
Strategic design of VO2 encased in N-doped carbon as an efficient electrocatalyst for the nitrogen reduction reaction in neutral and acidic media†
Ashis
Chhetri‡
ab,
Ashmita
Biswas‡
 c,
Sumana
Podder
ab,
Ramendra Sundar
Dey
*c and
Joyee
Mitra
*ab
c,
Sumana
Podder
ab,
Ramendra Sundar
Dey
*c and
Joyee
Mitra
*ab
aInorganic Materials and Catalysis Division, CSIR-Central Salt and Marine Chemicals Research Institute, Gijubhai Badheka Marg, Bhavnagar 364002, Gujarat, India. E-mail: joyeemitra@csmcri.res.in; joyeemitra@gmail.com
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
cInstitute of Nano Science and Technology, Sector 81, Mohali 140306, Punjab, India. E-mail: rsdey@inst.ac.in
First published on 25th March 2024
Abstract
Electrocatalytic nitrogen fixation to ammonia (NH3), a precursor for fertilizer production and a promising energy carrier, has garnered widespread interest as an environment-friendly and sustainable alternative to the energy-intensive fossil-feedstock-dependent Haber–Bosch process. The large-scale deployment of this process is contingent on the identification of inexpensive, Earth-abundant systems that can operate efficiently, irrespective of the electrolyte pH for the selective production of NH3. In this regard, we discuss the scalable synthesis of VO2 anchored on N-doped carbon (VO2@CN), and its applicability as a robust electrocatalyst for the nitrogen reduction reaction (NRR). Benefitting from the presence of exposed VO2, which presumably acts as the active site for nitrogen reduction, and its activity over a broad pH range (from acidic to neutral), VO2@CN exhibits a high NH3 yield of 0.31 and 0.52 μmol h−1 mgcat−1 and a maximum Faradaic efficiency (FE) of 67.9% and 61.9% at −0.1 V vs. RHE, under neutral and acidic conditions, respectively. The obscured reaction intermediates of the NRR were identified from in situ ATR-IR studies under both electrolyte conditions. Additionally, the high selectivity of the catalyst was ascertained from the absence of hydrazine production and the competing hydrogen evolution reaction (HER). However, ammonia production underwent a reduction over 12 h of continuous operation presumably owing to the leaching of catalyst under these electrolysis conditions, which was more pronounced in electrolytes with acidic pH. Overall, the present report unveils the performance of an earth-abundant vanadium oxide-based system as an efficient electrocatalyst for the NRR under acidic and neutral pH conditions.
Introduction
Ammonia (NH3) obtained from the reduction of nitrogen is perceived as a clean energy carrier and a replacement for gaseous hydrogen owing to its compression to the liquid state at a low pressure of ∼8 bar and room temperature and its high energy density in the liquid state.1,2 In addition, NH3 is vital in agriculture, in the production of fertilizers to feed the ever-increasing global population. The industrial production of NH3 relies on the Haber–Bosch process, which requires harsh temperature (400–600 °C) and pressure (20–40 MPa) conditions, has an energy efficiency of ∼65%, and accounts for about 2% of global energy consumption, releasing a large amount of CO2 into the atmosphere causing an increase in the global temperature.3–5 This emphasizes the necessity of developing a clean, sustainable alternative process for nitrogen fixation, which precludes the necessity of elevated temperatures and reduces the carbon footprint.6,7 Various routes for nitrogen fixation, including photochemical, electrochemical and biological approaches have been explored by the research community to arrive at environmentally safe and energy-efficient methods to artificially fix nitrogen.8–11 Among these approaches, the electrochemical NRR is considered a promising alternative to NH3 production due to its mild reaction conditions and environmentally friendly precursors of N2 and H2O, which can revolutionize green ammonia production when used in conjunction with renewable energy resources.5,12 Intensive efforts have been devoted worldwide to the design and investigation of catalysts for the selective production of NH3via the NRR with a high FE.13–19 It is extremely arduous to dissociate the inert dinitrogen molecule (bond energy of N2 is 941 kJ mol−1), and mechanistically, the NRR involves multiple proton-coupled electron-transfer steps.20 In addition, the utility of the process is limited by the competing hydrogen evolution reaction (HER), which has faster kinetics and operates under a similar potential window.The literature reveals a series of transition metal-based systems that have been reported to facilitate the NRR under ambient conditions.21–28 Of these, Mo, Fe, Rh and Ru are at the top of the volcano diagram, and hence, they show the best activity.23 However, the competing HER lowers their efficiency for ammonia production. Early transition metals including Zr, Ti, Sc, V etc., have been reported to bind N-adatoms more strongly compared to H-adatoms, thereby enhancing the production of NH3 compared to H2.23 Among these, vanadium is noteworthy, as it plays a vital role in the nitrogenase enzyme, responsible for replenishing bioavailable ammonia from atmospheric dinitrogen.29 Vanadium carbide, oxynitrides and phosphides have been extensively reported as electrocatalysts for the NRR mostly under neutral pH conditions,7,24,30,31 but vanadium oxides have been relatively less explored in this regard, despite the surmise that the NRR actually operates at O-supported vanadium Lewis acidic sites.27,28 DFT calculations indicate that N2 activation is energetically favoured on O-supported V centres by ∼18 kcal mol−1 compared to that on N-supported V centres.32 A survey of the literature disclosed that the coordination environment and oxophilicity of the metal are important in modulating the performance of electrocatalysts, as these properties affect the binding of N atoms and N2 molecules on the substrate and the stability of the intermediates culminating in the release of NH3.33 Catalysts with electron-deficient centres were reported to be beneficial for NRR performance enhancement.27 On the other hand, carbon-based nanomaterials have garnered widespread attention for electrocatalytic applications owing to their mechanical stability, superior electrical conductivity and enhanced surface areas. Doping of heteroatoms on the carbon surface creates defect sites and a redistribution of electrons to regulate the electron transfer and alters the sorption of reactants, intermediates and products on the catalyst surface. N-doped carbon substrates have been reported to facilitate N2 adsorption and N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N cleavage, though they show very mild electrocatalytic NRR activity.13,34 Though there are a couple of reports on the utility of vanadium oxide and N-doped carbon individually for the electrocatalytic NRR, their combined efficacy is yet to be explored for the NRR, to the best of our understanding.
N cleavage, though they show very mild electrocatalytic NRR activity.13,34 Though there are a couple of reports on the utility of vanadium oxide and N-doped carbon individually for the electrocatalytic NRR, their combined efficacy is yet to be explored for the NRR, to the best of our understanding.
Motivated by the above discussions, we present vanadium dioxide encased in an N-doped carbon support (VO2@CN) as an electrocatalyst for the NRR. A multidimensional structure due to a combination of vanadium dioxide and N-doped carbon is expected to improve the exposure of active sites and increase electrical conductivity, which might facilitate electrochemical nitrogen fixation. Considering the advantage of reactive Lewis acidic oxo-vanadium centres for efficient N2 adsorption and the stability of vanadium oxides over broad potential and pH ranges,35 we have explored VO2@CN as an electrocatalyst for the NRR under acidic and neutral electrolyte conditions. Facilitated by the effectual N2 adsorption, and stabilization of vanadium in multiple oxidation states to modulate multiple electron-transfer steps, VO2@CN exhibits a high NH3 yield of 0.31 and 0.52 μmol h−1 mgcat−1 and a maximum FE of 67.9% and 61.9% at −0.1 V vs. RHE, under neutral and acidic conditions, respectively. The complexities of the NRR could be minimized by the detection of the intermediates formed during the course of the reaction under both electrolyte conditions using in situ ATR-IR studies. This in turn paves the way for a better understanding of the reaction kinetics on the VO2@CN surface.
Results and discussion
Synthesis and structural characterization
A simple solution-processing route followed by carbonization (Fig. 1a) was employed to synthesize VO2@CN. First, vanadium pentoxide was dissolved in 3 M HNO3 to form VO2(NO3) species. The acidic solution of oxo-vanadium species was thoroughly mixed with a solution of melamine in methanol. The resultant precipitate was evaporated to dryness, where the oxo-vanadium ions were trapped by the melamine moieties, resulting in the formation of VO2(NO3)@melamine (referred to as VO2@mel hereafter). The reaction proceeded by carbonization under an inert atmosphere at 600 °C to yield the VO2@CN material. NH3 liberated during the thermolysis of melamine at elevated temperatures acted as a reducing agent to convert V+5 in VO2@mel to V+4 in VO2@CN.36 The coordination of dinitrogen to transition metal centres is usually facilitated by a lower oxidation state of the metal.37 In the case of vanadium, V+5 is the most stable oxidation state, while the lower oxidation states are often sensitive to air and moisture. The N-doped carbon matrix in VO2@CN not only aids in imparting stability to VO2, but it is expected to facilitate the NRR. It is envisaged that the VO2 component of the catalyst would bring about σ-donation and counter π-back donation interactions between the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of the catalyst active site and N2, as schematically illustrated in Fig. 1b. This interaction would help weaken the NN bond strength, activating the molecule towards a successful reduction reaction. It is to be noted that V+5 in the precursor VO2@mel cannot favour π-back donation to weaken the NN bond. The PXRD analysis of the as-synthesized material shows distinct peaks corresponding to vanadium dioxide, which can be assigned to the monoclinic phase of VO2 (JCPDS file no. 01-082-0661, space group P21/c) (Fig. 2).38 The presence of melamine in acidic solution prevents the fast aggregation of VO6 octahedra to favour the exclusive formation of the stable monoclinic VO2 (M) phase. The absence of any other phases in the as-prepared sample ascertains the purity of the VO2 (M) phase. The presence of N-doped carbon was supported by the appearance of a broad signal at ∼2θ = 23° corresponding to the 002 plane. The FTIR spectrum of VO2@CN (Fig. S2†) shows the presence of a sharp band at ∼435 cm−1 characteristic of the long-range stretching vibrations of the V–O–V bridge and at ∼684 cm−1 indicative of the packing of VO6 octahedra, respectively.39 The breathing mode of triazine can be observed from a peak at ∼813 cm−1. In addition, the incorporation of nitrogen into the carbon framework can be identified from an FTIR signal at ∼1372 cm−1.40 Broad bands at ∼3200 cm−1 are attributable to N–H stretching and hydrogen bonding in the VO2@CN sample. Raman spectroscopy was performed under the excitation wavelength of 532 nm, which revealed three peaks at 1373 cm−1, 1598 cm−1 and 2760 cm−1 corresponding to the D, G and 2D bands of N-doped carbon, respectively (Fig. S3†). The calculated ID/IG value of 1.13 indicated the introduction of nitrogen atoms into the carbon framework resulting in significant lattice distortion.41 TGA measurements reveal multiple weight loss phases until ∼450 °C indicative of the release of adhered water molecules and interlayer water molecules, respectively, followed by the decomposition of N-doped carbon layers beyond 600 °C (Fig. S4†).42
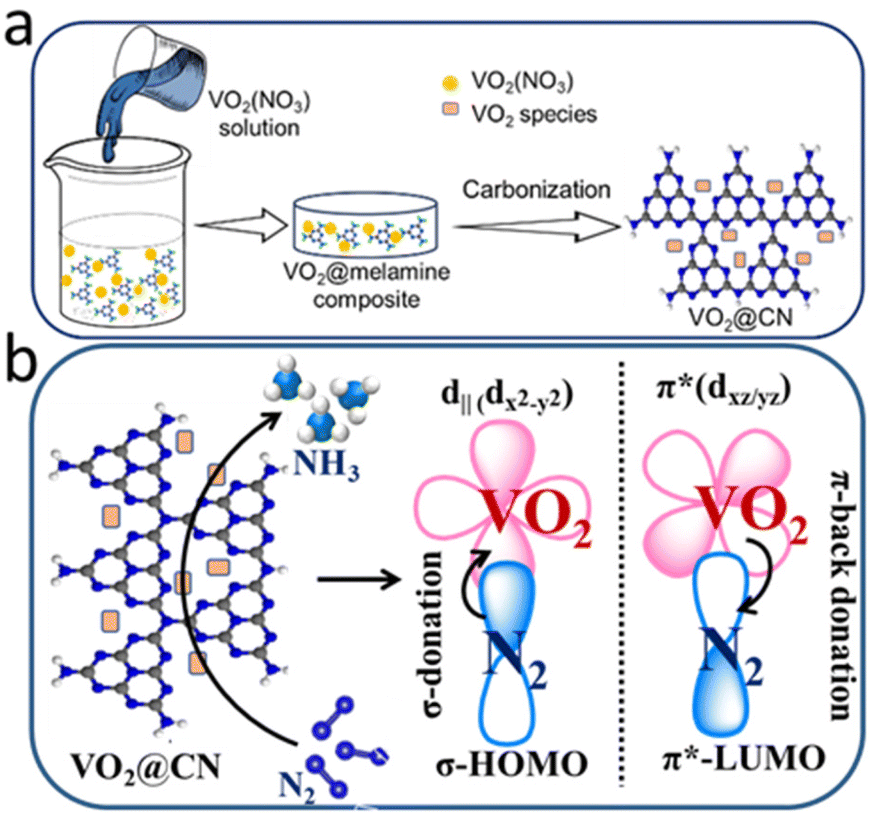 | ||
| Fig. 1 (a) Schematic representation of the synthesis of VO2@CN; (b) orbital interaction between N2 and the VO2 centre of the catalyst, responsible for N2 reduction to NH3 (NRR). | ||
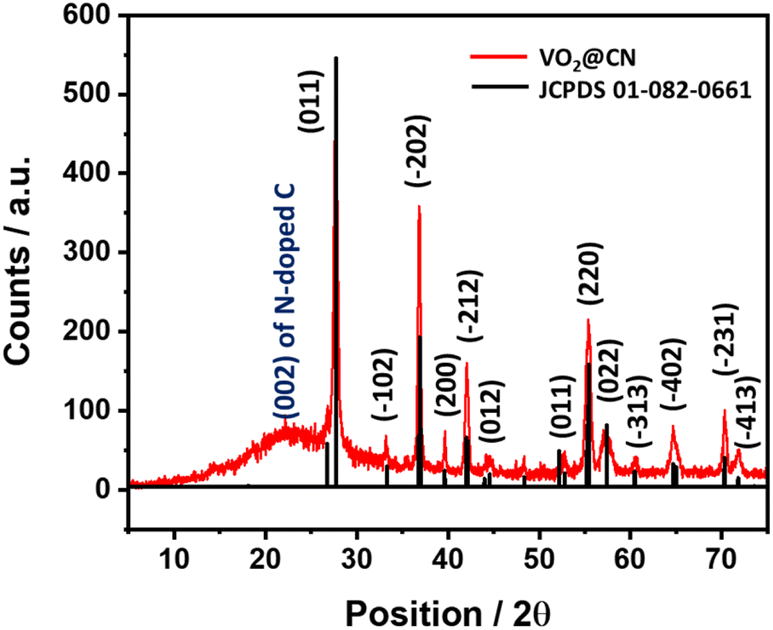 | ||
| Fig. 2 PXRD pattern of the as-synthesized VO2@CN indicating the presence of VO2 (M) and N-doped carbon. | ||
The microenvironment of VO2@CN and its precursor VO2@mel was investigated from X-ray photoelectron spectroscopy (XPS) measurements (Fig. 3a–c, ES1†). As shown in Fig. 3a, the V 2p3/2 spectrum exhibited a V+4 feature at binding energy 515.9 eV, and a feature at 516.8 eV indicative of the surface oxidation of vanadium to the V+5 state, which has been reported for vanadium oxide samples.43 A V 2p1/2 signal was observed at binding energy 523.8 eV. The O 1s spectrum revealed the presence of V–O bonds at 529.45 eV and V+4–O−2 bonds at 530.14 eV (Fig. 3b). Another peak at 531.77 eV indicated OH− on the VO2 surface.28 The N 1s spectrum of VO2@CN depicted the presence of three peaks at binding energies of 398.7 eV, 399.6 eV and 401.0 eV, which can be assigned to pyridinic N, pyrrolic N and graphitic N, respectively (Fig. 3c).27 In contrast, the analysis of the valence state of the precursor VO2@mel (Fig. S1†) revealed the prominent presence of the V+5 state in the V 2p XPS results (binding energy 516.9 eV for V 2p1/2). The N 1s XP spectrum of VO2@mel revealed the presence of three peaks ascribed to the ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of melamine (398.5 eV), exocyclic –NH2 of melamine (399.6 eV) and nitrate counter anion (406.0 eV), attributed to the presence of VO2(NO3) in VO2@mel.28
N of melamine (398.5 eV), exocyclic –NH2 of melamine (399.6 eV) and nitrate counter anion (406.0 eV), attributed to the presence of VO2(NO3) in VO2@mel.28
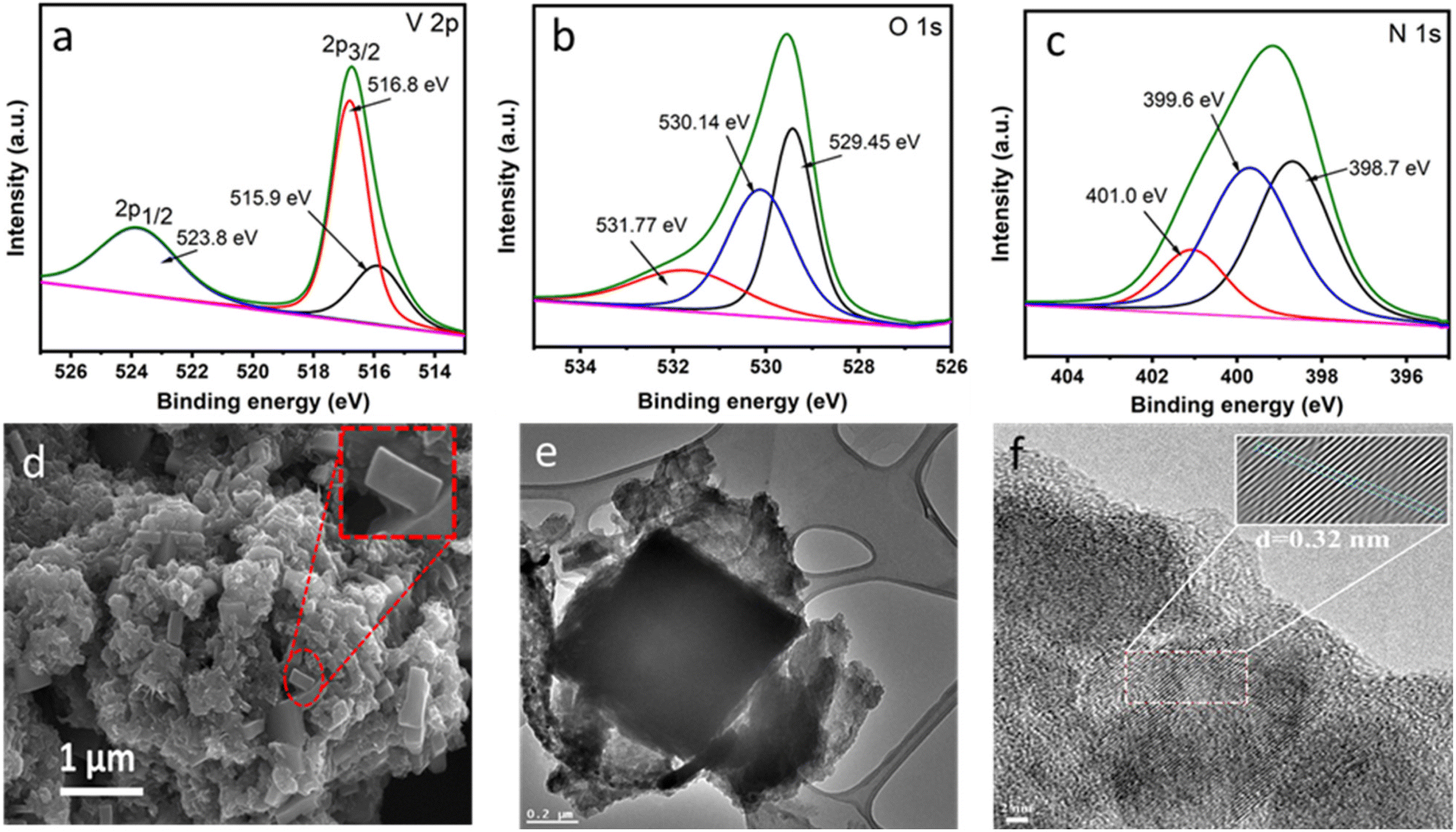 | ||
| Fig. 3 XPS spectra of VO2@CN: (a) V 2p, (b) O 1s and (c) N 1s. (d) FESEM image of VO2@CN indicating the presence of blocks of VO2 encapsulated in crumpled N-doped C layers. TEM and HRTEM images of VO2@CN are presented as (e) and (f), respectively. | ||
The scanning electron microscopy (SEM) images of VO2@CN revealed vanadium dioxide particles as rectangular block-shaped structures with a length of ∼500 nm amidst larger crumpled sheets of N-doped carbon, which did not show any significant alteration from the morphology of pristine N-doped carbon materials (Fig. 3d and S5†). The larger size of the VO2 rods could be attributed to the favourable grain growth of nanorods at elevated temperatures.38 The presence and uniform distribution of the constituent elements V, O, C and N was attested from the EDX and elemental mapping analysis (Fig. S6†). The transmission electron micrographs (TEM) of VO2@CN supported the presence of VO2 blocks encased in crumpled N-doped carbon layers (Fig. 3e). HRTEM confirmed the presence of VO2 encapsulated in N-doped carbon from the lattice fringes corresponding to a d-spacing value of 0.32 nm (Fig. 3f) indicative of the (011) plane of VO2 (M). The SAED pattern depicts the polycrystalline nature of the VO2@CN sample supporting the observation of N-doped carbon-encased VO2 blocks (Fig. S7†).
Electrochemical NRR performance of VO2@CN
Vanadium in various forms has been a popular candidate having abilities to reduce dinitrogen electrochemically.7,43 Mostly, vanadium nitrides are investigated for the NRR, but the involvement of lattice nitrogen and the unstable metal centre have prompted researchers to explore the oxy-nitride class of materials, where the oxygen-supported vanadium centres were proved to be energetically more favourable towards N2 scission as compared to the nitrogen supported ones.44 Investigation of V2O5 nanodots as an electrocatalyst for the NRR afforded a faradaic efficiency (FE) of 28.7%, attributable to the presence of oxygen vacancies that participated in the electron transfer to the V+5 state, to effectively generate V+4, thus underlining the importance of the V+4 species in electrocatalytic NRR.28 Moreover, vanadium-based systems were mostly found effective for the NRR at neutral pH, and have rarely been explored under acidic conditions in order to suppress the competing HER. Since the challenging part in the NRR is the bond polarization of N2 leading to its first protonation and subsequent conversion to ammonia, it forms the motivation for the catalyst (VO2@CN) synthesis in this work. All the potentials reported in this work were calibrated with respect to a reversible hydrogen electrode (RHE) following eqn (1) (ESI†). It was shown by Biswas et al. that the choice of the electrolyte is important as different catalysts respond differently in the presence of the ionic environment of the electrolyte (effect of the counter ions), and it crucially determines the nitrogen reduction performance of the catalyst.45,46 The ions from the electrolyte form a Stern layer across the electrode that mediates the adsorption of N2 and repels H+ from the electrode.47 It should be noted that the rationale behind avoiding the basic electrolyte is attributed to sample instability in the medium. Thus, in this study both acidic (0.1 M HCl) and neutral (0.1 M Na2SO4) electrolytes were explored, and a comparative study was accomplished. Before any electrochemical tests, the electrolyte solutions were degassed with Ar followed by N2 purging after passing the N2 feed gas through base and acid traps since contamination is unavoidable in the NRR and this calls for all precautionary measures to achieve reliable data. The electrochemical cell set-up used in this study is shown in Fig. S8.† As shown in Fig. S9,† the trap solutions were subjected to colorimetric tests for the detection of any dissolved NOx or NH4+ that would have been present in the feed gas. The UV-visible spectra for the N2-trapped solutions were overlaid with those of the bare ones, which indicated that there were negligible gaseous nitrogenous impurities in the feed gas itself.Thereafter, an elaborate study with chronoamperometry (CA) at different potentials was carried out in both the electrolytes for 1 h each (Fig. S10a and c†). The post-electrochemistry UV-visible studies with the electrolytes stained with colouring agents (indophenol blue method), depicted a maximum absorbance corresponding to the indophenol dye formed out of the produced ammonia at −0.1 V vs. RHE (Fig. S10b and d†). The material VO2@CN displayed a better ammonia yield rate (calculated using eqn (2), ESI†) in 0.1 M HCl as shown in the bar plot in Fig. 4a, while the FE (calculated using eqn (2), ESI†) in Fig. 4b was better in the case of Na2SO4. In fact, in the case of HCl, the FE showed a drastic reduction as we approached more closely towards a negative potential, owing to enhanced hydrogen evolution reaction (HER) kinetics. The acidic electrolyte with abundant proton sources helped to protonate the produced ammonia, which remained dissolved in the electrolyte in the form of NH4+ and ensured no loss as NH3 gas. However, at the same time, it also induced proton adsorption on the catalyst surface since the HER is a less energy-intensive process than the NRR, which caused the catalyst to incur a sacrificial FE over the entire potential range. With a maximum FE of 67.9% and 61.9% in Na2SO4 and HCl respectively at −0.1 V vs. RHE, the material was also proved to be selective towards ammonia production as there was no interference from side-product (hydrazine) formation (Fig. S10e and f†). This could be justified with respect to a very recent report by Huang et al. that experimentally and theoretically suggested that the NRR on V active sites could energetically prefer a distal pathway negating the chances of the formation of hydrazine, rightly corroborating our experimental results.48 Thus, the NRR on our active material of VO2@CN was kinetically facilitated yielding 0.31 and 0.52 μmol h−1 mgcat−1 of NH3 in Na2SO4 and HCl, respectively. The concentrations of ammonia in all cases were calculated using the standard calibration curves and the corresponding linear regression plot in the relevant acidic or neutral electrolytes (Fig. S11 and S12†). The interference from hydrazine formation was interpreted from the standard calibration and corresponding linear-fitted curves in both the electrolytes, as shown in Fig. S13.† It is noteworthy to mention that the bare Ti foil (pre-cleaned and used instantaneously in order to avoid any surface oxidation layer) showed an FE of 3.24% and 9.5% in 0.1 M Na2SO4 and 0.1 M HCl, respectively, as shown in the bar diagram in Fig. S14,† which indicated that there was only a slight interference of the substrate towards the NRR and it was the right choice to purposely use Ti foil as a substrate as it helped to elevate material performance.
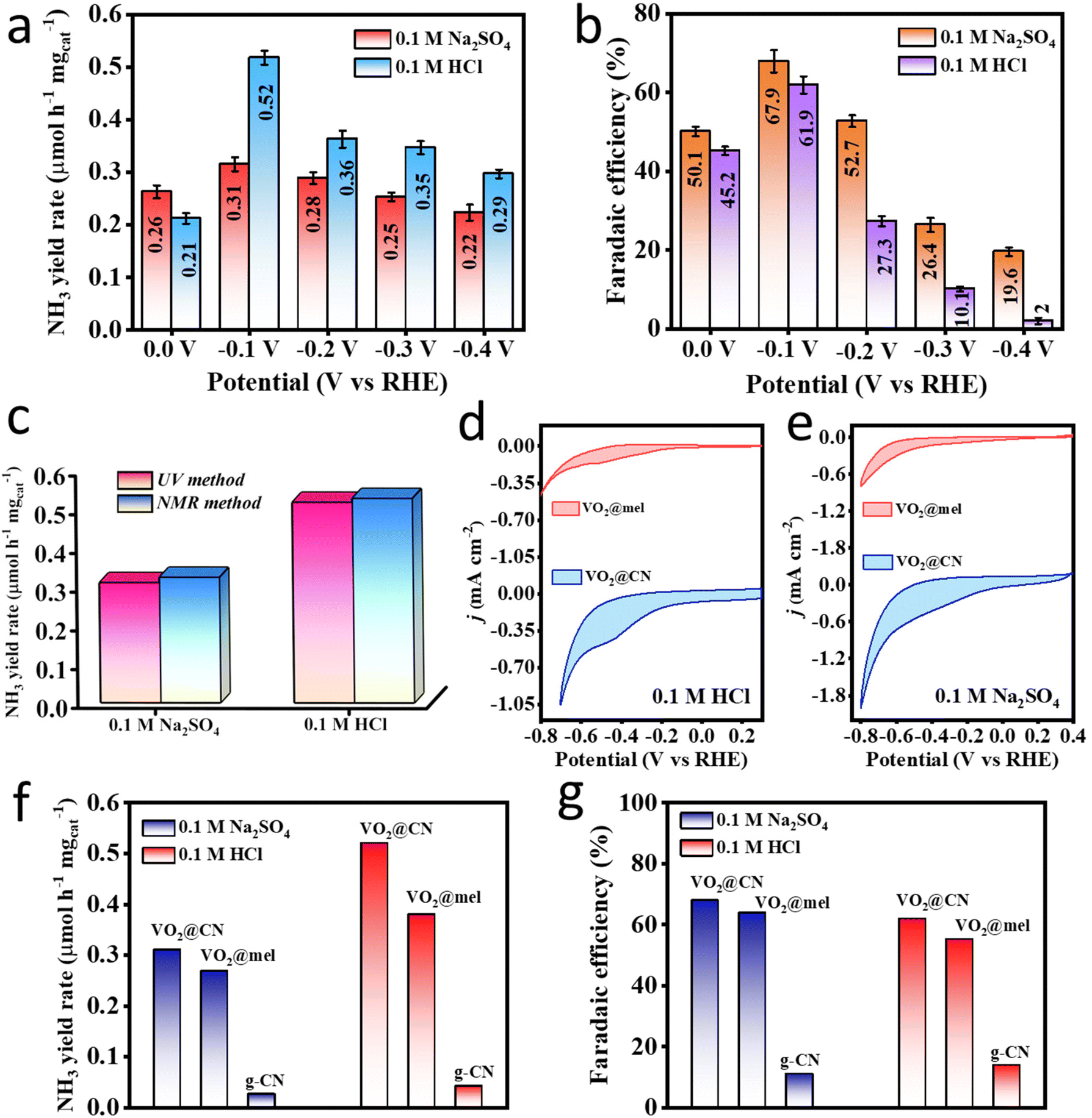 | ||
| Fig. 4 NRR performance comparison of the VO2@CN catalyst in both 0.1 M Na2SO4 and 0.1 M HCl electrolytes. (a and b) NH3 yield rate and FE of the catalyst over the wide-ranging potential window from 0.0 V to −0.4 V vs. RHE in the presence of continuous N2 purging; (c) quantitative analyses of NRR performance of VO2@CN using UV-visible and NMR methods; CV curves of VO2@CN and VO2@mel samples in (d) 0.1 M HCl and (e) 0.1 M Na2SO4 showing an enhancement in the current density upon graphitization of melamine due to increased conductivity; (f) NH3 yield rate and (g) FE comparison of VO2@CN with control samples like VO2@mel and g-CN in both acidic and neutral electrolyte media. The error bars represent the standard deviation values for three different data-sets. | ||
Since the nuclear magnetic resonance (NMR) method provides more accurate quantification, it was adopted to verify the NH3 yield rate and FE in acidic and neutral electrolytes. The details are provided in the ESI.† It is worth noting from Fig. S15† that, in both media, while there was no peak under Ar-purged conditions, a triplet appeared in the presence of N2 resembling 14NH4+ (coupling constant value of 52 Hz calculated using eqn (3) in the ESI†). Thereafter, a quantitative calculation using maleic acid as the standard (eqn (4) from the ESI†) revealed similar NH3 yield rates from both UV-visible and NMR methods, as shown in Fig. 4c, that justify the reliability of the experimental data. However, it is true that without control experiments it is not rational to claim the superiority of the catalyst. Thus, we took into consideration two control samples, i.e. the precursor VO2@mel, and g-CN for the study as the former would show the importance of a conducting carbon framework to anchor the active centres while the latter would justify the role of VO2 active sites of the catalyst for the NRR (detailed NRR studies and UV-visible data are shown in Fig. S16†). As expected, the CV curves at 10 mV s−1 in Fig. 4d and e in 0.1 M HCl and 0.1 M Na2SO4 respectively show an enhancement in the areal current as well as cathodic current density for VO2@CN as compared to VO2@mel signifying the enhancement of the electron conductivity due to the graphitization of melamine to the CN framework. However, the NH3 yield rate and FE for VO2@mel, although sound enough, were lower when compared to that of VO2@CN (Fig. 4f and g) because V was primarily in the V+5 oxidation state, which reduced the π-back donation to the LUMO of the N2 molecule; thus the cleavage of NN bond was relatively less. So, a long-term CA experiment would result in a loss of active sites from the catalyst surface. In contrast to this, the NRR performance of the g-CN sample was very low, being devoid of VO2 units (Fig. 4f and g). This clearly manifested the role of VO2 as the NRR active site with the simultaneous importance of the conjugated carbon–nitride framework in the pronounced electrocatalytic performance of our final material VO2@CN towards the NRR.
N bond polarization and the protonation steps that produce several obscured reaction intermediates on the catalyst surface. Out of these, the stable intermediates are detected in the in situ ATR-FTIR spectra including those attributed to peaks at 1307 and 1460 cm−1 observed in both electrolytes i.e., 0.1 M Na2SO4 and 0.1 M HCl (Fig. 5b and c), resembling H–N–H rocking and N–H bending, respectively.50 It is important to note that the peak corresponding to N–N stretching could be attributed to a stable H2N–NH2 intermediate on the catalyst surface and the corresponding peak appeared to be slightly red-shifted in HCl (1139 cm−1) than in the neutral electrolyte (1143 cm−1) suggesting the better extent of N–N bond polarization in the acidic medium and faster reaction kinetics. This also supported our claim for product (NH3) selectivity of the material as we did not obtain any trace of N2H4 from the UV-visible spectra. Furthermore, the abundance of protons in the acidic electrolyte activated the polarized N2 molecules giving a better yield rate for NH3 production with a comparatively faster kinetics. However, it is also noteworthy that the FTIR spectra in the acidic electrolyte were much more noisy owing to the simultaneous evolution of H2 (HER) on the catalyst surface along with the NRR, which although produced NRR intermediates and products, the current thus obtained contributed to the HER resulting in a sacrificial FE for the NRR in HCl rather than in Na2SO4. Thus, it is interesting to note that keeping all the electrochemical conditions the same, different electrolytes create different local electronic/ionic environments at the catalyst–electrolyte interface and variedly influence the NRR at the active site.
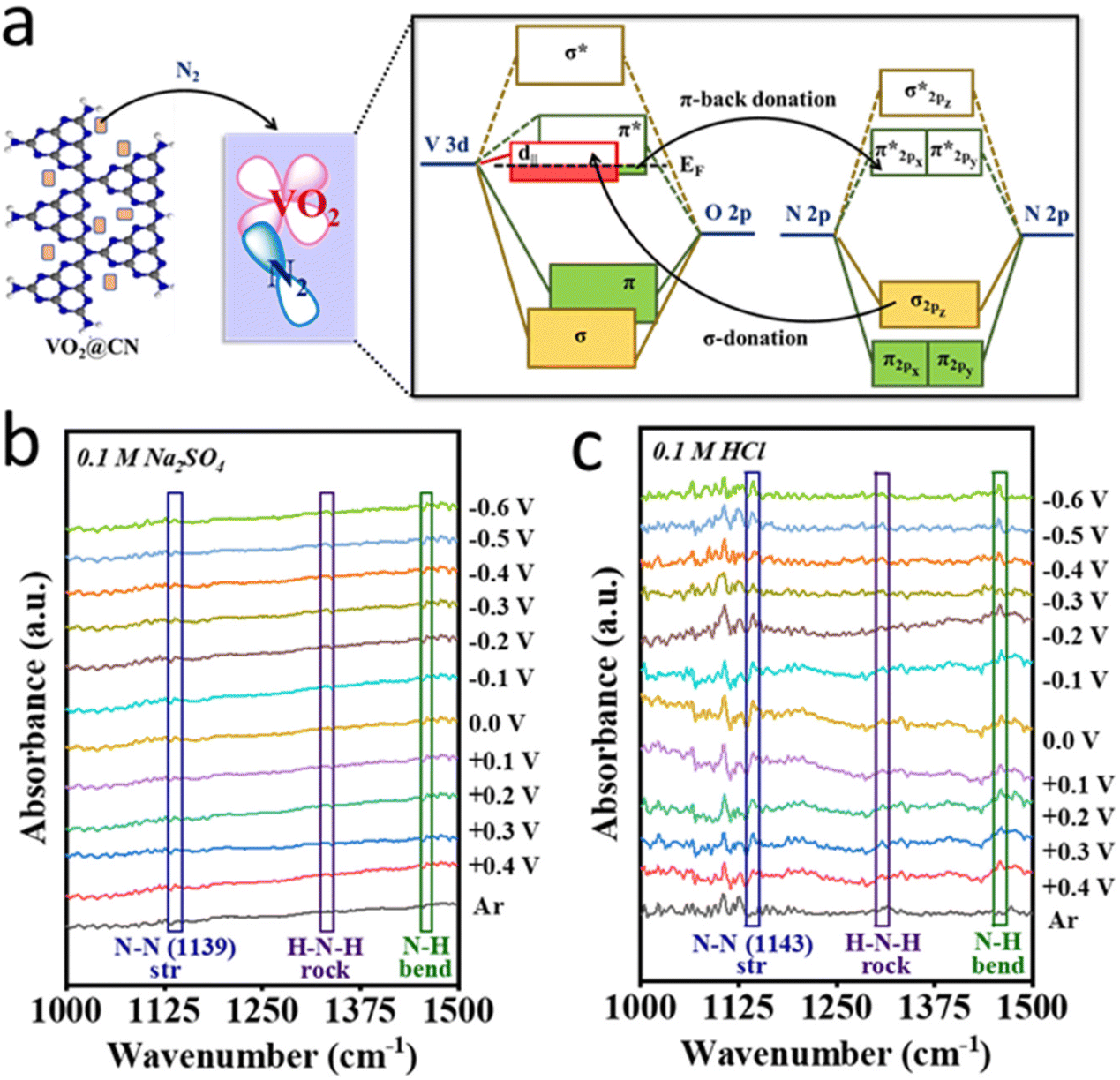 | ||
| Fig. 5 (a) Orbital interaction between the VO2 part of VO2@CN and N2 in terms of a molecular orbital diagram; unprocessed in situ ATR-FTIR spectra of the catalyst at different working potentials under N2- and Ar-purged conditions in (b) 0.1 M Na2SO4 and (c) 0.1 M HCl, respectively. | ||
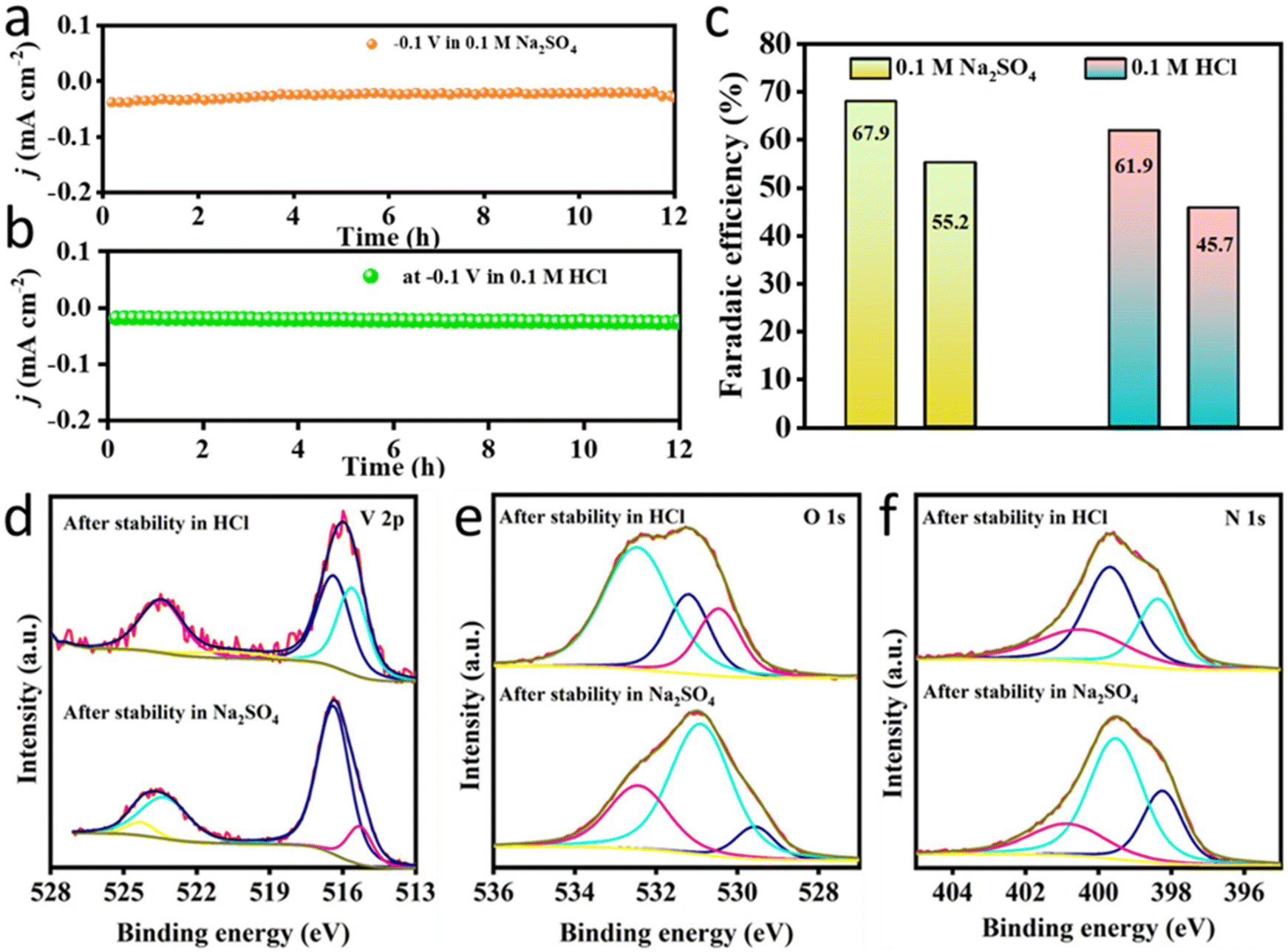 | ||
| Fig. 6 (a and b) CA response of VO2@CN during the NRR at −0.1 V vs. RHE in 0.1 M Na2SO4 and 0.1 M HCl, respectively, for a continuous 12 h period; (c) FE comparison of the catalyst after 1 and 12 h of NRR studies in both electrolytes; post-NRR XPS profiles for (d) V 2p, (e) O 1s and (f) N 1s, respectively, under HCl and Na2SO4 electrolyte conditions. | ||
XPS studies were conducted for VO2@CN samples after the electrochemical NRR stability experiments under HCl and Na2SO4, respectively, and compared with the pristine sample (Fig. 3a–c and 6d–f). The XPS data reveal minor alterations in the surface structure of the VO2@CN catalyst after electrochemical NRR. Under both pH conditions, the V 2p spectrum shifts towards a slightly lower binding energy, indicative of the removal of the surface oxidized V+5 (BE 516.4 eV in post-NRR samples vs. 516.8 eV in pristine) species that was observed in the pristine sample (Fig. 3a). In all cases, the pronounced V 2p3/2 maxima around the binding energy of 516 eV indicated V+4 as the major species (Fig. 6d).53 However, there is a reduction in the atomic percentage of vanadium in the post-NRR samples, which can be ascribed to the leaching of VO2 species in solution during the NRR. There is a significant increment in the post-NRR intensity of the O 1s signals around the binding energies of ∼531 eV and 532.4 eV ascribed to the prominence of oxide and adsorbed hydroxyl species, respectively (Fig. 6e). Unlike the case of the oxynitride species reported in the literature, the shift of the N 1s signal to lower binding energies (i.e. towards 396.5 eV)32 and any significant depletion in the N content of N-doped C were not observed in our case (Fig. 6f). There was no evidence of N-incorporation in VO2@CN from the N2-saturated electrolyte under NRR conditions, consistent with the literature reports.54 It should be mentioned here that the changes in the core level spectra of V 2p, O 1s and N 1s are induced by cathodic polarizations in HCl and Na2SO4 electrolytes, and not due to exposure under ambient conditions.55 The greater ease in protonation of the surface-bound N2 on vanadium oxide suggests that the increased acidity of the vanadium active site bound to the electronegative oxygen atoms and encapsulated in N-doped carbon stabilized the intermediates of the NRR and facilitated dinitrogen cleavage. This trend has been mentioned for other metals with high oxophilicity, including NbO2, which exhibited a high FE for the NRR.56 Thus, it can be concluded that there are several factors that rule the choice of electrolyte in the NRR. It is not a good practice to go with the catalyst performance for a 1 h potential-dependent study as the electrolyte might have a poisoning effect on the catalyst active site by either blocking or causing leaching of the active centre. This fact has relevance with respect to this study as it reveals that although the yield of NH3 was better in the acidic electrolyte, Na2SO4 stood out as a better contender in terms of FE and the stability study of the VO2@CN catalyst.
Experimental section
Synthesis of VO2@CN
4 g of melamine was added to a 250 mL round bottomed flask containing 50 mL of methanol. The solution was then stirred at room temperature for about 30 minutes. 300 mg of vanadium pentoxide dissolved in 20 mL of 3 M nitric acid was added to the solution containing melamine under continuous stirring. The reaction mixture was stirred for another hour under ambient conditions. The precipitate was evaporated to dryness.The obtained VO2@melamine (VO2@mel) was transferred into an alumina boat placed inside a tubular furnace and heated to 600 °C under continuous nitrogen flow for 3 h to obtain a black powder named VO2@CN.
Conclusions
In our quest to identify an inexpensive, Earth-abundant material as a universal-pH electrocatalyst for the NRR, we explored vanadium oxide encased in N-doped carbon, an Earth-abundant transition metal oxide that is synthesizable in bulk quantities. The data reported here designate VO2@CN as an efficient and selective electrocatalyst for nitrogen fixation with a high NH3 yield of 0.31 and 0.52 μmol h−1 mgcat−1 and a maximum FE of 67.9% and 61.9% at −0.1 V vs. RHE, under neutral and acidic conditions, respectively, without any observable incorporation of nitrogen into the vanadium oxide lattice. Furthermore, in situ ATR-FTIR studies at different potentials under N2 and Ar revealed the obscured reaction intermediates of the NRR and justified the differential activity trend of VO2@CN under both electrolyte conditions, owing to the possible impact of the competitive HER. In addition, the selectivity of the material was ascertained from the absence of side-products like hydrazine and suppression of the competing hydrogen evolution reaction. Our results demonstrate the competence of vanadium oxide on N-doped carbon as an electrocatalyst for the NRR under variable pH conditions, and substantiates oxophilicity as an important factor in designing electrocatalysts.Author contributions
AC: methodology, formal analysis, data curation (synthesis and characterization), and writing – original draft, AB: methodology, formal analysis, data curation (electrocatalytic NRR), and writing – original draft. SP: data curation (characterization) and writing – review and editing. RSD: funding acquisition, supervision (AB), and writing – review and editing. JM: funding acquisition, supervision (AC and SP), writing – original draft, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JM acknowledges DST SERB (SPG/2021/004430) and RSD acknowledges DST SERB (CRG/2020/005683) and BRE-BRNS (58/14/19/2023-BRNS) for financial assistance. Analytical support from AESD&CIF, CSMCRI and INST is gratefully acknowledged. CSMCRI PRIS No. 149/2023.References
- R. F. Service, Science, 2014, 345, 610–610 CrossRef CAS PubMed.
- W. Qiu, X. Y. Xie, J. Qiu, W. H. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef.
- M. H. Hasan, T. M. I. Mahlia, M. Mofijur, I. M. R. Fattah, F. Handayani, H. C. Ong and A. S. Silitonga, Energies, 2021, 14, 3732 CrossRef CAS.
- B. H. R. Suryanto, H. Du, D. Wang, J. Chen, A. N. Simonov and D. R. Macfarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- G.-F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L.-X. Ding and H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- J. Pan, H. A. Hansen and T. Vegge, J. Mater. Chem. A, 2020, 8, 24098 RSC.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- W. Ye, M. Arif, X. Fang, M. A. Mushtaq, X. Chen and D. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 28809–28817 CrossRef CAS PubMed.
- A. Good, Science, 2018, 359, 869–870 CrossRef CAS PubMed.
- M. Yang, T. Wei, J. He, Q. Liu, L. Feng, H. Li, J. Luo and X. Liu, Nano Res., 2024, 17, 1209–1216 CrossRef CAS.
- G. Zhang, G. Wang, Y. Wan, X. Liu and K. Chu, ACS Nano, 2023, 17, 21328–21336 CrossRef PubMed.
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- X. Han, C. S. Gerke, S. Banerjee, M. Zubair, J. Jiang, N. M. Bedford, E. M. Miller and V. S. Thoi, ACS Energy Lett., 2020, 5, 3237–3243 CrossRef CAS.
- U. K. Ghorai, S. Paul, B. Ghorai, A. Adaldar, S. Kapse, R. Thapa, A. Nagendra and A. Gain, ACS Nano, 2021, 15, 5230–5239 CrossRef CAS PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- M. Arif, G. Yasin, L. Luo, W. Ye, M. A. Mushtaq, X. Fang, X. Xiang, S. Ji and D. Yan, Appl. Catal., B, 2020, 265, 118559 CrossRef CAS.
- S. Chen, G. Qi, R. Yin, Q. Liu, L. Feng, X. Feng, G. Hu, J. Luo, X. Liu and W. Liu, Nanoscale, 2023, 15, 19577–19585 RSC.
- K. Chen, J. Xiang, Y. Guo, X. Liu, X. Li and K. Chu, Nano Lett., 2024, 24, 541–548 CrossRef CAS PubMed.
- J. Zhao, L. Zhang, X. Y. Xie, X. Li, Y. Ma, Q. Liu, W. H. Fang, X. Shi, G. Cui and X. Sun, J. Mater. Chem. A, 2018, 6, 24031–24035 RSC.
- H. Tao, C. Choi, L. X. Ding, Z. Jiang, Z. Han, M. Jia, Q. Fan, Y. Gao, H. Wang, A. W. Robertson, S. Hong, Y. Jung, S. Liu and Z. Sun, Chem, 2019, 5, 204–214 CAS.
- M. Nazemia, S. R. Panikkanvalappila and M. A. El-Sayed, Nano Energy, 2018, 49, 316–323 CrossRef.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- P. Chukwunenye, A. Ganesan, M. Gharaee, K. Balogun, F. Anwar, Q. Adesope, T. R. Cundari, F. D'Souza and J. A. Kelber, J. Mater. Chem. A, 2022, 10, 21401 RSC.
- R. Manjunatha, A. Karajić, V. Goldstein and A. Schechter, ACS Appl. Mater. Interfaces, 2019, 11, 7981–7989 CrossRef CAS PubMed.
- Z. Qiao, D. Johnson and A. Djire, Cell Rep., 2021, 2, 100438 CAS.
- L. Wang, Y. Liu, H. Wang, T. Yang, Y. Luo, S. Lee, M. G. Kim, T. T. T. Nga, C.-L. Dong and H. Lee, ACS Nano, 2023, 17, 7406–7416 CrossRef CAS PubMed.
- W. Li, Y. Ye, M. Jin, S. Zhang, C. Lin, C. Sun, Y. Zhang, G. Wang, C. Liang and H. Zhang, Chem. Eng. J., 2023, 451, 139494 CrossRef.
- Y. Hu, C. C. Lee and M. W. Ribbe, Dalton Trans., 2012, 41, 1118–1127 RSC.
- C. Zhang, D. Wang, Y. Wan, R. Lv, S. Li, B. Li, X. Zou and S. Yang, Mater. Today, 2020, 40, 18–25 CrossRef CAS.
- X. Li, L. Li, X. Ren, D. Wu, Y. Zhang, H. Ma, X. Sun, B. Du, Q. Wei and B. Li, Nano Res., 2020, 13, 2967–2972 CrossRef.
- A. Osonkie, A. Ganesan, P. Chukwunenye, F. Anwar, K. Balogun, M. Gharaee, I. Rashed, T. R. Cundari, F. D'Souza and J. A. Kelber, ACS Appl. Mater. Interfaces, 2022, 14, 531–542 CrossRef CAS PubMed.
- K. Balogun, A. Ganesan, P. Chukwunenye, M. Gharaee, Q. Adesope, S. Nemšák, P. S. Bagus, T. R. Cundari, F. D'Souza and J. A. Kelber, J. Phys.: Condens. Matter, 2023, 35, 333002 CrossRef PubMed.
- L.-H. Zhang, F. Yu and N. R. Shiju, ACS Sustainable Chem. Eng., 2021, 9, 7687–7703 CrossRef CAS.
- M. Privman and T. Hepel, J. Electroanal. Chem., 1995, 382, 137–144 CrossRef.
- A. Wen, Z. Cai, Y. Zhang and H. Liu, RSC Adv., 2022, 12, 13093–13103 RSC.
- D. Sellamnn, Angew. Chem., Int. Ed. Engl., 1974, 13, 639–649 CrossRef.
- J. Hou, Z. Wang, Z. Ding, Z. Zhang and J. Zhang, Sol. Energy Mater. Sol. Cells, 2018, 176, 142–149 CrossRef CAS.
- S. Liang, Q. Shi, H. Zhu, B. Peng and W. Huang, ACS Omega, 2016, 1, 1139–1148 CrossRef CAS PubMed.
- A. Misra, P. K. Tyagi, M. K. Singh and D. S. Misra, Diamond Relat. Mater., 2006, 15, 385–388 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- S.-Z. Huang, Y. Cai, J. Jin, Y. Li, X.-F. Zheng, H.-E. Wang, M. Wu, L.-H. Chen and B.-L. Su, J. Mater. Chem. A, 2014, 2, 14099–14108 RSC.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- F. D'Souza, A. Ganesan, A. Osonkie, P. Chukwunenye, I. Rashed, F. Anwar, M. Gharee, K. Balogun, T. R. Cundari and J. A. Kelber, ECS Meeting Abstracts, 2022, MA2022-01(45), 1893–1893.
- A. Biswas, S. Kapse, R. Thapa and R. S. Dey, Nano-Micro Lett., 2022, 14, 214 CrossRef CAS PubMed.
- A. Biswas, S. Kapse, B. Ghosh, R. Thapa and R. S. Dey, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204638119 CrossRef CAS PubMed.
- S. C. Jesudass, S. Surendran, J. Y. Kim, T.-Y. An, G. Janani, T.-H. Kim, J. K. Kim and U. Sim, Electrochem. Energy Rev., 2023, 6, 27 CrossRef CAS.
- W. Huang, L. Y. Peng, J. Zhang, C. Liu, G. Song, J. H. Su, W. H. Fang, G. Cui and S. Hu, J. Am. Chem. Soc., 2023, 145, 811–821 CrossRef CAS PubMed.
- N. B. Aetukuri, A. X. Gray, M. Drouard, M. Cossale, L. Gao, A. H. Reid, R. Kukreja, H. Ohldag, C. A. Jenkins, E. Arenholz, K. P. Roche, H. A. Dürr, M. G. Samant and S. S. P. Parkin, Nat. Phys., 2013, 9, 661–666 Search PubMed.
- S. Bhardwaj, S. K. Das, A. Biswas, S. Kapse, R. Thapa and R. S. Dey, Chem. Sci., 2023, 14, 8936–8945 RSC.
- R. Manjunatha, A. Karajić, H. Teller, K. Nicoara and A. Schechter, ChemCatChem, 2020, 12, 438–443 CrossRef CAS.
- Y. Abghoui and E. Skúlason, J. Phys. Chem. C, 2017, 121, 6141–6151 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- A. Ganesan, A. Osonkie, P. Chukwunenye, I. Rashed, T. R. Cundari, F. D'Souza and J. A. Kelber, J. Electrochem. Soc., 2021, 168, 026504 CrossRef CAS.
- A. Glaser, S. Surnev, F. P. Netzer, N. Fateh, G. A. Fontalvo and C. Mitterer, Surf. Sci., 2007, 601, 1153–1159 CrossRef CAS.
- L. Huang, J. Wu, P. Han, A. M. Al-Enizi, T. M. Almutairi, L. Zhang and G. Zheng, Small Methods, 2019, 3, 1800386 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00640b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |