
Efficient CO2 electroreduction to ethanol enabled by tip-curvature-induced local electric fields†
Jing
Zhou‡
ab,
Qianyue
Liang‡
ab,
Pu
Huang
ab,
Jing
Xu
c,
Tengfei
Niu
 a,
Yao
Wang
a,
Yuming
Dong
a and
Jiawei
Zhang
*ab
a,
Yao
Wang
a,
Yuming
Dong
a and
Jiawei
Zhang
*ab
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: jiawei.zhang@jiangnan.edu.cn
bInternational Joint Research Center for Photoresponsive Molecules and Materials, Jiangnan University, Wuxi 214122, China
cSchool of Food Science and Technology, Jiangnan University, Wuxi 214122, China
First published on 20th June 2024
Abstract
Electrocatalytic reduction of CO2 into multicarbon (C2+) products offers a promising pathway for CO2 utilization. However, achieving high selectivity towards multicarbon alcohols, such as ethanol, remains a challenge. In this work, we present a novel CuO nanoflower catalyst with engineered tip curvature, achieving remarkable selectivity and efficiency in the electroreduction of CO2 to ethanol. This catalyst exhibits an ethanol faradaic efficiency (FEethanol) of 47% and a formation rate of 320 μmol h−1 cm−2, with an overall C2+ product faradaic efficiency (FEC2+) reaching ∼77.8%. We attribute this performance to the catalyst's sharp tip, which generates a strong local electric field, thereby accelerating CO2 activation and facilitating C–C coupling for deep CO2 reduction. In situ Raman spectroscopy reveals an increased *OH coverage under operating conditions, where the enhanced *OH adsorption facilitates the stabilization of *CHCOH intermediates through hydrogen bonding interaction, thus improving ethanol selectivity. Our findings demonstrate the pivotal role of local electric fields in altering reaction kinetics for CO2 electroreduction, presenting a new avenue for catalyst design aiming at converting CO2 to ethanol.
Introduction
The transformation of CO2 into valuable chemicals through electrochemical reduction (CO2RR), powered by renewable energy, represents a pivotal strategy for achieving CO2 utilization and fostering sustainable development.1,2 Among various CO2RR products, ethanol stands out due to its high energy density (26.8 MJ kg−1) and significant market demand, capturing widespread research interest.3–5 Nevertheless, the selective production of ethanol poses considerable challenges due to the complex multi-electron–proton coupling transfer process.6–9 Copper (Cu) stands out as a remarkable electrocatalyst capable of converting CO2 into ethanol with significant efficiency.10–12 Ethanol and ethylene are the two primary multicarbon (C2+) products, with the pathway to ethylene often being thermodynamically favored.2,13,14 It is widely acknowledged that ethanol and ethylene share a common intermediate (*CHCOH).8,15,16 The divergent paths to these products are determined by the fate of *CHCOH: breaking the C–O bond leads to ethylene, whereas protonation of this intermediate yields ethanol. This critical branching point underscores the challenge of selectively directing CO2RR toward ethanol over ethylene,17–19 and highlights the great significance of catalyst optimization to favor the desired ethanol production pathway.14Quite recently, a fascinating, yet simple strategy to accelerate CO2 reduction through enriching the local concentration of CO2 at the active site leveraging the geometry-induced local electric field effects was proposed.20,21 It was believed that the local electric field induced by the shape architecture not only intensifies the local CO2 concentration but also lowers the energy barrier for the CO2RR, underscoring the critical role of the local electric field in manipulating the CO2RR reaction pathway.22,23 The local electric field has been demonstrated to be able to regulate ion concentrations, such as potassium and OH−, which play an essential role in determining the CO2RR selectivity.24,25 On the other hand, modifying the surface-bonded hydroxyl species (*OH) represents a pivotal advancement to regulate the CO2RR selectivity. Specifically, an optimal concentration of *OH can enhance the adsorption of intermediates like *COLFB, strengthening *CO binding and promoting its dimerization.26–28 This process notably improves the selectivity for C2+ products, including ethanol and ethylene. Importantly, surface-bonded hydroxyl species have also been demonstrated to enhance the selectivity of ethanol by creating a non-covalent interaction with *CHCOH intermediates, which inhibits its dehydration to suppress the competing ethylene production pathway, thus enhancing the selectivity of ethanol.29,30 Given the above, using the strong electric field to enrich surface bonded *OH may also availably modulate the ethanol selectivity for CO2RR.31–33 However, to our knowledge, the underlying principles of using local electric field to regulate ethanol selectivity has not been fully investigated.
In this contribution, aimed to further improve the ethanol selectivity in CO2RR, a novel CuO nanoflower catalyst with tip curvature was designed to introduce a strong local electric field to boost the CO2RR process. The as-crafted CuO nanoflower catalyst exhibited an ethanol faradaic efficiency (FEethanol) of 47% and a formation rate of 320 μmol h−1 cm−2, with an overall C2+ product faradaic efficiency (FEC2+) reaching ∼78%, underscoring its effectiveness in triggering the selective reduction of CO2. In situ electrochemical and spectroscopy characterization revealed the mechanism underpinning the selectivity: the tip architecture created a strong local electric field that enriched CO2 near the CuO nanoflower, enhancing *CO production and its subsequent coupling. Additionally, an enhanced *OH adsorption under operating conditions was also created under the strong local electric field,34,35 and surface-enriched *OH could facilitate the stabilization of *CHCOH intermediates through hydrogen bonding, thus improving ethanol selectivity.34,36 These findings highlight the pivotal role of local electric fields in selectively driving CO2 reduction to ethanol, and provide a path toward the rational design of ethanol-oriented catalysts.
Results and discussion
As a proof of concept, a solvent thermal method was utilized to craft a copper oxide-based catalyst with tip curvature. The detailed synthesis procedure is depicted in Fig. 1a. Cetyltrimethylammonium bromide (CTAB) which could be self-assembled into a micelle solution with a hydrophobic inner core in the aqueous solution is selected as the soft template to direct the growth of CuO. The CTAB micelle can direct the growth of CuO nanosheets, which then interconnect each other to form the flower-like CuO nanostructures. Initially, CTAB and Cu(NO3)2·3H2O were dissolved in a mixed solution of ethylene glycol and water (Vethylene glycol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vwater = 1:5). Subsequently, hexamethylenetetramine (HMT) was added to the mixture and then subjected to ultrasonication to achieve a homogeneous dispersion. After that, the mixture solution was transferred into a Teflon lined hydrothermal autoclave for the subsequent hydrothermal reaction. During this process, HMT undergoes hydrolysis to form ammonia and formaldehyde, which creates a base environment for the nucleation of copper species. After keeping at 115 °C for 4 hours, the reaction mixture was naturally cooled down to room temperature, the precipitants were collected by centrifugation, washed three times with ethanol and finally dried under vacuum at 60 °C for 8 h to afford CuO nanoflower catalyst with tip curvature.
:Vwater = 1:5). Subsequently, hexamethylenetetramine (HMT) was added to the mixture and then subjected to ultrasonication to achieve a homogeneous dispersion. After that, the mixture solution was transferred into a Teflon lined hydrothermal autoclave for the subsequent hydrothermal reaction. During this process, HMT undergoes hydrolysis to form ammonia and formaldehyde, which creates a base environment for the nucleation of copper species. After keeping at 115 °C for 4 hours, the reaction mixture was naturally cooled down to room temperature, the precipitants were collected by centrifugation, washed three times with ethanol and finally dried under vacuum at 60 °C for 8 h to afford CuO nanoflower catalyst with tip curvature.
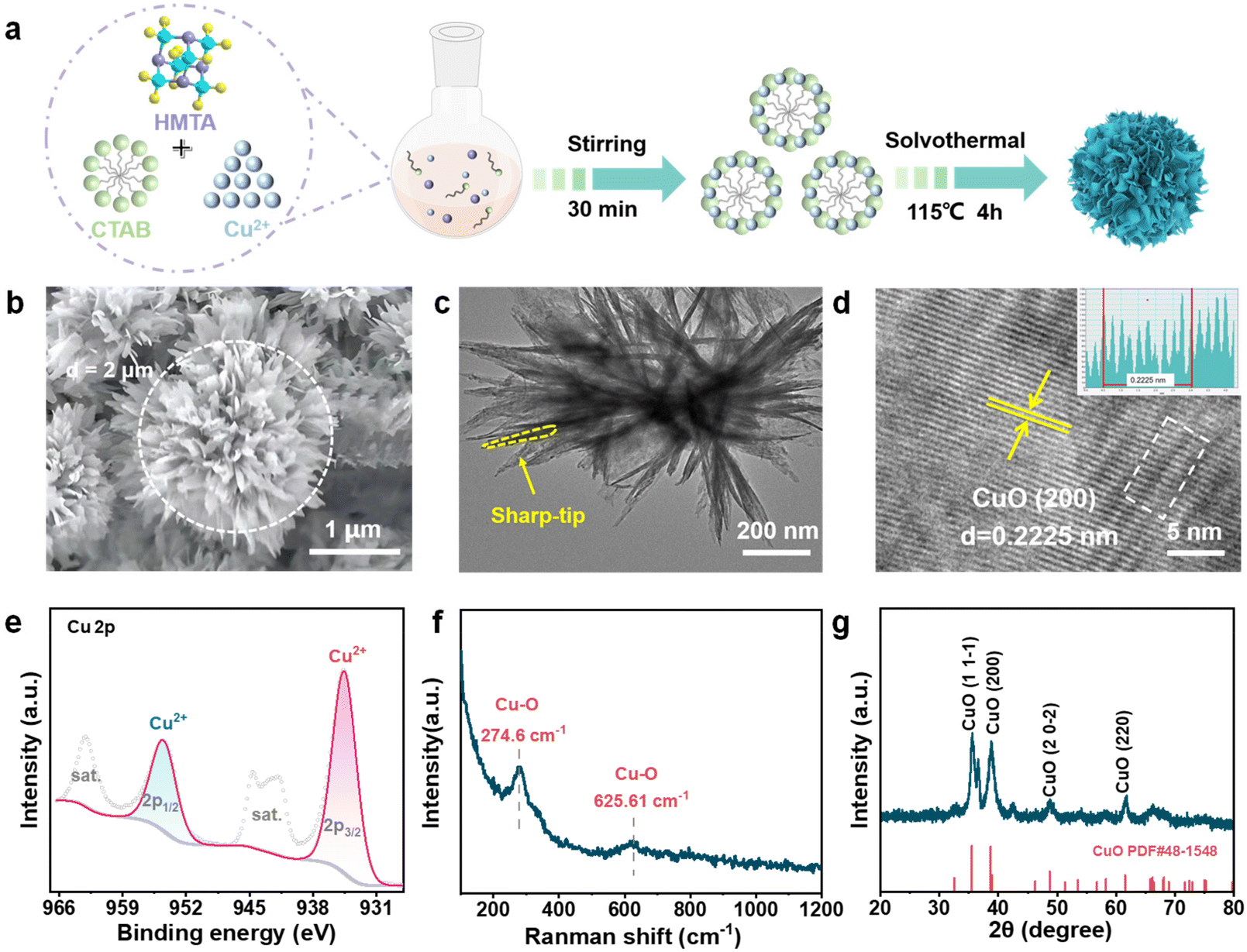 | ||
| Fig. 1 (a) The schematic diagram for preparing the CuO nanoflowers. (b) SEM, (c) TEM, (d) high-resolution TEM image of CuO nanoflowers. The inset is the result of lattice stripe spacing measurement by Digital Micrograph. (e) XPS spectra of Cu 2p from CuO nanoflowers. (f) Raman spectra of CuO nanoflowers. (g) XRD patterns of CuO nanoflowers. | ||
Scanning electron microscopy (SEM) and Transmission electron microscopy (TEM) were employed to directly observe the as-synthesized CuO nanoflower. As presented in Fig. 1b and Fig. S1,† a flower-like architecture featuring a high curvature petal-like structure stacked layer by layer was recognized. The nanoflower exhibited a homogeneous size distribution with a diameter of ∼2 μm. TEM image confirmed that the nanoflower architecture was composed of tip-shaped petals about 300 nm in length. It should be mentioned that the petals exhibit a nano-pyramids characteristic (Fig. 1c). As free electrons tend to migrate to the regions of the sharpest curvature on a charged electrode, a strong local electronic field is supposed to be created under electrochemical operating in our CuO flower catalyst. The high-resolution TEM (HRTEM) image reveals a distinct lattice fringe with a d spacing of 0.2225 nm, which could be assigned to the (200) plane of CuO (Fig. 1d), implying the formation of CuO-based catalysts. Subsequently, the surface property of the as-crafted CuO nanoflowers was studied by X-ray Photoelectron Spectroscopy (XPS) analysis. The XPS survey spectrum confirmed the coexistence of Cu and O on the CuO nanoflower (Fig. S2†). As shown in Fig. 1e, the high-resolution spectrum of Cu 2p valence state is characteristic of CuO materials where the representative peaks at 934.68 eV and 954.32 eV are attributed to Cu 2p3/2 and Cu 2p1/2, along with the satellite peaks at 944.08 eV and 963.28 eV, respectively.37 Correspondingly, in the O 1s spectra, the peaks at 530.28, 531.88, and 533.07 eV, which could be attributed to lattice oxygen, oxygen near the oxygen vacancy, and surface adsorbed oxygen, respectively, were observed, validating the presence of oxygen vacancy within the catalyst (Fig. S3†). The presence of oxygen vacancy is also beneficial for the adsorption and activation of CO2.35,38 Raman spectroscopy revealed prominent peaks at 274.6 and 625.6 cm−1, which could be assigned to the vibration of the Cu–O bond (Fig. 1f), agrees well with the XPS findings. X-ray diffraction (XRD) analysis further confirms the composition of CuO nanoflowers, with characteristic diffraction peaks observed at 35.6°, 38.8°, 48.8°, and 61.8°, corresponding to the (11−1), (200), (20−2), and (220) crystallographic planes of CuO PDF#48-1548, respectively (Fig. 1g). Given that above, the CuO nanoflower catalysts with tip curvature was successfully synthesized.
To validate the advantages of the tip-shaped architecture in manipulating the CO2RR process, the performance of the as-crafted CuO nanoflower was evaluated and benchmarked against commercially available Cu nanoparticles (NPs) featuring a smooth surface. All performance evaluations were conducted in a customized flow cell equipped with a gas-diffusion electrode (GDE) 1.0 M KOH electrolyte (versus the reversible hydrogen electrode (RHE) and no iR-compensation) (Fig. S4†). The gaseous products were quantitatively analyzed using online gas chromatography (GC), while the liquid phase products were detected by nuclear magnetic resonance (1H-NMR) (Fig. S5†). Initially, linear sweep voltammetry (LSV) curves were recorded electrolytes to give a primary investigation on the catalytic activity of Cu NPs and CuO nanoflowers electrodes for CO2RR (Fig. S6†), no less obvious current response was observed on CuO nanoflower and Cu NPs, possibly imposed by the limited OER performance of the counted electrode. For the Cu NPs catalyst, the current signal recorded under argon flow which was assigned to the competing hydrogen evolution reaction (HER) almost coincides with that collected under the CO2RR operating conditions, suggesting unsatisfactory selectivity toward CO2 reduction.39 By contrast, the current density assigned to CO2RR is larger than that of HER in the case of CuO nanoflower, implying a better CO2RR selectivity. Noteworthy, the current signal recorded on CuO nanoflower is higher than that on Cu NPs under the same bias, possibly attributed to the increased specific surface area of CuO nanoflower, which consequently leads to accelerated reaction kinetics.40 In addition, the LSV curve of CuO nanoflowers was also recorded in H-cell. As can be seen, the current density is below 50 mA cm−2 across the entire potential window, possibly due to the limited mass transfer of CO2 (Fig. S7†). Therefore, the flow cell was employed for subsequent CO2RR performance evaluation. To verify our assumption, the electrochemical active surface areas (ECSA) of these catalysts were estimated through the electrochemical double-layer capacitance (Cdl) measurements (Fig. S8a and b†). As expected, the ECSA of CuO nanoflower is ∼5.3 times higher than that of Cu NPs (Fig. S8c, and Table S1†), corroborating the increased exposure of active sites in the nanoflower-like architecture. Electrochemical impedance spectroscopy (EIS) was performed to probe charge transfer processes occurring at electrode/solution interfaces. As shown in Fig. S8d,† the CuO nanoflower electrode exhibited smaller semi-circle radii than Cu NPs, meaning the decreased charge transfer resistance.41 Thus, the strong local electric fields induced by the tip-shaped architecture contribute to the enhanced charge transfer kinetics.
Subsequently, the CO2RR selectivity over the CuO nanoflower catalyst was evaluated using the galvanostatic method, and the applied potential can be found in Fig. S9.† C1–C2 hydrocarbons/oxygenates and hydrogen were identified to be the major products over a wide current density ranging from 100 to 400 mA cm−2. Ethylene and ethanol were the major C2+ products, together with a tiny amount of acetate. As inferred from the product distribution plots (Fig. 2a and Fig. S10–S12†) the selectivity of H2 and CO on the CuO nanoflower are significantly lowered in contrast to Cu NPs across the observed potential window. This observation indicated that the unique architecture of CuO nanoflower could suppress the competing HER,37,42 and promote the C–C coupling process to form C2+ products simultaneously. Of note, the faradaic efficiency for C2+ products (FEC2+) of CuO nanoflower catalyst reached a remarkably high value of 77.8% at 150 mA cm−2, about 2.07 times higher than that of Cu NPs. Moreover, detailed product distribution confirmed an inversed relationship between FEC2+ and FECO over CuO nanoflower catalyst (Fig. 2b), consistent with previous findings that the formation of C2+ products originates from the coupling of *CO species. These findings demonstrated that the nanoflower structure holds the capability to promote the C–C coupling process.40 A detailed scrutinization of C2+ production composition indicated that ethanol is the dominant CO2RR product on the CuO nanoflower catalyst, with a remarkable FEethanol ∼47.1% (Fig. 2c). In contrast, ethylene is the major product in the case of Cu NPs (Fig. S13†), the maximum FEethanol of Cu NPs is only 19.4%. Fig. 2d depicts the partial current density of ethanol (jethanol) plotted against the applied potential, it was indicated that jethanol of CuO nanoflower is higher than that of Cu NPs across the entire potential range and peaks at 103 mA cm−2. Ethanol formation rates derived from jethanol were also calculated, and CuO nanoflower showcased the highest ethanol production rate of 320 μmol h−1 cm−2, 1.87 times higher than that of Cu NPs (Fig. S14†). Given the above, it could be concluded that the strong local electric field around the tip area in CuO nanoflower can increase CO2 accessibility of the active site, which consequently promotes the C–C coupling process by enabling a high *CO coverage. Additionally, this interesting local electric field can also induce an intrigue selectivity shift from ethylene to ethanol in CO2RR, possibly caused by the preferentially stabilized intermediates regarding the formation of ethanol in the local electronic fields (Fig. S15†). Considering that catalyst robustness is essential for practical applications, the long-term durability of CuO nanoflower in the membrane electrode assembly (MEA) cell under electrochemical operating conditions was also evaluated. The LSV recorded in the two-electrode MEA system showed that 200 mA cm−2 could be achieved under a cell voltage of −1.4 V (Fig. S16†), verifying the compact contact of our MEA device. As shown in Fig. 2e, the CuO nanoflower catalyst exhibited a relatively stable bias under a constant current density of 100 mA cm−2, with a negligible performance degradation within 35 hours,43 evidencing the excellent stability of CuO nanoflower under CO2RR operating conditions. An obvious potential change around 15 h seen in Fig. 2e was attributed to the cleaning of salt participates on GDE and refresh of the electrolyte. After the durability test, the morphology of CuO nanoflowers was well preserved (Fig. S17†), suggesting its excellent structure robustness under CO2RR operating conditions. The impedance of the spent CuO nanoflower was also evaluated, and the EIS results indicated a significantly smaller impedance and easier charge transfer process (Fig. S18†), which can be ascribed to the reduction of CuO species into conductive Cu. Stability of Cu NPs was also evaluated, and H2 became the main product after 13 hours of continuous operation (Fig. S19†).
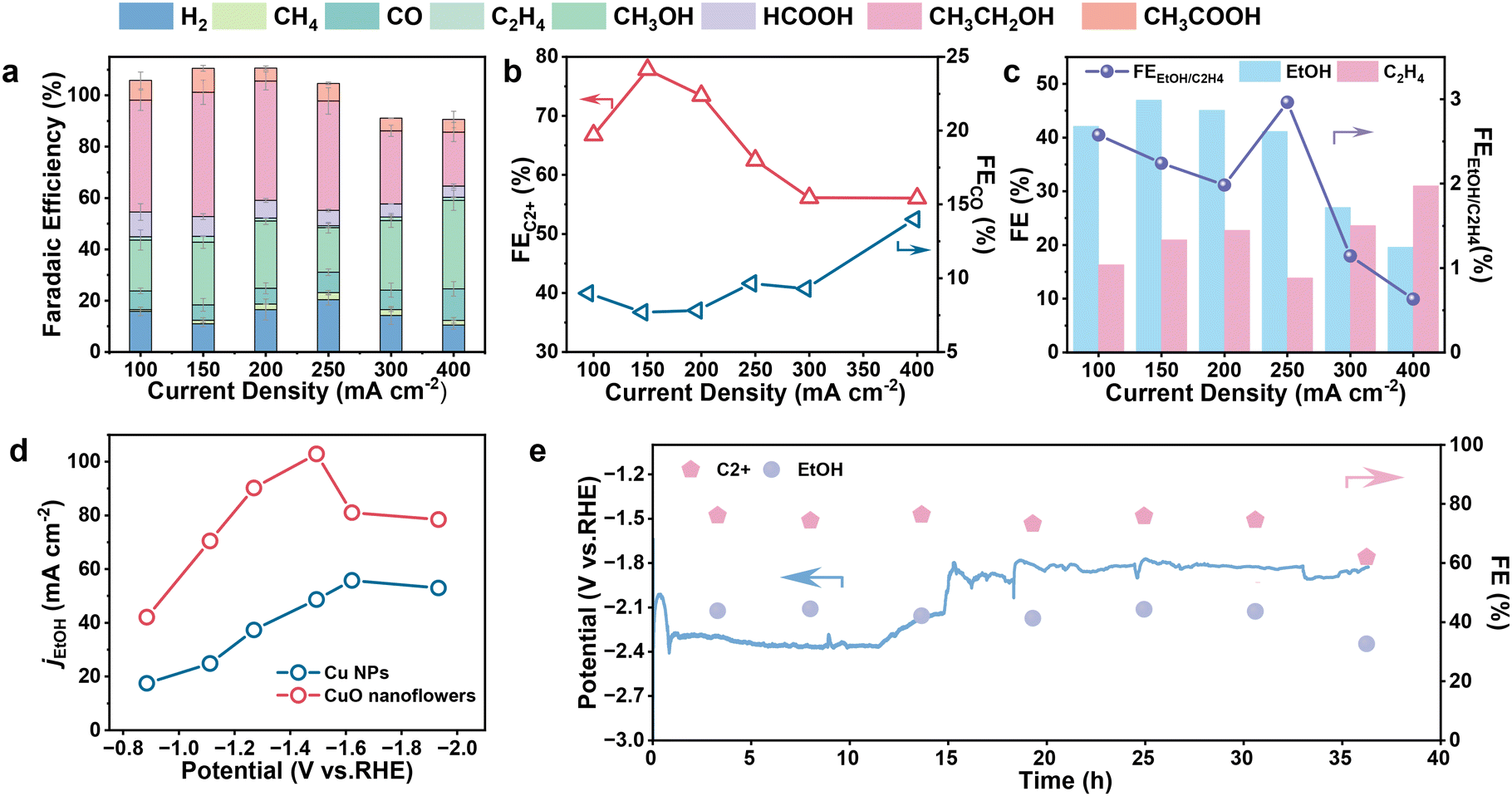 | ||
| Fig. 2 (a) Product distributions of CO2RR on CuO nanoflowers electrode. (b) The FEC2 and FECO of nanoflowers. (c) Ratio of FEEtOH to FEC2H4 and distribution of C2H4 and EtOH of CuO nanoflowers. (d) The jEtOH on CuO nanoflowers and Cu NPs. (e) Long-term stability of CuO nanoflowers electrode at 100 mA cm−2. | ||
Having established the excellent performance of CuO nanoflower toward CO2 to ethanol conversion, we then started to scrutinize the dynamic evolution of surface adsorptions during CO2RR and to elucidate the mechanism of boosted ethanol selectivity under the strong local electric field. Initially, in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was carried out (Fig. S20†). Potential dependent ATR-SEIRAS spectra figured out that the characteristics *CO infrared signal emerges at ∼2100 and ∼1830 cm−1, corresponding to linear (*COatop) and bridge-bonded *CO (*CObridge) which are extensively recognized as the active intermediate for subsequent reactions,44,45 respectively, exhibited a gradual shift to lower wave numbers upon negatively scanning the applied potential (Fig. 3a, and Fig. S21†). This redshift could be ascribed to the elongated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O dipoles due to the Stark tuning effect,46,47 also suggesting that an enhanced local electronic field can activate the key *CO intermediates for the subsequent C–C coupling process.48,49 Noteworthy, the intensity of *CO signal increases at first and then attenuates while increasing cathodic bias on both CuO nanoflower and Cu NPs, and peaking at ∼−0.7 VRHE. The enhanced *CO signal was ascribed to *CO accumulation under relatively low bias, and subsequent depletion could be explained by the accelerated C–C coupling.50 Given that all the ATR-SEIRAS spectra are recorded following the identical procedure, integration of the peak areas was employed for quantifying analysis. As concluded from the ATR-SEIRAS spectra, *CO intensity on CuO nanoflower is significantly higher than that on Cu NPs, meaning a higher *CO coverage on the CuO nanoflower catalyst. This could be attributed to the accelerated CO2 activation within the strong electronic fields in the CuO nanoflower catalyst. Since the higher *CO coverage is beneficial for C–C coupling, the relative higher C2+ product selectivity observed on the CuO nanoflower catalyst can thus be rationalized.51 Of note, peak associated with *COH was also detected at ∼1050 cm−1, implying the hydrogenation of *CO to form *COH is accelerated on the tip-shape architecture of CuO nanoflower. Since the asymmetric C–C coupling between *CO and *COH is more energetically favorable, the appearance of *COH evidenced a preferable microenvironment for the deep reduction of CO2.40 In addition, two key intermediates involved in CO2 to ethanol conversion, *OCCOH and *OC2H5, were also clearly distinguished at ∼1430 and 1120 cm−1. Integration of the *OC2H5 peak indicated a higher *OC2H5 intensity on CuO nanoflower than that of Cu NPs (Fig. 3b), in good agreement with the performance data that ethanol is the dominant product.
O dipoles due to the Stark tuning effect,46,47 also suggesting that an enhanced local electronic field can activate the key *CO intermediates for the subsequent C–C coupling process.48,49 Noteworthy, the intensity of *CO signal increases at first and then attenuates while increasing cathodic bias on both CuO nanoflower and Cu NPs, and peaking at ∼−0.7 VRHE. The enhanced *CO signal was ascribed to *CO accumulation under relatively low bias, and subsequent depletion could be explained by the accelerated C–C coupling.50 Given that all the ATR-SEIRAS spectra are recorded following the identical procedure, integration of the peak areas was employed for quantifying analysis. As concluded from the ATR-SEIRAS spectra, *CO intensity on CuO nanoflower is significantly higher than that on Cu NPs, meaning a higher *CO coverage on the CuO nanoflower catalyst. This could be attributed to the accelerated CO2 activation within the strong electronic fields in the CuO nanoflower catalyst. Since the higher *CO coverage is beneficial for C–C coupling, the relative higher C2+ product selectivity observed on the CuO nanoflower catalyst can thus be rationalized.51 Of note, peak associated with *COH was also detected at ∼1050 cm−1, implying the hydrogenation of *CO to form *COH is accelerated on the tip-shape architecture of CuO nanoflower. Since the asymmetric C–C coupling between *CO and *COH is more energetically favorable, the appearance of *COH evidenced a preferable microenvironment for the deep reduction of CO2.40 In addition, two key intermediates involved in CO2 to ethanol conversion, *OCCOH and *OC2H5, were also clearly distinguished at ∼1430 and 1120 cm−1. Integration of the *OC2H5 peak indicated a higher *OC2H5 intensity on CuO nanoflower than that of Cu NPs (Fig. 3b), in good agreement with the performance data that ethanol is the dominant product.
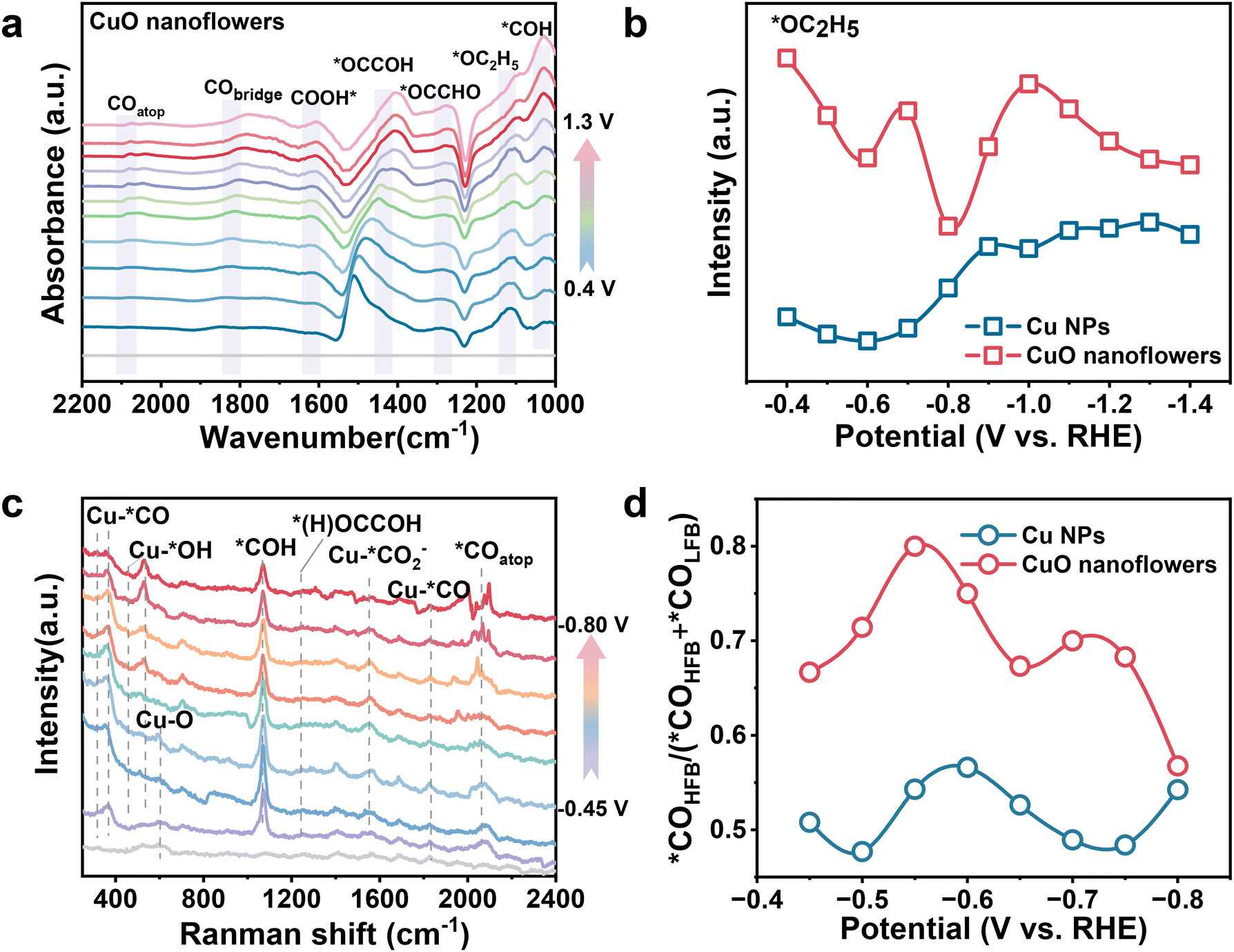 | ||
| Fig. 3 (a) In situ ATR–SEIRAS obtained during chronopotentiometry in a potential window −0.4 to −1.2 VRHE for CuO nanoflowers under CO2RR. (b) Potential dependence of ratio of *OC2H5 for CuO nanoflowers and Cu NPs. (c) The in situ Raman spectra over CuO nanoflowers catalyst at various applied potentials in 0.5 M KHCO3 electrolyte. (d) *COHFB/(*COHFB + *COLFB) ratio of CuO nanoflowers and Cu NPs. Raman shift of *CO is determined by the peak center of the fitted peak in Gaussian deconvolution of *CO signals. | ||
The in situ surface-enhanced Raman spectroscopy (SERS) (Fig. S22†) was also conducted to disclose clear spectral modes in CO2RR range from 300 to 2400 cm−1 (Fig. 3c and Fig. S23†), 1.0 M KOH was employed as the electrolyte. As presented, a characteristic band centered ∼625 cm−1 attributed to Cu–O stretching emerged under low overpotential and gradually disappeared upon negatively scanning the potential.46,52 This phenomenon indicates that CuO species in the nanoflower structure are reduced into metallic copper under CO2RR operating conditions. Moreover, characteristic bands centered at ∼300 and 388 cm−1 attributed to CO frustrated rotation and Cu–C stretching in Cu–*CO were identified. A bridge-bonded Cu–*CO band that appears at 1823 cm−1 was also detected, consistent with the ATR-SEIRAS results. Notably, two bands located at ∼425 and 545 cm−1 correspond to Cu–Oad or Cu–OH species were detected on CuO nanoflower and Cu NPs. The significantly increased intensity of these two peaks on the surface of CuO nanoflowers demonstrated that the strong electronic field induced by the tip-shape structure could accelerate *OH adsorption, thus suppressing the competing HER and stabilizing the key oxygen-containing intermediates for CO2 to ethanol conversion. *CO adsorption behaviors over CuO nanoflower and Cu NPs were also analyzed. Two kinds of CO stretching bands, namely low-frequency binding *CO (*COLFB) and high-frequency binding *CO (*COHFB), centered at ∼2040 cm−1 and 2080 cm−1, respectively, could be distinguished.46,53 Potential-dependent ratios of *COLFB and *COHFB were obtained by deconvoluting the *COatop Raman signal peaks (Fig. S24–S27†). Interestingly, the intensity of *COHFB at the CuO nanoflowers electrode was larger than that of *COLFB across the entire potential window, with the *COLFB being only 25% of the *COHFB at −0.55 VRHE. As the *COHFB is more active for subsequent C–C coupling to form C2+ products, the higher percentage of *COHFB on the CuO nanoflower also contributes to its high C2+ selectivity (Fig. 3d).51 Additionally, a strong vibration band appears ∼1070 cm−1 attributed to the stretching vibration of the *CHO species was also detected. This finding verified an asymmetric C–C coupling pathway on CuO nanoflower, by which *CO was hydrogenated to form *CHO and then coupled with *CO to form C2+ products. Furthermore, the experimental detection of the key intermediates (*(H)OCCOH, located at ∼1250 cm−1) allowed us to directly compare the C–C coupling capability.15 The stronger peak intensity of *(H)OCCOH species observed on the CuO nanoflower demonstrated its superior capability toward CO dimerization in comparison to Cu NPs, matching with its optimal selectivity for C2+ products.
To further verify the role of the *OH species in altering the reaction pathway. Coverage of *OH species on the catalyst surface was also determined using Cyclic Voltammetry (Fig. 4).33 A pair of peaks located in the underpotential region for CuO formation could be attributed to OH− adsorption/desorption, the intensity of these peaks could be regarded as important indicator for *OH coverage.36 As indicated, *OH coverage on CuO nanoflowers is higher than that of Cu NPs, exhibiting a positive correlation with ethanol selectivity in CO2RR. The enhanced *OH coverage could be ascribed to the electron deficiency around the base area of the tip due to the anisotropic electron movement caused by the “tip effect”, which consequently enriched OH− due to electrostatic interactions.54 It has been extensively accepted that CO2 conversion to ethylene and ethanol share the same *CH–COH intermediate, which prefers to accept one proton and then dehydrate to form *CCH, resulting in a more energetically preferable ethylene formation on a typical Cu surface. Given the high *OH coverage on CuO nanoflower and the observed excellent selectivity toward ethanol formation, it is reasonable to propose that *OH species adsorbed on the surface of CuO nanoflowers could stabilize the *CH–COH species through the hydron bonding interaction, inhibiting the process of dehydration process to form *CCH, thus improving the ethanol selectivity.
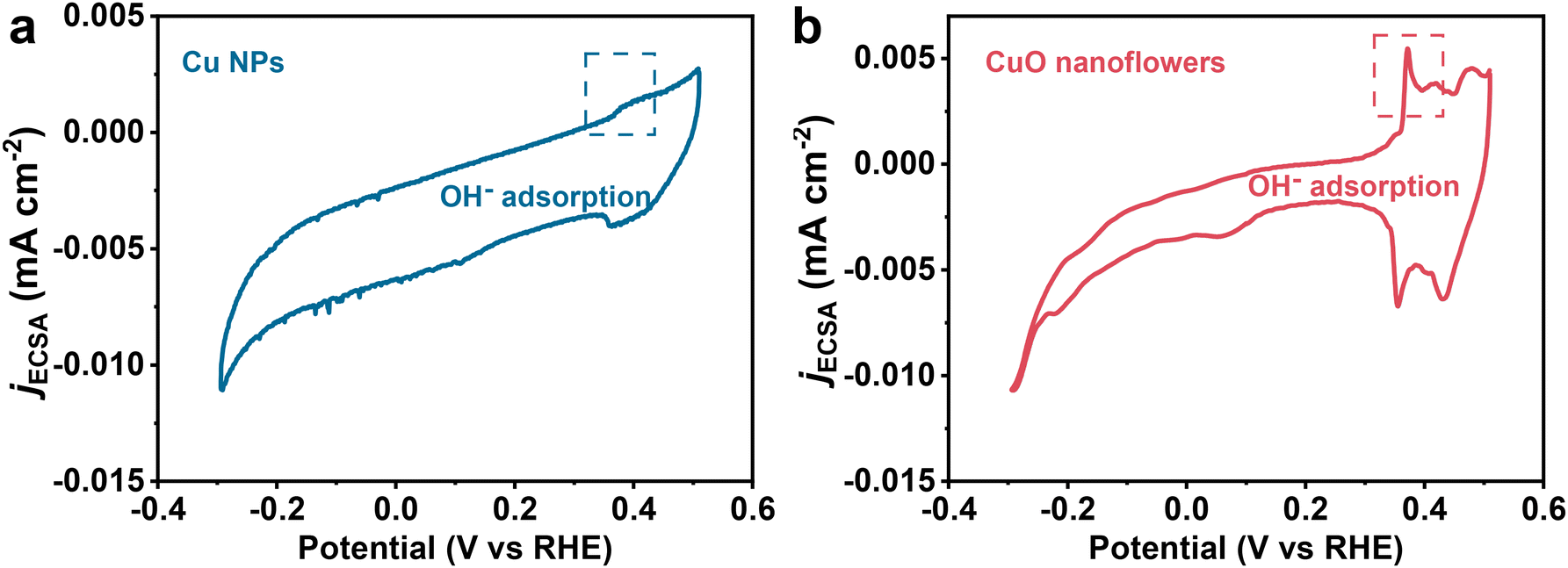 | ||
| Fig. 4 Cyclic voltammograms of (a) Cu NPs and (b) CuO nanoflowers in 0.1 M KOH (current density was normalized by ECSA), scanned to increasing anodic potentials, recorded at 50 mV s−1. OH− adsorption peaks are highlighted by a dashed rectangle. | ||
Conclusion
In conclusion, we have designed an excellent ethanol-oriented catalyst for CO2RR by capitalizing CuO nanoflower catalyst with tip curvatures. The sharp tip geometry induces a localized strong electric field, improving electron transport and ion concentration to regulate the reaction microenvironment in a kinetic way. The as-crafted CuO nanoflower exhibited a remarkable FEethanol of 47% and an ethanol formation rate of 325 μmol h−1 cm−2. Our findings indicated that the strong local electric field helps accelerate CO2 activation and subsequent C–C coupling to form deep reduction products. Moreover, the strong local electric field holds the capability to enhance the *OH coverage under CO2 operating, and the surface enriched *OH could stabilize the *CHCOH intermediates through the hydrogen bonding interaction and consequently increase ethanol selectivity. Beyond serving as a remarkable catalyst for the sustainable electrosynthesis of ethanol from CO2 and water, our findings offer valuable insights into favoring the ethanol formation pathway. This not only addresses the pressing need for ethanol production but also lays the groundwork for the rational design of catalysts optimized for ethanol synthesis.Conflicts of interest
There is no conflict to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21905089, No. 22305101), Startup Funding at Jiangnan University (No. 1045219032220100), and the Fundamental Research Funds for the Central Universities (No. JUSRP123020).References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- S. Chen, G. Qi, R. Yin, Q. Liu, L. Feng, X. Feng, G. Hu, J. Luo, X. Liu and W. Liu, Nanoscale, 2023, 15, 19577–19585 RSC.
- X. Hou, J. Ding, W. Liu, S. Zhang, J. Luo and X. Liu, Nanomaterials, 2023, 13, 309 CrossRef CAS PubMed.
- Y. Li, Y. Wei, W. He, Z. Tang, J. Xiong and Z. Zhao, Chin. Chem. Lett., 2023, 34, 108417 CrossRef CAS.
- S. Mou, Y. Li, L. Yue, J. Liang, Y. Luo, Q. Liu, T. Li, S. Lu, A. M. Asiri, X. Xiong, D. Ma and X. Sun, Nano Res., 2021, 14, 2831–2836 CrossRef CAS.
- J. Bi, P. Li, J. Liu, S. Jia, Y. Wang, Q. Zhu, Z. Liu and B. Han, Nat. Commun., 2023, 14, 2823 CrossRef CAS PubMed.
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O'Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CrossRef.
- S. Mou, T. Wu, J. Xie, Y. Zhang, L. Ji, H. Huang, T. Wang, Y. Luo, X. Xiong, B. Tang and X. Sun, Adv. Mater., 2019, 31, 1903499 CrossRef PubMed.
- L. Ji, L. Li, X. Ji, Y. Zhang, S. Mou, T. Wu, Q. Liu, B. Li, X. Zhu, Y. Luo, X. Shi, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2019, 59, 758–762 CrossRef PubMed.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- J. Liu, F. You, B. He, Y. Wu, D. Wang, W. Zhou, C. Qian, G. Yang, G. Liu, H. Wang, Y. Guo, L. Gu, L. Feng, S. Li and Y. Zhao, J. Am. Chem. Soc., 2022, 144, 12410–12420 CrossRef CAS PubMed.
- T. Wei, S. Zhang, Q. Liu, Y. Qiu, J. Luo and X. Liu, Acta Phys.-Chim. Sin., 2022, 202207026–202207020 Search PubMed.
- T. Wang, Q. Zhang, K. Lian, G. Qi, Q. Liu, L. Feng, G. Hu, J. Luo and X. Liu, J. Colloid Interface Sci., 2024, 655, 176–186 CrossRef CAS PubMed.
- Y. Cao, Z. Chen, P. Li, A. Ozden, P. Ou, W. Ni, J. Abed, E. Shirzadi, J. Zhang, D. Sinton, J. Ge and E. H. Sargent, Nat. Commun., 2023, 14, 2387 CrossRef CAS PubMed.
- Y. Dai, H. Li, C. Wang, W. Xue, M. Zhang, D. Zhao, J. Xue, J. Li, L. Luo, C. Liu, X. Li, P. Cui, Q. Jiang, T. Zheng, S. Gu, Y. Zhang, J. Xiao, C. Xia and J. Zeng, Nat. Commun., 2023, 14, 3382 CrossRef CAS PubMed.
- W. F. Xiong, D. H. Si, H. F. Li, X. Song, T. Wang, Y. B. Huang, T. F. Liu, T. Zhang and R. Cao, J. Am. Chem. Soc., 2024, 146, 289–297 CrossRef CAS PubMed.
- C. Peng, J. Ma, G. Luo, S. Yan, J. Zhang, Y. Chen, N. Chen, Z. Wang, W. Wei, T. K. Sham, Y. Zheng, M. Kuang and G. Zheng, Angew. Chem., Int. Ed., 2024, e202316907, DOI:10.1002/anie.202316907.
- Y. Luo, S. Chen, J. Zhang, X. Ding, B. Pan, L. Wang, J. Lu, M. Cao and Y. Li, Adv. Mater., 2023, 35, e2303297 CrossRef PubMed.
- Z. Z. Niu, F. Y. Gao, X. L. Zhang, P. P. Yang, R. Liu, L. P. Chi, Z. Z. Wu, S. Qin, X. Yu and M. R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed.
- H. Jiang, Z. Hou and Y. Luo, Angew. Chem., Int. Ed., 2017, 56, 15617–15621 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- L. Huang, Z. Liu, G. Gao, C. Chen, Y. Xue, J. Zhao, Q. Lei, M. Jin, C. Zhu, Y. Han, J. S. Francisco and X. Lu, J. Am. Chem. Soc., 2023, 145, 26444–26451 CrossRef CAS PubMed.
- W. Guo, S. Zhang, J. Zhang, H. Wu, Y. Ma, Y. Song, L. Cheng, L. Chang, G. Li, Y. Liu, G. Wei, L. Gan, M. Zhu, S. Xi, X. Wang, B. I. Yakobson, B. Z. Tang and R. Ye, Nat. Commun., 2023, 14, 7383 CrossRef PubMed.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- Y. Gao, Q. Wu, X. Liang, Z. Wang, Z. Zheng, P. Wang, Y. Liu, Y. Dai, M. H. Whangbo and B. Huang, Adv. Sci., 2020, 7, 1902820 CrossRef CAS PubMed.
- X. Yan, M. Zhang, Y. Chen, Y. Wu, R. Wu, Q. Wan, C. Liu, T. Zheng, R. Feng, J. Zhang, C. Chen, C. Xia, Q. Zhu, X. Sun, Q. Qian and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202301507 CrossRef CAS PubMed.
- W. Nie, G. P. Heim, N. B. Watkins, T. Agapie and J. C. Peters, Angew. Chem., Int. Ed., 2023, 62, e202216102 CrossRef CAS PubMed.
- J. Li, K. Xu, F. Liu, Y. Li, Y. Hu, X. Chen, H. Wang, W. Xu, Y. Ni, G. Ding, T. Zhao, M. Yu, W. Xie and F. Cheng, Adv. Mater., 2023, 35, e2301127 CrossRef PubMed.
- S. Yan, Z. Chen, Y. Chen, C. Peng, X. Ma, X. Lv, Z. Qiu, Y. Yang, Y. Yang, M. Kuang, X. Xu and G. Zheng, J. Am. Chem. Soc., 2023, 145, 26374–26382 CrossRef CAS PubMed.
- D. Wei, Y. Wang, C.-L. Dong, Z. Zhang, X. Wang, Y.-C. Huang, Y. Shi, X. Zhao, J. Wang, R. Long, Y. Xiong, F. Dong, M. Li and S. Shen, Angew. Chem., Int. Ed., 2023, 62, e202217369 CrossRef CAS PubMed.
- Y. Liu, H. Jiang and Z. Hou, Angew. Chem., Int. Ed., 2021, 60, 11133–11137 CrossRef CAS PubMed.
- J. Zhang, G. Zeng, S. Zhu, H. Tao, Y. Pan, W. Lai, J. Bao, C. Lian, D. Su, M. Shao and H. Huang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218987120 CrossRef CAS PubMed.
- H. L. Wu, L. Q. Huang, J. Timoshenko, K. Qi, W. S. Wang, J. F. Liu, Y. Zhang, S. K. Yang, E. Petit, V. Flaud, J. Li, C. Salameh, P. Miele, L. Lajaunie, B. R. Cuenya, D. W. Rao and D. Voiry, Nat. Energy, 2024, 9, 422–433 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2019, 3, 75–82 CrossRef.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- T. Zhao, J. Li, J. Liu, F. Liu, K. Xu, M. Yu, W. Xu and F. Cheng, ACS Catal., 2023, 13, 4444–4453 CrossRef CAS.
- L.-J. Zhu, D.-H. Si, F.-X. Ma, M.-J. Sun, T. Zhang and R. Cao, ACS Catal., 2023, 13, 5114–5121 CrossRef CAS.
- M. Filippi, T. Möller, L. Liang and P. Strasser, Energy Environ. Sci., 2023, 16, 5265–5273 RSC.
- J. Y. Kim, D. Hong, J. C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G. D. Lee, D. H. Nam and Y. C. Joo, Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- J. Cho, A. Medina, I. Saih, J. Il Choi, M. Drexler, W. A. Goddard, 3rd, F. M. Alamgir and S. S. Jang, Angew. Chem., Int. Ed., 2024, 63, e202320268 CrossRef CAS PubMed.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- R. E. Vos, K. E. Kolmeijer, T. S. Jacobs, W. van der Stam, B. M. Weckhuysen and M. T. M. Koper, ACS Catal., 2023, 13, 8080–8091 CrossRef CAS PubMed.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- W. Shan, R. Liu, H. Zhao, Z. He, Y. Lai, S. Li, G. He and J. Liu, ACS Nano, 2020, 14, 11363–11372 CrossRef CAS PubMed.
- E. P. Delmo, Y. Wang, Y. Song, S. Zhu, H. Zhang, H. Xu, T. Li, J. Jang, Y. Kwon, Y. Wang and M. Shao, J. Am. Chem. Soc., 2024, 146, 1935–1945 CrossRef CAS PubMed.
- C. Li, K. Yu, A. Bird, F. Guo, J. Ilavsky, Y. Liu, D. A. Cullen, A. Kusoglu, A. Z. Weber, P. J. Ferreira and J. Xie, Energy Environ. Sci., 2023, 16, 2977–2990 RSC.
- Z. Guo, P. Zhou, L. Jiang, S. Liu, Y. Yang, Z. Li, P. Wu, Z. Zhang and H. Li, Adv. Mater., 2023, e2311149, DOI:10.1002/adma.202311149.
- Y. Ouyang, L. Shi, X. Bai, C. Ling, Q. Li and J. Wang, ACS Catal., 2023, 13, 15448–15456 CrossRef CAS.
- M. Moradzaman and G. Mul, ChemElectroChem, 2021, 8, 1478–1485 CrossRef CAS.
- S. Y. Lee, J. Kim, G. Bak, E. Lee, D. Kim, S. Yoo, J. Kim, H. Yun and Y. J. Hwang, J. Am. Chem. Soc., 2023, 145, 23068–23075 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01173b |
| ‡ Jing Zhou and Qianyue Liang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |