
Narcissistic self-sorting in Zn(II) porphyrin derived semiconducting nanostructures†
Yelukula
Ramakrishna
ab,
Madarapu
Naresh
ab,
Madoori
Mrinalini
 *abc,
Nagadatta
Pravallika
a,
Priti
Kumari
a,
Botta
Bhavani
ab,
Lingamallu
Giribabu
ab and
Seelam
Prasanthkumar
*ab
*abc,
Nagadatta
Pravallika
a,
Priti
Kumari
a,
Botta
Bhavani
ab,
Lingamallu
Giribabu
ab and
Seelam
Prasanthkumar
*ab
aDepartment of Polymer & Functional Materials, CSIR-Indian Institute of Chemical Technology (IICT), Tarnaka, Hyderabad-500007, Telangana, India. E-mail: mabani1n@gmail.com; prasanth@iict.res.in
bAcademy of Scientific and Innovation Research (AcSIR), Ghaziabad-201 002, India
cMaterials Chemistry Department, CSIR-Institute of Minerals and Materials Technology (IMMT), Bhubaneswar – 751 013, Odisha, India
First published on 16th April 2024
Abstract
The narcissistic self-sorted phenomenon is explicitly attributed to the structural similarities in organic molecules. Although such relevant materials are rarely explored, self-sorted structures from macrocyclic π-conjugated-based p- and n-type organic semiconductors facilitate the increase of exciton dissociation and charge separation in bulk heterojunction solar cells. Herein, we report two extended π-conjugated derivatives consisting of zinc-porphyrin-linked benzothiadiazole acting as an acceptor (PB) and anthracene as a donor (PA). Despite having the same porphyrin π-conjugated core in PA and PB, variations in donor and acceptor moieties make the molecular packing form one-dimensional (1D) self-assembled nanofibers via H- and J-type aggregates. Interestingly, a dissimilar aggregate of PA and PB exists as a mixture (PA + PB), promoting narcissistic self-sorted structures. Electrochemical impedance investigation reveals that the electronic characteristics of self-sorting assemblies are influenced by the difference in electrostatic potentials for PA and PB, resulting in a transitional electrical conductivity of 0.14 S cm−1. Therefore, the design of such materials for the fabrication of effective photovoltaics is promoted by these extraordinary self-sorted behaviors in comparable organic π-conjugated molecules.
 Madoori Mrinalini | Dr Mrinalini received her M. Sc. in Chemistry from St Francis College for Women and secured the first rank with a gold medal. Subsequently, she enrolled in the AcSIR Ph.D. program under the joint supervision of Dr L. Giribabu and Dr S. Prasanthkumar in Supramolecular Chemistry at CSIR-IICT, Telangana, and was awarded the degree in 2021. Afterwards, she was selected as a Senior Project Associate at the CSIR-Institute of Minerals and Materials Technology (CSIR-IMMT), Odisha, India. Her research interests include designing stimuli-responsive organic semiconducting materials for sensors and energy devices and developing novel redox couples for artificial photosynthesis systems. |
Introduction
Constructing self-sorted nanostructures from synthetic organic molecules to mimic nature has garnered a lot of interest in electronics and biomedical applications.1–15 A versatile technique for identifying desirable products in numerous intricate networks and making it easier to assemble basic components into complex structures is self-sorting.16 Besides, attraction towards like or unlike molecules can be utilized in supramolecular systems to identify self-sorting; attraction towards like molecules promotes narcissistic self-sorting, whereas affinity towards unlike molecules encourages social self-sorting or co-assemblies.17,18 For example, synthetic peptides, oligophenyleneethylenes, perylene diimides, and bola amphiphiles have shown self-sorted structures until now.1 As is known, self-sorted structures usually follow either kinetic or thermodynamic pathways.19 By modulating kinetic and thermodynamic variables during self-assembly, S. J. George and co-workers have recently reported self-sorted, block, and random supramolecular polymers from cholesterol-linked naphthalene diimide (NDI)-based two-component systems.20 Upon studying several alkyl chains substituted at the periphery of perylene diimides, the Würthner group discovered that π-stacking modes of H- and J-aggregates tend to self-sort during copolymerization.21 Furthermore, narcissistic and social self-sorting phenomena were observed in similar OPE derivatives by altering concentration and solvent parameters.22 Ajayaghosh and co-workers also reported on the self-sorted co-axial alignment of p/n heterojunctions with strong photoconductivity, which was shown by combining two distinct structural p- and n-type organic semiconductors.23 As a result, most self-sorting assemblies were created by structural modifications or by optimizing the dynamics of assemblies.24–26 Although it is presently difficult to accomplish, the structural similarities between p- and n-type semiconductors envision narcissistic self-sorting, which is crucial for charge separation and exciton dissociation to create effective bulk heterojunction solar cells.27–30In order to facilitate long-range ordered assemblies at the macroscopic level, we are creating bulky π-conjugated systems that are appended with redox-active electron donor and acceptor moieties.31 In this context, porphyrin is selected as an extended π-conjugated system because of its 18 π electron structure, planarity, and aromaticity which facilitate the formation of well-defined self-assembled nanostructures. Additionally, its strong absorption in the visible to near-infrared range makes it a great photosensitizer for solar cells.32–34 On the other hand, anthracene and benzothiadiazole were taken into consideration as electron donor and acceptor moieties since both of them have exceptional oxidation and reduction capabilities.35,36 Thus, we designed two porphyrin derivatives which are linked with anthracene (PA)/benzothiadiazole (PB) and directional amide groups were added to the periphery of the moieties to support the hydrogen bonding interactions (Fig. 1a).37,38 Despite the structural similarities in PA and PB, it can be either social self-sorting or narcissistic self-sorting during mixing in the solution and aggregated states.39–42 Photophysical, morphological and impedance analyses reveal that PA and PB form H- and J-aggregates and their mixture displays a narcissistic self-sorting characteristic as a result of their assembly dynamics. Using π–π stacking and H-bonding interactions, long axial self-assembled nanofibers were subsequently obtained from PA and PB. In the mixture (PA + PB), self-sorting inhibits long axial growth, resulting in short nanorods with an average length of 1 μm to 5 μm (Fig. 1b).43 Comparing PA with PB, it is interesting to note that the former shows more negative potential, which aids in improving electrical conductivity on self-assembled nanofibers, while the latter shows transitional electrical conductivity, suggesting that aggregate dynamics control not only the self-sorted phenomenon but also tune their electronic properties at the nanoscale.
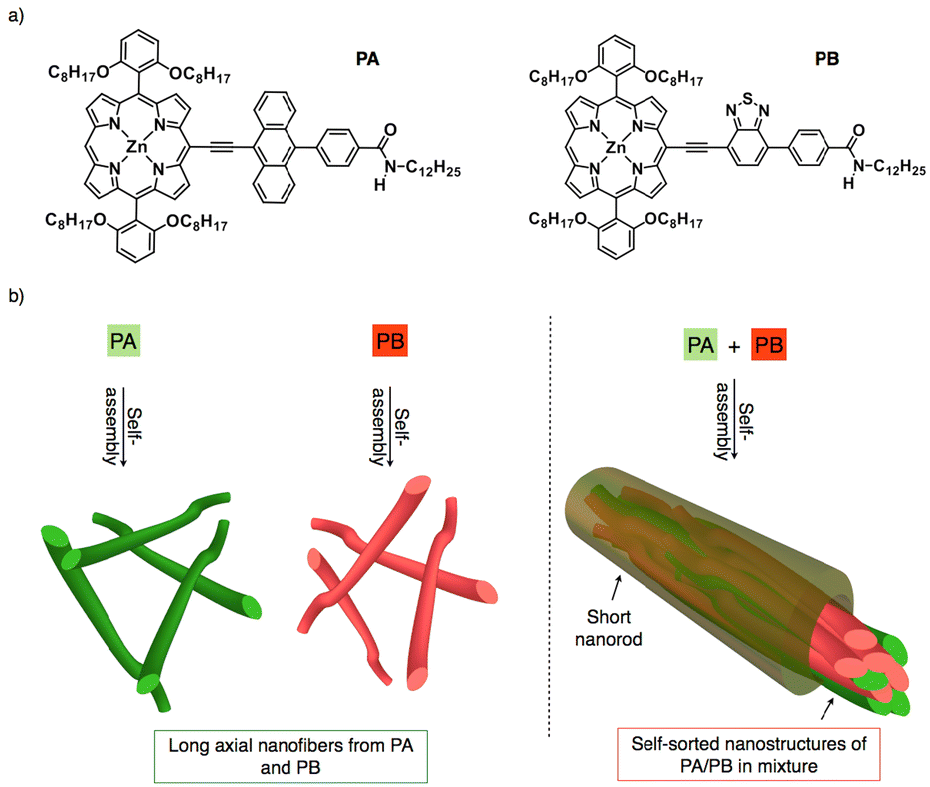 | ||
| Fig. 1 Molecular structures of Zn(II) metallated porphyrin derivatives comprising (a) anthracene functioning as an electron donor in PA and benzothiadiazole as an electron acceptor in PB. (b) Schematic representation of long axial nanofibers from PA and PB and self-sorted nanostructures embedded in short nanorods from PA + PB upon self-assembly. | ||
Results and discussion
PA and PB were synthesized by Sonogashira and peptide coupling reactions and characterized with 1H nuclear magnetic resonance (NMR), MALDI-TOF-mass spectra and Fourier transform-infrared (FT-IR) analyses (Fig. S1–S6†). Subsequently, photophysical properties were determined to ascertain the possibility of charge transfer from similar PA and PB while mixing in solution. UV-visible spectroscopic analyses of PA, PB, and PA + PB (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 M) were performed in chloroform with a concentration of 1 × 10−5 M at 25 °C. PA showed absorption bands at around 427 nm and 613 nm, whereas PB showed the same at 421 nm and 621 nm, which correspond to the Soret and Q-bands, respectively. PA + PB has demonstrated absorption bands similar to those of PA and PB in chloroform suggesting that both molecules exist as individual monomers in the solution state (Fig. S7†). Subsequently, fluorescence spectra also depict that PA and PB retain their individual characteristic features in PA + PB, which is conspicuous from the emission peaks (Fig. S15†). Two prominent emission peaks were observed for PA (626, 680 nm) and PB (634, 690 nm) at an excitation wavelength of 470 nm. However, the fluorescence intensity was quenched in PA + PB when compared to PA, indicating the possibility of an inter-electron transfer mechanism between the two molecules (Fig. S15a†). Furthermore, proton nuclear magnetic resonance (1H NMR) and nuclear Overhauser effect spectroscopy (NOESY) analyses were carried out to confirm the individuality of PA and PB in the mixed state. The resultant spectra reveal that the proton peaks in the aromatic and aliphatic regions of PA and PB match with the mixture without any additional chemical shifts, suggesting that the molecules exist independently in solution (Fig. S8–S11†). Subsequently, FT-IR analyses on PA, PB, and PA + PB were also performed in chloroform, revealing a broad band at 3392 cm−1, which represented the N–H stretching frequency, and bands at 2921 cm−1 and 2850 cm−1 for PA and PB, which represented C–H stretching frequencies. Whereas PA + PB also displayed similar frequencies, as PA and PB support the individual behaviour in the mixture in the solution state; hence, the charge transfer phenomenon can be ruled out from the similar PA and PB (Fig. S12†).
:1 M) were performed in chloroform with a concentration of 1 × 10−5 M at 25 °C. PA showed absorption bands at around 427 nm and 613 nm, whereas PB showed the same at 421 nm and 621 nm, which correspond to the Soret and Q-bands, respectively. PA + PB has demonstrated absorption bands similar to those of PA and PB in chloroform suggesting that both molecules exist as individual monomers in the solution state (Fig. S7†). Subsequently, fluorescence spectra also depict that PA and PB retain their individual characteristic features in PA + PB, which is conspicuous from the emission peaks (Fig. S15†). Two prominent emission peaks were observed for PA (626, 680 nm) and PB (634, 690 nm) at an excitation wavelength of 470 nm. However, the fluorescence intensity was quenched in PA + PB when compared to PA, indicating the possibility of an inter-electron transfer mechanism between the two molecules (Fig. S15a†). Furthermore, proton nuclear magnetic resonance (1H NMR) and nuclear Overhauser effect spectroscopy (NOESY) analyses were carried out to confirm the individuality of PA and PB in the mixed state. The resultant spectra reveal that the proton peaks in the aromatic and aliphatic regions of PA and PB match with the mixture without any additional chemical shifts, suggesting that the molecules exist independently in solution (Fig. S8–S11†). Subsequently, FT-IR analyses on PA, PB, and PA + PB were also performed in chloroform, revealing a broad band at 3392 cm−1, which represented the N–H stretching frequency, and bands at 2921 cm−1 and 2850 cm−1 for PA and PB, which represented C–H stretching frequencies. Whereas PA + PB also displayed similar frequencies, as PA and PB support the individual behaviour in the mixture in the solution state; hence, the charge transfer phenomenon can be ruled out from the similar PA and PB (Fig. S12†).
Nevertheless, due to the difference in electron density between porphyrin-linked anthracene and benzothiadiazole of PA and PB, these molecules tend to have a redox nature. Cyclic voltammetry of three samples was recorded in chloroform using three electrode systems and tetrabutyl ammonium perchlorate as a supporting electrolyte. The oxidation potentials of PA are 0.74, 0.83 V and that of PB is 1.22 V while their reduction potentials are −0.80 and −1.23 V, respectively. In contrast PA + PB showed 0.71 and −0.82 V, indicating the ease of oxidation relative to PA and PB alone, thereby promoting efficient electron transfer between PA and PB (Fig. S13†). Consequently, spectroelectrochemistry analyses of PA, PB, and PA + PB depict the absorption spectral changes during the redox process at an applied oxidation potential of 1.2 V (Fig. S14†). PA displayed a nominal decrease in intensity at 427 nm together with a red shift at Q-bands, while PB exhibits a significant bathochromic shift in the Soret and Q-band regions, signifying the stability of oxidation species. Similarly, the combination has shown a decrease in the intensity of the Soret band along with a considerable red shift revealing that PA and PB exist independently and uphold the stability of redox species to facilitate more effective electron transfer than individual molecules. Therefore, photophysical, spectroscopic, and electrochemical data emphasize that PA and PB appear as identical bulky π-systems but exist independently whilst mixing in the solution, prompting the nanoscopic investigation of self-sorting assemblies that could assist in developing efficient bulk heterojunction solar cells.15,23,44–47
Furthermore, utilizing the heating–cooling technique, the assemblies of PA, PB, and PA + PB were prepared in cyclohexane and examined by UV-visible absorption, emission spectroscopy, microscopy, PXRD and electrochemical impedance analyses. However, PA and PB were moderately soluble in cyclohexane and the suspension was dissolved by heating and subsequent cooling, which facilitated aggregate formation. The absorption spectrum of PA showed a hypsochromic shift (∼9 nm) in the Soret band together with a shoulder band at 650 nm. This large blue shift at the Soret band suggests that the molecules are organized into H-type aggregates. In contrast, assemblies of PB have shown an ∼3 nm bathochromic shift in the Soret band region, with significant quenching in the intensity, proposing that PB exists as J-type aggregates. Interestingly, PA and PB possess similarities in structures, but different types of aggregates that are distinct from each other may have an impact on co-assembly studies. Furthermore, aggregates of PA + PB were prepared by mixing PA and PB at a 1:1 molar ratio via the heating–cooling method. The absorption spectrum of PA + PB reveals a Soret band at 418 nm and Q-bands at 650–700 nm which remain unaltered, suggesting that PA and PB prefer narcissistic self-sorting over social aggregation in the mixed state (Fig. 2a). Consequently, temperature-dependent absorption studies confirmed that the difference in the thermodynamic stability of aggregates of PA and PB drives the self-sorting assemblies in PA + PB. Fig. 2d represents the temperature-dependent absorption spectra of PA + PB in cyclohexane as the temperature is raised by 10 °C increments from 20 °C to 70 °C. Upon increasing the temperature, the intensity of the Soret band at 418 nm increases gradually with a concomitant redshift to 423 nm and then moderately decreases in the Q-band region, indicating that the aggregates deteriorate for PA and are stable for PB until 70 °C. For comparison, assemblies of PA and PB were also studied at regulated temperatures, and it was observed that PA reached the monomeric state at 60 °C while the aggregates of PB remained stable at >70 °C (Fig. 2b and c). Subsequently, the fraction of aggregates (αagg) was calculated in the Soret band region (λmax: 420 nm) for PA, PB and PA + PB against variable temperatures revealing that PA has shown a sigmoidal curve which follows the isodesmic mechanism (Fig. 2e). On the other hand, PB and PA + PB represent a non-sigmoidal curve and abide by the co-operative mechanism which is different from PA.48–51 Alongside, the thermal stability of aggregates was found to be highest for PB followed by PA + PB and PA. Despite PA and PB having similar structures, they uphold individuality at the molecular level which results in narcissistic self-sorting assemblies, perhaps an intriguing phenomenon for organic solar cell applications.
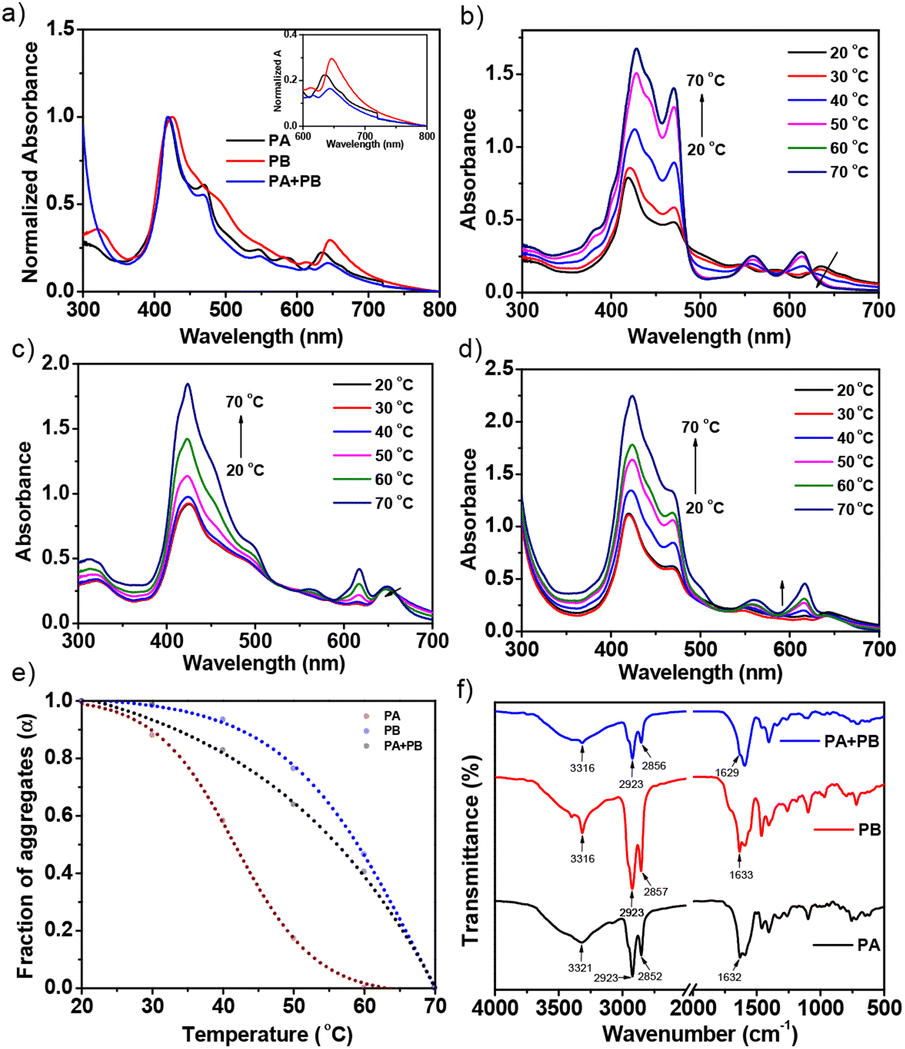 | ||
Fig. 2 (a) Normalized ultraviolet–visible absorption spectra of PA, PB, and PA + PB (1:1 M) in cyclohexane at a concentration of 1 × 10−5 M at 25 °C. The inset shows the conspicuous shift in the peaks of PA, PB, and PA + PB. (b–d) Temperature-dependent UV-visible absorption spectra of PA, PB and PA + PB assemblies in cyclohexane with an increment in temperature from 20 °C to 70 °C. (e) Fraction of aggregates (αagg) of PA, PB and PA + PB calculated from apparent absorption intensity at the Soret band: λ = 420 nm at different temperatures. (f) FT-IR analysis of PA, PB and PA + PB aggregates denoting the considerable stretching frequencies of N–H, C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H bending frequencies. O and N–H bending frequencies. | ||
In addition, temperature-controlled fluorescence spectra revealed that, in comparison with the solution state, the intensity of PA + PB was quenched more than PA and PB intensity, suggesting that intermolecular electron transfer favors the assembled state (Fig. S16†).26,52,53 Furthermore, a 440 nm LED source was used for excitation during lifetime analysis in both the aggregated and solution states. PA shows bi-exponential decay in cyclohexane and mono-exponential decay in chloroform (Fig. S17 and Table S1†). The resultant lifetime values are 5.72, 4.06, and 2.14 ns for PA, PB and PA + PB in chloroform, respectively. On the other hand, the measured bi-exponential photoluminescence decay values of PA recorded are 0.11 and 3.13 ns whereas those of PB are 6.5 and 5.8 ns. However, PA + PB showed lifetime values of 0.1 and 1.4 ns, suggesting that effective inter-electron transfer occurred between PA and PB, which is reflected in the lifetime values.
H-bonding plays a vital role during aggregation which assists in understanding the self-sorting behaviour of PA and PB in the mixture. According to FT-IR analysis, the aggregated samples have shown significant changes in stretching and bending frequencies when compared to the solution state (Fig. S12†). PA and PB depict that the N–H stretching frequency at 3322 cm−1, together with CO stretching and N–H bending, resulted in hierarchical nanostructures. Alongside, C–H stretching frequencies at 2850 and 2921 cm−1 observed in PA and PB indicate that the dodecyl chains were stretched to assist in interdigitating with other molecules through van der Waals interactions during assemblies. Likewise, stretching and bending frequencies of PA + PB match well with the individuals of PA and PB, suggesting that H-bonding and van der Waals interactions remain unaltered in co-assemblies, affirming the narcissistic self-sorting behavior (Fig. 2f).54–57
Having confirmed the self-sorting assemblies of PA and PB in a mixture, electron microscopy analyses were established to check the surface morphology of the three samples. Scanning electron microscopy (SEM) images of PA showed one-dimensional nanofibers with an average diameter of 50–100 nm and several micrometers in length (Fig. 3a). These nanofibers were formed by utilizing non-covalent interactions like π–π stacking from bulky π-conjugated structures, H-bonding with directional amide groups, and long alkyl chains assisting van der Waals interactions. Transmission electron microscopy (TEM) images also showed similar nanofibers to SEM (Fig. 3d). However, these nanofibers do not have considerable electron diffraction patterns. Likewise, PB formed nanofibers with an average size of 100 nm and several micrometers in length, respectively (Fig. 3b and e). Thus, the freshly prepared 10 μM solutions of PA and PB comprise metastable assemblies which reorganize to form long axial nanofibers to attain thermodynamic stability. Conversely, PA + PB (1:1 M) in cyclohexane illustrates short nanorod structures with an average length of 1–5 μm and a width of 0.2–0.7 μm via narcissistic self-sorting using π–π stacking and H-bonding interactions. As a result, the formation of co-assemblies containing two or more different molecules is promoted by thermodynamically favourable interactions (Fig. 3c, f and S18†). Moreover, the driving force for the obstruction of the co-assembly of the molecules is the hydrophobic collapse of their C12 (dodecyl) alkyl chain (hydrophobic interaction between the C12 alkyl chain and the solvent) that constricts the PA + PB to collapse, leading to the formation of short nanorods. Thus, these short nanorods embedded within the self-sorted assemblies of PA and PB hinder the extended growth of nanostructures. Besides, an ordered diffraction pattern was observed for the nanorods, suggesting that the PA + PB mixture is crystalline in nature. Moreover, the elemental composition of the self-sorted assemblies of PA and PB in the nanorod structure was investigated using high-angle annular dark field imaging in scanning transmission electron microscopy (HAADF-STEM). In PA + PB, nanorods were seen together with both elements corresponding to PA and PB; however, PB assemblies are more dominant and vivid than PA (Fig. 3i). Thus, elemental analysis also correlates well with our insights because the data corresponding to PB are more perceptible when compared to PA. Parallel to this, the elemental composition of PA- and PB-derived nanofibers was also recorded, and it was found that the elements present in both images signify the individual assemblies of PA and PB (Fig. 3g and h). Therefore, optical and microscopic data adhere to the narcissistic self-sorted assemblies of PA and PB, a remarkable phenomenon observed in redox active systems.
 | ||
| Fig. 3 (a) SEM and TEM images of an air-dried suspension of (a and d) PA. (b and e) PB. (c and f) PA + PB. Scanning transmission electron microscopy (HAADF-STEM) images of (g) PA, (h) PB and (i) PA + PB. | ||
To emphasize the molecular packing in individual and self-sorted assemblies, powder X-ray diffraction analyses were performed on PA, PB, and PA + PB (Fig. 4a and Fig. S19†). PA and PB displayed similar small and wide-angle diffraction peaks and the corresponding d-spacing values of 32.5 Å, 16.8 Å and 3.4 Å. Two intense diffraction peaks in wide angle regions of 32.5 Å and 16.8 Å represent the end-to-end molecular length and the dodecyl chain length. An intense peak was seen for both molecules because van der Waals interactions cause the alkyl chains to be stretched and interdigitated. A second broad peak that is perpendicular to the long axis of the nanofibers appears in a small angle region of 3.4 Å, indicating the π–π-stacking between the conjugated units.58–62 However, an additional intense peak at 8.5 Å was observed for PB, implying the repulsion of the lone pair on ‘S’ in benzothiadiazole units with other molecules, resulting in the slippage of molecular packing while being absent in PA. Thus, PA and PB exhibit two different molecular packings via H-/J-type aggregates using π–π stacking, H-bonding, and van der Waals interaction, leading to the hierarchical growth of one-dimensional nanofibers (Fig. 4b and c). On the other hand, the PA + PB mixture has shown similar diffraction peaks to PA and PB, indicating that both aggregate individually resulting in self-sorted behavior (Fig. 4a, b and d). Additionally, the peak at 4.1 Å is observed specifically in PA + PB, owing to the dodecyl chains which aid in interdigitating with other molecules via van der Waals interactions during the self-sorting process in the mixed state. Hence, the difference in the molecular packing of molecules that governs the dynamics of aggregates which facilitates the attainment of narcissistic self-sorted nanostructures in a mixture is an interesting route to develop bulk heterojunctions (Fig. 4d). Nevertheless, the electronic properties of PA + PB are also crucial for the application of photovoltaics. In this context, the energy levels of frontier orbitals and electric potentials are significant factors in evaluating the electronic properties of organic materials.
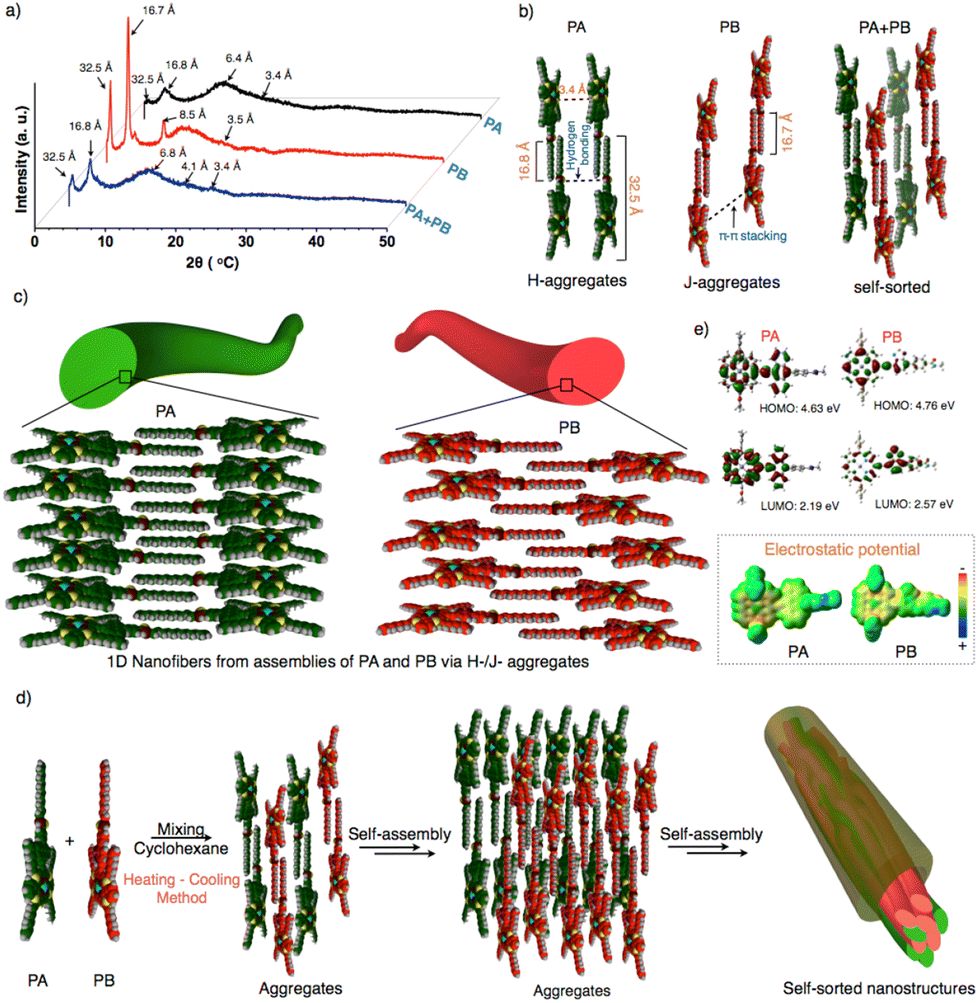 | ||
| Fig. 4 (a) Powder X-ray diffraction analysis of PA, PB and PA + PB. (b) Molecular packing of PA, PB, and PA + PB leading to H, J-aggregates and self-sorted from the corresponding d-spacing values for each length of the center to the end, π–π stacking, and H-bonding. (c) Schematic representation of the self-assembly in PA and PB, leading to the formation of 1D nanofibers. (d) Schematic illustration of the formation of self-sorted nanostructures by mixing PA + PBvia self-assembly. (e) Optimized structures and the corresponding HOMO/LUMO energy levels of PA and PB from DFT calculations. The electrostatic potential maps of PA and PB exhibit the charge distribution. | ||
Consequently, we performed density functional theory calculations to estimate the energy levels over PA and PB by using the functional basis set of B3lYP/6-31G (d,p). PA revealed the electron density distribution of anthracene at the HOMO and porphyrin at the LUMO but the difference is not much noticeable due to their similarity in oxidation potentials. The resultant HOMO and LUMO energy values were 4.63 and 2.19 eV, respectively. Subsequently, electrostatic potential (ESP) data have shown a uniform distribution of negative potentials throughout the system, revealing that PA performs better as an electron transporting material (Fig. 4e). Whereas in PB, the difference in electron density distribution is clearly seen because the HOMO is on porphyrin and the LUMO is on benzothiadiazole. As a result, PB possesses an enhanced redox-active nature, as proved by spectroelectrochemical spectra. However, the ESP image reveals that the positive and negative potential distributions on benzothiadiazole and porphyrin might prefer charge recombination or an intramolecular electron transfer process (Fig. 4e). Thus, significant variations in the electrical potential and energy levels of PA and PB may exhibit a greater impact on the electronic properties, as confirmed by a detailed investigation conducted on nanofibers and nanorods to analyze their conductivity at the bulk scale for the future generation of photovoltaics.
Electrochemical impedance spectroscopy was employed to estimate the bulk conductivity of self-assembled nanofibers and nanorods.63,64PA, PB and PA + PB aggregates were prepared and drop-cast on an etched indium tin oxide plate with dimensions measuring 1 × 1 mm (Fig. 5a). All the three samples have shown semicircular curves in which PA has a lower bulk resistance (Rb) than the other two samples due to the high negative electric potential of PA, and head-to-head stacking of molecules in nanofibers facilitates efficient intermolecular electron transfer that leads to a high specific conductivity of 4.07 S cm−1 at 25 °C. On the other hand, PB exhibits very low conductivity, attributed to the charge recombination and slippage of molecular stacking in nanofibers, which hampers electron flow and results in a lower specific conductivity of about 0.035 S cm−1. Thus, with the largest differences in the conductivity of PA and PB, mixed samples achieved transitional specific conductivity (0.14 S cm−1), which is higher than PB and lower than PA, suggesting that self-sorted assemblies aid in improving the intermolecular electron transfer from PA to PB. However, the resultant conductivity in self-sorted structures is relatively low because PB is more predominant than PA in terms of thermodynamic stability (Fig. 5b). Subsequently, to determine the electrical conductivity, temperature-dependent impedance analyses were carried out by raising the temperature from 25 °C to 85 °C. Fig. 5c and d represent the Nyquist plot of PA + PB at different temperatures, depicting a gradual increment in resistance as temperature rises (Table S2†).
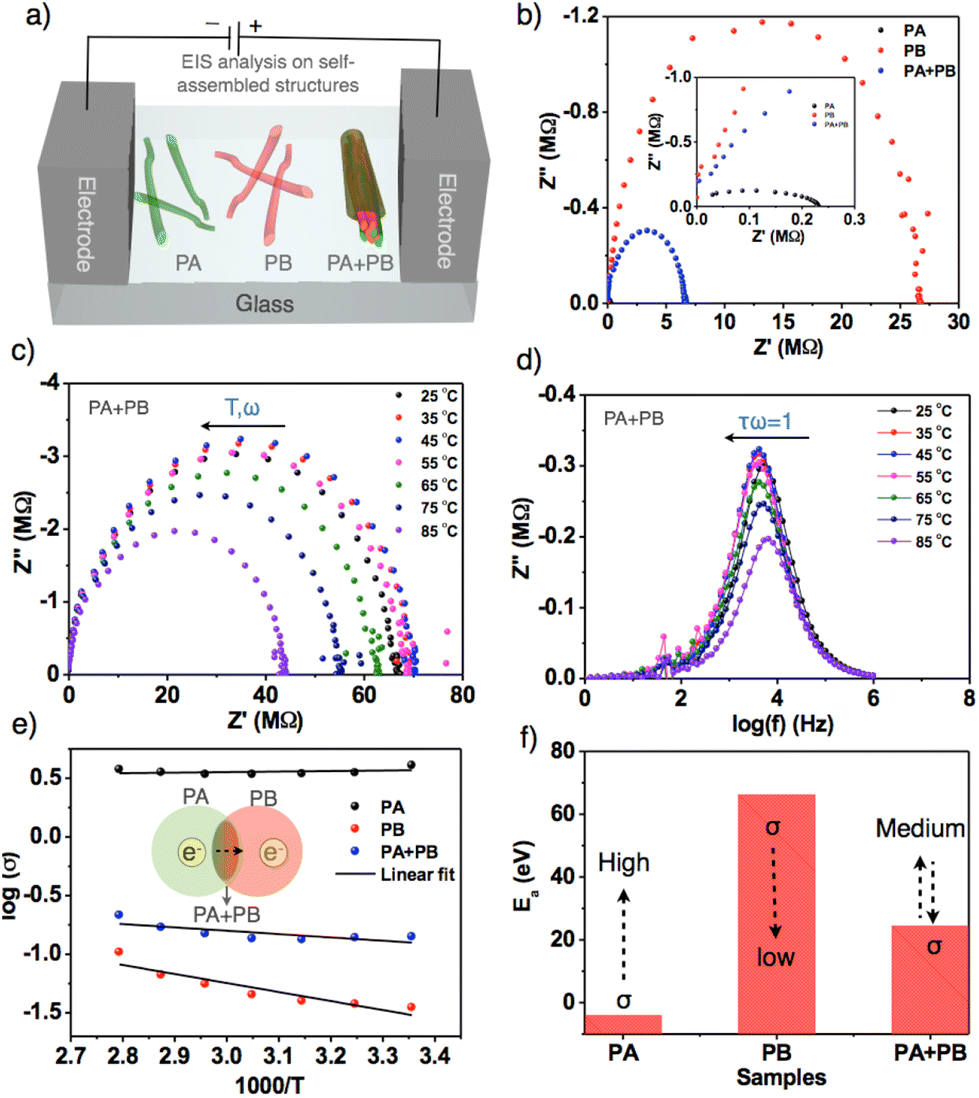 | ||
| Fig. 5 (a) Schematic illustration of the cell diagram for EIS analysis on each PA and PB derived nanofibers and PA + PB-based self-sorted structures. (b) Nyquist plot of PA, PB and PA + PB at 25 °C. The inset shows the low resistance area for three samples. (c) Temperature-dependent Nyquist plot of PA + PB aggregates with temperatures ranging from 25 °C to 85 °C. (d) Temperature-dependent logarithmic frequency (log(f)) vs. the imaginary impedance of PA + PB aggregates. (e) logσ vs. 1000/T graph for the determination of the electrical conductivity of PA, PB, and PA + PB. The inset shows the schematic representation of electronic flow from PA to PB in a mixture. (f) Activation energy (Ea) plot of PA, PB, and PA + PB. | ||
Furthermore, we computed 1000/T against logσ of PA + PB, revealing linear growth in conductivity that falls within the electrical conductivity regime (Fig. 5e). Similarly, PA and PB comply with electrical conductivity behavior; however, PA exhibits a linear relationship with conductivity due to efficient electron transport at controlled temperature, and the minor fluctuation in the linearity of PB was ruled by charge recombination (Fig. S20 and S21†).65 Later, activation energy (Ea) was calculated from the above graph, and it revealed that Ea is low for PA, indicating high conductivity as per the known inverse relationship between activation energy and conductivity. Whereas for PB and PA + PB, the values are high and medium, resulting in low and transitional conductivity (Fig. 5f). Alongside, the bulk capacitance was also estimated by meeting the specific condition ωRCb = 1 and the resultant specific capacitance values were in the range of 0.3–0.4 pF (Table S2†).63 Additionally, bulk relaxation time was evaluated from the graph logf vs. imaginary impedance. Therefore, thorough analysis of newly designed bulky π-conjugated systems demonstrated that molecular structures and aggregate dynamics can regulate the electrical conductivity of nanofibers and nanorods.
Conclusions
In conclusion, we demonstrated a rational approach towards narcissistic self-sorted assemblies by mixing two similar porphyrin appended anthracene and benzothiadiazole derivatives. Self-assembled nanofibers were formed via distinct H- and J-type aggregates and their mixture remained the same but resulted in self-sorted structures embedded in nanorods, as confirmed by optical, microscopy, and diffraction analyses. Electrostatic potentials suggest that the differences in electric potentials impact efficient electron transport, and charge recombination results in high and low electrical conductivity for PA and PB derived nanofibers. Thereby, self-sorted structures exhibit transitional conductivity through the intermolecular electron transfer from PA to PB. Consequently, similar macrocyclic π-conjugated linked donor and acceptor systems endow the self-sorted structures with controlled dynamics and remarkable electronic properties, promoting the development of novel π-conjugated redox active systems that possess the ability to improve the charge carrier separation and transport mechanism in bulk heterojunction solar cells.Author contributions
Y.R., M.M., and S.P. designed the study, analyzed the data and wrote the manuscript. Y.R. and M.N. performed the synthesis, conducted the optical, electrochemical, and conductivity studies and prepared the manuscript. N.P. and P.K. participated in the discussion of results and wrote the manuscript. B.B. performed the conductivity analyses. L.G. was involved in analyzing the data and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. thanks the Science and Engineering Research Board (SERB) – Startup Research Grant (SRG/2022/000132) for financial support. Y. R. and P. K. acknowledge the Council of Scientific and Industrial Research – Indian Institute of Chemical Technology (CSIR/IICT/MLP-0110) for fellowship support. B. B. thanks DST-Inspire for the fellowship. All the authors thank Dr Pratay Basak for EIS analysis support and the Director of CSIR-IICT for his support with IICT/Pubs./2023/400.References
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed.
- N. A. Campbell, J. B. Reece, M. R. Taylor, E. J. Simon and J. L. Dickey, Biology: Concepts and Connections, 6th ed, Benjamin/Cummings Publishing Company, San Francisco, CA, 2008 Search PubMed.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366 CrossRef CAS PubMed.
- J. Nelson, Structure and function in cell signalling, Wiley, Chichester, 2009 Search PubMed.
- H. Kar and S. Ghosh, Isr. J. Chem., 2019, 59, 881 CrossRef CAS.
- C. H. Chen, L. C. Palmer and S. I. Stupp, Soft Matter, 2021, 17, 3902 RSC.
- Q. Liu, B. Jin, Q. Li, H. Yang, Y. Luo and X. Li, Soft Matter, 2022, 18, 2484 RSC.
- H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165 CrossRef CAS PubMed.
- H. Lee, M. Lee, J. H. Hwang, I. Kim, E. Lee and W.-D. Jang, Nanoscale, 2023, 15, 18224–18232 RSC.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78 RSC.
- C. Rest, M. J. Mayoral and G. Fernández, Int. J. Mol. Sci., 2013, 14, 1541–1565 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS PubMed.
- Y. Lin, Y. Firdaus, F. H. Isikgor, M. I. Nugraha, E. Yengel, G. T. Harrison, R. Hallani, A. El-Labban, H. Faber, C. Ma, X. Zheng, A. Subbiah, C. T. Howells, O. M. Bakr, I. McCulloch, S. D. Wolf, L. S. Tsetseris and T. D. Anthopoulos, ACS Energy Lett., 2020, 5, 2935 CrossRef CAS.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jäckel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499 RSC.
- A. X. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831 CrossRef CAS PubMed.
- P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538 CrossRef CAS.
- A. Shivanyuk and J. Rebek, J. Am. Chem. Soc., 2002, 124, 12074 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606 CrossRef CAS PubMed.
- S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343 CrossRef CAS PubMed.
- M. J. Mayoral, C. Rest, J. Schellheimer, V. Stepanenko and G. Fernández, Chem. – Eur. J., 2012, 18, 15607 CrossRef CAS PubMed.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed., 2015, 54, 946 CrossRef CAS PubMed.
- J. van Herrikhuyzen, A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 10021 CrossRef CAS PubMed.
- A. Sandeep, V. K. Praveen, K. K. Kartha, V. Karunakaran and A. Ajayaghosh, Chem. Sci., 2016, 7, 4460 RSC.
- K. Sugiyasu, S. I. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863 CrossRef CAS.
- M. Madhu, R. Ramakrishnan, V. Vijay and M. Hariharan, Chem. Rev., 2021, 121, 8234 CrossRef CAS PubMed.
- O. Inganas, Adv. Mater., 2018, 30, 1800388 CrossRef PubMed.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821 CrossRef CAS PubMed.
- A. M. Castilla, E. R. Draper, M. C. Nolan, C. Brasnett, A. Seddon, L. L. E. Mears, N. Cowieson and D. J. Adams, Sci. Rep., 2017, 7, 8380 CrossRef PubMed.
- M. Mrinalini, M. Naresh, S. Prasanthkumar and L. Giribabu, J. Porphyrins Phthalocyanines, 2021, 25, 382 CrossRef CAS.
- J. M. Park, K.-I. Hong, H. Lee and W.-D. Jang, Acc. Chem. Res., 2021, 54, 2249 CrossRef CAS PubMed.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910 CrossRef CAS PubMed.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC.
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397 CrossRef CAS PubMed.
- B. Bhavani, M. Mrinalini, J. V. S. Krishna, P. Basak, L. Giribabu and S. Prasanthkumar, ACS Appl. Electron. Mater., 2021, 3, 176 CrossRef CAS.
- M. Lübtow, I. Helmers, V. Stepanenko, R. Q. Albuquerque, T. B. Marder and G. Fernández, Chem. – Eur. J., 2017, 23, 6198 CrossRef PubMed.
- S.-G. Chen, Y. Yu, X. Zhao, Y. Ma, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2011, 133, 11124–11127 CrossRef CAS PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. T. Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7168–7175 CrossRef CAS PubMed.
- J. P. Coelho, J. Matern, R. Q. Albuquerque and G. Fernández, Chem. – Eur. J., 2019, 25, 8960 CrossRef CAS PubMed.
- A. Sarkar, T. Behera, R. Sasmal, R. Capelli, C. Empereur-mot, J. Mahato, S. S. Agasti, G. M. Pavan, A. Chowdhury and S. J. George, J. Am. Chem. Soc., 2020, 142, 11528–11539 CrossRef CAS PubMed.
- M. F. Hagan and G. M. Grason, Rev. Mod. Phys., 2021, 93, 025008 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766 CrossRef CAS PubMed.
- T. Aida and E. W. Meijer, Isr. J. Chem., 2020, 60, 33 CrossRef CAS.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- S. Dhiman, A. Sarkar and S. J. George, RSC Adv., 2018, 8, 18913 RSC.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344 CrossRef CAS PubMed.
- S. Panja, B. Dietrich, A. J. Smith, A. Seddon and D. J. Adams, ChemSystemsChem, 2022, 4, e202200008 CrossRef CAS.
- T. Okada, H. Oohari and N. Mataga, Bull. Chem. Soc. Jpn., 1970, 43, 2750 CrossRef CAS.
- M. R. Molla, A. Das and S. Ghosh, Chem. – Eur. J., 2010, 16, 10084190 CrossRef PubMed.
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. Tang and Y. Cao, ACS Omega, 2020, 5, 8572–8578 CrossRef CAS PubMed.
- S. Prasanthkumar, A. Saeki, S. Seki and A. Ajayaghosh, J. Am. Chem. Soc., 2010, 132, 8866–8867 CrossRef CAS PubMed.
- A. Sikder, D. Ray, V. K. Aswal and S. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1606–1611 CrossRef CAS PubMed.
- E. Lee, J.-K. Kim and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 6375–6378 CrossRef CAS PubMed.
- A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148 CrossRef CAS PubMed.
- M. Kimura, H. Narikawa, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Chem. Mater., 2002, 14, 2711 CrossRef CAS.
- M. Kimura, T. Kuroda, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Langmuir, 2003, 19, 4825 CrossRef CAS.
- W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 9434 CrossRef CAS PubMed.
- S. Wang, J. Zhang, O. Gharbi, V. Vivier, M. Gao and M. E. Orazem, Nat. Rev. Methods Primers, 2021, 1, 41 CrossRef CAS.
- A. Ch. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162 CrossRef CAS PubMed.
- M. Naresh, P. S. Gangadhar, P. Kumari, N. Pravallika, Y. Ramakrishna, L. Giribabu and S. Prasanthkumar, ACS Appl. Electron. Mater., 2023, 5, 3603 CrossRef CAS.
- Y. Ramakrishna, M. Naresh, B. Bhavani and S. Prasanthkumar, Phys. Chem. Chem. Phys., 2023, 25, 24539 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00991f |
| This journal is © The Royal Society of Chemistry 2024 |