
Design and synthesis of nucleic acid nano-environment interactome-targeting small molecule PROTACs and their anticancer activity†‡
Sadiya
Tanga
 ab,
Arkadeep
Karmakar§
b,
Arpita
Hota§
b,
Paramita
Banerjee
c and
Basudeb
Maji
*b
ab,
Arkadeep
Karmakar§
b,
Arpita
Hota§
b,
Paramita
Banerjee
c and
Basudeb
Maji
*b
aAshoka University, Department of Chemistry, Rajiv Gandhi Education City, Sonipat, Haryana 131029, India
bBose Institute, Department of Biological Sciences, EN 80, Sector V, Bidhannagar, Kolkata 700091, West Bengal, India. E-mail: bmaji@jcbose.ac.in
cS N Bose National Centre for Basic Science, JD Block, Sector 3, Bidhannagar, Kolkata 700106, West Bengal, India
First published on 31st May 2024
Abstract
Targeted protein degradation through PROteolysis TArgeting Chimeras (PROTACs) is a relatively new modality in cellular interventions. The minimum requirement for PROTACs to function is forming a tertiary complex of the protein of interest (POI), E3 ligase, and the molecular glue PROTAC. Here, we propose a new approach to modulate the nano-environment interactome of a non-protein target through a plausible quaternary complex of interactome–biomolecule of interest (BOI)–PROTAC and E3 ligase. We report nucleic acid-targeting PROTAC (NA-TAC) molecules by conjugating DNA-binding and E3 ligase ligands. We demonstrate that NA-TACs can target the G-quadruplex DNA and induce elevated DNA damage and cytotoxicity compared to the conventional G-quadruplex binding ligands. Our new class of NA-TACs lays the foundation for small molecule-based non-protein targeting PROTACs for interactome and nanoenvironment mapping and nucleic acid-targeted precision medicines.
Introduction
Proteolysis-targeting chimeras (PROTACs) integrate a protein engager with an E3 ubiquitin ligase recruiter, offering potent therapeutic potential to address proteins that were once considered challenging targets.1 PROTACs target the protein of interest by binding with a non-active site, ensuring a unique approach to target engagement.2 The heightened potency is a direct result of their catalytic mechanism of action, setting them apart from conventional inhibitors.3 Furthermore, the formation of a ternary complex not only enhances selectivity but also introduces a stabilizing factor, as both ligands contribute to the stability of this complex, and with these distinctive features, PROTAC technology surpasses traditional inhibitor-based targeting of proteins.2 Since their initial report in 2001, the field of PROTACs has witnessed exponential growth with the development of a diverse array of PROTACs.1 This burgeoning technology has demonstrated promising outcomes across various targets, including nuclear receptors, kinases, misfolded proteins, epigenetic readers or erasers, and transcription factors.4 Various types of PROTAC have been devised to tackle the previously highlighted challenges, including small molecule-based, peptide-based, antibody-based, and nucleic acid-based ones.4c When it comes to targeting nucleic acids specifically, the development has primarily centered around nucleic acid-based PROTACs (NAP), which consist of nucleic acid sequences as target of interest binding moieties, a linker, and an E3 ligase binding moiety.4a,5 Based on NAPs, diverse strategies have emerged to target and degrade specific cellular components precisely. These approaches include TF-PROTACs, designed for the targeted degradation of transcription factors; RNA-PROTACs, tailored for the degradation of RNA-binding proteins; aptamer PROTACs, utilizing aptamers to achieve targeted degradation; G4-PROTACs, engineered to exploit G-quadruplex structures for selective degradation; and, most recently, TeloTACs, specialized chimeras designed for the targeted degradation of cellular components in telomeric regions.4a,c,5b,6 This report introduces small molecule-based, cell-permeable nucleic acid PROTACs (NA-TACs) designed for targeting nucleic acids (Fig. 1). For our demonstration, we have opted for a thoroughly characterized G4 binding ligand to serve as the nucleic acid binding moiety.5a Additionally, we selected CRBN as the E3 ligase, ensuring a comprehensive and robust proteolytic activity.7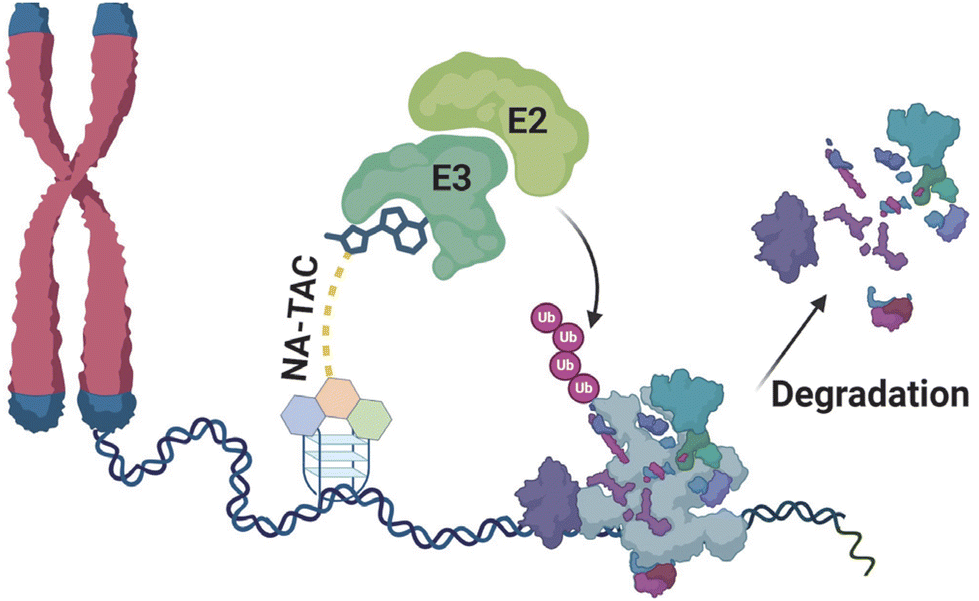 | ||
| Fig. 1 Schematic diagram depicting the possible function of nucleic acid-targeting PROTAC molecules (NA-TACs) in a cellular context. | ||
We hypothesized that the small-molecule PROTAC toolbox could be extended to target non-protein biomolecules and their interactomes. Unlike the recent TF-PROTACs or those with DNA/RNA as the PROTAC warhead, we developed a new class of PROTACs based on cell-permeable small molecules NA-TACs.4a,6a,8 We selected a higher-order DNA structure, G-quadruplex (G4), as the target for proof-of-principle.9 We sought to use a small molecule binder and investigate the possibility of targeting non-protein biomolecules like DNA and their cellular effects. We identified a well-characterized G-quadruplex DNA-binding ligand scaffold and systematically designed the PROTAC molecules (NA-TACs).10
Carbazole–benzimidazole ligands developed by Bhattacharya and co-workers have been shown to target G-quadruplex DNA with high affinity.9–11 In this work, we selected benzimidazole–carbazole as the G4 DNA binder and conjugated it with the E3 ligase ligand thalidomide to develop the NA-TAC molecules.12
As the linker plays a pivotal role in PROTAC function, we used two oxyethylene linkers and two different linking positions on thalidomide.13 We strategically incorporated PEG-based linkers to enhance flexibility and solubility.14 The molecules were further tested for their G4 DNA binding ability and cellular activity.
Initially, we developed four morpholine-benzimidazole-based NA-TAC molecules (P1–P4) and investigated their G4 DNA binding ability compared to their control ligand (C1) (Fig. 2A and ESI, Scheme A‡). CD spectroscopic studies confirmed the G4 DNA binding ability of all four PROTACs, prompting us to investigate the extent of stabilization (Fig. 2B, ESI Fig. S1‡). Subsequently, we performed thermal denaturation studies to identify the most efficient binder (Fig. 2C). Even though all the NA-TACs showed elevation in thermal stability, the P3 compound with the triethylene glycol linker attached at the 5th position on thalidomide exhibited higher thermal stabilization activity (Fig. 2C).15
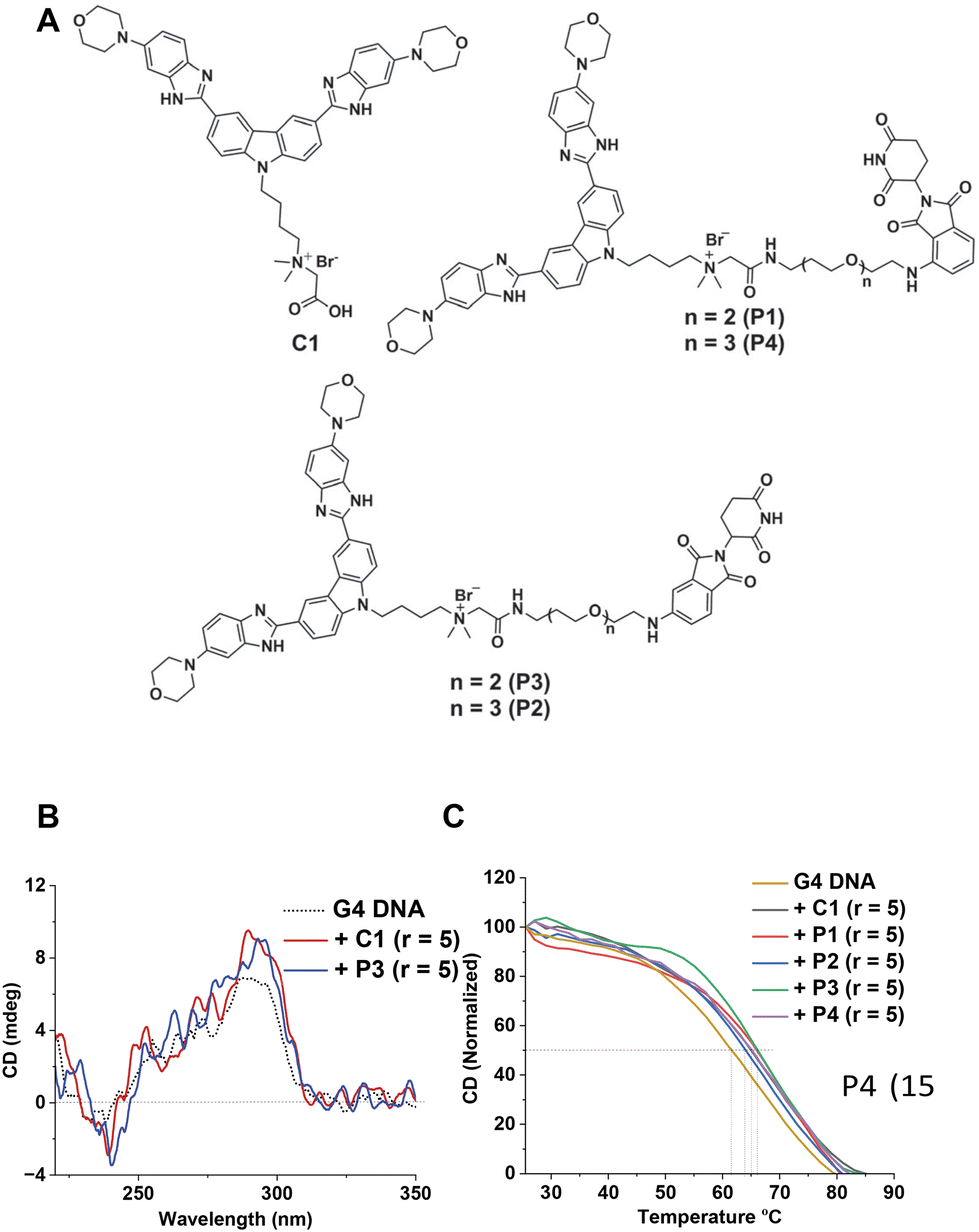 | ||
| Fig. 2 (A) Chemical structure of NA-TACs (P1–P4) and their control compound C1. (B) CD spectra of 3 μM Hum 21-mer G4 DNA alone, with compound C1 and with P3 (15 μM) in 10 mM Tris-HCl (pH 7.4) containing 0.1 M KCl and 0.1 mM EDTA. (C) CD melting profiles of the pre-formed Hum 21-mer G4 DNA alone, and G-quadruplex DNA incubated with ligands C1, P1, P2, P3, and P4 (15 μM) in 10 mM Tris-HCl (pH 7.4) containing 0.1 M KCl and 0.1 mM EDTA. | ||
To improve the activity further, we asked whether additional protonatable moieties on the G4 ligand could enhance the binding properties. We designed three additional compounds where the morpholine O is replaced by N-methyl, N-ethyl, and N-propyl (C1a, C1b, and C1c) (Fig. 3A). We performed molecular docking studies with the three designed molecules and investigated their G4 DNA binding activity to identify the most suitable candidate. The molecular docking suggests that all the molecules can stack efficiently on top of the G4 tetrad (Fig. S2‡). However, the binding energy decreases with increased length of the N-piperazine substituted alkyl chain (Fig. 3B). While methyl and ethyl substituted molecules (C1a and C1b) occupied almost the same binding pocket, the propyl-substituted molecule (C1c) occupied a different binding space to accommodate the longer side chain (Fig. S2‡). Subsequently, we prioritized the N-methylpiperazine-benzimidazole ligand and 5th position substituted thalidomide with triethyleneglycol linker to develop the additional NA-TAC molecule, P5, along with its control compound C2 (Fig. 4A, ESI page 2–35‡). We confirmed the efficient G4 DNA binding ability of C2 and P5 in both CD spectroscopic studies and thermal denaturation (Fig. 4B and C). However, we included all the PROTACs and their control molecules for further investigation.
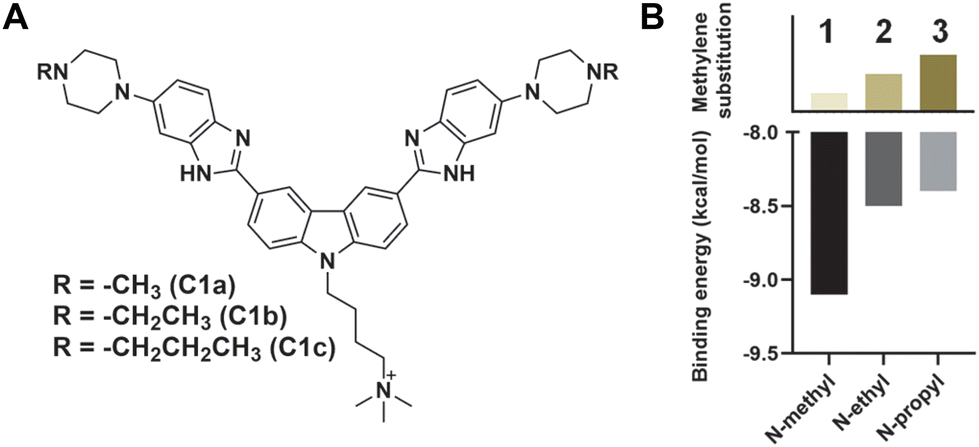 | ||
| Fig. 3 (A) Chemical structures of the C1 analogs and (B) their binding energy with K+-stabilized Hum 21-mer G4 DNA as predicted by AutoDock Vina.16 | ||
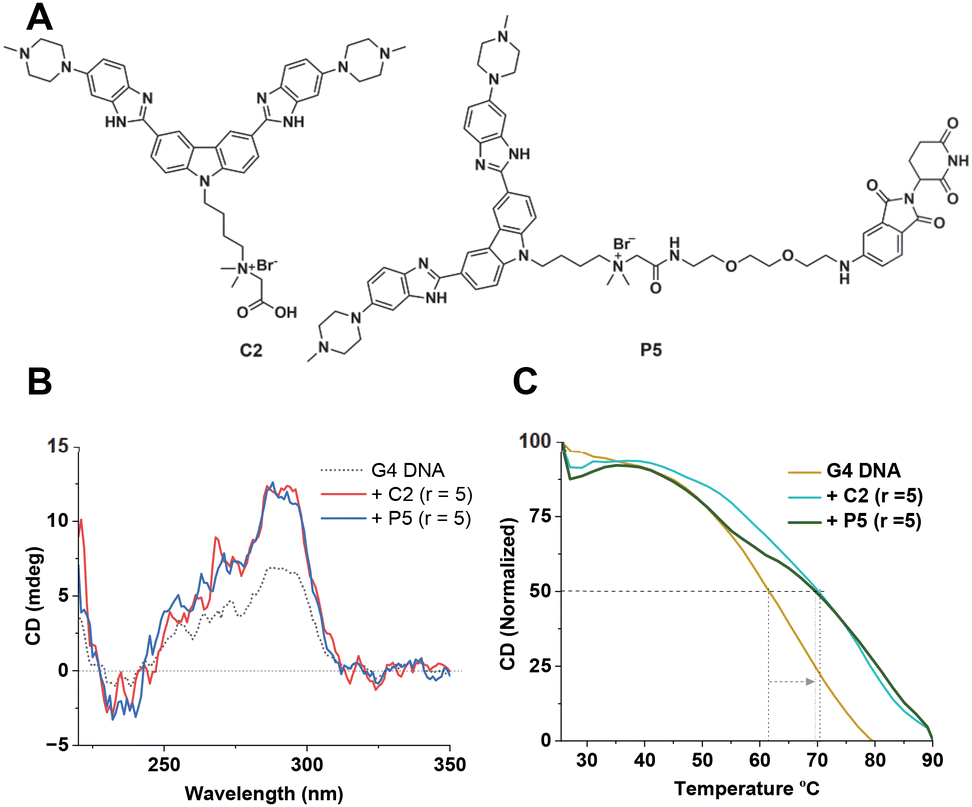 | ||
| Fig. 4 (A) Chemical structures of molecules C2 and P5. (B) CD spectra of 3 μM Hum 21-mer G4 DNA alone, and with compounds C2 and P3 (15 μM) in 10 mM Tris-HCl (pH 7.4) containing 0.1 M KCl and 0.1 mM EDTA. (C) CD melting profiles of the pre-formed Hum 21-mer G4 DNA alone, and G-quadruplex DNA incubated with ligands C2 and P5 (15 μM) in 10 mM Tris-HCl (pH 7.4) containing 0.1 M KCl and 0.1 mM EDTA. | ||
We undertook UV-Vis spectroscopic studies to evaluate the G4 DNA binding affinities of the compounds. All the molecules showed a significant hypochromic effect upon binding, indicating a possible stacking mode of binding with the G-tetrads (Fig. 5A and B, and ESI, Fig. S3‡). While the G4 ligands alone (C1 and C2) showed higher binding affinities, we observed a small reduction for their corresponding PROTAC molecules (Fig. 5C). Interestingly, N-methylpiperazine benzimidazole (C2) showed an almost 25-fold increase in binding affinity compared to the morpholine derivative (C1). However, upon linkage with the thalidomide moiety, the PROTAC molecule P5 showed a 10-fold decrease in the binding affinity. The effect on the binding affinity could be due to the presence of the non-interacting moiety, thalidomide. It is important to note that the PROTAC molecule P5 still showed higher binding affinity than any of the morpholine-benzimidazole derivative PROTACs (P1–P4).
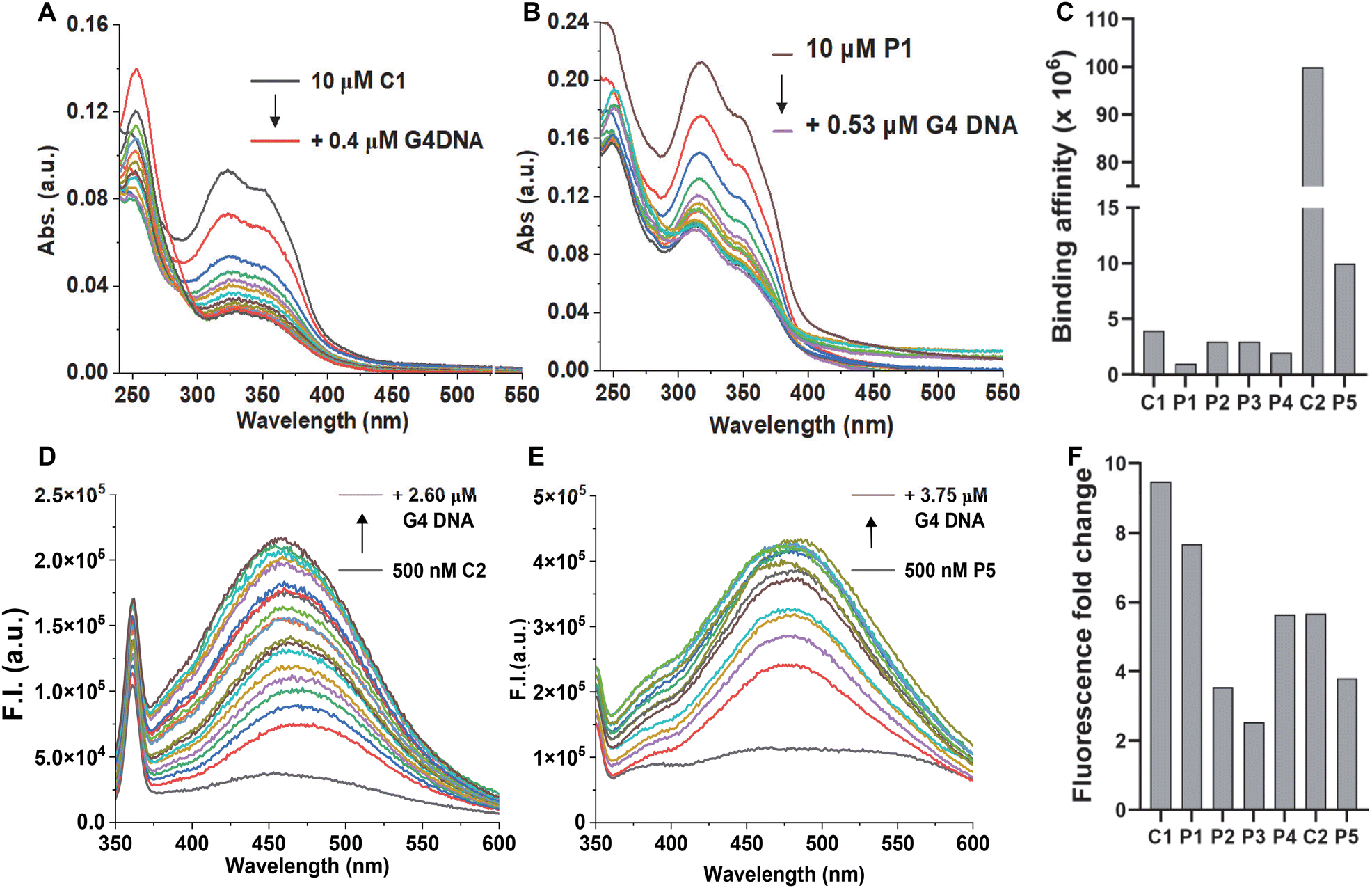 | ||
| Fig. 5 UV-Vis titration spectra of 10 μM ligands (A) C1 and (B) P1, and (C) binding affinities for all the molecules in 10 mM Tris-HCl (pH 7.4), 0.1 mM EDTA and 0.1 M KCl with 10 μM pre-formed Hum 21-mer G4 DNA. Fluorescence titration spectra of 0.5 μM ligands (D) C2 and (E) P5, and (F) fold-change for all the molecules in 10 mM Tris-HCl (pH 7.4), 0.1 mM EDTA and 0.1 M KCl with 10 μM pre-formed Hum 21-mer G4 DNA. | ||
As the benzimidazole scaffold is known to be intrinsically fluorescent, we employed fluorescence spectroscopic studies to probe the G4 DNA binding activities. All the molecules showed low fluorescence signals in the buffer due to the quenching by the aqueous medium. However, the molecules showed a sharp increase in the fluorescence signal upon adding G4 DNA (Fig. 5D–F, and ESI, Fig. S4‡). The recovery in the fluorescence signal indicates their association with G4 DNA and, thus, displacement of water molecules from their environment. Taken together, the fluorescence studies further confirmed the G4 DNA binding activities of the molecules. As the PROTAC molecules have two modules, we asked which moiety is primarily responsible for G4 DNA binding. As the control molecules (C1 and C2) showed strong binding in both UV-vis and fluorescence titrations (Fig. 5 and ESI, Fig. S3 and 4‡) we checked the binding activity of the thalidomide molecule (ESI Scheme E, 23‡) by monitoring its fluorescence. The thalidomide molecule alone showed a minimum change in the fluorescence intensity (∼1.2-fold), indicating a non-interacting nature with G4 DNA (Fig. S4‡). Furthermore, all the PROTAC molecules showed low binding affinity in UV-vis studies (Fig. 5C) and lower fluorescence change in fluorescence titrations (Fig. 5F) than their corresponding control molecules, indicating the non-interacting nature of the thalidomide moiety.
After confirming the G4 DNA binding activities of the molecules by various spectroscopic methods, we sought to unravel their cellular activities. We performed a cell viability assay to assess the compounds’ anti-proliferative activities in lung carcinoma epithelial cell line A549. As expected, none of the compounds showed prominent short-term toxicity up to 6 days (Fig. 6, and ESI, Fig. S5 and 6). However, the compounds started exhibiting anti-proliferative activity in the long term (Fig. 6, and ESI, Fig. S5 and 6). Both the control compounds, C1 and C2, showed moderate anti-proliferative activities against A549 cells. Interestingly, P3 showed higher anti-proliferative activity than its control compound C1 (Fig. 6, and ESI, Fig. S5 and 6). In addition, PROTAC P5 showed significantly higher toxicity than its control compound C2 (Fig. 6, and ESI, Fig. S5 and 6). The dose–time-dependent long-term cytotoxicity confirms the highest activity of the P5 compound with the best IC50 value of 4.13 μM (Fig. 6D–F). Even though some of the NAPs are reported to show better activity, they require delivery vehicles due to their cellular impermeability.6,8,17 The brightfield microscopy images also revealed the anti-proliferative activities of the NA-TAC molecules (Fig. 7). While the cells treated with DMSO for 14 days showed normal morphology with a highly proliferative nature (Fig. 7A), the control compound (C2) showed a retardation in cell growth (Fig. 7D). PROTAC P3 showed a dose-dependent high anti-proliferative activity compared to its control molecule C1 (Fig. 7B and E). Notably, PROTAC P5 exhibited severe toxicity and the highest anti-proliferative activity (Fig. 7C and F).
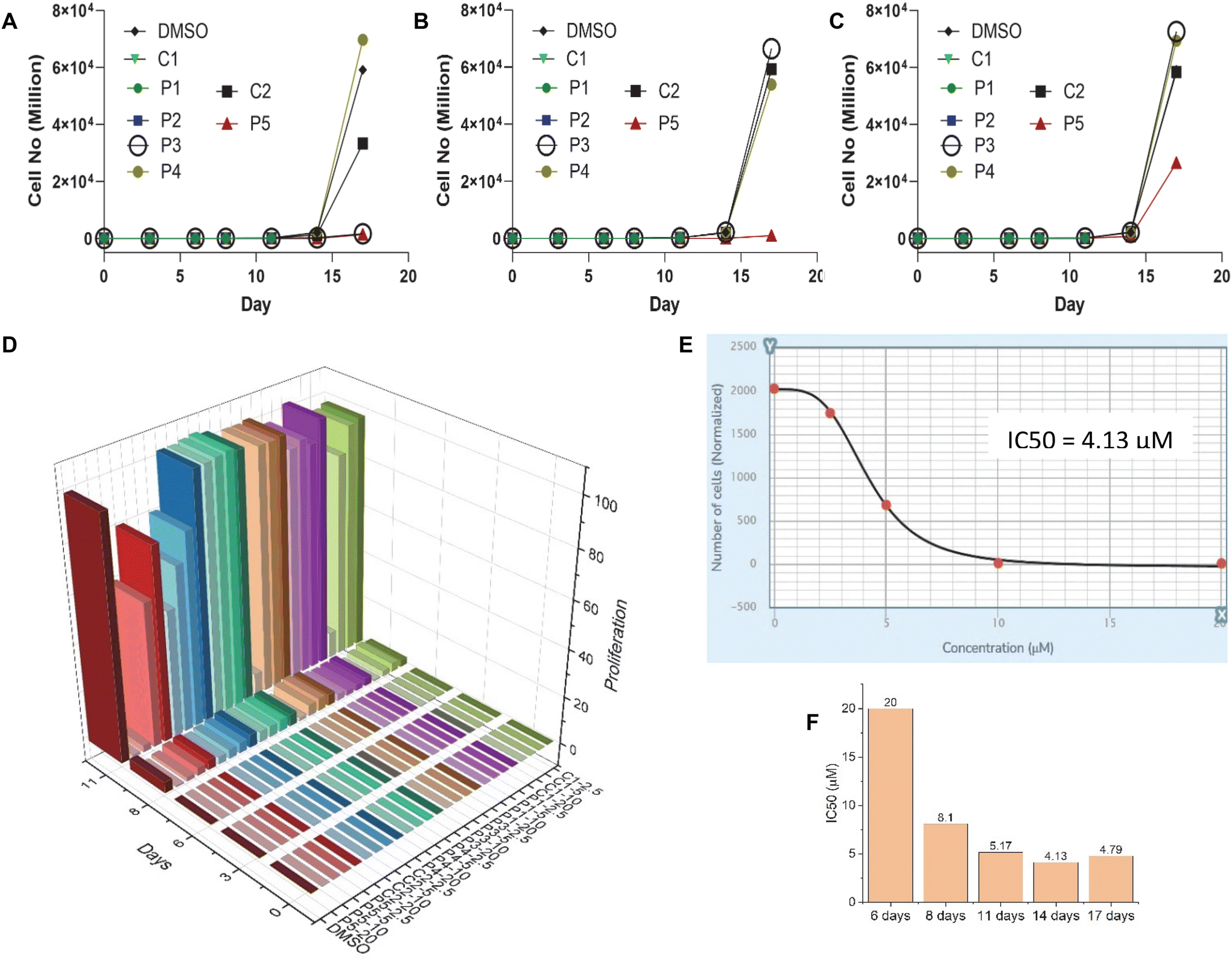 | ||
| Fig. 6 Effect of ligands on the A549 cell viability upon long-term exposure to different concentrations: (A) 20 μM, (B) 10 μM, and (C) 5 μM (up to 17 days), as analyzed by FACS. (D) Long-term cytotoxicity 3D plot showing the interplay of dose–time–antiproliferative activity. (E) IC50 value calculation from a dose–response antiproliferation plot in a 14-day long-term assay for P5. (F) IC50 values of P5 as evaluated in a long-term cytotoxicity assay for different incubation times. | ||
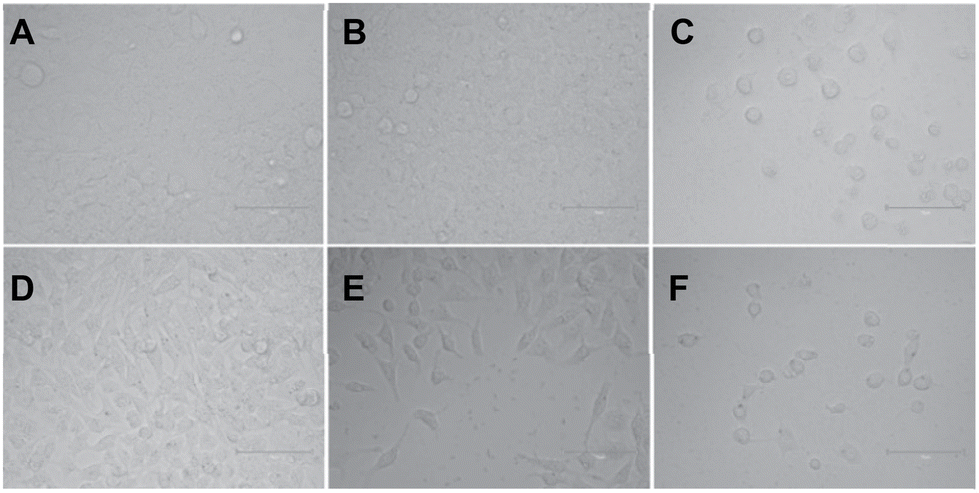 | ||
| Fig. 7 Representative bright field images of the cell morphology of A549 cells treated with (A) DMSO, (B) 10 μM P3, (C) 10 μM P5, (D) 20 μM C2, (E) 20 μM P3, and (F) 20 μM P5 after 14 days. | ||
After confirming the G4 DNA targeting ability of the PROTAC molecules and their anti-proliferative and cytotoxic activities, we wanted to investigate the cellular mechanism.11,18 We selected the most active P5 and its control compound, C2, for cell cycle analysis studies. The Annexin V/PI assay showed that while DMSO-treated cells remained healthy, the C2-treated A549 cells generated a small early and late apoptosis population (Fig. 8A and B). Interestingly, the effect was even more prominent for the PROTAC molecule P5 (Fig. 8C). This observation indicates that, most likely, the molecules induce a DNA damage-mediated apoptotic pathway.
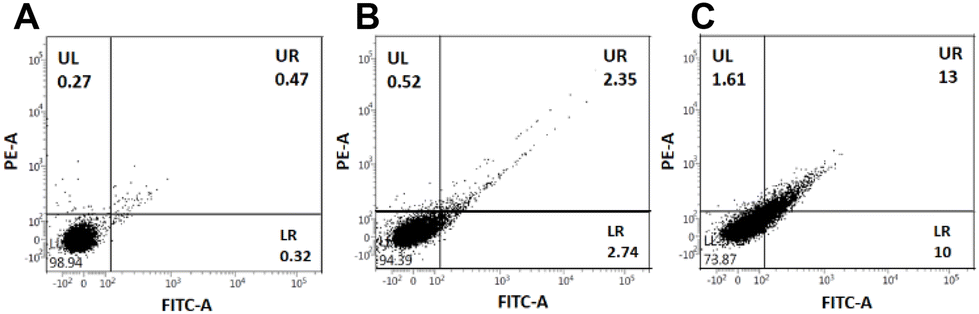 | ||
| Fig. 8 Representative dot plots for Annexin-V and PI staining of (A) untreated cells (A549) as a control and after incubation with 20 μM ligands (B) C2 and (C) PROTAC P5. The different stages of cells were assigned as alive cells (LL), early apoptotic cells (LR), late apoptotic cells (UR), and necrotic cells (UL). The depiction of the apoptotic cell population is made in the lower right quadrant (LR), which originated due to the Annexin V–FITC staining only. | ||
To investigate the effect of the compounds further, we performed fluorescence confocal microscopy imaging of the treated A549 cells and compared them with their untreated counterparts. While the DMSO-treated cells showed mostly intact and healthy nuclei with an average 5 μm size (Fig. 9A and B), the P3-treated cells showed significant depletion in the genomic material and nuclear fragmentation (Fig. 9C and D). PROTAC P5 showed an even more prominent phenotypic effect, resulting in severe cytotoxicity (Fig. 9E and F). Interestingly, upon prolonging the treatment time, P5 showed visual evidence of anti-proliferative activity in the residual cells (Fig. 9G–L). The microscopy images indicate that the nuclei are severely fragmented (Fig. 9G–J) or cells are trapped in the premitotic stage (Fig. 9K and L), leading to the combined anti-proliferative activity.19
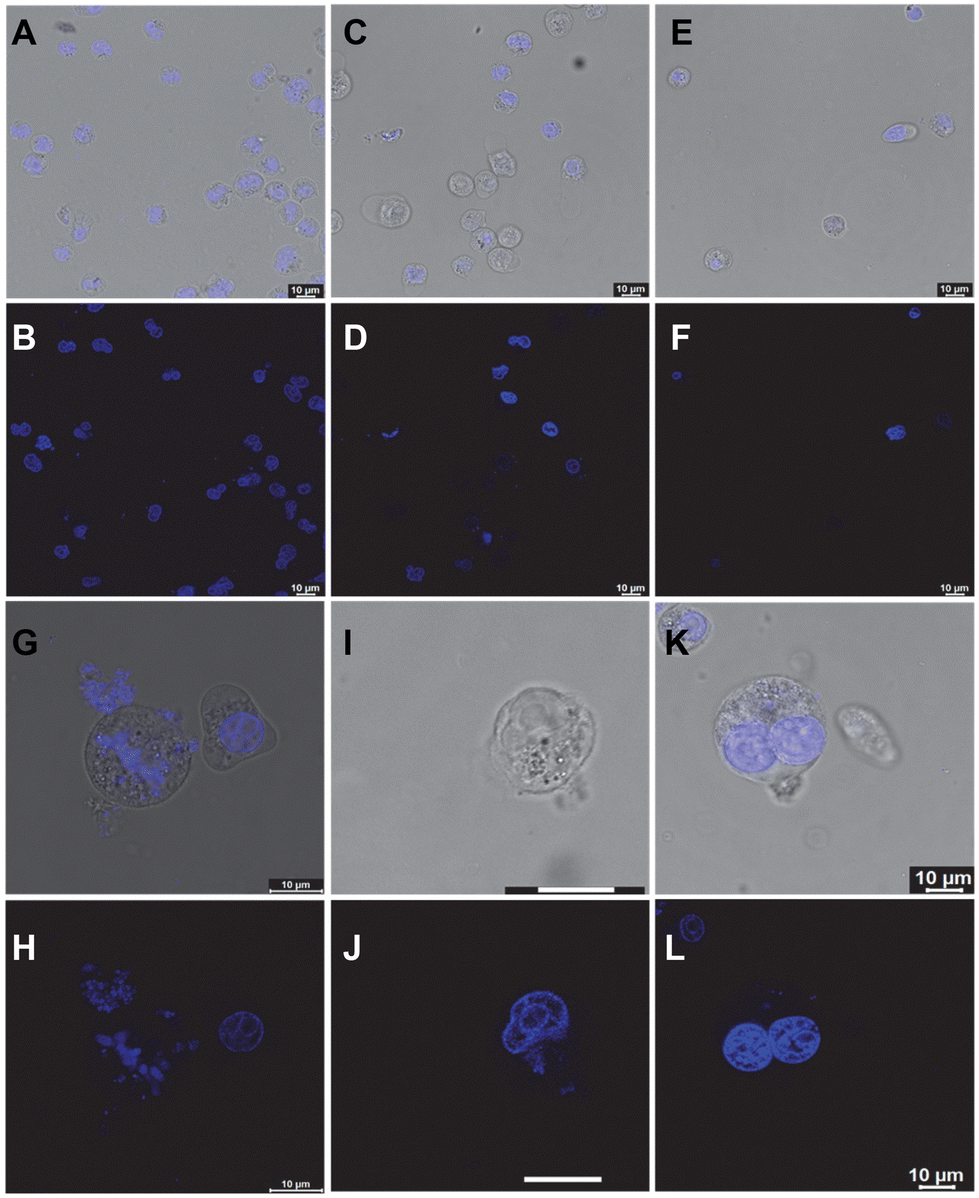 | ||
| Fig. 9 Confocal microscopy images of A549 cells treated with (A and B) DMSO, (C and D) 10 μM P3, and (E and F) 10 μM P5 for 8 days before transferring onto the imaging slide and counter-staining with DAPI. Panels G–L are representative high-resolution confocal images of A549 cells treated with 10 μM P5 for 11 days. | ||
Furthermore, to decipher the molecular mechanism of NA-TAC-induced apoptosis, we evaluated the genomic integrity of the treated cells. H2AX is a variant of one of the eukaryotic core histone proteins, named H2A, which remains tightly bound to the chromatin, indicating genome integrity.20 We wanted to check if the NA-TAC molecule exerts any change in the H2AX with the help of the immunofluorescence technique. H2AX is a 143 amino acid protein that gets phosphorylated at the Ser139 position by kinases like ATR, ATM, and DNA-PK and forms γ-H2AX in response to DNA damage and repair.21 These γ-H2AX, or DNA damage foci, were visualized using fluorescence confocal microscopy.22 Interestingly, the γ-H2AX intensity detected by immunofluorescence in A549 cells showed a dramatic increase in the foci signal from the nuclei of P5-treated cells (Fig. 10D–L), while no such signal was observed in the DMSO control (Fig. 10A–C). These observations further confirm the NA-TAC-induced substantial DNA damage resulting in programmed cell death.
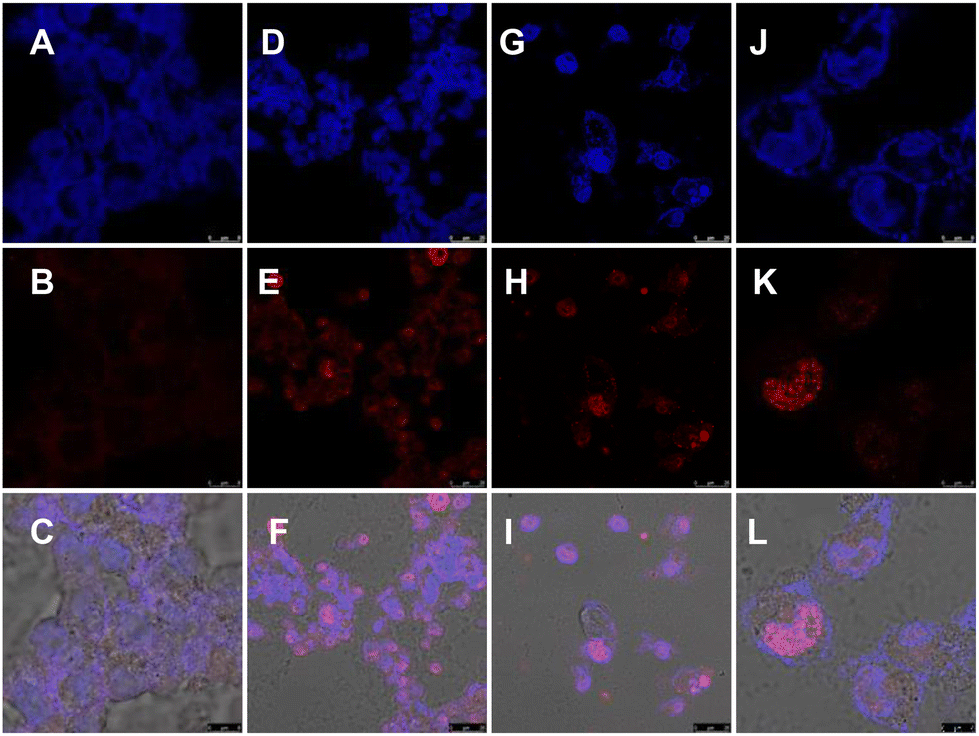 | ||
| Fig. 10 Representative fluorescence microscopy images of immunofluorescent γ-H2AX in A549 cells treated with (A–C) DMSO, (D–F) 10 μM P5, or (G–L) 20 μM P5. The top panels represent the DAPI channel, the middle panels represent the γ-H2AX channel, and the bottom panels represent the overlay of DAPI and γ-H2AX. | ||
Based on the spectroscopic studies, it is evident that the benzimidazole ligand of the PROTAC molecules interacts with the target G4 DNA (Fig. 5). On the other hand, thalidomide binding with E3 ligase cereblon is well characterized and documented. We asked whether a putative model for the ternary complex could be developed. Initially, we performed a molecular docking study of the PROTAC molecule P5 fragment with the potassium-stabilized human telomeric G-quadruplex DNA (PDB 1KF1).23 The docking study reveals that the benzimidazole–carbazole planner scaffold efficiently stacks over the G4 tetrad (Fig. 11A). The distance between the molecule and the G4 tetrad is around 3.9 Å, comparable with the stacking distances between the G4 tetrads (3.5 Å) (Fig. 11B). In addition, the N-atom of the benzimidazole residue makes a close contact of 2.5 Å, possibly due to the polar interaction (Fig. 11B). The protonatable side chain residues of the ligand approached the negatively charged grooves of the G4 DNA due to favorable ionic interactions (Fig. 11A and B). On the other hand, the interaction between the thalidomide module and cereblon is well understood, and several co-crystal structures are reported. We selected the human cereblon-bound thalidomide co-crystal structure (PDB 8OIZ) for the other end of the ternary complex.24 We developed a putative model where the benzimidazole ligand interacts with the G4 DNA (Fig. 11C) and the thalidomide ligand is bound to the cereblon protein (Fig. 11D) to form the plausible ternary complex (Fig. 11E).
 | ||
| Fig. 11 Computational analysis of the G DNA and molecular interaction. (A and B) Docked structure of C1a with 1KF1 showing the stacking mode of binding onto the G4 tetrad. (C) Modelling of the P5–1KF1 complex based on C1a–1KF1 docking. (D) Extracted model of the human cereblon bound thalidomide structure from crystal structure PDB 8OIZ. (E) Putative model of the ternary complex between cereblon, P5, and 1KF1. | ||
Conclusions
In summary, we introduced a new class of PROTAC molecules for targeting non-protein biomolecules. The molecules are systematically designed to target higher-order G-quadruplex DNA structures. We unambiguously demonstrate that the PROTAC molecules can bind with the G4 DNA and show anti-proliferative activity. Furthermore, we uncovered the cellular pathway and found that the PROTAC molecules induced DNA damage, nuclear fragmentation, and apoptosis. Importantly, even though the control molecules have higher G4 DNA binding activities, the PROTACs exhibited higher cellular activities across several orthogonal assays. These observations strongly suggest that our designed NA-TACs are not only targeting the G4 DNA and perturbing the associated cellular pathways but also possibly inducing additional proteolytic activities to the interactome through the thalidomide module. As the carbazole–benzimidazole ligands are capable of targeting both telomeric and genomic G4 DNA, the cellular effects are most possibly due to multiplexed activities.25 Our future studies will focus on deciphering each of these complex pathways to establish the field of G4-specific NA-TACs and precision medicine.Author contributions
All the authors have contributed to the manuscript. BM conceptualized the project. ST, AK, AH, and BM performed the experiments and processed the data. PB, AH, and BM performed the computational analysis and modeling. All the authors contributed to the manuscript writing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
ST, AK, and AH thank Ashoka University, CSIR, and UGC for financial support, respectively. BM thanks Bose Institute for financial support and CIF (Bose Institute) for the instrumental facility.References
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- R. Casement, A. Bond, C. Craigon and A. Ciulli, Methods Mol. Biol., 2021, 2365, 79–113 CrossRef CAS PubMed.
- K. Li and C. M. Crews, Chem. Soc. Rev., 2022, 51, 5214–5236 RSC.
- (a) J. Liu, H. Chen, H. Ü. Kaniskan, L. Xie, X. Chen, J. Jin and W. Wei, J. Am. Chem. Soc., 2021, 143, 8902–8910 CrossRef CAS PubMed; (b) X. Huang, F. Wu, J. Ye, L. Wang, X. Wang, X. Li and G. He, Acta Pharm. Sin. B, 2024, 2402–2427 CrossRef CAS PubMed; (c) L. Zhang, L. Li, X. Wang, H. Liu, Y. Zhang, T. Xie, H. Zhang, X. Li, T. Peng, X. Sun, J. Dai, J. Liu, W. Wu, M. Ye and W. Tan, Mol. Ther.–Nucleic Acids, 2022, 30, 66–79 CrossRef CAS PubMed; (d) J. Lu, Y. Huang, J. Huang, R. He, M. Huang, X. Lu, Y. Xu, F. Zhou, Z. Zhang and K. Ding, J. Med. Chem., 2022, 65, 2313–2328 CrossRef CAS PubMed; (e) M. Philpott, J. Yang, T. Tumber, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, T. Keates, I. Felletar, A. Ciulli, S. Knapp and T. D. Heightman, Mol. BioSyst., 2011, 7, 2899–2908 RSC.
- (a) B. Maji and S. Bhattacharya, Chem. Commun., 2014, 50, 6422–6438 RSC; (b) A. Ghidini, A. Cléry, F. Halloy, F. H. T. Allain and J. Hall, Angew. Chem., 2021, 133, 3200–3206 CrossRef.
- (a) K. M. Patil, D. Chin, H. L. Seah, Q. Shi, K. W. Lim and A. T. Phan, Chem. Commun., 2021, 57, 12816–12819 RSC; (b) Z. Wang, J. Liu, H. Chen, X. Qiu, L. Xie, H. Ü. Kaniskan, X. Chen, J. Jin and W. Wei, J. Am. Chem. Soc., 2023, 145, 10872–10879 CrossRef CAS PubMed.
- M. Girardini, C. Maniaci, S. J. Hughes, A. Testa and A. Ciulli, Bioorg. Med. Chem., 2019, 27, 2466–2479 CrossRef CAS PubMed.
- Y. Tong, Y. Lee, X. Liu, J. L. Childs-Disney, B. M. Suresh, R. I. Benhamou, C. Yang, W. Li, M. G. Costales, H. S. Haniff, S. Sievers, D. Abegg, T. Wegner, T. O. Paulisch, E. Lekah, M. Grefe, G. Crynen, M. Van Meter, T. Wang, Q. M. R. Gibaut, J. L. Cleveland, A. Adibekian, F. Glorius, H. Waldmann and M. D. Disney, Nature, 2023, 618, 169–179 CrossRef CAS PubMed.
- M. H. Kaulage, B. Maji, S. Pasadi, A. Ali, S. Bhattacharya and K. Muniyappa, Eur. J. Med. Chem., 2018, 148, 178–194 CrossRef CAS PubMed.
- B. Maji, K. Kumar, M. Kaulage, K. Muniyappa and S. Bhattacharya, J. Med. Chem., 2014, 57, 6973–6988 CrossRef CAS PubMed.
- B. Maji, K. Kumar, K. Muniyappa and S. Bhattacharya, Org. Biomol. Chem., 2015, 13, 8335–8348 RSC.
- T. Mori, T. Ito, S. Liu, H. Ando, S. Sakamoto, Y. Yamaguchi, E. Tokunaga, N. Shibata, H. Handa and T. Hakoshima, Sci. Rep., 2018, 8, 1294 CrossRef PubMed.
- X. Yang, Z. Wang, Y. Pei, N. Song, L. Xu, B. Feng, H. Wang, X. Luo, X. Hu, X. Qiu, H. Feng, Y. Yang, Y. Zhou, J. Li and B. Zhou, Eur. J. Med. Chem., 2021, 218, 113341 CrossRef CAS PubMed.
- V. Poongavanam, Y. Atilaw, S. Siegel, A. Giese, L. Lehmann, D. Meibom, M. Erdelyi and J. Kihlberg, J. Med. Chem., 2022, 65, 13029–13040 CrossRef CAS PubMed.
- A. Bricelj, Y. L. Dora Ng, D. Ferber, R. Kuchta, S. Müller, M. Monschke, K. G. Wagner, J. Krönke, I. A.-O. Sosič, M. A.-O. Gütschow and C. A.-O. Steinebach, ACS Med. Chem. Lett., 2021, 12, 1733 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 2010 CrossRef PubMed.
- (a) A. Ghidini, A. Cléry, F. Halloy, F. H. T. Allain and J. Hall, Angew. Chem., Int. Ed., 2021, 60, 3163–3169 CrossRef CAS PubMed; (b) K. T. G. Samarasinghe, S. Jaime-Figueroa, M. Burgess, D. A. Nalawansha, K. Dai, Z. Hu, A. Bebenek, S. A. Holley and C. M. Crews, Cell Chem. Biol, 2021, 28, 648–661 CrossRef CAS PubMed; (c) S. He, F. Gao, J. Ma, H. Ma, G. Dong and C. Sheng, Angew. Chem., Int. Ed., 2021, 60, 23299–23305 CrossRef CAS PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1955, 184, 39–51 CrossRef PubMed.
- D. Wengerodt, C. Schmeer, O. W. Witte and A. Kretz, in Cells, 2019, Vol. 8, p. 1546 Search PubMed.
- O. A. Sedelnikova, D. R. Pilch, C. Redon and W. M. Bonner, Cancer Biol. Ther., 2003, 2, 233–235 CrossRef CAS PubMed.
- (a) L. Mah, A. El-Osta and T. Karagiannis, Leukemia, 2010, 24, 679–686 CrossRef CAS PubMed; (b) A. Ivashkevich, C. E. Redon, A. J. Nakamura, R. F. Martin and O. A. Martin, Cancer Lett., 2012, 327, 123–133 CrossRef CAS PubMed.
- M. Poivre, M.-H. Antoine, K. Kryshen, A. Atsapkina, A. N. Shikov, L. Twyffels, A. Nachtergael, P. Duez and J. Nortier, Toxins, 2023, 15, 52 CrossRef CAS PubMed.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS PubMed.
- H. A.-O. Bouguenina, A. Scarpino, J. A. O'Hanlon, J. Warne, H. Z. Wang, L. C. Wah Hak, A. Sadok, P. C. McAndrew, M. Stubbs, O. A. Pierrat, T. Hahner, M. P. Cabry, Y. V. Le Bihan, C. Mitsopoulos, F. J. Sialana, T. I. Roumeliotis, R. Burke, R. L. M. van Montfort, J. Choudhari, R. Chopra, J. J. Caldwell and I. Collins, ChemBioChem, 2023, 24, e202300351 CrossRef CAS PubMed.
- (a) M. Kaulage, B. Maji, J. Bhat, Y. Iwasaki, S. Chatterjee, S. Bhattacharya and K. Muniyappa, J. Med. Chem., 2016, 59, 5035–5050 CrossRef CAS PubMed; (b) T. Hussain, D. Saha, G. Purohit, A. Kar, A. Kishore Mukherjee, S. Sharma, S. Sengupta, P. Dhapola, B. Maji, S. Vedagopuram, N. T. Horikoshi, N. Horikoshi, R. K. Pandita, S. Bhattacharya, A. Bajaj, J.-F. Riou, T. K. Pandita and S. Chowdhury, Sci. Rep., 2017, 7, 11541 CrossRef PubMed; (c) R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
Footnotes |
| † This work is dedicated to Prof. Santanu Bhattacharya on his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01006j |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |