
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization versus competing de-metalation, trans-metalation and (cyclo)-dehydrogenation of Pd porphyrins at a copper surface†
Stefania
Baronio
a,
Mattia
Bassotti
 b,
Francesco
Armillotta
ac,
Eleanor
Frampton
d,
Nikolay A.
Vinogradov
d,
Luca
Schio
e,
Luca
Floreano
e,
Alberto
Verdini
*be and
Erik
Vesselli
*aef
b,
Francesco
Armillotta
ac,
Eleanor
Frampton
d,
Nikolay A.
Vinogradov
d,
Luca
Schio
e,
Luca
Floreano
e,
Alberto
Verdini
*be and
Erik
Vesselli
*aef
aDepartment of Physics, University of Trieste, Trieste, Italy
bDepartment of Physics, University of Perugia, Perugia, Italy
cInstitute of Physics Ecole Polytechnique Fédérale de Lausanne (EPFL) Station 3, Lausanne, Switzerland
dMAX IV Laboratory, Lund, Sweden
eCNR-IOM – Istituto Officina dei Materiali, Area Science Park, Trieste, Italy. E-mail: evesselli@units.it; verdini@iom.cnr.it
fCenter for Energy, Environment and Transport Giacomo Ciamician, University of Trieste, Trieste, Italy
First published on 18th June 2024
Abstract
Metal-porphyrins are studied intensively due their potential applications, deriving from the variety of electronic and chemical properties, tunable by selecting metal centers and functional groups. Metalation, de- and trans-metalation processes are fundamental in this sense to investigate both the synthesis and the stability of these molecular building blocks. More specifically, Pd coordination in tetrapyrroles revealed to be potentially interesting in the fields of cancer therapy, drug delivery and light harvesting. Thus, we focused on the stability of palladium tetraphenyl porphyrins (PdTPPs) on a copper surface by means of combined spectroscopy and microscopy approaches. We find that PdTPPs undergo coverage-dependent trans-metalation accompanied by steric rearrangements already at room temperature, and fully trans-metalate to CuTPPs upon mild annealing. Side reactions such as (cyclo)-dehydrogenation and structural reorganization affect the molecular layer, with Pd–Cu alloying and segregation occurring at higher temperature. Instead, oxygen passivation of the Cu support prevents the metal-involving reactions, thus preserving the layer and increasing the chemical and temperature stability of the Pd porphyrins.
Introduction
Porphyrins have been extensively studied due to their ability to host a tetra-coordinated single metal atom center and self-assemble in 2D ordered layers at surfaces.1,2 The variety of different metal atoms that can be coordinated and the tunability offered by the substitutional groups make them versatile building blocks to create tailored materials with potential applications in different fields including gas sensing,3 catalysis (as single atom catalysts),4,5 light harvesting,6 and spintronics.7 While self-metalation at surfaces is a well-established method to synthesize metalated porphyrins at the solid-vacuum interface,8 trans-metalation, where the metal hosted by the macrocycle is exchanged with a different metal species, is a less common process. At variance with generally observed reactions in liquid environments, a few cases were reported recently, showing that it is possible to promote a redox metal exchange reaction also at a surface in vacuo, where the metal atom is ejected from the macrocycle, reduced, and replaced by a surface ad-atom that gets oxidized.9–13 The investigation of the surface reactions of porphyrins (including side-reactions like (cyclo)-dehydrogenation) is fundamental to yield comprehensive control over their structural and electronic properties.Attracted by their important potential role in the fields of cancer therapy,14 drug delivery,15 and photo-sensitizing in association with their long-lived triplet states,16 we focused on Pd porphyrins. In particular, after investigating the metalation process,17 we focus here on the de- and trans-metalation behavior of PdTPPs on a Cu(100) surface, by means of combined spectroscopic and microscopic approaches under ultra-high vacuum (UHV) conditions, exploiting synchrotron radiation X-ray Photoelectron Spectroscopy (XPS), Near-Edge X-ray Absorption Fine Structure spectroscopy (NEXAFS), and Scanning-Tunneling Microscopy (STM). The core levels reveal that the Pd atom is spontaneously ejected from the molecules already at room temperature (RT), at least for a (coverage dependent) fraction of the molecular population. Following, due to the unsaturated N bonds, coordination with the supporting copper metal surface occurs, similarly to previous cases of self-metalation of tetraphenyl porphyrins at the Cu(111) termination.18,19 Mild annealing further promotes this process, accompanied by a realignment of the molecule with respect to the surface main crystallographic axes, while the vacant site in the macrocycle cage is promptly filled by a Cu atom caught from the surface. NEXAFS and STM provide information on the adsorption geometries, proving flat adsorption of the tetrapyrroles. Two thermally-driven side reactions take also place, i.e. progressive (cyclo)-dehydrogenation and Pd migration into the bulk. Finally, we show that it is possible to prevent and control the de- and trans-metalation processes up to 500 K by pre-oxidation of the metal surface, e.g. by creating a decoupling (2√2 × 2√2)R45° O–Cu(100) layer. Annealing beyond 500 K causes instead oxygen reaction with the hydrogen terminations, again yielding degradation of the layer.
Experimental
The XPS and NEXAFS data were collected at the ALOISA CNR-IOM beamline at the Elettra synchrotron radiation facility in Trieste (Italy). STM measurements were instead performed at the STM Lab of the MAX IV synchrotron (Lund, Sweden). All experiments were performed in UHV, both setups having a base pressure in the low 10−10 mbar range, on two physically different Cu single crystals for the spectroscopy and microscopy measurements, respectively. The Cu(100) single crystals were cleaned by repeated cycles of Ar+ sputtering (2 keV) followed by annealing at 770 K, with residual adsorbed contaminants (O and CO) below the % ML range (Fig. S1†). Porphyrins were purchased from PorphyChem (Dijon-France – purity 98%) and deposited upon sublimation from a hot crucible held at 593 K with the sample kept at RT (unless otherwise specified). The evaporation rate was calibrated by means of a quartz microbalance and films were grown at typical rates of ∼0.2–0.3 Å min−1 at the sample surface. Following established recipes,7 the (2√2 × 2√2)R45° O–Cu(100) surface with 0.5 ML oxygen coverage was prepared by dosing 1200 L O2 (2 × 10−6 mbar O2 for 13.5 minutes) keeping the sample at 575 K. Surface symmetry and order were then confirmed by Reflection High Energy Electron Diffraction (RHEED). The core level measurements were performed at normal emission and grazing incidence (4.0°) with photon energies of 515 eV (overall resolution of ∼160 meV) for N and C 1s, Pd 3d and Cu 3p, and 660 eV (overall resolution of ∼190 meV) for O 1s and Cu 3p core levels, respectively. The bulk Cu 3p5/2 peak position (75.1 eV) was used as a reference for the alignment of the binding energy scale.20 N 1s spectra were fitted with Voigt profiles and superimposed on a linear background, while metallic Pd and Cu core levels were fitted with Doniach-Šunjić functions,21 convoluted with Gaussian envelopes to account for thermal and inhomogeneity broadening, and for the experimental resolution. The two lineshapes differ substantially in the asymmetry, which is associated with the non-adiabaticity of the photoemission process in metals, characterized by a continuum of states close to the Fermi level. NEXFAS measurements of the N and C K-edges were performed in partial electron yield (PEY) mode by means of a channeltron equipped with a grid polarized at negative bias to reject low energy secondary electrons (−250 V and −370 V for the C and K-edges, respectively). The polarization was changed from Transverse Magnetic (close to p-polarization) to Transverse Electric (s-polarization) by rotating the sample around the beam axis, with the incidence angle kept fixed at 6.0°.22,23 Reference NEXAFS spectra were measured on both Cu(100) and O–Cu(100) surfaces, which were subtracted to spectra measured on the molecular films for renormalization.23 Absolute photon energy calibration was obtained a posteriori from the characteristic absorption features of the I0 (drain current on last refocusing mirror) that are referenced to the gas phase absorption fine structure, as measured simultaneously with an in-line window-less gas ionization cell.24 The drain current was also used to take into account photon beam flux and fluctuations. All spectroscopy measurements were performed at RT, while annealing was performed by keeping the sample at the target temperature for a few seconds in the case of XPS and for 5 minutes for NEXAFS. The STM measurements were performed with a commercial VT UHV XA STM from Scienta-Omicron, GmbH, with electrochemically etched W tips purchased from Scienta-Omicron, GmbH. For these measurements, the samples were prepared in situ by evaporation of PdTPP molecules from a BN crucible onto a clean Cu(100) substrate at a rate of ∼0.01 ML min−1, calibrated with preliminary low-resolution STM imaging. During the PdTPP deposition, the substrate was held at room temperature (∼295 K), except for the measurement reported in Fig. S5,† where PdTPPs were deposited with the sample held at ∼100 K. For imaging, the copper substrate was cooled to 110 K. For the stepwise annealing series, the sample was placed on a manipulator in the preparation chamber (base pressure ∼ 1 × 10−10 mbar) and heated radiatively using a hot W filament mounted on a manipulator behind the sample. The temperature of the sample on the manipulator was monitored with a K-type thermocouple, spotwelded to the sample. The temperature in the STM scanning stage was estimated with a Pt/Ir thermocouple on the sample receiver. The raw STM data were processed with Gwyddion software.25 The image processing routine included global plane levelling, bow correction and sharpening for improved visual representation. In some of the images we have applied a polynomial distortion to the data to compensate for the effects of the thermal drift and piezo scanner creep.Results and discussion
Interaction with the bare metal surface
Fig. 1 reports the spectroscopic investigation of the stability and reactivity of a PdTPP monolayer on Cu(100), which was initially performed by measuring the temperature-related evolution of the Pd 3d5/2, N and C 1s core levels. The complete temperature-dependent sequence of the binding energy region covering the entire Pd 3d spin–orbit doublet is shown in panel (a) and was obtained upon linear annealing (0.1 K s−1) of the surface. Panel (b) depicts high energy resolution spectra of the full core level set that were measured at RT after stepwise annealing at the indicated temperature values in a separate experiment (the complete sequence is plotted in Fig. S2,† while Table S1† reports the relative intensities of each component for a quantitative comparison). In the Pd 3d5/2 spectrum of the as-prepared layer (RT), two components can be resolved at 335.9 and 338.6 eV, respectively. The latter, most intense peak (blue) is associated with Pd(II) coordinated by the four N atoms in the porphyrin macrocycle, as suggested in previous XPS measurements of Pd-metalated tetrapyrroles.12,26,27 Instead, the former component (green), at lower binding energy, is associated with Pd(0) surface atoms at the Cu termination.28–30 The presence of ejected single Pd atoms indicates that the porphyrins have undergone partial, spontaneous loss of the metal center already upon deposition on Cu(100) at RT. | ||
| Fig. 1 Evolution of a monolayer of PdTPP/Cu(100) as a function of temperature: (a) temperature-dependent Pd 3d core level spectra upon linear annealing; (b) N 1s, Pd 3d5/2 and C 1s core levels collected at RT after stepwise annealing to the indicated temperature values (corresponding cut-lines in (a) are shown for best clarity). | ||
The fraction of tetrapyrroles having lost the inner Pd metal atom grows with increasing annealing temperature, reaching a close-to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentration ratio of the two porphyrin species at 595 K for the monolayer case. Based on the extensive studies of Pd films at Cu surfaces and Pd–Cu alloys available in the literature,28–31 it is known that annealing promotes Pd dissolution in the Cu bulk with the initial creation of surface Pd–Cu alloys. Pd migration into the Cu bulk is confirmed also in our case where, after annealing at 725 K, the overall Pd 3d5/2 core level signal decreases, with a new component (grey) growing at 337.2 eV associated with bulk-like Pd atoms embedded in the first Cu layers. Interestingly, we notice from the temperature-dependent core level signal intensities (Fig. 1a, S2 and 3†) that the conversion reaction is promoted by both the thermalization process upon adsorption of the PdTPPs on the surface and by the surface temperature, with a critical role played by the surface coverage (Fig. S4†), yielding close to complete conversion in the zero-coverage limit (as it will be evident from the STM measurements). While thermalization contributes to the observed initial fraction of the tetrapyrroles having lost the inner Pd atom, surface temperature further promotes conversion upon heating. The temperature-dependent intensity behavior of the Pd core level components extracted from the data of Fig. 1a (plotted in Fig. S3†) allowed to evaluate the initial de-metalation energy barrier by means of the Redhead model equation,32 yielding a value of 1.5 eV for a porphyrin monolayer. In parallel, the integrated N and C 1s signal intensities remain unaffected by temperature, thus excluding desorption of the porphyrins. Herritsch et al. suggested that the reduction of the metal center in a porphyrin can occur also without de-metalation, since it could also be caused by an increased charge transfer from the surface induced by a geometrical distortion of the porphyrin.9 In our case, however, XPS clearly indicates that the new Pd(0) component stems from Pd adatoms. Thus, at this stage of our discussion, Pd-loss is initiated as a first step towards trans-metalation already at room temperature.
:1 concentration ratio of the two porphyrin species at 595 K for the monolayer case. Based on the extensive studies of Pd films at Cu surfaces and Pd–Cu alloys available in the literature,28–31 it is known that annealing promotes Pd dissolution in the Cu bulk with the initial creation of surface Pd–Cu alloys. Pd migration into the Cu bulk is confirmed also in our case where, after annealing at 725 K, the overall Pd 3d5/2 core level signal decreases, with a new component (grey) growing at 337.2 eV associated with bulk-like Pd atoms embedded in the first Cu layers. Interestingly, we notice from the temperature-dependent core level signal intensities (Fig. 1a, S2 and 3†) that the conversion reaction is promoted by both the thermalization process upon adsorption of the PdTPPs on the surface and by the surface temperature, with a critical role played by the surface coverage (Fig. S4†), yielding close to complete conversion in the zero-coverage limit (as it will be evident from the STM measurements). While thermalization contributes to the observed initial fraction of the tetrapyrroles having lost the inner Pd atom, surface temperature further promotes conversion upon heating. The temperature-dependent intensity behavior of the Pd core level components extracted from the data of Fig. 1a (plotted in Fig. S3†) allowed to evaluate the initial de-metalation energy barrier by means of the Redhead model equation,32 yielding a value of 1.5 eV for a porphyrin monolayer. In parallel, the integrated N and C 1s signal intensities remain unaffected by temperature, thus excluding desorption of the porphyrins. Herritsch et al. suggested that the reduction of the metal center in a porphyrin can occur also without de-metalation, since it could also be caused by an increased charge transfer from the surface induced by a geometrical distortion of the porphyrin.9 In our case, however, XPS clearly indicates that the new Pd(0) component stems from Pd adatoms. Thus, at this stage of our discussion, Pd-loss is initiated as a first step towards trans-metalation already at room temperature.
Detailed analysis and interpretation of the C and N 1s core level spectra are more challenging because of the presence of side reactions taking place in parallel with the Pd loss. Non-metalated 2HTPPs are known to display a double peak N 1s structure due to the presence of two inequivalent N species, pyrrolic –NH and iminic ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–, contributing spectroscopically at around 398 and 400 eV, respectively.33 On the contrary, M-TPPs (M is the central metal) have four chemically equivalent iminic N atoms, resulting in a single N 1s peak that is usually situated in between the –NH and N– spectral energy positions. Moreover, the energy of the metal-coordinated N feature can shift by as much as 1 eV, depending on the core metal species and on the interaction with the supporting substrate.13,19,33–36 Just to mention a few examples, it overlaps almost completely with the iminic N– peak at 398.4 eV for CuTPP/Cu(111) (and similar Cu-porphyrins)13,19,33 and just a few tenths of eV lower than the iminic nitrogen for M(II)TPP/TiO2 with M = Co, Ni, Cu, Zn,34 while it is positioned at about 399 eV for CoPc/Cu(111)35 and at 398.7 eV for FeTPP/Ag(111).36 In our case, after deposition at RT, a prominent N 1s feature is observed at 399 eV, with a minor shoulder found at 398.2 eV. Therefore, we can affirm that, when depositing a monolayer at RT, the PdTPPs mostly maintain their structure intact, while only a minor fraction loses the central Pd atom. Since self-metalation of 2HTPPs and other non-metalated porphyrins at Cu surfaces was already known to occur,18,19,37–39 a similar behavior could be expected also for de-metalated PdTPPs.9 Thus, we would expect a Cu atom to replace the ejected Pd in the macrocycle. In addition, the absence of the central hydrogen atoms, present in pristine empty porphyrins, allows skipping the first intermediate metalation steps described by Doyle et al.37 Röckert et al.18 performed a metalation experiment of 2HTPPs on Cu(111): they observed the growth of a N 1s component at 398.5 eV that shifts to 398.7 eV as metalation proceeds. In our case, the faint shoulder at 398.2 eV that we observe already at RT grows in intensity and progressively shifts up to 398.6 eV upon annealing. We associate this latter feature with the fraction of de-metalated porphyrins, which readily undergo trans-metalation into CuTPPs. The iminic nitrogen atoms interact with the Cu substrate, shifting the N 1s photoelectron line. Both N 1s components are affected by temperature-dependent shifts of the order of a few 100 s of meV, together with a significant gaussian broadening (from 0.7 eV to 0.9 eV) for the highest temperature values. This evolution is the result of a parallel stepwise (cyclo)-dehydrogenation reaction and of the consequent reconstruction of the carbon cage of the porphyrins for increasing temperature.40 Indeed, these spectroscopic changes of the N 1s components are paralleled by a corresponding evolution of the C 1s signal. Upon heating, the intensity of a feature at 284.4 ± 0.1 eV grows at the expense of the main RT peak located at 285.2 ± 0.1 eV (Fig. 1b and S2†). While the latter component is associated with the pristine molecules, the former unexpectedly parallels the trans-metalation steps. At high temperature these changes in the C 1s profile are known to witness the process of partial dehydrogenation and the fusion of the phenyl rings with the macrocycle, accompanied by the flattening of the phenyl rings. However, at low temperature it reflects the re-alignment of the porphyrins with respect to the main crystallographic axes (rotation by 45°) occurring upon trans-metalation (Fig. 2a and b). Overall, the entire process yields a stronger C–Cu interaction, contributing finally to the peak at 283.4 eV at the highest temperature (Fig. 2c).9,18 Furthermore, additional spectroscopic measurements reveal that the onset of the trans-metalation process is highly coverage-dependent (Fig. S4†). When a PdTPP multilayer (2–4 ML) is annealed at 475 K, the layers not in direct contact with the copper termination undergo desorption (Fig. S4a and b†),9 while only the PdTPP/Cu(100) undergo trans-metalation. In the specific case of a bilayer, the Pd(0) component is not observed below 475 K, but only at 555 K, after desorption of the second layer, when trans-metalation is suddenly complete. Thus, interestingly, multiple layers contribute in the stabilization of the pristine PdTPPs below the multilayer desorption temperature.
N–, contributing spectroscopically at around 398 and 400 eV, respectively.33 On the contrary, M-TPPs (M is the central metal) have four chemically equivalent iminic N atoms, resulting in a single N 1s peak that is usually situated in between the –NH and N– spectral energy positions. Moreover, the energy of the metal-coordinated N feature can shift by as much as 1 eV, depending on the core metal species and on the interaction with the supporting substrate.13,19,33–36 Just to mention a few examples, it overlaps almost completely with the iminic N– peak at 398.4 eV for CuTPP/Cu(111) (and similar Cu-porphyrins)13,19,33 and just a few tenths of eV lower than the iminic nitrogen for M(II)TPP/TiO2 with M = Co, Ni, Cu, Zn,34 while it is positioned at about 399 eV for CoPc/Cu(111)35 and at 398.7 eV for FeTPP/Ag(111).36 In our case, after deposition at RT, a prominent N 1s feature is observed at 399 eV, with a minor shoulder found at 398.2 eV. Therefore, we can affirm that, when depositing a monolayer at RT, the PdTPPs mostly maintain their structure intact, while only a minor fraction loses the central Pd atom. Since self-metalation of 2HTPPs and other non-metalated porphyrins at Cu surfaces was already known to occur,18,19,37–39 a similar behavior could be expected also for de-metalated PdTPPs.9 Thus, we would expect a Cu atom to replace the ejected Pd in the macrocycle. In addition, the absence of the central hydrogen atoms, present in pristine empty porphyrins, allows skipping the first intermediate metalation steps described by Doyle et al.37 Röckert et al.18 performed a metalation experiment of 2HTPPs on Cu(111): they observed the growth of a N 1s component at 398.5 eV that shifts to 398.7 eV as metalation proceeds. In our case, the faint shoulder at 398.2 eV that we observe already at RT grows in intensity and progressively shifts up to 398.6 eV upon annealing. We associate this latter feature with the fraction of de-metalated porphyrins, which readily undergo trans-metalation into CuTPPs. The iminic nitrogen atoms interact with the Cu substrate, shifting the N 1s photoelectron line. Both N 1s components are affected by temperature-dependent shifts of the order of a few 100 s of meV, together with a significant gaussian broadening (from 0.7 eV to 0.9 eV) for the highest temperature values. This evolution is the result of a parallel stepwise (cyclo)-dehydrogenation reaction and of the consequent reconstruction of the carbon cage of the porphyrins for increasing temperature.40 Indeed, these spectroscopic changes of the N 1s components are paralleled by a corresponding evolution of the C 1s signal. Upon heating, the intensity of a feature at 284.4 ± 0.1 eV grows at the expense of the main RT peak located at 285.2 ± 0.1 eV (Fig. 1b and S2†). While the latter component is associated with the pristine molecules, the former unexpectedly parallels the trans-metalation steps. At high temperature these changes in the C 1s profile are known to witness the process of partial dehydrogenation and the fusion of the phenyl rings with the macrocycle, accompanied by the flattening of the phenyl rings. However, at low temperature it reflects the re-alignment of the porphyrins with respect to the main crystallographic axes (rotation by 45°) occurring upon trans-metalation (Fig. 2a and b). Overall, the entire process yields a stronger C–Cu interaction, contributing finally to the peak at 283.4 eV at the highest temperature (Fig. 2c).9,18 Furthermore, additional spectroscopic measurements reveal that the onset of the trans-metalation process is highly coverage-dependent (Fig. S4†). When a PdTPP multilayer (2–4 ML) is annealed at 475 K, the layers not in direct contact with the copper termination undergo desorption (Fig. S4a and b†),9 while only the PdTPP/Cu(100) undergo trans-metalation. In the specific case of a bilayer, the Pd(0) component is not observed below 475 K, but only at 555 K, after desorption of the second layer, when trans-metalation is suddenly complete. Thus, interestingly, multiple layers contribute in the stabilization of the pristine PdTPPs below the multilayer desorption temperature.
 | ||
| Fig. 2 STM images of sub-monolayer PdTPP/Cu(100) collected at LN2 after RT deposition: (a) as-deposited and (b and c) after annealing at increasing temperature. Insets of ∼7 × 7 nm2 detail the observed nonequivalent porphyrin species. Lines put in evidence the molecular alignment with respect to the main crystallographic directions of the surface. Scale bar is 10 nm. Molecular pictorial models of the most representative surface species are reported on the right. Measuring parameters: (a) 42 × 42 nm2, Vbias = −2.01 V, It = 80 pA, (b) 40 × 40 nm2, Vbias = −2.03 V, It = 50 pA, inset Vbias = −2.02 V, It = 70 pA (c) 40 × 40 nm2, Vbias = −1.85 V, It = 100 pA. | ||
Summarizing the results obtained from the analysis of the XPS core level spectra, we can conclude already at this stage that due to the strong interaction between the PdTPPs and the Cu substrate, a small fraction of the Pd atoms is ejected from the macrocycle already at RT in the (close-to)-monolayer case, and the process is further promoted by annealing. The porphyrins are left with four unsaturated N bonds free to interact with the extremely reactive copper substrate, by distortion of the macrocycle and reduced adsorbate-substrate distance, and by successive metalation.18 XPS also reveals that there are other thermally driven processes occurring at this stage: Pd alloying with Cu and stepwise dehydrogenation with the consequent structural reconstruction of the porphyrins.
The picture depicted so far is complemented by the STM measurements (Fig. 2) that were conducted at LN2 after deposition at RT or after a 5-minutes annealing at increasing selected temperature values (panels a–c) as indicated by the labels. The measurements were performed in a sub-monolayer, very low coverage regime to facilitate the imaging of single molecules and to prevent their aggregation into islands. At low coverage, the lateral interactions are weak compared to the strong molecule-substrate forces.1,41 The insets depicted in each of the panels of Fig. 2 provide an enlarged view of the inequivalent species of porphyrins that could be clearly imaged and distinguished. Directly after deposition and, thus, at RT, (Fig. 2a), the dominant species (see Table S2 for imaging-related statistics†) displays a 2-fold symmetry with a bright macrocycle, while the phenyl lobes are somewhat dim (inset in Fig. 2a). The molecules are aligned with the high symmetry directions of the surface.9 A minority species (white circle in Fig. 2a) displays instead four visible lobes. These were attributed to the phenyl groups, coupled by a single less bright protrusion and a dim center. The asymmetric appearance of the phenyl groups is explained by their out-of-plane tilt angle,19,37 while the two-fold appearance of the macrocycle is attributed to the strong interaction with the substrate, inducing a typical saddle-shape geometrical distortion.9 The molecule appears to be rotated by 45° with respect to the main surface crystallographic axes. We associate this appearance with the still intact PdTPPs, being a rare species at RT in the zero-coverage limit, but dominant when depositing with the surface at LN2 temperature (see Fig. S5†). The majority species observed in STM at RT is associated instead to CuTPPs,18,42 indicating that the 45° rotation represents a crucial step in the trans-metalation process. Since when PdTPPs are deposited at LN2 temperature (Fig. S4†) metastable PdTPPs can be observed, the role of a “hot precursor” state has to be taken into account, favoring diffusion along with thermalization,43 thus overcoming the activation barriers for rotation and trans-metalation.44 The apparent rotation of the molecule occurring upon trans-metalation accounts in principle also for the coverage dependence of the process, which is hindered at high packing densities, possibly in association with steric barriers. This is evident also from the coverage-dependent XPS data shown in Fig. S4e.† Finally, the two majority species observed in STM after annealing to 425 and 525 K (Fig. 2b and c) are associated with the trans-metalated CuTPPs at different dehydrogenation stages, respectively. The molecules, shown in greater detail in the inset of Fig. 2b, show 4-fold symmetric appearance, 45° back-rotation and no bright protrusion at the center, as expected for CuTPPs.9,18 Their apparent height of ∼250 pm is considerably larger than that expected for saddle-type CuTPPs as in Fig. 2a (∼140 pm), which suggests a yet stronger distortion of the molecule. Such a 4-lobe symmetric shape has been previously reported for intermediate (0.35–0.50 ML) molecular coverages and was attributed to a porphyrin molecule in the second layer bridging two molecules in the first layer.45 However, our STM data unambiguously point that such tetraphenyl porphyrin conformation can appear at lower coverages in the absence of molecules underneath, as seen in Fig. 2b. We believe therefore that in our case (0.1–0.2 ML) upon annealing to 425 K such representation of porphyrin molecules can be explained by the pyrrolic moieties aligning with the macrocycle, while the tilt angle of the phenyl rings increases, as calculated by Diller et al.19 At this stage in this low coverage range dehydrogenation should not be favored yet, as the annealing temperature is still below the 520 K threshold reported for free-base TPPs on Cu(111).18 Further annealing accompanied by a partial dehydrogenation of the metal-porphyrins leads to a dramatic structural reorganization of the organic cage (Fig. 2c), as first described by Di Santo et al.46 and more recently by Herritsch et al.9 The phenyl groups bend towards the macrocycle ring and form unreversible C–C bonds, stabilizing the molecular structure, in agreement with both our C 1s spectra and the literature.9,18 Upon reaching the temperature of ∼525 K the dehydrogenation is almost complete. This contributes to a stronger interaction of the molecules with the substrate, inducing the observed torsional distortion of the organic cage. The inset in Fig. 2c shows partially dehydrogenated porphyrins distorted in a spiral-like shape. NEXAFS provides insight into the intramolecular bonding angles and directions, helping in the comprehension of the molecular adsorption geometry. Fig. 3 shows the N and C K-edges measured at RT before and after a 5-minutes annealing to 560 K. For best insight, we can compare these spectra to the N and C edges of self-metalated CuTPP and NiTPP molecules at Cu surfaces already reported in the literature.19,37,47 Similarly, after deposition at RT of the PdTPPs on Cu(100), both N and C edges show a pronounced dichroic behavior (bottom curves in the respective panels of Fig. 3). The N K-edge (top) displays two intense π resonances in the region below 404 eV, at 399 and 401 eV, associated with transitions from the 1s level to inequivalent LUMOs.19,47 These spectra display a significant distortion with respect to those of isolated metal(II)-porphyrins (e.g. multilayer ones),47 where the LUMO appears to be quenched and, as a consequence of electron injection, the higher energy unoccupied orbitals are shifted (and partially filled) to lower energy.48 The spectral shape as well as the peaks positions are almost identical to the NiTPP/Cu(100) spectra.47 As a consequence, the residual intensity of the lowest energy feature in s-pol may be interpreted as a distortion of the corresponding molecular orbital by rehybridization, due to the charge transfer from the metal substrate. This is compatible with the two-fold appearance of the porphyrins’ species imaged by STM in Fig. 2a and b. We observe also a clear evolution in the N edge spectra upon trans-metalation. However, the convolution of this effect with the lineshape changes associated with the dehydrogenation and re-alignment of the molecular plane makes it less diagnostic at this level. Concerning the carbon edge (bottom panel), we observe again a major change of the resonances with respect to those of isolated molecules, as reported in the literature. However, the charge transfer mostly affects the molecular orbitals localized on the pyrrolic carbon atoms (partially filled and shifted to lower energy). Thus, we can associate the features at lower energy (∼285 eV) and the isolated resonance at ∼287.5 eV with π*-symmetry molecular orbitals, while the sharpest resonance at 286 eV corresponds to the π* orbitals of the phenyl rings, which are practically unaffected by charge transfer, in good agreement with the NiTPP/Cu(100) case.47 The latter phenyl ring resonance is very intense in s-pol, confirming that the peripheral groups tend to bend out of plane upon adsorption at the Cu surface. Another pronounced resonance is observed at 287.5 eV, also in agreement with previous cases, but with no straightforward assignment.19,47 Upon annealing, the s-pol intensities of the π resonances are strongly quenched, confirming the progressively growing interaction strength with the substrate and the flattening of the molecular planes.
 | ||
| Fig. 3 N and C K-edge of a monolayer PdTPP/Cu(100), as-deposited and post-annealing in the lower and upper part of the graphs, respectively. Different colors refer to different polarizations of the photon beam with respect to the incidence plane. | ||
Preventing trans-metalation
So far, we demonstrated that PdTPPs on Cu(100) spontaneously de- and trans-metalate to CuTPPs, with dehydrogenation occurring upon heating. Considering the importance of Pd-containing tetrapyrroles addressed in the introduction, we investigated a possible stabilization mechanism. To this purpose, based on the literature, we exploited pre-oxidation of the copper substrate to hinder trans-metalation.7,47,49,50 Thus, we deposited the PdTPPs on the (2√2 × 2√2)R45°-O/Cu(100) termination, corresponding to 0.5 ML of atomic oxygen obtained by dissociative chemisorption of O2.51 In the XPS core level analysis (Fig. 4) we find that at RT the N 1s and Pd 3d5/2 spectra show a single sharp component, as expected for the pristine PdTPPs. Neither de-metalation, nor dehydrogenation occur at this surface upon adsorption, thus preventing reactivity during the adsorption-related thermalization process. The binding energies of the two features are slightly different compared to the case of the bare Cu surface. The different molecule-surface interaction induces line positions of 399.0 eV (−400 meV shift) for N–Pd and 338.1 eV (−500 meV shift) for Pd(II), respectively. Surface pre-oxidation is enough to maintain the structure intact up to 500 K (Fig. 4, S6 and Table S1†), with no observable changes in the core level spectra. Higher temperatures are required to promote reactivity on the CuO termination, triggering dehydrogenation and oxygen titration (O 1s spectra in Fig. 4 and S6†). At this stage, direct interaction of the molecules with the bare metallic copper substrate becomes accessible, as in the previous case. Pd loss starts above 500 K, again accompanied by the de- and trans-metalation steps, as witnessed by the evolution of the corresponding Pd 3d5/2, C and N 1s spectra (Fig. 4 and S6†). NEXAFS spectra collected at RT (Fig. S7†) after deposition of PdTPPs on the oxygen-covered Cu surface show the same resonances that were identified in the case of the direct molecule–Cu interaction, but with sharper profiles and stronger dichroic behavior, indicating weaker molecule–substrate interaction and flat adsorption.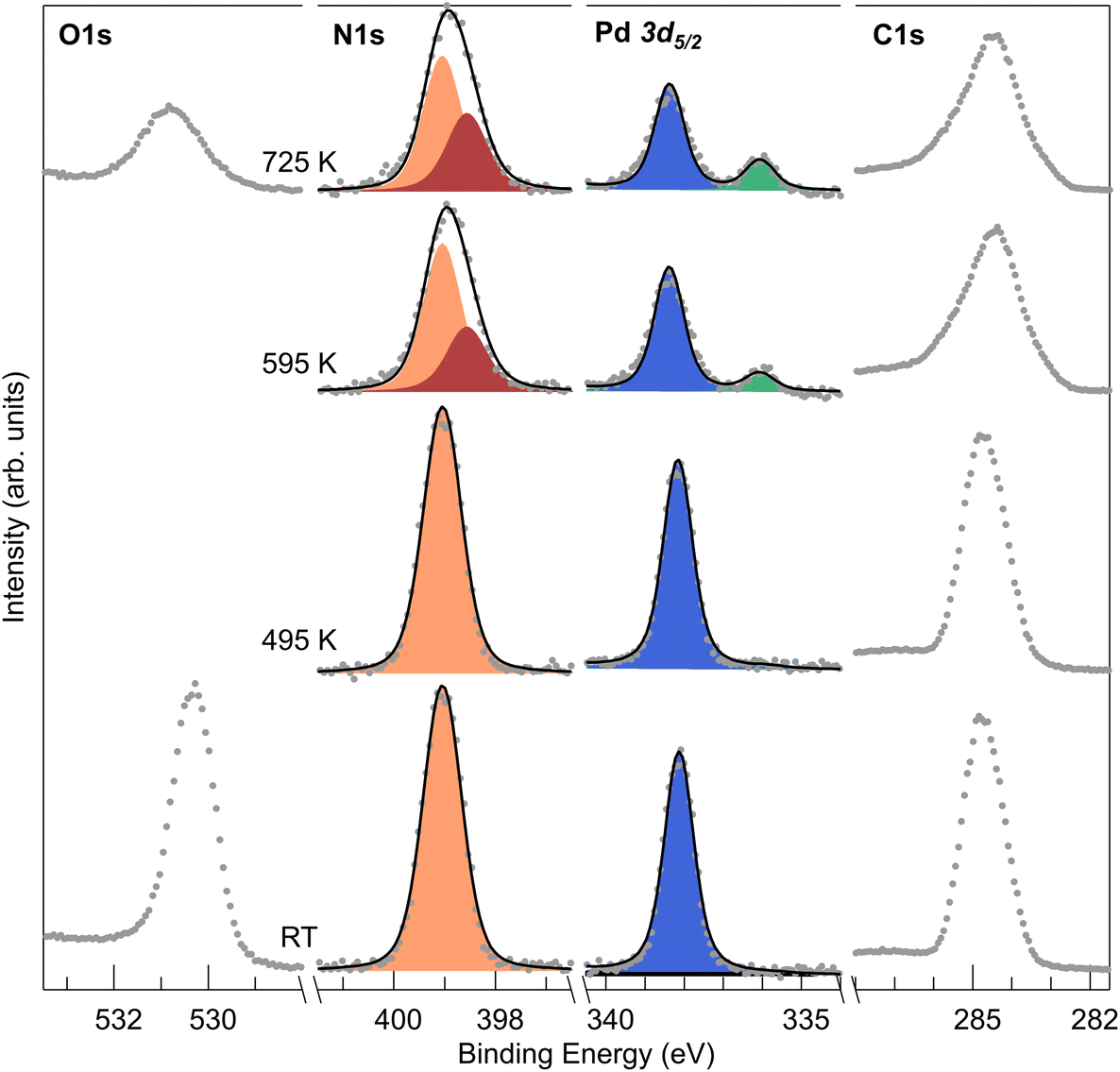 | ||
| Fig. 4 XPS measurements of a monolayer PdTPP/CuO. Evolution of N 1s, Pd 3d5/2 and C 1s measured at room temperature after annealing at the indicated T values. | ||
Conclusions
In conclusion, by means of spectroscopy and microscopy experimental approaches we have characterized the stability and reactivity of PdTPPs on the Cu(100) surface. While Pd porphyrins tend to be reactive already at and beyond room temperature, stabilization up to about 500 K of the pristine molecular form can be obtained by introducing a decoupling atomic oxygen layer. On the contrary, the strong molecule–metal surface interaction promotes de- and trans-metalation processes paralleled by the progressive dehydrogenation. The latter process yields a strong, direct C–Cu interaction and the deformation of the porphyrin backbone.Data availability
Data for this article, including raw microscopy metadata, are available at the OSF Home repository at https://osf.io/hvxnb/?view_only=fe42b052a66f457d917c33c0d3e97583.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. V. and E. V. acknowledge funding from the project “FERMAT – Fast ElectRon dynamics in novel hybrid organic-2D MATerials” funded by the MUR Progetti di ricerca di Rilevante Interesse Nazionale (PRIN) Bando 2017 – grant 2017KFY7XF. S. B. and E. V. acknowledge financial support by Unione Europea – Next Generation EU through project PRIN2022 XXJNRS “2DOrNotToBe”.References
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef.
- E. Vesselli, JPhys Mater., 2020, 3, 022002 CrossRef CAS.
- Z.-W. Hao, M.-M. Dong, R.-Q. Zhang, C.-K. Wang and X.-X. Fu, Phys. Chem. Chem. Phys., 2021, 23, 11852–11862 RSC.
- B. Wurster, D. Grumelli, D. Hötger, R. Gutzler and K. Kern, J. Am. Chem. Soc., 2016, 138, 3623–3626 CrossRef CAS.
- F. Armillotta, D. Bidoggia, S. Baronio, P. Biasin, A. Annese, M. Scardamaglia, S. Zhu, B. Bozzini, S. Modesti, M. Peressi and E. Vesselli, ACS Catal., 2022, 12, 7950–7959 CrossRef CAS.
- G. B. Bodedla, V. Piradi, M. Imran, J. Zhao, X. Zhu and W.-Y. Wong, J. Mater. Chem. A, 2023, 11, 1473–1481 RSC.
- H. M. Sturmeit, I. Cojocariu, A. Windischbacher, P. Puschnig, C. Piamonteze, M. Jugovac, A. Sala, C. Africh, G. Comelli, A. Cossaro, A. Verdini, L. Floreano, M. Stredansky, E. Vesselli, C. Hohner, M. Kettner, J. Libuda, C. M. Schneider, G. Zamborlini, M. Cinchetti and V. Feyer, Small, 2021, 17, 2104779 CrossRef CAS.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS.
- J. Herritsch, S. R. Kachel, Q. Fan, M. Hutter, L. J. Heuplick, F. Münster and J. M. Gottfried, Nanoscale, 2021, 13, 13241–13248 RSC.
- D. Hötger, P. Abufager, C. Morchutt, P. Alexa, D. Grumelli, J. Dreiser, S. Stepanow, P. Gambardella, H. F. Busnengo, M. Etzkorn, R. Gutzler and K. Kern, Nanoscale, 2018, 10, 21116–21122 RSC.
- G. Fratesi, D. Paoloni, L. Persichetti, L. Camilli, A. Caporale, A. Baby, D. Cvetko, G. Kladnik, A. Morgante and A. Ruocco, Inorg. Chim. Acta, 2024, 559, 121790 CrossRef CAS.
- K. Shen, B. Narsu, G. Ji, H. Sun, J. Hu, Z. Liang, X. Gao, H. Li, Z. Li, B. Song, Z. Jiang, H. Huang, J. W. Wells and F. Song, RSC Adv., 2017, 7, 13827–13835 RSC.
- C. M. Doyle, J. P. Cunniffe, S. A. Krasnikov, A. B. Preobrajenski, Z. Li, N. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2014, 50, 3447 RSC.
- J. Deng, H. Li, M. Yang and F. Wu, Photochem. Photobiol. Sci., 2020, 19, 905–912 CrossRef CAS.
- Y. Yu, Q. Xu, S. He, H. Xiong, Q. Zhang, W. Xu, V. Ricotta, L. Bai, Q. Zhang, Z. Yu, J. Ding, H. Xiao and D. Zhou, Coord. Chem. Rev., 2019, 387, 154–179 CrossRef CAS.
- Y. Gao, V. Piradi, X. Zhu and S. K. So, ACS Appl. Energy Mater., 2022, 5, 4916–4925 CrossRef CAS.
- F. Armillotta, E. D'Incecco, M. Corva, M. Stredansky, J.-J. Gallet, F. Bournel, A. Goldoni, A. Morgante, E. Vesselli and A. Verdini, Angew. Chem., Int. Ed., 2021, 60, 25988–25993 CrossRef CAS.
- M. Röckert, M. Franke, Q. Tariq, S. Ditze, M. Stark, P. Uffinger, D. Wechsler, U. Singh, J. Xiao, H. Marbach, H.-P. Steinrück and O. Lytken, Chem. – Eur. J., 2014, 20, 8948–8953 CrossRef.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS.
- F. Sedona, M. Di Marino, D. Forrer, A. Vittadini, M. Casarin, A. Cossaro, L. Floreano, A. Verdini and M. Sambi, Nat. Mater., 2012, 11, 970–977 CrossRef CAS.
- S. Doniach and M. Sunjic, J. Phys. C: Solid State Phys., 1970, 3, 285–291 CrossRef CAS.
- G. Bavdek, A. Cossaro, D. Cvetko, C. Africh, C. Blasetti, F. Esch, A. Morgante and L. Floreano, Langmuir, 2008, 24, 767–772 CrossRef CAS.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- L. Floreano, G. Naletto, D. Cvetko, R. Gotter, M. Malvezzi, L. Marassi, A. Morgante, A. Santaniello, A. Verdini, F. Tommasini and G. Tondello, Rev. Sci. Instrum., 1999, 70, 3855–3864 CrossRef CAS.
- D. Nečas and P. Klapetek, Open Phys., 2012, 10, 181–188 CrossRef.
- K. S. Lokesh and A. Adriaens, Dyes Pigm., 2013, 96, 269–277 CrossRef CAS.
- X. Zhang, L. Hou, A. Cnossen, A. C. Coleman, O. Ivashenko, P. Rudolf, B. J. van Wees, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2011, 17, 8957–8964 CrossRef CAS.
- J. P. Reilly, C. J. Barnes, N. J. Price, R. A. Bennett, S. Poulston, P. Stone and M. Bowker, J. Phys. Chem. B, 1999, 103, 6521–6532 CrossRef CAS.
- G. W. Graham, P. J. Schmitz and P. A. Thiel, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 3353–3359 CrossRef CAS.
- T. D. Pope, K. Griffiths and P. R. Norton, Surf. Sci., 1994, 306, 294–312 CrossRef CAS.
- J. Loboda-Cackovic, M. Mousa and J. Block, Vacuum, 1995, 46, 89–96 CrossRef CAS.
- P. Woodruff, Modern Techniques of Surface Science, Cambridge University Press, Cambridge, 2016, vol. 41 Search PubMed.
- J. Xiao, S. Ditze, M. Chen, F. Buchner, M. Stark, M. Drost, H. P. Steinrück, J. M. Gottfried and H. Marbach, J. Phys. Chem. C, 2012, 116, 12275–12282 CrossRef CAS.
- L. Schio, G. Bavdek, C. Grazioli, C. Gutiérrez Bolaños, A. Goldoni, A. Vittadini, M. Tormen and L. Floreano, Appl. Surf. Sci., 2023, 616, 156548 CrossRef CAS.
- E. Annese, J. Fujii, I. Vobornik and G. Rossi, J. Phys. Chem. C, 2011, 115, 17409–17416 CrossRef CAS.
- F. Buchner, K. Flechtner, Y. Bai, E. Zillner, I. Kellner, H.-P. Steinrück, H. Marbach and J. M. Gottfried, J. Phys. Chem. C, 2008, 112, 15458–15465 CrossRef CAS.
- C. M. Doyle, S. A. Krasnikov, N. N. Sergeeva, A. B. Preobrajenski, N. A. Vinogradov, Y. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2011, 47, 12134 RSC.
- R. González-Moreno, C. Sánchez-Sánchez, M. Trelka, R. Otero, A. Cossaro, A. Verdini, L. Floreano, M. Ruiz-Bermejo, A. García-Lekue, J. Á. Martín-Gago and C. Rogero, J. Phys. Chem. C, 2011, 115, 6849–6854 CrossRef.
- A. Verdini, P. Shinde, G. L. Montanari, S. T. Suran-Brunelli, M. Caputo, G. Di Santo, C. A. Pignedoli, L. Floreano, D. Passerone and A. Goldoni, Chem. – Eur. J., 2016, 22, 14672–14677 CrossRef CAS.
- F. Bischoff, A. Riss, G. S. Michelitsch, J. Ducke, J. V. Barth, K. Reuter and W. Auwärter, J. Am. Chem. Soc., 2021, 143, 15131–15138 CrossRef CAS.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- F. Albrecht, F. Bischoff, W. Auwärter, J. V. Barth and J. Repp, Nano Lett., 2016, 16, 7703–7709 CrossRef CAS.
- J. Harris and B. Kasemo, Surf. Sci. Lett., 1981, 105, L281–L287 CAS.
- H. Marbach and H.-P. Steinrück, Chem. Commun., 2014, 50, 9034–9048 RSC.
- M. Röckert, S. Ditze, M. Stark, J. Xiao, H.-P. Steinrück, H. Marbach and O. Lytken, J. Phys. Chem. C, 2014, 118, 1661–1667 CrossRef.
- G. Di Santo, S. Blankenburg, C. Castellarin-Cudia, M. Fanetti, P. Borghetti, L. Sangaletti, L. Floreano, A. Verdini, E. Magnano, F. Bondino, C. A. Pignedoli, M.-T. Nguyen, R. Gaspari, D. Passerone and A. Goldoni, Chem. – Eur. J., 2011, 17, 14354–14359 CrossRef CAS.
- I. Cojocariu, H. M. Sturmeit, G. Zamborlini, A. Cossaro, A. Verdini, L. Floreano, E. D'Incecco, M. Stredansky, E. Vesselli, M. Jugovac, M. Cinchetti, V. Feyer and C. M. Schneider, Appl. Surf. Sci., 2020, 504, 144343 CrossRef CAS.
- A. Calabrese, L. Floreano, A. Verdini, C. Mariani and M. G. Betti, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115446 CrossRef.
- A. Picone, D. Giannotti, A. Brambilla, G. Bussetti, A. Calloni, R. Yivlialin, M. Finazzi, L. Duò, F. Ciccacci, A. Goldoni, A. Verdini and L. Floreano, Appl. Surf. Sci., 2018, 435, 841–847 CrossRef CAS.
- X. Yang, I. Krieger, D. Lüftner, S. Weiß, T. Heepenstrick, M. Hollerer, P. Hurdax, G. Koller, M. Sokolowski, P. Puschnig, M. G. Ramsey, F. S. Tautz and S. Soubatch, Chem. Commun., 2018, 54, 9039–9042 RSC.
- P. Hofmann, R. Unwin, W. Wyrobisch and A. M. Bradshaw, Surf. Sci., 1978, 72, 635–644 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00699b |
| This journal is © The Royal Society of Chemistry 2024 |