
Ordered mesoporous carbon with binary CoFe atomic species for highly efficient oxygen reduction electrocatalysis†
Fengying
Pan‡
a,
Ziyan
Shen‡
a,
Xianjun
Cao
a,
Yuxia
Zhang
b,
Cheng
Gong
a,
Jinhu
Wu
a,
Jinqiang
Zhang
*c,
Hao
Liu
 *c,
Xiaowei
Li
*d and
Yufei
Zhao
*ac
*c,
Xiaowei
Li
*d and
Yufei
Zhao
*ac
aJoint International Laboratory on Environmental and Energy Frontier Materials, School of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, P. R. China. E-mail: yufei-zhao@shu.edu.cn
bSchool of Materials Science and Engineering, Shijiazhuang Tiedao University, Shijiazhuang 050043, P. R. China
cCentre for Clean Energy Technology, Faculty of Science, University of Technology Sydney, Broadway, Sydney, NSW 2007, Australia. E-mail: Jinqiang.Zhang@uts.edu.au; Hao.liu@uts.edu.au
dSchool of Environmental and Chemical Engineering, Organic Compound Pollution Control Engineering, Ministry of Education, Shanghai University, Shanghai 200444, P. R. China. E-mail: lixiaowei419@shu.edu.cn
First published on 4th April 2024
Abstract
The exploration of powerful, efficient and precious metal-free electrocatalysts for facilitating the sluggish kinetics of the oxygen reduction reaction (ORR) is a crucial endeavor in the development and application of energy conversion and storage devices. Herein, we have rationally designed and synthesized bimetallic CoFe species consisting of CoFe nanoparticles and atomically dispersed dual atoms anchored on an ordered mesoporous carbon matrix (CoFe/NC) as highly efficient ORR electrocatalysts. The pyrolyzation temperature for CoFe/NC plays a vital role in regulating the morphology and composition of both the carbon matrix and CoFe species. The optimized CoFe/NC-750 exhibits a favorable ORR performance in 0.1 M KOH with a high half-wave potential (E1/2) of 0.87 V vs. RHE, excellent tolerance to methanol and remarkable durability (no obvious decrease in E1/2 value after 3000 cycles), all of which are superior to the performance of commercial Pt/C. Experimental measurements and density functional theory (DFT) calculations reveal that the improved ORR performance of CoFe/NC-750 is mainly attributed to the electronic structure of atomically dispersed Fe active sites modulated by the surrounding CoFe alloys and Co single atoms, which accelerates the dissociation and reduction of intermediate OH* species and promotes the ORR process.
 Yufei Zhao | Yufei Zhao is an Associate Professor at Shanghai University. She received her Ph.D. dual-degree from Beijing Institute of Technology in 2017 and University of Technology Sydney, in 2018. After receiving her Ph.D., she worked as a Postdoctoral Research Associate at the University of New South Wales, University of Technology Sydney and South China University of Technology. Her research interest focuses on the synthesis and application of atomically dispersed catalysts, high-entropy materials, and noble metal alloys for electrocatalytic conversion reactions, including water splitting, fuel cells, and CO2 reduction reactions. |
1 Introduction
Proton-exchange membrane fuel cell and metal–air batteries have aroused widespread interest in providing clean energy as a promising alternative to fossil fuels for remarkable energy conversion efficiency, high mass energy density as well as near-zero emissions.1 Their inherent defects of sluggish cathode oxygen reduction reaction (ORR) kinetics, coupled with exorbitant cost, scarcity and poor durability of platinum-group metals with excellent ORR catalytic performance, have caused great resistance to their widespread commercial application.2–5 For decades, massive efforts have been made to explore non-precious metal catalysts as candidates to replace Pt-based composites for the ORR, such as metal oxides, nitrides, carbides, hydroxides, and sulfides.6,7 Nevertheless, the utilization efficiency of metal atoms is relatively low and the catalytic mechanism of such electrocatalysts have been limited due to their heterogeneous properties.Recently, nitrogen-stabilized transition metal species (TM–NX) supported by carbon substrates have been proved to be one of the most promising electrocatalyst alternatives owing to their maximized metal atom utilization and tunable electronic properties.8–11 Among them, Fe–N–C electrocatalysts have exhibited high catalytic activity with Fe–N coordination bonds as ORR active sites.12,13 It has been demonstrated that their ORR activity can be further boosted by altering their electronic structure, which highly affects the binding energy with the ORR intermediates.14 For instance, the introduction of other types of heteroatoms (e.g., S, P, B) can tune and enhance the kinetic activity of catalytic sites (M–S1N3, P/Fe–N–C) by regulating their electron-withdrawing/donating properties.15,16 Bimetallic sites incorporating another metal species endowed with synergistic properties have great potential to accelerate ORR kinetics. The intrinsic ORR activity of the active metal species of SACs follows the order of Fe > Co > Cu > Mn > Ni.17–19 Therefore, incorporating Co species into Fe–N–C electrocatalysts and forming adjacent metal atoms, greatly improves the structural adjustability. The chemical interaction between Fe and Co atoms leads to a unique electronic structure and optimizes the ORR performance. For instance, FeN3–CoN3 active sites highly promote the cleavage of O–O bonds by bridge-cis adsorption of O-containing intermediates in the ORR process. In addition, the insertion of atomic clusters into atomically dispersed M–N–C substrates (e.g., FeAC@FeSA–N–C, Fe–ACSA@NC, Co–ACSA@NC, and Ni–ACSA@NC) inspired electron redistribution between the metal centers in M–N–C and the carbon support, regulating the bonding length of M–N to promote the desorption of intermediates during the ORR process.20–22 Furthermore, the introduction of nanoparticles near the atomically dispersed M–N–C (M/M–NX) is also promising to optimize the ORR performance.23–25 For instance, M/M–NX composite sites (e.g., M/FeCo–SAs–N–C and FeN3/Fe NPs) enhanced the oxygen adsorption capability and prolonged the length of the O–O bond, making the O–O bond easily fracture and thereby enhance ORR catalytic activity.26,27 Apart from collaborating with M–N–C, the incorporated heteroatoms, clusters or nanoparticles may also provide additional active sites or protect the M–N–C active site from poisoning,28,29 further boosting the catalytic activity and stability. However, an in-depth understanding of the interaction between the active sites and reactants to accelerate the ORR kinetics is still heavily hampered by the heterogeneity of the structure and composition of the electrocatalysts,6,27 while establishing a definitive correlation between the atomic structure and the ORR catalytic performance remains a challenge.30,31
Herein, we designed a template-assisted method to prepare highly efficient ORR electrocatalysts composed of an ordered mesoporous carbon and bimetallic CoFe species. The pyrolysis temperature for CoFe/NC highly regulates the properties of the carbon matrix and CoFe species. The optimized CoFe/NC-750 exhibits an ordered porous architecture, and the coexistence of atomically dispersed Co/Fe atoms and CoFe alloys, which ensure efficient mass transport and modulation of active sites for the ORR. As a result, CoFe/NC-750 exhibited an excellent ORR performance under alkaline conditions with a high E1/2 value, excellent tolerance to methanol and superior durability, outperforming those of commercial Pt/C and other prepared comparison samples. The boosted ORR performance of CoFe/NC-750 may mainly be contributed to the active sites of atomically dispersed Fe species modulated by the surrounding Co atoms and CoFe alloys, which lowers the energy barrier in the rate-determining step (OH* → OH−), thus accelerating the ORR process.
2 Results and discussion
2.1 Fabrication and characterization
The synthesis procedure of CoFe/NC-750 is depicted in Fig. 1a. The SBA-15 hard template was prepared based on the reported methods.32 Then, the metal–organic ligand complexes were in situ formed in the pores of SBA-15 with the incorporation of organic ligand 2-methylimidazole, Co2+ and Fe3+ nodes. The bimetallic CoFe species (CoFe nanoparticles and atomically dispersed Co/Fe atoms) anchored on the mesoporous carbon matrix (CoFe/NC-750) was achieved after pyrolysis at 750 °C, hard template removal, and acid treatment to remove the accessible metal nanoparticles.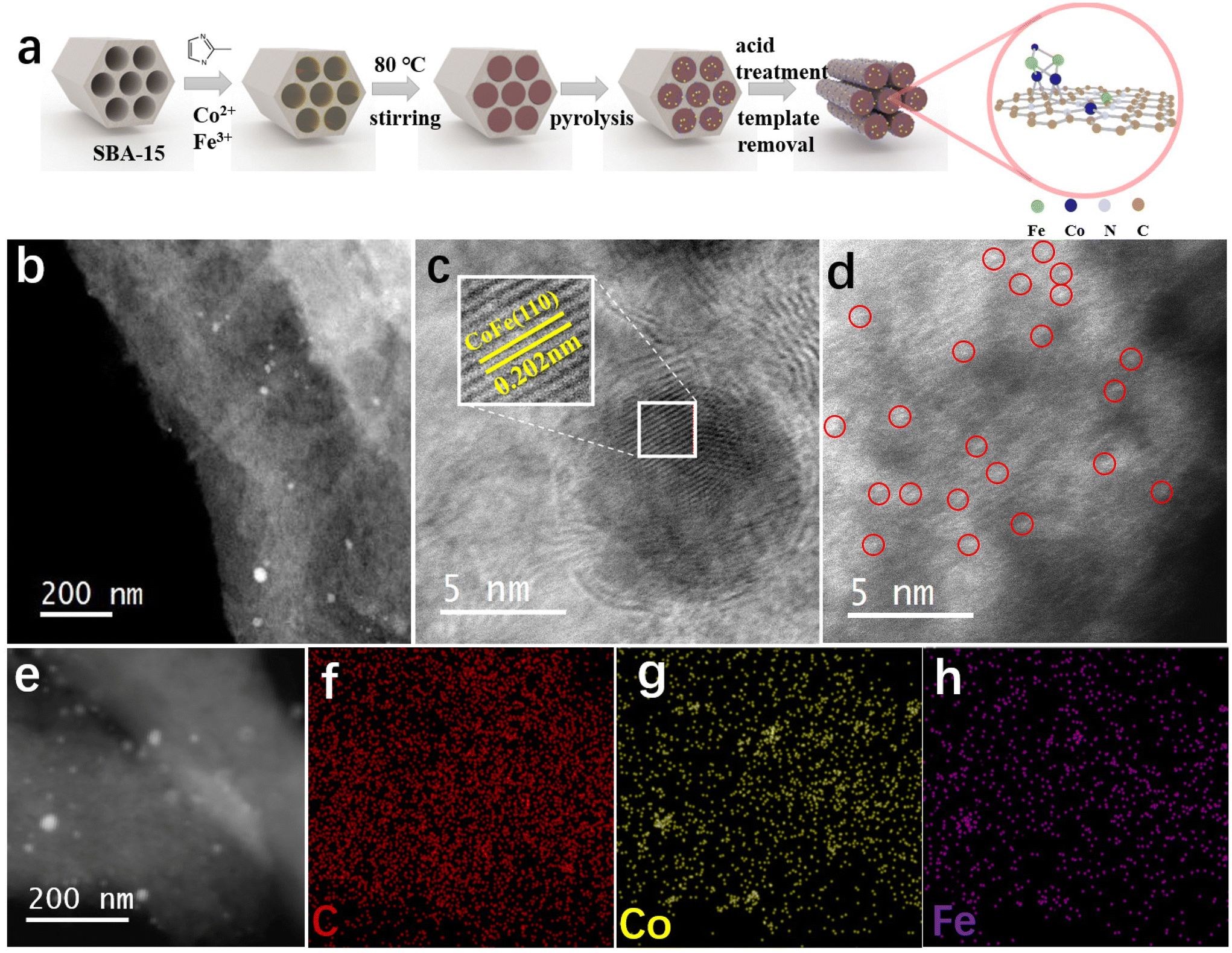 | ||
| Fig. 1 Schematic of the synthesis procedure and morphology characterization of the catalysts. (a) Schematic of the synthesis of CoFe/NC. (b–d) HAADF-STEM, high-resolution TEM and HAADF-STEM images of CoFe/NC-750. (e–h) Elemental mapping of CoFe/NC-750. | ||
The ordered mesoporous structure of CoFe/NC-750 derived from the reverse replication of SBA-15 (SEM image in Fig. S1†) has been confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. S2†). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image in Fig. 1b further shows that CoFe/NC-750 inherits the rod-like structure with nanoparticles scattered on its surface. The high resolution HAADF-STEM image in Fig. 1c reveals the presence of a well-crystallized nanoparticle structure with lattice fringes of 0.202 nm, corresponding to the (110) facet of the bcc CoFe alloy.33 Meanwhile, numerous bright dots have been observed on the carbon support as shown in Fig. 1d, attributed to the atomically dispersed Co or Fe species. Thus, we have successfully prepared CoFe nanoparticles and CoFe atomically dispersed atoms co-existing on the carbon matrix. The energy dispersion spectrum (EDS) element mapping (Fig. 1e–h) combined with the electron energy loss spectroscopy (EELS) elemental mapping (Fig. S3†) further validate the uniform distribution of C, N, Co, and Fe elements and the presence of nano-aggregated CoFe metal alloy nanoparticles. The Co and Fe loading on the carbon matrix in CoFe/NC-750 are 5.42 wt% and 1.80 wt%, respectively, derived from inductively coupled plasma optical emission spectrometry (ICP-OES) measurements.
CoFe/NCs pyrolyzed at different temperatures have also been prepared as comparison materials to investigate the critical role of temperature, denoted as CoFe/NC-T (T = 700 °C, 750 °C, 800 °C, and 850 °C). As shown in Fig. 2a, at a lower temperature of 700 °C, it is obvious to observe the nanoparticles due to their fast formation properties. With the increase of temperature to 750 °C, the co-existence of small nanoparticles and dual atoms has been observed (Fig. 1b–d). When the temperature is further increased to 800 °C, the carbon surface becomes much rougher (Fig. 2a). Many carbon nanotubes (CNTs) are formed due to the catalytic properties of CoFe alloys at higher temperature. More of the organic ligand 2-methylimidazole served as the C source for the growth of CNTs in the presence of metallic alloys at a temperature of 850 °C (forming interconnected CNTs), and eventually destroyed the original ordered structure of CMK-3. Moreover, many fewer nanoparticles were observed for CoFe/NC-800 and CoFe/NC-850, which may be attributed to the quick movement of the metal species at high temperatures, reducing the aggregation properties. The above phenomena indicate that the temperature indeed plays a significant role in the unique morphology and composition. The results of N2 adsorption/desorption isotherms shown in Fig. S5† reveal that CoFe/NC-750 with an ordered mesoporous structure and small nanoparticles exhibited the largest specific surface area of 2109.3 m2 g−1 among all prepared CoFe/NC samples (566.1 m2 g−1 for CoFe/NC-700, 1487.2 m2 g−1 for CoFe/NC-800 and 874.6 m2 g−1 for CoFe/NC-850), which is favorable for mass transport, more exposed active sites and the adsorption/desorption of intermediates.34
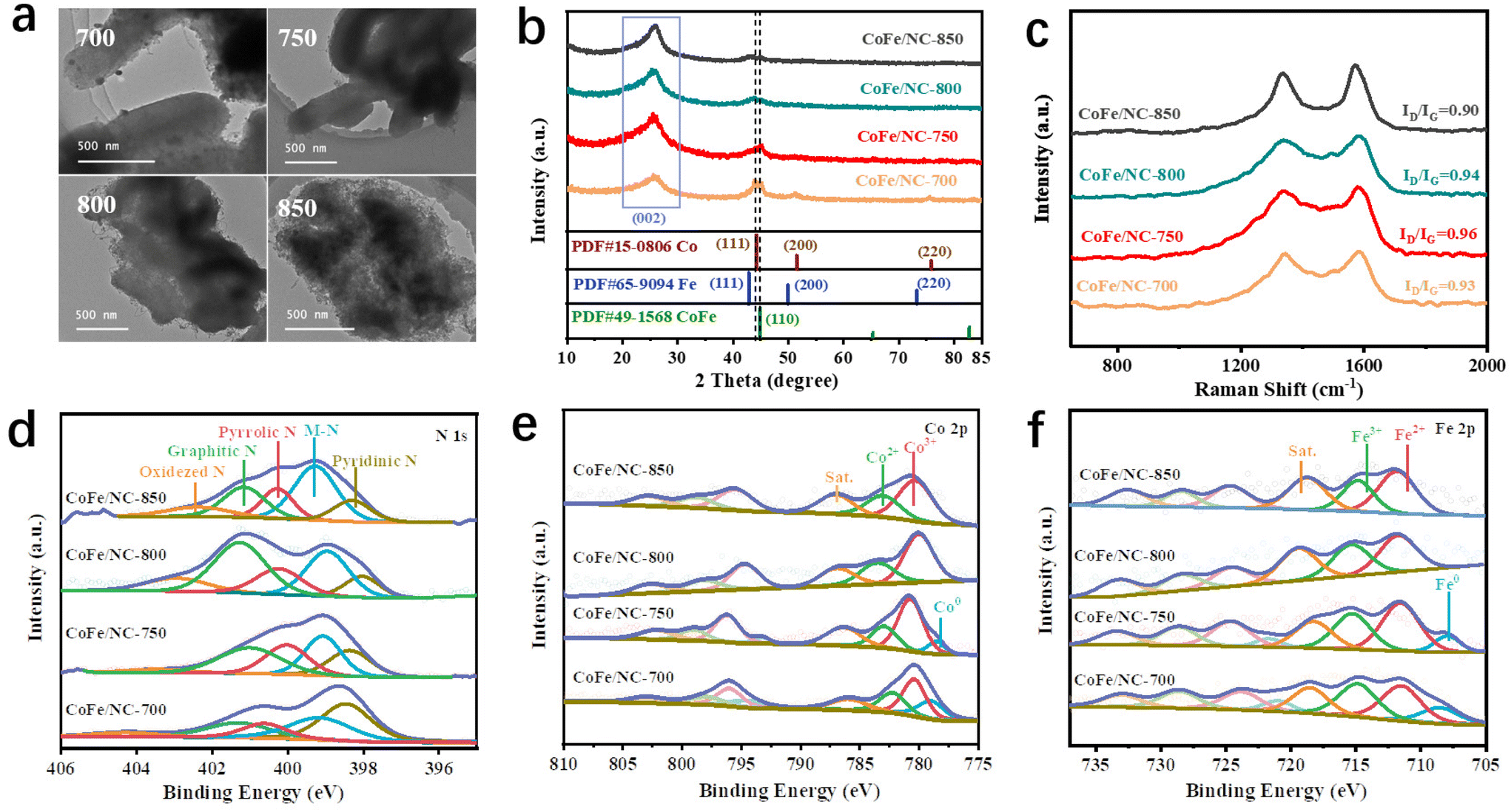 | ||
| Fig. 2 Structural characterization of the catalysts. (a) TEM images of CoFe/NC-T (T = 700, 750, 800, 850 °C). (b and c) XRD patterns and Raman spectra of CoFe/NC-T. Deconvoluted (d) N 1s, (e) Co 2p and (f) Fe 2p XPS spectra of CoFe/NC-T. | ||
The composition and crystal structure of CoFe/NCs have been investigated using X-ray diffraction (XRD). As displayed in Fig. 2b, the dominant diffraction peak at 26° for all samples corresponds to the (002) planes of graphitic carbon (JCPDS no. 41-1487).12 Two sharp peaks at around 45° have been observed for CoFe/NCs-700, belonging to the (111) plane of Co and the (110) plane of the CoFe alloy (JCPDS no. 49-1568).33 CoFe/NC-750 exhibited one peak at 45°, corresponding to the (110) plane of the CoFe alloy. In contrast, the peaks for metallic species were almost indiscernible for both CoFe/NC-800 and CoFe/NC-850. The XRD results are consistent with the TEM images shown in Fig. 2a. Fig. 2c shows the Raman spectra of CoFe/NC, and the characteristic peaks at around 1345 and 1585 cm−1 are dictated by the D-band of sp3 defects and the G-band of the sp2 graphite carbon, respectively.35,36 CoFe/NC-750 exhibits an ID/IG intensity ratio of 0.96, which is a little higher than those of CoFe/NC-700 (0.93), CoFe/NC-800 (0.94) and CoFe/NC-850 (0.90). The increased ID/IG ratio suggests that the carbon lattice of CoFe/NC-750 possesses more defects, which may provide additional active sites for the ORR process.
The chemical and electronic states of the as-prepared catalysts have been investigated using X-ray photoelectron spectroscopy (XPS) measurements. The XPS survey spectra in Fig. S6† show that all CoFe/NC samples are composed of C, N, O, Fe and Co elements. The high-resolution N 1s segments of CoFe/NCs shown in Fig. 2d are deconvoluted into five peaks centered at approximately ∼398, ∼399, ∼400, ∼401 and ∼402 eV, which are attributed to pyridinic N, metal-coordinated nitrogen (M–N), pyrrolic N, graphitic N and oxidized N, respectively, revealing the successful incorporation of N doping in CoFe/NC.37,38 The presence of M–N reveals the coordination of the N atom with the Fe or Co atom.39 Moreover, the intensities of M–N peaks change significantly with temperature. CoFe/NC-750, CoFe/NC-800 and CoFe/NC-850 exhibited much higher peak intensity compared to that of CoFe/NC-700, indicating that many more Co/Fe species were stabilized by N atoms on increasing the temperature, which is consistent with the TEM/XRD results that metal species move faster with increasing temperature, eventually changing from particles to single atoms.40 Analogously, the peak intensity of graphitic N exhibits a similar trend to that of M–N, indicating high temperature in favor of the formation of graphitic N. The high-resolution Co 2p spectra of CoFe/NC-700 and CoFe/NC-750 (Fig. 2e) show two distinct peaks, belonging to Co 2p3/2 and Co 2p1/2. These peaks can be further deconvoluted into four pairs of peaks located at ∼778/∼793 eV, ∼781/∼796 eV, ∼783/∼799 eV, and ∼786/∼802 eV, corresponding to Co 2p3/2 and Co 2p1/2 of metallic Co (Co0), Co3+, Co2+ and the satellite peaks, respectively,41 confirming the coexistence of atomic and metallic states of Co species in both CoFe/NC-700 and CoFe/NC-750. Moreover, the peak intensity of metallic Co decreased from that of CoFe/NC-700 to CoFe/NC-750. No corresponding peak of Co0 was observed in spectra for either CoFe/NC-800 or CoFe/NC-850, further demonstrating the transition from the metal state (Co0) to the atomic state (Co3+/Co2+) at high temperatures. The results of high-resolution Fe 2p spectra in Fig. 2f show a similar trend to that of Co 2p results, revealing the coexistence of the metallic (∼707/∼720 eV) and atomic state of Fe (Fe2+(∼711/∼722 eV) and Fe3+(∼715/∼728 eV)) in CoFe/NC-700 and CoFe/NC-750,42 and merely atomic states of Fe in CoFe/NC-800 and CoFe/NC-850. The above results suggest that the temperature plays an important role in promoting the transformation of the metallic species into atomically dispersed metal species during CoFe preparation, which has great influence on the morphology and composition of CoFe species and thus on the ORR catalytic capability.
2.2 ORR electrocatalytic performance
The electrocatalytic ORR performance of the as-prepared materials was critically investigated by linear sweep voltammetry (LSV) using a glassy rotating disk electrode (RDE) in O2-saturated 0.1 M KOH solution with a rotation speed of 1600 rpm. As depicted in Fig. 3a and b, and Fig. S7,† CoFe/NC-750 displays high Eonset and E1/2 values of 1.02 V and 0.87 V vs. RHE, respectively, which are much better than those of Pt/C (Eonset = 0.98 V and E1/2 = 0.86 V) and CoFe/NCs (Eonset = 0.97 V, 0.96 V, 0.95 V for CoFe/NC-700, CoFe/NC-800, CoFe/NC-850 and E1/2 = 0.84 V, 0.85 V, 0.84 V for CoFe/NC-700, CoFe/NC-800, CoFe/NC-850, respectively). Meanwhile, the kinetic current density (JK) of CoFe/NC-750 is 8.76 mA cm−2 at a potential of 0.85 V vs. RHE and the Tafel slope is 83 mV dec−1, which is superior to the comparison samples (JK = 3.85 mA cm−2, 5.32 mA cm−2, 4.07 mA cm−2, and 5.3 mA cm−2 for CoFe/NC-700, CoFe/NC-800, CoFe/NC-850, and Pt/C, and Tafel slope = 103 mV dec−1, 93 mV dec−1, 95 mV dec−1, and 90 mV dec−1 for CoFe/NC-700, CoFe/NC-800, CoFe/NC-850, and Pt/C, respectively). The above results demonstrate the enhanced electrocatalytic ORR activity and improved kinetics for CoFe/NC-750, which are ascribed to the ordered mesoporous structure promoting mass/charge transfer, and CoFe species offering ORR active sites. The LSV curves of CoFe/NC-750 were achieved at the speed range of 400 to 2025 rpm and the corresponding Koutecky–Levich (K–L) plots are shown in Fig. 3c. The approximately parallel fitting line properties (Fig. 3c, inset) reveal the first-order ORR kinetics of CoFe/NC-750.43,44 Moreover, the electron transfer number and hydrogen peroxide (H2O2) yield of CoFe/NC-750 during the ORR were scrutinized by using rotating ring-disk electrode (RRDE) tests. The results in Fig. 3d show that the electron transfer number is close to 4, and the H2O2 yield is lower than 4% throughout the whole potential window, confirming the four-electron ORR pathway of CoFe/NC-750. Compelling tolerance of CoFe/NC-750 to methanol was investigated by adding methanol to the electrolyte during the chronoamperometry measurement, when only less than 3% current attenuation was observed, demonstrating the intrinsic methanol tolerance capability. In contrast, a significant decay for commercial Pt/C has been observed under the same conditions (Fig. 3e). The durability of CoFe/NC-750 was assessed by the accelerated cyclic voltammetry test (ADT) and chronoamperometric test. The results in Fig. 3f show the LSV curves before and after ADT of 3000 cycles, manifesting slight deviation of LSV curves and no obvious decay in E1/2, which reveals the excellent ORR stability of CoFe/NC-750. In addition, the chronoamperometric result presented in Fig. S8† shows that the current density of CoFe/NC-750 maintains 99.9% of its initial value after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000s, further demonstrating excellent stability properties during the ORR process.
000s, further demonstrating excellent stability properties during the ORR process.
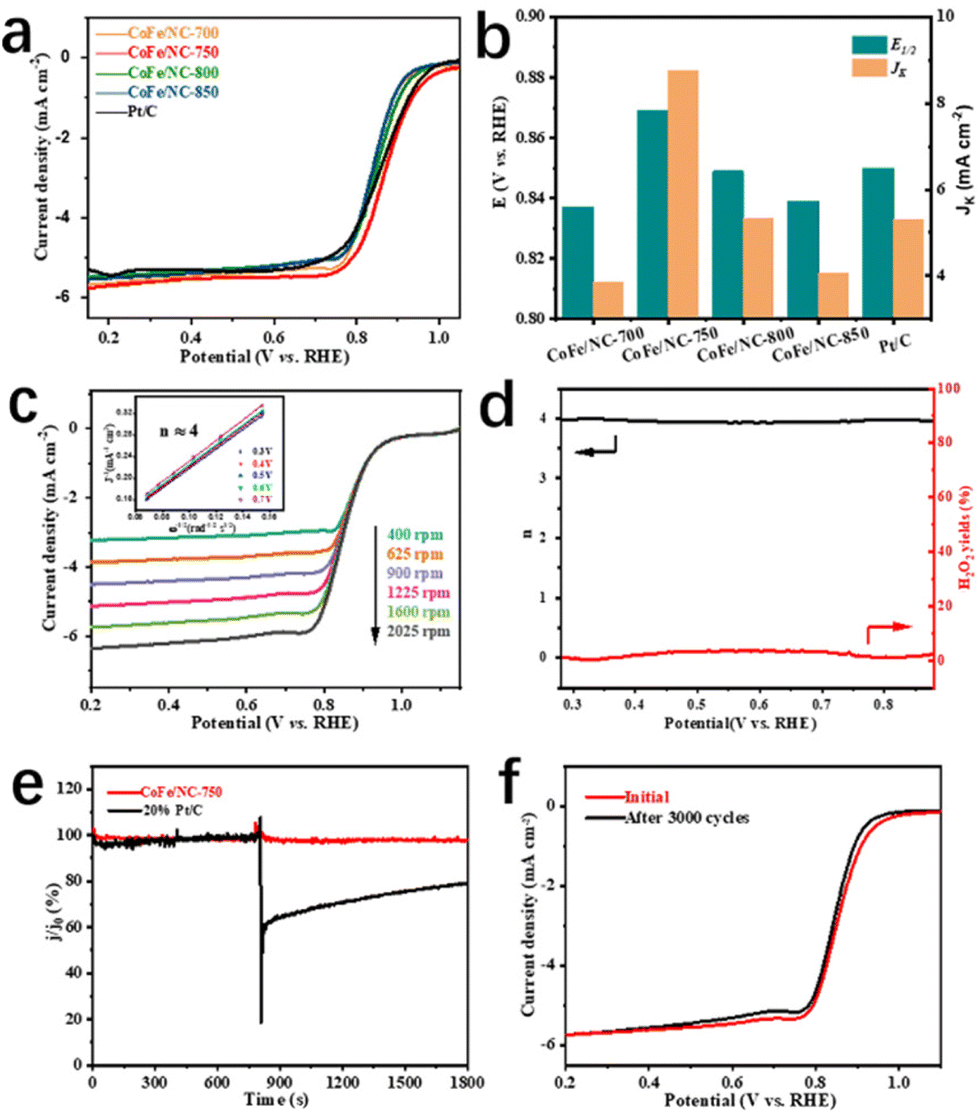 | ||
| Fig. 3 Electrocatalytic ORR performance of the catalysts. (a) LSV polarization curves of CoFe/NC-T (T = 700, 750, 800, and 850 °C) and Pt/C in O2-saturated 0.1 M KOH solution with a rotation rate of 1600 rpm. (b) The comparison of E1/2 and JK at 0.85 V of CoFe/NC-T and Pt/C. (c) LSV curves of CoFe/NC-750 at different rotation rates (inset: the corresponding K–L plot). (d) Electron transfer number and H2O2 yield of CoFe/NC-750 from RRDE measurement. (e) Chronoamperometric measurements of a methanol crossover test with CoFe/NC-750 and Pt/C. (f) Stability result for CoFe/NC-750. | ||
The superior ORR catalytic performance of CoFe/NC-750 over CoFe/NCs achieved at other temperatures suggests that the ordered mesoporous structure and CoFe species play essential roles. We further prepared single metal species-based materials through the same synthesis procedure, namely Co/NC-750 and Fe/NC-750, to investigate the advantages of the bimetallic species compared with the single metallic species in improving ORR activity. SEM images in Fig. S9a and S9b† show that Co/NC-750 and Fe/NC-750 maintain a rod-like morphology similar to that of CoFe/NC-750. The structural properties of these materials are further characterized by XRD and Raman measurements. As illustrated in Fig. 4a, the XRD patterns of Co/NC-750 and Fe/NC-750 exhibit dominant characteristic peaks of the graphitic carbon (002) plane at around 26°. No obvious peaks related to metallic Co or Fe species have been observed for Co/NC-750 and Fe/NC-750, which is a different phenomenon from that of CoFe/NC-750, indicating that the solely metallic Co or Fe species may be easily removed by acidic treatment. The Raman spectra in Fig. 4b show a higher ID/IG intensity ratio of CoFe/NC-750, compared to that of Co/NC-750 and Fe/NC-750. Furthermore, N2 adsorption/desorption isotherm results (Fig. S5, S9c and S9d†) reveal that CoFe/NC-750 possesses a higher BET surface area in comparison with Co/NC-750 and Fe/NC-750, contributing to the exposure of more active sites and increased ORR kinetics activity. The ORR test results presented in Fig. 4c and d and Fig. S10† reveal that CoFe/NC-750 exhibits superior Eonset, E1/2 and JK values to those of Co/NC-750 and Fe/NC-750, indicating that the synergistic effect of Co and Fe species contributed to the enhanced electrocatalytic ORR activity. In addition, as shown in Fig. S10,† CoFe/NC-750 exhibits the smallest Tafel slope (83 mV dec−1) compared to those of Co/NC-750 and Fe/NC-750, further demonstrating the faster ORR kinetics of CoFe/NC-750 originating from the presence of bimetallic species.44,45
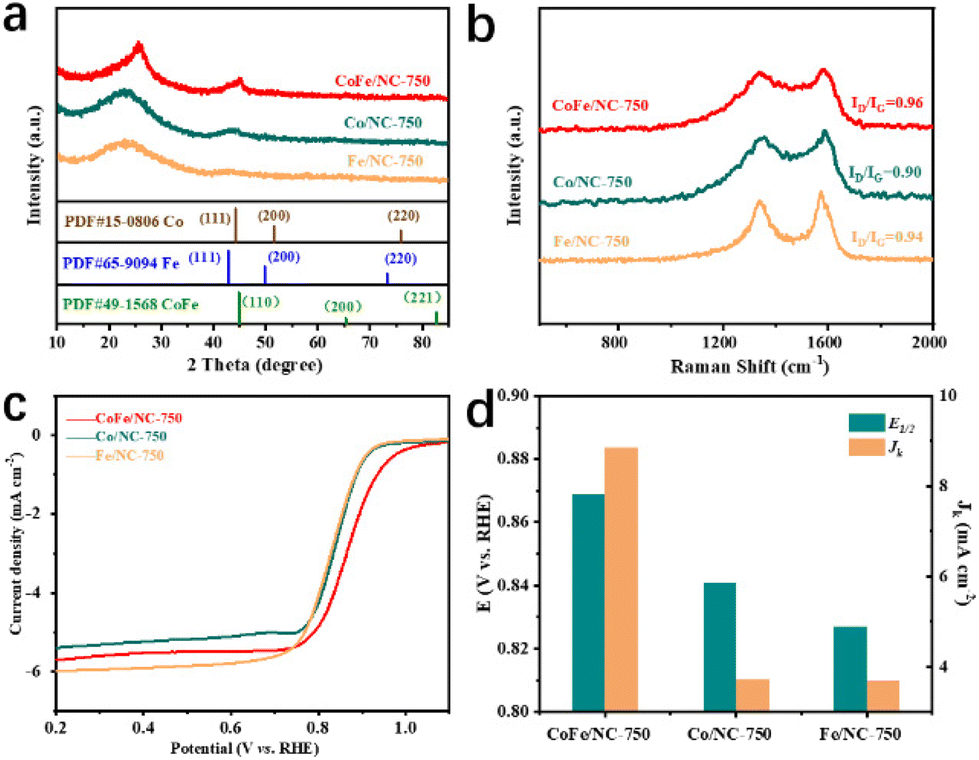 | ||
| Fig. 4 (a and b) XRD patterns and Raman spectra of CoFe/NC-750, Co/NC-750 and Fe/NC-750. (c and d) LSV polarization curves and comparison of E1/2 and JK at 0.85 V vs. RHE of CoFe/NC-750, Co/NC-750 and Fe/NC-750. | ||
2.3 DFT
DFT calculations were performed to understand the electronic structure and catalytic processes of the as-prepared CoFe/NC-750, further elucidating the prominence of its ORR activity. We have constructed two models, CoFe/NC with CoFe alloys and dual atom sites and CoFe-dual atoms/NC with CoFe merely as dual atom sites (Fig. 5a and Fig. S11a†). As demonstrated in Fig. 5a, CoFe/NC-750 delivers a 4-electron pathway of the ORR process, thus the optimized configurations of the OOH*, O*, and OH* intermediates were investigated. The Gibbs free energy diagram in Fig. 5b shows that the final step of OH* reduction is the rate-determining step (RDS) for both CoFe-dual atoms/NC and CoFe/NC. CoFe/NC exhibited a lower energy barrier of −0.526 eV for OH* reduction compared to that of CoFe-dual atoms/NC (−0.477 eV), demonstrating that CoFe/NC is more favorable for the ORR process, which may originate from the electronic structure regulation of the Fe single atom sites from the CoFe nanoparticles. We further employed the Fe atoms from the alloys as the active sites for OH* adsorption. The results presented in Fig. S11b† show that the energy barrier of the RDS was increased, indicating that the CoFe nanoparticles in CoFe/NC are promoters rather than the catalytic active sites to boost the ORR process. Furthermore, the projected density of states (PDOS) of the active sites of Fe atoms in CoFe-dual atoms/NC and CoFe/NC are illustrated in Fig. 5c. The calculated d-band center (εd) of Fe atoms (−2.012 eV) in CoFe/NC is slightly lower than that in CoFe-dual atoms/NC (−1.970 eV), promoting the dissociation of OH* and thus accelerating the ORR process. This further demonstrates that the incorporation of CoFe nanoparticles modulates the electronic structure of Fe active sites for a highly efficient ORR.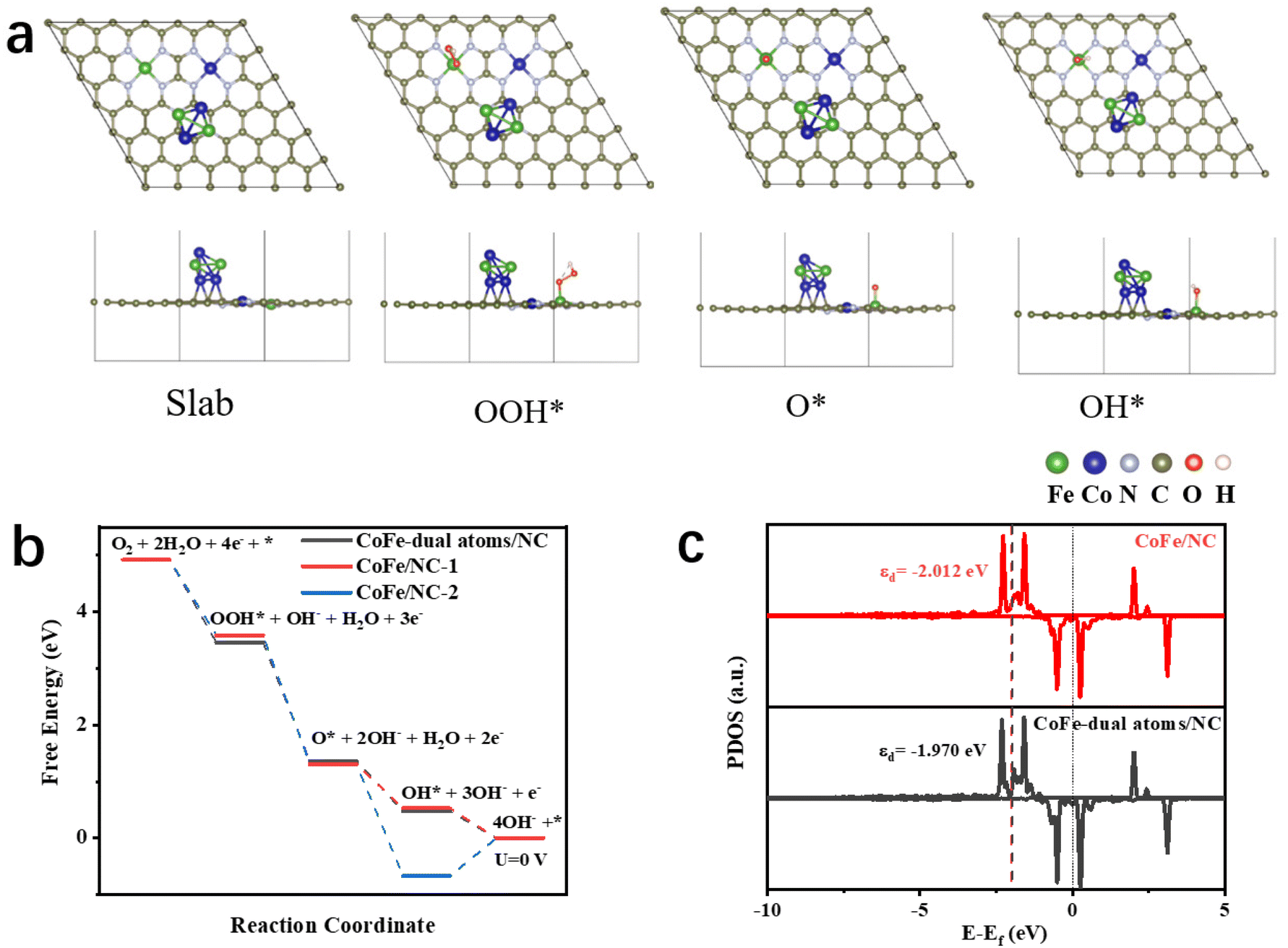 | ||
| Fig. 5 DFT calculation results of CoFe/NC-750. (a) The reaction scheme with the intermediates in the ORR process on CoFe/NC. (b) Free-energy paths of ORR intermediates on CoFe-dual atoms/NC, CoFe/NC-1 and CoFe/NC-2. (c) The PDOS of d orbitals of active Fe atoms on CoFe/NC and CoFe-dual atoms/NC. | ||
3 Conclusions
In summary, we have developed a template-assisted method for preparing ordered mesoporous carbon-supported CoFe species (CoFe/NC) as highly efficient ORR catalysts. The optimized temperature ensures the successful fabrication of the ordered mesoporous carbon and the co-existence of dual metal atoms/alloys. The ordered mesoporous structure promotes the mass/charge transfer and the synergistic effect of the CoFe bimetallic species modulates the active sites for a boosted ORR. CoFe/NC-750 exhibits an excellent ORR performance with high E1/2 of 0.87 V vs. RHE in 0.1 M KOH, and no obvious attenuation after 3000 cycles, which is superior behavior to that of commercial Pt/C. The DFT calculations illustrated that the superior ORR performance originated from the electronic structure modulation of the catalytic active sites (atomically dispersed Fe sites) by CoFe alloy nanoparticles, which accelerates the dissociation and reduction of OH* and promotes the ORR process. Therefore, this study provides a promising strategy for designing highly efficient electrocatalysts for boosting the ORR process and other energy conversion applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Y. Z. acknowledges the support from the National Natural Science Foundation of China (22209103). All authors are grateful for support from the “Joint International Laboratory on Environmental and Energy Frontier Materials” and the “Innovation Research Team of High-Level Local Universities in Shanghai”. H. L. acknowledges financial support from the Australian Research Council (FT180100705) and support from CSIRO “International Hydrogen Research Collaboration Program-RESEARCH FELLOWSHIPS”. J. Z. acknowledges support from the UTS Chancellor's Research Fellowships.References
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- D. U. Lee, J.-Y. Choi, K. Feng, H. W. Park and Z. Chen, Adv. Energy Mater., 2014, 4, 1301389 CrossRef.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- X. Cao, J. Huo, L. Li, J. Qu, Y. Zhao, W. Chen, C. Liu, H. Liu and G. Wang, Adv. Energy Mater., 2022, 12, 2202119 CrossRef CAS.
- S. D. Bhoyate, J. Kim, F. M. de Souza, J. Lin, E. Lee, A. Kumar and R. K. Gupta, Coord. Chem. Rev., 2023, 474, 214854 CrossRef CAS.
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- J. Zhang, Y. Zhao, C. Chen, Y. C. Huang, C. L. Dong, C. J. Chen, R. S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- J. Huo, Z. Shen, X. Cao, L. Li, Y. Zhao, H. Liu and G. Wang, Small, 2022, 18, e2202394 CrossRef PubMed.
- J. Huo, X. Cao, Y. Tian, L. Li, J. Qu, Y. Xie, X. Nie, Y. Zhao, J. Zhang and H. Liu, Nanoscale, 2023, 15, 5448–5457 RSC.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W. C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, e1800588 CrossRef PubMed.
- Y. Zhao, Z. Shen, J. Huo, X. Cao, P. Ou, J. Qu, X. Nie, J. Zhang, M. Wu, G. Wang and H. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308349 CrossRef CAS PubMed.
- Y. Tan, Z. Zhang, Z. Lei, L. Yu, W. Wu, Z. Wang and N. Cheng, Appl. Catal., B, 2022, 304, 121006 CrossRef CAS.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Mullen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- L. Osmieri, A. H. A. Monteverde Videla, P. Ocón and S. Specchia, J. Phys. Chem. C, 2017, 121, 17796–17817 CrossRef CAS.
- X. Ao, W. Zhang, Z. Li, J. G. Li, L. Soule, X. Huang, W. H. Chiang, H. M. Chen, C. Wang, M. Liu and X. C. Zeng, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS PubMed.
- H. Huang, D. Yu, F. Hu, S. C. Huang, J. Song, H. Y. Chen, L. L. Li and S. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CrossRef CAS PubMed.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- Y. Cheng, H. Song, J. Yu, J. Chang, G. I. N. Waterhouse, Z. Tang, B. Yang and S. Lu, Chin. J. Catal., 2022, 43, 2443–2452 CrossRef CAS.
- C. Xu, C. Guo, J. Liu, B. Hu, J. Dai, M. Wang, R. Jin, Z. Luo, H. Li and C. Chen, Energy Storage Mater, 2022, 51, 149–158 CrossRef.
- Z. Wang, C. Zhu, H. Tan, J. Liu, L. Xu, Y. Zhang, Y. Liu, X. Zou, Z. Liu and X. Lu, Adv. Funct. Mater., 2021, 31, 2104735 CrossRef CAS.
- S. H. Yin, J. Yang, Y. Han, G. Li, L. Y. Wan, Y. H. Chen, C. Chen, X. M. Qu, Y. X. Jiang and S. G. Sun, Angew. Chem., Int. Ed., 2020, 59, 21976–21979 CrossRef CAS PubMed.
- S. N. Zhao, J. K. Li, R. Wang, J. Cai and S. Q. Zang, Adv. Mater., 2022, 34, e2107291 CrossRef PubMed.
- Y. Li, H. Huang, S. Chen, X. Yu, C. Wang and T. Ma, Nano Res., 2019, 12, 2774–2780 CrossRef CAS.
- S. Hu, W. Ni, D. Yang, C. Ma, J. Zhang, J. Duan, Y. Gao and S. Zhang, Carbon, 2020, 162, 245–255 CrossRef CAS.
- S. Saifi, G. Dey, J. Karthikeyan, R. Kumar, D. Bhattacharyya, A. S. K. Sinha and A. Aijaz, Inorg. Chem., 2023, 62, 8200–8209 CrossRef CAS PubMed.
- Y. Pan, X. Ma, M. Wang, X. Yang, S. Liu, H. C. Chen, Z. Zhuang, Y. Zhang, W. C. Cheong, C. Zhang, X. Cao, R. Shen, Q. Xu, W. Zhu, Y. Liu, X. Wang, X. Zhang, W. Yan, J. Li, H. M. Chen, C. Chen and Y. Li, Adv. Mater., 2022, 34, e2203621 CrossRef PubMed.
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed.
- K. Kim, K. Min, Y. Go, Y. Lee, S. E. Shim, D. Lim and S.-H. Baeck, Appl. Catal., B, 2022, 315, 121501 CrossRef CAS.
- X. Cheng, Y. Wang, Y. Lu, L. Zheng, S. Sun, H. Li, G. Chen and J. Zhang, Appl. Catal., B, 2022, 306, 121112 CrossRef CAS.
- X. Duan, W. Tian, H. Zhang, H. Sun, Z. Ao, Z. Shao and S. Wang, ACS Catal., 2019, 9, 7494–7519 CrossRef CAS.
- K. Wang, C. Qiu, Z. Wang, Q. Chen, J. Pan, J. Li, M. Wu, H. Dong, K. Shi and Q. Liu, Carbon, 2023, 214, 118333 CrossRef CAS.
- L. Song, J. Zhang, S. Sarkar, C. Zhao, Z. Wang, C. Huang, L. Yan and Y. Zhao, Chem. Eng. J., 2022, 433, 133686 CrossRef CAS.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. V. Voorhis, J. T. Miller and Y. Surendranath, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- W. Xue, Q. Zhou, X. Cui, J. Zhang, S. Zuo, F. Mo, J. Jiang, X. Zhu and Z. Lin, Angew. Chem., Int. Ed., 2023, 62, e202307504 CrossRef CAS PubMed.
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- F. Shi, K. Zhu, X. Li, E. Wang, X. Zhu and W. Yang, J. Energy Chem., 2021, 61, 327–335 CrossRef CAS.
- W. Zhang, Y. P. Chen, L. Zhang, J. J. Feng, X. S. Li and A. J. Wang, J. Colloid Interface Sci., 2022, 626, 653–661 CrossRef CAS PubMed.
- X. Guo, W. Zhang, J. Shi, M. Duan, S. Liu, J. Zhang, Y. Liu, S. Xiong and Q. Kong, Nano Res., 2021, 15, 2092–2103 CrossRef.
- X. Guo, J. Shi, M. Li, J. Zhang, X. Zheng, Y. Liu, B. Xi, X. An, Z. Duan, Q. Fan, F. Gao and S. Xiong, Angew. Chem., Int. Ed., 2023, 62, e202314124 CrossRef CAS PubMed.
- T. Tang, Y. Wang, J. Han, Q. Zhang, X. Bai, X. Niu, Z. Wang and J. Guan, Chin. J. Catal., 2023, 46, 48–55 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00175c |
| ‡ Fengying Pan and Ziyan Shen made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2024 |