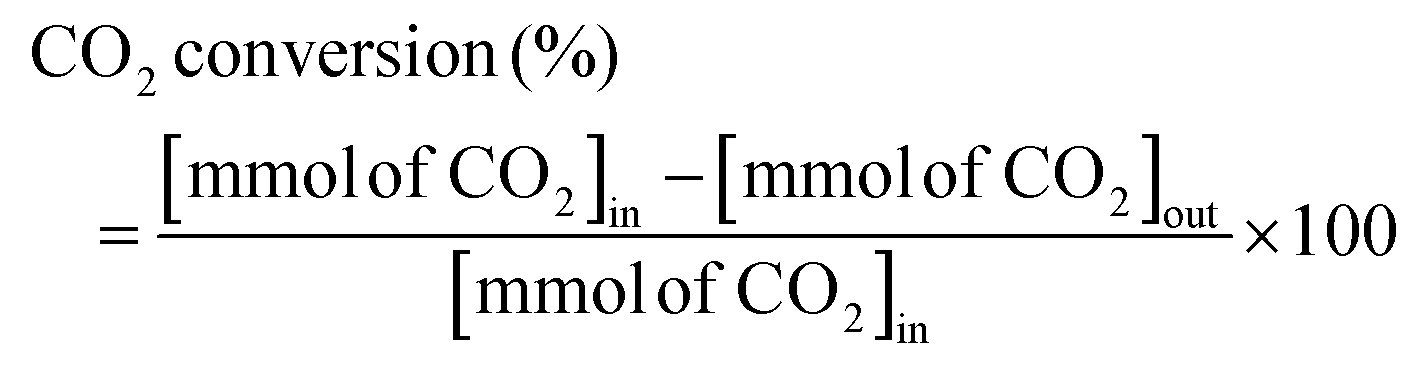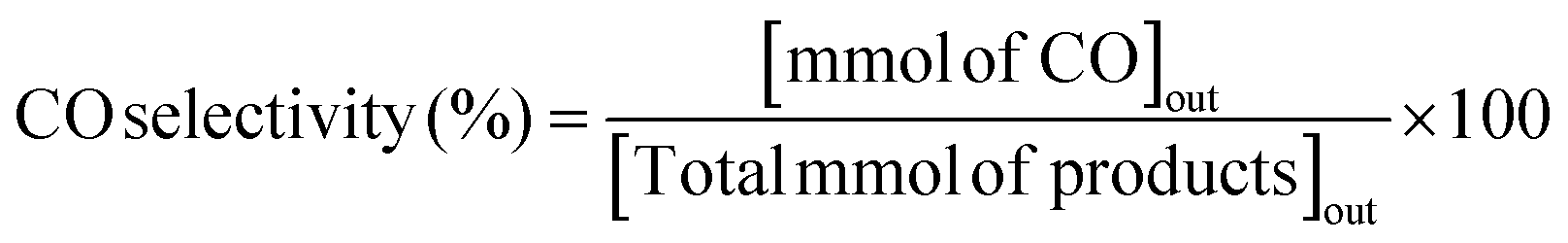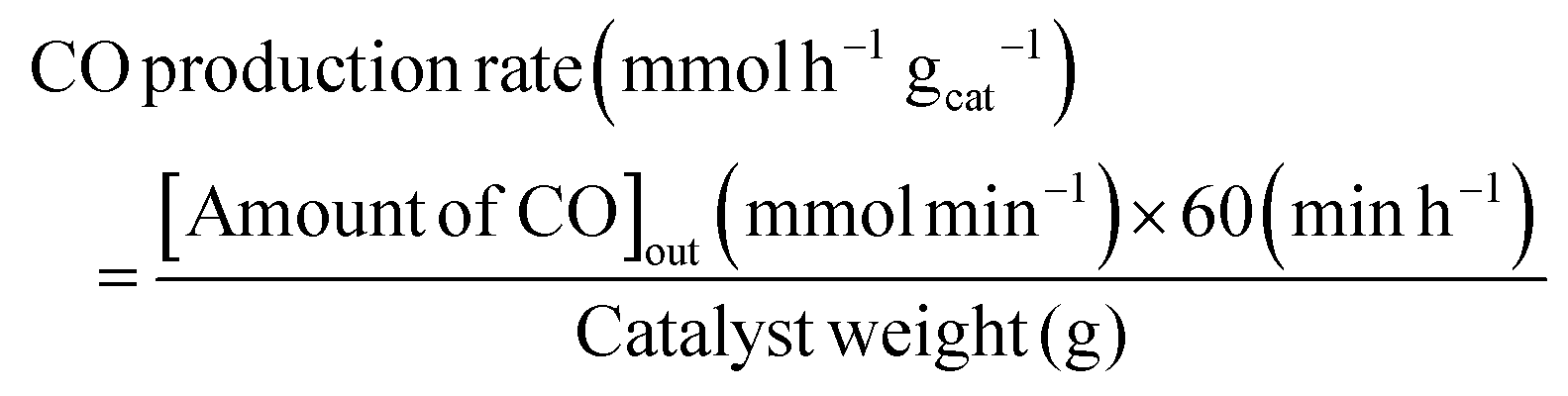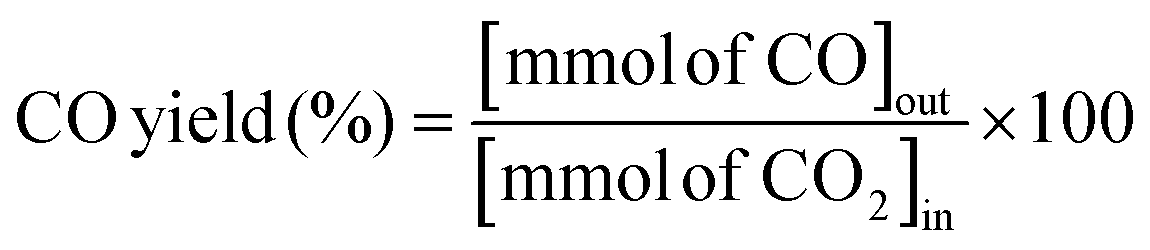DOI:
10.1039/D4NR01185F
(Paper)
Nanoscale, 2024,
16, 14066-14080
Polyoxometalate-HKUST-1 composite derived nanostructured Na–Cu–Mo2C catalyst for efficient reverse water gas shift reaction†
Received
18th March 2024
, Accepted 28th June 2024
First published on 29th June 2024
Abstract
Transforming CO2 to CO via reverse water–gas shift (RWGS) reaction is widely regarded as a promising technique for improving the efficiency and economics of CO2 utilization processes. Moreover, it is also considered as a pathway towards e-fuels. Cu-oxide catalysts are widely explored for low-temperature RWGS reactions; nevertheless, they tend to deactivate significantly under applied reaction conditions due to the agglomeration of copper particles at elevated temperatures. Herein, we have synthesized homogeneously distributed Cu metallic nanoparticles supported on Mo2C for the RWGS reaction by a unique approach of in situ carburization of metal–organic frameworks (MOFs) using a Cu-based MOF i.e. HKUST-1 encapsulating molybdenum-based polyoxometalates. The newly derived Na–Cu–Mo2C nanocomposite catalyst system exhibits excellent catalytic performance with a CO production rate of 3230.0 mmol gcat−1 h−1 with 100% CO selectivity. Even after 250 h of a stability test, the catalyst remained active with more than 80% of its initial activity.
Introduction
Carbon dioxide (CO2) anthropogenic emissions have drawn immense worldwide interest in recent years due to their possible environmental, social, and economic implications, with a focus on global warming. Therefore, CCUS (carbon capture, utilization, and sequestration) technologies have been developed to utilize CO2 as a carbon pool to synthesize value-added compounds and to mitigate CO2 emissions globally.1 A key component of C1 chemistry is carbon monoxide (CO), which is specifically produced by the reverse water–gas shift (RWGS) reaction (eqn (1)).2 When coupled with renewable hydrogen (H2), the produced CO serves as a feedstock for the production of e-fuels via the Fischer–Tropsch (FT) process or methanol synthesis.3| | 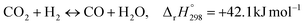 | (1) |
As an endothermic process, the RWGS reaction frequently requires a high temperature to promote the equilibrium conversion which limits the catalyst's stability.2 Although Pd- and Pt-based noble metal catalysts have been utilized extensively for this process, their practical uses are restricted due to their high cost and low catalytic efficiency.4 Cu-based catalysts’ high activity, selectivity, and cost-effectiveness make them the most preferred non-noble metal catalysts for this process (Table 1). The conventional Cu-oxide catalysts, on the other hand, at elevated temperatures suffer from severe agglomeration of supported Cu particles leading to rapid deactivation under typical operating conditions.5 This is due to the low Tammann temperature of the Cu particle, which causes sintering at higher temperatures.6 Therefore, the primary impediment to their industrial implementation is their poor stability.
Table 1 A brief quantitative comparison of the previously reported RWGS catalysts with this work
| Catalysts |
CO2 conversion (%) |
CO selectivity (%) |
CO yield (%) |
CO production rate (mmol h−1 gcat−1) |
Stability test (h) |
Ref. |
| CuO/γ-Al2O3 |
60 |
100 |
60 |
— |
80 |
21
|
| 0.25Fe0.75Cu |
37 |
100 |
37 |
134.3 |
48 |
22
|
| FeCu/CeAl |
42 |
100 |
42 |
102.2 |
48 |
23
|
| DFNS–TiO2–Cu10 |
10 |
99.8 |
9.98 |
5350 |
200 |
24
|
| CuSiO–I |
9.8 |
100 |
9.8 |
2585 |
45 |
25
|
| Cu-2D-SiO2-850r |
10 |
100 |
10 |
296 |
54 |
26
|
| K–Co/CeO2 (1/10) |
37 |
100 |
37 |
2478 |
NR |
27
|
| 15CuCe |
60 |
100 |
60 |
2466 |
230 |
28
|
| Cu/Al2O3 |
47 |
100 |
47 |
2097 |
40 |
29
|
| Cu/β-Mo2C |
40 |
100 |
40 |
1786 |
40 |
12
|
| CuSiO/CuOx |
17.8 |
100 |
17.8 |
114 |
24 |
30
|
| Cu/Al2O3 |
50 |
100 |
50 |
22 |
NR |
31
|
| Co/Mo2C |
9.5 |
100 |
9.5 |
51 |
36 |
32
|
|
Na–Cu–Mo
2
C
|
5.9
|
100
|
5.9
|
3230
|
250
|
This work
|
|
Na–Cu–Mo
2
C
|
74
|
100
|
74
|
57.8
|
100
|
This work
|
One of the main challenges to developing effective and durable RWGS catalysts is finding a suitable support material that can substantially improve Cu dispersion while impeding particle agglomeration during the reaction. Transition-metal carbide (TMC) supports are an intriguing option that have attracted a lot of attention in recent years since they are economical, non-toxic and non-hazardous and exhibit near noble metal-like behavior.7 Furthermore, the unique catalytic properties of TMCs, which facilitate H2 dissociation and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond scission in addition to improving the dispersion and activation of small metal particles, possibly promote the RWGS activity.8 Among the different TMCs, Mo2C has emerged as one of the most promising supports that may be used in RWGS reactions because of these characteristics and its low cost.9 Mo2C has been commonly seen in β-Mo2C, which has a hexagonal closed packed (hcp) structure, and α-Mo2C, which has a face-centered cubic (fcc) structure.10 However, the β-Mo2C support is commonly used for RWGS reactions due to its relatively higher stability and CO2 conversion rate.11,12 To further enhance the CO selectivity and RWGS rate, alkali metals i.e., Na, K, Cs, are also employed in conjunction with Cu–Mo2C catalysts. One potential effect of integrating alkali metals is an increased CO2 adsorption capacity, accelerating electron transfer and improving interactions with CO2 molecules.13 Xu et al. demonstrated that K incorporation on Cu/β-Mo2C resulted in a significant increase in CO2 conversion compared to Cu/β-Mo2C.14 Similarly, Zhang et al. reported that the Cs incorporation enhanced the CO2 conversion and CO selectivity in Cu/β-Mo2C catalysts.15
O bond scission in addition to improving the dispersion and activation of small metal particles, possibly promote the RWGS activity.8 Among the different TMCs, Mo2C has emerged as one of the most promising supports that may be used in RWGS reactions because of these characteristics and its low cost.9 Mo2C has been commonly seen in β-Mo2C, which has a hexagonal closed packed (hcp) structure, and α-Mo2C, which has a face-centered cubic (fcc) structure.10 However, the β-Mo2C support is commonly used for RWGS reactions due to its relatively higher stability and CO2 conversion rate.11,12 To further enhance the CO selectivity and RWGS rate, alkali metals i.e., Na, K, Cs, are also employed in conjunction with Cu–Mo2C catalysts. One potential effect of integrating alkali metals is an increased CO2 adsorption capacity, accelerating electron transfer and improving interactions with CO2 molecules.13 Xu et al. demonstrated that K incorporation on Cu/β-Mo2C resulted in a significant increase in CO2 conversion compared to Cu/β-Mo2C.14 Similarly, Zhang et al. reported that the Cs incorporation enhanced the CO2 conversion and CO selectivity in Cu/β-Mo2C catalysts.15
Recently published results suggest that MOFs (Metal–Organic Frameworks) are interesting precursors for the synthesis of nano-porous carbides as heterogeneous catalysts.16 The MOF-assisted synthesis can endow the active metals in the derived materials with greater stability than the original MOFs.17–20 Moreover, the organic linker-coordinated metal clusters may partially or completely prevent the pyrolysis-induced agglomeration of the resulting metal nanoparticles.33 Moreover, the large surface area of the materials ensures the efficient diffusion of reactants and products from active sites.34 The North East Normal University-5, or NENU-5 MOF is a bimetallic MOF in which the pores of a Cu-based MOF [HKUST-1; Cu3(BTC)2(H2O)3] enclose a Keggin type Mo-based polyoxometalate (POM) (H3PMo12O40). Mo interacts with the carbonaceous ligands to form MoCx, and Cu2+ is reduced to Cu metallic particles during the carbonization of NENU-5 in an inert atmosphere, producing uniformly dispersed Cu metallic particles on the MoCx support, i.e. Cu–MoCx.35 This MOF-assisted synthesis of MoCx avoids the necessity for a CH4 + H2 gas mixture for molybdenum phase carburization and the pre-reduction step for Cu2+ reduction to the Cu metallic phase.11 The resultant material can be utilized directly as a heterogeneous catalyst or after removing the Cu metallic particles to produce only MoCx as a heterogeneous catalyst. Wu et al. produced porous MoCx nano-octahedra by eliminating Cu particles from Cu–MoCx generated from NENU-5 MOF for the Hydrogen Evolution Reaction (HER).35 Zhang et al. doped Pt over NENU-5 MOF followed by carbonization to form a Mo2C-supported Pt–Cu nanoalloy heterostructure for the same purpose.36 Based on these presumptions, we attempted for the first time to synthesize a NENU-5 MOF derived Na–Cu–Mo2C nanocomposite catalyst for the RWGS reaction. This approach generated high surface area porous nano-structured highly dispersed Cu metallic particles over the Mo2C support.
Experimental
Synthesis of an octahedron NENU-5 MOF assisted nanostructured porous Na–Cu–Mo2C catalyst
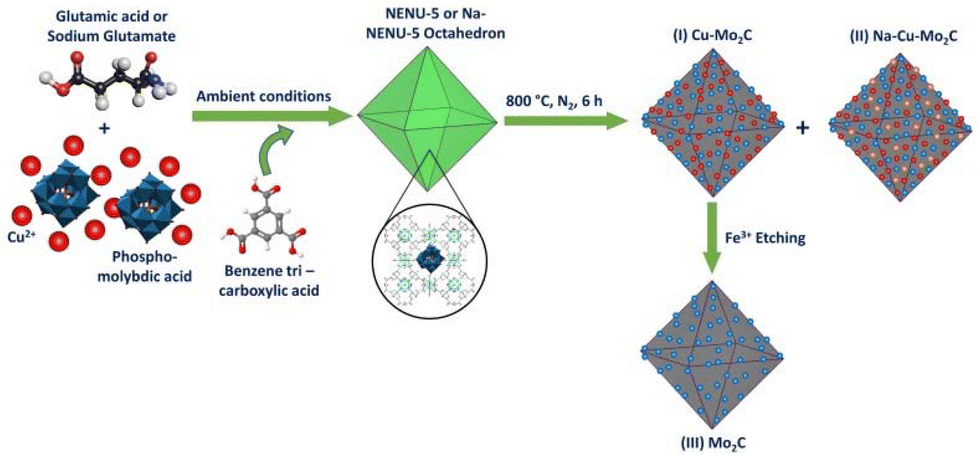
The overall synthesis procedure to prepare a nanostructured porous Na–Cu–Mo2C catalyst for an efficient RWGS reaction is illustrated in Scheme 1. Here, two types of NENU-5 MOF were prepared by a direct precipitation method, one with glutamic acid and the second with a sodium glutamate modulator. First, a 40 ml aqueous solution of 1 mmol copper(II) acetate monohydrate (0.2 g) (TCI, >95%), 0.3 g of phosphomolybdic acid hydrate (CDH), and 0.5 mmol L-glutamic acid (0.073565 g) (Thermo Scientific, 99%) or 0.5 mmol monosodium glutamate (0.08455 g) (Loba Chemie, 99%) was prepared. After that, a solution of 40 ml ethanol (Fisher Chemical) and 0.67 mmol 1,3,5-benzene tricarboxylic acid (BTC) (0.1408 g) (TCI, >98%) was added dropwise to that solution at room temperature to form a green precipitate. After 14 h of stirring, the material was washed with ethanol and dried at 70 °C to obtain pure octahedron NENU-5 and Na-NENU-5 MOF. The obtained NENU-5 and Na-NENU-5 MOF were then pyrolyzed in a tubular furnace in the presence of N2 at 800 °C for 6 h to obtain Cu–Mo2C and Na–Cu–Mo2C catalysts. The Cu–Mo2C catalyst was disseminated in 0.1 M FeCl3·3H2O solution to remove the Cu particles and produce the third catalyst, named Mo2C. The catalysts were then directly tested for the RWGS reaction.
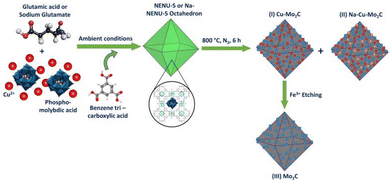 |
| | Scheme 1 Synthesis scheme for the nano-structured porous Na–Cu–Mo2C catalyst. | |
Catalyst characterization
X-ray diffraction (XRD) patterns were acquired using a PROTO AXRD® Benchtop Powder diffractometer. Raman spectra were collected with a Horiba LabRAM HR Evolution Raman Spectrometer using a 532 nm laser as an excitation source. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM) images were captured using the JEOL JEM-2100 instrument. SEM images were captured using a Quanta 200 F, M/s FEI instrument. X-ray Photoelectron Spectroscopy (XPS) was carried out on a Thermo Scientific NEXSA XPS spectrometer. Thermogravimetric analysis (TGA) was carried out using a PerkinElmer TGA 4000 thermogravimetric analyzer. N2O titration analysis was performed using a Micrometrics® Autochem II 2920 instrument. CO2 Temperature-Programmed Desorption (CO2 TPD) experiments were carried out on a Micrometrics® Autochem II 2920 instrument. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis was performed on the Teledyne Leeman Labs Prodigy7 instrument. The FTIR spectra of the catalysts were obtained on a Spectrum Two, PerkinElmer instrument using a pellet form of the catalysts diluted with potassium bromide. N2 adsorption–desorption isotherms at 93 K were obtained using the Micromeritics ASAP 2020 Surface Area & Porosity Analyzer and the BET equation. In situ CO2 DRIFT spectra were obtained using the PerkinElmer Inc. in Massachusetts, USA. The other details of characterization techniques and procedures are given in the ESI.†
Catalytic activity tests
The RWGS reaction was conducted to evaluate the catalyst's activity using a 4 mm diameter vertical fixed bed continuous-flow reactor. Using a layer of quartz wool (4 mm diameter) in the center of the reactor, 0.02 g of the catalyst was loaded to form a catalyst bed. To monitor the reactor system's temperature, a thermocouple was placed directly beneath the catalyst bed. A temperature differential of 50 °C and a gas space hour velocity (GHSV) of 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ml gcat−1 h−1 were the conditions under which all activity tests were conducted. As an internal standard to monitor the effluent volume and determine the reactant conversion and product selectivity, N2 was also fed into the reactor along with the reactant. The Na–Cu–Mo2C catalyst was also tested with various H2:CO2 ratios (other parameters were the same as the activity test) to maximize the rate at which CO was produced (for 3:1, H2:CO2:N2 vol% was 60:20:20; for 2:1, H2:CO2:N2 vol% was 60:30:10; and for 1:1, H2:CO2:N2 vol% was 45:45:10). The activity of the Na–Cu–Mo2C catalyst was also compared with that of the benchmark Cu/ZnO/Al2O3 catalyst (HiFUEL W220) from 350–600 °C as well as its stability for 20 h at 600 °C with 300000 ml gcat−1 h−1 GHSV (H2:CO2 ratio 1). Subsequently, a high space velocity test (3000000 ml gcat−1 h−1) with a 1:1 H2:CO2 ratio was carried out using 0.002 g of Na–Cu–Mo2C catalyst in the same reactor at 350–600 °C with a 50 °C temperature differential. The Na–Cu–Mo2C catalyst was also subjected to a 250 h stability test at 600 °C using the same reaction conditions. Finally, the activity of the Na–Cu–Mo2C catalyst was also analyzed in a wider temperature range from 300–800 °C with a relatively lower GHSV (10000 ml gcat−1 h−1) (H2:CO2 ratio 4), and further stability was analyzed at 800 °C for 100 h. Following 60 minutes of reaction at each temperature, the composition of the effluent gas was analyzed and quantified using an online Agilent 7890B Refinery Gas Analyser gas chromatograph. A Molsieve 5A column was used to separate the gases H2, CO2, N2, CH4, and CO which were then measured using a thermal conductivity detector. All of the gas cylinders used in the activity test were acquired from SIGMA GAS SERVICES (SGS) and were at least 99% pure. The following formulas were used to calculate the CO2 conversion, CO selectivity, CO production rate, and CO yield:
000 ml gcat−1 h−1 were the conditions under which all activity tests were conducted. As an internal standard to monitor the effluent volume and determine the reactant conversion and product selectivity, N2 was also fed into the reactor along with the reactant. The Na–Cu–Mo2C catalyst was also tested with various H2:CO2 ratios (other parameters were the same as the activity test) to maximize the rate at which CO was produced (for 3:1, H2:CO2:N2 vol% was 60:20:20; for 2:1, H2:CO2:N2 vol% was 60:30:10; and for 1:1, H2:CO2:N2 vol% was 45:45:10). The activity of the Na–Cu–Mo2C catalyst was also compared with that of the benchmark Cu/ZnO/Al2O3 catalyst (HiFUEL W220) from 350–600 °C as well as its stability for 20 h at 600 °C with 300000 ml gcat−1 h−1 GHSV (H2:CO2 ratio 1). Subsequently, a high space velocity test (3000000 ml gcat−1 h−1) with a 1:1 H2:CO2 ratio was carried out using 0.002 g of Na–Cu–Mo2C catalyst in the same reactor at 350–600 °C with a 50 °C temperature differential. The Na–Cu–Mo2C catalyst was also subjected to a 250 h stability test at 600 °C using the same reaction conditions. Finally, the activity of the Na–Cu–Mo2C catalyst was also analyzed in a wider temperature range from 300–800 °C with a relatively lower GHSV (10000 ml gcat−1 h−1) (H2:CO2 ratio 4), and further stability was analyzed at 800 °C for 100 h. Following 60 minutes of reaction at each temperature, the composition of the effluent gas was analyzed and quantified using an online Agilent 7890B Refinery Gas Analyser gas chromatograph. A Molsieve 5A column was used to separate the gases H2, CO2, N2, CH4, and CO which were then measured using a thermal conductivity detector. All of the gas cylinders used in the activity test were acquired from SIGMA GAS SERVICES (SGS) and were at least 99% pure. The following formulas were used to calculate the CO2 conversion, CO selectivity, CO production rate, and CO yield:| | 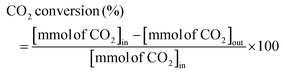 | (2) |
| | 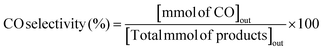 | (3) |
| | 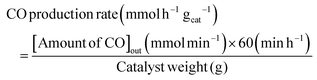 | (4) |
| | 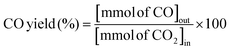 | (5) |
The subscripts “in” and “out” stand for mmol min−1 of a given species at the reactor's input and output during the reaction, respectively.
Results and discussion
The characterization of the as-synthesized NENU-5 MOF
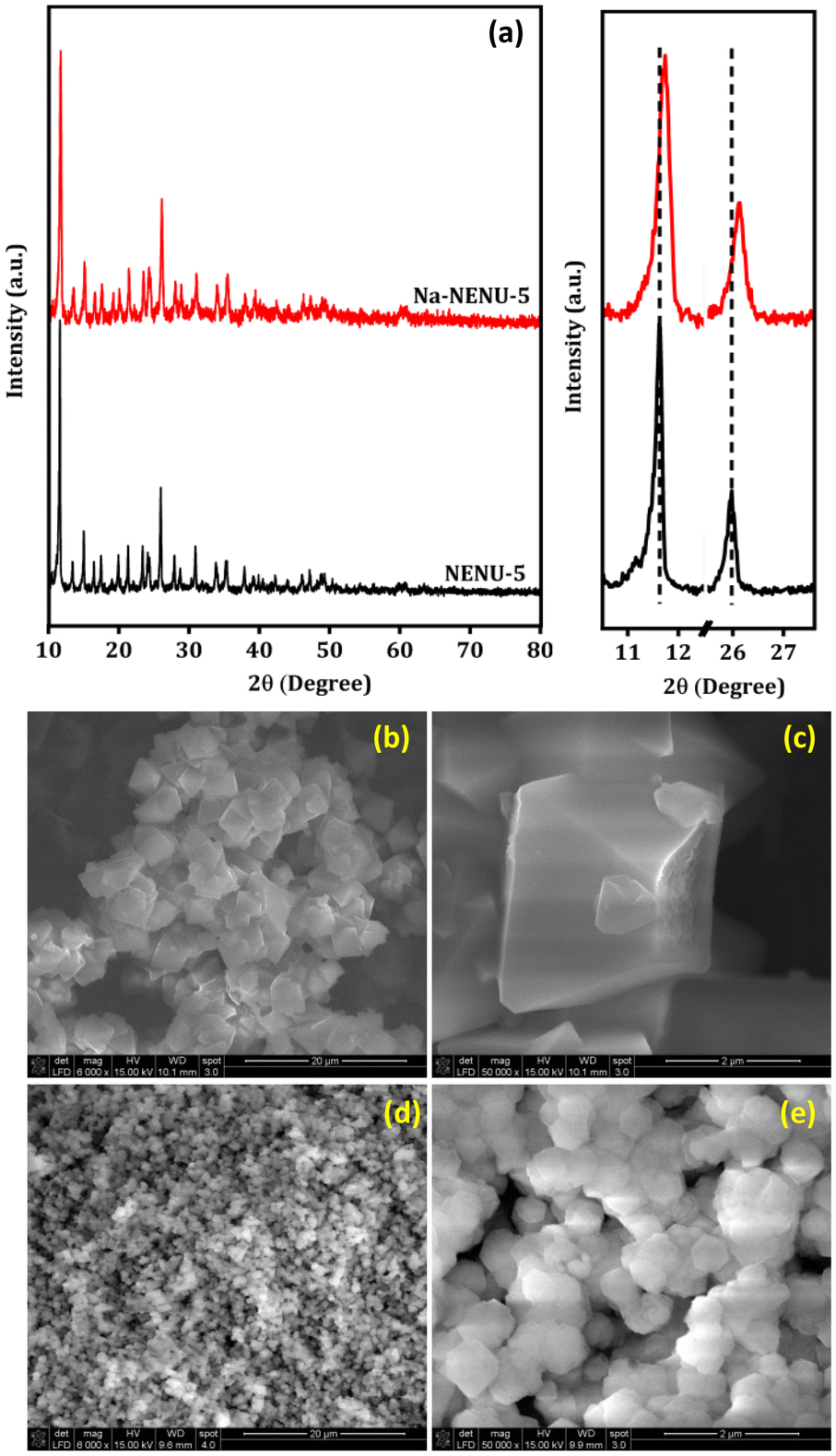
The X-ray diffraction patterns of the NENU-5 and Na-NENU-5 are shown in Fig. 1(a) which confirm the excellent crystallinity of the as-synthesized samples and the diffraction pattern matched well with previously published works.35–37 There is a positive shift and broadening of the diffraction peaks for Na-NENU-5, demonstrating a slight change in the crystal structure and a lower crystallite size compared to the NENU-5. The calculated crystallite size for the most intense peak (2θ ∼ 11.7°) of Na-NENU-5 and NENU-5 MOF is 30.4 and 68.4 nm. In the NENU-5 (Fig. 1b and c), the SEM images confirmed a conventional octahedron morphology with particle sizes of around ∼2–3 μm; however in the Na-NENU-5 (Fig. 1d and e), the particle size significantly decreased and was in the range of ∼400–500 nm. The SEM energy-dispersive X-ray spectroscopy (EDX) investigation of NENU-5 and Na-NENU-5 shows the existence of well-distributed component elements (Fig. S1 and S2†). The effect of a decrease in particle size in MOFs is also clearly visible in the N2 adsorption–desorption isotherms. As displayed in Fig. S3,† the BET surface area of Na-NENU-5 (410 m2 g−1) was larger than that of the NENU-5 (352 m2 g−1), with a characteristic hysteresis loop demonstrating the porous nature of the MOFs.
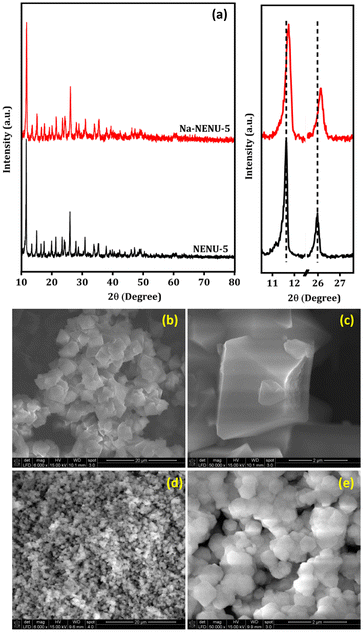 |
| | Fig. 1 XRD pattern (a) and SEM images of NENU-5 (b and c), and Na-NENU-5 MOF (d and e). | |
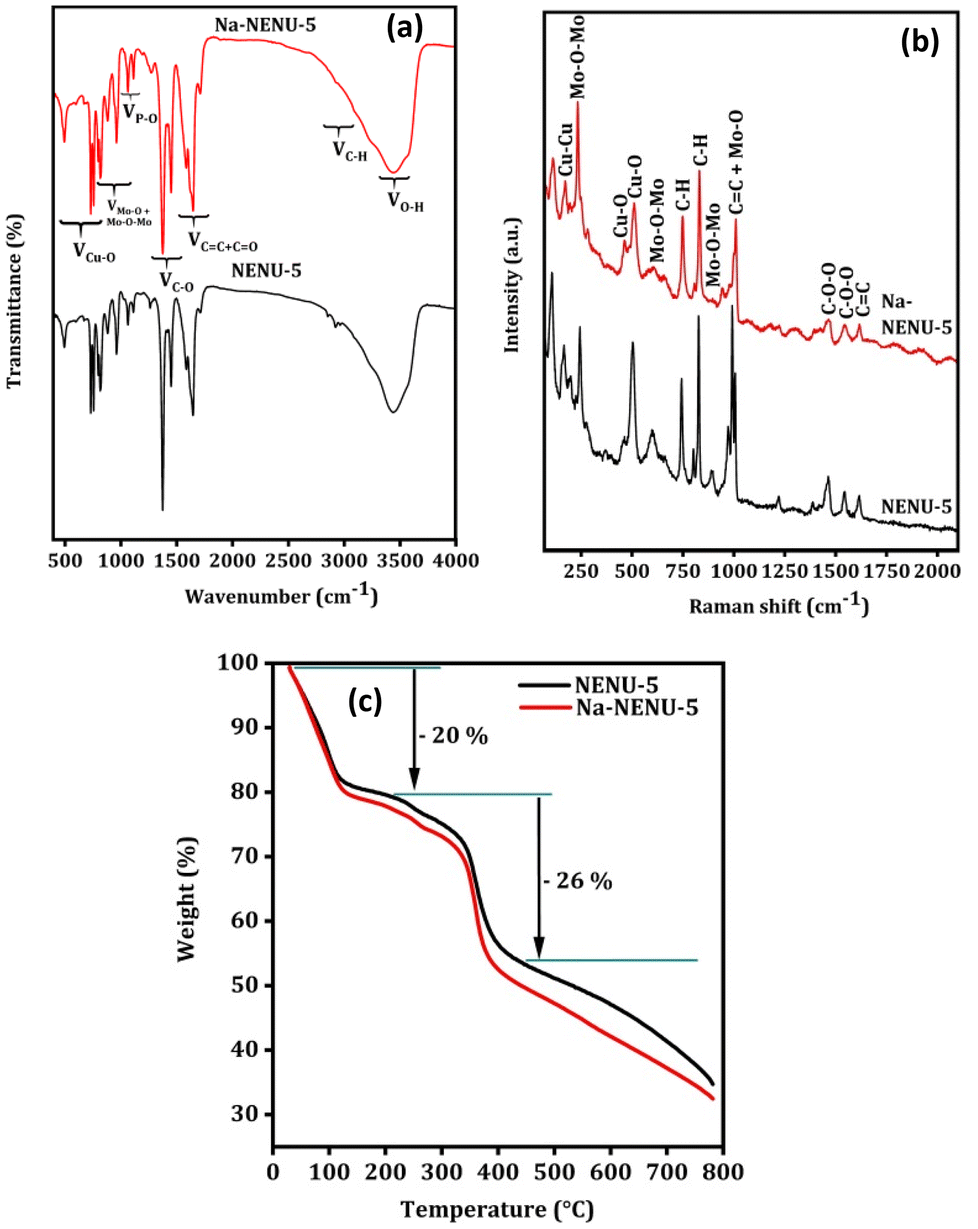
The FTIR spectrum was used to further analyze the existence of various functional groups in the synthesized MOFs. As shown in Fig. 2(a), the characteristic bands at 1106, 1065, and 1032 cm−1 can be attributed to the P–O bonds and those at 955, 890, 812, and 790 cm−1 can be attributed to various modes of Mo–O or Mo–O–Mo bonds, supporting the presence of a Keggin type Mo-based polyoxometalate.38–40 Similarly, the IR bands at 1373 cm−1 for C–O bonds, at 1450 and 1555 cm−1 for CO bonds, and at 1645 cm−1 for CC bonds in the BTC ligand of HKUST-1 were observed.41 The IR bands at 760, 730, and 494 cm−1 can be attributed to the Cu–O bonds in HKUST-1.42,43 The C–H stretching of the methylene group in BTC shows small bands near 2922 cm−1 along with a broad-band centered near 3450 cm−1 for O–H vibration.38 As can be seen in the Raman spectra in Fig. 2(b) of MOFs, the Raman bands at 1000, 973, 890, 603, and 233 cm−1 are due to different modes of Mo–O and Mo–O–Mo bonds of a Keggin type Mo-based polyoxometalate.38 The other Raman bands can be attributed to the different vibrational modes of functional groups present in the HKUST-1, whose Raman shift matched well with previously published works.44,45 This overall provided additional evidence in support of the Keggin type Mo-based polyoxometalate's successful incorporation into the HKUST-1 framework. The thermal stability of the MOFs was further compared in a N2 atmosphere and results are shown in Fig. 2(c). Both MOFs exhibited comparable weight loss and patterns, with a 20% loss between 50–200 °C attributed to the removal of water or solvent molecules and a 26% loss between 200–400 °C due to complete decomposition of the organic framework.46
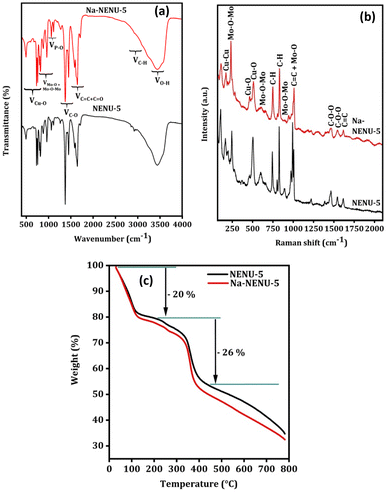 |
| | Fig. 2 FT IR spectra (a), Raman spectra (b), and TGA analysis (c) of NENU-5 and Na-NENU-5. | |
The characterization of the pyrolyzed MOFs
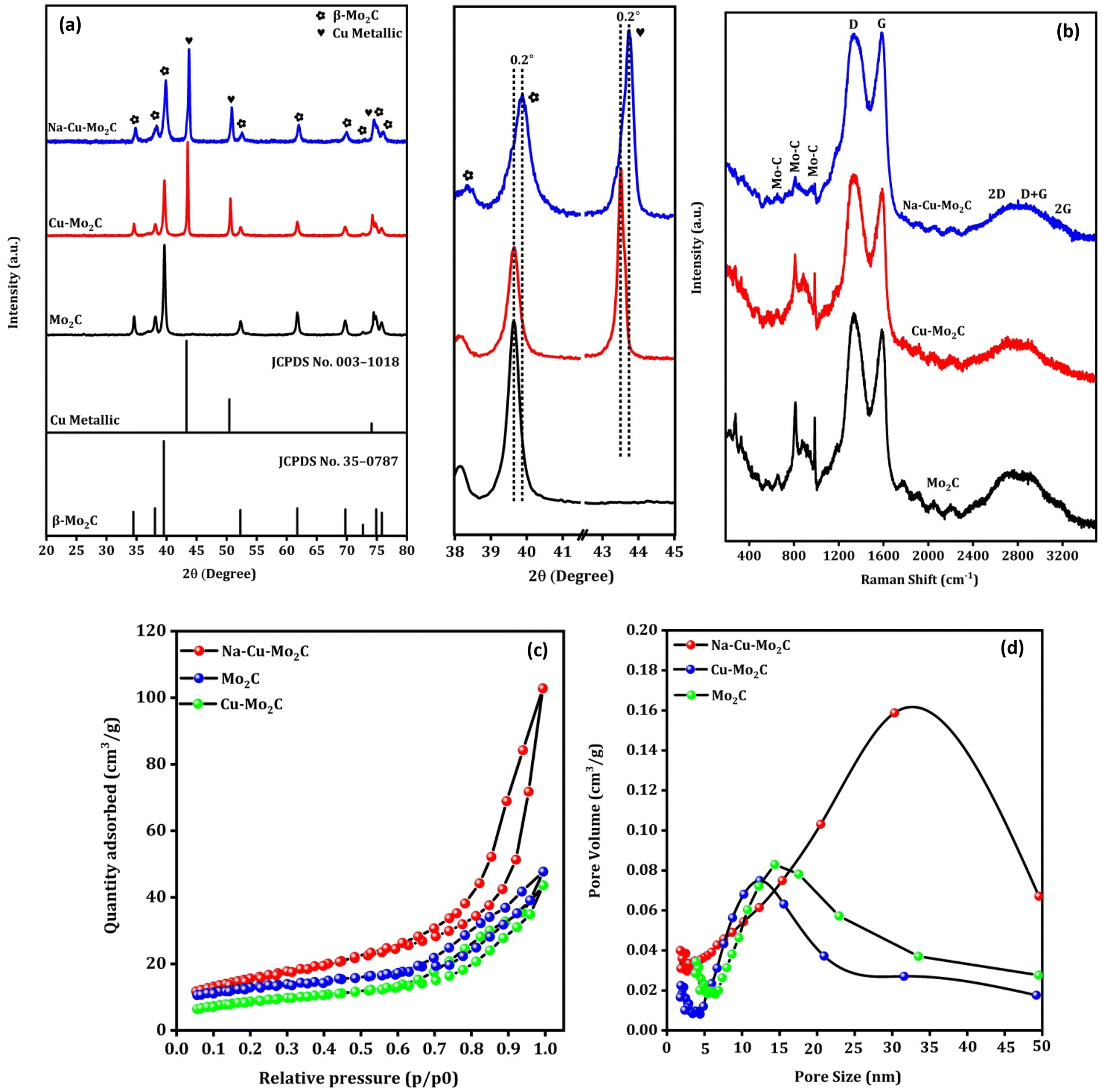
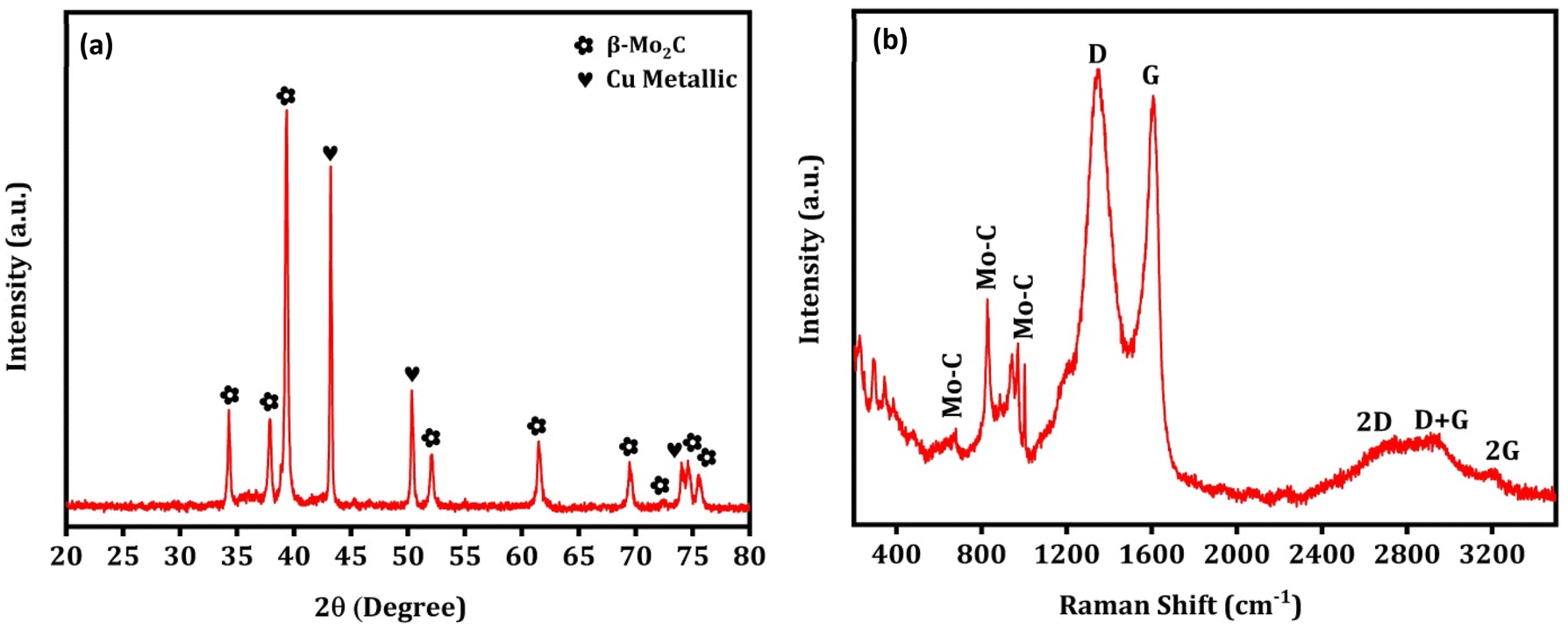
Fig. 3(a) shows the XRD pattern of the prepared catalysts. The characteristic diffraction peaks at 2θ = 34.5°, 38.0°, 39.5°, 52.3°, 61.8°, 69.8°, 74.9°, and 75.9° can be attributed to the (100), (002), (101), (102), (110), (103), (112) and (201) crystal planes of hexagonal β-Mo2C (JCPDS no. 35-0787), respectively.47 The formation of the Mo2C phase after pyrolysis of Mo salts and carbon sources in this temperature range is consistent with previously published works.48,49 The diffraction peaks at 2θ = 43.3°, 50.4°, and 74.1° can be assigned to the (111), (200) and (220) crystal planes of the Cu metallic phase (JCPDS no. 003-1018).50 Overall, the analysis showed that the bulk of the catalysts is composed of the Mo2C and Cu metallic phases. The diffraction peaks of the Cu metallic phase disappeared completely in the Mo2C, indicating that Cu particles were successfully removed from the catalyst. This was further supported by ICP-OES analysis, where Cu wt% decreased from 21.3 to 0.25 from Cu–Mo2C to Mo2C (Table 2). It is worth mentioning that the positively shifted and broader diffraction peaks of the Mo2C and Cu phases in the Na–Cu–Mo2C catalyst imply a slight change in the crystal structure and a smaller crystallite size of these phases in Na–Cu–Mo2C when compared with the Cu–Mo2C catalyst. It also demonstrates that the structural changes observed in the original MOFs were inherited in the catalysts even after the pyrolysis. The crystallite size of both phases was determined using the most intense peaks (2θ ∼ 39.5° for Mo2C and 43.3° for the Cu phase), and the crystallite size of Mo2C is 25.1 nm, 23.1 nm, and 15.4 nm for Mo2C, Cu–Mo2C, and Na–Cu–Mo2C, respectively. The crystallite size of Cu was calculated and found to be 38.9 nm for Cu–Mo2C and 29.3 nm for Na–Cu–Mo2C. Fig. 3(b) displays the Raman spectra of the synthesized catalysts, confirming the formation of the β-Mo2C phase. The Raman bands at 656, 812, and 986 cm−1 can be attributed to the Mo–C stretching of the β-Mo2C phase.51 Additionally, the D and G bands at 1350 and 1580 cm−1 correspond to the stretching of the disordered graphitic carbon and sp2 bonded carbon atoms, respectively confirming the presence of the carbon species.51,52 In N2 adsorption–desorption isotherms, the BET surface area for Cu–Mo2C, Mo2C, and Na–Cu–Mo2C was 31 m2 g−1, 35 m2 g−1, and 56 m2 g−1, respectively (Fig. 3c). The pore size distribution for Cu–Mo2C, Mo2C, and Na–Cu–Mo2C was around 12 nm, 14 nm, and 32 nm, respectively, indicating the porous nature of the catalysts (Fig. 3d). N2O titration analysis is an effective method to estimate the Cu metal dispersion (DisCu) as reported previously.53,54 Based on the following reactions during the N2O titration method followed by H2 consumption during H2 pulse reduction, we can estimate that the DisCu ∝ H2 consumed after N2O titration:
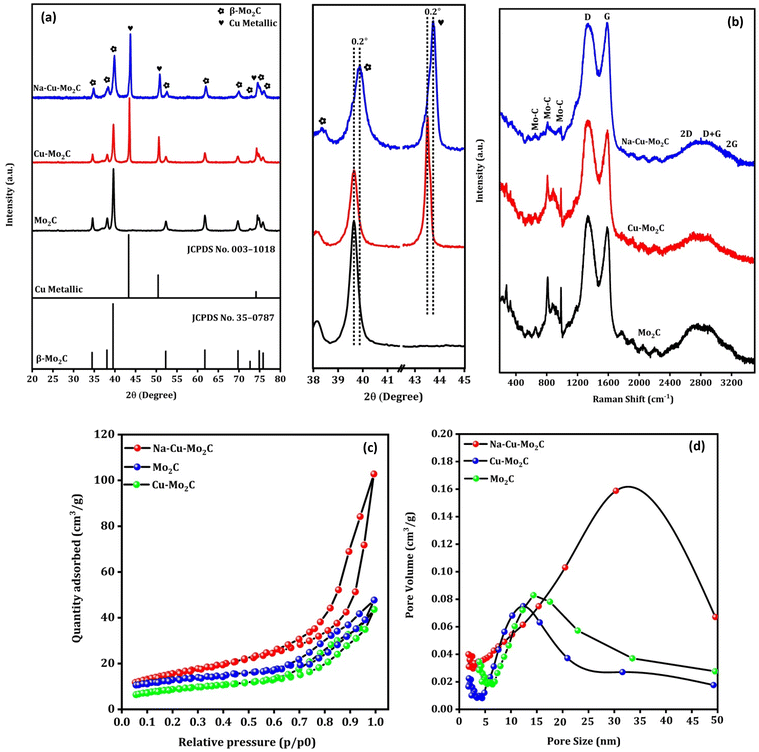 |
| | Fig. 3 XRD pattern (a), Raman spectra (b), N2 adsorption–desorption isotherms (c), and pore size distribution (d) of the derived catalysts. | |
Table 2 Surface composition, ICP-OES elemental analysis, physiochemical properties, crystallite size and H2 consumption during H2 pulse reduction after N2O titration of MOF derived catalysts
| Catalysts |
Relative surface composition by XPS (atomic%) |
ICP-OES elemental analysis (wt%) |
Physicochemical properties |
Crystallite size (nm) |
H2 consumption after N2O titration (mmol g−1) |
| Cu |
Mo |
C |
Na |
Cu |
Mo |
Surface area (m2 g−1) |
Pore size (nm) |
Cu |
Mo |
| Mo2C |
0.2 |
12.8 |
87.0 |
— |
0.25 |
55.2 |
35 |
14 |
— |
25.1 |
0.001 |
| Cu–Mo2C |
6.4 |
18.4 |
75.2 |
— |
21.3 |
40.2 |
31 |
12 |
38.9 |
23.1 |
0.022 |
| Na–Cu–Mo2C |
5.9 |
16.3 |
76.6 |
1.2 |
20.5 |
38.3 |
56 |
32 |
29.3 |
15.4 |
0.036 |
As a result, the amount of H2 consumed following the N2O titration analysis in eqn (6) can be used as a benchmark to assess the DisCu in catalysts. As tabulated in Table 2, the H2 consumption for the Mo2C, Cu–Mo2C, and Na–Cu–Mo2C was 0.001, 0.022, and 0.036 mmol g−1, respectively. In another way, it indicates that the Cu dispersion for the derived catalysts follows the order Mo2C < Cu–Mo2C < Na–Cu–Mo2C.
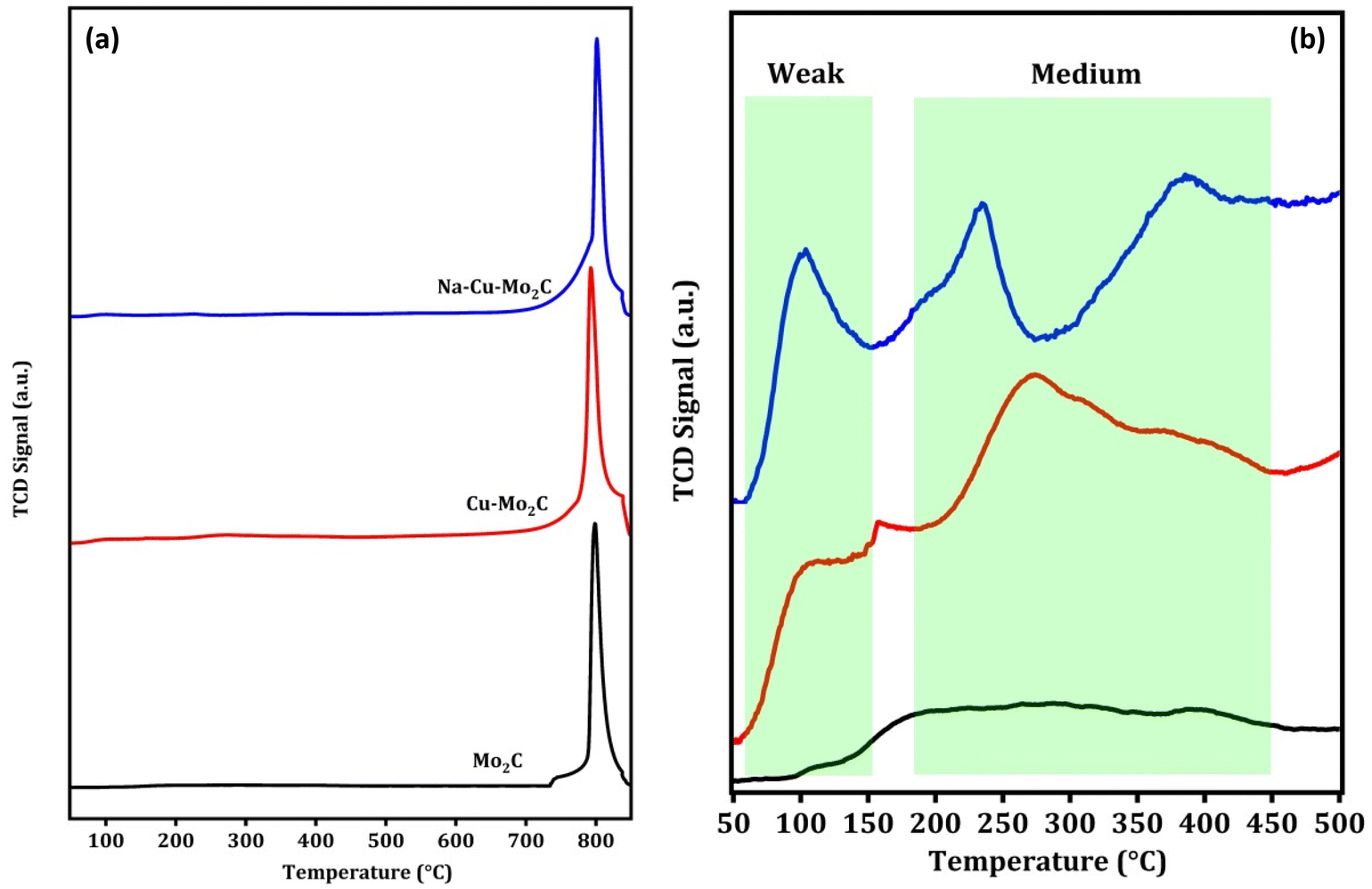
CO2 adsorption properties are an essential characteristic of the CO2 conversion processes that can be directly related to the activity of the catalysts. CO2 TPD experiments were carried out to investigate the CO2 adsorption properties of the catalysts and the results are shown in Fig. 4(a and b). As observed in Fig. 4a, all catalysts exhibit one prominent CO2 desorption peak between 750–850 °C, which is attributed to CO2 desorption from strong basic sites (>500 °C). This suggests that all of the catalysts are highly saturated with strong basic sites. The significant difference in CO2 TPD results can be seen in the graph below 500 °C, as shown in Fig. 4b. The desorption peaks can be divided into two regions, weak basic sites (<150 °C) and medium basic sites (150–450 °C). There are no discernible desorption peaks for the Mo2C catalyst in this region. Nevertheless, in Cu–Mo2C, the presence of Cu results in an increase in the CO2 desorption peaks in this region, which was the consequence of the formation of additional weak and medium basic sites. Furthermore, in Na–Cu–Mo2C, the inclusion of Na not only considerably enhanced the area of desorption peaks, but also demonstrated additional desorption peaks in this region. The formation of additional basic sites in the presence of Na is consistent with previously reported works.13,55 As a result, we can infer the following sequence of increases in CO2 adsorption ability: Mo2C < Cu–Mo2C < Na–Cu–Mo2C.
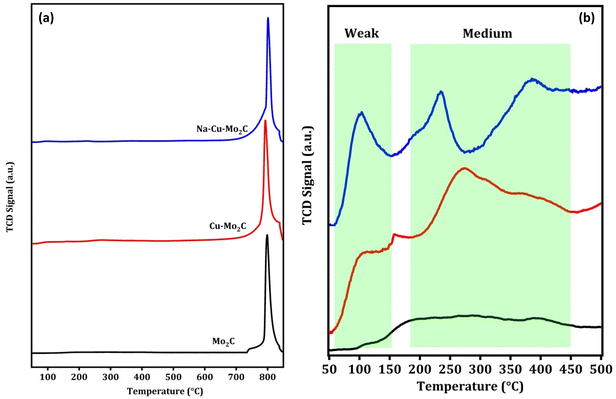 |
| | Fig. 4 CO2 TPD analysis of the derived catalysts (a) and below 500 °C (b). | |
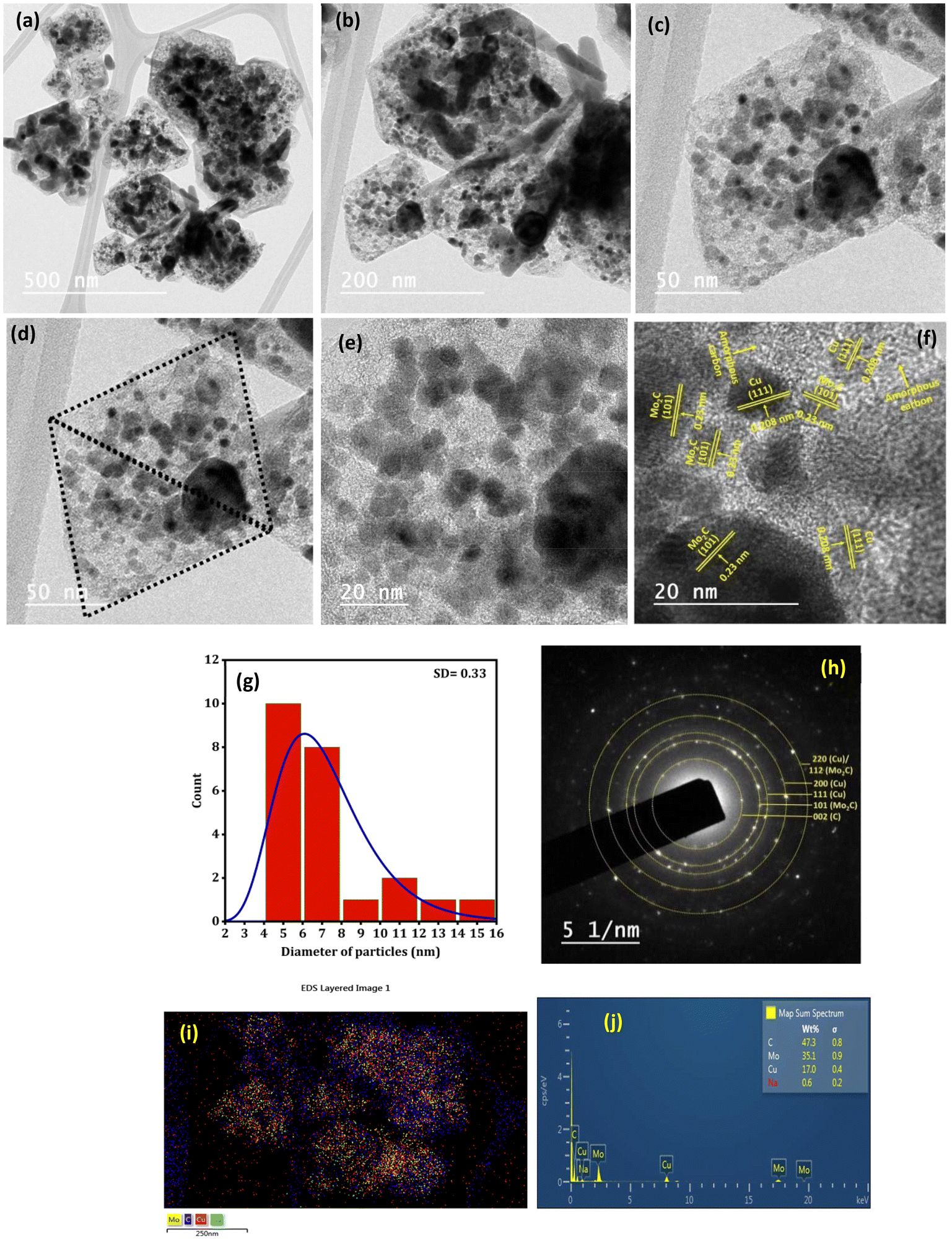
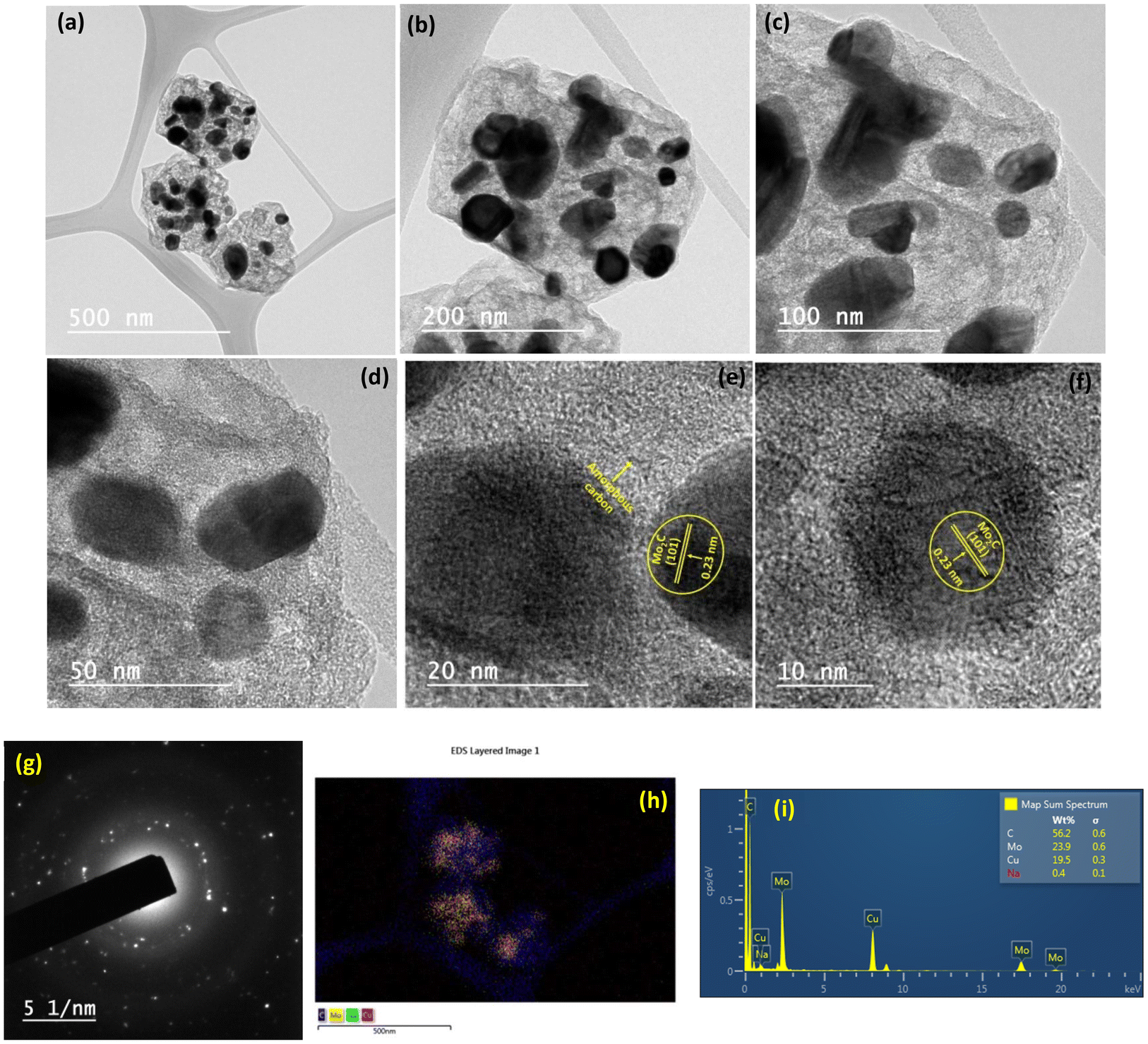
Fig. 5(a–f) depict the TEM images of the Na–Cu–Mo2C catalyst for investigating detailed nanostructured information. After pyrolysis of Na-NENU 5, the octahedral shape of the particles is well retained as a diamond-shaped outline of the octahedron can be seen in the TEM images. A more in-depth examination of the octahedron shape reveals that it consists of nano-particles of about 5–7 nm [Fig. 5(g)]. The high-resolution TEM (HRTEM) image of these nano-particles displays lattice fringes of Mo2C and Cu metallic phases with interplanar distances of 0.23 nm and 0.208 nm for the (101) and (111) crystal planes, respectively. A thin layer of amorphous carbon can be seen between these particles, which is also supported by the Raman spectra of the catalysts. The formation of this layer is significant because it can prevent the agglomeration of the Mo2C and Cu particles during the carburization process.35 The selected area electron diffraction (SAED) pattern indicates that these nanoparticles are both single crystalline and polycrystalline, which correspond to Mo2C and Cu phases [Fig. 5(h)]. The EDX elemental mappings as shown in Fig. 5(i, j) and S4† further demonstrate the presence and uniform distribution of Na, Cu, Mo, and C elements.
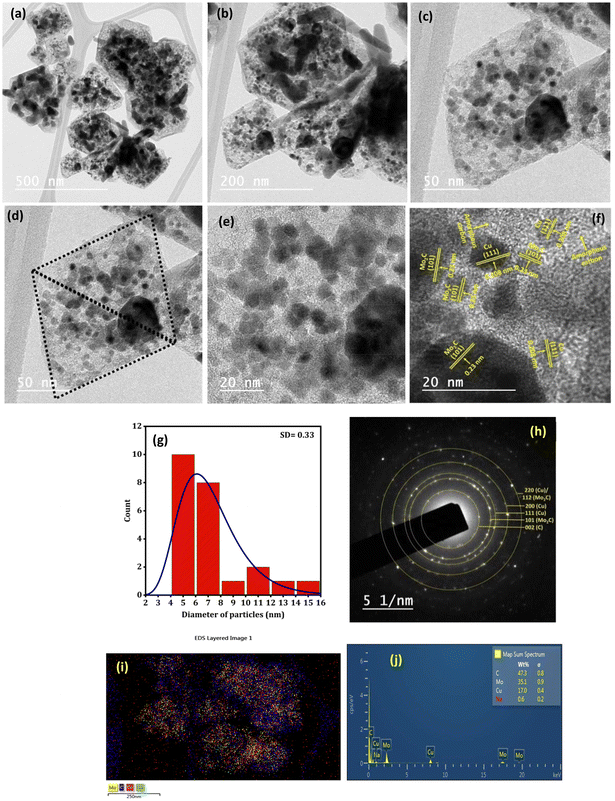 |
| | Fig. 5 TEM analysis of the Na–Cu–Mo2C catalyst; TEM images (a–f), (g) particle size distribution of image (f), SAED pattern (h) and EDX/elemental mapping (i and j). | |
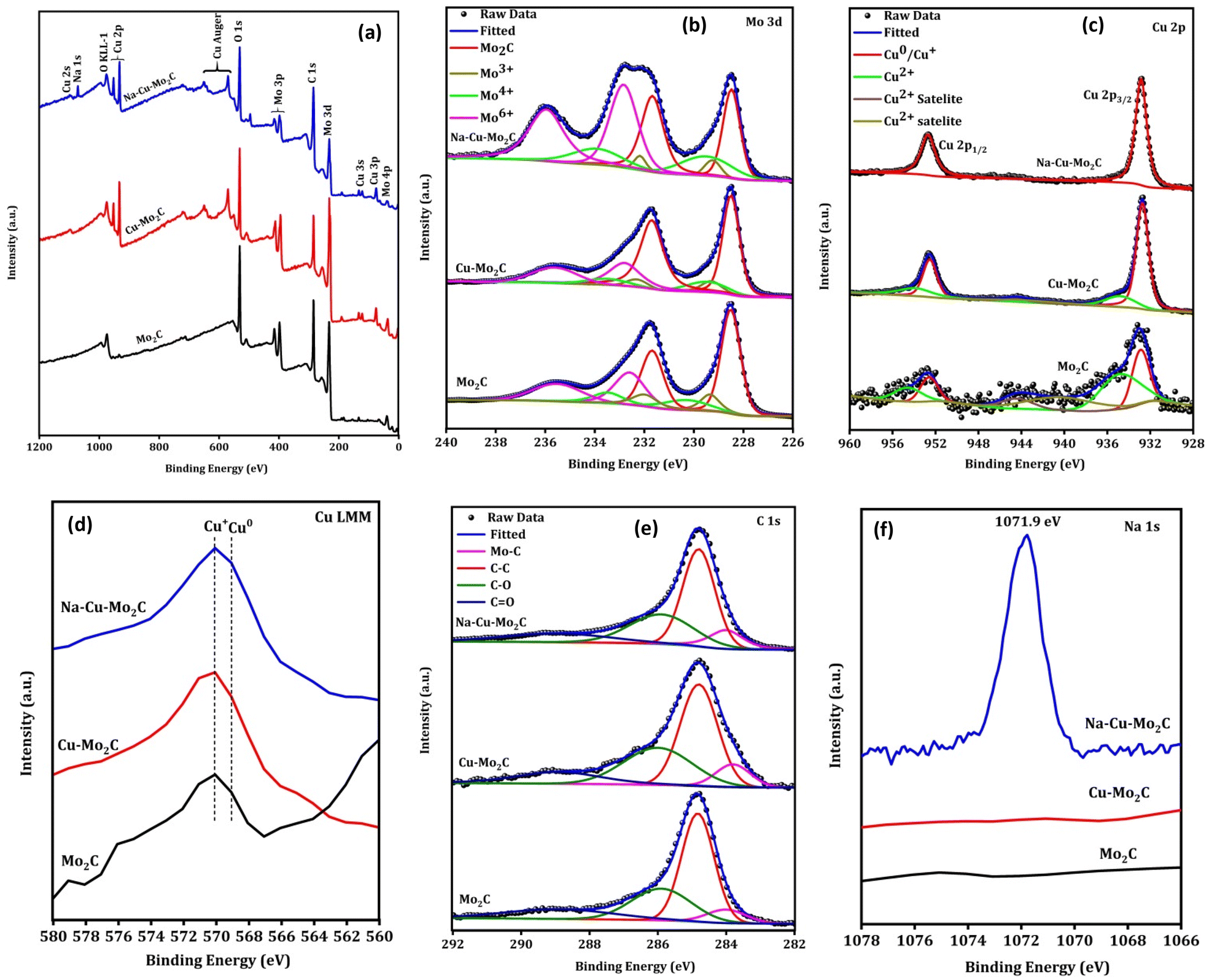
The surface elemental composition and valence states of elements present in the catalysts were examined using X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum of catalysts implies the existence of the respective elements [Fig. 6(a)]. In the Mo 3d XPS spectra in Fig. 6(b), the fitting of the peaks suggests the presence of the Mo–C, Mo3+, Mo4+, and Mo6+ oxidation states with a binding energy near 228.5, 229.2, 229.5, and 232.9 eV for Mo 3d5/2 spin–orbital coupling.56,57 The presence of various oxidation states other than Mo–C species can be attributed to the oxidation of Mo–C owing to exposure to air, as previously reported.56 In the Cu 2p XPS spectrum (Fig. 6c), the two prominent peaks at 952.8 and 932.8 eV can be attributed to the 2p3/2 and 2p1/2 for the Cu0 or Cu+ oxidation state.58 The additional small peaks at 956 and 934.7 eV with satellite peaks near 944.3 eV can be assigned to the Cu2+ oxidation state.58 Since it is challenging to distinguish between the Cu0 and Cu+ species during deconvolution because of their comparable binding energies, the Cu LMM Auger area at around 568 eV was also examined to provide information about a particular chemical state of the Cu element (Fig. 6d). In the Cu LMM Auger, the peak location at 570 and 569 eV corresponds to the Cu+ and Cu0 oxidation states, respectively, confirming the coexistence of both phases in the surface of all catalysts.59 The existence of a Cu+ oxidation state in catalysts, albeit Cu2O does not appear in the XRD peak, could be attributed to the surface oxidation of Cu0 resulting from air exposure. Importantly, as can be seen from the XPS survey spectrum and Cu 2p XPS peak, the XPS peak of the Cu species significantly decreased for Mo2C catalysts in comparison to those for Cu–Mo2C and Na–Cu–Mo2C, indicating the successful removal of Cu species concurrent with the XRD pattern. As shown in Fig. 6e, the C 1s spectra also deconvoluted to four major peaks at 283.9 (Mo–C), 284.8 (C–C), 285.9 (C–O), and 288.9 eV (CO).60 The existence of Na in Na–Cu–Mo2C is further verified from the Na 1s XPS, which shows a prominent peak at 1071.9 eV for the Na species (Fig. 6f).61
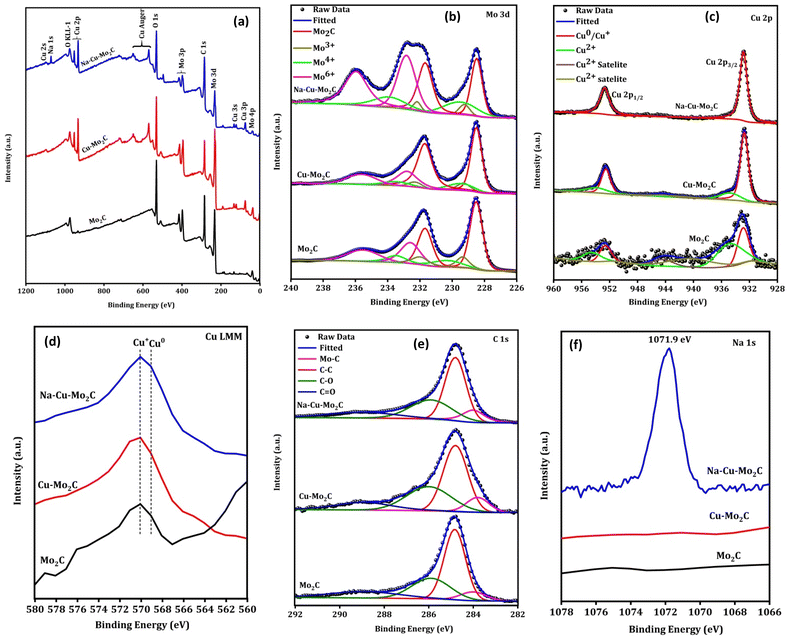 |
| | Fig. 6 XPS survey spectra (a), high-resolution Mo 3d (b), Cu 2p XPS (c), Cu Auger LMM (d), C 1s (e) and Na 1s (f) of the derived catalysts. | |
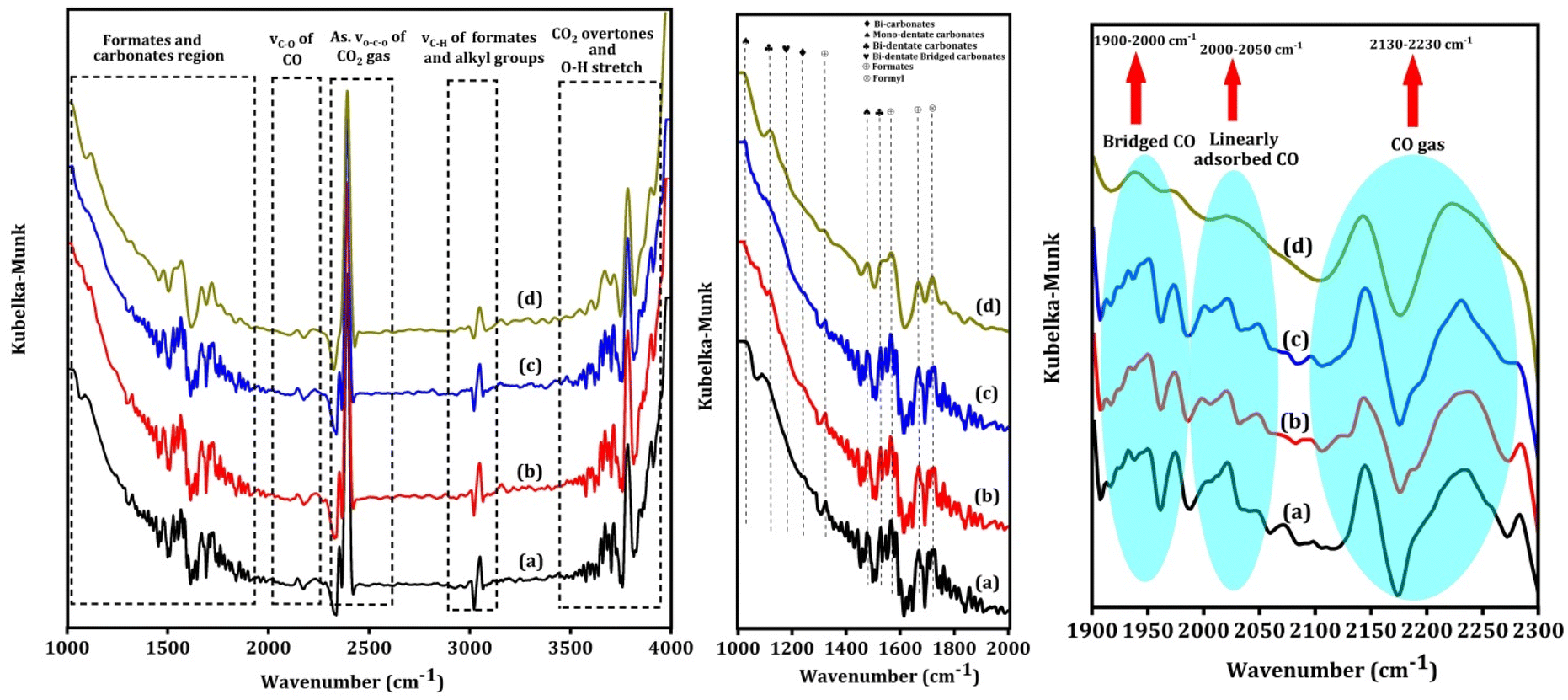
Fig. 7 shows the in situ CO2 DRIFT spectra over the Na–Cu–Mo2C catalyst at 550 °C with 5 ml min−1 of CO2 + 20 ml min−1 H2 flow. The strong bands between 2200–2450 cm−1 are associated with the asymmetric vibrations of the gas phase CO2 molecule's O–C–O bond62 and O–H stretching vibrations of –OH groups, combination bands, and CO2 overtones are responsible for the bands observed after 3500 cm−1. The bicarbonates, mono- or bi-dentate carbonates, and bi-dentate bridging carbonate species exhibit both symmetric and asymmetric vibrations of C–O bonds, which account for the broad peaks observed between 1300–1450, 1470–1700, and 980–1080 cm−1, respectively.62–64 The strong wide bands that appear between 1900–2000 cm−1, 2000–2050 cm−1, and 2130–2230 cm−1 correspond to the stretching vibration of the C–O bond of bridged CO, linearly adsorbed CO, and gaseous CO, respectively.65 The strong bands near 1320, 1570, and 1670 cm−1 correspond to the symmetric and asymmetric vibration of the C–O bonds of formates species.62,66 A band near 1720 cm−1 can be attributed to the symmetric and asymmetric vibration of C–O bonds of formyl species.66 The associative mechanism and redox mechanism are the two pathways that the RWGS reaction follows. In the associative mechanism surface adsorbed species interact with each other to form possible intermediates i.e., formates, formyl, carbonates, etc. The intermediates then decompose to form CO and H2O molecules.67,68 However, the redox mechanism proceeds without any possible intermediates. The appearance of the formate and formyl species in the spectra indicates that the RWGS reaction over the Na–Cu–Mo2C catalyst follows the associative mechanism.
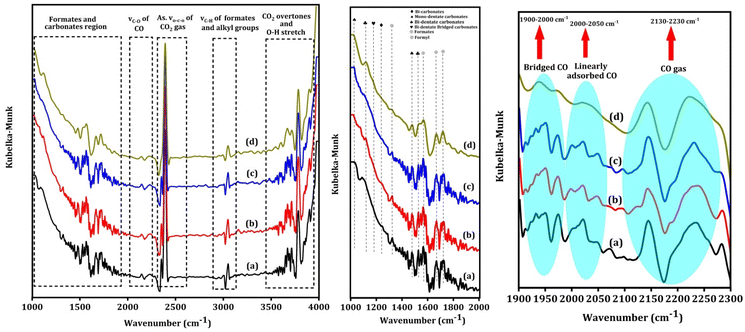 |
| | Fig. 7
In situ CO2 DRIFT spectra over the Na–Cu–Mo2C catalyst, at 550 °C after 5 ml min−1 CO2 + 20 ml min−1 H2 flow for 60 min (a) 120 min (b), 180 min (c) and 240 min (d). | |
Catalytic results of the pyrolyzed MOF for the RWGS reaction
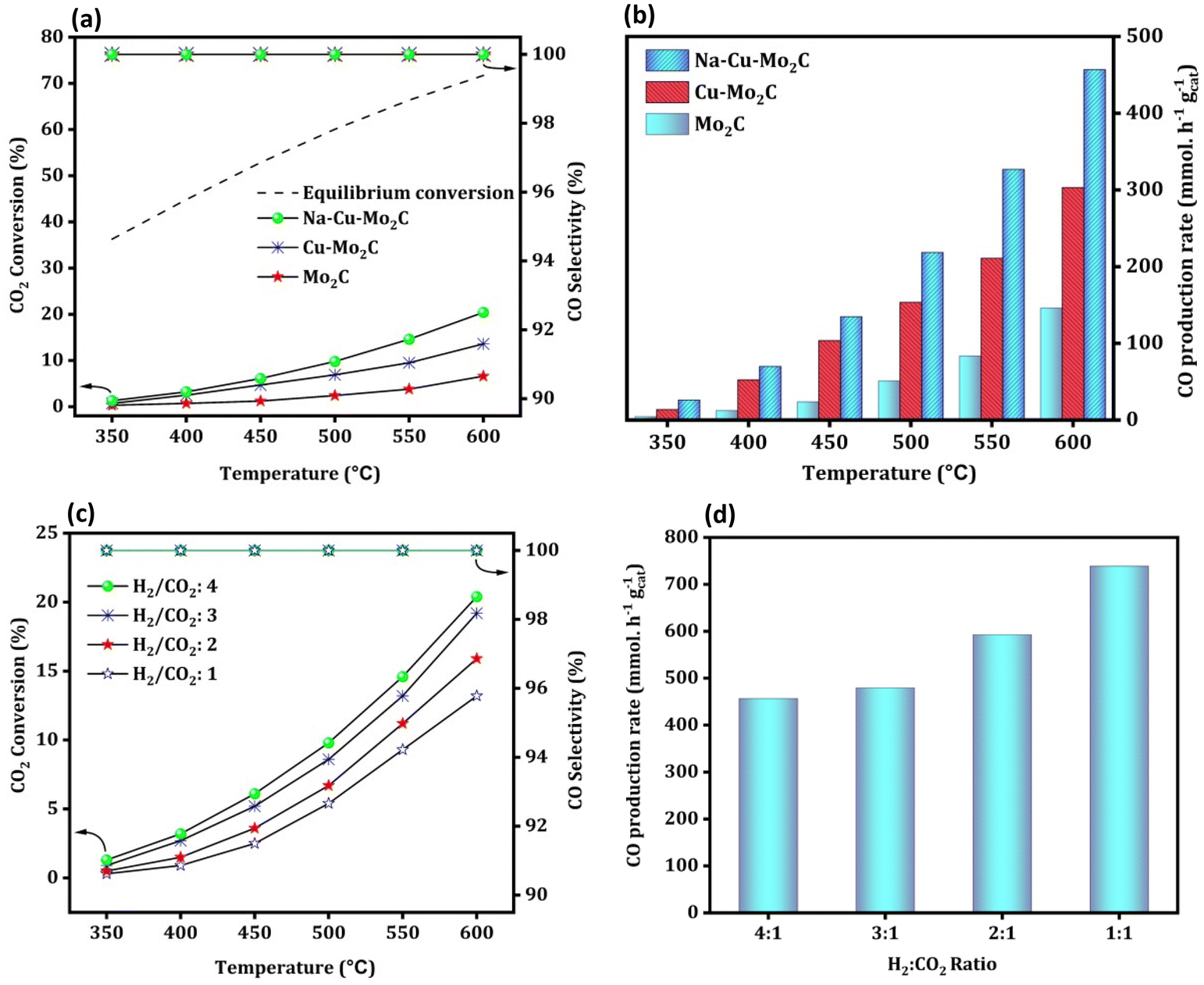
Fig. 8(a) depicts the activity of the catalysts for the RWGS reaction at atmospheric pressure and reaction temperatures ranging from 350 to 600 °C with 50 °C increments and 300000 ml gcat−1 h−1 of GHSV. The CO2 conversion of all catalysts increased with temperature, as predicted from reaction thermodynamics. At 350 °C, the activity of the catalysts is so low that it is not possible to compare it in terms of CO2 conversion. However, with temperature, CO2 conversion increases in the order Mo2C < Cu–Mo2C < Na–Cu–Mo2C for all screened temperatures with 100% CO selectivity. The catalysts, Mo2C, Cu–Mo2C, and Na–Cu–Mo2C converted 6.6, 13.6, and 20.4% CO2 at 600 °C, respectively (the enlarged portion of Fig. 8a shown in Fig. S5†). The CO production rate for Mo2C, Cu–Mo2C, and, Na–Cu–Mo2C is 146.0, 303.2, and 456.7 mmol gcat−1 h−1 at 600 °C [Fig. 8(b)]. The further optimization of the Na–Cu–Mo2C catalyst was also carried out with different H2:CO2 ratios to increase the CO production rate. With an H2:CO2 ratio of 4, 3, 2, and 1, CO2 conversion over Na–Cu–Mo2C was 20.4, 19.2, 16.0, and 13.2% with 100% CO selectivity at 600 °C, respectively. At the same temperature, the CO production rate increased to 456.7, 478.0, 593.2, and 739.2 mmol gcat−1 h−1 with a decrease in the H2:CO2 ratio from 4 to 1 [Fig. 8(c and d)].
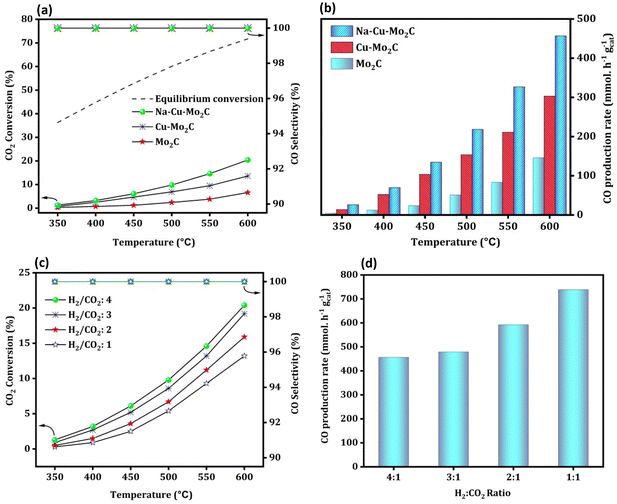 |
| | Fig. 8 RWGS activity of the derived catalysts; CO2 conversion, CO selectivity, and CO production rate (a and b); the effect of different H2:CO2 ratios over the Na–Cu–Mo2C catalyst on CO2 conversion and CO production rate (c and d). | |
We have also compared the results of the Na–Cu–Mo2C catalyst with the benchmark Cu/ZnO/Al2O3 catalyst (HiFUEL W220). The reaction was conducted between a temperature of 350–600 °C having 300000 ml gcat−1 h−1 GHSV (H2:CO2 ratio 1). The reaction was further proceeded for 20 h at 600 °C to compare the stability of both catalyst systems. As shown in Fig. S6,† the Cu/ZnO/Al2O3 catalyst is more efficient at all temperatures; nevertheless, this catalyst was severely deactivated during the stability test. Within 20 h, the Cu/ZnO/Al2O3 catalyst lost 44% of its original activity (conversion reached approximately 14% from 24% at 600 °C after 20 h). In contrast, the Na–Cu–Mo2C catalyst remained stable for 20 h without any substantial deactivation. Both catalysts, however, remained 100% selective for the CO formation during activity comparison and the stability test.
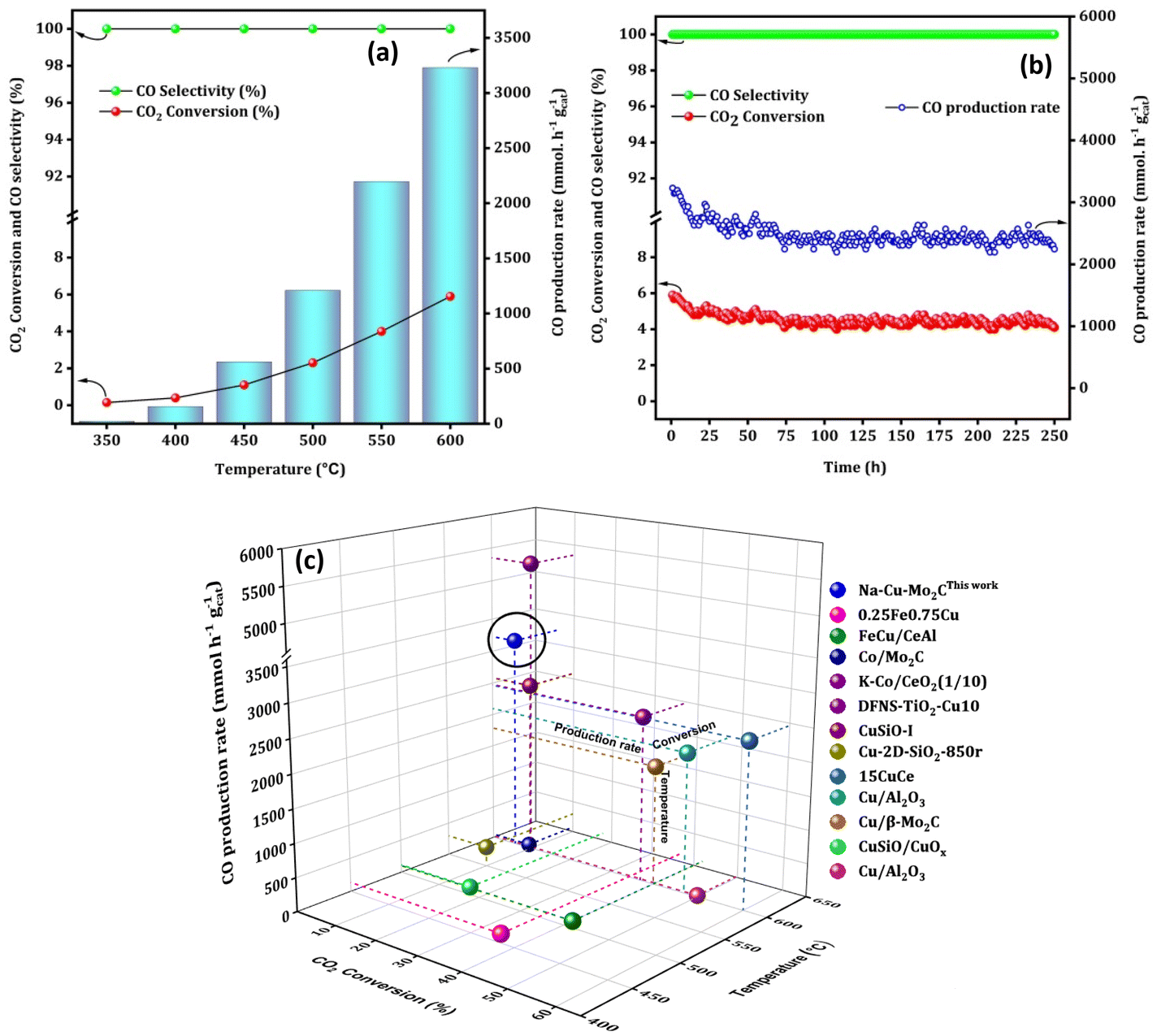
To maximize the CO formation rate, we performed an additional reaction over the Na–Cu–Mo2C catalyst at a relatively higher GHSV (3000000 ml gcat−1 h−1) with a 1:1 H2:CO2 ratio [Fig. 9(a)]. Although CO2 conversion decreased at higher GHSV, there is a significant increase in CO production rate at each screening temperature as compared to the lower GHSV test. The CO production rate reached up to 3230.0 mmol gcat−1 h−1 with 5.9% CO2 conversion at 600 °C [Fig. 9(b)]. Under similar reaction conditions, a stability test of the Na–Cu–Mo2C catalyst was also conducted at 600 °C for 250 h of reaction time. As shown in Fig. 9(c), the CO production rate and CO2 conversion initially slightly decreased and then remained at an average value of 2590.0 mmol gcat−1 h−1 and 4.7% throughout the reaction, respectively. This reveals that the Na–Cu–Mo2C catalyst maintains 80% of its initial activity after 250 h of reaction time, demonstrating excellent stability.
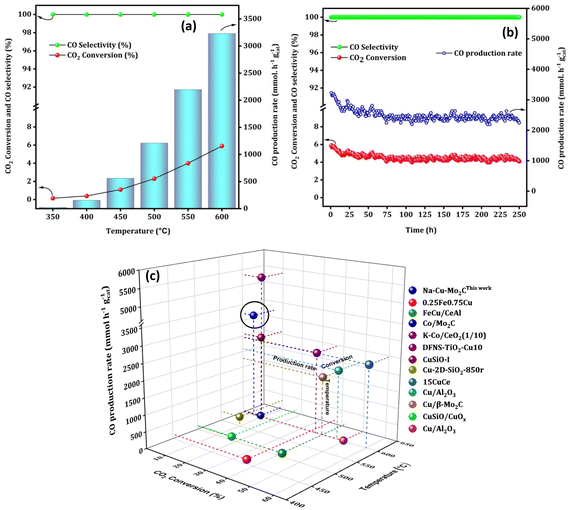 |
| | Fig. 9 The high space velocity test (a), stability test at 600 °C (b) over the Na–Cu–Mo2C catalyst and comparison with the best reported Fe, Co and Cu based catalysts (c). | |
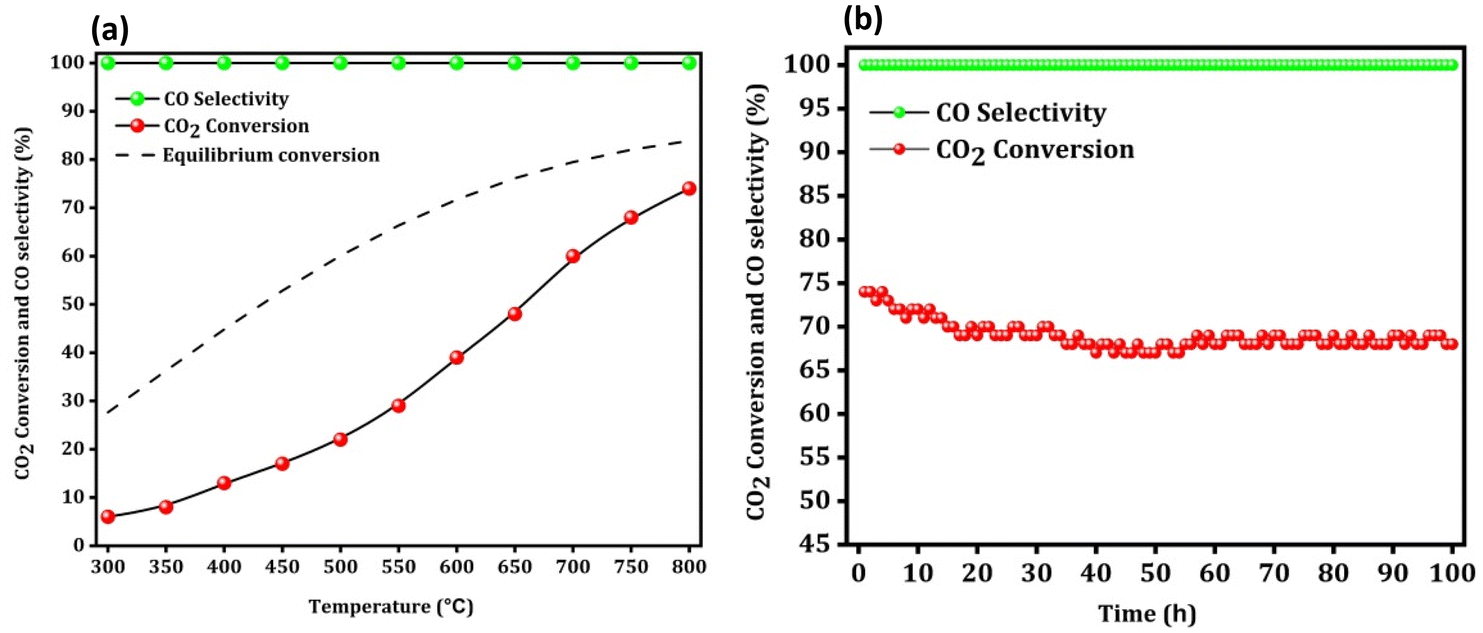
Finally, a low space velocity test (10000 ml gcat−1 h−1) (H2:CO2 ratio 4) with a wide temperature range (300–800 °C) was also conducted to increase the CO2 conversion to a practical and reasonable value. As shown in Fig. 10a, the CO2 conversion at 300 °C, 400 °C, 500 °C, 600 °C, 700 °C and 800 °C was around 6, 13, 22, 39, 60, and 74%, respectively.
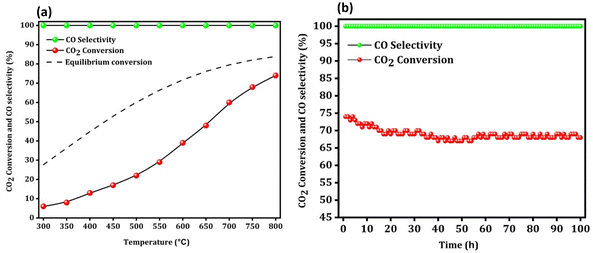 |
| | Fig. 10 The low space velocity test with a wide temperature range (a) and the stability test at 800 °C (b) over the Na–Cu–Mo2C catalyst. | |
Remarkably, despite having a low space velocity, the Na–Cu–Mo2C catalyst is 100% selective for CO production at all screened temperatures. At 800 °C, 74% CO2 conversion and 100% CO selectivity resulted in a 74% CO yield. Under these harsh reaction conditions (high temperature, high CO2 conversion, and high CO concentration), we further proceeded to check any catalyst deactivation at 800 °C (Fig. 10b). We observed an initial slight decrease in the CO2 conversion up to 20 h of reaction, after which the conversion remained steady between 68–70%. However, the reaction was performed for 100 h, and at that time CO2 conversion was 68%, indicating that the catalyst lost just 8% of its initial activity.
Interestingly, the catalysts are active after pyrolysis due to the in situ formation of the active phases, i.e. Mo2C formed by the interaction of Mo with the carbonaceous ligand simultaneously reduced the Cu2+ to Cu metallic phase. This is a significant benefit since this approach eliminates the need for a CH4 + H2 combination for molybdenum phase carburization and an H2 pre-reduction step for Cu2+ reduction to the Cu metallic phase, resulting in major process efficiencies in a real industrial application. Moreover, the catalyst Na–Cu–Mo2C outperformed most of the previously reported Fe, Cu, and Co-based catalyst systems with its exceptional stability over 250 h and CO production rate of 3230.0 mmol gcat−1 h−1 (Fig. 9c and Table S1†). Importantly, Na promotion is not done in a traditional separate step and instead remains in the Na–Cu–Mo2C catalysts as a salt residue after the synthesis.
Characterization of the used Na–Cu–Mo2C catalyst after a 100 h stability test at 800 °C
To decipher the changes in the catalyst under these severe reaction conditions, the Na–Cu–Mo2C catalyst that was employed underwent further characterization using XRD, Raman, and TEM/HR-TEM analyses following a 100 h stability test at 800 °C. As shown in Fig. 11a, the XRD pattern of the catalyst shows the presence of the β-Mo2C and Cu metallic phase with diffraction peaks matching well with their respective JCPDS file no. as displayed in Fig. 3a. Similarly, Raman spectra of the catalyst show the bands near 656, 812, and 986 cm−1 for the Mo–C stretching of the β-Mo2C phase with the D and G bands near 1350 and 1580 cm−1 for the carbon species (Fig. 11b). These results demonstrate that the chemical composition of the catalyst remains unchanged even after the 100 h stability test. The catalyst was also examined using TEM/HR-TEM analysis to figure out the change in its nanostructure (Fig. 12a–i). As illustrated in Fig. 12a, the octahedral shape is still apparent and its constituent particles are embedded firmly within the amorphous carbon. However, as shown in Fig. 12b–d, we observed an increase in the particle size inside the octahedral shape compared to the particle size in the fresh Na–Cu–Mo2C catalyst. This increase in the particle size can be the reason for the initial decrease in the CO2 conversion up to 20 h of reaction. This initial increase in particle size is inevitable under the reaction conditions because the reaction was carried out at 800 °C, which was also the pyrolysis temperature of the MOFs. The EDX elemental mappings reveal a good distribution of all constituent elements, and the quantitative data obtained were comparable to the fresh Na–Cu–Mo2C catalyst (Fig. 12h, i and Fig. S7†). Overall, the characterization findings of the Na–Cu–Mo2C catalyst after a 100 h stability test at 800 °C demonstrate that the chemical composition and morphology of the catalyst remained unchanged.
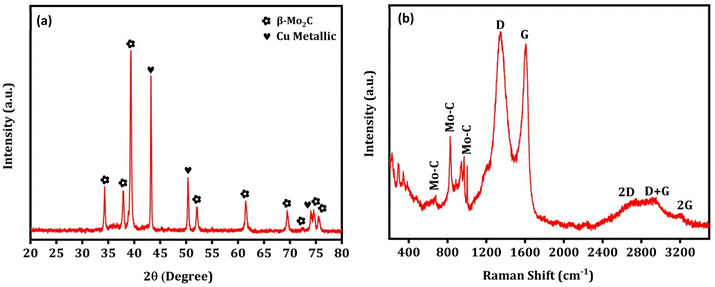 |
| | Fig. 11 The XRD pattern (a) and Raman spectra (b) after the stability test for 100 h at 800 °C over the Na–Cu–Mo2C catalyst. | |
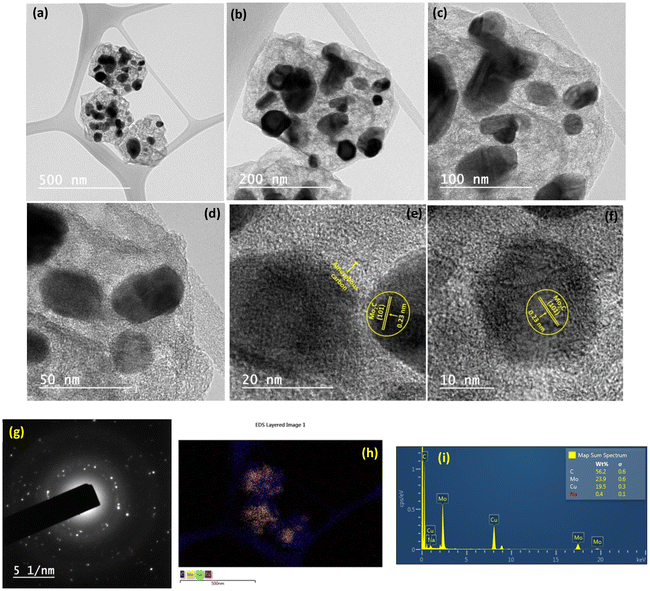 |
| | Fig. 12 TEM analysis of the Na–Cu–Mo2C catalyst after the 100 h stability test at 800 °C; TEM images (a–f), SAED pattern (g) and EDX/elemental mapping (h and i). | |
Conclusions
We reported a successful synthesis of a new NENU-5 MOF-derived nanocomposite catalyst having Mo2C as a support with Cu nanoparticles as an active site for CO2 transformation to CO. The in situ carburization of the NENU-5 MOF generated partially sintered Cu particles which exhibited remarkable stability with a prolonged activity. The Na–Cu–Mo2C nanocomposite exhibited outstanding stability up to 250 h studied reaction time while retaining its 80% initial activity. This catalyst displayed an exceptional CO production rate of 3230.0 mmol gcat−1 h−1 without any side product formation at 600 °C temperature, superior performance to the majority of the reported copper-based catalyst systems.
Data availability
All data obtained in this study are included in this article.
Author contributions
Gaje Singh: writing – original draft, visualization, validation, methodology, investigation, data curation, and conceptualization. Satyajit Panda: formal analysis, investigation, and data curation. Siddharth Sapan: formal analysis, investigation, and data curation. Jogender Singh: investigation, conceptualization, and data curation. Pranay Rajendra Chandewar: formal analysis, investigation, and data curation. Ankush Biradar: writing – review & editing and supervision. Debaprasad Shee: writing – review & editing and supervision. Ankur Bordoloi: writing – review & editing, writing – original draft, validation, supervision, resources, methodology, investigation, funding acquisition, formal analysis, and conceptualization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Gaje Singh sincerely acknowledges UGC New Delhi, India for providing the fellowship. Ankur Bordoloi gratefully acknowledges CSIR-India (HCP-48) for a research grant. The Director, CSIR-IIP is gratefully acknowledged for his kind encouragement. The authors appreciate the analytical science division (ASD) CSIR-IIP for providing their analytical service.
References
- X. Yu, C. O. Catanescu, R. E. Bird, S. Satagopan, Z. J. Baum, L. M. Lotti Diaz and Q. A. Zhou, ACS Omega, 2023, 8, 11643–11664 CrossRef CAS.
- M. L. T. Triviño, N. C. Arriola, Y. Seok Kang and J. Gil Seo, Chem. Eng. J., 2024, 150369 CrossRef.
- M. Alsunousi and E. Kayabasi, Int. J. Hydrogen Energy, 2024, 54, 1169–1178 CrossRef CAS.
- Q. Zhang, M. Bown, L. Pastor-Pérez, M. S. Duyar and T. R. Reina, Ind. Eng. Chem. Res., 2022, 61, 12857–12865 CrossRef CAS PubMed.
- M. L. Phey Phey, T. A. Tuan Abdullah, U. F. Md Ali, M. Y. Mohamud, M. Ikram and W. Nabgan, RSC Adv., 2023, 13, 3039–3055 RSC.
- Q. Imtiaz, A. Armutlulu, F. Donat, M. A. Naeem and C. R. Müller, ACS Sustainable Chem. Eng., 2021, 9, 5972–5980 CrossRef CAS.
- Y. C. Kimmel, X. Xu, W. Yu, X. Yang and J. G. Chen, ACS Catal., 2014, 4, 1558–1562 CrossRef CAS.
- Q. Zhang, L. Pastor-Pérez, Q. Wang and T. Ramirez Reina, J. Energy Chem., 2022, 66, 635–646 CrossRef CAS.
- K. P. Reddy, S. Dama, N. B. Mhamane, M. K. Ghosalya, T. Raja, C. V. Satyanarayana and C. S. Gopinath, Dalton Trans., 2019, 48, 12199–12209 RSC.
- Z. Tišler, R. Velvarská, L. Skuhrovcová, L. Pelíšková and U. Akhmetzyanova, Materials, 2019, 12, 415 CrossRef.
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y.-W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912–918 CrossRef CAS.
- G. Singh, D. Khurana, T. S. Khan, I. K. Ghosh, B. Chowdhury, A. Y. Khodakov and A. Bordoloi, Appl. Surf. Sci., 2023, 616, 156401 CrossRef CAS.
- J. Xu, X. Gong, R. Hu, Z. Liu and Z. Liu, Mol. Catal., 2021, 516, 111954 CrossRef CAS.
- Q. Zhang, L. Pastor-Pérez, W. Jin, S. Gu and T. R. Reina, Appl. Catal., B, 2019, 244, 889–898 CrossRef CAS.
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC.
- Y. Zhu, K. Yue, C. Xia, S. Zaman, H. Yang, X. Wang, Y. Yan and B. Y. Xia, Nano-Micro Lett., 2021, 13, 137 CrossRef CAS.
- G. Singh, S. Panda, J. Gahtori, P. Rajendra Chandewar, P. Kumar, I. K. Ghosh, A. Biradar, D. Shee and A. Bordoloi, ACS Sustainable Chem. Eng., 2023, 11, 11181–11198 CrossRef CAS.
- J. Gahtori, C. L. Tucker, T. S. Khan, C. de Sá Codeço, T. Rocha and A. Bordoloi, ACS Appl. Mater. Interfaces, 2022, 14, 38905–38920 CrossRef CAS PubMed.
- J. Gahtori, G. Singh, J. Kaishyop, C. P. Rajendra, C. L. Tucker, T. S. Khan, D. Shee and A. Bordoloi, Fuel Process. Technol., 2023, 244, 107719 CrossRef CAS.
- A. Zakharova, M. W. Iqbal, E. Madadian and D. S. A. Simakov, ACS Appl. Mater. Interfaces, 2022, 14, 22082–22094 CrossRef CAS.
- L. Yang, L. Pastor-Pérez, J. J. Villora-Pico, A. Sepúlveda-Escribano, F. Tian, M. Zhu, Y.-F. Han and T. Ramirez Reina, ACS Sustainable Chem. Eng., 2021, 9, 12155–12166 CrossRef CAS.
- L. Yang, L. Pastor-Pérez, J. J. Villora-Pico, S. Gu, A. Sepúlveda-Escribano and T. R. Reina, Appl. Catal., A, 2020, 593, 117442 CrossRef CAS.
- R. Belgamwar, R. Verma, T. Das, S. Chakraborty, P. Sarawade and V. Polshettiwar, J. Am. Chem. Soc., 2023, 145, 8634–8646 CrossRef CAS.
- R. Jin, J. Easa and C. P. O'Brien, ACS Appl. Mater. Interfaces, 2021, 13, 38213–38220 CrossRef CAS PubMed.
- S. Wang, K. Feng, D. Zhang, D. Yang, M. Xiao, C. Zhang, L. He, B. Yan, G. A. Ozin and W. Sun, Adv. Sci., 2022, 9, 2104972 CrossRef CAS PubMed.
- L. Wang, H. Liu, Y. Chen, R. Zhang and S. Yang, Chem. Lett., 2013, 42, 682–683 CrossRef CAS.
- H.-X. Liu, S.-Q. Li, W.-W. Wang, W.-Z. Yu, W.-J. Zhang, C. Ma and C.-J. Jia, Nat. Commun., 2022, 13, 867 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, M. Kılıç, C. J. Baranowski, L. Artiglia, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, ACS Catal., 2019, 9, 6243–6251 CrossRef CAS.
- Y. Yu, R. Jin, J. Easa, W. Lu, M. Yang, X. Liu, Y. Xing and Z. Shi, Chem. Commun., 2019, 55, 4178–4181 RSC.
- S. Choi, B.-I. Sang, J. Hong, K. J. Yoon, J.-W. Son, J.-H. Lee, B.-K. Kim and H. Kim, Sci. Rep., 2017, 7, 41207 CrossRef CAS.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- L. Hu, W. Li, L. Wang and B. Wang, EnergyChem, 2021, 3, 100056 CrossRef CAS.
- Z. Chen, J. Chen and Y. Li, Chin. J. Catal., 2017, 38, 1108–1126 CrossRef CAS.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS.
- C. Zhang, Q. Liu, P. Wang, J. Zhu, D. Chen, Y. Yang, Y. Zhao, Z. Pu and S. Mu, Small, 2021, 17, 2104241 CrossRef CAS.
- N. M. Mahmoodi, M. Oveisi and E. Asadi, J. Cleaner Prod., 2019, 211, 198–212 CrossRef CAS.
- R. Chilivery and R. K. Rana, ACS Appl. Mater. Interfaces, 2017, 9, 3161–3167 CrossRef CAS.
- I. Nalumansi, G. Birungi, B. Moodley and E. Tebandeke, Orient. J. Chem., 2020, 36, 607–612 CrossRef CAS.
- N. Al-zaqri, A. Alsalme, S. F. Adil, A. Alsaleh, S. G. Alshammari, S. I. Alresayes, R. Alotaibi, M. Al-Kinany and M. R. H. Siddiqui, J. Saudi Chem. Soc., 2017, 21, 965–973 CrossRef CAS.
- A. Yang, P. Li and J. Zhong, RSC Adv., 2019, 9, 10320–10325 RSC.
- L. Guo, J. Du, C. Li, G. He and Y. Xiao, Fuel, 2021, 300, 120955 CrossRef CAS.
- M. Jin, X. Qian, J. Gao, J. Chen, D. K. Hensley, H. C. Ho, R. J. Percoco, C. M. Ritzi and Y. Yue, Inorg. Chem., 2019, 58, 8332–8338 CrossRef CAS.
- M. Todaro, A. Alessi, L. Sciortino, S. Agnello, M. Cannas, F. M. Gelardi and G. Buscarino, J. Spectrosc., 2016, 2016, e8074297 Search PubMed.
- D. Mohanadas, M. A. A. Mohd Abdah, N. H. N. Azman, T. B. S. A. Ravoof and Y. Sulaiman, Sci. Rep., 2021, 11, 11747 CrossRef CAS PubMed.
- S.-Y. Jiang, W.-W. He, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Inorg. Chem., 2018, 57, 6118–6123 CrossRef CAS.
- J. Ni, Z. Ruan, S. Zhu, X. Kan, L. Lu and Y. Liu, ChemElectroChem, 2019, 6, 5958–5966 CrossRef CAS.
- W.-F. Chen, C.-H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943–951 RSC.
- L. F. Pan, Y. H. Li, S. Yang, P. F. Liu, M. Q. Yu and H. G. Yang, Chem. Commun., 2014, 50, 13135–13137 RSC.
- Y.-P. Huang, C.-W. Tung, T.-L. Chen, C.-S. Hsu, M.-Y. Liao, H.-C. Chen and H. Ming Chen, Nanoscale, 2022, 14, 8944–8950 RSC.
- S. Hussain, D. Vikraman, A. Feroze, W. Song, K.-S. An, H.-S. Kim, S.-H. Chun and J. Jung, Front. Chem., 2019, 7 DOI:10.3389/fchem.2019.00716.
- S. Sameer, G. Singh, J. Gahtori, R. Goyal, I. K. Ghosh, N. Barrabes and A. Bordoloi, J. Environ. Chem. Eng., 2022, 10, 108988 CrossRef CAS.
- Y. Zhao, Z. Guo, H. Zhang, B. Peng, Y. Xu, Y. Wang, J. Zhang, Y. Xu, S. Wang and X. Ma, J. Catal., 2018, 357, 223–237 CrossRef.
- N. Ma, X. Wang, X. Liu, N. Li and Y. Liu, Ind. Eng. Chem. Res., 2023, 62, 5533–5542 CrossRef CAS.
- B. Liang, H. Duan, T. Sun, J. Ma, X. Liu, J. Xu, X. Su, Y. Huang and T. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 925–932 CrossRef CAS.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., 2014, 126, 6525–6528 CrossRef.
- J. A. Schaidle, A. C. Lausche and L. T. Thompson, J. Catal., 2010, 272, 235–245 CrossRef CAS.
- C. Zhang, P. Wang, W. Li, Z. Zhang, J. Zhu, Z. Pu, Y. Zhao and S. Mu, J. Mater. Chem. A, 2020, 8, 19348–19356 RSC.
- J. Jang, S. Zhu, E. P. Delmo, T. Li, Q. Zhao, Y. Wang, L. Zhang, H. Huang, J. Ge and M. Shao, EcoMat, 2023, 5, e12334 CrossRef CAS.
- S. Zhu, C. Wang, H. Shou, P. Zhang, P. Wan, X. Guo, Z. Yu, W. Wang, S. Chen, W. Chu and L. Song, Adv. Mater., 2022, 34, 2108809 CrossRef CAS.
- M. Kalapsazova, R. Stoyanova, E. Zhecheva, G. Tyuliev and D. Nihtianova, J. Mater. Chem. A, 2014, 2, 19383–19395 RSC.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- A. Bansode, B. Tidona, P. R. von Rohr and A. Urakawa, Catal. Sci. Technol., 2013, 3, 767–778 RSC.
- L. F. Bobadilla, V. Garcilaso, M. A. Centeno and J. A. Odriozola, ChemSusChem, 2017, 10, 1193–1201 CrossRef CAS.
- J. Tian, P. Zheng, T. Zhang, Z. Han, W. Xu, F. Gu, F. Wang, Z. Zhang, Z. Zhong, F. Su and G. Xu, Appl. Catal., B, 2023, 339, 123121 CrossRef CAS.
- N. Podrojková, V. Sans, A. Oriňak and R. Oriňaková, ChemCatChem, 2020, 12, 1802–1825 CrossRef.
- G. Singh, J. Kaishyop, Md J. Gazi, V. K. Shrivastaw, M. Shah, I. K. Ghosh, T. S. Khan and A. Bordoloi, Chem. Eng. J., 2024, 494 DOI:10.1016/j.cej.2024.153205.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 bd,
Debaprasad
Shee
c and
Ankur
Bordoloi
bd,
Debaprasad
Shee
c and
Ankur
Bordoloi