
Enhancing the electronic structure of Ni-based electrocatalysts through N element substitution for the hydrogen evolution reaction†
Yibin
Yang
 a,
Xin
Jin
b,
Fangyang
Zhan
*c and
Yang
Yang
*d
a,
Xin
Jin
b,
Fangyang
Zhan
*c and
Yang
Yang
*d
aChemical Pollution Control Chongqing Applied Technology Extension Center of Higher Vocational Colleges, Chongqing Industry Polytechnic College, Chongqing 401120, PR China
bCollege of Physics and Electronic Engineering, Chongqing Normal University, Chongqing 401331, P. R. China
cInstitute for Structure and Function & Department of Physics, Chongqing University, Chongqing 400044, P. R. China. E-mail: zhan_fyang@cqu.edu.cn
dKey Laboratory of Chemical Additives for China National Light Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science & Technology, 710021 Xi'an, China. E-mail: yyang399@sust.edu.cn
First published on 23rd May 2024
Abstract
The weak orbital coupling between Ni3N and H2O, caused by its interstitial structure and attenuated Ni–N interaction, is attributed to the high unoccupied d orbital energy of Ni3N. Consequently, the kinetics for water dissociation in the HER are slow. In this study, we effectively lowered the energy state of vacant d orbitals in Ni3N, which resulted in an exceptionally efficient HER. The as-synthesized Ni3N catalyst demonstrates an overpotential of 135 mV when subjected to a current density of 10 mA cm−2. The refined structural characterization suggests that the introduction of oxygen results in a reduction in electron densities surrounding the Ni sites. Furthermore, DFT calculations provide additional evidence that the electrocatalyst of Ni3N generates a greater number of lowest unoccupied orbitals (LUMOs) and improved alignment, thereby enhancing the adsorption and splitting of water. The notion of orbital-regulated electronic levels on Ni sites introduces a distinctive methodology for the systematic development of catalysts used in hydrogen evolution and other applications.
Introduction
In recent years, the electrochemical hydrolysis method has gained increasing attention owing to its high efficiency and environmentally friendly nature, making it a promising approach for generating high-purity hydrogen, and electrocatalytic water splitting undoubtedly has the potential to decelerate the consumption of conventional fossil fuels and alleviate the burden of environmental pollution.1–4 However, the hydrogen evolution reaction (HER) requires a high overpotential,1,5–9 which inevitably limits the application of hydrogen production in industrial practice. Currently, Pt-based electrocatalysts are widely recognized as active electrocatalysts for the HER because of their optimal Pt–H bond strength and Gibbs free energy (ΔG) to form the Pt–H bond.10–13 However, the widespread application of these resources is impeded by their exorbitant cost. Therefore, the development of platinum-free electrocatalysts, which are both cost-effective and exhibit superior HER performance, is of paramount importance. This is a critical step towards realizing large-scale hydrogen production.14–18Over the past several years, significant progress has been accomplished in the quest for cost-effective and efficient electrocatalysts for the HER.9,19–22 This success is attributed to the relentless efforts and comprehensive research conducted in this field. Transition metal oxides,23–26 sulfides15,19,27 and carbides28–32 have been developed and widely used as alternatives to precious metal electrocatalysts for the HER in acid and/or alkaline solutions. However, the major drawback of these electrocatalysts lies in their inadequate electron conductivity, which significantly impedes the transport of electrons from the surface of the electrocatalyst to the supporting electrode. Furthermore, the inadequate electron conductivity occasionally results in the formation of Schottky barriers at both the electrocatalyst-electrolyte and the electrocatalyst-support electrode interface. Consequently, additional overpotential is required to overcome these energy barriers, resulting in a decrease in energy conversion efficiency. Hence, researchers have identified two effective approaches for designing active catalysts within these systems. The initial approach involves the incorporation of additional active sites for the HER and the induction of structural disorders. Numerous studies have been dedicated to enhancing the density of electrocatalytic active sites via forming defects. The second approach focuses on improving the electrical conductivity of electrocatalysts. Considerable research has been conducted to investigate the utilization of conducting materials as catalyst supports in order to improve the electrical conductivity of the electrocatalysts. With the aforementioned problems of electrocatalysts, significant efforts are dedicated to enhancing the performance of the HER by improving both of these factors.
Recently, transition metal nitrides (TMNs) have emerged as highly efficient electrocatalysts for hydrogen production.18,28,33–41 This is primarily due to their metallic nature, which enables enhanced electrocatalytic activity. The remarkable catalytic performance of these TMNs within the HER domain can be ascribed to their distinctive electronic configuration. This unique structure promotes optimal proton adsorption onto the nitride surface throughout the electrocatalytic reaction. The incorporation of catalysts with high metallicity significantly enhances electron transport in the HER process, thereby promoting the overall activity of HER. Recently, Zhang et al.34 have successfully developed a 3D porous nickel molybdenum nitride material that demonstrates exceptional performance in HER. Additionally, they presented an efficient water electrolysis device, which is based on hierarchical TiN@Ni3N nanowire arrays.34 Theoretical investigations suggest that Ni3N possesses intrinsic metallic properties and has excellent electrical conductivity.36 In addition, the disordered atomic structure of nanosheets that are only a few atoms thick can offer a greater number of active sites. Accordingly, it is anticipated that 2D Ni3N nanosheets will exhibit exceptional electrocatalytic activity. Specifically, uncovering the exact active site location of Ni3N is expected to serve as an effective protocol for enhancing the activity of HER and designing electrocatalysts for HER. Among many materials, Ni3N is enormously favored by researchers because of its excellent conductivity. However, the water dissociation efficiency of Ni3N is insufficient, while the energy barrier for HER is prohibitively elevated. The markedly inferior performance of Ni3N in comparison to Pt can be attributed to primary factors, thereby presenting substantial opportunities for enhancement. The coupling effect of the interface is instrumental in augmenting the electrocatalytic activity. This enhancement is achieved through the regulation of the local electronic structure at the interface, which in turn adjusts the orbital overlap among various atoms. Ultimately, this process modifies the strength of adsorption–desorption between the electrocatalysts and the reaction intermediates. Therefore, integrating an appropriate element into Ni3N to establish a composite, characterized by a robust interface coupling effect, harbors significant potential for augmenting the electrocatalytic activity.
In this study, we present the design and synthesis of 3D Ni3N/NF to improve the performance of HER in 1.0 M KOH. Benefiting from the robust chemical coupling and synergistic effects at the interface of the electrocatalysts, the Ni3N/NF hybrid electrode demonstrates exceptional catalytic performance for HER. The single NiO electrode is surpassed by achieving an overpotential of a mere 135 mV at a current density of 10 mA cm−2 in a 1 M KOH solution, demonstrating a reduction of 177 mV. Moreover, the utilization of 3D metallic Ni3N nanosheets can lead to further enhancements in carrier concentration and electrical conductivity, as demonstrated by DFT calculations. Specifically, the Ni site on the Ni3N surface exhibits the lowest ΔGH* value of 1.63 eV. This can be attributed to the coupling effect between the Ni site and N atoms around the Ni site, making these sites the most active for the HER in Ni3N. The Ni3N nanosheets we have developed and studied, which exhibit excellent hydrogen evolution reaction activity under the guidance of a supervisor, hold significant potential for future applications in hydrogen production technologies.
Results and discussion
The Ni(OH)2/NF nanosheets array, shown in Fig. 1a, was directly grown on Ni foam using a hydrothermal method. Subsequently, the Ni(OH)2/NF was calcined under NH3 atmosphere at 400 °C to obtain Ni3N/NF. Fig. 1b illustrates the XRD patterns of the as-synthesized Ni(OH)2, NiO, and Ni3N nanosheets scraped down from NF. The XRD patterns of Ni3N/NF (JCPDS Card No. 10-0280) are represented by the green line in Fig. 1b. The diffraction peaks observed, specifically (110), (002), (112), (300), (111), (200), (220), and (113), exhibit a sequence of Bragg reflections. These reflections align harmoniously with the established patterns of Ni3N and nickel foam, thereby confirming their consistency. The diffraction peaks identified at 44.4°, 51.8°, and 76.3° correspond respectively to the (111), (200), and (220) planes of the nickel foam (Ni, JCPDS Card No. 01-1258). Therefore, the successful synthesis of Ni3N/NF is confirmed. Additionally, NiO/NF (violet line) and Ni(OH)2/NF (blue line) were also successfully prepared. Fig. 1c demonstrates the scanning electron microscopy (SEM) image of the three-dimensional open porous structure of the NF. The SEM images in Fig. 1d and e show the vertical growth of nanosheets of Ni(OH)2 and Ni3N on the NF substrate, respectively. The nanosheet morphology is further revealed in the transmission electron microscopy (TEM) image shown in Fig. S1.† The formation of the crystallized phase of Ni3N is confirmed in Fig. S2.† Additionally, the high-resolution transmission electron microscopy (HRTEM) image presented in Fig. S3† exhibits distinct lattice fringes with an interplanar spacing of 0.24 nm, which is indicative of the (110) plane of the nickel nitride (Ni3N) nanosheets. Furthermore, the selected area electron diffraction (SAED) patterns, as illustrated in Fig. S4,† reveal diffraction rings that are indicative of the (001) and (110) planes of the Ni3N nanosheets. These observations are in agreement with the X-ray diffraction (XRD) patterns, specifically the green line in Fig. 1b, confirming the crystallographic planes of the Ni3N nanosheets. In order to ascertain the spatial distribution of individual elements within the nickel foam, energy dispersive X-ray spectroscopy (EDS) elemental mapping analysis was conducted. Fig. 1f clearly illustrates a homogeneous distribution of Ni and N elements, with no discernible segregation. This suggests the successful synthesis of Ni3N on the NF substrate. However, the presence of oxygen in the samples is indicated by EDS (Fig. 1f), which could be attributed to oxide or absorbed oxygen species. | ||
| Fig. 1 (a) A schematic of the synthesis process of Ni3N; (b) XRD patterns of as-prepared Ni(OH)2/NF, NiO/NF and Ni3N/NF; (c) SEM image of nickel foam; (d and e) SEM images of as-prepared Ni(OH)2/NF, NiO/NF and Ni3N/NF nanosheets; (f) EDX patterns of Ni3N/NF. | ||
X-ray photoelectron spectroscopy (XPS) was utilized to examine the influence of non-metallic elements on the surface conditions and electronic configuration of the freshly synthesized NiO, Ni3N, and Ni(OH)2 specimens. The XPS survey spectra, as depicted in Fig. 2a, indicate that the samples display analogous survey spectra. However, an exception is noted in the form of a minor peak, which is ascribed to the existence of the N element in the Ni3N sample. The high-resolution Ni 2p spectra of Ni(OH)2, as depicted in Fig. 2b, exhibit two peaks. These peaks, with binding energies at 855.8 eV and 873.7 eV, correspond to the Ni2+ 2p3/2 and Ni2+ 2p1/2 states, respectively. This observation aligns with findings from a prior study.42 Compared with Ni(OH)2, the peak positions of NiO (855.9 eV and 853.6 eV) and Ni3N (854.9 eV and 852.9 eV) in the electrocatalysts are shifted to lower energies, elucidating that O and N in the electrocatalysts figure electron-rich centres and the Ni atoms in the NiO and Ni3N receive electron transfers from O and Ni, respectively. Especially, the peak position of Ni atoms in Ni3N is lower than that of the Ni atoms in NiO and Ni(OH)2, indicating the Ni atoms in Ni3N receive more electrons than the Ni atoms in NiO and Ni(OH)2. Additionally, the XPS O 1s profiles in Ni(OH)2 can typically be deconvoluted into two dominating characteristic peaks at 533.6 eV and 532.1 eV, which corresponded to the absorbed O species, e.g., water molecules and surface hydroxyl groups, respectively (Fig. 2c). In comparison with the peak position of O atoms in Ni(OH)2, the peak position of the O atoms in NiO (533.1 eV and 530.8 eV) peaks also exhibits an obvious negative shift, suggesting that the O atoms in NiO receive more electrons than the O atoms in Ni(OH)2. The high-resolution N 1s spectrum of Ni3N exhibits a peak at 398.2 eV, which corresponds to the bound N element (Fig. 2d). In comparison with the XPS peak of N 1s in N2 at 406 eV,43 the electron is transferred from the N atom to the Ni atom in Ni3N. In other words, the charge is transferred from N-sites to Ni-sites in Ni3N, and the number of electrons on the Ni-site in the electrocatalysts is increased, which would affect the adsorption energy and energetic barrier of dissociation of active species for HER.
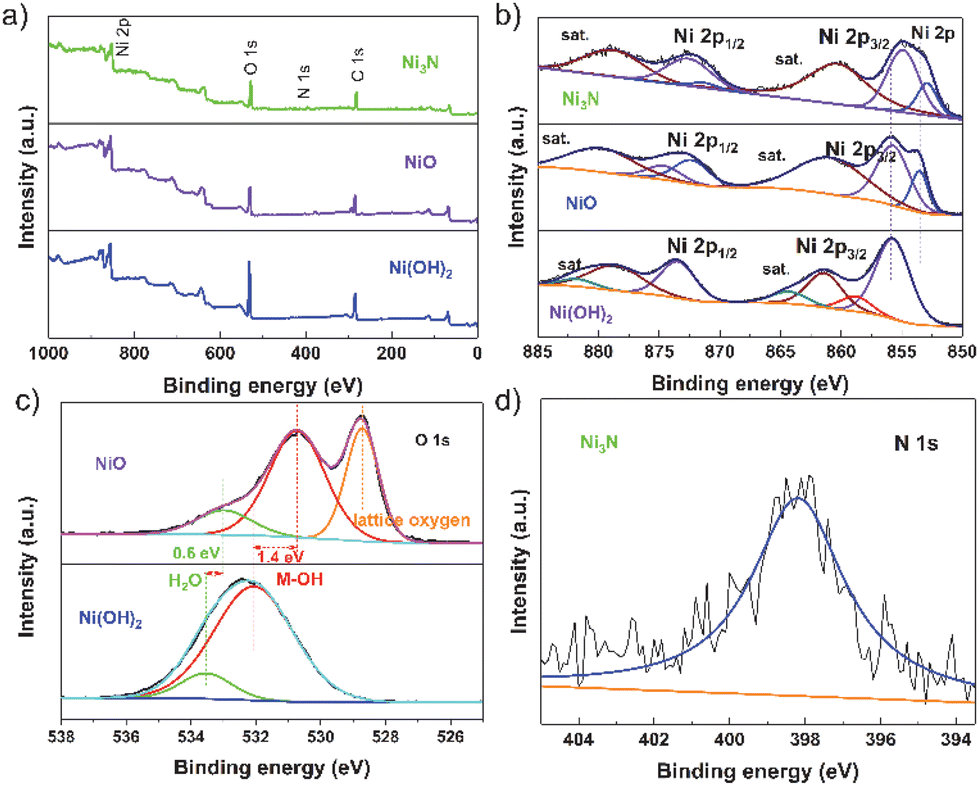 | ||
| Fig. 2 (a) The XPS survey spectra of Ni3N, NiO and Ni(OH)2. The high-resolution XPS spectra of (b) Ni and (c) O in the NiO and Ni(OH)2. (d) the XPS spectra of N in Ni3N. | ||
To assess the electrocatalytic ability to evolve hydrogen, we performed a detailed characterization of electrocatalyst samples with a surface area of 1 × 1 cm2. Comparative analyses were carried out to measure the electrocatalytic activities of bare nickel foam (NF), nickel nitride on nickel foam (Ni3N/NF), nickel oxide on nickel foam (NiO/NF), and nickel hydroxide on nickel foam (Ni(OH)2/NF). The cathodic polarization curves of HER on the electrocatalysts are exhibited in Fig. 3a. The Ni3N/NF electrocatalyst necessitated an overpotential of merely 135 mV to attain an electrocatalytic current density of 10 mA cm−2. This value is substantially inferior to the overpotentials required by the bare nickel foam (298 mV), nickel oxide/nickel foam (NiO/NF, 177 mV), and nickel hydroxide/nickel foam (Ni(OH)2/NF, 147 mV), highlighting the enhanced efficiency of the Ni3N/NF catalyst. This indicates that Ni3N/NF exhibits remarkable catalytic activity towards HER, with a lower onset potential compared to NF, NiO/NF, and Ni(OH)2/NF.
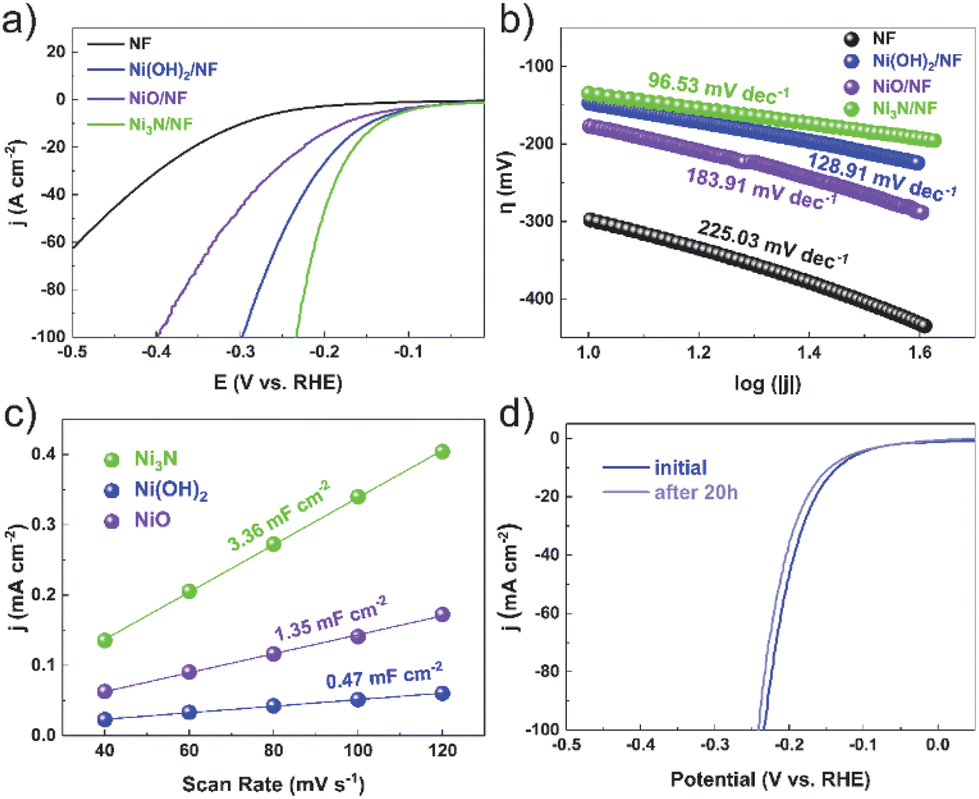 | ||
| Fig. 3 (a) Polarization curves of Ni3N, NiO and Ni(OH)2 and Pt/C electrocatalysts. (b) The corresponding Tafel plots of the three catalysts. (c) The Cdl calculations of Ni3N, NiO and Ni(OH)2. (d) Polarization curves of Ni3N/NF in 1 m KOH before and after 20 h under a certain potential (at −0.15 V vs. RHE). | ||
The kinetic parameters of the electrocatalytic reaction were determined by analyzing the polarization curves of the HER, which describe the relationship between overpotential and current density (Fig. 3b). The Tafel slope value of Ni3N/NF was found to be 96.53 mV dec−1, which is less than that of bare NF (225.03 mV dec−1), Ni(OH)2/NF (128.91 mV dec−1), and NiO/NF (183.91 mV dec−1). This indicates that Ni3N/NF exhibits a faster kinetic HER process, leading to an improved HER performance.
The electrochemical double-layer capacitance (Cdl) is directly proportional to the electrochemical active surface area (ECSA). Consequently, the ECSA was estimated by calculating the Cdl. This was achieved by recording cyclic voltammetry curves within a potential window devoid of redox reactions (Fig. S6–S8†). Ni3N/NF had much larger Cdl (3.36 mF cm−2) than Ni(OH)2/NF (0.47 mF cm−2) and NiO/NF (1.35 mF cm−2), showing more electrocatalytic active sites on Ni3N/NF. Furthermore, electrochemical Impedance spectroscopy (EIS) is utilized to investigate the interfacial characteristics of electrocatalytic materials. As shown in Fig. S9,† the electron-transfer resistance exhibited by Ni3N/NF is significantly lower compared to that of Ni(OH)2/NF and NiO/NF. This observation is consistent with the outcomes derived from LSV analyses. The long-term stability is an essential parameter for assessing catalyst performance. Before and after testing at the potential of −0.15 V (vs. RHE) for 20 h, the polarization curves for Ni3N/NF exhibit a comparable LSV profile to the initial curve, with an insignificant decrease in current density, as shown in Fig. 3d.
In order to elucidate the intrinsic correlation between the improved electrocatalytic performance and the influence of non-metal elements in catalyzing the hydrogen evolution process (Fig. 4), we conducted calculations to determine the adsorption strength of H atoms on the Ni site in Ni3N and NiO (Fig. 4a and b). The adsorption strength of H atoms on the Ni site in Ni3N (i.e., Ni3N: ΔE*H = E*H − E* = −2.03 eV) was found to be relatively weaker compared to that of NiO (i.e., NiO: ΔE*H = E*H − E* = −4.28 eV). However, the desorption energy of H atoms on the Ni site in Ni3N (i.e., Ni3N: ΔE*H = E* − E*H = 2.03 eV) was lower than that of H atoms on the Ni site in NiO (i.e., NiO: ΔE*H = E*H − E* = 4.28 eV), indicating that H atoms on the Ni site in Ni3N are more easily desorbed than those on the Ni site in NiO. Furthermore, we have also calculated the Gibbs free energy variation (ΔG) for each fundamental reaction step on the Ni site in Ni3N and NiO during the HER process (Fig. 4c). The Gibbs free energy difference value of the Ni sites in Ni3N (ΔG2 = 1.63 eV) was lower than that of NiO (ΔG1 = 1.72 eV) during the desorption of hydrogen atoms, suggesting that the catalytic activity of Ni3N is greater than that of NiO. In order to delve into the intricacies of the doping effect, we conducted an analysis of the charge density difference plots pertaining to *H adsorption. This was done with the objective of qualitatively illustrating the interaction between *H radicals and Ni3N and NiO. As depicted in Fig. 4d and e, the charge accumulation on the Ni-site radical is more pronounced on Ni3N (1.94 eV) compared to NiO (1.54 eV), while the charge redistribution for the Ni site on Ni3N is almost identical to that on NiO.
 | ||
| Fig. 4 Schematic slab models of (a) Ni3N and (b) NiO. (c) Free energy diagram for the HER on three considered models at zero potential. The charge density difference plots for *H on (d) Ni3N and (e) NiO with an isosurface value of 0.003 e Å−3. Yellow and cyan contours represent electron accumulation and depletion of O-intermediate species, respectively. Electron localization function (ELF) for (f) Ni3N and (g) NiO. | ||
It is noteworthy that there is a greater transfer of charge from the N atom to the Ni-site in Ni3N compared to the charge transfer from the N atom to the Ni-site in NiO. Specifically, when Ni3N interacts with an *H molecule, the Ni-site in Ni3N exhibits a higher affinity for accepting electrons compared to the Ni-site in NiO, leading to the effective activation of Ni-sites in Ni3N for the HER. Fig. S10† illustrates that the bandgap of NiO, which is approximately 1.48 eV in the total density of states (TDOS), is larger than that of Ni3N (which is about 0 eV). Consequently, the conductivity of Ni3N is higher than that of NiO. Fig. S11† depicts the projected density of states (PDOS) for the d band of Ni in both Ni3N and NiO. The d-band of the Ni atom in Ni3N is visibly narrower compared to the d-band of the Ni atom in NiO, indicating the electron localization of the d orbitals of the Ni atom in Ni3N. The d-band center of the Ni atom in Ni3N (−2.78 eV) is higher than that of the Ni atom in NiO (−3.42 eV), suggesting that the electron localization function (ELF) identifies the localization of electrons around N atoms on the (001) surface of Ni3N, while the distribution is uniform within Ni3N (Fig. 4f). Additionally, the electron distribution around Ni atoms is relatively enhanced, indicating the effective activation of Ni sites. As previously reported, the manipulation of electron localization can be utilized to engineer the energetics of reaction intermediates for electrocatalysis.
Our DFT calculation results display that the improved electrocatalytic performance of Ni3N primarily stems from the electron-rich interfacial region, which facilitates electron transfer from N-site to Ni-site, leading to charge accumulate on the Ni-site in the Ni3N and negatively shift the d-band centre of the Ni atom in Ni3N. Consequently, this promotes the adsorption H2O to form *H. Moreover, the charge accumulation on the Ni-site radical is more pronounced on Ni3N compared to NiO for the HER, suggesting the charge transfer for the Ni site on Ni3N is superior to that on NiO. Our results suggest the notion of orbital-regulated the electronic levels on Ni-site introduces a distinctive methodology for the systematic development of catalysts used for hydrogen evolution in the future.
Conclusions
In conclusion, we have successfully demonstrated the utilization of metallic 3D Ni3N nanosheets as an exceptional electrode material. Our research presents an innovative yet straightforward approach to enhance the kinetics of water dissociation in Ni3N, utilizing the electron redistribution in d orbitals induced by oxygen. Compared to the HER activity of NiO, the fabricated Ni3N demonstrates a higher overpotential, measuring 135 mV when operating at a current density of 10 mA cm−2. XPS analysis indicates that oxygen dopants, possessing a higher electron-pulling capacity, generate additional unoccupied states in the Ni sites. Moreover, DFT calculations elucidate that Ni3N can also depress the d-band center of Ni sites, thereby catalyzing hydrogen evolution and consequently activating and expediting water dissociation. This innovative concept of electron-regulated electrocatalysis could furnish substantial insights for catalyst design, not only for HER but also for other applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the fundamental research funds for the Natural Science Foundation of Chongqing (cstc2021jcyj-msxmX0626) and the Science and Technology Research Program of Chongqing Municipal Education Commission (KJZD-K202203201).References
- X. Jin, J. Li, Y. Cui, X. Liu, X. Zhang, J. Yao and B. Liu, Inorg. Chem., 2019, 58, 11630–11635 CrossRef CAS PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, e1806296 CrossRef PubMed.
- A. Wu, Y. Xie, H. Ma, C. Tian, Y. Gu, H. Yan, X. Zhang, G. Yang and H. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS.
- P. F. Guo, Y. Yang and B. Zhu, et al., Heterostructural NiFeW disulfideand hydroxide dual-trimetallic core-shell nanosheets for synergistically effective water oxidation, Carbon Energy, 2024, e532, DOI:10.1002/cey2.532.
- R. R. Chen, G. Chen, X. Ren, J. Ge, S. J. H. Ong, S. Xi, X. Wang and Z. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 25884–25890 CrossRef CAS PubMed.
- R. R. Chen, Y. Sun, S. J. H. Ong, S. Xi, Y. Du, C. Liu, O. Lev and Z. J. Xu, Adv. Mater., 2020, 32, e1907976 CrossRef PubMed.
- W. H. Lee, M. H. Han, Y. J. Ko, B. K. Min, K. H. Chae and H. S. Oh, Nat. Commun., 2022, 13, 605 CrossRef CAS PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- H. Sun, L. Li, Y. Chen, H. Kim, X. Xu, D. Guan, Z. Hu, L. Zhang, Z. Shao and W. Jung, Appl. Catal., B, 2023, 325, 122388 CrossRef CAS.
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Nat. Commun., 2018, 9, 4531 CrossRef PubMed.
- M. Wang, W. Ma, Z. Lv, D. Liu, K. Jian and J. Dang, J. Phys. Chem. Lett., 2021, 12, 1581–1587 CrossRef CAS PubMed.
- Y. Yang, Q. Dai, L. Shi, Y. Liu, T. T. Isimjan and X. Yang, J. Phys. Chem. Lett., 2022, 13, 2107–2116 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, J. Li, X. Jin, Y. Li, Q. Qian, Y. Wang, A. El-Harairy, Z. Li, Y. Zhu, H. Zhang, M. Cheng, S. Zeng and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 3881–3890 CrossRef CAS PubMed.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, e2003297 CrossRef PubMed.
- T. Wu, Y. Sun, X. Ren, J. Wang, J. Song, Y. Pan, Y. Mu, J. Zhang, Q. Cheng, G. Xian, S. Xi, C. Shen, H. J. Gao, A. C. Fisher, M. P. Sherburne, Y. Du, J. W. Ager, J. Gracia, H. Yang, L. Zeng and Z. J. Xu, Adv. Mater., 2023, 35, e2207041 CrossRef PubMed.
- Y. Yang, P. Li, X. Zheng, W. Sun, S. X. Dou, T. Ma and H. Pan, Chem. Soc. Rev., 2022, 51, 9620–9693 RSC.
- A. R. Zeradjanin, G. Polymeros, C. Toparli, M. Ledendecker, N. Hodnik, A. Erbe, M. Rohwerder and F. La Mantia, Phys. Chem. Chem. Phys., 2020, 22, 8768–8780 RSC.
- H. Zhang, J. Wang, F. Qin, H. Liu and C. Wang, Nano Res., 2021, 14, 3489–3496 CrossRef CAS.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- J. Hao, K. Wu, C. Lyu, Y. Yang, H. Wu, J. Liu, N. Liu, W. M. Lau and J. Zheng, Mater. Horiz., 2023, 10, 2312–2342 RSC.
- H. Jung, S. Choung and J. W. Han, Nanoscale Adv., 2021, 3, 6797–6826 RSC.
- D. Guan, H. Xu, Q. Zhang, Y.-C. Huang, C. Shi, Y.-C. Chang, X. Xu, J. Tang, Y. Gu, C.-W. Pao, S.-C. Haw, J.-M. Chen, Z. Hu, M. Ni and Z. Shao, Adv. Mater., 2023, 35, 2305074 CrossRef CAS PubMed.
- X. Li, H. Zhang, X. Li, Q. Hu, C. Deng, X. Jiang, H. Yang and C. He, Nano Res., 2023, 16, 2245–2251 CrossRef CAS.
- J. Lu, P.-J. Deng, A. Chen, C. Yang, H. Zhu and H.-P. Liang, J. Mater. Chem. A, 2023, 11, 2452–2459 RSC.
- J. T. Ren, L. Wang, L. Chen, X. L. Song, Q. H. Kong, H. Y. Wang and Z. Y. Yuan, Small, 2023, 19, 2206196 CrossRef CAS PubMed.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- D. Tian, S. R. Denny, K. Li, H. Wang, S. Kattel and J. G. Chen, Chem. Soc. Rev., 2021, 50, 12338–12376 RSC.
- C. Yang, K. Shen, R. Zhao, H. Xiang, J. Wu, W. Zhong, Q. Zhang, X. Li and N. Yang, Adv. Funct. Mater., 2022, 32, 2108167 CrossRef CAS.
- C. Yang, R. Zhao, H. Xiang, J. Wu, W. Zhong, W. Li, Q. Zhang, N. Yang and X. Li, Adv. Energy Mater., 2020, 10, 2002260 CrossRef CAS.
- G.-Q. Yu, B.-Y. Huang, X. Chen, D. Wang, F. Zheng and X.-B. Li, J. Phys. Chem. C, 2019, 123, 21878–21887 CrossRef CAS.
- Y. Yu, J. Zhou and Z. Sun, Adv. Funct. Mater., 2020, 30, 2000570 CrossRef CAS.
- Y. Fang, D. Sun, S. Niu, J. Cai, Y. Zang, Y. Wu, L. Zhu, Y. Xie, Y. Liu, Z. Zhu, A. Mosallanezhad, D. Niu, Z. Lu, J. Shi, X. Liu, D. Rao, G. Wang and Y. Qian, Sci. China: Chem., 2020, 63, 1563–1569 CrossRef CAS.
- D. Gao, J. Zhang, T. Wang, W. Xiao, K. Tao, D. Xue and J. Ding, J. Mater. Chem. A, 2016, 4, 17363–17369 RSC.
- X. Gao, X. Liu, W. Zang, H. Dong, Y. Pang, Z. Kou, P. Wang, Z. Pan, S. Wei, S. Mu and J. Wang, Nano Energy, 2020, 78, 105355 CrossRef CAS.
- X. Gao, W. Zang, X. Li, Z. Wang, L. Zheng and Z. Kou, Chem. Eng. J., 2023, 451, 138698 CrossRef CAS.
- M. Li, X. Wu, K. Liu, Y. Zhang, X. Jiang, D. Sun, Y. Tang, K. Huang and G. Fu, J. Energy Chem., 2022, 69, 506–515 CrossRef CAS.
- S. Niu, Y. Fang, J. Zhou, J. Cai, Y. Zang, Y. Wu, J. Ye, Y. Xie, Y. Liu, X. Zheng, W. Qu, X. Liu, G. Wang and Y. Qian, J. Mater. Chem. A, 2019, 7, 10924–10929 RSC.
- B. Zhang, J. Wang, J. Liu, L. Zhang, H. Wan, L. Miao and J. Jiang, ACS Catal., 2019, 9, 9332–9338 CrossRef CAS.
- D. Zhang, H. Li, A. Riaz, A. Sharma, W. Liang, Y. Wang, H. Chen, K. Vora, D. Yan, Z. Su, A. Tricoli, C. Zhao, F. J. Beck, K. Reuter, K. Catchpole and S. Karuturi, Energy Environ. Sci., 2022, 15, 185–195 RSC.
- P. Zhou, G. Zhai, X. Lv, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Appl. Catal., B, 2021, 283, 119590 CrossRef CAS.
- X. Jin, J. Li, Y. Cui, X. Liu, K. Wang, Y. Zhou, W. Yang, X. Zhang, C. Zhang, X. Jiang and B. Liu, Int. J. Hydrogen Energy, 2019, 44, 5739–5747 CrossRef CAS.
- D. Shah, S. Bahr, P. Dietrich, M. Meyer, A. Thißen and M. R. Linford, Surf. Sci. Spectra, 2019, 26, 014023 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01071j |
| This journal is © The Royal Society of Chemistry 2024 |