
Asymmetric contact-induced selective doping of CVD-grown bilayer WS2 and its application in high-performance photodetection with an ultralow dark current†
Abdul Kaium
Mia
 a,
M.
Meyyappan
a and
P. K.
Giri
*ab
a,
M.
Meyyappan
a and
P. K.
Giri
*ab
aCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India. E-mail: giri@iitg.ac.in
bDepartment of Physics, Indian Institute of Technology Guwahati, Guwahati 781039, India
First published on 28th March 2024
Abstract
Two-dimensional (2D) transition metal dichalcogenides (TMDs) are excellent candidates for high-performance optoelectronics due to their high carrier mobility, air stability and strong optical absorption. However, photodetectors made with monolayer TMDs often exhibit a high dark current, and thus, there is a scope for further improvement. Herein, we developed a 2D bilayer tungsten disulfide (WS2) based photodetector (PD) with asymmetric contacts that exhibits an exceptionally low dark current and high specific detectivity. High-quality and large-area monolayer and bilayer WS2 flakes were synthesized using a thermal chemical vapor deposition system. Compared to conventional symmetric contact electrodes, utilizing metal electrodes with higher and lower work functions relative to bilayer WS2 aids in achieving asymmetric lateral doping in the WS2 flakes. This doping asymmetry was confirmed through the photoluminescence spectral profile and Raman mapping analysis. With the asymmetric contacts on bilayer WS2, we find evidence of selective doping of electrons and holes near the Ti and Au contacts, respectively, while the WS2 region away from the contacts remains intrinsic. When compared with the symmetric contact case, the dark current in the WS2 PD with asymmetric (Au, Ti) contact decreases by an order of magnitude under reverse bias with a concomitant increase in the photocurrent, resulting in an improved on/off ratio of ∼105 and overall improved device performance under identical illumination conditions. We explained this improved performance based on the energy band alignment showing a unidirectional charge flow under light illumination. Our results indicate that the planar device structure and compatibility with current nanofabrication technologies can facilitate its integration into advanced chips for futuristic low-power optoelectronic and nanophotonic applications.
Introduction
There is an ever-growing demand for advancements in the development of energy-efficient optoelectronic devices with low power consumption1–3 for diverse applications ranging from optoelectronics to point-of-care usage.4–6 2D layered materials such as graphene, transition metal dichalcogenides (TMDs), and MXenes have attracted attention due to their capability to scale down electronic devices to the atomic level.7–9 Among all 2D materials, semiconducting TMDs, characterized by their tunable bandgap, planar structure, and high carrier mobility, have been explored for a diverse range of applications from photodetectors (PDs) and biosensors to energy storage devices.10–12 The formation of P–N junctions through heterostructures involving TMDs such as MoS2/WSe2,13 WS2/WSe2,14 WS2/MoS2,15 and MoSe2/WS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 has been explored for enhanced photovoltaics and electronic switching applications. There is a lack of direct and precisely adjustable doping methods in 2D TMD semiconductors when compared to silicon technology. As a result, alternative attempts have been made to achieve precise asymmetric control of doping in 2D TMDs. These methods encompass local electrostatic gating,17 chemical doping,18 carrier adjustment through plasma treatment19 and carrier injection using uneven metal contacts.20,21
16 has been explored for enhanced photovoltaics and electronic switching applications. There is a lack of direct and precisely adjustable doping methods in 2D TMD semiconductors when compared to silicon technology. As a result, alternative attempts have been made to achieve precise asymmetric control of doping in 2D TMDs. These methods encompass local electrostatic gating,17 chemical doping,18 carrier adjustment through plasma treatment19 and carrier injection using uneven metal contacts.20,21
WS2 is a member of the TMD family and has been identified as an excellent candidate for optoelectronic applications due to its high stability, high carrier mobility and potential for mass production.22,23 There have been reports on WS2-based photodetectors in the visible spectrum using monolayer, few layer and multilayer WS2.24–30 However, the performance of these devices is not up to the standards of typical commercial photodetectors. For instance, Perea-López et al. observed a low responsivity on the order of μA W−1 in multilayer WS2 PDs within the visible range.24 By passivating the sulfur vacancy sites, the responsivity of few-layered WS2 was enhanced to a few mA W−1 to 5.7 A W−1; however, the on/off ratio dropped by a few orders. This decrease was caused by the increase in dark current due to the electron doping effect.25,31 The photoresponse was improved by many orders and reached ∼108 in a 3-terminal field effect transistor (FET) configuration but remains low in two terminal devices.26 The responsivity of monolayer WS2 PDs in two-terminal devices was enhanced by incorporating plasmonic nanoparticles.32 The incorporation of plasmonic gold nanoparticles enhanced the visible absorption, improving the responsivity and on/off ratio. Thus, one can enhance the WS2 PD performance by doping. The formation of a Schottky junction with WS2 improves the performance in contrast to the Ohmic junctions33 by reducing the dark current and facilitating charge separation due to the built-in potential.20 2D WS2 synthesized by chemical vapor deposition (CVD) exhibits high stability with a low defect density and high crystalline quality, and it usually exhibits good mobility at room temperature.34 Interestingly, the carrier mobility in 2D WS2 is layer-dependent. Specifically, bilayer WS2 displays mobility that is twice that of monolayer WS2, and it exhibits enhanced photo-absorption due to a higher thickness, underscoring the significance of using bilayer WS2 in photodetection applications.35 Chemical doping on the WS2 surface yields improved photodetection performance. However, indigenous asymmetric doping through an asymmetric contact-based photodetector using 2D WS2 has not been explored yet.
Herein, we report an asymmetric contact-based bilayer WS2 photodetector in a planar configuration showing significantly enhanced performance. We synthesized uniform large-area high-quality monolayer and bilayer WS2 on a SiO2/Si substrate by a CVD process. The WS2 flakes were transferred from the growth substrate to a dry SiO2/Si substrate using a polymethyl methacrylate (PMMA)-assisted wet transfer method, ensuring that the optoelectronic properties remained intact. We conducted comparative analyses between symmetric and asymmetric contacts to differentiate the impact of asymmetric contacts on the photodetection performance. The asymmetric contacts lead to the creation of two lateral junctions in WS2, resulting in a p–n–n+ type junction. This configuration significantly reduces the dark current under reverse bias and enhances the on/off ratio by an order of magnitude with a peak responsivity of 1.2 A W−1.
Experimental section
Synthesis of WS2 flakes by CVD
WS2 flakes were synthesized by thermal CVD using a two-zone muffle furnace. The schematic representation of the experimental setup is shown in Fig. 1. Growth details can be found in the ESI.† Additionally, the temperature profile and Ar flow throughout the experiment are displayed in Fig. S1.† We synthesized uniform large-area monolayer and bilayer WS2 flakes through meticulous adjustment of growth conditions, as shown in Fig. 1(a) and (c). Fig. 1(b and d) display the atomic force microscopy (AFM) images indicating the respective thickness profiles of the as-grown WS2 flakes. | ||
| Fig. 1 Schematic representation of the CVD setup used for the synthesis of 2D WS2 flakes. (a) Optical image of the monolayer WS2 flakes on SiO2/Si (90 nm oxide) wafer showing uniform large area coverage. (b) AFM image of a monolayer WS2 flake with the thickness profile showing a monolayer thickness of 0.75 nm. (c) Optical image of bilayer WS2 flakes on SiO2/Si (280 nm oxide) wafer showing uniform large area coverage. (d) AFM image of a bilayer WS2 flake with the thickness profile showing a bilayer thickness of 1.65 nm. | ||
Transfer of WS2 flakes and device fabrication
The WS2 flakes were transferred from the growth substrate to a dry SiO2/Si substrate by a PMMA-assisted wet transfer process in two different ways, as discussed in the ESI.† The substrate was coated with PMMA using a spin coater at 3000 rpm for 30 seconds. Following this, the PMMA-coated wafer was immersed in a 1.0 M NaOH solution for 3 hours, resulting in the detachment of the PMMA film with the WS2 flakes from the substrate, as illustrated in Fig. S2.† The film was washed multiple times in DI water to reduce the amount of impurities. It was subsequently transferred onto a dry SiO2/Si wafer, and the top PMMA layer was eliminated by immersing it in acetone overnight. The optical image in Fig. S3† shows the WS2 flakes following a successful transfer in two different ways. Fig. S3(a and b)† shows the presence of cracks and mechanical damage to the transferred WS2 flakes, while Fig. S3(c and d)† demonstrates that the flakes remain intact without any signs of mechanical damage. The same transfer process as Fig. S3(c and d)† was employed for transferring WS2 on TEM grids for field emission transmission electron microscopy (FETEM) characterization.PD devices were fabricated using a conventional UV lithography technique, and the metal contacts were deposited via an e-beam evaporation system. First, the photoresist (PR) S1813 (MICROPOSIT™) was coated using a spin coater. For symmetric contact-based PD, both metal pads were patterned simultaneously by a direct UV laser writing process on a single flake, followed by metal deposition and lift-off. The detailed fabrication process for asymmetric contact-based PDs is presented in the Results and discussion section. Titanium (Ti) contact was patterned on one side of a flake by selective opening via UV lithography followed by 45 nm Ti deposition and lift-off. The Au electrode was patterned following the same steps after focusing on the exact flake. Finally, the devices were heated at 90 °C for 10 minutes to remove the residual solvents.
Characterization
The thickness of the WS2 flakes was measured using atomic force microscopy (AFM) (Cypher, Oxford Instruments) imaging in non-contact tapping mode. A Titan Themis 300 kV (FEI, now Thermo Fisher) was used for FETEM analysis. The chemical composition and oxidation states of WS2 were analyzed using an X-ray photoelectron spectroscopy (XPS) system (ULVAC, PHI). The Raman spectra of WS2 were recorded using a high-resolution micro-Raman spectrometer (LabRam HR800, Jobin–Yvon) with excitation wavelengths of 532 nm and 488 nm with a 100× objective having a laser spot size of ∼1 μm. The same instrument was used to capture the optical images and photoluminescence (PL) spectra. UV–Vis absorption spectra of the samples were recorded using a spectrophotometer (PerkinElmer, Lamda 950). An FETEM (JEOL, 2100F) operated at 200 kV was used to perform energy-dispersive X-ray spectroscopy (EDS) for the transferred WS2. All the electrical characterization studies were performed in a shielded DC probe station integrated with a source meter (Keithley 4200 SCS) with external laser assembly and function generators.Results and discussion
A schematic of the CVD setup for the growth of monolayer and bilayer WS2 is shown in Fig. 1 (top panel). The uniform contrast of the WS2 flakes observed in the optical images depicted in Fig. 1(a) and (c) indicates the precise control of monolayer and bilayer WS2 synthesis by CVD. The AFM height profile analysis reveals that the monolayer WS2 has a thickness of ∼0.75 nm, while the bilayer is ∼1.65 nm, as shown in Fig. 1(b) and (d), respectively.36 The WS2 flakes were synthesized at an extremely high temperature of 1000 °C, which impacted the SiO2 dielectric layer. Consequently, the WS2 flakes, in their as-grown state, were transferred from the original growth substrate to a SiO2/Si substrate with a 300 nm dry oxide layer (Namkang Hi-Tech CO., Ltd) for subsequent device fabrication.The Raman spectrum of the as-grown bilayer WS2 flake is displayed in Fig. 2(a). Several phonon modes are observed for 2D WS2 when excited with a 532 nm laser as shown in Fig. 2(a). The two characteristic Raman modes, A1g and E2g, corresponding to out-of-plane and in-plane vibrations of W and S atoms, are observed at 418.8 cm−1 and 355.1 cm−1, respectively, for bilayer WS2. Other observed optical phonon modes are labeled in the deconvoluted spectra in Fig. 2(a).37 Charge doping and layer numbers immensely influence the out-of-plane A1g mode, and the in-plane mode E2g is sensitive to the induced strain.38 The separation between these two modes is characteristic of layer thickness, providing an indirect way to know layer numbers.39 Since the transferred WS2 was used for PD fabrication, the quality of transferred WS2 flakes was analyzed with the help of Raman spectroscopy. The fitted Raman spectra of monolayer and bilayer WS2 both before and after transfer are shown in Fig. S4.† The distinct Raman modes after transfer indicate seamless transfer to the device substrate without compromising its quality.40 The E2g mode experiences a blue shift of 1.1 cm−1 after the transfer of WS2, which is consistent with the anticipated reduction in strain following the transfer process.40,41 The lattice mismatch and difference of thermal expansion coefficients between SiO2 and WS2 cause significant strain for the high-temperature grown WS2 flakes directly on SiO2/Si substrates, which is subsequently reduced due to the transfer process.42 The 532 nm laser excites various phonon modes, and the peak fitting can notoriously be error-prone. Hence, the Raman spectra obtained using a 532 nm laser are not ideal for finding the characteristic mode separation. When excited with a 488 nm laser, only intense in-plane and out-of-plane characteristic Raman modes are observed along with a low-intensity 2LA mode, as shown in Fig. S5.† Hence, the separation of the A1g and E2g modes was measured using 488 nm laser excitation. Monolayer WS2 exhibits a separation of ∼61.4 cm−1, which increases to ∼62.8 cm−1 for the bilayer.39 The shift of the A1g mode for the bilayer is the indication of a higher electronic charge density for bilayer WS2.
 | ||
| Fig. 2 (a) Deconvoluted Raman spectra of bilayer WS2 under 532 nm laser excitation. (b) XPS survey spectra of CVD-grown bilayer WS2. (c) The high-resolution core binding energy spectrum of sulfur (S). (d) The high-resolution core binding energy spectrum of tungsten (W). | ||
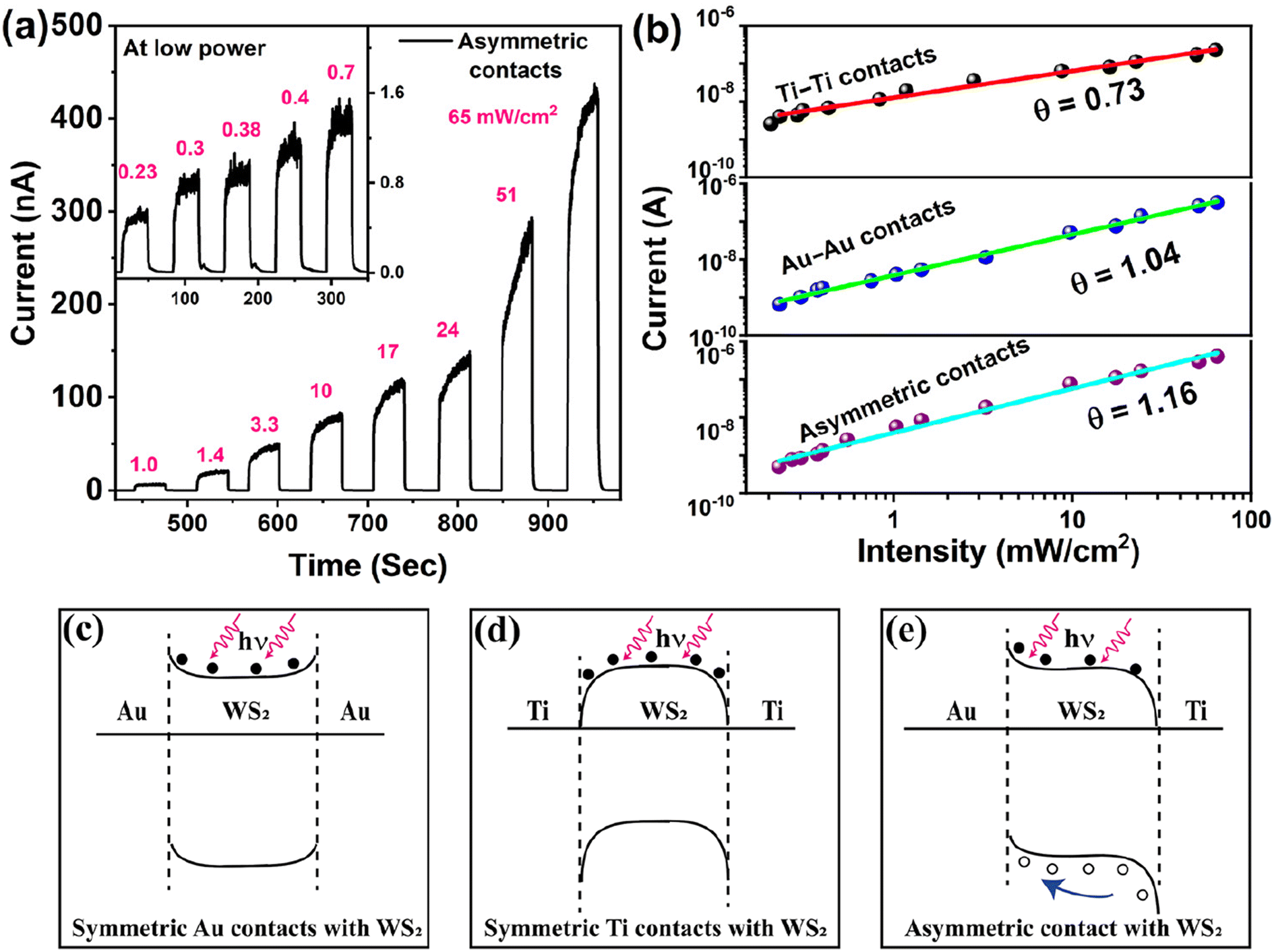
The chemical composition and stoichiometry of the WS2 flakes were analyzed using XPS spectra. The presence of two characteristic peaks of tungsten and sulfur in the survey spectra of Fig. 2(b) confirms the synthesis of WS2 using transition metal oxide and chalcogen precursors. All the peak positions were measured with respect to the carbon peak at 284.8 eV. The high-resolution binding energy spectrum of S 2p in Fig. 2(c) shows a doublet at 162.9 eV and 164.1 eV, corresponding to S 2p3/2 and S 2p1/2, respectively.43 These two peaks correspond to the S2− reduction state of sulfur, confirming WS2 synthesis. Furthermore, we noticed a minor peak at 163.3 eV (with a spectral weight of 3.0%), corresponding to sulfur vacancy defects.44,45 This observation underscores the excellent growth quality of WS2 achieved through CVD. In the high-resolution XPS spectra of W 4f, we observed two prominent peaks centered at 33.3 eV and 35.4 eV in Fig. 2(d), corresponding to the W 4f7/2 and W 4f5/2, respectively, associated with W4+ oxidation states. These findings confirm the presence of tungsten in the W4+ oxidation state, providing evidence for the successful synthesis of the semiconducting 2H WS2 phase.43 Furthermore, there are two low-intensity peaks at 36.2 eV and 38.3 eV, which correspond to the W4f7/2 and W4f5/2 orbitals, respectively, and indicate the presence of the W6+ oxidation state.43,46 The substrate was placed just above the WO3 powder for the CVD growth of WS2. The Raman spectra recorded on the WS2 flakes using the micro-Raman set-up showed no signature of WO3. Thus, the minute presence of the W6+ oxidation state can be attributed to the presence of WO3 precursors at a few places on the substrate. The curve fitted within the XPS spectrum was employed for determining the S to W ratio. The S to W ratio was calculated using the formula, 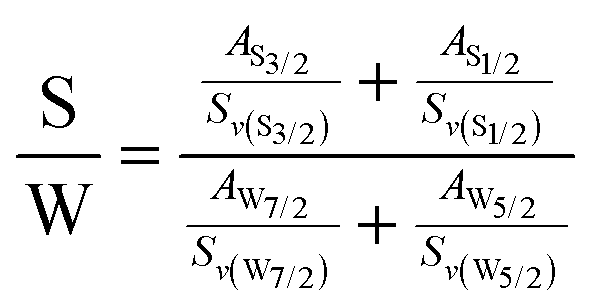 , where Ax is the area under the fitted curve for the x-orbital and Svx is the corresponding sensitivity factor,47 yielding a value of 2.04, which closely aligns with the anticipated value of 2.
, where Ax is the area under the fitted curve for the x-orbital and Svx is the corresponding sensitivity factor,47 yielding a value of 2.04, which closely aligns with the anticipated value of 2.
The FETEM images of the CVD-grown WS2 flakes are displayed in Fig. 3. Fig. 3(a) shows a single free-standing monolayer WS2. The flake underwent folding during the transfer from the growth substrate to the TEM grid. Fig. 3(b) illustrates the presence of hexagonal bright spots in the reciprocal space when performing Selected Area Electron Diffraction (SAED) on a monolayer WS2. The diffraction spots were labeled and the lattice parameter was observed to be approximately 0.295 nm from the SAED pattern, which aligns with previous reports.48,49Fig. 3(c) displays the high-resolution TEM (HRTEM) image of a monolayer WS2, revealing an interplanar spacing of 0.26 nm associated with the 〈100〉 crystallographic plane of hexagonal WS2.50 The composition was additionally determined through EDS elemental mapping using TEM. The EDS analysis reveals the existence of 40.0% tungsten and 60.0% sulfur, resulting in a sulfur-to-tungsten ratio of 1.5, suggesting the presence of sulfur vacancies. XPS analysis determined a sulfur-to-tungsten ratio of 2.04, suggesting the creation of vacancy sites due to the high-energy electron beam during the prolonged EDS examination in FETEM.51
 | ||
| Fig. 3 TEM images of transferred WS2 flakes. (a) Freestanding monolayer WS2 flake, folded during the transfer process. (b) Hexagonal diffraction (SAED) pattern on monolayer WS2. (c) HRTEM image with lattice planes; the inset shows an interplanar spacing of 0.26 nm. (d) EDS elemental mapping of WS2 flakes. (e) and (f) Elemental mapping of tungsten and sulfur, respectively. | ||
The UV-Vis absorption spectrum in Fig. 4(a) shows two strong excitonic absorption peaks.52 The A and B excitonic absorption peaks that arise from direct bandgap transition at K points are found to be at 622 nm and 514 nm, respectively, for monolayer WS2.53 The spin–orbit coupling-induced splitting in the valence band at K point gives rise to an energy gap of approximately 0.42 eV between those peaks. There is a redshift in the absorption peaks for the bilayer, which are found at 628 nm and 518 nm, corresponding to A and B excitonic absorption, respectively. Besides the redshift, there is a substantial rise in absorption intensity that can be attributed to the increased thickness in bilayer WS2, implying higher absorption. Moreover, two faint absorption peaks exist at 453 nm and 415 nm, denoted as C and D in the absorption spectrum. These peaks result from optical transitions between the peaks of the density of states in the valence and conduction bands.53,54
 | ||
| Fig. 4 (a) UV-visible absorption spectra of monolayer and bilayer WS2 flakes. (b) PL emission spectra of monolayer and bilayer WS2 under 532 nm laser excitation. The inset shows the bar diagram of the relative PL intensity for monolayer and bilayer WS2. (c) Deconvoluted PL emission spectrum for monolayer WS2. (d) Excitation wavelength dependent photo I–V characteristics of the WS2 PD for symmetric Au contacts. | ||
Fig. 4(b) shows a comparison of the PL emission spectra for monolayer and bilayer WS2. When the thickness of 2D WS2 is reduced to a monolayer, there is a transition from an indirect to a direct bandgap, resulting in significantly enhanced PL emission.55 The low-dimension monolayer WS2 exhibits high exciton binding energy at room temperature, aiding strong PL emission.56 This heightened PL at room temperature serves as a distinguishing characteristic that sets monolayers apart from other morphologies.57 When excited using a 532 nm laser, the bilayer WS2 PL exhibits an almost 99% reduction in intensity compared to monolayer WS2, accompanied by a shift towards longer wavelengths from 630 nm to 638 nm. The bar diagram in the inset of Fig. 4(b) compares the intensity of PL emission of monolayer and bilayer WS2. The laser excitation also generates trions and defect-bound excitons, which can be attributed to excess carrier generation and defects induced by local heating.58,59 The PL spectrum of monolayer WS2 was deconvoluted to find contributions from neutral exciton (Å), trion (A−), and defect-bound excitons (Xd), as shown in Fig. 4(c). The WS2 monolayer has high trion binding energy in the range of 30–40 meV at room temperature, leading to strong trion emission at room temperature.60 We observed a neutral exciton (Å) peak at 630.0 nm along with a trion (A−) peak at 641.0 nm and a defect-bound exciton (Xd) peak at 656.0 nm. The low spectral weight of defect-bound excitons indicates high-quality growth of WS2 by CVD, as revealed by Raman, XPS and FETEM analyses. The PL spectra of the transferred monolayer WS2 on SiO2/Si were compared with those of as-grown monolayer WS2. The transferred WS2 on the SiO2/Si substrate exhibits a 25.7 meV blue shift in the neutral exciton from 630.0 nm to 621.3 nm, as depicted in Fig. S6.† The blue shift observed can be ascribed to a decrease in strain during wet transfer processes, which was confirmed through Raman spectroscopy.61 There is an increase in trion formation from 24.0% to 32.5% after transfer, which is attributed to interfacial charge at the interface and solvent processing.42
A suitable energy photon can modulate the carrier concentration in the semiconducting 2H phase WS2, and hence the conductivity by photoinduced carrier generation. Fig. 4(d) displays the I–V characteristics under dark and light (various wavelength laser excitation) conditions. There is a minimal change in current with respect to dark conditions when exposed to 808 nm laser illumination, while the current increases by several orders of magnitude under 640 nm, 533 nm, 450 nm and 405 nm laser illumination. The bandgap of monolayer and bilayer WS2 is approximately 2.0 eV. However, the energy of the 808 nm laser excitation is only 1.5 eV, significantly lower than the bandgap of bilayer WS2. Consequently, the 808 nm laser excitation cannot generate excess electron–hole pairs in bilayer WS2, resulting in no change in current upon excitation using an 808 nm laser. The excitation energy for lasers with wavelengths other than 808 nm approaches or exceeds the bandgap of bilayer WS2, which leads to the generation of excess electron–hole pairs, thus increasing the current. This aligns with the UV-Vis absorption data, which shows no absorption beyond 700 nm but significant absorption below 650 nm. These observations confirm that the current enhancement results from carrier generation in the WS2 bilayer after suitable wavelength laser illumination, enabling its function as a photodetector.
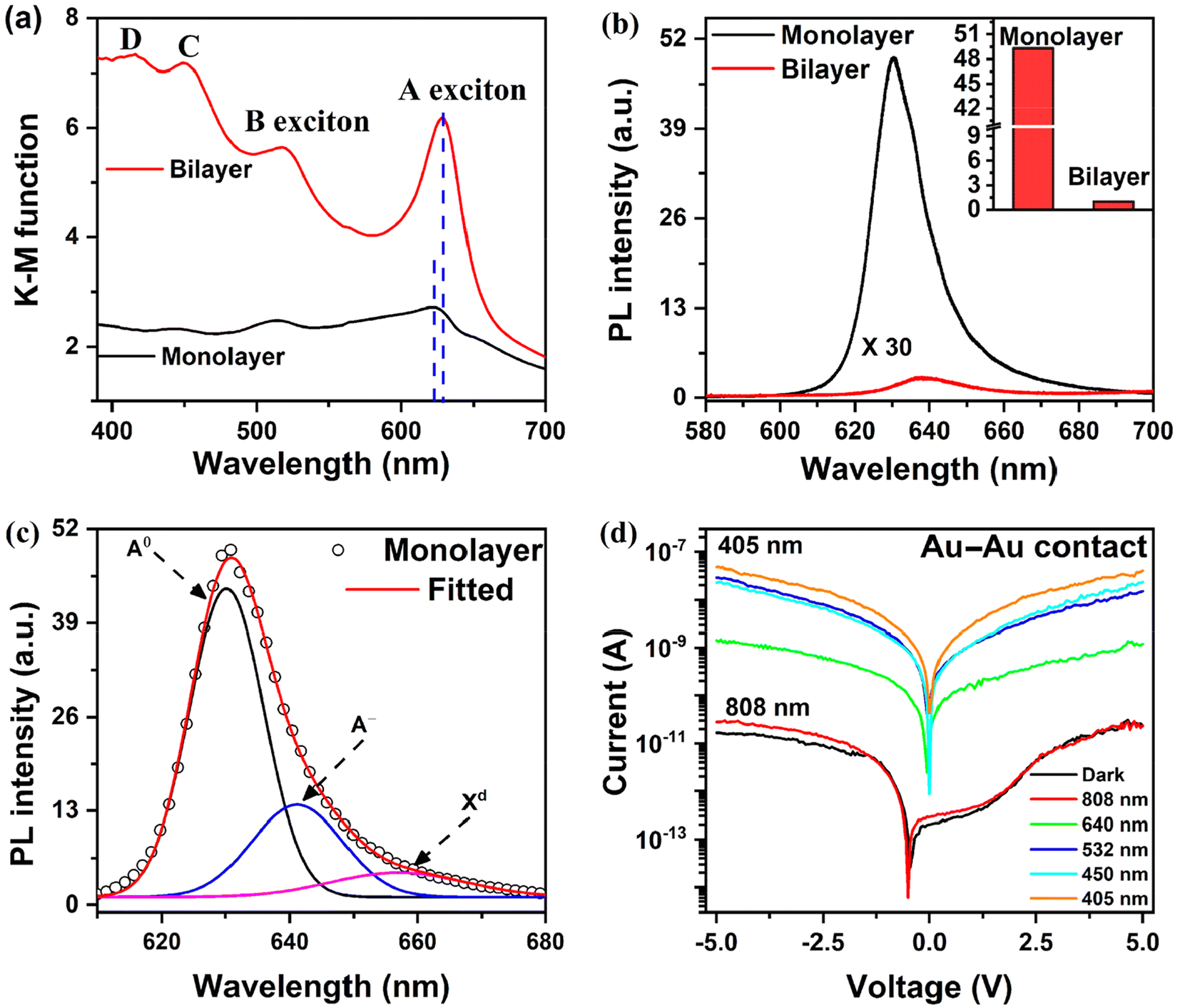
Fig. 5(a) shows a schematic representation of the fabrication process for asymmetric contacts based WS2 PD. Lateral electronic charge doping in WS2 was examined using Raman mapping and the PL spectrum of the monolayer WS2. As mentioned previously, the monolayer WS2 exhibits remarkable PL emission at room temperature when exposed to 532 nm laser excitation. External charge doping significantly affects its PL emission, leading to significant changes in neutral and trion contributions.62 Following the formation of asymmetric contacts, the PL emission spectra after contact deposition on transferred WS2 at various locations are depicted in Fig. 5(b). The inset shows the relative change in the ratio of the intensities of the trion peak to the neutral exciton peak for three different locations. The PL spectra show a low trion contribution near the Au contact, dropping from 32.1% to 19.5%. This decrease in the trion formation can be attributed to the electron transfer from the WS2 monolayer to Au due to band alignments, which is analogous to p-doping in WS2. The electron transfer from WS2 to Au decreases the electron concentration in WS2, resulting in reduced trion formation compared to the transferred monolayer WS2. We observed an increase in the trion contribution in the vicinity of the Ti contact from 32.1% to 38.8%. This indicates an increase in electron density due to electron transfer from Ti to WS2, which is analogous to n-type doping to WS2 by the Ti electrode. Thus, we can selectively introduce electron and hole doping near the Ti and Au contacts, respectively, while the central region of the WS2 flake remains intrinsic. Hence, a p–n–n+ type junction is formed with WS2 due to the asymmetric metal contacts.
 | ||
| Fig. 5 (a) Schematic representation of the fabrication process of an asymmetric contact based WS2 PD. (b) PL spectra of monolayer WS2 at different locations after the fabrication of the asymmetric contact based WS2 PD on transferred monolayer WS2. The inset shows a bar chart of the variation of the trion to neutral exciton intensity ratio for the corresponding positions. (c) Spatial profile of the Raman A1g mode of WS2 at different locations after fabricating a symmetric Ti contact based PD; the inset shows the optical image of the symmetric Ti contacts with mapping points (dots). (d) Spatial profile of the Raman A1g mode of WS2 at different locations after fabricating an asymmetric contact based PD; the inset shows the optical image of the asymmetric contacts with mapping points (dots). | ||
For further validation, we conducted Raman line mapping of WS2 flakes after forming Ti–Ti and Ti–Au asymmetric contacts, as shown in Fig. 5(c) and (d), respectively. As the A1g Raman mode is responsive to charge density, the doping effect is expected to be reflected in the A1g mode spatial profile. Fig. 5(c) illustrates the Raman mapping of the A1g mode following symmetric Ti–Ti contacts. The inset displays an optical image of the device, highlighting the measurement points. Examining the spectra reveals a more pronounced A1g mode near the contacts, contrasted with a weaker signal at the central region. The electron doping near the Ti contact gives rise to stronger electron–phonon coupling and hence a stronger A1g mode, whereas the effect of doping from the Ti electrode at the center is minimum and hence weak vibration, thus confirming n-type doping by the Ti electrode. The A1g Raman mode mapping after the formation of asymmetric contact is displayed in Fig. 5(d). The inset displays an optical image of the device, highlighting the measurement points. The mapping shows that the A1g mode intensity increases toward the Ti contacts, verifying the enhanced electron–phonon coupling. No Raman signal is detected at the point above the metal contacts, indicating that the metal pads fully absorb the laser light. The A1g mode intensity is supposed to decrease near the Au metal pads as the electron density decreases, but the plasmonic effect of gold, when excited with a 532 nm laser, increases the Raman intensity,32 and hence a significant reduction in intensity is not observed. Thus, mapping the A1g mode also confirms the asymmetric doping in WS2, forming a p–n–n+ type junction.
The current–voltage (I–V) characteristics under dark and light with different intensities (405 nm laser) are shown in Fig. 6. The Ti pad was grounded and the Au contact pad was biased with a voltage sweep from −5 V to +5 V during the I–V measurements of the asymmetric contact WS2 PD. The illumination intensity-dependent I–V characteristics with symmetric Ti–Ti and Au–Au contacts are shown in Fig. 6(a) and (b), respectively. The monolayer and bilayer WS2 have a work function of 4.9 eV and are generally an n-type semiconducting material.44 Since Ti has a work function of 4.3 eV, there is no potential barrier between the Ti–WS2 junction, resulting in a symmetric I–V characteristic, as shown in Fig. 6(a). In contrast, Au has a higher work function of 5.1 eV than n-type WS2, forming a Schottky junction giving asymmetric I–V characteristics.63Fig. 6(c) displays the I–V characteristics of the WS2 PD with asymmetric Ti–Au contacts, revealing a significant reduction in dark current by an order of magnitude. The formation of a p–n–n+ junction along the WS2 flakes creates two depletion regions, allowing the unidirectional flow of carriers (electrons), resulting in a lower dark current under reverse bias conditions. Excess electron–hole pairs are generated under 405 nm laser illumination, and hence, there is an increase in current. The on/off ratios of the devices at a low laser intensity of 0.23 mW cm−2 under 5 V reverse bias were ∼35, ∼42, and ∼180 for Ti–Ti, Au–Au, and Ti–Au (asymmetric) contacts, respectively. Thus, asymmetric contacts help to obtain a higher current on/off ratio. The formation of two distinct inhomogeneous junctions contributes to reduced dark current and improved separation of photocarriers under illumination. Note that the WS2 PD with symmetric Ti contacts exhibits an on/off ratio of 4.5 × 103 under 405 nm laser illumination at 65 mW cm−2. This ratio is enhanced to 1.83 × 104 when using symmetric Au contacts, a result attributed to the built-in potential created through charge transfer. The on/off ratio increases further to 1.38 × 105 with the Ti–Au asymmetric configuration due to one order reduction of dark current under reverse bias. We have compared the dark current of symmetric Au–Au contact and asymmetric Ti–Au contact based WS2 PDs in Fig. 6(d). It is evident that the dark current for the asymmetric contact under reverse bias is very low when compared to the Au–Au contacts. Selective doping assisted by asymmetric metal contacts acts as a Schottky/p–n junction type diode, enabling the directional flow of carriers as understood by the band alignments and I–V characteristics. The I–V characteristics of asymmetric Ti–Au contacts under dark conditions, displayed in Fig. 6(d), clearly reveal the rectifying behavior of the asymmetric junction and, thus, a directional flow of carriers. The inset of Fig. 6(d) shows the optical microscopy images with symmetric Ti–Ti and asymmetric Ti–Au contacts on the actual device fabricated on a WS2 flake through lithography.
 | ||
| Fig. 6 Dark and photo I–V characteristics of the WS2 PD. (a–c) I–V characteristics of WS2-based PDs under 405 nm laser illumination at different intensities corresponding to symmetric Ti–Ti, symmetric Au–Au and asymmetric (Ti–Au) contact-based systems, respectively. (d) A comparison of the dark currents for symmetric Au–Au contacts and asymmetric Ti–Au contacts; the inset shows optical images of PDs with symmetric (top left) and asymmetric (bottom right) contacts. | ||
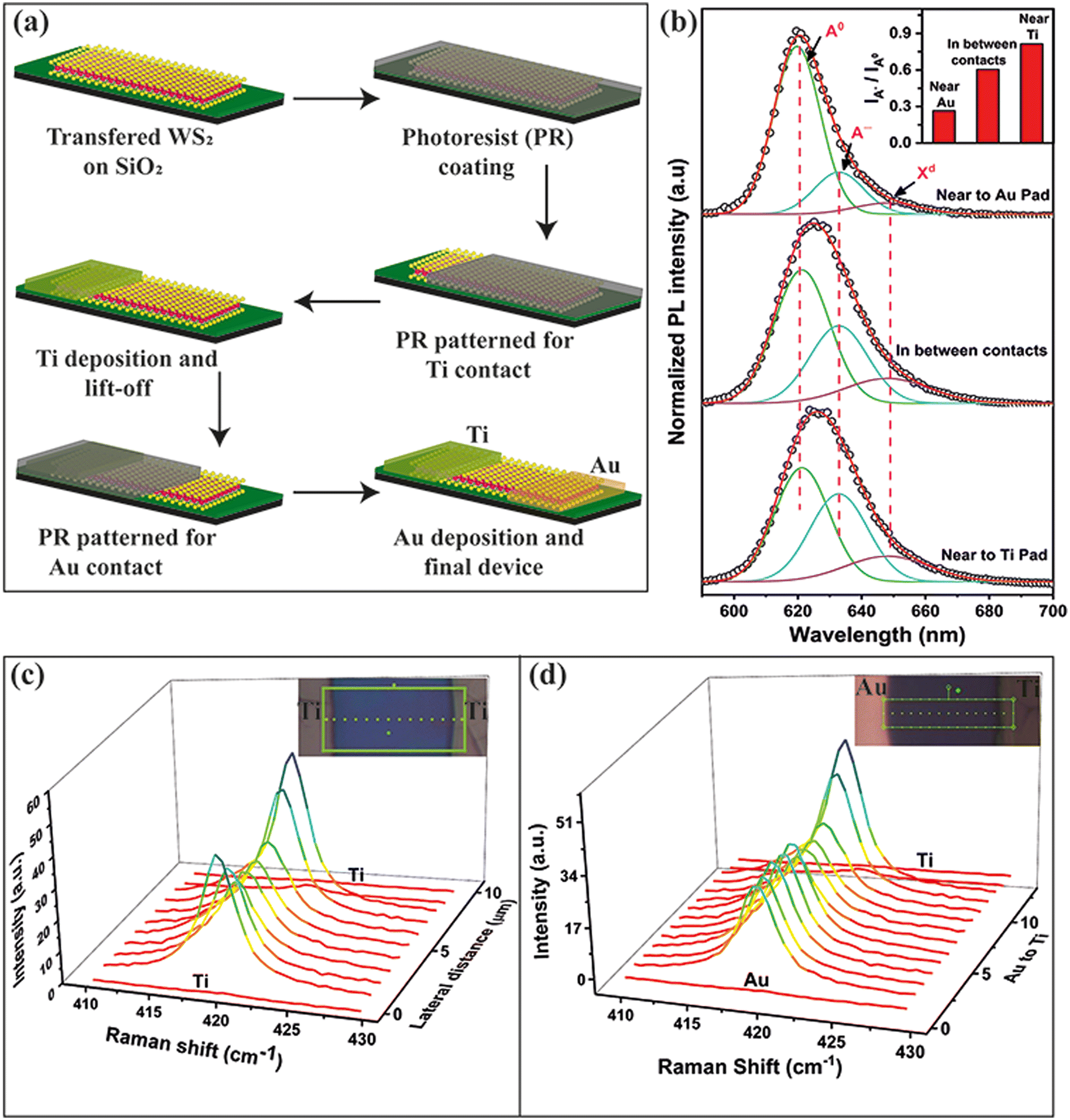
Next, we investigated the power-dependent photoresponse of the WS2 PD using a 405 nm pulse laser. The photoresponse was measured over the 0.23 to 65 mW cm−2 intensity range under a 5 V reverse bias. The photoresponse of the asymmetric contact-based bilayer WS2 PD is shown in Fig. 7(a). The photo response is depicted in Fig. S7(a)† for the device utilizing symmetric Ti contacts. A minimal dark current of 31 pA was observed without laser illumination, which progressively increases to 230 nA with laser illumination at an intensity of 65 mW cm−2 for symmetric Ti contacts. The inset of Fig. S7(a)† displays the photoresponse at low illumination intensity. The calibration curve was constructed to correlate the laser intensity with power using Iph = APθ, where Iph signifies the photocurrent at a laser intensity of P, A is a proportionality constant, and θ denotes the exponential constant, which has an ideal value of 1, signifying the linear response of the PD. The slope of the logarithmic plot determines the θ value, which was found to be 0.73 for the photodetector with Ti contacts, as shown in Fig. 7(b). The deviation from the ideal θ value of 1 signifies a reduced photocurrent than anticipated, which can be ascribed to Auger recombination and trapping of photoelectrons by defects and trap sites at the interface.64–66 The photo response under identical illumination conditions for the WS2 photodetector with Au contact is depicted in Fig. S7(b).† The built-in potential established by the Schottky junction at the Au–WS2 interface reduces the dark current threefold to 11 pA and elevates the photocurrent to 316 nA, thereby contributing to the enhancement of the on/off ratio. The calibration curve in Fig. 7(b) demonstrates a θ value of 1.04, suggesting that the built-in potential aids in charge separation, contributing to an elevated photocurrent by minimizing the carrier recombination.67 Turning our attention to the asymmetric configuration, the two junctions along WS2 behaving like a p–n–n+ configuration due to the asymmetric doping (as explained later) confine the carrier flow along one direction and diminish the dark current by an order of magnitude, reducing it from 31 pA to 3 pA under 5 V reverse bias, while concurrently increasing the photocurrent to 415 nA under 65 mW cm−2 leading to a significant on/off ratio of 1.38 × 105. The θ value remains nearly at unity while the photocurrent exhibits an enhancement. This reduction in dark current, coupled with the amplification in photocurrent, is ascribed to the built-in potential at the p–n–n+ junction, facilitating more efficient charge separation.21 The improvement in performance achieved with asymmetric contacts can be understood through careful examination of the band alignment.
 | ||
| Fig. 7 (a) Photocurrent response under pulsed 405 nm laser illumination at different intensities for WS2-based PDs with asymmetric Ti–Au contacts. (b) The photoresponse calibration curve as a function of illumination intensity for three different configurations of the PDs. Band diagrams of the WS2 based PDs under illumination conditions at zero bias for (c) symmetric Au–Au contacts, (d) symmetric Ti–Ti contacts, and (e) asymmetric Au–Ti contacts with WS2. | ||
The I–V characteristics of monolayer WS2 with asymmetric contact are shown in Fig. S8.† We have observed a rectifying nature from the I–V characteristics, as shown in Fig. S8(a).† The reverse bias current remains 86 fA (instrument limit 1 fA) under dark conditions, while the forward current is 1 pA. The reverse bias current changes to 1.6 nA under the illumination of a 405 nm laser at an intensity of 65 mW cm−2 while the forward bias current is at 17 nA, thus giving an on–off ratio of ∼104. The inset of Fig. S8(a)† shows the magnified view of the I–V characteristics. The illumination current is minimal at zero bias, indicating the potential for self-biased photodetectors. The self-bias photoresponse generated using a 405 nm laser with varying intensities is shown in Fig. S8(b)† with the calibration curve in the inset. From the fitted curve, we found the slope θ ∼ 0.38, indicating that the majority of photo-generated carriers are trapped before reaching the contact pads. When subjected to an external bias of 0.5 V, the slope rises to 0.63, leading to improved separation of photo-generated carriers, as shown in Fig. S8(c and d).†
The band diagram of WS2 with three different contact configurations under illumination conditions at zero bias is shown in Fig. 7(c–e). WS2 is an n-type semiconducting material with a bandgap of ∼2.0 eV, and its Fermi level is at 4.9 eV with reference to the vacuum.9 The energy band positions of different layers and the band diagram of each metal/WS2 junction under zero bias and under dark conditions are shown in Fig. S9.† The work functions of Au and Ti are 5.1 eV and 4.3 eV, respectively. In contrast, the generation of excess carriers by 405 nm laser excitation moves the Fermi level of WS2 upward via photoinduced carrier doping.68 This leads to a change in its work function. The band alignment for the symmetric Au–Au contacts with WS2 is shown in Fig. 7(c). The junction between Au and WS2 under dark conditions creates a potential barrier of 0.2 eV (Vbi = Φm − EF) due to electron transfer from WS2 to Au, thereby limiting electron movement through this junction. One of the two WS2–Au junctions is always forward biased during the voltage sweep from −5 V to +5 V, thus allowing the carrier flow. The junction at the WS2–Ti interface allows electron movement without any potential barrier because Ti's work function is lower than that of n-type WS2, as shown in Fig. 7(d). Now, referring to the asymmetric contacts with WS2, a potential barrier is present only at the Au/WS2 junction but no barrier at the Ti/WS2 configuration, hence blocking the carrier flow under reverse bias only. Beyond this metal–semiconductor junction, there are two other semiconductor–semiconductor (homo) junctions along WS2 due to asymmetric contact-induced inhomogeneous doping. The proximity of Au results in an effect equivalent to p-type doping in WS2 near the Au contact, while WS2 away from the Au contact remains intrinsic. This leads to creating a p–n junction in the WS2 region near the Au contact. Similarly, electrons are transferred from Ti to WS2 near the Ti contact due to band alignments, increasing the electron concentration close to the Ti contact and forming an n–n+ junction. These two junctions further restrict the movement of carriers, leading to an order of magnitude decrease in dark current from 31 pA to 3.0 pA under 5 V reverse bias. There are opposite doping types only near the metal contacts, while WS2 remains unaffected in the region away from the junction. Therefore, these two junctions are equivalent to a p–n–n+ configuration, as shown in Fig. S9(e).† Note that as-grown 2D WS2 is intrinsically n-type, usually due to the substrate effect. Under dark conditions, electrons are the majority carriers in WS2. In the case of symmetric contacts (e.g., Au/WS2/Au), under biasing, one of the two junctions always gets forward biased and the device dark current is thus high. In contrast, for asymmetric contacts (Ti/WS2/Au), due to the doping and Fermi level alignment, the band bending is such that the current flow is unidirectional, similar to a p–n junction. Under reverse bias (under dark conditions), the barrier height at the Au/WS2 junction increases, reducing the electron flow and consequently lowering the dark current. Conversely, under forward bias, the barrier height decreases, promoting smoother electron flow and resulting in a higher dark current, as evidenced in Fig. 6(d). The applied reverse bias not only reduces the carrier flow under dark conditions but also facilitates faster separation of photo-generated carriers in the depletion regions due to the high electric field across the junction. Electrons are collected at the Ti contacts, while holes are collected at the Au contacts. This enhances the photocurrent and consequently, the improved on/off ratio.
The photodetector performance parameters are characterized by the following quantities: responsivity (R), detectivity (D*) and external quantum efficiency (EQE). Responsivity pertains to the generation of photocurrent under unit power illumination, while detectivity indicates the minimum light intensity that a photodetector can discern. The following formulae were used to compute these metrics:69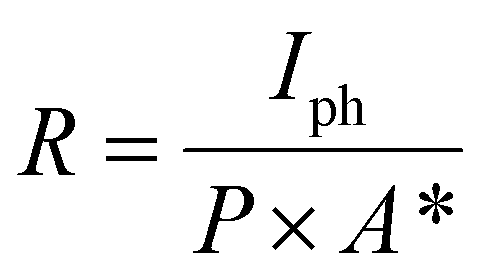 ;
; 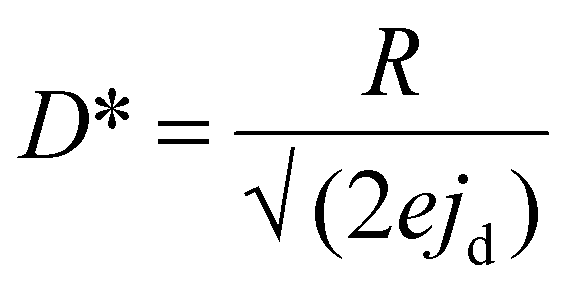 and
and 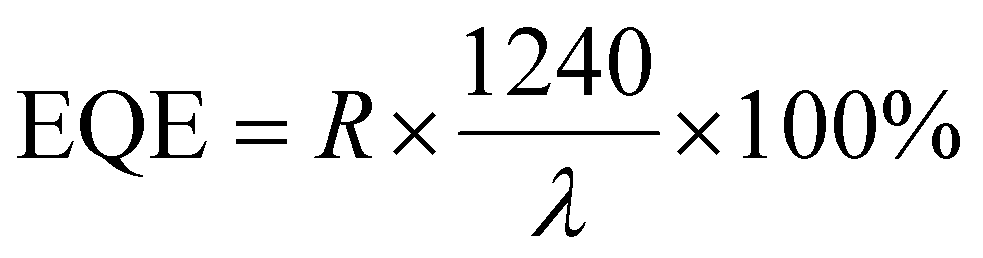 . Here, Iph represents the photocurrent, P is the intensity of the incident laser and A* denotes the effective device area, specifically the area of the WS2 flake situated between the contact pads. e is the electronic charge, jd is the dark current density and λ is the wavelength of light used for the photoresponse measurements. The variation of the responsivity with incident laser intensity is shown in Fig. 8(a). A maximum responsivity of 0.3 A W−1 is seen at low light intensity for the symmetric Ti contact. However, recombination causes a reduction in photocurrent at more intense levels, which subsequently decreases the responsivity, reaching a value of 0.05 A W−1 at 65 mW cm−2, as shown in Fig. 8(a). The responsivity spans from 0.5 to 1.2 A W−1 across the spectrum for the remaining two configurations. The symmetric Au contact exhibits superior responsivity at intensities below 1 mW cm−2. The asymmetric contact-based PD, however, displays an increased responsivity with a peak value of 1.2 A W−1 for intensities exceeding this 1.0 mW cm−2. This is due to the fact that the photocurrent for the PDs with asymmetric contacts increases at a higher power compared to the two devices with symmetric contacts. The detectivity is influenced by both the responsivity and the dark current density. Fig. 8(b) illustrates the changes in detectivity with variation of the incident intensity. The detectivity in the case of asymmetric contact sees an enhancement factor of 5 compared to the other two devices with symmetric contacts due to the dark current reducing by an order. The photodetector with asymmetric contacts boasts the highest detectivity, registering at 2.3 × 1012 Jones. In contrast, the other two have detectivities of 1.2 × 1011 and 4.6 × 1011 Jones, respectively. The EQE at 65 mW cm−2 intensity of 405 nm laser illumination was 162%, 287%, and 345%, corresponding to Ti–Ti, Au–Au, and Ti–Au asymmetric contacts.
. Here, Iph represents the photocurrent, P is the intensity of the incident laser and A* denotes the effective device area, specifically the area of the WS2 flake situated between the contact pads. e is the electronic charge, jd is the dark current density and λ is the wavelength of light used for the photoresponse measurements. The variation of the responsivity with incident laser intensity is shown in Fig. 8(a). A maximum responsivity of 0.3 A W−1 is seen at low light intensity for the symmetric Ti contact. However, recombination causes a reduction in photocurrent at more intense levels, which subsequently decreases the responsivity, reaching a value of 0.05 A W−1 at 65 mW cm−2, as shown in Fig. 8(a). The responsivity spans from 0.5 to 1.2 A W−1 across the spectrum for the remaining two configurations. The symmetric Au contact exhibits superior responsivity at intensities below 1 mW cm−2. The asymmetric contact-based PD, however, displays an increased responsivity with a peak value of 1.2 A W−1 for intensities exceeding this 1.0 mW cm−2. This is due to the fact that the photocurrent for the PDs with asymmetric contacts increases at a higher power compared to the two devices with symmetric contacts. The detectivity is influenced by both the responsivity and the dark current density. Fig. 8(b) illustrates the changes in detectivity with variation of the incident intensity. The detectivity in the case of asymmetric contact sees an enhancement factor of 5 compared to the other two devices with symmetric contacts due to the dark current reducing by an order. The photodetector with asymmetric contacts boasts the highest detectivity, registering at 2.3 × 1012 Jones. In contrast, the other two have detectivities of 1.2 × 1011 and 4.6 × 1011 Jones, respectively. The EQE at 65 mW cm−2 intensity of 405 nm laser illumination was 162%, 287%, and 345%, corresponding to Ti–Ti, Au–Au, and Ti–Au asymmetric contacts.
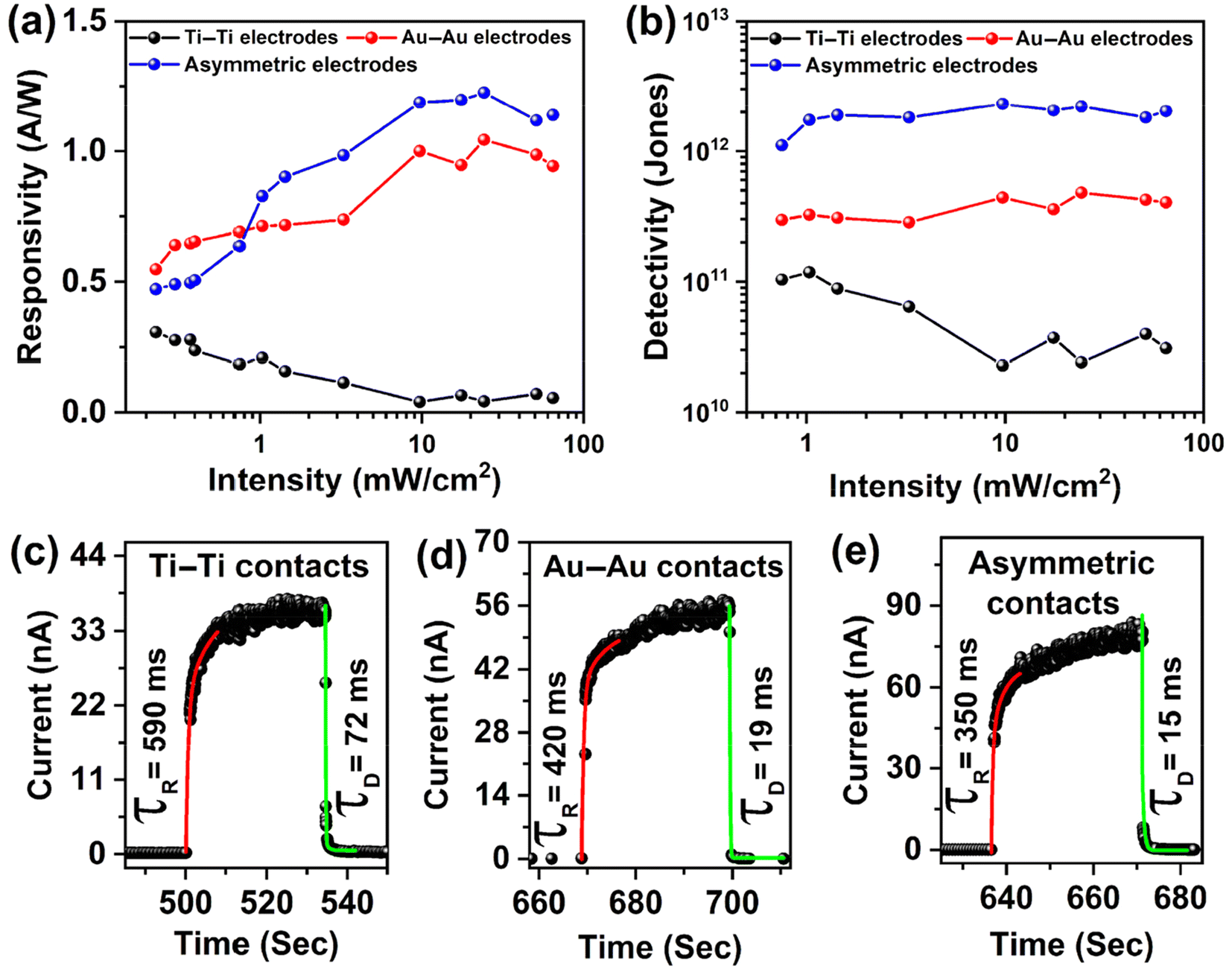 | ||
| Fig. 8 (a) A comparison of the responsitivity of bilayer WS2 PDs for different metal electrode combinations. (b) A comparison of the detectivity of bilayer WS2 PDs based on different metal electrode combinations. (c–e) Temporal photocurrent responses and time constants of symmetric Ti–Ti and Au–Au and asymmetric Ti–Au contact based bilayer WS2 PDs. | ||
The response time of the WS2 PDs with three different configurations is displayed in Fig. 8(c–e). The time response was measured using the Keithley 4200 SCS source meter which has a slow response in the low current (10−12 A) range. The dark current here is very low (few pA) with the photocurrent in the order of nA. Using the exponential fitting function, we have fitted one response peak at an intensity of 10 mW cm−2 under 405 nm laser illumination. The response and decay times of the devices, which are limited by the measuring instrument response time, are tabulated in Table 1. The response time decreases when moving from symmetric Ti contacts to asymmetric contact-based WS2 PDs, which is attributed to the faster charge separation aided by the built-in potential at the p–n–n+ junction. These values are faster with respect to earlier reports on PDs with monolayer WS270 as shown in Table 2 and the observed on/off ratio of 1.38 × 105 is the highest for two-terminal WS2 photodetectors though higher values are possible with FET configurations.26
| Device configuration | On/off ratio | Response time (ms) | Decay time (ms) | EQE (%) |
|---|---|---|---|---|
| Symmetric Ti contacts | 4.5 × 103 | 590 | 72 | 162 |
| Symmetric Au contacts | 1.83 × 104 | 420 | 19 | 287 |
| Asymmetric contacts | 1.38 × 105 | 350 | 15 | 345 |
| Material | Wavelength (nm) | On/off ratio | Responsivity (A/W) | Detectivity (Jones) | Response time | Ref. |
|---|---|---|---|---|---|---|
| WS2 nanotubes | 633 | 391 | 3.14 | — | 228 μs/330 μs | 71 |
| Few layer WS2 | 514 | — | 92 × 10−6 | — | 5.3 ms | 24 |
| Multilayer WS2 | 633 | 25 | 5.7 | — | 20 ms | 25 |
| Monolayer WS2 | 405 | 103 | 290 | 52 × 1014 | 0.2 s/0.07 s | 70 |
| Few layer WS2 | 532 | 108 | 2.97 | 1010 | — | 26 |
| Multilayer WS2 | 532 | — | 0.51 | 2.7 × 109 | 4.1 s/4.4 s | 28 |
| Multilayer WS2 | 365 | — | 53.4 | 1.22 × 1011 | — | 29 |
| Bilayer WS2 | 457 | 1.4 × 104 | 3 × 103 | 5 × 1012 | 59 μs/87 μs | 30 |
| Monolayer WS2 | 532 | 8 × 103 | 5 × 10−3 | 4.9 × 109 | 560 μs | 31 |
| Few layer WS2 | 590 | 103 | 1050 | — | 100 ms/200 ms | 32 |
| Multilayer WS2 | 405 | 104 | 3.45 | 4.94 × 1011 | 7.8/37.3 ms | 33 |
| Bilayer WS2 | 520 nm | — | 2.5 × 10−3 | 3.55 × 108 | 2.6 ms/1.5 ms | 27 |
| Bilayer WS2 | 405 nm | 105 | 1.2 | 2.2 × 1012 | 0.35 s/15 ms | This work |
Conclusion
We have successfully synthesized large-area monolayer and bilayer WS2 flakes via CVD. We were able to achieve selective lateral electron/hole doping by employing metal–semiconductor–metal junctions using two different metal electrodes with different work functions, leading to the establishment of a p–n–n+ type configuration on the WS2 flakes. The formation of a p–n–n+ junction for asymmetric contact-based devices enables the directional flow of carriers. The low work function metal, Ti, facilitates the seamless movement of carriers without any potential barrier, resulting in mirror-symmetric I–V characteristics and an on/off ratio of 103 under 405 nm laser illumination. In contrast, the high work function metal, Au, establishes a potential barrier at the WS2–Au junction, hindering the free movement of carriers and resulting in rectifying I–V characteristics. The built-in potential aids in the enhanced separation of photo-generated carriers, thereby increasing the on/off ratio to 104 under the same illumination conditions. On the other hand, utilizing asymmetric metal contacts on WS2 flakes resulted in a significant increase in the rectification ratio and an on/off ratio of 1.38 × 105 under identical conditions. The detectivity in the case of asymmetric contacts also improved by an order of magnitude by reducing the dark current, rising from 1011 to 1012 Jones. These results are significant for the future development of high-performance planar photodetectors using ultrathin 2D materials.Author contributions
AKM designed and performed all the experiments, MM helped with the analysis of the results and writing of the manuscript and PKG supervised the project and helped with the design of the study, interpretation of the results, and writing of the manuscript.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
We acknowledge the financial support from SERB (Grant Number CRG/2021/006397) and MEITY (Grant No. 5(9)/2022-NANO (VOL-II)) for carrying out part of this work. A part of this research work was accomplished using characterization facilities at CeNSE, Indian Institute of Science Bengaluru, funded by the Ministry of Education (MoE), Ministry of Electronics and Information Technology (MeitY), and Nano mission, Department of Science and Technology (DST), Government of India.References
- S. Aftab, H. H. Hegazy and M. Z. Iqbal, Nanoscale, 2023, 15, 3651–3665 RSC.
- S. Lee, J. Kim, H. Kwon, D. Son, I. S. Kim and J. Kang, Nano Energy, 2023, 110, 108379 CrossRef CAS.
- S. Kawabata, S. Bai, K. Obata, G. Miyaji and K. Sugioka, Int. J. Extreme Manuf., 2023, 5, 015004 CrossRef.
- H. Ezoji and M. Rahimnejad, in Handbook of Nanobioelectrochemistry: Application in Devices and Biomolecular Sensing, ed. U. P. Azad and P. Chandra, Springer Nature, Singapore, 2023, pp. 43–68 Search PubMed.
- S. Paul, M. T. Hossain, A. K. Mia and P. K. Giri, ACS Appl. Nano Mater., 2021, 4, 12527–12540 CrossRef CAS.
- G. Marappan, A. K. Mia, K. Puspharaj, S. Vaidyanathan, Y. Kawazoe, Y. Sivalingam and V. J. Surya, Surf. Interfaces, 2024, 44, 103648 CrossRef CAS.
- J. Zheng, L. Wang, R. Quhe, Q. Liu, H. Li, D. Yu, W.-N. Mei, J. Shi, Z. Gao and J. Lu, Sci. Rep., 2013, 3, 1314 CrossRef PubMed.
- F. Wu, H. Tian, Y. Shen, Z. Hou, J. Ren, G. Gou, Y. Sun, Y. Yang and T.-L. Ren, Nature, 2022, 603, 259–264 CrossRef CAS PubMed.
- S. B. Desai, S. R. Madhvapathy, A. B. Sachid, J. P. Llinas, Q. Wang, G. H. Ahn, G. Pitner, M. J. Kim, J. Bokor, C. Hu, H.-S. P. Wong and A. Javey, Science, 2016, 354, 99–102 CrossRef CAS PubMed.
- S. Susarla, A. Kutana, J. A. Hachtel, V. Kochat, A. Apte, R. Vajtai, J. C. Idrobo, B. I. Yakobson, C. S. Tiwary and P. M. Ajayan, Adv. Mater., 2017, 29, 1702457 CrossRef PubMed.
- A. K. Mia, M. Meyyappan and P. K. Giri, Biosensors, 2023, 13, 169 CrossRef CAS PubMed.
- D. M. Soares, S. Mukherjee and G. Singh, Chem. – Eur. J., 2020, 26, 6320–6341 CrossRef CAS PubMed.
- A. Nourbakhsh, A. Zubair, M. S. Dresselhaus and T. Palacios, Nano Lett., 2016, 16, 1359–1366 CrossRef CAS PubMed.
- H. Kim, J. Kim, I. Uddin, N. A. N. Phan, D. Whang and G.-H. Kim, ACS Appl. Electron. Mater., 2023, 5, 913–919 CrossRef CAS.
- Y. Gong, J. Lin, X. Wang, G. Shi, S. Lei, Z. Lin, X. Zou, G. Ye, R. Vajtai, B. I. Yakobson, H. Terrones, M. Terrones, B. K. Tay, J. Lou, S. T. Pantelides, Z. Liu, W. Zhou and P. M. Ajayan, Nat. Mater., 2014, 13, 1135–1142 CrossRef CAS PubMed.
- Y. Meng, T. Wang, C. Jin, Z. Li, S. Miao, Z. Lian, T. Taniguchi, K. Watanabe, F. Song and S.-F. Shi, Nat. Commun., 2020, 11, 2640 CrossRef CAS PubMed.
- Y. Wu, D. Li, C.-L. Wu, H. Y. Hwang and Y. Cui, Nat. Rev. Mater., 2023, 8, 41–53 CrossRef.
- Y. Wang, A. Slassi, M.-A. Stoeckel, S. Bertolazzi, J. Cornil, D. Beljonne and P. Samorì, J. Phys. Chem. Lett., 2019, 10, 540–547 CrossRef CAS PubMed.
- A. Azcatl, X. Qin, A. Prakash, C. Zhang, L. Cheng, Q. Wang, N. Lu, M. J. Kim, J. Kim, K. Cho, R. Addou, C. L. Hinkle, J. Appenzeller and R. M. Wallace, Nano Lett., 2016, 16, 5437–5443 CrossRef CAS PubMed.
- C. Zhou, S. Zhang, Z. Lv, Z. Ma, C. Yu, Z. Feng and M. Chan, npj 2D Mater. Appl., 2020, 4, 1–9 CrossRef.
- Y. Li, J. Xiao, X. Cao, Z. Gu and W. Zhang, Adv. Funct. Mater., 2023, 33, 2213385 CrossRef CAS.
- C. Lan, C. Li, J. C. Ho and Y. Liu, Adv. Electron. Mater., 2021, 7, 2000688 CrossRef CAS.
- A. Dodda, D. Jayachandran, A. Pannone, N. Trainor, S. P. Stepanoff, M. A. Steves, S. S. Radhakrishnan, S. Bachu, C. W. Ordonez, J. R. Shallenberger, J. M. Redwing, K. L. Knappenberger, D. E. Wolfe and S. Das, Nat. Mater., 2022, 21, 1379–1387 CrossRef CAS PubMed.
- N. Perea-López, A. L. Elías, A. Berkdemir, A. Castro-Beltran, H. R. Gutiérrez, S. Feng, R. Lv, T. Hayashi, F. López-Urías, S. Ghosh, B. Muchharla, S. Talapatra, H. Terrones and M. Terrones, Adv. Funct. Mater., 2013, 23, 5511–5517 CrossRef.
- N. Huo, S. Yang, Z. Wei, S.-S. Li, J.-B. Xia and J. Li, Sci. Rep., 2014, 4, 5209 CrossRef CAS PubMed.
- J. Sun, Z. Lin, X. Jia, H. Li, C. Song, F. Pan, L. Fang, J. Zhang and Y. Wang, Mater. Today Phys., 2023, 35, 101133 CrossRef CAS.
- S. Zhao, Y. Zhang, S. Wang, D. Zhao, H. Kang, J. Li, R. Xiao, Z. Kong, Z. Chen, Y. Sui, J. Wang, Y. Chen and G. Yu, Mater. Lett., 2022, 317, 132103 CrossRef CAS.
- J. D. Yao, Z. Q. Zheng, J. M. Shao and G. W. Yang, Nanoscale, 2015, 7, 14974–14981 RSC.
- L. Zeng, L. Tao, C. Tang, B. Zhou, H. Long, Y. Chai, S. P. Lau and Y. H. Tsang, Sci. Rep., 2016, 6, 20343 CrossRef CAS PubMed.
- R. Yang, S. Feng, J. Xiang, Z. Jia, C. Mu, F. Wen and Z. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 42001–42010 CrossRef CAS PubMed.
- C. Lan, Z. Zhou, Z. Zhou, C. Li, L. Shu, L. Shen, D. Li, R. Dong, S. Yip and J. C. Ho, Nano Res., 2018, 11, 3371–3384 CrossRef CAS.
- Y. Liu, W. Huang, W. Chen, X. Wang, J. Guo, H. Tian, H. Zhang, Y. Wang, B. Yu, T.-L. Ren and J. Xu, Appl. Surf. Sci., 2019, 481, 1127–1132 CrossRef CAS.
- W. Gao, S. Zhang, F. Zhang, P. Wen, L. Zhang, Y. Sun, H. Chen, Z. Zheng, M. Yang, D. Luo, N. Huo and J. Li, Adv. Electron. Mater., 2021, 7, 2000964 CrossRef CAS.
- F. Reale, P. Palczynski, I. Amit, G. F. Jones, J. D. Mehew, A. Bacon, N. Ni, P. C. Sherrell, S. Agnoli, M. F. Craciun, S. Russo and C. Mattevi, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- D. Ovchinnikov, A. Allain, Y.-S. Huang, D. Dumcenco and A. Kis, ACS Nano, 2014, 8, 8174–8181 CrossRef CAS PubMed.
- B. Chakraborty, A. Bera, D. V. S. Muthu, S. Bhowmick, U. V. Waghmare and A. K. Sood, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 161403 CrossRef.
- M. R. Molas, K. Nogajewski, M. Potemski and A. Babiński, Sci. Rep., 2017, 7, 5036 CrossRef PubMed.
- M. W. Iqbal, K. Shahzad, G. Hussain, M. K. Arshad, R. Akbar, S. Azam, S. Aftab, T. Alharbi and A. Majid, Mater. Res. Express, 2019, 6, 115909 CrossRef CAS.
- A. Berkdemir, H. R. Gutiérrez, A. R. Botello-Méndez, N. Perea-López, A. L. Elías, C.-I. Chia, B. Wang, V. H. Crespi, F. López-Urías, J.-C. Charlier, H. Terrones and M. Terrones, Sci. Rep., 2013, 3, 1755 CrossRef.
- J. T. Mlack, P. Masih Das, G. Danda, Y.-C. Chou, C. H. Naylor, Z. Lin, N. P. López, T. Zhang, M. Terrones, A. T. C. Johnson and M. Drndić, Sci. Rep., 2017, 7, 43037 CrossRef CAS PubMed.
- Y. Wang, C. Cong, W. Yang, J. Shang, N. Peimyoo, Y. Chen, J. Kang, J. Wang, W. Huang and T. Yu, Nano Res., 2015, 8, 2562–2572 CrossRef CAS.
- K. M. McCreary, A. T. Hanbicki, S. Singh, R. K. Kawakami, G. G. Jernigan, M. Ishigami, A. Ng, T. H. Brintlinger, R. M. Stroud and B. T. Jonker, Sci. Rep., 2016, 6, 35154 CrossRef CAS PubMed.
- K. M. McCreary, A. T. Hanbicki, G. G. Jernigan, J. C. Culbertson and B. T. Jonker, Sci. Rep., 2016, 6, 19159 CrossRef CAS PubMed.
- A. Bora, S. Paul, M. T. Hossain and P. K. Giri, J. Phys. Chem. C, 2022, 126, 12623–12634 CrossRef CAS.
- A. K. Mia, A. Bora, M. T. Hossain, S. Sinha and P. K. Giri, J. Mater. Chem. B, 2023, 11, 10206–10217 RSC.
- K. Mullapudi, R. Addou, C. L. Dezelah, D. F. Moser, R. K. Kanjolia, J. H. Woodruff and J. F. Conley, Chem. Mater., 2023, 35, 4649–4659 CrossRef CAS.
- F. Lavini, A. Calò, Y. Gao, E. Albisetti, T.-D. Li, T. Cao, G. Li, L. Cao, C. Aruta and E. Riedo, Nanoscale, 2018, 10, 8304–8312 RSC.
- Y. Guan, H. Yao, H. Zhan, H. Wang, Y. Zhou and J. Kang, RSC Adv., 2021, 11, 14085–14092 RSC.
- G. Zhang, C. Wang, B. Yan, B. Ning, Y. Zhao, D. Zhou, X. Shi, S. Chen, J. Shen, Z. Xiao and H. Zhao, J. Mater. Sci.: Mater. Electron., 2022, 33, 22560–22572 CrossRef CAS.
- C. S. Rout, P. D. Joshi, R. V. Kashid, D. S. Joag, M. A. More, A. J. Simbeck, M. Washington, S. K. Nayak and D. J. Late, Sci. Rep., 2013, 3, 3282 CrossRef PubMed.
- H. Shi, B. Zou, Z. Li, M. Luo and W. Wang, Beilstein J. Nanotechnol., 2019, 10, 1434–1442 CrossRef CAS PubMed.
- A. K. Mishra, K. V. Lakshmi and L. Huang, Sci. Rep., 2015, 5, 15718 CrossRef CAS PubMed.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P.-H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef CAS PubMed.
- S. Sharma, S. Bhagat, J. Singh, R. C. Singh and S. Sharma, J. Mater. Sci., 2017, 52, 11326–11336 CrossRef CAS.
- C. Ernandes, L. Khalil, H. Almabrouk, D. Pierucci, B. Zheng, J. Avila, P. Dudin, J. Chaste, F. Oehler, M. Pala, F. Bisti, T. Brulé, E. Lhuillier, A. Pan and A. Ouerghi, npj 2D Mater. Appl., 2021, 5, 1–7 CrossRef.
- B. Zhu, X. Chen and X. Cui, Sci. Rep., 2015, 5, 9218 CrossRef PubMed.
- H. R. Gutiérrez, N. Perea-López, A. L. Elías, A. Berkdemir, B. Wang, R. Lv, F. López-Urías, V. H. Crespi, H. Terrones and M. Terrones, Nano Lett., 2013, 13, 3447–3454 CrossRef PubMed.
- S. Tongay, J. Suh, C. Ataca, W. Fan, A. Luce, J. S. Kang, J. Liu, C. Ko, R. Raghunathanan, J. Zhou, F. Ogletree, J. Li, J. C. Grossman and J. Wu, Sci. Rep., 2013, 3, 2657 CrossRef PubMed.
- R. Kesarwani, K. B. Simbulan, T.-D. Huang, Y.-F. Chiang, N.-C. Yeh, Y.-W. Lan and T.-H. Lu, Sci. Adv., 2022, 8, eabm0100 CrossRef CAS PubMed.
- A. Chernikov, T. C. Berkelbach, H. M. Hill, A. Rigosi, Y. Li, B. Aslan, D. R. Reichman, M. S. Hybertsen and T. F. Heinz, Phys. Rev. Lett., 2014, 113, 076802 CrossRef CAS PubMed.
- T. Chowdhury, D. Paul, D. Nechiyil, G. M. A. K. Watanabe, T. Taniguchi, G. V. P. Kumar and A. Rahman, 2D Mater., 2021, 8, 045032 CrossRef CAS.
- N. Peimyoo, W. Yang, J. Shang, X. Shen, Y. Wang and T. Yu, ACS Nano, 2014, 8, 11320–11329 CrossRef CAS PubMed.
- H. Huang, Y. Sheng, Y. Zhou, Q. Zhang, L. Hou, T. Chen, R.-J. Chang and J. H. Warner, ACS Appl. Nano Mater., 2018, 1, 6874–6881 CrossRef CAS.
- C. Zhou, S. Raju, B. Li, M. Chan, Y. Chai and C. Y. Yang, Adv. Funct. Mater., 2018, 28, 1802954 CrossRef.
- J. Ghosh, L. P. L. Mawlong, M. G. B. A. J. Pattison, W. Theis, S. Chakraborty and P. K. Giri, J. Mater. Chem. C, 2020, 8, 8917–8934 RSC.
- L.-H. Zeng, Q.-M. Chen, Z.-X. Zhang, D. Wu, H. Yuan, Y.-Y. Li, W. Qarony, S. P. Lau, L.-B. Luo and Y. H. Tsang, Adv. Sci., 2019, 6, 1901134 CrossRef CAS PubMed.
- B. Ezhilmaran, A. Patra, S. Benny, M. R. Sreelakshmi, S. V. Bhat and C. S. Rout, J. Mater. Chem. C, 2021, 9, 6122–6150 RSC.
- X. H. Wang, J. Q. Ning, Z. C. Su, C. C. Zheng, B. R. Zhu, L. Xie, H. S. Wu and S. J. Xu, RSC Adv., 2016, 6, 27677–27681 RSC.
- A. Bora, L. P. Mawlong and P. K. Giri, ACS Appl. Electron. Mater., 2021, 3, 4891–4904 CrossRef CAS.
- R. K. Prasad, K. Ghosh, P. K. Giri, D.-S. Kim and D. K. Singh, ACS Appl. Electron. Mater., 2023, 5, 3634–3640 CrossRef CAS.
- C. Zhang, S. Wang, L. Yang, Y. Liu, T. Xu, Z. Ning, A. Zak, Z. Zhang, R. Tenne and Q. Chen, Appl. Phys. Lett., 2012, 100, 243101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06118c |
| This journal is © The Royal Society of Chemistry 2024 |