
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable “snow lantern-like” aggregates of silicon nanoparticles suitable as a drug delivery platform†
Hennie Marie
Johnsen
 *ab,
Seyedmehdi
Hossaini Nasr
c,
Ricardo
De Luna
c,
Werner
Filtvedt
b,
Michael J.
Sailor
c,
Jo
Klaveness
a and
Marianne
Hiorth
a
*ab,
Seyedmehdi
Hossaini Nasr
c,
Ricardo
De Luna
c,
Werner
Filtvedt
b,
Michael J.
Sailor
c,
Jo
Klaveness
a and
Marianne
Hiorth
a
aDepartment of Pharmacy, University of Oslo, Sem Sælands vei 3, 0371 Oslo, Norway
bNacamed AS, Oslo Science Park, Gaustadalléen 21, 0349 Oslo, Norway
cDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA, USA
First published on 18th April 2024
Abstract
Nanomedicine is a growing field where development of novel organic and inorganic materials is essential to meet the complex requirements for drug delivery. This includes biocompatibility, suitability for surface modifications, biodegradability, and stability sufficient to carry a drug payload through various tissues for the desired timespan. Porous silicon nanoparticles (pSi NP) are shown to have several beneficial traits in drug delivery in addition to a porous structure to maximize drug loading. The conventional synthesis of pSi NP using electrochemical etching is costly, time-consuming and requires large quantities of highly toxic hydrofluoric acid (HF). As such this research attempted a novel method to address these limitations. Mesoporous silicon nanoparticles were prepared by centrifugal Chemical Vapor Deposition (cCVD) without the use of HF. This process generated aggregates consisting of multiple primary particles fused into each other, similar to snowballs fused together in a snow-lantern (snowball pyramid). Our results demonstrated that the cCVD Si particles were versatile in terms of surface chemistry, colloidal stability, degradability, minimization of acute in vitro toxicity, and modulation of drug release. Dynamic light scattering, scanning electron microscopy, and cryogenic nitrogen adsorption isotherm measurements confirmed the overall size (210 nm), morphology, and pore size (14–16 nm) of the prepared materials. Agglomeration in phosphate-buffered saline (PBS) was minimized by PEGylation by a two-step grafting procedure that employed a primary amine linker. Finally, the release rate of a model drug, hydrocortisone, was evaluated with both PEGylated and pristine particles. Conclusively, these snow-lantern cCVD Si particles do indeed appear suitable for drug delivery.
1 Introduction
Ever since Paul Ehrlich introduced the concept of the “magic bullet” in 1907,1 researchers have strived to develop drug delivery platforms to enhance drug effectiveness and minimize side effects for improving human health. As opposed to nonspecific distribution of drug molecules, nanomedicines have been designed using various organic and inorganic materials to improve directional drug transport. However, the visions for nanomedicine have yet to be realized in clinical translation due to the complex biological barriers. In addition to requirements of biocompatibility and high drug capacity, the various routes of administration set directions for rational design of optimized drug delivery materials. For instance, after intravenous administration the particles must avoid uptake by the mononuclear phagocyte system and overcome hemorheological limitations, tissue penetration, and possibly cell membrane internalization and endosomal escape.2 The particles should be biodegradable while displaying sufficient stability to deliver their active ingredient in a manner appropriate for the specific therapeutic indication. Emphasis on overcoming specific biological barriers also allows for accommodating individual variations for development of personalized or precision nanomedicine.3The base material, particle size, shape, surface charge and coating will determine the in vivo fate of nanomedicines. For instance, spherical and larger particles have been shown to ensure longer circulation time while rod-shaped particles penetrate blood vessels and tumor tissue more readily. Smaller and positively charged particles more easily traverse mucosal membranes.3 Active targeting or biomimicry can be used to avoid clearance, prolong circulation as well as adding specific targeting to diseased tissue.2 The properties of the particle surface are of high importance to surmount several barriers, and access to facile surface modifications of nanoparticles can be beneficial. Additionally, precise control over drug release kinetics can be achieved by various surface modifications.4 To meet the demands of complex biological barriers a comprehensive toolset in nanomedicine design is essential. The biological complexity might complicate the nanoparticle design. However, simplicity is important for achieving clinical translation in terms of scale-up, mass-production and regulatory approval. Therefore, ongoing discovery of novel base nanomaterials suitable for drug delivery serves as a catalyst in advancing the field of nanomedicine.
Mesoporous inorganic nanomaterials, like porous silicon nanoparticles (pSi NP), provide new possibilities in nanomedicine compared to the widely used organic materials due to their versatile surface chemistries and their intrinsic properties that can enable new functions such as imaging.5,6 In addition to drug delivery, pSi NPs have potential applications in several fields including catalysis, energy storage, sensor technology and chemical separations. Therefore, extensive efforts have been directed to fabrication methods for nanostructured Si materials to yield suitable size, shape, pore morphology and surface chemistry for the intended applications. These methods include top-down approaches such as photolithography7 or electrochemical etching of Si wafers8 as well as bottom-up approaches such as solution chemistry,9–11 solid-state synthesis12 and Chemical Vapor Deposition (CVD).13 One of the most important pSi NPs synthesis routes involves electrochemical etching of crystalline silicon wafers using hydrofluoric acid (HF)-containing electrolytes. Parallel pores form perpendicular to the wafer surface, forming a mesoporous layer. Ultrasonic power is then employed to fracture the mesoporous silicon wafer and form nanoparticles. The nanoparticles of the desired size can be gathered using a series of centrifugation and washing steps.
Over 20 years of extensive research into the use of pSi NPs in biomedical applications have identified methods that generate biocompatible and biodegradable material, by dissolution into orthosilicic acid under physiological conditions.5,14 The surface area and pore dimensions can be adjusted during synthesis by varying the current density to accommodate different sizes of payloads, from small molecular drugs to larger biologics. Several animal studies have demonstrated biocompatibility of pSi NPs as well as therapeutic effects of the drug-loaded particles.15,16 Chemical grafting of functional and targeting groups on the porous Si surface enables control over the particles’ fate in vivo, while surface polyethylene glycol (PEG) groups increase circulation time by protecting against immune system recognition.17,18 Nevertheless, pSi NP-based therapeutics can face challenges for clinical translation due to the complex and costly fabrication route, hazardous chemistry involving HF, and high material losses during particle size refining.
This work presents an alternative approach for pSi NP production using centrifugal Chemical Vapor Deposition (cCVD). This scalable one-step method does not use HF or other hazardous etching chemistries. The method yielded Si particles that have the ability to produce therapeutic levels of hydrogen gas19 and have shown promising biocompatibility measures.20 The cCVD process allows control over synthesis parameters such as temperature and growth time to regulate particle properties like primary and total particle size.21,22 A means to achieve porous particles by controlled aggregation of small primary particles into larger “snow lantern-like” particles using the cCVD process, and its potential as a drug delivery material, is documented in the present study. Surface modifications were applied to wafer-derived and cCVD Si particles, to compare the applicability to the two materials. Finally, the suitability of cCVD Si particles for drug delivery was assessed by biodegradation, biocompatibility and drug loading studies.
2 Materials and methods
2.1 Materials
Aggregated cCVD Si particles were obtained from Dynatec Engineering AS. Reference porous silicon flakes (L634 formulation, prepared by electrochemical anodization of single-crystalline wafers, porosity: 80 ± 10%, pore size: 10–20 nm) were purchased from a commercial source, TruTag Technologies, inc (https://trutags.com/). These flakes were then processed into particles as described below. For stability and degradability studies, albumin from human serum (≥96%, Sigma), ethanol and isopropanol (prima, Antibac), methanol (VWR), N,N-dimethylformamide (DMF, Thermo Scientific), dimethyl sulfoxide (DMSO, Fisher Chemical) and phosphate buffered saline (PBS) pH 7.4 tablets (ionic concentration ∼0.15 M, Sigma-Aldrich) were all used as-received, with no further purification. For the cellular studies, Dulbecco's Modified Eagle Media (DMEM), containing 4.5 g L−1D-glucose supplemented by 10% fetal bovine serum (FBS), 1% Penicillin–Streptomycin and 0.5% L-glutamine and the required supplements were obtained from Gibco. Black 96-well plates with transparent bottoms were obtained from Corning Inc. AlamarBlue reagent was purchased from Invitrogen, Thermo Scientific. For particle oxidation and coating, hydrogen peroxide solution (H2O2 35%) and sodium tetraborate decahydrate (Na2B4O7 × 10 H2O) were obtained from Merck. 3-Aminopropyl-dimethylethoxysilane (APDMES) was purchased from Oakwood chemical. 2,2-Dimethoxy-1,6-diaza-2-silacyclooctane (DMDASCP, 10% in cyclohexane) was purchased from Gelest Inc. mPEG-silane (5k Da) and maleimide (MAL)-PEG-silane (5k Da) were obtained from Creative PEGWorks. mPEG-succinimidyl valerate (SVA, 5k Da) and MAL-PEG-SVA (Mw 5k) were obtained from Laysan Bio Inc. For drug loading and release studies hydrocortisone (HC) and acetonitrile were purchased from Sigma-Aldrich, High-Performance Liquid Chromatography (HPLC) grade water from Thermo Scientific and trifluoroacetic acid from Merck.2.2 Nanoparticles synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g and pressure of approximately 1 bar.
000 g and pressure of approximately 1 bar.
The complex thermal SiH4 decomposition process includes formation of different higher order silane species (SixHy) that eventually grow into polymeric chains and rings, and the rings finally stack into complexes.26,27 At this stage, the reactions are exothermic and irreversible. To grow stable aggregate particles, the reaction parameters were adjusted to maximize formation of a large number of nuclei, such that each particle would compete for the same precursor gas and thus be limited in size. The temperature was maintained sufficiently low to allow the formation of many intermediate SiHx species at the surface of the particles to enhance the tendency of the particles to adhere to other particles during this stage. This resulted in many particle-to-particle collisions and eventually aggregated particles. It was important to terminate the process at a consistent growth time, using a centripetal acceleration of at least 1000 g, such that the aggregates achieved a narrow size distribution. These cCVD Si particles were used as obtained and handled in ambient air unless otherwise noted.
A cCVD Si particle batch of primary (SEM) and aggregate (DLS) particle size of approximately 30 and 210 nm, respectively, were used for all the experiments herein except for the cell viability studies. A particle batch of identical chemical composition but with approximately 60 and 350 nm primary and aggregate particle size, respectively, was used for the cytotoxicity studies. The cCVD particles were used as obtained and after exposure to ambient air unless otherwise noted.
000 rpm for 10 min and the supernatant was discarded. The remaining pellet was redispersed by ultrasonication in ethanol and the process was repeated. Then, the pellet was re-suspended in ethanol and centrifuged at 2500 rpm for 5 min. The pellet resulting from this step was discarded. The nanoparticles remaining in the supernatant at this stage were of the desired size, and they were stored in ethanol until further use.
2.3 Oxidation and surface modifications
The cCVD Si particles were oxidized by one of the following methods to perform surface chemistry via surface silanol groups: one month exposure to ambient air, chemical oxidation by two-day immersion in either H2O2:EtOH (1:1) or 2 mM Na2B4O7, followed by water and ethanol wash, or by heat treatment (50 °C min−1 up to 700 °C for 90 min). TruTag Si particles were oxidized by two hours immersion in H2O2. Immersion in H2O2 was the standard oxidation treatment used unless otherwise noted.
To functionalize the surface of Si cCVD NPs with amine groups, different chemistries were tested. For APDMES grafting, 1 mL EtOH and 15 μL of the APDMES solution was added to 1 mg particles before redispersion by ultrasonication followed by two hours of vortex shaking. For DMDASCP grafting, 800 μL DMF and 200 μL of DMDASCP solution was added to 1 mg particles before redispersion and placing on a spinning wheel overnight. All samples were then washed with water and ethanol. As a control, particles prepared from TruTag were aminated using the same chemistries.
The surface of the cCVD Si NPs was decorated with PEG using two different approaches, (a) direct PEGylation through the reaction of PEG-silane with Si and (b) by amide bond formation between an amine-functionalized Si (APDMES or DMDASCP) and SVA-PEG moieties. For each method mPEG and MAL-PEG were used. The PEG chemicals (10 mg for 1 mg Si) were dissolved in 1:9 DMSO:EtOH before adding to the Si samples. The samples were then redispersed and left on a spinning wheel overnight before washing with water and ethanol. The same procedures were applied to TruTag NP as a control study.
2.4 Characterization
2.5 Stability evaluations
The colloidal stability of pristine cCVD Si particles was studied in PBS and in other conditions including 4% albumin solutions, at room temperature or 37 °C and with or without magnetic stirring (∼200 rpm). After 5 hours or 4 days, the particle samples (5 mg mL−1) were diluted with water and either shaken by hand or ultrasonication treated before size was measured by DLS (as described in 2.4.1). A 24-hour stability study of pristine and PEGylated cCVD Si particles (mPEG and DMDASCP-mPEG) was conducted in water and PBS at 200 μg mL−1. The dispersions were placed on a spinning wheel and samples were withdrawn after 30 min, 2, 4, 8, and 24 hours and diluted in the same medium before size analysis by DLS. No ultrasonication treatment was used before measurement. The shelf-life stability of dry powder in room temperature and ambient air was measured monthly over 4 months by DLS (as described in 2.4.1). FTIR spectrum was captured for fresh cCVD Si particles and after one month storage in room temperature.2.6 In vitro biodegradation assay
In vitro dissolution of cCVD Si particles was investigated in PBS pH 7.4 with a concentration of 100 μg mL−1, temperature of 37 °C and under constant stirring at 100 rpm. Samples were withdrawn after 30 min, 1, 2, 4, 8 and 24 hours and centrifuged (15 min at 14k rpm) to remove remaining particles. The supernatants were analyzed for dissolved silicon content using inductively coupled plasma-optical emission spectrometry (ICP-OES, Ultima2, Horiba Scientific, Jobin Yvon). Quantification of silicon was done by comparing the signal at λ = 251 nm to a standard curve. The experiment was done in triplicate.2.7 In vitro cytotoxicity studies
The AlamarBlue bioassay was used for in vitro cytotoxicity studies of non-coated cCVD Si particles of primary and aggregate size of approximately 60 and 350 nm. Autoclave treatment (121 °C for 15 min) was used for sterilizing the particles before added to the cells. The pancreatic ductal adenocarcinoma murine cell line KPC was maintained in DMEM cell culture medium and incubated in 37 °C and 5% CO2 atmosphere. For viability studies, the cells were seeded at a density of 3000 cells per well in 96 well plates. Growth medium containing the particles of concentrations from 62.5 to 500 μg mL−1 (n = 8 for each concentration) were added the cells, and then incubated before measuring viability after 24 and 48 hours. The plates were rinsed twice with PBS, then 100 μL AlamarBlue solution and 900 μL growth medium was added to each well followed by two hours incubation. The fluorescence was measured with the spectrophotometer SpectraMax i3x (Molecular Devices) with excitation at 560 nm and detection at 590 nm. Cell viability was calculated by dividing the fluorescence intensity of the treated wells by the fluorescence intensity of the control wells. Assay interference between the Si particles and AlamarBlue assay components was tested to verify no interference with the viability results (data not shown), which has also been described for Si NPs in literature.28 Immediately after viability measurements (48 h), optical bright field microscopy images were taken for assessing visual changes in cell density and morphology using an inverted light optical microscope (Eclipse TS100 (Nikon)) and a camera (Zyla 4.2 sCMOS (Andor, Oxford Instruments)) with a 40× magnification lens.2.8 cCVD NP drug encapsulation study
:1 Si:HC in DMF (HC concentration below 30 mg ml−1) followed by drying under vacuum overnight at 60 °C. The following day, the samples were washed three times with cold water followed by drying under vacuum overnight at 60 °C. Drug loading was quantified by extraction of HC by immersion of loaded particles in methanol overnight, in three parallels. HPLC analyses of the samples were compared to a standard curve and %mass load (%M.l.) was calculated by dividing the mass of HC with the total mass of the loaded particles (×100%). As a reference sample of the free drug, HC was treated the same way by dissolving in DMF (without particles) and dried overnight.
3. Results
3.1 Characterization of stable snow lantern-like particles
The cCVD method generated Si aggregate “snow lantern” particles composed of smaller “snowballs”, as shown by the particle characteristics. Si particles obtained from this method displayed surface areas ranging from 9 to 90 m2 g−1. The particle size (hydrodynamic diameter) typically varied between 150 nm and 500 nm. The particle batch studied herein for surface modification, stability and drug delivery properties exhibited a monodisperse size distribution with hydrodynamic diameter of 210 nm, a surface charge of −37 mV, and an amorphous (Fig. SI1†) and mesoporous structure with BET surface area of 60 m2 g−1 (Table 1). The nitrogen sorption isotherm data included in Fig. SI1† indicates large mesopores, as would be expected for interparticle spaces. SEM images (Fig. 1) revealed a spherical morphology of the primary particles and a more irregular shape for the aggregated particles. The calculated particle diameter based on the surface area was 43 nm (ESI section 1.1 and Table SI1†), smaller than the observed DLS size of the aggregate particles, but larger than the SEM primary particle size (approximately 20–40 nm). Aggregated particles of 200 nm (DLS) and primary particle size of 20–40 nm (SEM) correspond to aggregation of about 5 × 5 × 5 primary particles. As such these data confirm that the studied cCVD particles consist of aggregates and possess a mesoporous structure.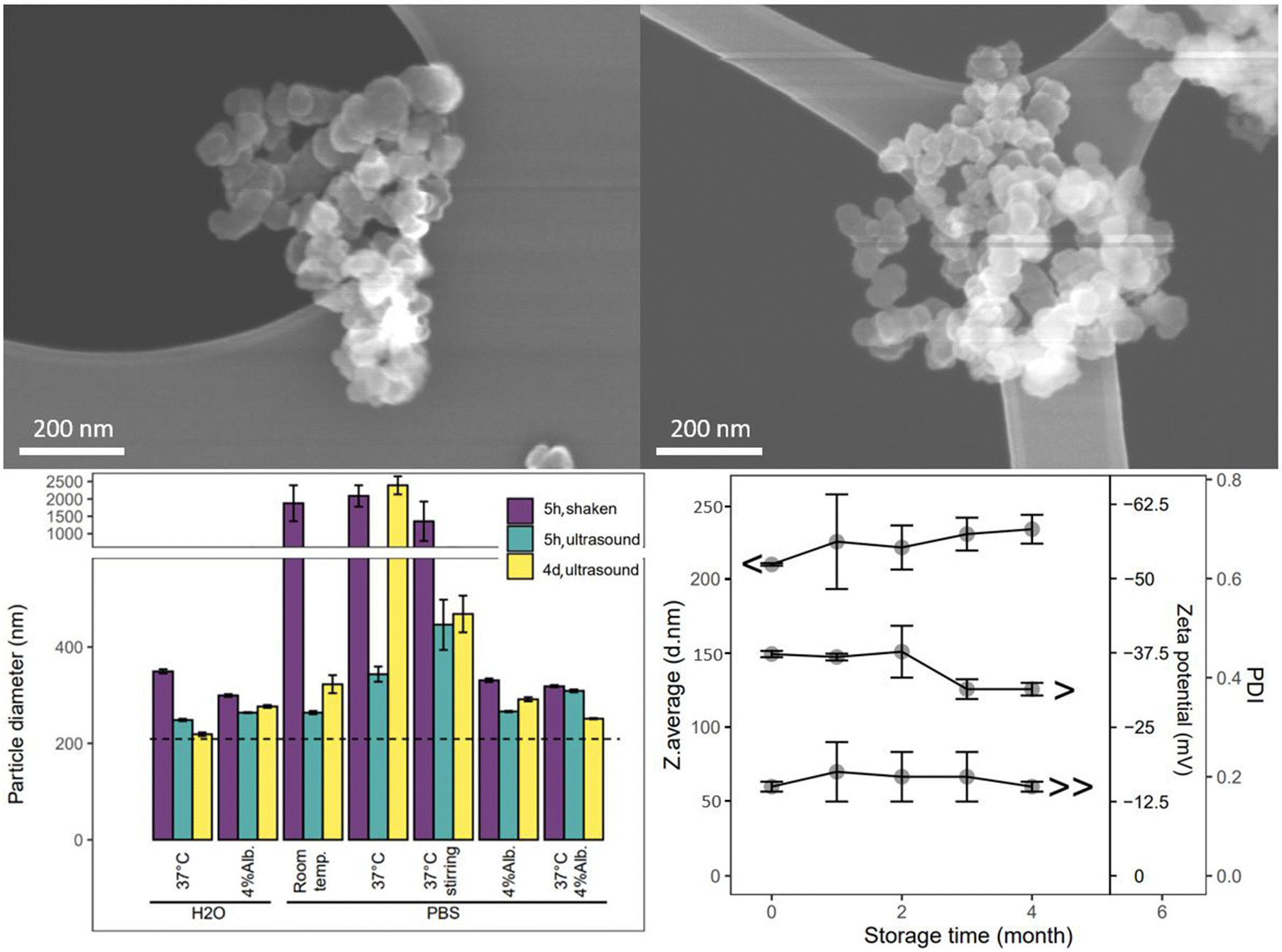 | ||
| Fig. 1 SEM images of aggregate cCVD Si particles made up of solid spherical primary particles (top). Colloidal stability of the particles in various aqueous media measured by particle size/DLS, measured after hand shaking or ultrasonication treatment after 5 hours (5h) and 4 days (4d) incubation (bottom left). Dashed line indicates control sample in water (diameter = 210 nm and PDI = 0.2). Alb. = human serum albumin. Storage stability of cCVD Si particles (bottom right) by DLS analyses measured monthly after storage in ambient air (top line – Z.average, middle line – zeta potential, bottom line – PDI). Averages of at least three measurements are shown with error bars showing standard deviations. | ||
| S.d. = standard deviation. BJH = Barrett–Joyner–Halenda (pore volume analysis). | |
|---|---|
| Hydrodynamic diameter ± s.d. | 210 ± 0.9 nm |
| Polydispersity index ± s.d. | 0.18 ± 0.01 |
| Zeta potential | −37.3 ± 0.5 mV |
| BET surface area | 59.8 m2 g−1 |
| Pore size adsorption | 13.7 nm |
| Pore size desorption | 16.4 nm |
| BJH pore volume | 0.32 cm3 g−1 |
The SEM images showed larger aggregated particles compared to DLS measurements, likely due to assumptions of spherical particles in DLS analysis and agglomeration (reversible) of the aggregate particles (irreversible) during SEM sample preparation. SEM was done on dry powder and DLS in suspension, thus the imaged particles likely contained multiple aggregate particles of the single particles observed in DLS.
The aggregated cCVD particles formed a stable colloid, maintaining their integrity and not disassembling into primary particles, when dispersed in aqueous solutions with variations in salts, temperature, and protein content (Fig. 1), as well as in organic solvents like ethanol, isopropanol, and dimethylformamide (data not shown). An increase in particle size and size distribution (PDI shown in Fig. SI2†) was seen in PBS solutions. This was further increased at 37 °C and under stirring. The observed size increase is attributed to particle agglomeration, and the clumping of particles could be partially reversed by ultrasonication. The tendency to agglomerate in these media was reduced by addition of 4% albumin. Furthermore, a shelf-life study of the cCVD Si particles in ambient air showed minimal changes in particle size, size distribution and surface charge over a period of 4 months (Fig. 1). The particles remained stable when stored at room temperature, although the formation of native oxide over time might have contributed to a slight increase in particle size and a change in zeta potential from −37 mV to −30 mV.
3.2 Surface chemistry of snow-lantern particles
Chemical modification of the particle surface to introduce functional groups suitable for various applications was successfully carried out using silane chemistry on surface silanol groups. The required oxidation of the particle surface was done using different techniques, resulting in different degrees of surface oxidation. The FTIR spectrum (Fig. SI3†) showed that all the chemically and native oxidized samples had at least one of the following peaks, corresponding to the Si–O stretching modes: 1180, 1060 and 795 cm−1. Freshly prepared pristine cCVD particles had a peak at 2100 cm−1 from Si–H stretching for the hydrogen-terminated surface, which was absent in the surface oxidized samples. The non-oxidized cCVD particles possessed a lower level of surface oxide, in comparison to the non-oxidized TruTag particles. The Raman spectrum (Fig. SI4†) showed similar trends as the FTIR, with Si–Si lattice modes visible for all samples. The Trutag particles displayed a Raman shift of ∼520 cm−1, corresponding to the lattice mode of crystalline Si, while the cCVD Si particles displayed a broader and slightly red-shifted band assigned to amorphous silicon.29,30 Oxidation caused a reduction in intensity of the Si–Si lattice signal and/or an increase in intensity of the Si–O stretching band at ∼400 cm−1 for the Na2B4O7, H2O2 and heat-oxidized cCVD particles, as compared to the non-oxidized material.To illustrate functionalization possibilities and control surface charge, a primary amine group was introduced to both non-oxidized and oxidized Si particles. The two reagents used for amination, APDMES and DMDASCP, gave 0.4% and 1.9% organic content (weight%, Fig. SI5 & Table SI2†) and both resulted in a change in zeta potential which confirmed alterations in surface chemistry. Prior to amination, the surface charge of cCVD Si particles ranged from −22 to −32 mV, while for TruTag particles, it varied from −38 to −43 mV, as depicted in Fig. 2. After APDMES grafting, a drastic change in zeta potential occurred, resulting in surface charges ranging from −4 to 21 mV. DMDASCP grafting yielded even higher surface charges, between 33 and 43 mV due to higher surface coverage.
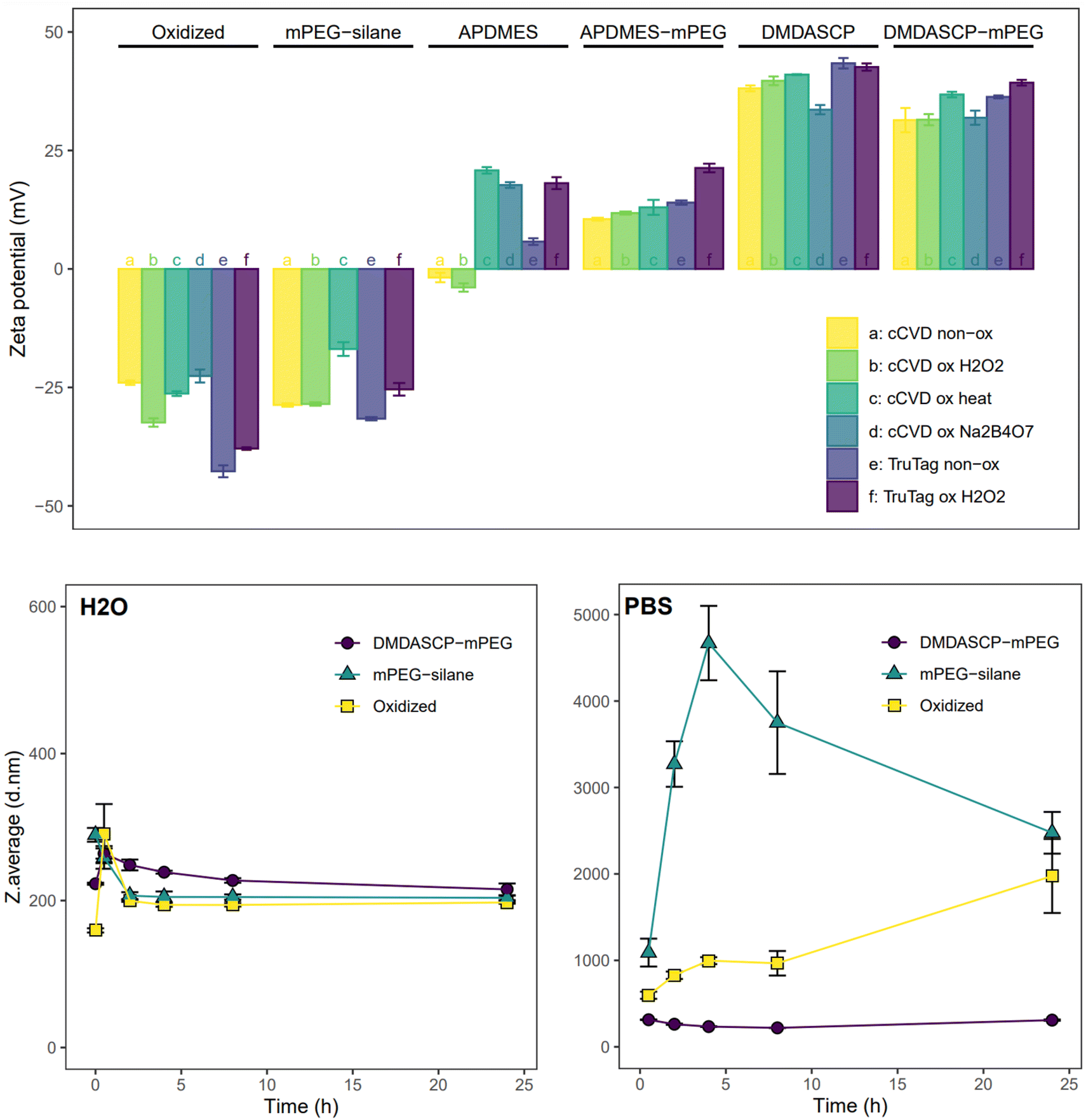 | ||
| Fig. 2 Zeta potential (top) and colloidal stability of chemically modified particles in H2O (bottom left) and PBS (bottom right). Zeta potential of the particles modified by oxidation, amination (APDMES or DMDASCP) or PEGylation (one- or two-step). Stability shown by hydrodynamic diameter (bottom) measured by DLS in either water (left) or PBS (right). All the data are shown as an average of three measurements with error bars showing standard deviations. | ||
The three approaches used for PEG grafting intended for biomedical applications resulted in varying surface coverage. TGA showed a total organic content of 0.4%, 2.9% and 7.9% for H2O2 oxidized one-step mPEG-silane, two-step APDMES-mPEG and DMDASCP-mPEG samples, respectively (Fig. SI5 & Table SI2†). Following mPEG-silane grafting, the surface charge approached neutrality but still ranged between −17 and −32 mV for all particles. APDMES-mPEG particles exhibited a narrower charge range of 11 to 21 mV, while DMDASCP-mPEG modification yielded higher surface charges between 31 and 39 mV. FTIR measurements showed a peak around 2900 cm−1 for the PEGylated samples which corresponds to C–H stretching (Fig. SI6†). This peak was larger for DMDASCP-mPEG than for mPEG-silane modified sample, confirming a higher PEGylation coverage for the former. In water, the colloidal stability of the unmodified (H2O2 oxidized), the one-step mPEG-silane and the two-step DMDASCP-mPEG modified particles remained consistent when observed over a 24-hour period. However, both the unmodified and mPEG-silane modified particles quickly agglomerated in PBS, whereas the DMDASCP-mPEG modified particles maintained stability over 24 hours (Fig. 2). PDI data displayed similar trends (Fig. SI7†). Improved colloidal stability observed in DMDASCP-mPEG modified particles confirmed successful surface grafting. Successful grafting of other functional groups through one- or two-step conjugation, such as the PEG-MAL, was confirmed by observed changes in particle charge (Fig. SI8†).
3.3 Snow-lantern particles as carrier for drug delivery application
The cCVD Si particles hold potential for drug delivery applications based on the biodegradability and biocompatibility assessment. In PBS solution at 100 μg ml−1, the particles displayed chemical degradation over 24 hours (Fig. 3). After 1, 2, and 8 hours in PBS pH 7.4, more than 50%, 75%, and nearly 95% of the Si content had degraded, respectively. In terms of in vitro toxicity response, cCVD Si particles with a hydrodynamic diameter of 350 nm (consisting of 60 nm primary particles) exhibited low toxicity at concentrations of 125 μg ml−1 and below (>88% cell viability). After 24 hours, the cell viability remained above 80% for all tested concentrations. However, after 48 hours, the viability decreased to 69% and 66% for concentrations of 250 μg mL−1 and 500 μg mL−1, respectively (Fig. 3). Microscopy images showed no change in cell morphology after 48 hours, but the cell density correlated with the viability data (data not shown). The images also suggested that reduced viability at higher concentrations may be attributed to physical adsorption of particles to the cell membrane, which could potentially impede vital cell mechanisms.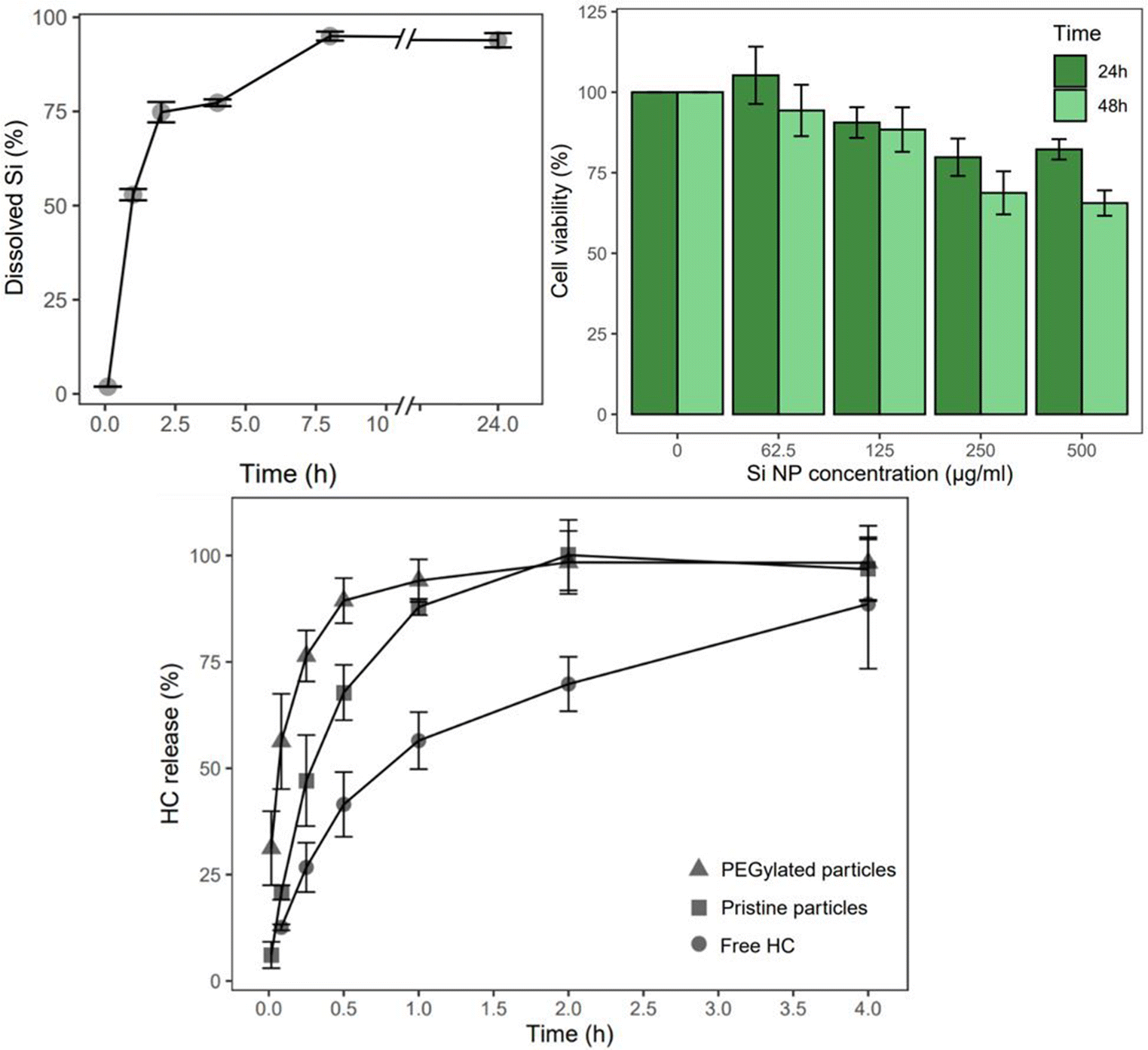 | ||
| Fig. 3 Assessment of the drug delivery properties of the cCVD Si particles. In vitro degradation kinetics in PBS pH 7.4 at 37 °C, by ICP measurement of dissolved Si (top left, n = 3). Cell viability assay on KPC cell line using the AlamarBlue reagent (top right, n = 8). Cumulative release of HC from pristine and PEGylated (DMDASCP-mPEG) cCVD Si particles and dissolution rate of free HC in PBS at pH 7.4 and 37 °C (bottom, n = 3). | ||
The HC loaded particles (Table 2), both pristine and DMDASCP-mPEG modified, had a similar particle size and size distribution compared to before loading (Table 1) but possessed different release properties. The two-step PEGylation method via DMDASCP exhibited high surface grafting, improved dispersibility in PBS, and was therefore utilized in HC release studies. The zeta potential remained unchanged for the pristine particles but was lower for the PEGylated particles after loading compared to before loading. A mass load of 31% was achieved for the pristine particles and 27% for the PEGylated particles. In terms of drug release, both types of particles demonstrated a faster release compared to the dissolution of the free drug (statistically significant <4 h). An increased release rate was seen for the PEGylated particles compared to the pristine particles (statistically significant <1 h) (Fig. 3). At one hour, both particle types achieved a release of 88%, compared to free drug dissolution of the same amount after 4 hours. After 4 hours, the free HC dissolved slowly and after 24 hours a maximum of 96 ± 5.2% was achieved.
| Si-HC | Si-PEG-HC | |
|---|---|---|
| D = hydrodynamic diameter. Z.p. = Zetapotential. S.d. = standard deviation. | ||
| D (nm) ± s.d. | 196 ± 0.8 | 216 ± 4.0 |
| PDI ± s.d. | 0.24 ± 0.01 | 0.26 ± 0.03 |
| Z.p. (mV) ± s.d. | −37.0 ± 2.4 | 11.8 ± 0.7 |
| %M.l. ± s.d. | 31 ± 6.0 | 27 ± 13.5 |
4 Discussion
The main finding of this study describes biocompatible and stable, yet degradable, aggregated Si particles with a porous snow-lantern structure produced by cCVD method with the ability to add different surface modalities, load drugs and alter the release rate. The present part moves on to discuss these findings in three different sections. The first section is about the cCVD aggregate particles’ inherent properties. The second section discusses surface chemistry and the possibilities therein. The third and final section evaluates the drug delivery applicability of this material.4.1 Stable aggregate snow-lantern particles
Herein, we introduced a novel etching-free synthesis method of porous silicon nanoparticles with high surface area using the one-step cCVD approach. This fabrication method allows for tailoring particle properties and producing a large quantity of homogeneous material. Porosity was created by smaller primary particles connected in a controlled process to form stable aggregated particles. A high surface area is crucial for various applications of porous nanomaterials, particularly for drug carriers requiring significant drug adsorption. The surface area of the aggregate cCVD particles can be controlled by adjusting the primary particle size. With 10 nm primary particles, the aggregate surface area could reach >200 m2 g−1 (Table SI1†), which is a typical value seen for pSi NPs prepared from electrochemical etch in HF electrolytes.The primary particles were connected by covalent bonds formed during synthesis, enabling their stability in dispersed solutions. High surface charge and ionic repellant interactions kept the particles stable in water. Stability was compromised in PBS due to charge neutralization by salts.31 However, addition of 4% albumin to the PBS solution improved stability possibly due to the hydrophobic nature of albumin and the hydrophobic–hydrophobic interactions forming a coating around the particles, promoting dispersion by steric hindrance. This demonstrates the potential of coating, whether through adsorption or covalent bonding, to enhance particle stability. On the other hand, as seen from albumin's affinity for the cCVD particle surface, the absorption of biomolecules and protein corona formation can affect the nanoparticles’ fate in vivo as well as affecting the function of the biomolecules. Cellular responses primarily reflect the nanoparticle surface, and thus the dynamic protein corona, rather than the nanomaterial itself. However, there is a potential in controlling or limiting the protein corona by particle coatings, such as PEGylation.32,33
4.2 Surface chemistry
The cCVD particles were shown to be suitable for chemical modification by forming covalent bonds over oxidized Si groups. Oxidation was confirmed by FTIR and Raman and covalent coating with primary amine groups was confirmed by TGA and a change in surface charge from negative to positive. DMDASCP grafting exhibited higher density of amine groups and correspondingly higher zeta potential compared to APDMES grafting, consistent with literature findings that cyclic silane chemistry achieves greater reaction efficiency and surface coverage than monoethoxysilanes.34 Even mild air oxidation was sufficient for DMDASCP grafting.Blood circulation time, protein corona formation and elimination through reticuloendothelial system (RES) of nanomedicines can be improved by different PEGylation chemistries.35 PEG is also used to control dispersibility for other applications. Steric repulsive interactions unaffected by charge screening were introduced by PEG coating, stabilizing the particles in PBS similarly to the effect of albumin. Several techniques for PEGylation of silicon and silica surfaces are discussed in literature.36–38 In this study, the two-step PEGylation method with DMDASCP-mPEG was successful and achieved particle stabilization in PBS. The one-step PEGylated particles were not stable in PBS, due to low surface coverage. Method optimization of the one-step approach, however, may improve its grafting coverage. Despite the wide clinical use of PEG in drug-polymer conjugates and nanomedicines as well as in numerous consumer products, concerns have been raised about immunogenic and hypersensitive side-effects upon repeated exposure.39–41 Thus, there is a need for new alternative coatings that can retain the beneficial effects of PEGylation in nanomedicine.42
The surface modification techniques established herein can be utilized to incorporate different functionalities to the cCVD Si particles’ surface. Terminal amine groups serve as versatile conjugation sites for grafting various compounds or facilitating electrostatic interactions and non-covalent adsorption, due to the positive charge. The PEG-MAL terminus allows for conjugation with thiol and cysteine-containing molecules, making it suitable for peptide or protein conjugations like for instance the targeting ligand iRGD.43 The surface chemistries tested herein appear to be equally effective for the cCVD and TruTag particles. Therefore, one can assume that the wide variety of chemistries developed for electrochemically etched pSi can be successfully applied to cCVD Si particles.
4.3 Drug delivery
The cCVD particle characteristics discussed in the two sections above make them suitable for use as nanocarriers in drug delivery in at least four aspects. They are colloidally stable and have porous structure that can be loaded with different payloads. Versatile chemical modifications of the cCVD Si particles allow for incorporation of bioactive molecules. Additionally, the cCVD particles were shown to be non-toxic and degraded in physiologic conditions at 100 μg mL−1 and below. These in vitro studies suggest safety of the cCVD Si particles without surface modifications, similar to the properties of electrochemically etched pSi NP.5 Varying toxicity for different surface modifications of porous silicon materials has been documented.20,44,45 Furthermore, depending on the administration route, extensive in vivo studies and clinical trials of an eventual final nanomedicine product are needed to guarantee clinical safety.46The release rate of drugs from nanocarriers depends on several factors including the degradation rate of the carrier material, the surface area available for adsorption and dissolution, the drug solubility and the hydrophilic-lipophilic, electrostatic, or other interactions between the drug and carrier. The HC release was faster from loaded particles compared to free dissolution. This shows the potential of the cCVD Si NPs to increase the dissolution rate of poorly water-soluble drugs, similarly to observations made with electrochemically etched pSi NP and other poorly soluble drugs.47,48 A slower release rate was seen from the non-PEGylated particles which indicates that HC has a stronger affinity to the unmodified particle surface. The change in zeta potential of PEGylated particles after loading may indicate surface adsorption rather than pore loading. Additionally, improved dispersibility observed for PEGylated particles in PBS could provide a larger exposed surface area resulting in a faster release of the drug payload. These findings suggest that PEGylation and other coatings can modify drug release kinetics from cCVD Si particles.
5 Conclusion
Controlled aggregated snow lantern-like Si nanoparticles were successfully prepared from the cCVD method. These particles hold promise as drug carriers, being stable aggregates with a mesoporous structure, biocompatible, degradable at physiological conditions, and versatile for surface modifications. SEM and BET revealed sub-50 nm primary particle size, while DLS confirmed aggregate size of 210 nm. Surface area measurements supported aggregate formation and showed porosity. The cCVD synthesis parameters can be tuned to control the particle surface area and porosity which can be useful for fitting different drug payloads. A variety of different chemical modifications were applied on the oxidized cCVD Si particle surface. Surface grafting with primary amine groups was achieved using APDMES and DMDASCP, with higher grafting observed with the latter. Two-step PEGylation via DMDASCP gave 7.9 weight% organic content and ensured colloidal stability in PBS. Impregnation loading of a model drug, hydrocortisone, gave a mass load of 27–31%. The release rate from the loaded particles was enhanced compared to free hydrocortisone. Thus, the cCVD Si particles have a potential for use in drug delivery.Author contributions
HMJ: conceptualization, formal analysis, investigation, writing – original draft, visualization. SHN: methodology, writing – review & editing. WF: conceptualization, writing – review & editing. RD: formal analysis, investigation, visualization. MJS: resources, writing – review & editing, supervision. JK: conceptualization, methodology, writing – review & editing. MH: methodology, writing – review & editing, supervision.Conflicts of interest
JK, WF and HMJ are inventors of a patent application with the title “Silicon particles for drug delivery”. The assignee of the patent application is Nacamed AS. WF and JK have shares in Nacamed AS and are consultants for the company. MJS is a scientific founder (SF), member of the Board of Directors (BOD), Advisory Board (AB), Scientific Advisory Board (SAB), acts as a paid consultant (PC) or has an equity interest (EI) in the following: Aivocode, Inc (AB, EI); Bejing ITEC Technologies (SAB, PC); Lisata Therapeutics (EI); Illumina (EI), Impilo Therapeutics (SAB, EI), Matrix Technologies (EI); Precis Therapeutics (SF, BOD, EI), Quanterix (EI), Spinnaker Biosciences, Inc. (SF, BOD, EI); TruTag Technologies (SAB, EI); and Well-Healthcare Technologies (SAB, PC). Although one or more of the grants that supported this research has been identified for conflict-of-interest management based on the overall scope of the project and its potential benefit to the companies listed, the research findings included in this publication may not necessarily relate to their interests. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies.Acknowledgements
This research was supported by the Research Council of Norway under the NANO2021 program, project number 313954. This research was also partially supported by NSF through the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC), DMR-2011924. The authors also acknowledge the use of facilities and instrumentation supported by NSF through the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC), DMR-2011924. The authors thank Samson Yuxiu Lai for contributing with SEM analysis, Magnus Kristoffersen for facilitating ICP-OES analyses and Magnus Sørby for contributing with XRD analyses. The SEM and XRD work were carried out at the Institute for Energy Technology (IFE), Norway. The ICP-OES analyses were carried out at the University of Oslo, Department of Chemistry, Norway. The cell viability studies were conducted at the Norwegian University of Science and Technology (NTNU), Department of Physics, Norway. The particle surface modification experiments, the BET, TGA, Raman and FTIR analyses were performed in the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC) Materials Characterization Facility at the University of California San Diego, USA. The remaining work was carried out at the University of Oslo, Department of Pharmacy, Norway.References
- F. Winau, O. Westphal and R. Winau, Paul Ehrlich—in search of the magic bullet, Microbes Infect., 2004, 6(8), 786–789 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33(9), 941–951 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20(2), 101–124 CrossRef CAS PubMed.
- A. Z. Mirza and F. A. Siddiqui, Nanomedicine and drug delivery: a mini review, Int. Nano Lett., 2014, 4, 94 CrossRef.
- J. H. Park, L. Gu, G. Von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Biodegradable luminescent porous silicon nanoparticles for in vivo applications, Nat. Mater., 2009, 8(4), 331–336 CrossRef CAS PubMed.
- H. A. Santos, E. Mäkilä, A. J. Airaksinen, L. M. Bimbo and J. Hirvonen, Porous silicon nanoparticles for nanomedicine: preparation and biomedical applications, Nanomedicine, 2014, 9(4), 535–554 CrossRef CAS PubMed.
- B. Godin, C. Chiappini, S. Srinivasan, J. F. Alexander, K. Yokoi, M. Ferrari, P. Decuzzi and X. Liu, Discoidal porous silicon particles: fabrication and biodistribution in breast cancer bearing mice, Adv. Funct. Mater., 2012, 22(20), 4225–4235 CrossRef CAS PubMed.
- Z. Qin, J. Joo, L. Gu and M. J. Sailor, Size control of porous silicon nanoparticles by electrochemical perforation etching, Part. Part. Syst. Charact., 2014, 31(2), 252–256 CrossRef CAS.
- S. Semlali, B. Cormary, M. L. De Marco, J. Majimel, A. Saquet, Y. Coppel, M. Gonidec, P. Rosa and G. L. Drisko, Effect of solvent on silicon nanoparticle formation and size: a mechanistic study, Nanoscale, 2019, 11(11), 4696–4700 RSC.
- B. M. Nolan, T. Henneberger, M. Waibel, T. F. Fässler and S. M. Kauzlarich, Silicon Nanoparticles by the Oxidation of [Si4] 4−-and [Si9] 4−-Containing Zintl Phases and Their Corresponding Yield, Inorg. Chem., 2015, 54(1), 396–401 CrossRef CAS PubMed.
- M. Dasog, Z. Yang, S. Regli, T. M. Atkins, A. Faramus, M. P. Singh, E. Muthuswamy, S. M. Kauzlarich, R. D. Tilley and J. G. Veinot, Chemical insight into the origin of red and blue photoluminescence arising from freestanding silicon nanocrystals, ACS Nano, 2013, 7(3), 2676–2685 CrossRef CAS PubMed.
- S. K. Bux, M. Rodriguez, M. T. Yeung, C. Yang, A. Makhluf, R. G. Blair, J. P. Fleurial and R. B. Kaner, Rapid solid-state synthesis of nanostructured silicon, Chem. Mater., 2012, 22(8), 2534–2540 CrossRef.
- S. W. Chung, J. Y. Yu and J. R. Heath, Silicon nanowire devices, Appl. Phys. Lett., 2000, 76, 2068–2070 CrossRef CAS.
- S. H. C. Anderson, H. Elliott, D. J. Wallis, L. T. Canham and J. J. Powell, Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions, Phys. Status Solidi A, 2003, 197(2), 331–335 CrossRef CAS.
- T. Tanaka, L. S. Mangala, P. E. Vivas-Mejia, R. Nieves-Alicea, A. P. Mann, E. Mora, H. D. Han, M. Shahzad, X. Liu, R. Bhavane, J. Gu, J. R. Fakhoury, C. Chiappini, C. Lu, K. Matsuo, B. Godin, R. L. Stone, A. M. Nick, G. Lopez-Berestein, A. K. Sood and M. Ferrari, Sustained small interfering RNA delivery by mesoporous silicon particles, Cancer Res., 2010, 70(9), 3687–3696 CrossRef CAS PubMed.
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z. G. She, D. Kim, A. P. Mann, T. Molder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy, Nat. Biomed. Eng., 2018, 2(2), 95–103 CrossRef CAS PubMed.
- B. Godin, J. Gu, R. E. Serda, R. Bhavane, E. Tasciotti, C. Chiappini, X. Liu, T. Tanaka, P. Decuzzi and M. Ferrari, Tailoring the degradation kinetics of mesoporous silicon structures through PEGylation, J. Biomed. Mater. Res., Part A, 2010, 94(4), 1236–1243 CrossRef PubMed.
- D. Lumen, S. Näkki, S. Imlimthan, E. Lambidis, M. Sarparanta, W. Xu, V. P. Lehto and A. J. Airaksinen, Site-specific 111In-radiolabeling of dual-PEGylated porous silicon nanoparticles and their in vivo evaluation in murine 4T1 breast cancer model, Pharmaceutics, 2019, 11(12), 686 CrossRef CAS PubMed.
- H. M. Johnsen, W. Filtvedt, M. Hiorth and J. Klaveness, Silicon nanoparticles for oral administration of molecular hydrogen, Int. J. Pharm., 2022, 629, 122371 CrossRef CAS PubMed.
- D. Lumen, S. Wang, E. Mäkilä, S. Imlimthan, M. Sarparanta, A. Correia, C. W. Haug, J. Hirvonen, H. A. Santos, A. J. Airaksinen, W. Filtvedt and J. Salonen, Investigation of silicon nanoparticles produced by centrifuge chemical vapor deposition for applications in therapy and diagnostics, Eur. J. Pharm. Biopharm., 2021, 158, 254–265 CrossRef CAS PubMed.
- G. M. Wyller, T. Preston, T. T. Mongstad, H. Klette, Ø. Nordseth, D. Lindholm, W. O. Filtvedt and E. S. Marstein, Thermal decomposition of monosilane observed in a free space reactor, 32nd European Photovoltaic Solar Energy Conference and Exhibition (EU PVSEC XXXII 2BO), 2016, 2, 294–299 Search PubMed.
- G. M. Wyller, T. J. Preston, H. Klette, Ø. Nordseth, T. T. Mongstad, W. O. Filtvedt and E. S. Marstein, Critical nucleation concentration for monosilane as function of temperature observed in a free space reactor, Energy Procedia, 2016, 92, 904–912 CrossRef CAS.
- W. O. Filtvedt and J. Filtvedt, Reactor and method for production of silicon by chemical vapor deposition, PCT patent application, WO2013048258, 2013 Search PubMed.
- W. O. Filtvedt, Method for producing silicon particles for use as anode material in lithium ion rechargeable batteries, use of a rotating reactor for the method and particles produced by the method and a reactor for operating the method, PCT patent application, WO2018052318, 2019 Search PubMed.
- W. O. Filtvedt, J. Klaveness and H. M. Johnsen, Silicon particles for drug delivery, PCT patent application, WO2022223822, 2022 Search PubMed.
- G. M. Wyller, T. J. Preston, S. G. Anjitha, M. O. Skare and E. S. Marstein, Combination of a millimeter scale reactor and gas chromatography-mass spectrometry for mapping higher order silane formation during monosilane pyrolysis, J. Cryst. Growth, 2020, 530, 125305 CrossRef CAS.
- G. M. Wyller, T. J. Preston, H. Klette, T. Mongstad and E. S. Marstein, Identification of higher order silanes during monosilane pyrolysis using gas chromatography-mass spectrometry, J. Cryst. Growth, 2018, 498, 315–327 CrossRef CAS.
- K. J. Ong, T. J. MacCormack, R. J. Clark, J. D. Ede, V. A. Ortega, L. C. Felix, M. Dang, G. Ma, H. Fenniri, J. Veinot and G. G. Goss, Widespread nanoparticle-assay interference: implications for nanotoxicity testing, PLoS One, 2014, 9(3), e90650 CrossRef PubMed.
- R. S. Dariani and Z. Ahmadi, Study of porous silicon structure by Raman scattering, Optik, 2013, 124(22), 5353–5356 CrossRef CAS.
- L. Z. Liu, X. L. Wu, Z. Y. Zhang, T. H. Li and P. K. Chu, Raman investigation of oxidation mechanism of silicon nanowires, Appl. Phys. Lett., 2009, 95(9), 093109 CrossRef.
- D. Takács, M. Tomšič and I. Szilagyi, Effect of Water and Salt on the Colloidal Stability of Latex Particles in Ionic Liquid Solutions, Colloids Interfaces, 2022, 6, 2 CrossRef.
- I. Lynch and K. A. Dawson, Protein–nanoparticle interactions, Nano Today, 2008, 3(1), 40–47 CrossRef CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Biomolecular coronas provide the biological identity of nanosized materials, Nat. Nanotechnol., 2012, 7(12), 779–786 CrossRef CAS PubMed.
- D. Kim, J. M. Zuidema, J. Kang, Y. Pan, L. Wu, D. Warther, B. Arkles and M. J. Sailor, Facile surface modification of hydroxylated silicon nanostructures using heterocyclic silanes, J. Am. Chem. Soc., 2016, 138(46), 15106–15109 CrossRef CAS PubMed.
- C. F. Adhipandito, S. H. Cheung, Y. H. Lin and S. H. Wu, Atypical renal clearance of nanoparticles larger than the kidney filtration threshold, Int. J. Mol. Sci, 2021, 22(20), 11182 CrossRef CAS PubMed.
- L. Adumeau, C. Genevois, L. Roudier, C. Schatz, F. Couillaud and S. Mornet, Impact of surface grafting density of PEG macromolecules on dually fluorescent silica nanoparticles used for the in vivo imaging of subcutaneous tumors, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861(6), 1587–1596 CrossRef CAS PubMed.
- M. P. Schwartz, F. Cunin, R. W. Cheung and M. J. Sailor, Chemical modification of silicon surfaces for biological applications, Phys. Status Solidi A, 2005, 202(8), 1380–1384 CrossRef CAS.
- K. Ma, D. Zhang, Y. Cong and U. Wiesner, Elucidating the mechanism of silica nanoparticle PEGylation processes using fluorescence correlation spectroscopies, Chem. Mater., 2016, 28(5), 1537–1545 CrossRef CAS.
- B. M. Chen, T. L. Cheng and S. R. Roffler, Polyethylene glycol immunogenicity: theoretical, clinical, and practical aspects of anti-polyethylene glycol antibodies, ACS Nano, 2021, 15(9), 14022–14048 CrossRef CAS PubMed.
- N. d'Avanzo, C. Celia, A. Barone, M. Carafa, L. Di Marzio, H. A. Santos and M. Fresta, Immunogenicity of polyethylene glycol based nanomedicines: mechanisms, clinical implications and systematic approach, Adv. Theranostic, 2020, 3(3), 1900170 CrossRef.
- M. Ibrahim, E. Ramadan, N. E. Elsadek, S. E. Emam, T. Shimizu, H. Ando, Y. Ishima, O. H. Elgarhy, H. A. Sarhan, A. K. Hussein and T. Ishida, Polyethylene glycol (PEG): The nature, immunogenicity, and role in the hypersensitivity of PEGylated products, J. Controlled Release, 2022, 351, 215–230 CrossRef CAS PubMed.
- Y. W. Kong and E. C. Dreaden, PEG: Will it come back to you? polyethelyne glycol immunogenicity, COVID vaccines, and the case for new PEG derivatives and alternatives, Front. Bioeng. Biotechnol., 2022, 10, 879988 CrossRef PubMed.
- J. Joo, X. Liu, V. R. Kotamraju, E. Ruoslahti, Y. Nam and M. J. Sailor, Gated luminescence imaging of silicon nanoparticles, ACS Nano, 2015, 9(6), 6233–6241 CrossRef CAS PubMed.
- E. Secret, K. Smith, V. Dubljevic, E. Moore, P. Macardle, B. Delalat, M. L. Rogers, T. G. Johns, J. O. Durand, F. Cunin and N. H. Voelcker, Antibody–functionalized porous silicon nanoparticles for vectorization of hydrophobic drugs, Adv. Healthcare Mater., 2013, 2(5), 718–727 CrossRef CAS PubMed.
- M. A. Shahbazi, M. Hamidi, E. M. Mäkilä, H. Zhang, P. V. Almeida, M. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, The mechanisms of surface chemistry effects of mesoporous silicon nanoparticles on immunotoxicity and biocompatibility, Biomaterials, 2013, 34(31), 7776–7789 CrossRef CAS PubMed.
- C. Domingues, A. Santos, C. Alvarez-Lorenzo, A. Concheiro, I. Jarak, F. Veiga, I. Barbosa, M. Dourado and A. Figueiras, Where is nano today and where is it headed? A review of nanomedicine and the dilemma of nanotoxicology, ACS Nano, 2022, 16(7), 9994–10041 CrossRef CAS PubMed.
- J. Salonen, L. Laitinen, A. M. Kaukonen, J. Tuura, M. Björkqvist, T. Heikkilä, K. Vähä-Heikkilä, J. Hirvonen and V. P. Lehto, Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs, J. Controlled Release, 2005, 108(2–3), 362–374 CrossRef CAS PubMed.
- K. L. Jarvis, T. J. Barnes and C. A. Prestidge, Surface chemistry of porous silicon and implications for drug encapsulation and delivery applications, Adv. Colloid Interface Sci., 2012, 175, 25–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05655d |
| This journal is © The Royal Society of Chemistry 2024 |