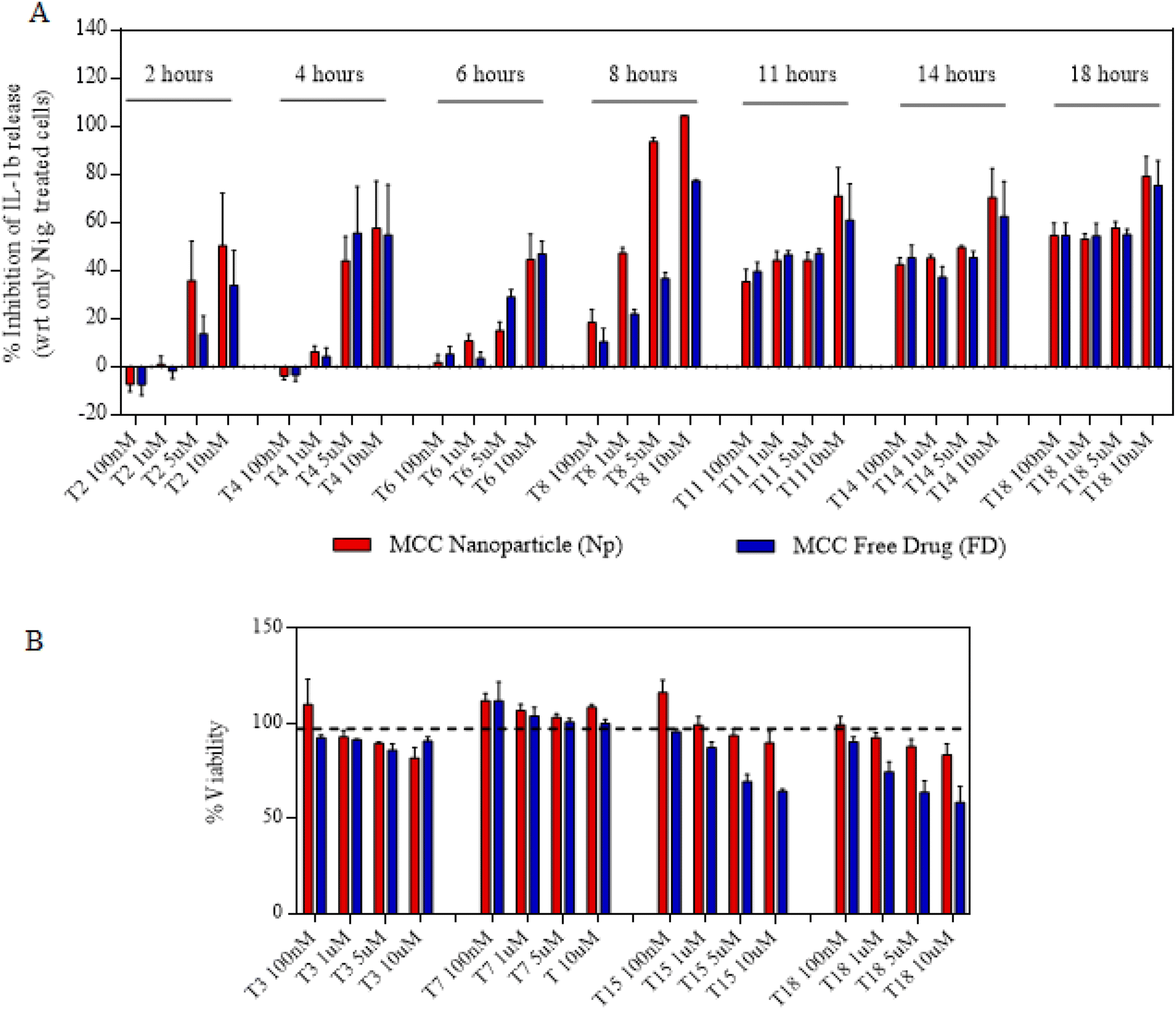
We next proceeded towards examining if the nanoparticles are as efficacious as that of free drugs and their kinetics as well as response at different time points and concentrations. In order to do that, we first primed the immortalized macrophages for 2 h and then treated them with either free drugs or nanoparticles for a maximum of 4 h (Fig. 3A). After 4 h, the cells were replaced with fresh LPS media and allowed to incubate for the next 14 h. This treatment was further proceeded towards signal 2 nigericin at different time points starting from 0 h to 18 h immediately after the LPS priming. The indicated time points of 0, 2, 4, 6, 8, 11, 14 and 18 represent the total nanoparticle treatment and incubation time points at which nigericin was added to different wells in order to activate the inflammasome. The assays were performed such that all the different time point treatments end at the same time so as to add nigericin to all the wells exactly at one time point. For this, the initial set up or treatments were performed at different time points so as to ensure these variations as mentioned above. After 1 h of nigericin treatment, the supernatant was collected and tested for IL-1β levels using ELISA. Before nigericin treatment, the cells were subjected to the MTT assay so as to determine the cytotoxicity of the nanoparticles and free drugs in order to determine if the IL-1β inhibition is solely via the drug's activity. Both Fig. 3C and4A show that at later time points, even 100 nM of nanoparticles show a similar response to that of free drugs. Even if at initial time points free drugs seem to be more efficacious in IL-1β inhibition at certain concentrations, there is no significant difference in the activity at later time points. Moreover, Fig. 3B and 4B show that free drugs are toxic at higher concentration and later time points in the case of both MCC-950 and DSR, whereas all the nanoparticles maintain the cell viability of 80–90%, which makes them more efficient than free drugs due to the sustained release of drugs, thus reducing their toxicity. After determining the single-drug nanoparticle efficacy over the respective free drug, we next proceeded towards examining the activity of dual-drug nanoparticles.
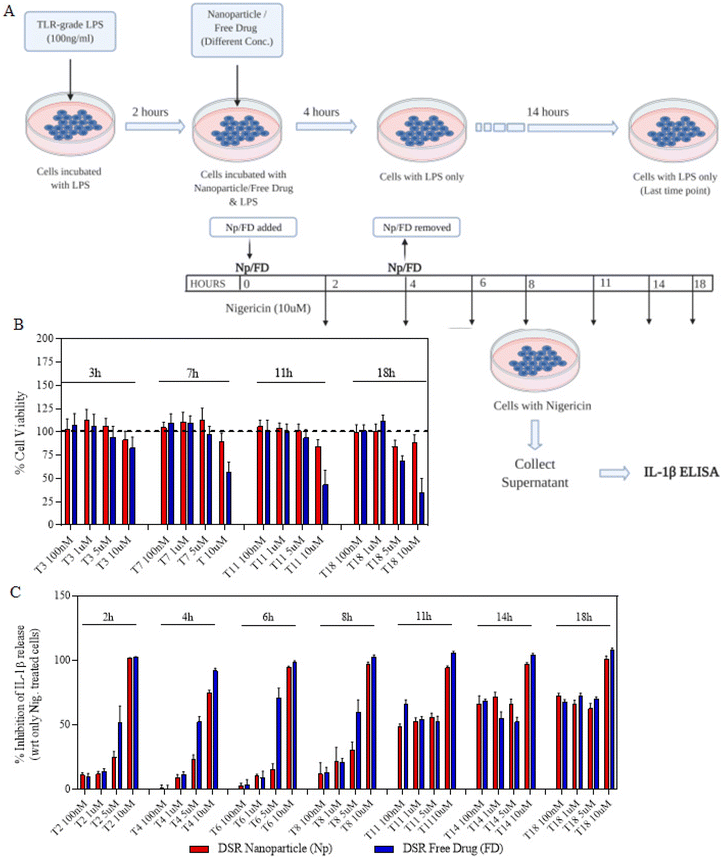 |
| | Fig. 3 Activity and cytotoxicity of DSR Nps. (A) Schematic illustration of sequence and time points for different treatments of a single drug or nanoparticles; including LPS, nanoparticles or free drugs, followed by nigericin. (B) Percentages of cell viability of the cells treated with different concentrations of DSR Nps or free drugs for different time durations. (C) Concentration and time-wise percentage inhibition in IL-1β stimulation after treating the cells with either DSR Nps or DSR-free drugs. Data shown in (B) and (C) are the mean ± S.E.M. (n = 3). | |
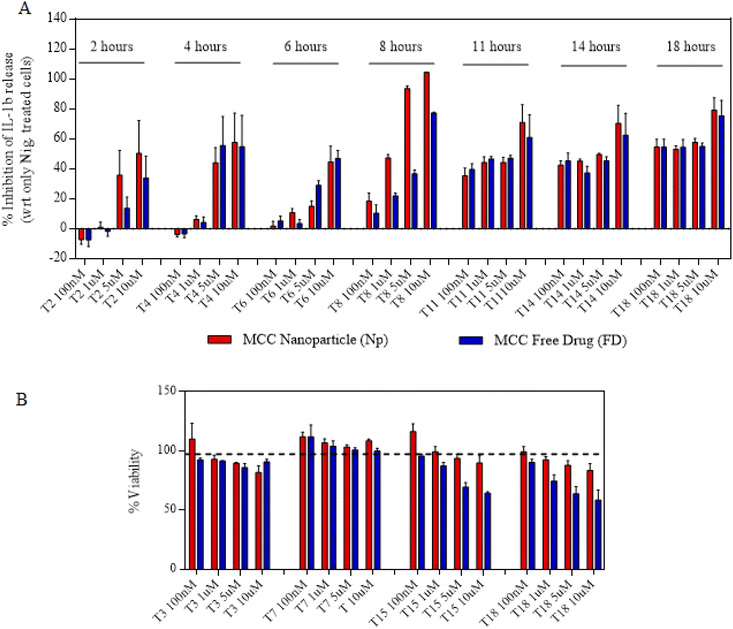 |
| | Fig. 4 Activity and cytotoxicity of MCC Nps. (A) Concentration and time-wise percentage of inhibition in IL-1β stimulation after treating the cells with either MCC Nps or MCC-free drugs. (B) Percentages of cell viability of the cells treated with different concentrations of MCC Nps or free drugs for different time durations. Data shown in (A) and (B) are the mean ± S.E.M. (n = 3). | |
Dual-drug nanoparticles show a synergistic inhibition in IL-1β release in nigericin-treated iBMDMs
Next, we examined the efficacy of MCC–DSR dual-Nps with respect to the dual-free drug and single-drug Nps at different time points ranging from 4 h to 36 h. The time points represent the total time of 4 h nanoparticle treatment and incubation afterwards. For example, 8 h time point means that the cells were incubated with nanoparticles or free drugs for 4 h and then incubated in LPS media for another 4 h. Similarly, 36 h represents 4 h nanoparticle or free drug treatment and 32 h of incubation in LPS media for sustained release. We utilized 344 nM of MCC950 and 1 μM of DSR for all our in vitro experiments by keeping these doses consistent in all the platforms, including the dual-Nps, single-drug Nps and via free drugs. Fig. 5A plots the percentage inhibition in IL-1β released by different treatment groups with respect to the nigericin-treated cells. It shows that even though there is not a significant difference among all the treatment groups until 20 h, MCC–DSR Nps show greater inhibition in IL-1β starting from 24 h. At 36 h, dual-Nps show significantly higher response shown by inhibiting IL-1β release, followed by MCC Nps, dual-free drugs and DSR Nps, thus pointing towards the greater efficacy of the dual-drug nanoparticles due to synergistic properties of both the drugs. To reconfirm this response, we next evaluated the expression of different inflammasome-associated proteins50 in the cells treated with different free drugs and nanoparticles for 24 h (4 h treatment and 20 h incubation) as shown in Fig. 5B using western blotting. We determined the protein expression in both the cell lysate and supernatant. We observed no significant difference in pro-caspase-1 expression in the cell lysate, but in the supernatant both MCC–DSR Nps and MCC Nps showed significant inhibition in active capsase-1 expression, confirming their better efficacy than the respective free-drug treatments. Additionally, we observed a remarkable inhibition in active GSDMD released into the supernatant of MCC FD-, MCC Np- and MCC–DSR Np-treated cells, again proving their efficiency in inflammasome inhibition. However, we were not able to observe much difference in DSR Nps but were able to observe some inhibition in DSR FD, which might be due to the particular time point we are using for western blotting. We also imaged and quantified the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) speck formation in iBMDMs, which can be used as a simple upstream readout for inflammasome activation.49,51–55 The minimal speck generation upon dual-drug Np treatment (Fig. S4A and S4B†) indicated that the ASC proteins are dispersed in the cytosol and thus correlated with lower inflammasome activation. This further corroborates our previous findings by exhibiting a significant decrease in NLRP3 inflammasome activation due to the combined effect of the dual-drug nanoparticles.
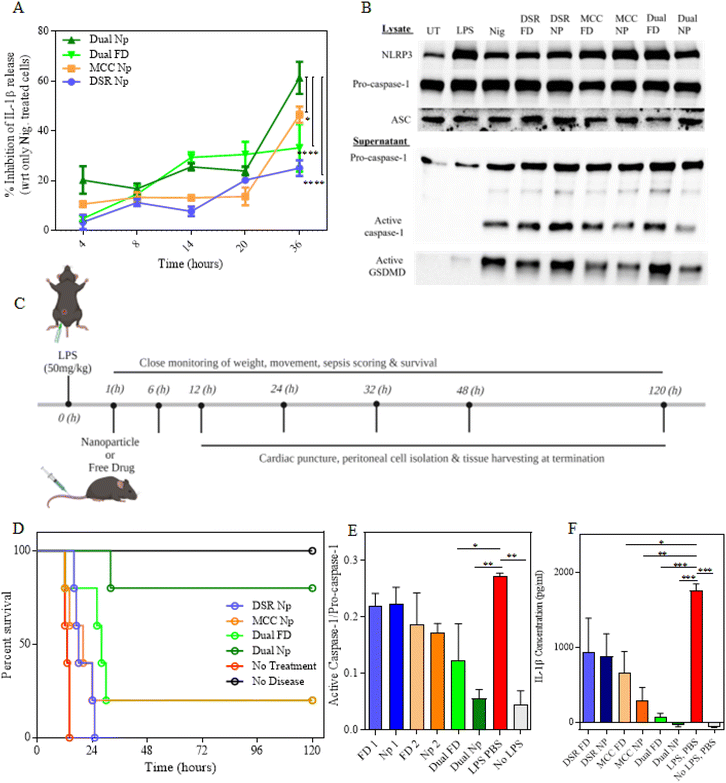 |
| | Fig. 5
In vitro and In vivo efficacy of dual-drug MCC–DSR Nps. (A) Graph plots of the percentage of IL-1β inhibition obtained from indicated treatment groups, including dual-Nps, dual-FD, MCC Nps and DSR Nps. Data shown are the mean ± S.E.M. (n = 3). Statistical analysis was performed by two-way ANOVA and Dunnett's multiple comparison test. *p < 0.05, ***p < 0.001. (B) Representative western blotting images of different treatment groups exhibiting the expression of NLRP3 signaling-associated proteins in the cell lysate and supernatant. (C) Schematics representing the in vivo animal trial timeline, displaying all the time points and sequence of different treatments and related assays. (D) Graph plots of the survival curves of indicated treatment groups for a duration of 120 h. (E) Graph plots of the quantitation of the expression of active caspase-1 normalized to pro-caspase-1 observed via simple protein Wes analysis. Data shown are the mean ± S.E.M. (n = 3). Statistical analysis was performed by ordinary one-way ANOVA and Dunnett's multiple comparison test. *p < 0.05, **p < 0.01. (F) Graph plots of serum IL-1β cytokine levels in different treatment groups. Data shown are the mean ± S.E.M. (n = 3). Statistical analysis was performed by ordinary one-way ANOVA and Dunnett's multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001. | |
Dual-drug nanoparticles induce complete protection against LPS-induced sepsis
We next examined the in vivo efficacy of dual-drug nanoparticles in an LPS peritonitis sepsis model. For this, the mice were intraperitoneally injected with 50 mg kg−1 LPS to induce sepsis systemically (Fig. 5C). After 1 h of LPS injection, these mice were intravenously injected with either free drugs or nanoparticles and then closely monitored for survival. The doses for MCC950 were 0.4 mg per mouse (∼2 mg kg−1) and that of DSR was 0.8 mg per mouse (∼4 mg kg−1), delivered either via nanoparticles or as free drugs. Fig. 5D shows the survival curve for all the treatment groups. We observed that all the mice without any treatment died within 12 h to 18 h; however, the survival was extended to 24 h when the mice were treated with DSR Nps. Moreover, one out of five mice managed to survive and recover in both the treatments with MCC Nps as well as dual-FD, with dual-FD showing enhanced survival for other mice when compared with just MCC Nps. The dual-Nps showed the maximum survival out of all the treatment groups where four out of five mice survived and fully recovered until the later time point, 120 h. On day 5, all these four mice completely recovered from septic shock and were able to regain their normal weight and movement. All the mice were subjected to peritoneal cell isolation, cardiac puncture and tissue harvesting after their sacrifice. All the mice were closely monitored for their posture, mobility, appearance, and weight loss; and they were all humanely sacrificed as per IACUC guidelines. After sacrificing the mice, the heart, lungs, liver, kidneys, and spleen were removed and sectioned for hematoxylin & eosin (H&E) staining to assess treatment biosafety as well as the presence of peritonitis from the kidney and spleen samples. Fig. S5† shows the H&E biosafety results, exhibiting a completely normal histology in PBS without the LPS group, as well as the MCC FD and Np groups. The other treatment groups also exhibited a healthy histology but with some interesting findings. For example, for the PBS LPS, DSR FD, and DSR Np groups, there was a high presence of neutrophils in the lungs but only in the surrounding tissue, not in the alveolar sacks, meaning that there is no evidence of pneumonia which could result in lung inflammation. In the dual-Np group and slightly less prevalent in the dual-FD group, the spleen exhibited a blue follicle surrounded by normal red tissues with some acute immune cell infiltration (blue spots), indicating the tail-end of an ameliorating inflammatory response. However, in total, the H&E biosafety results exhibit largely normal tissue morphologies and do not provide evidence for any significant toxicities from the treatment groups.
The recovered mice were sacrificed on day 5. The isolated peritoneal cells were immediately lysed using RIPA buffer enriched with 1× protease inhibitor and subjected to protein isolation. Later, the protein was quantified using the BCA assay and an equal amount of protein was loaded and quantified using protein simple Wes. Fig. 5E shows the protein expression of p20 caspase-1 normalized to pro-caspase-1 in the peritoneal cells isolated from the mice treated with the indicated free drugs and nanoparticles. The graph shows significantly higher activation of the caspase-1 enzyme in the LPS-treated mice without any drug treatment. This expression is inhibited by around 40% in the MCC + DSR-free drug treatment group and 80% in the mice treated with MCC–DSR Nps, which is remarkably greater than the response obtained from single-drug treatments. Moreover, the dual-drug liposome treatment group showed almost equivalent levels of active capsase-1 as that of the no disease control mice (without any LPS injection), proving that these mice were fully recovered from sepsis. We further examined all the treatment groups for serum IL-1β levels and observed a significantly higher level of cytokine (∼1800 pg ml−1) in LPS-injected mice without any drug treatment (Fig. 5F). These were sequentially inhibited by treatment with different free drugs and nanoparticles, with a 20-fold reduction upon treating the mice with the dual-drug Nps, showing no serum IL-1β similar to that of no disease control. Additionally, the dual-Nps also showed a significant reduction in IL-6 secretion (Fig. S2†) without a consequential change in TNFα release (Fig. S3†), suggesting their potential efficacy in the simultaneous blockade of multiple cytokines, thereby mitigating the cytokine storm often accompanied by sepsis. We also performed sectioning of peritoneal lining tissues to assess the presence of peritonitis by H&E staining and assessment by pathologists. Fig. S6† depicts selected H&E stains from the sections taken from the peritonitis-positive mainly splenic mice as well as some liver peritoneum of the septic mice, shown in 10× and 40× magnifications to depict the characteristics of immune cell infiltration in peritonitis. The H&Es from each sample were also pathologically assessed for peritonitis in both spleen/kidney sections (Fig. S6A†), which serve as the most representative for peritoneal lining sections, and in the heart/lungs/liver (Fig. S7B†) of the septic mice tissues. Of particular note is the presence of acute or chronic peritonitis in all groups of the spleen/kidney samples, but no presence of peritonitis is detected in the PBS without LPS group (negative control), as well as the dual-FD sample II and dual-Np groups, suggesting the amelioration of sepsis and peritonitis in these tissues, providing further evidence for the effectiveness of the MCC–DSR drug combo, including in the nanoparticle system. Overall, all the in vivo and ex vivo assays support the fact that MCC–DSR Nps enable a synergistic response, which offers complete protection against LPS-mediated sepsis.
Conclusions
This study successfully demonstrated the stable synthesis of MCC–DSR dual-drug nanoparticles of less than 200 nm hydrodynamic diameter and neutral surface charge. These nanoparticles showed remarkable stability in PBS for about 30 days and in serum for about 2 days. The combination nanoparticles displayed a significantly higher efficiency than an equivalent amount of free-drug combinations as well as individual-drug nanoparticles in vitro, as evidenced by significant IL-1β inhibition after 24 h of total treatment. Western blot analysis further corroborated these results, revealing a substantial inhibition in active caspase-1 and active GSDMD expression after 24 h (4 h treatment and 20 h incubation) dual-nanoparticle treatment in contrast to no inhibition after free-drug treatment. Notably, even a mere 1 μM of DSR was able to induce sufficient inhibition when delivered along with 344 nM of MCC due to the synergistic response. This dose is around 10–20 times lower than the previously employed doses of DSR-free drugs to achieve in vitro pyroptosis inhibition. Furthermore, our study unveiled a remarkably high in vivo efficiency in an LPS-mediated sepsis model, with dual-drug Nps inducing a complete protection against sepsis even when exposed to a very high LPS dose of 50 mg kg−1. The co-delivery of MCC and DSR via combination nanoparticles not only greatly enhanced the survival rates of the mice, but also facilitated their recovery from severe septic shock to completely normal states. This response not only outperforms other platforms tested simultaneously in this study, but also surpasses the efficacy of high doses of free drugs in previous studies.28,29 In this study, we employed doses of the GSDMD inhibitor and the NLRP3 inhibitor that were 3–10 times lower than those in previous studies, achieving superior results compared to individual drug treeatments.30–32 In conclusion, our results establish that this platform not only offers a synergistic inflammasome inhibitory response, which greatly reduces the required individual drug doses, but also provides a safe delivery system, thereby mitigating toxicity and enabling sustained release overtime, and ultimately enhancing drug retention. This study serves as a proof of principle for combinatorial approaches that have the potential to overcome the existing limitations of inflammasome-inhibiting drugs, which have thus far encountered barriers to clinical translation.
Materials and methods
Materials
All the reagents were procured from commercial suppliers including ThermoFisher, InvivoGen, Tocris, Avanti, Adipogen, Biolegend, BioRad, Cell Signaling Technology, Biorad and Sigma Aldrich. Nanoparticle components, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, Cat# 850375P) and 3-phosphoethanolamine-N-[amine (polyethylene glycol)-2000] (DSPE-PEG 2000 amine, Cat# 880128P) were purchased from Avanti Polar Lipids. The disulfiram drug, also known as bis(diethylthiocarbamyl) disulfide (Cat# 97-77-8), was obtained from Millipore Sigma. MCC-950 sodium (CP-456773 Sodium) was purchased from Selleckchem. For cell-based assays, all the immortalized bone-marrow-derived macrophages (iBMDMs) including the cell lines expressing ASC-citrine were generously gifted by Kate Fitzgerald, UMass Chan Medical School, Worcester. Cell culture medium components such as Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and penicillin–strep. were ordered from Gibco Life Technologies. For inflammasome stimulation, ultrapure lipopolysaccharide from Escherichia coli (Ultrapure LPS, E. coli 0111:B4, Cat# tlrl-3pelps) and nigericin sodium salt (Cat# 431210) were obtained from InvivoGen and Tocris Bioscience, respectively. All the western primary and secondary antibodies were procured from Adipogen Life Sciences, Biolegend, Abcam, Santa Cruz Biotechnology and Cell Signaling Technology. Other reagents like RIPA buffer, NP-40 lysis buffer, HALT protease, phosphatase single-use EDTA-free 100× cocktail, J60015 4× Laemmli SDS reducing sample buffer, NucBlue live ready probes reagent (Hoechst 33342), and LysoTracker Red DND-99 were purchased from ThermoFisher Scientific. Moreover, some kits like IL-1β mouse uncoated ELISA and Pierce BCA protein assay were also obtained from ThermoFisher Scientific. Wes reagents were procured from protein simple.
Detailed methods
Synthesis of MCC Nps, DSR Nps and MCC–DSR Nps.
Both the single- and dual-drug particles were synthesized using two lipids, DOPC {1,2-dioleoyl-sn-glycero-3-phosphocholine} and DSPE-PEG (2000) amine 204 at 60 mol percentage and 30 mol percentage, respectively, by the thin lipid film hydration method. Individual or a combination of two drugs were physically encapsulated, accounting for a total of 10 mol percentage. While preparing the thin film, both the co-lipids and disulfiram were dissolved in dichloromethane, whereas MCC-950 was dissolved in methanol. The film was obtained in a round bottom flask using a rotavapor, which was then hydrated in PBS for 1.5 h at 60 °C to obtain drug-encapsulated self-assembled liposomes. The unencapsulated free drug was filtered using a G-25 Sephadex column. Later, the eluted liposomes were extruded using a 200 nm polycarbonate filter membrane to obtain nanoparticles of desired size. The drug encapsulation efficiency was evaluated using UV-Vis spectrophotometry, and the size and zeta potential were determined using a Malvern Zetasizer Nano ZS90.
Characterization of nanoparticles
For stability studies, the nanoparticles were incubated either in PBS at 4 °C or in 10% human serum at room temperature for an extended duration of time. PBS stability was used for storage conditions, whereas serum stability was used for physiological conditions. While incubating in the respective buffers, the nanoparticles were assessed for their size and surface charge at indicated time points. Additionally, their morphology and size were confirmed via cryo-TEM performed by the UMASS Chan Medical School Core Facility.
Cellular uptake of FITC-encapsulated nanoparticles
For internalization studies, nanoparticles were encapsulated with FITC at 5 mol percentage to obtain fluorescent nanoparticles for internalization studies. For concentration dependent studies, the immortalized macrophages were incubated with fluorescent nanoparticles ranging from 0.1 to 10 mM for 4 hours, and the fluorescence intensity was recorded using flow cytometry. For time-dependent studies, the cells were incubated with fluorescent nanoparticles for different time points of 0 h, 2 h, 4 h and 8 h, followed by NucBlue staining in order to stain the nuclei. Furthermore, the cells were imaged using a Crest V2 spinning disk and the fluorescence intensity was measured using an NIS Elements V6.2.
Cell culture, NLRP3 stimulation and drug treatment
Immortalized macrophages were utilized for all the in vitro assays. They were cultured in complete DMEM containing 10% FBS and 1% pen–strep. For NLRP3 stimulation, the cells were first primed using ultrapure LPS for 2 h in order to activate signal 1 and then they were treated with 10 mM nigericin for ½ to 1 h for signal 2. Free drug or nanoparticle treatments were performed in between signal 1 and signal 2 for indicated time points.
LPS-induced peritonitis sepsis model
All the animal protocols were approved by IACUC (Institutional Animal Care Use Committees) at the University of Massachusetts, Amherst. Female C57BL/6 mice of age 6–8 weeks were procured from Charles River and housed in a pathogen-free environment under 12 h light or dark cycles. For establishing the LPS peritonitis sepsis model, the mice were first injected with pure LPS intraperitoneally. This was followed by intravenous I.V. injection of nanoparticles or free drugs after 1 h of LPS injection. For injections, the nanoparticles and MCC-950-free drugs were reconstituted in ultrapure PBS, whereas disulfiram was reconstituted in sesame oil. After all the injections, the mice were very closely monitored for weight, mobility, posture, appearance, and survival. All the mice were humanely sacrificed by following the rules set in the animal protocol, in compliance with the IACUC. Immediately after sacrificing the mice, their blood was collected with cardiac puncture and peritoneal cells were collected by rinsing the peritoneal cavity with ice-cold PBS containing 3% FBS. Peritoneal cells were then lysed using RIPA buffer to isolate the total protein. Additionally, all the tissues were harvested and fixed in formalin. All the animal procedures were performed in accordance with the institutional guidelines for using and careful handling of laboratory animals of the University of Massachusetts, Amherst, MA, USA and all the procedures were approved by the Animal Ethics Committee of the University of Massachusetts Amherst.
ELISA
IL-1β ELISA was performed according to the manufacturer's protocol using the ThermoFisher IL-1 beta mouse uncoated ELISA kit. In vitro assays were performed in the cell supernatant by diluting them with 1× ELISPOT, whereas for in vivo assays, isolated serum was utilized. For all the ELISAs, the sample was incubated overnight at 4 °C to allow maximum binding.
Immunoblotting
For both in vitro and in vivo assays, protein was isolated by cell lysis using RIPA buffer enriched with 1× protease inhibitor. For in vitro, both the cell lysate and supernatant were used for immunoblotting. For in vivo, total protein was isolated from peritoneal cells immediately after their isolation. The protein was quantified using the BCA assay and an equal amount of protein was loaded onto gels. For in vitro samples, we performed regular western protein analysis using 10% polyacrylamide gels and wet transfer on a PVDF membrane. Later, the membrane was blocked in 5% skim milk and stained with the respective primary and secondary antibodies prepared in TBST, whereas for in vivo, we chose to use protein simple Wes, due to the very limited sample quantity. The manufacturer's protocol was utilized for loading the plate and carrying out the assay.
Hematoxylin & eosin (H&E) staining for biosafety/toxicity
Tissues from the vital organs (liver, spleen, kidneys, lungs and heart) were collected at the termination, which were then fixed in 10% neutral buffered formalin for 24 h, decanted and stored in 70% ethanol for a long time at 4 degree Celsius. The samples were then transported to Biospecimen Resource and Molecular Analysis Facility (BRaMA), PVLSI, UMass Chan Medical-Baystate, where they were paraffin embedded, sectioned at 5 μM thickness and H&E stained. Analysis was performed by the residents from the Department of Pathology, UMass Chan Medical School at Bay State Health, Springfield, MA.
Statistical analysis
GraphPad Prism 10 was utilized to analyze statistics. For two groups comparison, two-tailed unpaired t-test was utilized, whereas for multiple groups, ordinary one-way or two-way ANOVA was performed followed by Dunnett's or Sidak's multiple comparisons. The results were represented as either mean ± S.E.M. (standard error of mean) or mean ± s.d. (standard deviation). A p-value less than 0.05 was considered as significant.
Author contributions
A. K. conceived the idea and supervised the research; D. N. and A. K. designed the experiments; D. N., M. D., J. F. III and A. P. performed the experiments; D. N., M. D., J. F. III and A. K. wrote the paper and received comments from all the authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This work was financially supported by the National Science Foundation CAREER Award (2142917) to A. K. This work was supported in part by a fellowship from the University of MassachusettsUniversity of Massachusetts to J. Förster III as part of the Biotechnology Training Program (National Research Service Award T32 GM135096). We would like to thank the Biophysical Characterization Core at the Institute for Applied Life Sciences (IALS), the University of Massachusetts Amherst, for lending their expertise with regard to characterization experiments. We would also like to thank the Light Microscopy Core Facility at the University of Massachusetts Amherst for their help and consultation while performing confocal imaging. All institutional and national guidelines for the care and use of laboratory animals were followed and approved by the University of Massachusetts Amherst Institutional Use and Care of Animals Committee. All the visual figures were created with Biorender.com.
References
- T. van der Poll, M. Shankar-Hari and W. J. Wiersinga, The Immunology of Sepsis, Immunity, 2021, 54(11), 2450–2464, DOI:10.1016/j.immuni.2021.10.012.
- M. J. Delano and P. A. Ward, Sepsis-Induced Immune Dysfunction: Can Immune Therapies Reduce Mortality?, J. Clin. Invest., 2016, 126(1), 23–31, DOI:10.1172/JCI82224.
- D. De Backer, M. Cecconi, J. Lipman, F. Machado, S. N. Myatra, M. Ostermann, A. Perner, J.-L. Teboul, J.-L. Vincent and K. R. Walley, Challenges in the Management of Septic Shock: A Narrative Review, Intensive Care Med., 2019, 45(4), 420–433, DOI:10.1007/s00134-019-05544-x.
- L. Ulloa, M. Brunner, L. Ramos and E. A. Deitch, Scientific and Clinical Challenges in Sepsis, Curr. Pharm. Des., 2009, 15(16), 1918–1935 CrossRef CAS PubMed.
-
M. J. Schultz, M. W. Dünser, A. M. Dondorp, N. K. J. Adhikari, S. Iyer, A. Kwizera, Y. Lubell, A. Papali, L. Pisani, E. D. Riviello, D. C. Angus, L. C. Azevedo, T. Baker, J. V. Diaz, E. Festic, R. Haniffa, R. Jawa, S. T. Jacob, N. Kissoon, R. Lodha, I. Martin-Loeches, G. Lundeg, D. Misango, M. Mer, S. Mohanty, S. Murthy, N. Musa, J. Nakibuuka, A. S. Neto, N. H. Mai, B. N. Thien, R. Pattnaik, J. Phua, J. Preller, P. Povoa, S. Ranjit, D. Talmor, J. Thevanayagam and C. L. Thwaites, Current Challenges in the Management of Sepsis in ICUs in Resource-Poor Settings and Suggestions for the Future, in Sepsis Management in Resource-limited Settings, ed. A. M. Dondorp, M. W. Dünser and M. J. Schultz, Springer International Publishing, Cham, 2019, pp. 1–24. DOI:10.1007/978-3-030-03143-5_1.
- M. J. Delano and P. A. Ward, The Immune System's Role in Sepsis Progression, Resolution and Long-Term Outcome, Immunol. Rev., 2016, 274(1), 330–353, DOI:10.1111/imr.12499.
- R. S. Hotchkiss, C. M. Coopersmith, J. E. McDunn and T. A. Ferguson, Tilting toward Immunosuppression, Nat. Med., 2009, 15(5), 496–497, DOI:10.1038/nm0509-496.
- X. Shi, S. Tan and S. Tan, NLRP3 Inflammasome in Sepsis (Review), Mol. Med. Rep., 2021, 24(1), 1–8, DOI:10.3892/mmr.2021.12153.
- J. S. Boomer, J. M. Green and R. S. Hotchkiss, The Changing Immune System in Sepsis, Virulence, 2014, 5(1), 45–56, DOI:10.4161/viru.26516.
- F. Steinhagen, S. V. Schmidt, J.-C. Schewe, K. Peukert, D. M. Klinman and C. Bode, Immunotherapy in Sepsis - Brake or Accelerate?, Pharmacol. Ther., 2020, 208, 107476, DOI:10.1016/j.pharmthera.2020.107476.
- L. G. Danielski, A. D. Giustina, S. Bonfante, T. Barichello and F. Petronilho, The NLRP3 Inflammasome and Its Role in Sepsis Development, Inflammation, 2020, 43(1), 24–31, DOI:10.1007/s10753-019-01124-9.
- D. C. Cornelius, O. K. Travis, R. W. Tramel, M. Borges-Rodriguez, C. H. Baik, M. Greer, C. A. Giachelli, G. A. Tardo and J. M. Williams, NLRP3 Inflammasome Inhibition Attenuates Sepsis-Induced Platelet Activation and Prevents Multi-Organ Injury in Cecal-Ligation Puncture, PLoS One, 2020, 15(6), e0234039, DOI:10.1371/journal.pone.0234039.
- K. Mao, S. Chen, M. Chen, Y. Ma, Y. Wang, B. Huang, Z. He, Y. Zeng, Y. Hu, S. Sun, J. Li, X. Wu, X. Wang, W. Strober, C. Chen, G. Meng and B. Sun, Nitric Oxide Suppresses NLRP3 Inflammasome Activation and Protects against LPS-Induced Septic Shock, Cell Res., 2013, 23(2), 201–212, DOI:10.1038/cr.2013.6.
- K. V. Swanson, M. Deng and J. P.-Y. Ting, The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics, Nat. Rev. Immunol., 2019, 19(8), 477–489, DOI:10.1038/s41577-019-0165-0.
- J. Xu and G. Núñez, The NLRP3 Inflammasome: Activation and Regulation, Trends Biochem. Sci., 2023, 48(4), 331–344, DOI:10.1016/j.tibs.2022.10.002.
- J. Xu and G. Núñez, The NLRP3 Inflammasome: Activation and Regulation, Trends Biochem. Sci., 2023, 48(4), 331–344, DOI:10.1016/j.tibs.2022.10.002.
- Y. He, H. Hara and G. Núñez, Mechanism and Regulation of NLRP3 Inflammasome Activation, Trends Biochem. Sci., 2016, 41(12), 1012–1021, DOI:10.1016/j.tibs.2016.09.002.
- D. Nandi, N. S. S. Farid, H. A. R. Karuppiah and A. Kulkarni, Imaging Approaches to Monitor Inflammasome Activation, J. Mol. Biol., 2022, 434(4), 167251, DOI:10.1016/j.jmb.2021.167251.
- X. Zhang, A. Xu, J. Lv, Q. Zhang, Y. Ran, C. Wei and J. Wu, Development of Small Molecule Inhibitors Targeting NLRP3 Inflammasome Pathway for Inflammatory Diseases, Eur. J. Med. Chem., 2020, 185, 111822, DOI:10.1016/j.ejmech.2019.111822.
- A. Zahid, B. Li, A. J. K. Kombe, T. Jin and J. Tao, Pharmacological Inhibitors of the NLRP3 Inflammasome, Front. Immunol., 2019, 10, 2538 CrossRef PubMed , https://doi.org/10.3389/fimmu.2019.02538.
- G. E.-S. Batiha, A. I. Al-Gareeb, D. Rotimi, O. S. Adeyemi and H. M. Al-kuraishy, Common NLRP3 Inflammasome Inhibitors and Covid-19: Divide and Conquer, Sci. Afr., 2022, 18, e01407, DOI:10.1016/j.sciaf.2022.e01407.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, Nano Based Drug Delivery Systems: Recent Developments and Future Prospects, J. Nanobiotechnol., 2018, 16(1), 71, DOI:10.1186/s12951-018-0392-8.
- H. Wang, Y. Zhou, Q. Sun, C. Zhou, S. Hu, C. Lenahan, W. Xu, Y. Deng, G. Li and S. Tao, Update on Nanoparticle-Based Drug Delivery System for Anti-Inflammatory Treatment, Front. Bioeng. Biotechnol., 2021, 9, 630352, DOI:10.3389/fbioe.2021.630352.
- D. Placha and J. Jampilek, Chronic Inflammatory Diseases, Anti-Inflammatory Agents and Their Delivery Nanosystems, Pharmaceutics, 2021, 13(1), 64, DOI:10.3390/pharmaceutics13010064.
- Sepsis and septic shock | Nature Reviews Disease Primers. https://www.nature.com/articles/nrdp201645 (accessed 2023-09-30).
- Inflammation and Coagulation: Implications for the Septic Patient | Clinical Infectious Diseases | Oxford Academic. https://academic.oup.com/cid/article/36/10/1259/307497 (accessed 2023-09-30).
- S. Fujishima, Organ Dysfunction as a New Standard for Defining Sepsis, Inflammation Regener., 2016, 36(1), 24, DOI:10.1186/s41232-016-0029-y.
- A. S. Cross and S. M. Opal, A New Paradigm for the Treatment of Sepsis: Is It Time To Consider Combination Therapy?, Ann. Intern. Med., 2003, 138(6), 502–505, DOI:10.7326/0003-4819-138-6-200303180-00016.
- K. Upadhyay, B. Hiregoudar, E. Meals, B. K. English and A. J. Talati, Combination Therapy with Ampicillin and Azithromycin Improved Outcomes in a Mouse Model of Group B Streptococcal Sepsis, PLoS One, 2017, 12(7), e0182023, DOI:10.1371/journal.pone.0182023.
- G. Vazquez-Grande and A. Kumar, Optimizing Antimicrobial Therapy of Sepsis and Septic Shock: Focus on Antibiotic Combination Therapy, Semin. Respir. Crit. Care Med., 2015, 36(1), 154–166, DOI:10.1055/s-0034-1398742.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, Nano Based Drug Delivery Systems: Recent Developments and Future Prospects, J. Nanobiotechnol., 2018, 16(1), 71, DOI:10.1186/s12951-018-0392-8.
- S. A. A. Rizvi and A. M. Saleh, Applications of Nanoparticle Systems in Drug Delivery Technology, Saudi Pharm. J., 2018, 26(1), 64–70, DOI:10.1016/j.jsps.2017.10.012.
- S. Gelperina, K. Kisich, M. D. Iseman and L. Heifets, The Potential Advantages of Nanoparticle Drug Delivery Systems in Chemotherapy of Tuberculosis, Am. J. Respir. Crit. Care Med., 2005, 172(12), 1487–1490, DOI:10.1164/rccm.200504-613PP.
- A. P. Perera, R. Fernando, T. Shinde, R. Gundamaraju, B. Southam, S. S. Sohal, A. A. B. Robertson, K. Schroder, D. Kunde and R. Eri, MCC950, a Specific Small Molecule Inhibitor of NLRP3 Inflammasome Attenuates Colonic Inflammation in Spontaneous Colitis Mice, Sci. Rep., 2018, 8, 8618, DOI:10.1038/s41598-018-26775-w.
- Engineering precision nanoparticles for drug delivery | Nature Reviews Drug Discovery. https://www.nature.com/articles/s41573-020-0090-8 (accessed 2023-09-14).
- A. Ramesh, S. Kumar, D. Nandi and A. Kulkarni, CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages, Adv. Mater., 2019, 31(51), e1904364, DOI:10.1002/adma.201904364.
- C. Hald Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, The Role of Lipid Components in Lipid Nanoparticles for Vaccines and Gene Therapy, Adv. Drug Delivery Rev., 2022, 188, 114416, DOI:10.1016/j.addr.2022.114416.
- J. B. Strachan, B. P. Dyett, Z. Nasa, C. Valery and C. E. Conn, Toxicity and Cellular Uptake of Lipid Nanoparticles of Different Structure and Composition, J. Colloid Interface Sci., 2020, 576, 241–251, DOI:10.1016/j.jcis.2020.05.002.
- R. C. Coll, A. A. B. Robertson, J. J. Chae, S. C. Higgins, R. Muñoz-Planillo, M. C. Inserra, I. Vetter, L. S. Dungan, B. G. Monks, A. Stutz, D. E. Croker, M. S. Butler, M. Haneklaus, C. E. Sutton, G. Núñez, E. Latz, D. L. Kastner, K. H. G. Mills, S. L. Masters, K. Schroder, M. A. Cooper and L. A. J. O'Neill, A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases, Nat. Med., 2015, 21(3), 248–255, DOI:10.1038/nm.3806.
- T. van der Heijden, E. Kritikou, W. Venema, J. van Duijn, P. J. van Santbrink, B. Slütter, A. C. Foks, I. Bot and J. Kuiper, NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice—Brief Report, Arterioscler. Thromb. Vasc. Biol., 2017, 37(8), 1457–1461, DOI:10.1161/ATVBAHA.117.309575.
- L. V. Walle, I. B. Stowe, P. Šácha, B. L. Lee, D. Demon, A. Fossoul, F. V. Hauwermeiren, P. H. V. Saavedra, P. Šimon, V. Šubrt, L. Kostka, C. E. Stivala, V. C. Pham, S. T. Staben, S. Yamazoe, J. Konvalinka, N. Kayagaki and M. Lamkanfi, MCC950/CRID3 Potently Targets the NACHT Domain of Wild-Type NLRP3 but Not Disease-Associated Mutants for Inflammasome Inhibition, PLoS Biol., 2019, 17(9), e3000354, DOI:10.1371/journal.pbio.3000354.
- A. P. Perera, R. Fernando, T. Shinde, R. Gundamaraju, B. Southam, S. S. Sohal, A. A. B. Robertson, K. Schroder, D. Kunde and R. Eri, MCC950, a Specific Small Molecule Inhibitor of NLRP3 Inflammasome Attenuates Colonic Inflammation in Spontaneous Colitis Mice, Sci. Rep., 2018, 8, 8618, DOI:10.1038/s41598-018-26775-w.
- J. J. Hu, X. Liu, S. Xia, Z. Zhang, Y. Zhang, J. Zhao, J. Ruan, X. Luo, X. Lou, Y. Bai, J. Wang, L. R. Hollingsworth, V. G. Magupalli, L. Zhao, H. R. Luo, J. Kim, J. Lieberman and H. Wu, FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation, Nat. Immunol., 2020, 21(7), 736–745, DOI:10.1038/s41590-020-0669-6.
- L. Sborgi, S. Rühl, E. Mulvihill, J. Pipercevic, R. Heilig, H. Stahlberg, C. J. Farady, D. J. Müller, P. Broz and S. Hiller, GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death, EMBO J., 2016, 35(16), 1766–1778, DOI:10.15252/embj.201694696.
- C. Wang, T. Yang, J. Xiao, C. Xu, Y. Alippe, K. Sun, T.-D. Kanneganti, J. B. Monahan, Y. Abu-Amer, J. Lieberman and G. Mbalaviele, NLRP3 Inflammasome Activation Triggers Gasdermin D-Independent Inflammation, Sci. Immunol., 2021, 6(64), eabj3859, DOI:10.1126/sciimmunol.abj3859.
- W. He, H. Wan, L. Hu, P. Chen, X. Wang, Z. Huang, Z.-H. Yang, C.-Q. Zhong, J. Han and D. Gasdermin, Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion, Cell Res., 2015, 25(12), 1285–1298, DOI:10.1038/cr.2015.139.
- H. Zhang, Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation, Methods Mol. Biol., 2017, 1522, 17–22, DOI:10.1007/978-1-4939-6591-5_2.
- L. Xu, X. Wang, Y. Liu, G. Yang, R. J. Falconer and C.-X. Zhao, Lipid Nanoparticles for Drug Delivery, Adv. NanoBiomed Res., 2022, 2(2), 2100109, DOI:10.1002/anbr.202100109.
- Nanoreporter for Real-Time Monitoring of Inflammasome Activity and Targeted Therapy – PubMed. https://pubmed.ncbi.nlm.nih.gov/36603165/(accessed 2023-10-02).
- J. Sand, E. Haertel, T. Biedermann, E. Contassot, E. Reichmann, L. E. French, S. Werner and H.-D. Beer, Expression of Inflammasome Proteins and Inflammasome Activation Occurs in Human, but Not in Murine Keratinocytes, Cell Death Dis., 2018, 9(2), 1–14, DOI:10.1038/s41419-017-0009-4.
- A. Stutz, G. L. Horvath, B. G. Monks and E. Latz, ASC Speck Formation as a Readout for Inflammasome Activation, Methods Mol. Biol., 2013, 1040, 91–101, DOI:10.1007/978-1-62703-523-1_8.
- A. Nagar, T. Rahman and J. A. Harton, The ASC Speck and NLRP3 Inflammasome Function Are Spatially and Temporally Distinct, Front. Immunol., 2021, 12, 752482 CrossRef CAS PubMed , https://doi.org/10.3389/fimmu.2021.752482.
- D. Nandi, M. Shivrayan, J. Gao, J. Krishna, R. Das, B. Liu, S. Thayumanavan and A. Kulkarni, Core Hydrophobicity of Supramolecular Nanoparticles Induces NLRP3 Inflammasome Activation, ACS Appl. Mater. Interfaces, 2021, 13(38), 45300–45314, DOI:10.1021/acsami.1c14082.
- J. F. Iii, D. Nandi and A. Kulkarni, mRNA-Carrying Lipid Nanoparticles That Induce Lysosomal Rupture Activate NLRP3 Inflammasome and Reduce mRNA Transfection Efficiency, Biomater. Sci., 2022, 10(19), 5566–5582, 10.1039/D2BM00883A.
- M. Debnath, J. I. Förster, A. Ramesh and A. Kulkarni, Protein Corona Formation on Lipid Nanoparticles Negatively Affects the NLRP3 Inflammasome Activation, Bioconjugate Chem., 2023, 34(10), 1766–1779, DOI:10.1021/acs.bioconjchem.3c00329.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abcd
*abcd