
PSMA-targeted dendrimer as an efficient anticancer drug delivery vehicle for prostate cancer†
Anubhav
Dhull
a,
Jing
Wei‡
b,
Anunay James
Pulukuri‡
a,
Anu
Rani
a,
Rishi
Sharma
 a,
Nooshin
Mesbahi
a,
Hosog
Yoon
a,
Emily A.
Savoy
a,
Sylvia
Xaivong Vi
a,
Kenneth John
Goody
a,
Clifford E.
Berkman
a,
Boyang Jason
Wu
b and
Anjali
Sharma
*a
a,
Nooshin
Mesbahi
a,
Hosog
Yoon
a,
Emily A.
Savoy
a,
Sylvia
Xaivong Vi
a,
Kenneth John
Goody
a,
Clifford E.
Berkman
a,
Boyang Jason
Wu
b and
Anjali
Sharma
*a
aDepartment of Chemistry, College of Arts and Sciences, Washington State University, Pullman, WA, USA. E-mail: anjali.sharma@wsu.edu
bDepartment of Pharmaceutical Sciences, College of Pharmacy and Pharmaceutical Sciences, Washington State University, Spokane, WA, USA
First published on 1st March 2024
Abstract
Prostate cancer (PCa) is the second leading cause of cancer-related deaths among men in the United States. Although early-stage treatments exhibit promising 5-year survival rates, the treatment options for advanced stage disease are constrained, with short survival benefits due to the challenges associated with effective and selective drug delivery to PCa cells. Even though targeting Prostate Specific Membrane Antigen (PSMA) has been extensively explored and is clinically employed for imaging and radio-ligand therapy, the clinical success of PSMA-based approaches for targeted delivery of chemotherapies remains elusive. In this study, we combine a generation 4 hydroxy polyamidoamine dendrimer (PD) with irreversible PSMA ligand (CTT1298) to develop a PSMA-targeted nanoplatform (PD-CTT1298) for selective intracellular delivery of potent chemotherapeutics to PCa. PD-CTT1298-Cy5 exhibits a PSMA IC50 in the nanomolar range and demonstrates selective uptake in PSMA (+) PCa cells via PSMA mediated internalization. When systemically administered in a prostate tumor xenograft mouse model, PD-CTT1298-Cy5 selectively targets PSMA (+) tumors with significantly less accumulation in PSMA (−) tumors or upon blocking of the PSMA receptors. Moreover, the dendrimer clears rapidly from the off-target organs limiting systemic side-effects. Further, the conjugation of an anti-cancer agent, cabozantinib to the PSMA-targeted dendrimer translates to a significantly enhanced anti-proliferative activity in vitro compared to the free drug. These findings highlight the potential of PD-CTT1298 nanoplatform as a versatile approach for selective delivery of high payloads of potent chemotherapeutics to PCa, where dose related systemic side-effects are a major concern.
 Anjali Sharma | Dr Anjali Sharma is leading a “Translational Nanomedicines Research Laboratory” at Washington State University (WSU). She joined the chemistry faculty at WSU in 2022 after completing her PhD in Materials Chemistry from McGill University and postdoctoral training in nanomedicine at the Center for Nanomedicine at Johns Hopkins University. The interdisciplinary research in The Sharma Lab is focused on developing rationally designed and clinically translatable novel nanostructures for selective delivery of therapeutics to cellular and sub-cellular locations. The major focus of the group is to design these nanostructures to cross various biological barriers for targeted treatment of unmet medical needs in cancer, brain, and ocular diseases. Dr Sharma has published over 40 scientific articles and is the inventor on >15 patents and disclosures. |
Introduction
Prostate cancer (PCa) stands as one of the most prevalent forms of cancer and significantly contributes to male mortality worldwide.1 Although early-stage treatments show promising 5-year survival rates, the advanced disease has poor prognosis.2 Current standard of care for PCa involves androgen deprivation therapy,3,4 but most patients benefit from it for >24 months developing advanced disease.5,6 Treatment options for advanced PCa patients remain limited due to challenges associated with effective and selective drug delivery to PCa cells. Most chemotherapies are distributed throughout the body and often induce severe dose-dependent adverse effects owing to their off-target absorption within healthy organs and tissues. Hence, there exists a pressing requirement to develop platforms capable of selective intracellular delivery of potent chemotherapeutics to PCa cells, while exhibiting no or minimal side effects.Significant progress has been made in the realm of the management of PCa, particularly focusing on prostate-specific membrane antigen (PSMA) targets for imaging and therapy.7–9 PSMA is a transmembrane glycoprotein that is highly expressed on prostatic epithelium and PCa cells, especially in advanced or metastatic stages, while showing reduced levels of expression on normal cells.10–12 Because of its high expression in tumors, it has become an appealing target for the creation of targeted chemotherapeutic agents and radio imaging tracers designed to identify and locate suspected metastases. Targeting PSMA is used clinically for imaging and radio-ligand therapy.13,14 Recently, FDA approved the first PSMA-targeted radioligand therapy, 177Lu-PSMA-617 (Pluvicto™).15 The major hurdle in the clinical success of small molecule PSMA probes and inhibitors is their poor pharmacokinetic profiles that limits their effectiveness. Pluvicto™ is guided by a reversible PSMA inhibitor (ACUPA), for treating PSMA-positive (PSMA+) advanced PCa.16 It has been reported that the PSMA ligands exhibiting an irreversible mode of binding (such as CTT1298) demonstrate enhanced internalization in PSMA (+) cells in contrast to ligands with a reversible mode of binding.17–20 Utilizing irreversible PSMA ligands coupled with a nanotechnology-based approach may be a potential strategy to improve their pharmacokinetic profile and provide selective intracellular delivery of chemotherapeutic drugs to PCa cells.
Dendrimers are promising nanocarriers with proven potential to improve the pharmacokinetics, safety, and efficacy of bioactive ligands and therapeutic molecules.21–23 Polyamidoamine (PAMAM) dendrimers have been extensively studied for cancer imaging, diagnosis, and treatment applications, and are particularly well suited for multitasking such as simultaneous delivery of drugs/genes with imaging or delivery of a combination of drugs.24,25 Generation 4 hydroxyl polyamidoamine dendrimers (PAMAM-G4-OH) have garnered significant recognition for their exceptional pharmacokinetic characteristics, positioning them as promising nanocarriers within the field of targeted drug delivery. PAMAM-G4-OH dendrimers (PD) have been widely explored for targeted treatment of inflammation and are currently undergoing clinical trials (NCT03500627, NCT04321980, NCT05387837).26–32 PD platforms have also been explored for targeted delivery of drugs to tumor-associated macrophages for the treatment of glioblastoma with reported positive outcomes.33–35 Utilizing PD-based drug delivery for potent chemotherapy and immunotherapy has proven to enhance drug effectiveness while mitigating dose-related toxicity and systemic side effects. Systemic PD-drug conjugates exhibit targeted and sustained drug release within intracellular and intratumoral environments, demonstrating localized modulation of tumor immune response in animal models of brain tumors.33–35 However, to the best of our knowledge, a PD platform has never been previously explored for the treatment or diagnosis of PCa. Although a few other dendrimer-PSMA agents have been evaluated for targeting and drug delivery, these were based on cationic (positively charged) amine-terminating PAMAM-G5-NH2 dendrimers.36,37 The cationic dendrimers generally exhibit high toxicity and have limited clinical potential.38–40
In this report, we combine PSMA targeting via an irreversible PSMA ligand (CTT1298) with a neutral PAMAM-G4-OH (PD) dendrimer to rationally develop a novel systemic PSMA-targeted nanoplatform (PD-CTT1298) for selective intracellular delivery of potent chemotherapeutics to tumor cells. Due to the reported safety of the PD platform in preclinical and clinical studies, we opted to use this dendrimer to develop our PSMA-targeting nanoplatform.30–32,34 We here present the synthesis and characterization of PSMA-targeted dendrimer and dendrimer-cabozantinib conjugates via a highly efficient strain promoted azide–alkyne click (SPAAC) reaction, along with their in vitro and in vivo evaluation in PCa models.
Experimental section
Synthesis of PD-CTT1298-Cy5 and PD-CTT1298-Cabo conjugates
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (solvent A: 0.1% TFA and 5% ACN in water; solvent B: 0.1% TFA in ACN), gradually increased to 50:50 (A:B) at 20 minutes, 10:90 (A:B) at 38 minutes, and finally returned to 90:10 (A:B) at 40 minutes. A flow rate of 1 mL min−1 was maintained during the run. The dendrimers and drug were detected at 205, 210 or 305 nm. The Cy5 labeled conjugate was detected at 650 nm. The size and zeta potential distribution of dendrimers were determined using a Malvern Zetasizer Pro Blue (Malvern Panalytical) instrument. The samples for size distribution were dissolved in milli Q H2O at a concentration of 0.1 mg mL−1 and the samples for zeta potential distribution were dissolved in 10 mM sodium chloride solution at a concentration of 0.2 mg mL−1. Samples were measured in triplicate and averaged to provide the reported sizes and zeta potential.
:10 (solvent A: 0.1% TFA and 5% ACN in water; solvent B: 0.1% TFA in ACN), gradually increased to 50:50 (A:B) at 20 minutes, 10:90 (A:B) at 38 minutes, and finally returned to 90:10 (A:B) at 40 minutes. A flow rate of 1 mL min−1 was maintained during the run. The dendrimers and drug were detected at 205, 210 or 305 nm. The Cy5 labeled conjugate was detected at 650 nm. The size and zeta potential distribution of dendrimers were determined using a Malvern Zetasizer Pro Blue (Malvern Panalytical) instrument. The samples for size distribution were dissolved in milli Q H2O at a concentration of 0.1 mg mL−1 and the samples for zeta potential distribution were dissolved in 10 mM sodium chloride solution at a concentration of 0.2 mg mL−1. Samples were measured in triplicate and averaged to provide the reported sizes and zeta potential.
Synthesis of compound 3. To a stirring of solution of CTT1298 (1) (0.27 g, 1.0 eq., 0.35 mmol) and KHCO3 (0.047 g, 1.3 eq., 0.46 mmol) in ddH2O (1.5 mL) was added a solution of DBCO-C6-NHS (0.1 g, 0.232 mmol) in THF (1.5 mL) dropwise. The reaction was stirred at ambient temperature for 3 hours, solvent was removed under reduced pressure and the resulting residue was purified via reverse-phase flash chromatography (100% ddH2O → 10% MeOH in ddH2O) to yield a white solid in 60% yield.
1H NMR (600 MHz, D2O) δ 7.69 (d, 1H), 7.55–7.40 (m, 6H), 7.34 (d, 1H), 5.11 (d, 1H), 4.17 (dd, 1H), 4.08 (dd, 1H), 3.82 (d, 1H), 3.74 (m, 2H), 3.49 (m, 1H), 3.11 (m, 2H), 2.38–2.06 (m, 10H), 2.01–1.79 (m, 6H), 1.71–1.58 (m, 5H), 1.5 (m, 2H), 1.36–1.14 (m, 6H); 13C NMR (101 MHz, D2O) δ 183.0, 181.5, 179.2, 178.4, 176.3, 176.1, 175.7, 174.9, 150.8, 147.6, 131.9, 129.1, 129.0, 128.8, 128.4, 128.1, 127.0, 125.6, 122.3, 121.5, 114.6, 107.9, 64.4, 64.4, 56.6, 55.4, 55.4, 54.8, 39.1, 35.6, 35.2, 34.0, 33.7, 32.3, 32.0, 31.9, 28.1, 28.0, 27.8, 26.8, 26.7, 25.7, 24.9, 24.4, 24.2; 31P NMR (162 MHz, D2O) δ 7.39. HRMS (MALDI) calcd [M − H]+ for C42H54N5O15P: 898.3276, found 898.3288
Synthesis of compound 5. PAMAM-G4-OH (PD) (1.0 g, 1.0 eq., 0.07 mmol) was dissolved in Anhy. DMF (5 mL). Azido Hexanoic Acid (77 mg, 7.0 eq., 0.49 mmol) was dissolved in anhy. DMF (5 mL) and was activated by adding EDC·HCl (134.5 mg, 10.0 eq., 0.7 mmol) and stirred for 15 minutes. It was then added dropwise to the solution of PD under continuous stirring followed by the addition of DMAP (38.5 mg, 5 eq., 0.32 mmol). The reaction mixture was stirred for 24 hours at room temperature. The reaction completion was confirmed by the shift in the retention time of the chromatogram in HPLC. The dialysis was performed using a 1 kDa dialysis membrane in deionized water for 24 h. The aqueous solution was lyophilized to afford PD-azide (5) in 82% yield.
1H NMR (500 MHz, DMSO) δ 8.15–7.73 (m, D-internal amide H), 4.76 (bs, D-OH), 4.04 (t, D-ester –CH2), 3.50–3.25 (m, D-CH2), 3.22–3.03 (m, D-CH2), 2.77–2.59 (m, D-CH2), 2.49–2.40 (m, D-CH2), 2.35–2.13 (m, D-CH2), 1.62–1.50 (linker –CH2), 1.39–1.32 (linker –CH2).
Synthesis of compound 6. To solution of PD-azide dendrimer (5) (20.0 mg, 1.0 eq., 0.0012 mmol) in DI Water (150 μL) was added DBCO C6 CTT1298 (3) (5.5 mg, 4.0 eq., 0.005 mmol), and the reaction mixture was stirred for 24 hours at room temperature. Reaction progress was tracked with HPLC. Upon completion, the dialysis was performed using a 1 kDa dialysis membrane in DI Water for 12 h. The aqueous solution was lyophilized to afford PD-CTT1298 (6) in 92% yield.
1H NMR (500 MHz, D2O) δ 7.72–7.13 (m, DBCO H), 4.18–3.92 (m, D-CH2 and ligand H), 3.73–3.50 (m, D-CH2 and ligand H), 3.45–3.14 (m, D-CH2 and ligand H), 3.08–2.97 (m, linker-CH2), 2.89–2.67 (m, D-CH2), 2.66–2.49 (D-CH2), 2.48–2.24 (m, D-CH2), 2.25–2.10 (m, ligand H), 2.06–1.93 (m, ligand H), 1.90–1.70 (m, ligand H), 1.62–1.35 (m, linker –CH2 and ligand H), 1.34–1.10 (m, linker –CH2 and ligand H).
31P NMR (202 MHz, D2O) δ 7.31.
Synthesis of compound 7. PD-CTT1298 (6) (19.0 mg, 1.0 eq., 0.0001 mmol) was dissolved in DI Water (150 μL) and stirred. Cy5-DBCO (2.77 mg, 3.0 eq., 0.003 mmol) was added to the dendrimer solution and the stirring mixture was left for 48 hours at room temperature. The completion of reaction was tracked using HPLC. Upon completion, the dialysis was performed using a 1 kDa dialysis membrane in DI Water for 12 h. The aqueous solution was lyophilized to afford compound PD-CTT1298-Cy5 (7) in 81% yield.
1H NMR (500 MHz, DMSO) δ 8.39–8.20 (m, Cy5 H), 8.14–7.05 (m, D-internal amide H, DBCO H, and Cy5 H), 6.59–6.49 (m, Cy5 H), 6.33–6.19 (m, Cy5 H), 5.86–5.73 (m, Cy5 H), 5.45–5.32 (m, Cy5 H), 4.94–4.47(m, D-OH and ligand H), 4.37–3.84 (D-ester-CH2 and ligand H), 3.21–2.97 (m, D-CH2 and ligand H), 2.84–2.59 (m, D-CH2), 2.37–1.99 (m, D-CH2 and ligand H), 1.80–1.61 (m, ligand H and Cy5 H), 1.60–1.43 (m, linker –CH2 and ligand H), 1.41–1.10 (m, linker –CH2 Cy5 H, and ligand H).
Synthesis of compound 10. To a stirred solution of 4-amino-phenol 9 (874 mg, 8.0 mmol, 1.2 eq.) was added sodium hydride (60% dispersion in mineral oil, 400 mg, 10.0 mmol, 1.5 eq.) in dry N,N-dimethylformamide (10 mL) at 0 °C. After 30 minutes, 7-benzyloxy-4-chloro-6-methoxy-quinoline 8 (2.0 g, 6.67 mmol, 1.0 eq.) was added and reaction mixture was stirred at 80 °C for 16 h. The progress of the reaction was monitored with TLC. Crude reaction mixture was diluted with water and extracted with ethyl acetate (50 mL × 4). The combined organic layer was dried over Na2SO4, filtered, and the solvent was evaporated. The final product was purified with silica gel chromatography (0–5% MeOH–DCM) to yield 2.2 g (yield = 92%) of 10 as yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.42 (d, J = 5.2 Hz, 1H), 7.49–7.57 (m, 3H), 7.47 (s, 1H), 7.41–7.46 (m, 2H), 7.34–7.41 (m, 1H), 6.89–6.97 (m, 2H), 6.63–6.71 (m, 2H), 6.37 (d, J = 5.3 Hz, 1H), 5.30 (s, 2H), 5.17 (s, –NH2). 13C NMR (125 MHz, DMSO-d6) δ 161.4, 151.7, 149.7, 149.3, 147.1, 146.5, 143.8, 137.0, 128.9, 128.4, 128.4, 122.2, 115.5, 115.3, 109.6, 102.7, 99.8, 70.2, 56.1.
Synthesis of compound 12. Compounds 10 (2.0 g, 5.37 mmol, 1.0 eq.) and 11 (1.8 g, 8.05 mmol, 1.5 eq.) were dissolved in dry DMF (8 mL). DIPEA (2.0 mL, 10.74 mmol, 2.0 eq.) and HATU (3.0 g, 8.05 mmol, 1.5 eq.) were added to the solution, and the mixture was stirred overnight at room temperature. EtOAc (100 mL) was added, washed with water and brine, dried over Na2SO4. After filtration and condensation, the residue was purified by silica gel chromatography (0–100% EtOAc–hexane) to yield 2.8 g (yield = 90%) of compound 12 as brown solid.; 1H NMR (500 MHz, DMSO-d6) δ 10.04 (s, –NH), 10.27 (s, –NH), 8.65 (d, J = 6.0 Hz, 1H), 7.82 (d, J = 9.0 Hz, 2H), 7.62–7.69 (m, 2H), 7.51–7.58 (m, 4H), 7.45 (t, J = 7.5 Hz, 2H), 7.39 (t, J = 7.3 Hz, 1H), 7.31 (d, J = 9.0 Hz, 2H), 7.16 (t, J = 8.9 Hz, 2H), 6.66 (d, J = 6.0 Hz, 1H), 5.36 (s, 2H), 4.00 (s, 3H), 1.47–1.54 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ 168.6, 168.6, 162.1, 158.7 (d, J = 238.7 Hz), 152.9, 150.4, 149.4, 147.7, 137.3, 136.6, 135.6 (d, J = 2.6 Hz), 129.0, 128.6, 128.5, 122.8 (d, J = 7.6 Hz), 122.6, 121.7, 115.6 (d, J = 22 Hz), 115.4, 107.3, 103.5, 100.1, 70.6, 56.4, 32.1, 15.8.
Synthesis of compound 13. The compound 12 (1.5 g, 2.59 mmol) was dissolved in 6 mL of TFA and mixture was stirred at 60 °C for 30 minutes and concentrated. The mixture was basified to pH 7 with saturated aqueous NaHCO3 and extracted with ethyl acetate (50 mL × 3). The combined organic extracts were washed with brine and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated to afford a residue, which was purified by silica gel chromatography (0–10% MeOH–DCM) to yield 1.1 g (yield = 87%) of 13 as yellow solid.; 1H NMR (500 MHz, DMSO-d6) δ 10.19 (s, –NH), 10.16 (s, –OH), 10.07 (s, –NH), 8.40 (d, J = 5.2 Hz, 1H), 7.75 (d, J = 9.0 Hz, 2H), 7.59–7.68 (m, 2H), 7.49 (s, 1H), 7.28 (s, 1H), 7.19–7.25 (m, 2H), 7.15 (t, J = 8.9 Hz, 2H), 6.36 (d, J = 5.2 Hz, 1H), 3.94 (s, 3H), 1.48 (s, 4H). 13C NMR (125 MHz, DMSO-d6) δ 168.6, 168.6, 160.4, 158.7 (d, J = 239 Hz), 151.2, 150.00, 149.5, 149.1, 147.0, 136.7, 135.6 (d, J = 2.7 Hz), 122.9 (d, J = 7.8 Hz), 122.6, 121.6, 115.5 (d, J = 23 Hz), 115.0, 111.2, 102.8, 99.7, 79.6, 56.1, 32.0, 15.8. LCMS (ESI) calcd [M + H]+ for C27H22FN3O5: 488.1622, found 488.1032.
Synthesis of compound 15. To a mixture of dibenzocyclooctyne acid 14 (150 mg, 0.49 mmol, 1.2 eq.) and compound 13 (200 mg, 0.41 mmol, 1.0 eq.) in 10 mL of dry DCM was added EDC·HCl (118 mg, 0.61 mmol, 1.5 eq.), DIPEA (0.1 mL, 0.61 mmol, 1.5 eq.) and DMAP (5.0 mg, 0.04 mmol, 0.1 eq.). The reaction mixture was stirred for 1 h at room temperature. Progress was monitored with TLC analysis. EtOAc (50 mL) was added, washed with water and brine, dried over Na2SO4. After filtration and condensation, the residue was purified by silica gel chromatography (0–10% MeOH–DCM) to yield 180 mg (yield = 57%) of Cabo-DBCO (15) as white solid.
1H NMR (500 MHz, DMSO-d6) δ 10.26 (s, –NH), 10.12 (s, –NH), 8.60 (d, J = 5.1 Hz, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.64–7.79 (m, 5H), 7.50–7.62 (m, 4H), 7.46 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.25–7.33 (m, 3H), 7.20 (t, J = 8.8 Hz, 2H), 6.58 (d, J = 5.1 Hz, 1H), 5.14 (d, J = 14.1 Hz, 1H), 3.88 (s, 3H), 3.73 (d, J = 14.0 Hz, 1H), 2.65–2.91 (m, 3H), 1.98–2.10 (m, 1H), 1.53 (s, 4H). 13C NMR (125 MHz, DMSO-d6) δ 170.8, 170.7, 168.6, 168.5, 160.7, 158.7 (d, J = 238.6 Hz), 151.7, 150.6, 150.1, 149.6, 148.7, 145.2, 143.6, 137.1, 135.6 (d, J = 2.7 Hz), 132.8, 130.1, 129.4, 128.8, 128.5, 128.2, 127.3, 125.6, 122.9, 122.8 (d, J = 7.8 Hz), 122.6, 122.1, 122.0, 121.7, 119.7, 115.5 (d, J = 23 Hz), 114.8, 108.4, 104.6, 100.9, 56.6, 55.5, 55.4, 32.0, 29.8, 29.3, 15.8. LCMS (ESI) calcd [M + H]+ for C46H35FN4O7: 774.2568, found 775.1006.
Synthesis of compound 16. PAMAM-G4-OH (PD) dendrimer 4 (1.0 g, 1.0 eq., 0.07 mmol) was dissolved in Anhy. DMF (5 mL). Azido hexanoic Acid (154 mg, 14.0 eq., 0.98 mmol) was dissolved in Anhy. DMF (5 mL) and was activated by adding EDC·HCl (268.5 mg, 20.0 eq., 1.4 mmol) and stirred for 15 minutes. It was then added dropwise to the G4-PAMAM dendrimer solution under continuous stirring followed by addition of DMAP (77 mg, 9.0 eq., 0.63 mmol) and the reaction mixture was stirred for 24 hours at room temperature. The reaction completion was confirmed by the shift in the retention time of the chromatogram in HPLC. The dialysis was performed using a 1 kDa dialysis membrane in deionized water for 24 h. The aqueous solution was lyophilized to afford PD-azide (16) in 86% yield.
1H NMR (500 MHz, DMSO) δ 8.15–7.68 (m, D-internal amide H), 4.81–4.61 (m, D-OH), 4.05–3.95 (m, D-ester –CH2), 3.50–3.23 (m, D-CH2), 3.18–2.98 (m, D-CH2), 2.92–2.85 (D-CH2), 2.78–2.55 (m, D-CH2), 2.46–2.36 (m, D-CH2), 2.35–1.99 (m, D-CH2), 1.58–1.46 (linker –CH2), 1.36–1.25 (linker –CH2).
Synthesis of compound 17. To a solution of PD-azide 16 (20 mg, 0.0013 mmol, 1.0 eq.) in DMF (100 μL) in 2 mL vial, was added solution of compound 15 (7.8 mg, 0.010 mmol, 8.0 eq.) dissolved in DMF (100 μL). The reaction mixture was stirred at RT for 3 h. The reaction completion was confirmed by the shift in the retention time of the chromatogram in HPLC. Upon completion, the compound was purified with TFF 3 kDa dialysis membrane. The product was lyophilized to afford PD-Cabo (17) in 88% yield.
White fluffy Solid, 1H NMR (500 MHz, DMSO) δ 10.14 (s, Cabo-amide H), 9.99 (s, Cabo-amide H), 8.46 (s, Cabo-Ar H), 8.06–6.96 (m, D-internal amide H, and Ar H), 6.45 (s, Cabo-Ar H), 6.0–5.73 (m, DBCO-CH2), 4.66 (bs, D-OH), 4.53–4.08 (m, linker and Cabo H), 3.97–3.73 (m, D-ester-CH2 and Cabo DBCO H), 3.85–3.31 (m, D-CH2 and Cabo H), 3.11–2.91 (m, D-CH2), 2.71–2.52 (m, D-CH2 and Cabo H), 2.40–2.27 (m, D-CH2 and Cabo H), 2.26–2.02 (m, D-CH2 and Cabo H), 1.52–1.10 (m, D-linker H and Cabo H).
13C NMR (125 MHz, DMSO-d6) δ 173.2, 172.0, 171.7, 170.8, 170.3, 168.6, 160.7, 159.6, 157.7, 150.7, 150.0, 149.6, 145.2, 144.2, 143.7, 142.6, 137.1, 135.6, 134.3, 131.2, 130.3, 122.9, 122.8, 122.6, 122.0, 121.7, 119.7, 115.5, 115.4, 115.0, 104.6, 100.9, 62.9, 60.3, 56.6, 52.6, 50.9, 50.0, 41.8, 40.5, 38.0, 37.3, 33.6, 33.6, 32.0, 29.3, 29.1, 28.4, 26.0, 25.4, 24.3, 24.1, 15.8.
Synthesis of compound 18. To a solution of PD-Cabo17 (20 mg, 0.0009 mmol, 1.0 eq.) in deionised water (100 μL) in a 2 mL glass vial was added solution of compound 3 (2.8 mg, 0.0028 mmol, 3.0 eq.) dissolved in DI water (50 μL). The completion of reaction was tracked using HPLC. Upon completion, the product was lyophilized to afford PD-CTT1298-Cabo (18) in 91% yield.
White fluffy Solid, 1H NMR (500 MHz, DMSO-d6) δ 10.2–10.0 (m, Cabo-amide H), 8.55–7.05 (m, D-internal amide H and Cabo and DBCO Ar H), 6.51 (s, Cabo-Ar H) 6.06–5.73 (DBCO –CH2), 5.16–4.59 (m, D-OH, ligand H), 4.53–3.69 (ligand H, linker H, and D-ester –CH2), 3.41–2.79 (m, D-CH2, ligand H, and Cabo H), 2.76–2.55 (m, D-CH2 and Cabo H), 2.47–2.33 (m, D-CH2, Cabo H, and ligand H), 2.32–2.01 (m, ligand H), 1.63–0.78 (m, D-linker H and Cabo H). 13C NMR (125 MHz, DMSO-d6) δ 0.84–1.48 (m, 107H), 1.48–1.71 (m, 75H), 2.26 (s, 250H), 2.40–2.54 (m, 112H), 2.69 (s, 238H), 3.02–3.21 (m, 193H), 3.25–3.36 (m, 50H), 3.36–3.54 (m, 167H), 3.84–4.09 (m, 57H), 4.14–4.67 (m, 59H), 4.70–5.32 (m, 60H), 5.86 (d, J = 39.1 Hz, 6H), 6.02 (dd, J = 53.3, 17.4 Hz, 16H), 6.58 (s, 8H), 7.20 (t, 29H), 7.25–7.53 (m, 62H), 7.53–7.75 (m, 63H), 7.74–7.99 (m, 49H), 7.99–8.47 (m, 89H), 8.56–8.62 (m, 8H), 10.12–10.35 (m, 16H). 31P NMR (202 MHz, D2O) δ 7.34. MALDI-ToF: theoretical: 25.5 kDa; obtained: 24.1 kDa.
Drug release studies
In vitro drug release studies were conducted under plasma conditions (phosphate-buffered saline, PBS, pH 7.4) and intra-tumoral conditions (citrate buffer, pH 5.5, containing esterase). PD-CTT1298-Cabo conjugate was dissolved at a concentration of 1 mg mL−1 in each respective buffer and underwent incubation at 37 °C with continuous shaking to replicate physiological conditions. At specific time intervals, samples were withdrawn, promptly quenched with an equivalent volume of methanol, and subsequently stored at −20 °C until further analysis. The released drug was then analyzed using HPLC, and the extent of drug release was determined by comparing it to the standard curve established for free drug (Cabo-OH) on the HPLC system.In vitro studies
000 events were collected per sample. The Mean Fluorescence Intensity (MFI) was acquired and normalized. For the blocking experiments, the same procedure was repeated except the cells were incubated with CTT 1057 (500 nM) for 30 min prior to the addition of PD-CTT1298-Cy5 dendrimer. For the dose response experiment, the cells were incubated with different concentrations (0.1 nM–50 nM) of the dendrimer for 1 H.
Animal studies and in vivo fluorescence imaging
The animal studies received prior approval from the Washington State University (WSU) IACUC and complied with IACUC recommendation. Male 5-week-old athymic nude mice were purchased from the Jackson Laboratory and housed in the animal research facility at WSU Spokane. The control (PSMA−) PC3 and PSMA overexpressing (PSMA+) PC3-PIP cell lines were cultured in RPMI medium containing 10% fetal bovine serum and 1% penicillin–streptomycin. 2 × 106 PC-3 cells were mixed 1:1 with Matrigel (BD Biosciences) and injected subcutaneously into the flank of the nude mice to allow tumor development. For in vivo fluorescence imaging, mice growing PSMA+ PC-3 PIP tumors at a size of 150–200 mm3 were randomly divided into two groups to receive intravenous injection of a blocking agent (100 μL injection volume) or not, 30 min prior to imaging (n = 3 for each group). Then all tumor-bearing mice were intravenously injected with PD-CTT1298-Cy5 at the dose of 20 mg kg−1 (100 μL injection volume) and subjected to whole-body fluorescence imaging at 1, 6, 24, and 48 H after injection using an IVIS SpectrumCT In Vivo Imaging System (PerkinElmer). At the endpoint, the mice were sacrificed and dissected to obtain tumors and vital organs for ex vivo fluorescence imaging and quantification. The fluorescence intensities in regions of interest (ROI) were calculated using the Indigo software equipped with the IVIS instrument. Whole blood was drawn from the heart and placed in a heparin-coated tube. Tissues were snap-frozen in liquid nitrogen and stored at −80 °C.
Ex vivo dendrimer quantification via fluorescence spectroscopy
Ex vivo quantification of dendrimers was carried out by thawing the frozen organs (tumor, heart, lungs, liver, kidneys, spleen, and brain) gradually on ice and weighing them. Known amounts of tissues from each organ were taken, weighed, and homogenized with stainless steel beads in methanol at a concentration of 1 mL/100 mg of tissue using a tissue homogenizer. The homogenized samples were then centrifuged at 4 °C, and the clear supernatant was transferred to Eppendorf tubes and stored at −80 °C in dark. For fluorescence quantification, the thawed supernatants were centrifuged again, and fluorescence intensity of the supernatant was measured using a Horiba Fluoromax spectrofluorophotometer. The fluorescence intensity for Cy5 (λex = 645 nm, λem = 662 nm) was determined. These fluorescence intensity values were converted to dendrimer concentrations using calibration curves of PD-CTT1298-Cy5 at different slit widths.PSMA IC50 for PD-CTT1298
The routine determination of IC50, as most recently described in our laboratory,41,42 for PD-CTT1298 was achieved using concentrations of 160, 80, 40, 20, and 10 nM. Results are presented as mean ± standard error of the mean (SE).Statistical analysis
The statistical analyses were performed using Student's 2-tailed t-test with unequal variances.Results and discussion
Synthesis of fluorescently labelled PD-CTT1298
By combining PSMA targeting through irreversible ligand (CTT1298) with a nanotechnology-based approach employing dendrimers, we have precisely engineered systemic PSMA-targeting dendrimers (PD-CTT1298) for enabling targeted intracellular delivery of potent chemotherapeutic agents to tumor cells. It is worth noting that while a few PSMA-targeted dendrimers have been assessed for targeting and drug delivery, these prior endeavours were based on cationic (positively charged) dendrimers.36,37 However, the toxicity concerns of cationic dendrimers due to their interactions with negatively charged cell membranes, has impeded their clinical translation.38–40 On the contrary, neutral PAMAM-G4-OH dendrimers (PD), which are non-cytotoxic, and biocompatible, have emerged as promising candidates for target-specific drug delivery applications. Moreover, PDs exhibit excellent water solubility making them favourable nanocarriers for drug delivery applications. The molecular and structural confirmations of PAMAM dendrimers have been extensively studied in literature using molecular dynamics simulations, which suggest that the generation 4 PAMAM dendrimers, in the presence of a favorable solvent, like water, assume spherical shape, with branches stretched out and surface groups protruding outside.43,44 Water is a choice of solvent for drug delivery applications. Even though these molecular simulation studies were conducted on amine-terminating PAMAM dendrimers, we expect similar configuration for hydroxyl-terminating PAMAM dendrimers in water, where terminal amines are replaced with hydroxyl groups keeping the backbone same. We chose CTT1298 as the PSMA-targeting agent due to its high-affinity and irreversible binding to PSMA (IC50 19 nM), that translated to extensive internalization in PSMA (+) tumor cells.18,19 However, its highly charged nature due to the presence of multiple carboxylate groups leads to rapid renal clearance, that can be avoided via dendrimer conjugation by increasing its blood circulation time. Moreover, dendrimer also provides a platform for the attachment of high payloads of therapeutic molecules combing drug delivery with targeting. We opted to attach 3 molecules of CTT1298 on the periphery of dendrimers based on the following rationale. An analogue of CTT1298 known as TG97 was used to deliver the enzyme yeast cytosine deaminase (yCD) to PSMA-positive cells.45 In addition, three molecules of CTT54 (another CTT1298 analogue) were used to deliver Cy5-streptavidin to PSMA-positive cells.46 Both of these examples confirm that highly-potent small molecule ligands to PSMA can deliver large molecular cargo. Moreover, CTT1298 has several carboxylic acids and a phosphonic acid group. Attachment of large number of ligands may lead to a negatively charged dendrimer conjugate that may show non-specific uptake, as previously reported for negatively charged nanoparticles.47,48 To achieve PSMA specific targeting with minimal accumulation of dendrimer at off-target organs, we opted to attach only ∼3 targeting ligands. The conjugation of CTT1298 targeting ligand on the surface of PD was achieved employing highly efficient and robust Strain-Promoted Alkyne Azide Cycloaddition (SPAAC) reactions. The selectivity of SPAAC enables controlled reactions, precise ligand, or drug loading, minimizing unwanted byproducts, and its ability to operate under mild conditions, including physiological temperatures, ensures its applicability to a wide range of applications.49 Its versatility in conjugating different molecules, from ligands to drugs and imaging agents, underscores its utility in creating targeted drug delivery systems, imaging probes, and facilitating biomolecular labelling.50The synthesis of PD-CTT1298-Cy5 was initiated with the modification of CTT1298 (1) to bring dibenzocyclooctyne (DBCO) group to participate in SPAAC reaction (Fig. 1A). This was achieved by reacting compound 1 with DBCO-NHS ester (2) to obtain DBCO-C6-CTT1298 (3). The presence of DBCO protons in the aromatic region along with the ligand protons confirmed the product formation (Fig. 1B, blue spectrum). Next, the hydroxyl groups on PD (4) were partially modified through a reaction with azido hexanoic acid via Steglich esterification (Fig. 1A), resulting in a partially azide-terminated dendrimer (5) with approximately six periphery azides, confirmed by the appearance of linker protons between δ 1–2 ppm and dendrimer-ester methylene protons at δ 4.7 ppm in 1H NMR (Fig. 1B, black spectrum). Subsequently, the PD-Azide (5) and DBCO-C6-CTT1298 (3) were conjugated via SPAAC reaction in deionized (DI) water. SPAAC facilitated the conjugation of the ligand onto the dendrimer surface in the desired equivalents to yield PD-CTT1298 (6) with ∼3 ligand molecules attached, confirmed by the comparative integration of aromatic protons from the ligand between δ 7 and 8 ppm and linker protons from dendrimer in the aliphatic region (Fig. 1B, pink spectrum). The HPLC chromatogram exhibited a shift in retention time, transitioning from 9.2 minutes for PD-azide to 9.6 minutes for PD-CTT1298 upon the conjugation of the PSMA targeting ligand (Fig. 2A). The HPLC purity level of PD-CTT1298 exceeded 99% (Fig. 2A). The size and zeta potential distribution of PD-CTT1298 was analyzed using dynamic light scattering (DLS). The hydrodynamic radius of PD-CTT1298 was found to be 4.2 ± 0.2 nm and the zeta potential distribution was −6 mV (Fig. 2B and C). To evaluate whether CTT1298 conjugated to the dendrimer retained activity as an inhibitor of PSMA, the PSMA-IC50 was evaluated using our previously published procedure.41,42 The IC50 of PD-CTT1298-Cy5 was still in nM range (20.26 nM), suggesting the retention of PSMA targeting ability upon dendrimer conjugation (Fig. 2C and S8†).
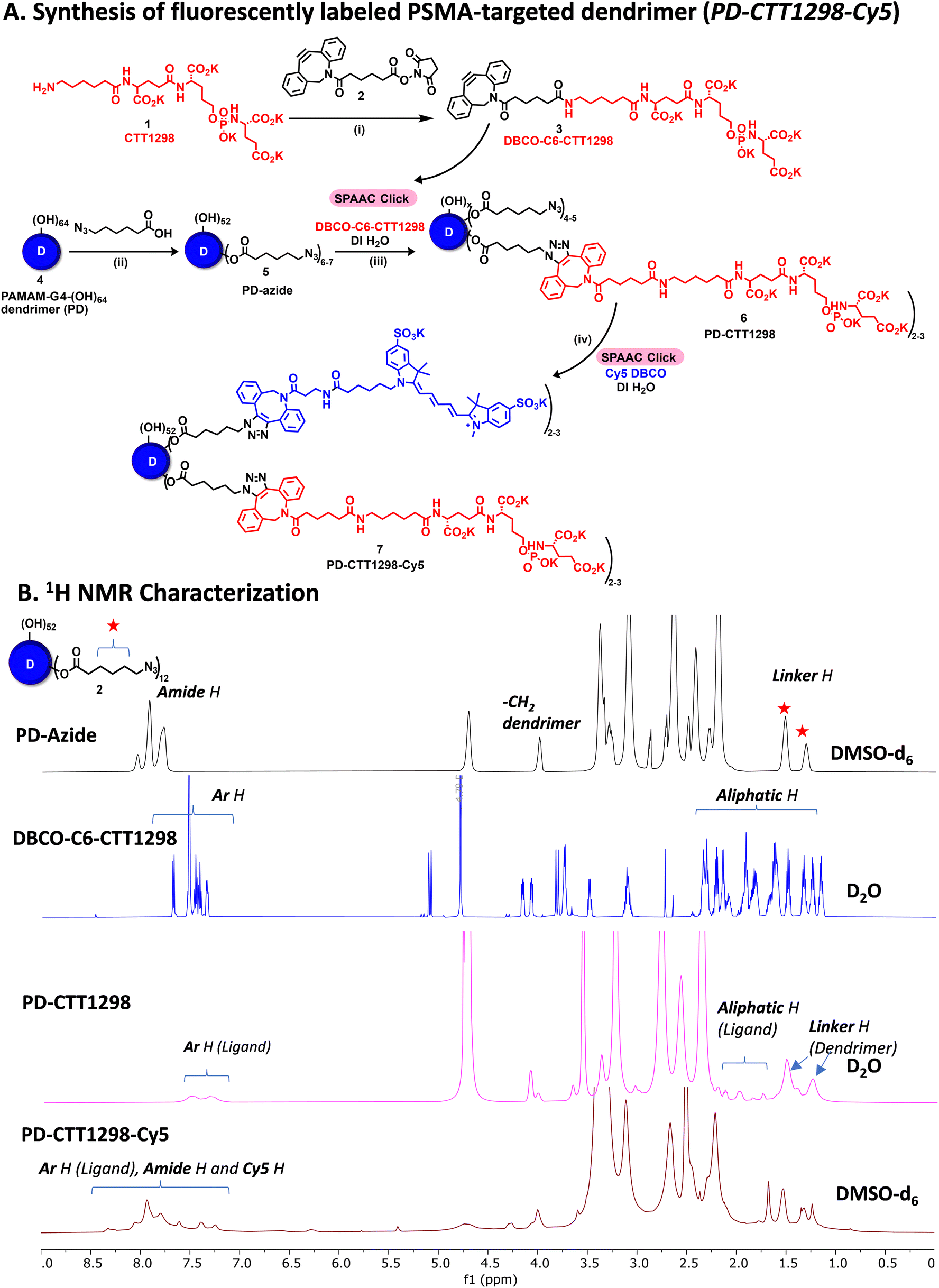 | ||
| Fig. 1 Preparation and structural elucidation of PD-CTT1298-Cy5 conjugate. (A) Schematic representation for the synthesis of fluorescently labelled PSMA targeted dendrimer via SPAAC reactions. Reagents and conditions: (i) KHCO3, ddH2O, THF, RT, 3 H, 60%; (ii) 6-azido-hexanoic acid, EDC·HCl, DMAP, Anhy. DMF, RT, 24 H, 82%; (iii) DI H2O, 12 H, RT, 92%; (iii) DI H2O, 48 H, RT, 81%. (B) 1H NMR spectra showing characteristic proton signals of CTT1298, Cy5 and PD at each step of the synthesis. | ||
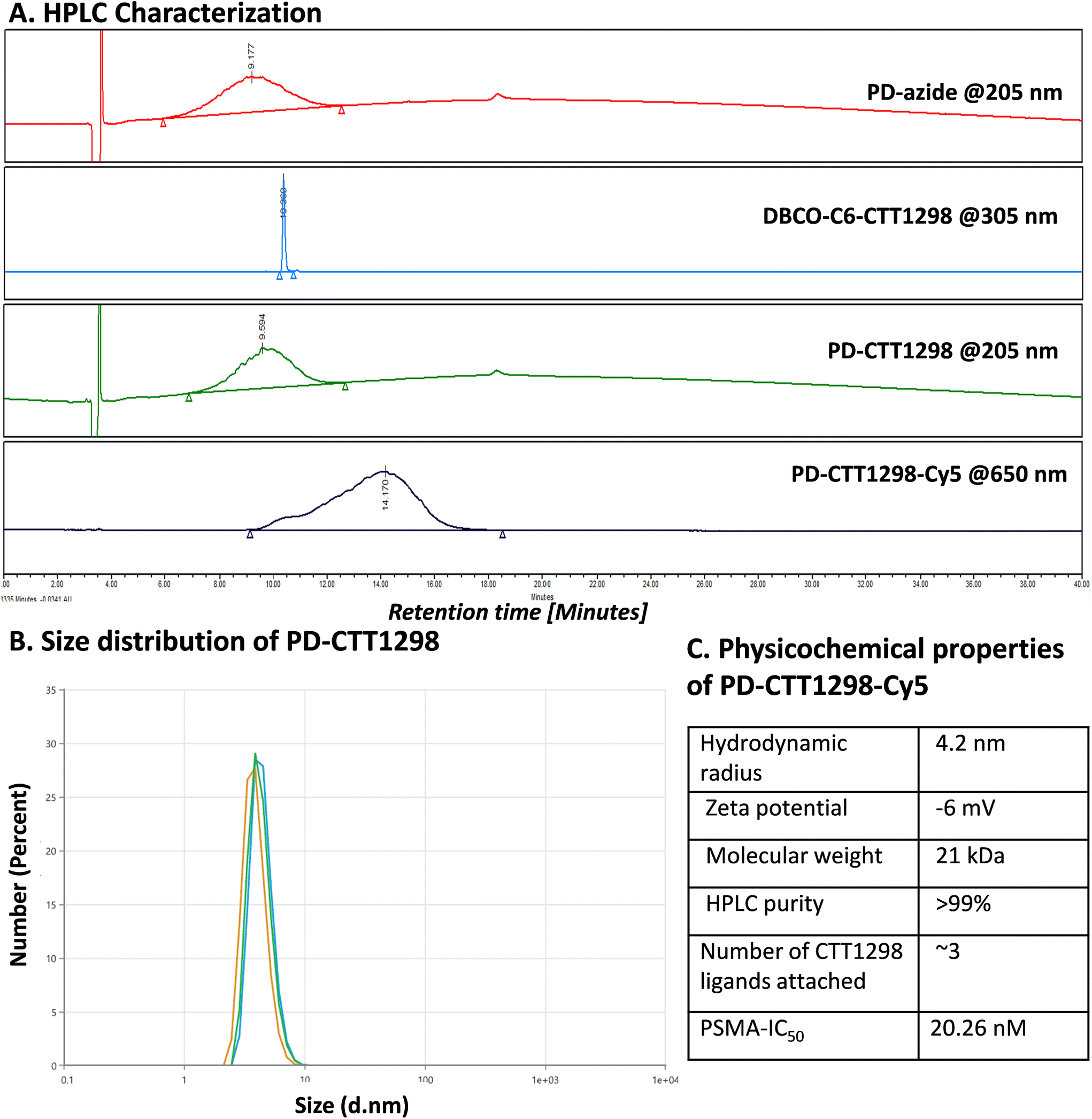 | ||
| Fig. 2 Physicochemical characterization of PD-CTT1298-Cy5 conjugate and intermediates. (A) HPLC chromatograms showing purity and shift in the retention time at different steps of synthesis and showing peak at 650 nm after successful conjugation of Cy5. (B) Hydrodynamic radius of PD-CTT1298 analyzed by dynamic light scattering (DLS) in triplicates. (C) Physicochemical properties of PD-CTT1298-Cy5 dendrimer. Size and zeta potential are presented for non-fluorescent PD-CTT1298. | ||
To further investigate the in vitro and in vivo PCa cell uptake and organ biodistribution of PD-CTT1298 via confocal and fluorescence spectroscopy, a near-infrared dye cyanine 5 (Cy5) was introduced at its surface. Subsequent SPAAC reaction of PD-CTT1298 with Cy5-DBCO yielded the final fluorescent dendrimer PD-CTT1298-Cy5 (7). Confirmation of Cy5 attachment was achieved through the observation of Cy5 protons in the 1H NMR spectrum (Fig. 1B). Using the proton integration method, the calculation indicated the attachment of approximately two Cy5 molecules on the dendrimer surface (Fig. 1B, red spectrum). PD-CTT1298-Cy5 demonstrated a purity exceeding 98% in HPLC, with the chromatogram showing a significant shift in retention time from 9.6 to 14.2 minutes upon Cy5 conjugation (Fig. 2A). All the intermediates and final conjugates were characterized using NMR and Mass spectroscopy and the purity was analyzed using HPLC (ESI Fig. S1–S7†).
PD-CTT1298-Cy5 demonstrates selective uptake in PSMA (+) PC3-PIP cells via PSMA mediated internalization
Next, to investigate the selective uptake of the PD-CTT1298-Cy5 dendrimer in PSMA (+) cells, cell uptake experiments were performed under various conditions (Fig. 3). We first analyzed the qualitative uptake of PD-CTT1298-Cy5 in PSMA (+) and PSMA (−) cells using confocal microscopy. The PSMA (+) PC3-PIP and PSMA (−) PC3 cells when incubated with 50 μg·mL−1 (2.5 μM) of the PD-CTT1298-Cy5, revealed the selective uptake of PSMA-targeted PD-CTT1298-Cy5 in PSMA (+) cells (Fig. 3A). A negligible uptake was observed in PSMA (−) PC3 cells. We next evaluated the quantitative uptake and mechanism of uptake of dendrimers using flow cytometry. At 1 μg mL−1 (50 nM) concentration, selective uptake was observed in PSMA (+) PC3-PIP cells over 2 H (Fig. 3B-a). As expected, in PC3-PIP cells, the Mean Fluorescence Intensity (MFI) increased significantly over the time points due to their high PSMA expression. A significant change in MFI (∼150-fold increase) was observed in as little as 30 minutes (Fig. 3B-a) when compared to the 0 H and blank samples. Minimal uptake of the PD-CTT1298-Cy5 dendrimer was observed in PC3 cells, which express little to no PSMA. The uptake was significantly more in PSMA (+) cells compared to PSMA (−) cells at all time-points. The histograms obtained also showed a 2-log shift in the PSMA (+) cells (Fig. 3B-d) compared to PSMA (−) cells (Fig. 3B-e), which was anticipated based of their PSMA expression. The results clearly demonstrate that the uptake of PD-CTT1298-Cy5 corelates with the PSMA expression on the cells, suggesting that the nanoplatform could serve as an effective means for targeted drug delivery to PSMA-positive prostate cancer cells.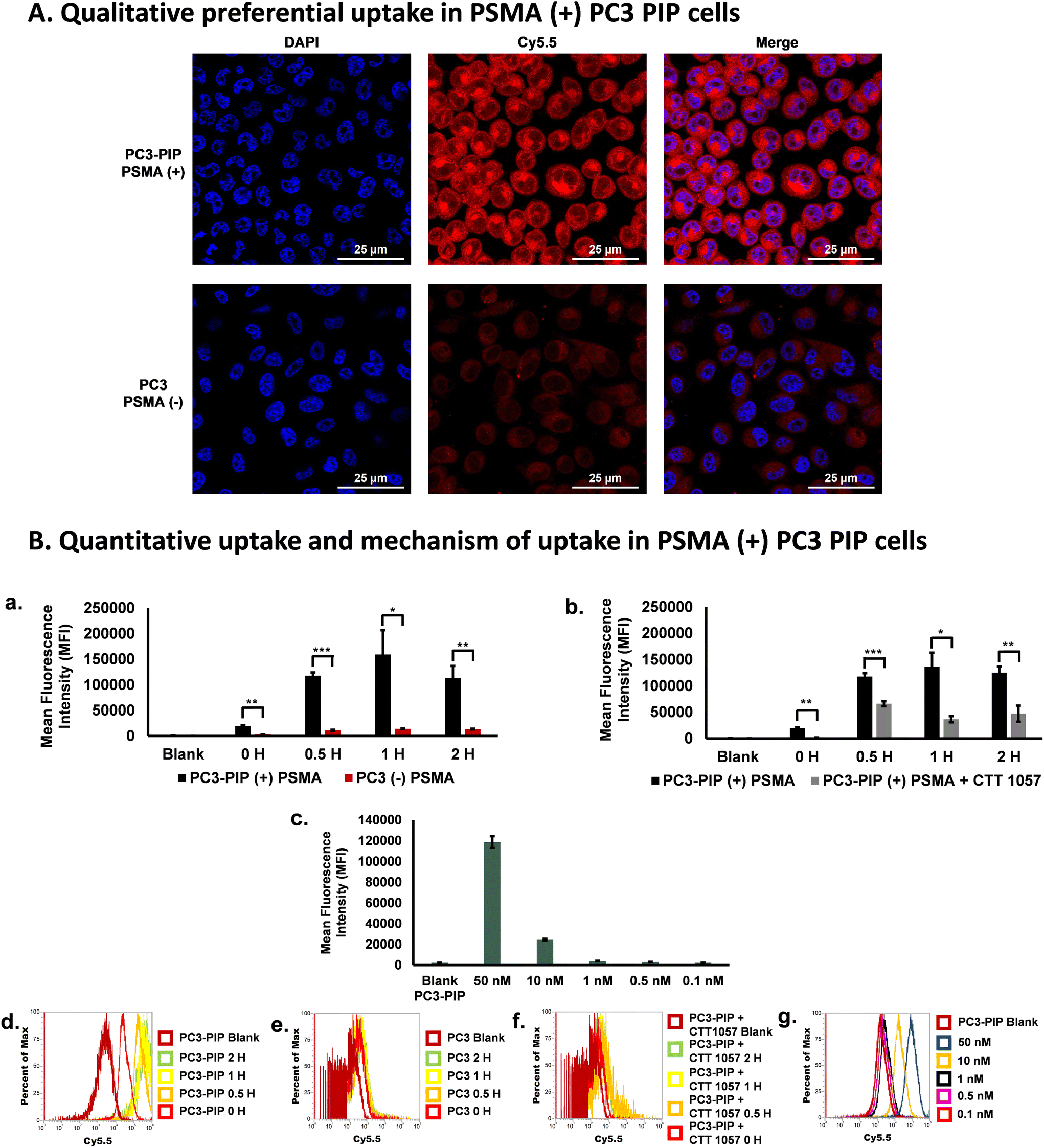 | ||
| Fig. 3 Selective uptake observed with CTT-1298-Cy5 in PSMA (+) PC3-PIP cells. (A) Confocal microscopy images from PSMA (+) PC3-PIP and PSMA (−) PC3 cells incubated with 50 μg mL−1 (2.5 μM) of the fluorescently labeled PSMA targeted dendrimer, PD-CTT1298-Cy5. (B) Selective uptake of PD-CTT1298-Cy5 observed with 1 μg mL−1 (50 nM) in PSMA (+) PC3-PIP cells. (B-a) Mean Fluorescence Intensities (MFI) of PC3-PIP and PC3 cells loaded with PD-CTT1298-Cy5 dendrimer over 2 H. (B-b) MFI of PC3-PIP cells incubated with and without a PSMA irreversible inhibitor, CTT 1057 (blocking agent), prior to loading with PD-CTT1298-Cy5 dendrimer over 2 H. (B-c) Dose response at 1 H with PSMA (+) PC3-PIP cells. (B-d) Histogram of PC3-PIP uptake via Cy5.5 channel over 2 H. (B-e) Histogram of PC3 uptake via Cy5.5 channel over 2 H. (B-f) Histogram via Cy5.5 channel at 1 H time point for dose response studies in PC3-PIP cells. (B-g) Histogram of PC3-PIP cells with and without CTT1057 via Cy5.5 channel over 2 H. Data representative of experiments performed in triplicate. The p-values were calculated between the PC3-PIP cells and PC3 cells in the uptake experiments including with and without the addition of CTT 1057 at each time point in the blocking experiments with * p < 0.05, ** p < 0.01 and *** p < 0.001. | ||
To study any non-specific uptake of the PD-CTT1298-Cy5 in PSMA (+) cells and to further confirm the mechanism of uptake via PSMA receptors, a blocking experiment was performed at different time points in the presence of a potent irreversible PSMA inhibitor (CTT 1057).18,19 In the blocking experiment, a drastic decrease in the uptake of the PD-CTT1298-Cy5 was observed when PC3-PIP cells were incubated with a PSMA irreversible inhibitor with nanomolar affinity, CTT 1057 (Fig. 3B-b and B-f). A significant decrease in MFI was observed at all time points when the cells were incubated with CTT 1057. A dose response study was performed at 1 H with different concentrations of PD-CTT1298-Cy5 (Fig. 3B-c and B-g). The selective uptake was dose dependent with the highest concentration tested (50 nM) resulting in the highest MFI. All together, these in vitro results clearly suggested that the PD-CTT1298-Cy5 was selectively targeted and taken up by cancer cells that expressed high levels of PSMA and internalized rapidly intracellularly following binding to PSMA.
Qualitative and quantitative tumor and organ biodistribution of systemically administered PD-CTT1298-Cy5 in a PCa tumor xenograft mouse model
We further explored the in vivo tumor-targeting potential and biodistribution of CTT-1298-Cy5 in a human PC3 (PSMA−) and PC3-PIP (PSMA+) tumor xenograft model. To study the mechanism of uptake, the tumor uptake and biodistribution was also studied in PC3-PIP (PSMA+) tumor xenograft model in the presence of a blocking agent, CTT1057. Upon intravenous administration of PD-CTT1298-Cy5 dendrimers in PSMA (−) and PSMA (+) tumor-bearing mice, Cy5.5 fluorescence signals arising from the tumors and mouse vital organs were measured and compared at different time points by the in vivo imaging system (IVIS) imaging. The fluorescence signal accumulated in the mouse organs in general decreased more rapidly with time than that in tumors across all three groups and appeared to be invisible starting 24 H post-injection. This clearly suggests the rapid clearance of dendrimers from off-target organs and tissues, which is highly desired for targeted delivery of potent chemotherapeutic agents where systemic side-effects are a major concern. Furthermore, while most other nanoparticles when systemically administered show unwanted accumulation of up to 80% in the liver,51 the PD-CTT1298-Cy5 takes advantage of its small size (∼4 nm) in the range of renal filtration and clears intact through kidneys as demonstrated earlier for PD nanoplatform.30,32PD-CTT1298-Cy5 is selectively targeted to PSMA (+) PC3-PIP tumor and did not show accumulation in the mice with PSMA (−) PC3 tumors. The lack of discernible fluorescence signal since 6 H post-injection suggested rapid clearance from PSMA (−) tumors. However, in PSMA (+) tumor, PD-CTT1298-Cy5 demonstrated stable and intense fluorescence signal starting 6 H post-injection which was retained up to 48 H and was dimmed out by prior uptake of a blocking agent, CTT1057, further confirming the mechanism of uptake through PSMA receptors (Fig. 4A, B, and S29†). In line with the in vivo observations, the endpoint ex vivo fluorescence imaging further indicated a high level of fluorescence signal in PSMA (+) tumors, which tended to be lower in PSMA (+) tumors with pre-treatment of a blocking agent and absent in PSMA (−) tumors (Fig. 4C, D and S29†). This was further confirmed by the quantitative uptake of PD-CTT1298-Cy5 in tumor tissues from all three groups using fluorescence spectroscopy. While the dendrimer uptake in PSMA (+) tumor group was ∼15% of the injected dose (ID), it decreased to ∼4% upon PSMA blocking, and was <2% in PSMA (−) tumor group (Fig. 5A), correlating to the ex vivo fluorescence imaging by IVIS.
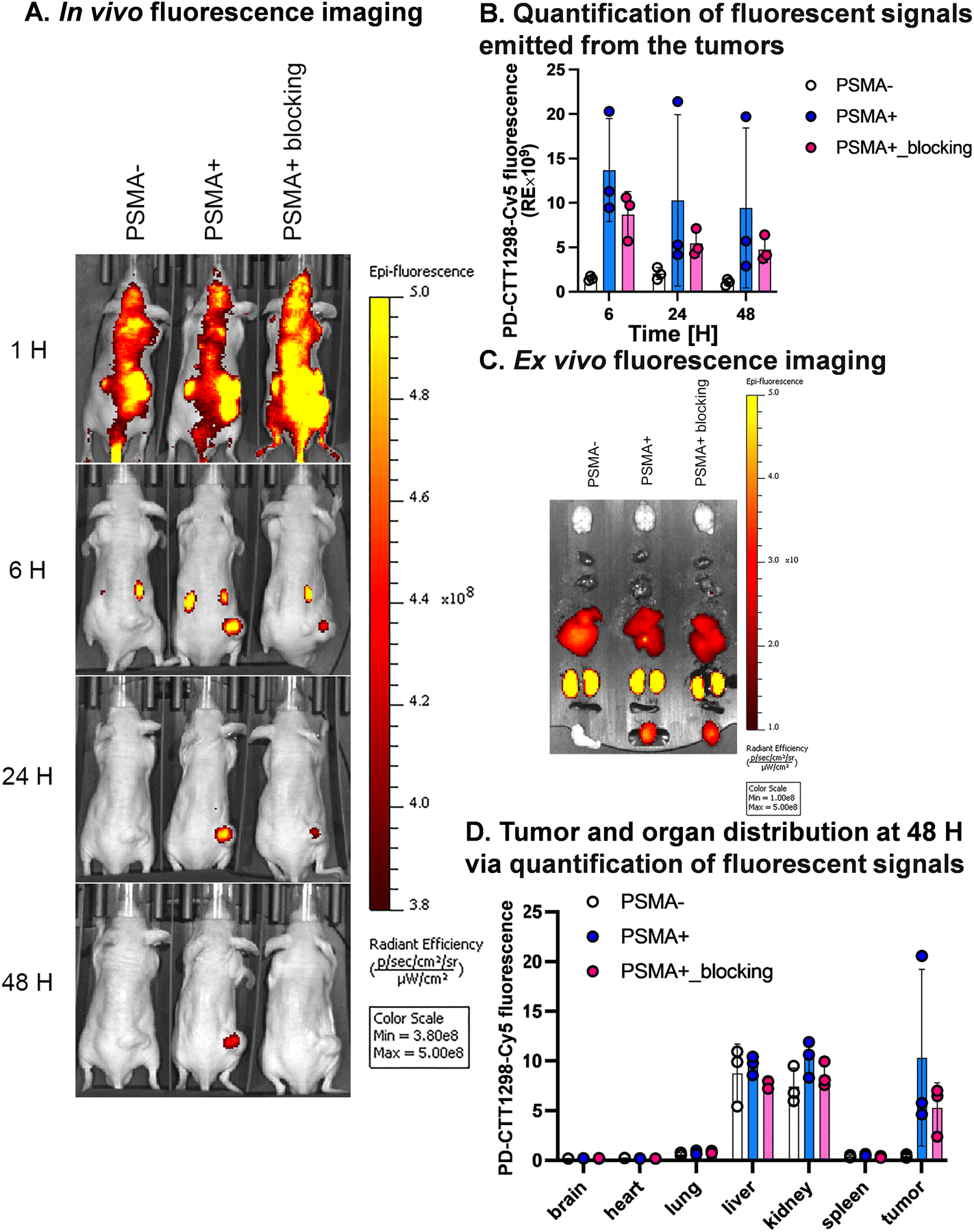 | ||
| Fig. 4 In vivo and ex vivo fluorescence imaging of PSMA-targeted dendrimer for tumor targeting and whole-body biodistribution. (A) Whole-body in vivo fluorescence images of mice subcutaneously growing (1) PSMA (−) PC3 tumors, (2) PSMA (+) PC3-PIP tumors, and (3) PSMA (+) PC3-PIP tumors with prior uptake of a blocking agent at 1 H, 6 H, 24 H, and 48 H after intravenous injection of PD-CTT1298-Cy5 (n = 3 mice per group). (B) Quantitative analysis of in vivo fluorescence signals from the tumors at 6 H, 24 H, and 48 H (n = 3). (C) Ex vivo fluorescence images of vital organs from representative mice of individual groups at 48 H post-injection of PD-CTT1298-Cy5. The order of organs from top to bottom is as follows: brain, heart, lungs, liver, kidneys, spleen, and tumor. (D) Quantitative analyses of ex vivo fluorescence signals from the organ tissues and tumors (n = 3). The data was presented as mean ± S.D. | ||
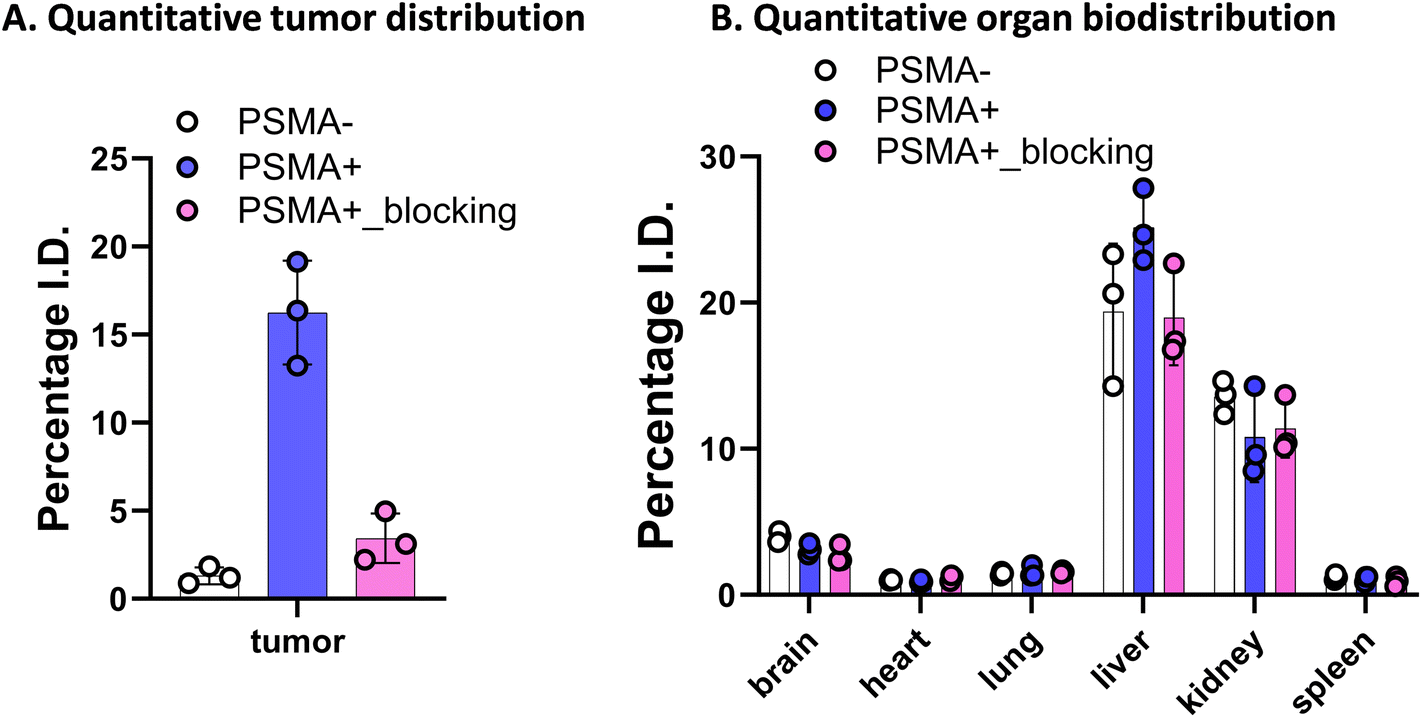 | ||
| Fig. 5 Quantitative tumor and organ biodistribution of PD-CTT1298-Cy5 at 48 H after systemic administration in PC3-PIP tumor xenograft mouse model. Quantitative biodistribution of PD-CTT1298-Cy5 in (A) tumour, and (B) all major organs at 24 h time point (n = 3). The data was obtained through fluorescence spectroscopy of homogenized tissue extracts containing PD-CTT1298-Cy5 and reported as a percentage of the injected dose in total organ. | ||
A significant challenge in the clinical application of nanomedicine-based therapeutics is their potential undesired accumulation in off-target organs. We next examined the ex vivo qualitative and quantitative distribution of PD-CTT1298-Cy5 in key organs, including the heart, lungs, liver, spleen, and kidneys at 48 H via IVIS and fluorescence spectroscopy (Fig. 4C, D, and 5B). The PD-CTT1298-Cy5 showed minimum accumulation in the vital organs including brain, heart, lungs, and spleen. The PD-CTT1298-Cy5 levels in the heart, lungs, spleen, and brain were found to be less than 5% ID in all groups, indicating the rapid clearance of the dendrimer from these organs by the 48 H time point (Fig. 5B). This was consistent with the ex vivo imaging of these organs (Fig. 4C and D). The ex vivo imaging showed some accumulation in liver and kidneys (Fig. 4C and D). In line with ex vivo imaging results, the tissue quantification suggested ∼20% and ∼10% ID of PD-CTT1298-Cy5 in liver and kidneys respectively (Fig. 5B). Although there is some non-specific liver uptake, the fluorescence signal in kidneys is rather expected due to the renal clearance mechanism of PD-CTT1298-Cy5.
To summarize these biodistribution results, (1) the uptake of PD-CTT1298-Cy5 was significantly higher in the tumor of the PSMA (+) group compared to both the PSMA (+) plus blocking and PSMA (−) groups, suggesting the uptake of PD-CTT1298-Cy5 in the tumor through PSMA mediated targeting; (2) PD-CTT1298-Cy5 cleared rapidly from other organs, indicating the targeted delivery of PD-CTT1298-Cy5 to the prostate tumor, and (3) there was a significant presence of PD-CTT1298-Cy5 in the tumor region of the PSMA (+) group at 48 H post-administration, suggesting the long-lasting sustained retention of PD-CTT1298-Cy5 in the prostate tumor regions. Collectively, these results suggested that PD-CTT1298-Cy5 dendrimers target PSMA (+) prostate tumors with preferential specificity making it a potential platform for the targeted delivery of potent chemotherapeutic agents to prostate cancer with positive PSMA expression.
Synthesis and characterization of PSMA-targeted dendrimer cabozantinib conjugate
To further investigate if PD-CTT1298-Cy5 can be utilized to deliver chemotherpautic agents for the treatment of PCa, we developed its drug conjugate with cabozantinib (Cabo). Cabo is a multi-tyrosine kinase inhibitor and is approved as a single agent for renal cell and hepatocellular carcinoma in the USA and Europe.52–55 Unfortunately, its PCa clinical trial (Phase III) was terminated due to significant negative effects at tolerated doses.56–59 However, in preclinical testing, Cabo exhibited significant inhibition of advanced PCa tumor progression.60–62 Cabo in combination with immunotherapy is now a standard treatment in metastatic renal cancer, and its efficacy is being tested in PCa.63 Therefore, we hypothesized that the PD-CTT1298 mediated intracellular delivery of Cabo may enhance its efficacy and reduce the negative side effects.The synthesis of PSMA-targeted dendrimer cabozantinib conjugate (PD-CTT1298-Cabo) began with the synthesis of DBCO modified Cabo (Cabo-DBCO) (Fig. 6A-I). The DBCO modification was carried out at position 7, on the solvent exposed site of Cabo. It has been previously reported that the modification at position 7 of Cabo, did not alter the activity towards c-Met.64 To synthesize Cabo-DBCO, we first synthesized 7-demethylated cabozantinib intermediate (Cabo-OH; 13) followed a previously published protocol,65 with slight modifications. The reaction initiated by treating 7-(benzyloxy)-4-chloro-6-methoxyquinoline (8) with 4-aminophenol (9) in the presence of NaH in DMF, resulting in the formation of 4-((7-(benzyloxy)-6-methoxyquinolin-4-yl)oxy)aniline (10) in 92% yield. The desired product formation was confirmed through the observation of a proton NMR signal of –NH2 at δ 5.1 ppm (Fig. S9†). Subsequently, the condensation of the amino intermediate (10) with 1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxylic acid (11) was carried out using HATU and DIEPA in dichloromethane, yielding compound 12. The structure of coupling product was validated by the appearance of two amide (–NH) proton peaks at δ 10.05 and 10.23 ppm, along with a cyclopropane ring proton singlet at δ 1.48 ppm (Fig. S11†). Finally, the removal of the benzyl protecting group at the 7th position was accomplished through treatment with trifluoroacetic acid at 60 °C for 30 minutes, resulting in the formation of the 7-demethylated cabozantinib intermediate (13) in 87% yield. The completion of the deprotection was evident from the 1H NMR, which showed the disappearance of benzyl (–OBn) proton signals in the aromatic region and its corresponding methylene (–OCH2) protons in the aliphatic region. Additionally, a hydroxyl peak at δ 10.16 ppm (Fig. S13†) further confirmed the structure of the intermediate. The mass spectra analysis also provided the confirmation of the successful formation of intermediate 13 (Fig. S15†). Further, the synthesis of compound 18, Cabo-DBCO, was achieved through the coupling of DBCO acid with Cabo-OH (13) in the presence of EDC-DMAP. The confirmation of the desired structure was established by the disappearance of the hydroxyl protons of Cabo-OH at δ 10.16 ppm. Simultaneously, the emergence of distinctive peaks corresponding to the DBCO ring's –CH2 protons at δ 3.73 ppm (d, J = 14.0 Hz, 1H) and 5.14 ppm (d, J = 14.1 Hz, 1H), along with the DBCO aromatic protons in the 1H NMR spectrum, validated the successful synthesis of Cabo-DBCO (Fig. 6B and S16†).
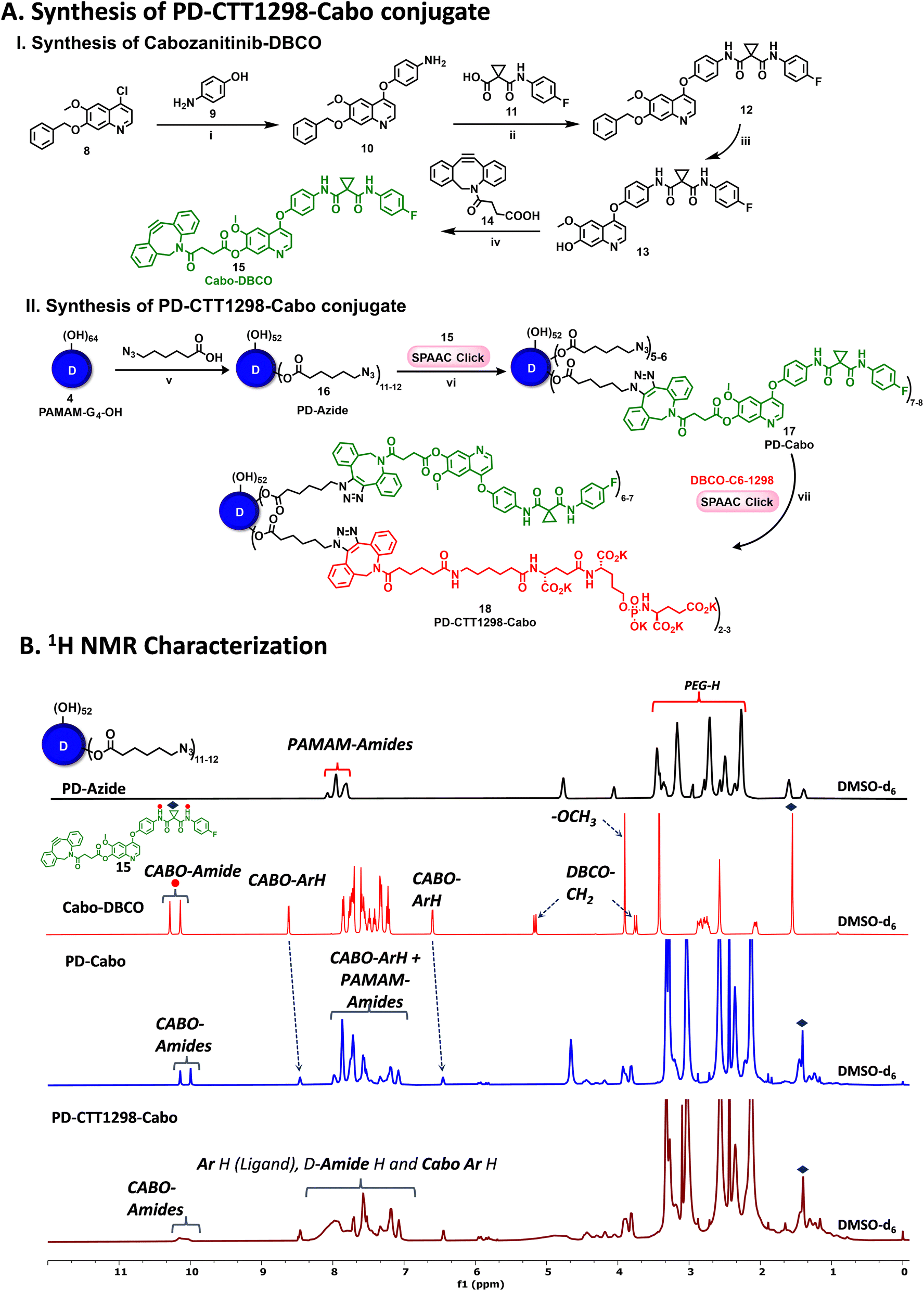 | ||
| Fig. 6 Synthesis and characterization of PD-CTT1298-Cabo conjugate. (A) Schematic representation of the conjugation of Cabo and PSMA ligand on PAMAM-G4-OH to synthesize PD-CTT1298-Cabo conjugate; reagents and conditions: (i) NaH, DMF, 80 °C, 16 h, 92%; (ii) HATU, DIPEA, DMF, RT, 16 h, 90%; (iii) TFA, 60 °C, 30 min, 87%; (iv) DBCO acid, EDC·HCl, DMAP, DCM, 2 h, RT, 57%; (v) 6-azido-hexanoic acid, EDC·HCl, DMAP, anhy. DMF, RT, 24 h, 86%; (vi) DMF, 3 h, RT, 88%; (vii) DI water, 3 h, RT, 91% (B) 1H NMR spectra of PD-CTT1298-Cabo dendrimer and intermediates representing the appearance and disappearance of characteristic protons. | ||
On the other hand, PD was modified to bring ∼12 azide groups to obtain PD-azide (16), which was reacted with Cabo-DBCO (15) using SPAAC reaction in DMF to obtain PD-Cabo conjugate 17 (Fig. 6A-II). The SPAAC reaction exhibited rapid kinetics and enabled the conjugation of an exact equivalent of Cabo without the need for any additional reagents. The reaction progress was monitored by HPLC, revealing a distinct shift in the chromatogram (10.95 to 10.82 min) as Cabo was successfully linked to the dendrimer surface (Fig. 7A). After dialysis purification and freeze-drying, the PD-Cabo was obtained in 88% yield. The successful conjugation of Cabo onto the dendrimer was verified using 1H NMR, where the emergence of characteristic Cabo peaks was observed alongside the dendrimer protons. 1H NMR confirmed the attachment of approximately 8 drug molecules per dendrimer corresponding to ∼16 weight percent drug loading (Fig. 6B and S21†). Our previous work on PD platform for targeted drug delivery applications demonstrates that the dendrimer platform retains its targeting capabilities and can successfully deliver the drugs to targeted intracellular locations when the drug loading is in the limit of 20 weight percent.30,34,66 The purity of PD-Cabo conjugate was ∼99% by HPLC (Fig. 7A and S23†). After the attachment of Cabo to the dendrimer surface, the conjugation of the PSMA ligand, CTT1298, onto the dendrimer was carried through SPAAC chemistry. The Compound 17 was treated with the DBCO-C6-CTT1298 in DI water at room temperature for 3 h, resulting in the synthesis of PD-CTT1298-Cabo (18). The HPLC chromatogram exhibited a shift from 10.82 to 10.58 min (Fig. 7A), and the appearance of additional protons in the 1H NMR spectrum indicated the successful incorporation of the ligand onto the dendrimer (Fig. 6B). Furthermore, a signal at δ 7.34 ppm in the 31P NMR spectrum validated the presence of the PSMA ligand on the dendrimer surface (Fig. S25†). Utilizing the proton integration method, the number of attached CTT1298 molecules on the dendrimer surface was calculated, suggesting the attachment of approximately three molecules of CTT1298. The HPLC analysis indicated a purity of PD-CTT1298-Cabo exceeding 98% (Fig. 7A and S27†). All the intermediates and final conjugates were characterized using NMR and mass spectroscopy, and HPLC techniques (Fig. S9–S27†). The physicochemical properties of PD-CTT1298-Cabo are presented in Fig. 7C. While both Cabo and Cabo-OH demonstrate poor aqueous solubility, dendrimer conjugation significantly improves the water solubility. The aqueous solubility of PD-CTT1298-Cabo is ∼100 mg mL−1 which translates to ∼16 mg mL−1 for Cabo-OH. The hydrodynamic radius of PD-CTT1298-Cabo is 4.40 ± 0.07 nm and zeta potential distribution is −2.0 ± 0.7 mV as analysed by the DLS (Fig. 7C and S28†). Cabo conjugation did not have much effect on the size and zeta potential of the PD-CTT1298 dendrimer, which was important to maintain the targeting potential of dendrimer intact. We further assessed the comparative c-Met inhibition activities (IC50) of PD-CTT1298-Cabo versus Cabo and Cabo-OH. The PD-CTT1298-Cabo conjugate exhibited a nanomolar c-Met inhibitory activity (IC50: 0.423 nM; Fig. S30†) that was better than both Cabo-OH (IC50: 26.3 nM; Fig. S30†) and Cabo (IC50: 1.3 nM)67 which could be due to the multivalency effect of dendrimers. These data confirm that Cabo is still active when conjugated in this manner.
 | ||
| Fig. 7 Characterization of PD-CTT1298-Cabo conjugates. (A) HPLC traces of intermediates and PD-CTT1298-Cabo conjugate demonstrating a shift in retention time at each synthetic step. (B) In vitro drug release profile of PD-CTT1298-Cabo conjugate at plasma and intra-tumoral conditions at 37 °C. (C) Table showing the physicochemical properties of PD-CTT1298-Cabo conjugate. (D) In vitro efficacy of PD-CTT1298-Cabo in comparison with free Cabo and Cabo-OH over 72 H in PC3-PIP PSMA (+) cells. Cell viability (%) was calculated using the luminescence values obtained from the controls used in the experiment. Data representative of experiments performed in triplicate. p-values were calculated between Cabo, Cabo-OH, and the PD-CTT1298-Cabo dendrimer at each concentration tested with * p < 0.05, ** p < 0.01 and *** p < 0.001. | ||
In vitro drug release study from PD-CTT1298-Cabo conjugate under physiological conditions
Next, we carried out an in vitro drug release study from the conjugate, examining conditions that mimic both the extracellular environment (physiological pH, PBS buffer at pH 7.4) and intratumoral conditions (pH 5.5, carboxyl-esterase) (Fig. 7B). Cabo-OH is linked to the dendrimer through an ester bond via its hydroxyl group at position, 7 allowing for pH and esterase-responsive release. This design facilitates controlled release in both intracellular and intratumor environments, effectively restricting drug exposure beyond the boundaries of the prostate tumor. In PBS buffer at pH 7.4, we observed a <40% drug release in about 2 weeks. Notably, only 10% of the drug was released within the first 8 hours. However, under intracellular conditions, the PD-CTT1298-Cabo conjugate demonstrated a gradual and sustained release of the drug over 2 weeks. Around 15% drug was released in first 8 hours, gradually increasing to ∼35% in 48 hours, reaching approximately 80% drug release over a 12-day period (Fig. 7B). This sustained intracellular release profile is favourable for delivering Cabo-OH through PD-CTT1298-Cabo conjugate to prostate cancer cells for prostate cancer treatment.PD-CTT1298-Cabo conjugate improves the anti-proliferative activity of free Cabo and Cabo-OH
To study the efficacy of the PD-CTT1298-Cabo conjugate in comparison to Cabo and Cabo-OH, a cell viability experiment was performed (Fig. 7D). PC3-PIP PSMA (+) cells were incubated with different concentrations of the compounds along with 10% DMSO which served as a control. Treatments were administered at equivalent drug bases. The cells were incubated for 72 H and the luminescence of the viable cells was measured using the Cell-Titer-Glo Luminescent Cell Viability assay. As expected, the 10% DMSO led to almost no viable cells present. Both dendrimer-Cabo conjugate (PD-CTT1298-Cabo) and free drugs (Cabo & Cabo-OH) exhibited dose dependent efficacy. At the highest concentration tested (1000 μg mL−1), the PD-CTT1298-Cabo was significantly more potent than free Cabo & Cabo-OH. The same results were observed at 100 μg mL−1. However, at lower concentrations the dendrimer conjugate was comparable to the free drugs. There was no significant difference observed between the dendrimer and the free drugs with the highest percentage of viable cells present after 72 H. Overall, the dendrimer was more effective at inducing apoptosis in PSMA (+) cells compared to the free drugs. PD-CTT1298-Cabo exhibited a lower IC50 of 9.83 ± 1.11 μg mL−1 compared to 24.83 ± 2.27 μg mL−1 of Cabo (p = 0.00012 free Cabo vs. PD-CTT1298-Cabo), and 77.30 ± 4.35 μg mL−1 of Cabo-OH (p = 0.0013 Cabo-OH vs. PD-CTT1298-Cabo) indicating stronger anti-proliferative activity. Given the findings from biodistribution and this in vitro experiment, we hypothesize that the in vivo targeting of PD-CTT1298-Cabo to prostate tumor could lead to markedly enhanced therapeutic outcomes for treating prostate cancer.Conclusions
In this study, we have developed a PSMA-targeted dendrimer platform using a highly efficient SPAAC chemistry, for PSMA (+) prostate cancer specific targeted drug delivery. We show that the conjugation of an irreversible PSMA inhibitor, CTT-1298 to a dendrimer retains its PSMA-binding ability in the nanomolar range. The PSMA targeted dendrimer specifically localizes in PSMA (+) prostate cancer cells in vitro and in a prostate tumor in vivo in a PC-3 PIP tumor xenograft mouse model. While the dendrimer remains in the tumor for 48 hours, it clears rapidly from the peripheral organs limiting systemic side-effects. We further developed a PSMA-targeted dendrimer cabozantinib conjugate, PD-CTT1298-Cabo, that exhibited a nanomolar c-Met inhibitory activity. Further, dendrimer based PSMA targeting, and triggered and sustained intra-tumoral drug release significantly improved the anti-proliferative activity of PD-CTT1298-Cabo in PSMA (+) prostate cancer cells. These results indicate that utilizing PSMA-targeted dendrimer-based delivery may improve efficacy and widen the therapeutic window of cabozantinib and other chemotherapeutic agents. This approach may be particularly beneficial in addressing concerns associated with dose-related systemic toxicities of chemotherapies for the treatment of prostate cancer.Conflicts of interest
The authors (AS and CEB) have pending patent relating to the PD-CTT1298 dendrimer technology.Acknowledgements
The animal work was supported by NIH/NCI grants R37CA233658 and R01CA258634 (to B.W.).References
- O. W. Brawley, Trends in prostate cancer in the United States, J. Natl. Cancer Inst. Monogr., 2012, 2012(45), 152–156 CrossRef PubMed.
- L. Bubendorf, A. Schöpfer, U. Wagner, G. Sauter, H. Moch, N. Willi, T. C. Gasser and M. J. Mihatsch, Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients, Hum. Pathol., 2000, 31(5), 578–583 CrossRef CAS PubMed.
- C. D. Fahrenholtz, F. G. Rick, M. I. Garcia, M. Zarandi, R.-Z. Cai, N. L. Block, A. V. Schally and K. L. Burnstein, Preclinical efficacy of growth hormone-releasing hormone antagonists for androgen-dependent and castration-resistant human prostate cancer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(3), 1084–1089 CrossRef CAS PubMed.
- J. B. Tennakoon, Y. Shi, J. J. Han, E. Tsouko, M. A. White, A. R. Burns, A. Zhang, X. Xia, O. R. Ilkayeva, L. Xin, M. M. Ittmann, F. G. Rick, A. V. Schally and D. E. Frigo, Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch, Oncogene, 2014, 33(45), 5251–5261 CrossRef CAS PubMed.
- W. W. Scott, M. Menon and P. C. Walsh, Hormonal therapy of prostatic cancer, Cancer, 1980, 45(7 Suppl), 1929–1936 CrossRef CAS PubMed.
- R. W. Ross, W. Xie, M. M. Regan, M. Pomerantz, M. Nakabayashi, T. J. Daskivich, O. Sartor, M. E. Taplin, P. W. Kantoff and W. K. Oh, Efficacy of androgen deprivation therapy (ADT) in patients with advanced prostate cancer: association between Gleason score, prostate-specific antigen level, and prior ADT exposure with duration of ADT effect, Cancer, 2008, 112(6), 1247–1253 CrossRef PubMed.
- S. Boinapally, H.-H. Ahn, B. Cheng, M. Brummet, H. Nam, K. L. Gabrielson, S. R. Banerjee, I. Minn and M. G. Pomper, A prostate-specific membrane antigen (PSMA)-targeted prodrug with a favorable in vivo toxicity profile, Sci. Rep., 2021, 11(1), 7114 CrossRef CAS PubMed.
- H. Chen, P. Cai, Y. Feng, Z. Sun, Y. Wang, Y. Chen, W. Zhang, N. Liu and Z. Zhou, In vitro and in vivo comparative study of a novel 68Ga-labeled PSMA-targeted inhibitor and 68Ga-PSMA-11, Sci. Rep., 2021, 11(1), 19122 CrossRef CAS PubMed.
- C. Baun, J. H. Dam, M. G. Hildebrandt, J. D. Ewald, B. W. Kristensen, V. S. Gammelsrød, B. B. Olsen and H. Thisgaard, Preclinical evaluation of [58mCo]Co-DOTA-PSMA-617 for Auger electron therapy of prostate cancer, Sci. Rep., 2023, 13(1), 18837 CrossRef CAS PubMed.
- S. Mannweiler, P. Amersdorfer, S. Trajanoski, J. A. Terrett, D. King and G. Mehes, Heterogeneity of prostate-specific membrane antigen (PSMA) expression in prostate carcinoma with distant metastasis, Pathol. Oncol. Res., 2009, 15(2), 167–172 CrossRef CAS PubMed.
- A. Ghosh and W. D. Heston, Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer, J. Cell. Biochem., 2004, 91(3), 528–539 CrossRef CAS PubMed.
- D. J. Bacich, J. T. Pinto, W. P. Tong and W. D. Heston, Cloning, expression, genomic localization, and enzymatic activities of the mouse homolog of prostate-specific membrane antigen/NAALADase/folate hydrolase, Mamm. Genome, 2001, 12(2), 117–123 CrossRef CAS PubMed.
- FDA Approves Pluvicto/Locametz for Metastatic Castration-Resistant Prostate Cancer, J. Nucl. Med., 2022, 63(5), 13N Search PubMed.
- K. De Man, N. Van Laeken, V. Schelfhout, W. P. Fendler, B. Lambert, K. Kersemans, S. Piron, N. Lumen, K. Decaestecker, V. Fonteyne, L. Delrue, F. De Vos and P. Ost, 18F-PSMA-11 Versus 68Ga-PSMA-11 Positron Emission Tomography/Computed Tomography for Staging and Biochemical Recurrence of Prostate Cancer: A Prospective Double-blind Randomised Cross-over Trial, Eur. Urol., 2022, 82(5), 501–509 CrossRef CAS PubMed.
- U. Hennrich and M. Eder, [(177)Lu]Lu-PSMA-617 (Pluvicto(TM)): The First FDA-Approved Radiotherapeutical for Treatment of Prostate Cancer, Pharmaceuticals (Basel), 2022, 15(10), 1292 CrossRef CAS PubMed.
- FDA approves Pluvicto for metastatic castration-resistant prostate cancer, FDA, 2022 Search PubMed.
- J. R. Nedrow-Byers, A. L. Moore, T. Ganguly, M. R. Hopkins, M. D. Fulton, P. D. Benny and C. E. Berkman, PSMA-targeted SPECT agents: Mode of binding effect on in vitro performance, Prostate, 2013, 73(4), 355–362 CrossRef CAS PubMed.
- S. Dannoon, T. Ganguly, H. Cahaya, J. J. Geruntho, M. S. Galliher, S. K. Beyer, C. J. Choy, M. R. Hopkins, M. Regan, J. E. Blecha, L. Skultetyova, C. R. Drake, S. Jivan, C. Barinka, E. F. Jones, C. E. Berkman and H. F. VanBrocklin, Structure-Activity Relationship of (18)F-Labeled Phosphoramidate Peptidomimetic Prostate-Specific Membrane Antigen (PSMA)-Targeted Inhibitor Analogues for PET Imaging of Prostate Cancer, J. Med. Chem., 2016, 59(12), 5684–5694 CrossRef CAS PubMed.
- T. Ganguly, S. Dannoon, M. R. Hopkins, S. Murphy, H. Cahaya, J. E. Blecha, S. Jivan, C. R. Drake, C. Barinka, E. F. Jones, H. F. VanBrocklin and C. E. Berkman, A high-affinity [(18)F]-labeled phosphoramidate peptidomimetic PSMA-targeted inhibitor for PET imaging of prostate cancer, Nucl. Med. Biol., 2015, 42(10), 780–787 CrossRef CAS PubMed.
- J. Nedrow, J. Latoche, K. Day, J. Modi, T. Ganguly, D. Zeng, B. Kurland, C. Berkman and C. Anderson, Targeting PSMA with a 64Cu-labeled phosphoramidate inhibitor for PET/CT imaging of variant PSMA-expressing xenografts in a mouse model of prostate cancer, Mol Imaging Biol, 2016, 18(3), 402–410 CrossRef CAS PubMed.
- A. M. Caminade, Dendrimers, an Emerging Opportunity in Personalized Medicine?, J. Pers. Med., 2022, 12(8), 1334 CrossRef PubMed.
- S. Mignani, X. Shi, J. Rodrigues, H. Tomas, A. Karpus and J.-P. Majoral, First-in-class and best-in-class dendrimer nanoplatforms from concept to clinic: Lessons learned moving forward, Eur. J. Med. Chem., 2021, 219, 113456 CrossRef CAS PubMed.
- A. Dhull, C. Yu, A. H. Wilmoth, M. Chen, A. Sharma and S. Yiu, Dendrimers in Corneal Drug Delivery: Recent Developments and Translational Opportunities, Pharmaceutics, 2023, 15(6), 1591 CrossRef CAS PubMed.
- F. Abedi-Gaballu, G. Dehghan, M. Ghaffari, R. Yekta, S. Abbaspour-Ravasjani, B. Baradaran, J. Ezzati Nazhad Dolatabadi and M. R. Hamblin, PAMAM dendrimers as efficient drug and gene delivery nanosystems for cancer therapy, Appl. Mater. Today, 2018, 12, 177–190 CrossRef PubMed.
- P. Tarach and A. Janaszewska, Recent Advances in Preclinical Research Using PAMAM Dendrimers for Cancer Gene Therapy, Int. J. Mol. Sci., 2021, 22(6), 2912 CrossRef CAS PubMed.
- F. Zhang, Z. Zhang, J. Alt, S. P. Kambhampati, A. Sharma, S. Singh, E. Nance, A. G. Thomas, C. Rojas, R. Rais, B. S. Slusher, R. M. Kannan and S. Kannan, Dendrimer-enabled targeted delivery attenuates glutamate excitotoxicity and improves motor function in a rabbit model of cerebral palsy, J. Controlled Release, 2023, 358, 27–42 CrossRef CAS PubMed.
- K. Liaw, R. Sharma, A. Sharma, S. Salazar, S. Appiani La Rosa and R. M. Kannan, Systemic dendrimer delivery of triptolide to tumor-associated macrophages improves anti-tumor efficacy and reduces systemic toxicity in glioblastoma, J. Controlled Release, 2021, 329, 434–444 CrossRef CAS PubMed.
- R. Sharma, S. P. Kambhampati, Z. Zhang, A. Sharma, S. Chen, E. I. Duh, S. Kannan, M. O. M. Tso and R. M. Kannan, Dendrimer mediated targeted delivery of sinomenine for the treatment of acute neuroinflammation in traumatic brain injury, J. Controlled Release, 2020, 323, 361–375 CrossRef CAS PubMed.
- R. Sharma, K. Liaw, A. Sharma, A. Jimenez, M. Chang, S. Salazar, I. Amlani, S. Kannan and R. M. Kannan, Glycosylation of PAMAM dendrimers significantly improves tumor macrophage targeting and specificity in glioblastoma, J. Controlled Release, 2021, 337, 179–192 CrossRef CAS PubMed.
- R. Sharma, A. Sharma, S. P. Kambhampati, R. R. Reddy, Z. Zhang, J. L. Cleland, S. Kannan and R. M. Kannan, Scalable synthesis and validation of PAMAM dendrimer-N-acetyl cysteine conjugate for potential translation, Bioeng. Transl. Med., 2018, 3(2), 87–101 CrossRef CAS PubMed.
- A. M. Gusdon, N. Faraday, J. S. Aita, S. Kumar, I. Mehta, H. A. Choi, J. L. Cleland, K. Robinson, L. D. McCullough, D. K. Ng, R. M. Kannan and S. Kannan, Dendrimer nanotherapy for severe COVID-19 attenuates inflammation and neurological injury markers and improves outcomes in a phase2a clinical trial, Sci. Transl. Med., 2022, 14(654), eabo2652 CrossRef CAS PubMed.
- S. Kannan, H. Dai, R. S. Navath, B. Balakrishnan, A. Jyoti, J. Janisse, R. Romero and R. M. Kannan, Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model, Sci. Transl. Med., 2012, 4(130), 130ra46 Search PubMed.
- K. Liaw, R. Sharma, A. Sharma, S. Salazar, S. Appiani La Rosa and R. M. Kannan, Systemic dendrimer delivery of triptolide to tumor-associated macrophages improves anti-tumor efficacy and reduces systemic toxicity in glioblastoma, J. Controlled Release, 2021, 329, 434–444 CrossRef CAS PubMed.
- A. Sharma, K. Liaw, R. Sharma, T. Spriggs, S. Appiani La Rosa, S. Kannan and R. M. Kannan, Dendrimer-Mediated Targeted Delivery of Rapamycin to Tumor-Associated Macrophages Improves Systemic Treatment of Glioblastoma, Biomacromolecules, 2020, 21(12), 5148–5161 CrossRef CAS PubMed.
- K. Liaw, R. Reddy, A. Sharma, J. Li, M. Chang, R. Sharma, S. Salazar, S. Kannan and R. M. Kannan, Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma, Bioeng. Transl. Med., 2021, 6(2), e10205 CrossRef CAS PubMed.
- B. Huang, J. Otis, M. Joice, A. Kotlyar and T. P. Thomas, PSMA-Targeted Stably Linked “Dendrimer-Glutamate Urea-Methotrexate” as a Prostate Cancer Therapeutic, Biomacromolecules, 2014, 15(3), 915–923 CrossRef CAS PubMed.
- W. G. Lesniak, S. Boinapally, S. R. Banerjee, B. Behnam Azad, C. A. Foss, C. Shen, A. Lisok, B. Wharram, S. Nimmagadda and M. G. Pomper, Evaluation of PSMA-Targeted PAMAM Dendrimer Nanoparticles in a Murine Model of Prostate Cancer, Mol. Pharm., 2019, 16(6), 2590–2604 CrossRef CAS PubMed.
- G. Thiagarajan, K. Greish and H. Ghandehari, Charge affects the oral toxicity of poly(amidoamine) dendrimers, Eur. J. Pharm. Biopharm., 2013, 84(2), 330–334 CrossRef CAS PubMed.
- A. S. Chauhan, N. K. Jain and P. V. Diwan, Pre-clinical and behavioural toxicity profile of PAMAM dendrimers in mice, Proc. R. Soc. A, 2010, 466(2117), 1535–1550 CrossRef CAS.
- C. F. Jones, R. A. Campbell, A. E. Brooks, S. Assemi, S. Tadjiki, G. Thiagarajan, C. Mulcock, A. S. Weyrich, B. D. Brooks, H. Ghandehari and D. W. Grainger, Cationic PAMAM dendrimers aggressively initiate blood clot formation, ACS Nano, 2012, 6(11), 9900–9910 CrossRef CAS PubMed.
- C. R. Ley, N. R. Beattie, S. Dannoon, M. Regan, H. VanBrocklin and C. E. Berkman, Synthesis and evaluation of constrained phosphoramidate inhibitors of prostate-specific membrane antigen, Bioorg. Med. Chem. Lett., 2015, 25(12), 2536–2539 CrossRef CAS PubMed.
- F. P. Olatunji, E. A. Savoy, M. Panteah, N. Mesbahi, A. Abbasi, C. M. Talley, C. L. Lovingier, L. A. Caromile and C. E. Berkman, Prostate-Specific Membrane Antigen-Targeted Turn-on Probe for Imaging Cargo Release in Prostate Cancer Cells, Bioconjugate Chem., 2021, 32(11), 2386–2396 CrossRef CAS PubMed.
- P. K. Maiti, T. Çaǧın, S.-T. Lin and W. A. Goddard, Effect of Solvent and pH on the Structure of PAMAM Dendrimers, Macromolecules, 2005, 38(3), 979–991 CrossRef CAS.
- P. K. Maiti, T. Çaǧın, G. Wang and W. A. Goddard, Structure of PAMAM Dendrimers: Generations 1 through 11, Macromolecules, 2004, 37(16), 6236–6254 CrossRef CAS.
- S. E. Martin, T. Ganguly, G. R. Munske, M. D. Fulton, M. R. Hopkins, C. E. Berkman and M. E. Black, Development of Inhibitor-Directed Enzyme Prodrug Therapy (IDEPT) for Prostate Cancer, Bioconjugate Chem., 2014, 25(10), 1752–1760 CrossRef CAS PubMed.
- T. Liu, J. R. Nedrow-Byers, M. R. Hopkins, L. Y. Wu, J. Lee, P. T. Reilly and C. E. Berkman, Targeting prostate cancer cells with a multivalent PSMA inhibitor-guided streptavidin conjugate, Bioorg. Med. Chem. Lett., 2012, 22(12), 3931–3934 CrossRef CAS PubMed.
- S. Ahn, E. Seo, K. Kim and S. J. Lee, Controlled cellular uptake and drug efficacy of nanotherapeutics, Sci. Rep., 2013, 3(1), 1997 CrossRef PubMed.
- X. Shi, T. P. Thomas, L. A. Myc, A. Kotlyar and J. J. R. Baker, Synthesis, characterization, and intracellular uptake of carboxyl-terminated poly(amidoamine) dendrimer-stabilized iron oxide nanoparticles, Phys. Chem. Chem. Phys., 2007, 9(42), 5712–5720 RSC.
- K. Li, D. Fong, E. Meichsner and A. Adronov, A Survey of Strain-Promoted Azide–Alkyne Cycloaddition in Polymer Chemistry, Chem. – Eur. J., 2021, 27(16), 5057–5073 CrossRef CAS PubMed.
- E. Kim and H. Koo, Biomedical applications of copper-free click chemistry: in vitro, in vivo, and ex vivo, Chem. Sci., 2019, 10(34), 7835–7851 RSC.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination, J. Controlled Release, 2016, 240, 332–348 CrossRef CAS PubMed.
- G. K. Abou-Alfa, T. Meyer, A.-L. Cheng, A. B. El-Khoueiry, L. Rimassa, B.-Y. Ryoo, I. Cicin, P. Merle, Y. Chen, J.-W. Park, J.-F. Blanc, L. Bolondi, H.-J. Klümpen, S. L. Chan, V. Zagonel, T. Pressiani, M.-H. Ryu, A. P. Venook, C. Hessel, A. E. Borgman-Hagey, G. Schwab and R. K. Kelley, Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma, N. Engl. J. Med., 2018, 379(1), 54–63 CrossRef CAS PubMed.
- N. Agarwal, U. Vaishampayan, M. Green, F. di Nucci, P. Y. Chang, C. Scheffold and S. Pal, 872P – Phase Ib study (COSMIC-021) of cabozantinib in combination with atezolizumab: Results of the dose escalation stage in patients (pts) with treatment-naïve advanced renal cell carcinoma (RCC), Ann. Oncol., 2018, 29, viii308 CrossRef.
- S. J. D. Vecchio and R. J. Ellis, Cabozantinib for the Management of Metastatic Clear Cell Renal Cell Carcinoma, J. Kidney Cancer VHL, 2018, 5(4), 1–5 CrossRef PubMed.
- T. K. Choueiri, T. Powles, M. Burotto, B. Escudier, M. T. Bourlon, B. Zurawski, V. M. Oyervides Juárez, J. J. Hsieh, U. Basso, A. Y. Shah, C. Suárez, A. Hamzaj, J. C. Goh, C. Barrios, M. Richardet, C. Porta, R. Kowalyszyn, J. P. Feregrino, J. Żołnierek, D. Pook, E. R. Kessler, Y. Tomita, R. Mizuno, J. Bedke, J. Zhang, M. A. Maurer, B. Simsek, F. Ejzykowicz, G. M. Schwab, A. B. Apolo and R. J. Motzer, Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma, N. Engl. J. Med., 2021, 384(9), 829–841 CrossRef CAS PubMed.
- N. Agarwal, A. Azad, J. Carles, S. Chowdhury, B. McGregor, A. S. Merseburger, S. Oudard, F. Saad, A. Soares, F. Benzaghou, Y. Kerloeguen, A. Kimura, N. Mohamed, A. Panneerselvam, F. Wang and S. Pal, A phase III, randomized, open-label study (CONTACT-02) of cabozantinib plus atezolizumab versus second novel hormone therapy in patients with metastatic castration-resistant prostate cancer, Future Oncol., 2022, 18(10), 1185–1198 CrossRef CAS PubMed.
- D. C. Smith, M. R. Smith, C. Sweeney, A. A. Elfiky, C. Logothetis, P. G. Corn, N. J. Vogelzang, E. J. Small, A. L. Harzstark, M. S. Gordon, U. N. Vaishampayan, N. B. Haas, A. I. Spira, P. N. Lara Jr., C. C. Lin, S. Srinivas, A. Sella, P. Schöffski, C. Scheffold, A. L. Weitzman and M. Hussain, Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial, J. Clin. Oncol., 2013, 31(4), 412–419 CrossRef CAS PubMed.
- M. Smith, J. D. Bono, C. Sternberg, S. L. Moulec, S. Oudard, U. D. Giorgi, M. Krainer, A. Bergman, W. Hoelzer, R. D. Wit, M. Bögemann, F. Saad, G. Cruciani, A. Thiery-Vuillemin, S. Feyerabend, K. Miller, N. Houédé, S. Hussain, E. Lam, J. Polikoff, A. Stenzl, P. Mainwaring, D. Ramies, C. Hessel, A. Weitzman and K. Fizazi, Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1, J. Clin. Oncol., 2016, 34(25), 3005–3013 CrossRef CAS PubMed.
- E. M. Basch, M. Scholz, J. S. de Bono, N. Vogelzang, P. de Souza, G. Marx, U. Vaishampayan, S. George, J. K. Schwarz, E. S. Antonarakis, J. M. O'Sullivan, A. R. Kalebasty, K. N. Chi, R. Dreicer, T. E. Hutson, A. C. Dueck, A. V. Bennett, E. Dayan, M. Mangeshkar, J. Holland, A. L. Weitzman and H. I. Scher, Cabozantinib Versus Mitoxantrone-prednisone in Symptomatic Metastatic Castration-resistant Prostate Cancer: A Randomized Phase 3 Trial with a Primary Pain Endpoint, Eur. Urol., 2019, 75(6), 929–937 CrossRef CAS PubMed.
- M. P. Labrecque, L. G. Brown, I. M. Coleman, H. M. Nguyen, D. W. Lin, E. Corey, P. S. Nelson and C. Morrissey, Cabozantinib can block growth of neuroendocrine prostate cancer patient-derived xenografts by disrupting tumor vasculature, PLoS One, 2021, 16(1), e0245602 CrossRef CAS PubMed.
- M. T. Haider, K. D. Hunter, S. P. Robinson, T. J. Graham, E. Corey, T. N. Dear, R. Hughes, N. J. Brown and I. Holen, Rapid modification of the bone microenvironment following short-term treatment with Cabozantinib in vivo, Bone, 2015, 81, 581–592 CrossRef CAS PubMed.
- H. M. Nguyen, N. Ruppender, X. Zhang, L. G. Brown, T. S. Gross, C. Morrissey, R. Gulati, R. L. Vessella, F. Schimmoller, D. T. Aftab and E. Corey, Cabozantinib inhibits growth of androgen-sensitive and castration-resistant prostate cancer and affects bone remodeling, PLoS One, 2013, 8(10), e78881 CrossRef CAS PubMed.
- F. Scirocchi, C. Napoletano, A. Pace, H. Rahimi Koshkaki, A. Di Filippo, I. G. Zizzari, M. Nuti and A. Rughetti, Immunogenic Cell Death and Immunomodulatory Effects of Cabozantinib, Front. Oncol., 2021, 11, 755433 CrossRef CAS PubMed.
- T. Chen, L. S. Zhuo, P. F. Liu, W. R. Fang, Y. M. Li and W. Huang, Discovery of 1,6-naphthyridinone-based MET kinase inhibitor bearing quinoline moiety as promising antitumor drug candidate, Eur. J. Med. Chem., 2020, 192, 112174 CrossRef CAS PubMed.
- A. A. Sachkova, D. V. Andreeva, A. S. Tikhomirov, A. M. Scherbakov, D. I. Salnikova, D. V. Sorokin, F. B. Bogdanov, Y. D. Rysina, A. E. Shchekotikhin, E. S. Shchegravina and A. Y. Fedorov, Design, Synthesis and In Vitro Investigation of Cabozantinib-Based PROTACs to Target c-Met Kinase, Pharmaceutics, 2022, 14(12), 2829 CrossRef CAS PubMed.
- A. Sharma, K. Liaw, R. Sharma, Z. Zhang, S. Kannan and R. M. Kannan, Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics, Theranostics, 2018, 8(20), 5529–5547 CrossRef CAS PubMed.
- B. Qi, F. Wang, H. He, M. Fan, L. Hu, L. Xiong, G. Gong, S. Shi and X. Song, Identification of (S)-1-(2-(2,4-difluorophenyl)-4-oxothiazolidin-3-yl)-3-(4-((7-(3-(4-ethylpiperazin-1-yl)propoxy)-6-methoxyquinolin-4-yl)oxy)-3,5-difluorophenyl)urea as a potential anti-colorectal cancer agent, Eur. J. Med. Chem., 2022, 239, 114561 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data of all the compounds, IC50 curves, and IVIS images in tumor xenograft mouse model are provided. See DOI: https://doi.org/10.1039/d3nr06520k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |