
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanosized Chevrel phases for dendrite-free zinc–ion based energy storage: unraveling the phase transformations†
Amr
Elgendy
abc,
Athanasios A.
Papaderakis
ab,
Andinet
Ejigu
 ab,
Katharina
Helmbrecht
de,
Ben F.
Spencer
f,
Axel
Groß
de,
Alex S.
Walton
af,
David J.
Lewis
*g and
Robert A. W.
Dryfe
*ab
ab,
Katharina
Helmbrecht
de,
Ben F.
Spencer
f,
Axel
Groß
de,
Alex S.
Walton
af,
David J.
Lewis
*g and
Robert A. W.
Dryfe
*ab
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: robert.dryfe@manchester.ac.uk; Tel: +44 (0) 161-306-4522
bHenry Royce Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cEgyptian Petroleum Research Institute, 11727, Cairo, Egypt
dInstitute of Theoretical Chemistry, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
eHelmholtz Institute Ulm (HIU) for Electrochemical Energy Storage, Helmholtzstraße 11, 89081 Ulm, Germany
fPhoton Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
gDepartment of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.lewis-4@manchester.ac.uk; Tel: +44 (0) 161-306-3561
First published on 20th June 2024
Abstract
The nanoscale form of the Chevrel phase, Mo6S8, is demonstrated to be a highly efficient zinc-free anode in aqueous zinc ion hybrid supercapacitors (ZIHSCs). The unique morphological characteristics of the material when its dimensions approach the nanoscale result in fast zinc intercalation kinetics that surpass the ion transport rate reported for some of the most promising materials, such as TiS2 and TiSe2. In situ Raman spectroscopy, post-mortem X-ray diffraction, Hard X-ray photoelectron spectroscopy, and density functional theory (DFT) calculations were combined to understand the overall mechanism of the zinc ion (de)intercalation process. The previously unknown formation of the sulfur-deficient Zn2.9Mo15S19 (Zn1.6Mo6S7.6) phase is identified, leading to a re-evaluation of the mechanism of the (de)intercalation process. A full cell comprised of an activated carbon (YEC-8A) positive electrode delivers a cell capacity of 38 mA h g−1 and an energy density of 43.8 W h kg−1 at a specific current density of 0.2 A g−1. The excellent cycling stability of the device is demonstrated for up to 8000 cycles at 3 A g−1 with a coulombic efficiency close to 100%. Post-mortem microscopic studies reveal the absence of dendrite formation at the nanosized Mo6S8 anode, in stark contrast to the state-of-the-art zinc electrode.
1. Introduction
Electrochemical systems, including batteries, supercapacitors, and hybrid devices (also known as “supercapatteries”), are set to play an increasing role in storage of electricity generated from renewable sources such as solar, wind, and tidal inputs.1–3 Lithium-ion batteries (LIB) continue to dominate the energy storage market, due to their high energy density.4 There are, however, increasing concerns about the sustainability of LIBs: the use of highly flammable organic electrolytes leads to safety challenges. Additionally, the uneven distribution of lithium resources has encouraged the development of post-LIB systems.4 One promising alternative is the use of zinc-based energy storage devices with mild aqueous electrolytes for grid-level energy storage. These devices have the advantages of simple manufacturing, high theoretical capacity (820 mA h g−1), abundant and low-cost raw materials, as well as higher intrinsic safety.5,6 In recent years, there has been significant progress towards the development of cathode materials that deliver high energy density in zinc-based energy storage devices. In particular, manganese-based materials, various organic (e.g., quinone derivatives, conductive polymers and dyes) as well as inorganic (e.g., Prussian blue analogues) hosts and vanadium-based ionic conductors (e.g., NASICON) have been reported as highly efficient cathodes for zinc-ion batteries (ZIBs).2,7,8 In parallel, several types of activated carbon have been effectively used as cathodes in zinc-ion hybrid supercapacitors (ZIHSCs). However, all of these devices require a bulk zinc electrode as anode to compensate for the issues of dendrite growth and corrosion, which lead to low columbic efficiency during the zinc plating/striping process.7 As a result, the actual energy and power densities of most zinc-ion batteries and supercapacitors are significantly lower than their theoretical values. In addition, aqueous zinc-ion energy storage devices often exhibit poor cycling stability due to the high rate of hydrogen gas evolution produced by the parasitic process of water splitting over the zinc metal anode.9 Consequently, it becomes evident that the use of metallic zinc anodes poses serious challenges. Currently, there are ongoing efforts to suppress the formation of Zn dendrites by using electrolyte additives, establishing an artificial solid electrolyte interphase (SEI) layer, and alloying Zn with other transition metals such as Cu.10,11 However, these approaches have their own drawbacks, namely increased battery cost and decreased specific and volumetric energy density in the case of electrolyte additives, instability issues of the deposited film when using an artificial SEI layer and reduced specific capacity for Zn-alloy anodes due to the decrease of the Zn active constituents upon alloying.Based on the acquired knowledge from lithium metal batteries, it is of paramount importance to develop a zinc metal-free anode that can reversibly intercalate/de-intercalate Zn ions and hence potentially address the Zn dendrite issue.12 Ideal intercalation anodes should have low intercalation potential vs. Zn2+/Zn, be electrochemically stable during charging/discharging, have good electronic conductivity, and high capacity. Currently, there are few materials that have shown the ability to intercalate Zn ions. These include layered-2D titanium disulfide (TiS2) with an intercalation potential of 0.4 V vs. Zn2+/Zn in aqueous electrolyte,13 TiSe2 with a sloping discharge plateau at +0.35 V vs. Zn2+/Zn and a discharge capacity of ∼63 mA h g−1 at 0.3 A g−1,7 as well as hexagonal MoO3 that exhibits a discharge potential of ∼0.37 V (vs. Zn2+/Zn) and a discharge capacity of 80 mA h g−1 at 0.5 A g−1.14 However, potential zinc-ion-free anodes are not limited to inorganic materials; organic hosts such as carbonyl-rich molecules (perylenetetracarboxylic dianhydride or PTCDA, and perylenetetracarboxylic diimide or PTCDI) demonstrate promising characteristics as anodes.15 There are major disadvantages associated with these materials, including their significantly lower electrical conductivity compared to the inorganic alternatives, limited cycling stability, high discharge potential (vs. Zn2+/Zn) and/or low discharge capacity, which severely limit their use in practical systems.
As a host material for different mono/multivalent ions, Chevrel phases (CPs), MxMo6T8 (M = metal; T = S, Se, or Te), are appealing because of their characteristic chemical intercalation properties for lithium, sodium, magnesium, calcium, manganese, iron, cobalt, nickel, and zinc.16–18 CPs exhibits a relatively low discharge potential of 0.32 V vs. Zn2+/Zn, being the lowest reported among Zn-free anode candidates. However, the low cycle stability, possibly due to the micro-sized structure of the prepared material, impedes its wider use.19 Recently, our group reported the facile, rapid and highly controllable synthesis of nanostructured CPs. The unique structural characteristics of the prepared material in combination with its high electrical conductivity renders it a promising anode material, exhibiting fast lithium ion (de)intercalation kinetics and high cycling stability.20,21
Inspired by the compelling physicochemical properties of the nano-sized Mo6S8, we explore its applicability as a Zn-free negative electrode for ZIHSC. It is worth mentioning that to the best of our knowledge, there are no previous reports on the use of CPs as an anode material in ZIHSC. On this basis, a complete and systematic physicochemical study of CPs electrochemistry in zinc electrolytes has been conducted. Most notably, the formation of the sulfur-deficient phase Zn2.9Mo15S19 has been identified, providing novel insights into the long-standing debate of Zn-ion trapping. With this knowledge, we have developed a ZIHSC with a nano-sized structure (de)intercalation battery-type anode and activated carbon (AC) double layer capacitance-type cathode using a 2 M ZnSO4 aqueous electrolyte. When compared with the traditional hybrid supercapacitor where zinc metal serves as the anode, the developed device has the following merits: (a) the use of a Zn-free anode prevents dendrite formation during the charge/discharge process and thus increases the coulombic efficiency while simultaneously reducing safety risks, (b) fast zinc ion diffusion into the Mo6S8 cavities as evidenced by means of the galvanostatic intermittent titration technique (GITT) renders the Mo6S8 anode compatible with the AC cathode, (c) the use of aqueous ZnSO4 electrolyte is cost-effective and “eco-friendly”, in contrast to the organic electrolytes commonly used. The Mo6S8 showed a multistep zinc ion (de)intercalation process with an average discharge potential of ∼0.32 V vs. Zn2+/Zn, and reversible discharge capacity of ∼66 mA h g−1 at the current density of 0.1 A g−1. As a result, the assembled ZIHSC achieves a high capacity of ∼38 mA h g−1 (0.2 A g−1) and a superior energy density of 43.8 W h kg−1. Notably, the power density remains as high as 2500 W kg−1 and the energy density is maintained at 25 W h kg−1 when the current density is increased to 2 A g−1. Moreover, the device shows extended cycle life of up to 8000 cycles at a moderate current density of 3 A g−1. Furthermore, microscopic studies of the cycled electrode revealed the complete absence of zinc dendrites.
2. Experimental section and methods
2.1. Materials and methods
Molybdenum(0) powder (1–5 μm, ≥99.9%), molybdenum(0) hexacarbonyl (98%), tetraethylthiuram disulfide (≥97%), zinc sulfate heptahydrate (≥99%) and zinc chloride(≥98%) were purchased from Sigma-Aldrich. Copper(II) dibutyldithiocarbamate (>98%) was purchased from Tokyo Chemical Industry (UK) Ltd. The activated carbon (AC, grade YEC-8 A) was purchased from Fuzhou Yihuan Carbon Co., Ltd (China). Ultrapure water (resisitivity = 18.2 MΩ cm, Milli-Q Direct 8) was used in all electrochemical studies. All the chemicals were used without further treatment.2.2. Synthesis of Cu2Mo6S8 nanocubes
The synthesis of Cu2Mo6S8 was carried out according to our previous reports and is schematically illustrated in Fig. S1.†20,21 Briefly, stoichiometric amounts of tetrakis(diethyldithiocarbamato) molybdenum(IV) (1.4 g, 2 mmol), Mo (0.2 g, 2 mmol), and copper(II) dibutyldithiocarbamate, (0.5 mg, 1.3 mmol) powders were thoroughly mixed using a pestle and mortar. All powders were then compressed into pellet form, with a diameter of 11 mm, using a hydraulic press at 20 tonnes to ensure better homogeneity and avoid phase separation. The pellet was centred in a ceramic boat inside a tube furnace and the system was purged with argon gas for 20 minutes. Finally, the temperature was gradually increased to 800 °C (3 °C min−1) and kept under Ar for 7 hours at this temperature. Subsequently, the prepared Cu2Mo6S8 powder was dispersed and stirred for 10 h in 6 M HCl solution (Fisher Scientific, Inc.). The solution was purged with oxygen to facilitate Cu leaching. The solution was centrifuged, and the resulting material was washed several times with de-ionized water and dried at 80 °C overnight to obtain Mo6S8. Furthermore, it is worth mentioning that copper can be successfully leached electrochemically from the neat Chevrel phase (a detailed process can be found in the ESI (Fig. S2†)).2.3. Material characterisation
The structure of the prepared materials was investigated by powder X-ray diffraction (P-XRD) using an XRD5-PANalytical X'Pert Pro diffractometer with Cu Kα radiation (λ = 1.5406 Å) operating at 40 kV. The (2θ) data were recorded in the range of 10° to 80°. The morphology of the active materials was analysed with a field-emission scanning electron microscope (FE-SEM, Quanta 650) equipped with energy-dispersive X-ray (EDX) at 15 kV. The sample was dried and then placed over a conductive carbon tape substrate. The Raman spectra were recorded using a Renishaw inVia Confocal microscope with a 523 nm excitation laser operated at a power of 0.274 mW with a grating of 1800 lines per mm and 50× objective. The in situ Raman cell was purchased from ECC-Opto-Std (EL-Cell GmbH, Hamburg, Germany). The cell comprised of a free-standing Mo6S8 film (positive electrode) pressed on a titanium foil substrate that contained a small hole in its middle (diameter ca. 1 mm). A glass membrane (7 mm diameter) soaked with 2 M ZnSO4 electrolyte was used as a separator and zinc metal as a counter and reference electrode. The laser beam was shone through a small hole in the centre of the Ti substrate that was covered with a thin glass window on the back of the free-standing Mo6S8 film. The Raman spectra were collected at various voltages during the charging/discharging process at 1 mV s−1. Hard X-ray Photoelectron Spectroscopy (HAXPES) was performed using monochromated Ga Kα metal jet X-ray radiation (9252 eV, 3.57 mA emission at 250 W, micro-focussed to 50 μm) and an EW-4000 high voltage electron energy analyser (HAXPES-Lab, Scienta Omicron GmbH); the instrument has a base vacuum pressure of 5 × 10−10 mbar.22,23 The entrance slit width used was 1.5 mm, and the pass energies used for survey and core level spectra were 500 and 100 eV respectively, with total energy resolutions of 2.0 and 0.6 eV respectively, as measured using the FWHM of the Au 4f7/2 core level on a clean gold reference sample.22 The HAXPES instrument also has a monochromated Al Kα X-ray source (1486 eV, 20 mA emission at 300 W) for surface-sensitive XPS at the same sample position. Charge neutralisation for insulating samples is achieved using a low energy electron flood source as required (FS40A, PreVac). Binding energy (BE) scale calibration was performed using Au 4f7/2 at 84 eV of a clean gold reference sample, otherwise C 1s at 284.8 eV BE, if the flood source was used. Analysis and curve fitting was performed using Voigt-approximation peaks using CasaXPS.24 Core level relative sensitivity factors for HAXPES quantification were calculated according to previous reports.23,252.4. Electrochemical characterisation
The zinc storage performance of the prepared Mo6S8 was tested in a three-electrode configuration. The working electrode was prepared by mixing 9 mg of active material with 1 mg of carbon black (“Super P”, supplied by Alfa Aesar, 99+% metals basis). The mixed powder was then dispersed in 1 ml of a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/ethanol mixture, followed by addition of 45 μL of Nafion® solution. The suspension was sonicated for 1 h to yield a homogenous ink. A specific amount of this ink was drop-cast on a polished glassy carbon (GC) substrate with a geometric area of 0.0707 cm2. The total mass loading of each electrode was approximately 1.08 mg cm−2. For electrochemical testing, Ag/AgCl (3.5 M KCl) and Pt mesh were used as the reference and counter electrodes, respectively. 2 M ZnSO4(aq.) bubbled, with N2 gas for 30 min, was used as the electrolyte. Note that although the three-electrode measurements were conducted using an Ag/AgCl reference electrode, for ease of comparison of three-electrode and coin cell data, all potentials are reported with respect to Zn2+/Zn (both scales are given in the relevant graphs in the ESI†).
:1 water/ethanol mixture, followed by addition of 45 μL of Nafion® solution. The suspension was sonicated for 1 h to yield a homogenous ink. A specific amount of this ink was drop-cast on a polished glassy carbon (GC) substrate with a geometric area of 0.0707 cm2. The total mass loading of each electrode was approximately 1.08 mg cm−2. For electrochemical testing, Ag/AgCl (3.5 M KCl) and Pt mesh were used as the reference and counter electrodes, respectively. 2 M ZnSO4(aq.) bubbled, with N2 gas for 30 min, was used as the electrolyte. Note that although the three-electrode measurements were conducted using an Ag/AgCl reference electrode, for ease of comparison of three-electrode and coin cell data, all potentials are reported with respect to Zn2+/Zn (both scales are given in the relevant graphs in the ESI†).
For ZIHSC device assembly, the negative electrode, Mo6S8, was prepared by mixing the active material, “Super P” and polytetrafluoroethylene binder (60 wt% in water, Aldrich) in a weight ratio of 8.0:1.0:1.0 to obtain a homogeneous free-standing slurry with the help of ethanol. Then the film was evenly pressed onto a stainless steel grid (grade 316) and used as the anode. For the positive electrode, activated carbon (YEC-8A), “Super P”, and polyvinylidene fluoride were mixed in 2 ml of N-Methyl-2-pyrrolidone (NMP) with a mass ratio of 8.0:1.0:1.0 to obtain a homogenous slurry. After that, the slurry was coated on a titanium substrate and dried at 100 °C overnight for further use. The electrochemical performance of the AC electrodes was tested in half-cell devices, i.e., the AC was used as the working electrode, 2 M ZnSO4 was used as the aqueous electrolyte, and zinc plate (14 mm) was used as the counter and reference electrode. In a similar design, the galvanostatic intermittent titration technique (GITT) was used to study the solid-state diffusion of zinc ions into the Mo6S8 crystal structure. The measurements were performed at a current density of 0.1 A g−1 for 15 s and a relaxation of 150 s. The ZIHSC was assembled in a CR2032-type coin cell using a hydraulic crimper (MSK-160D) with a Mo6S8 anode (3–5.8 mg cm−2), AC cathode (5.3–9.7 mg cm−2), and glass membrane used as the separator. In all electrochemical experiments, cyclic voltammetry (CV) measurements were carried out with Metrohm PGSTAT302N potentiostat, equipped with the FRA32 module and controlled via Nova 1.11 software. The galvanostatic charge/discharge tests (GCD) were recorded with a voltage range from 0 V to 1.6 V at different current densities, and the cyclic stability was carried at a current density of 3 A g−1 for 8000 cycles. Both GCD, and GITT measurements were performed on a Battery Test System (BaSyTec GmbH, Germany) at room temperature.
2.5. Computational details
The properties of the CP and similar structures were studied using periodic density functional theory,26–28 as implemented in the Vienna Ab initio Simulation Package.29–31 To account for exchange and correlation, the generalized gradient approximation in the formulation of Perdew, Burke and Ernzerhof (PBE)32 has been used. The electron-core interactions are represented by the Projector Augmented Wave (PAW)33 method. Calculations were first performed for the Mo6S8 unit cell of the CP with the Brillouin zone sampled using a 4 × 4 × 4 k-point grid. For other unit cells the k-point grid was scaled accordingly in each lattice direction to achieve the same density. The Zn2.9Mo15S19 was analyzed in its symmetry-reduced unit cell, which contains two formula units. The electronic structure was converged to 1 × 10−5 eV, applying a plane-wave cutoff energy of 450 eV. All formation energies with respect to the insertion of the Zn atoms are determined with respect to the Zn metal bulk energy. Hence a positive formation energy means that the insertion is not stable with respect to the formation of the bulk metal.3. Results and discussion
3.1. Synthesis of Mo6S8
The prepared Mo6S8 phase was confirmed with powder X-ray diffraction. Fig. S3† shows that the diffraction pattern of the prepared Mo6S8 is well matched with the standard Mo6S8 (ICSD#252376). Moreover, the presence of a strong peak at 13.8° associated with the (101) plane and the minimal appearance of the peak at 14.4° being indexed to MoS2, confirms the high purity of the synthesised Mo6S8 compound. These values are in good agreement with previous reports,18,34 demonstrating that the material was successfully synthesized. Full characterisation of the Mo6S8 prepared with the molecular precursor approach, including XRD, Rietveld refinement, Raman spectroscopy, and morphological characterisation via SEM and EDX can be found in our previous work.20,213.2. Storage behavior of zinc ions in Mo6S8
The electrochemical properties of zinc intercalation into Mo6S8 were thoroughly investigated in a three-electrode system. The Mo6S8 material shows two distinguishable quasi-reversible reduction and oxidation peaks related to the zinc intercalation/deintercalation process (Fig. 1(a and b)) according to the following equations:19| xZn2+ + 2xe− + Mo6S8 ⇌ ZnxMo6S8 0 ≤x ≤ 2 | (1) |
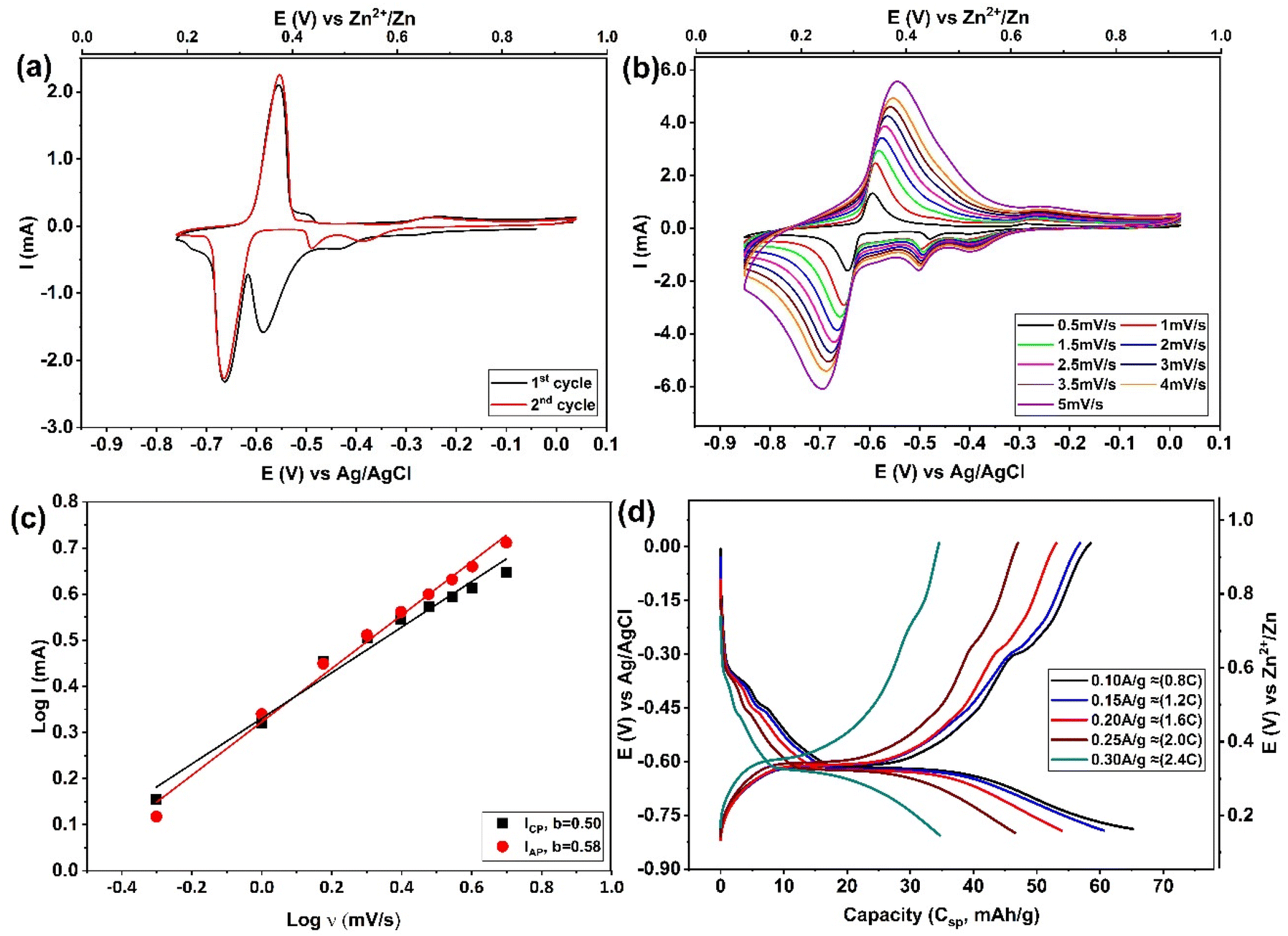 | ||
| Fig. 1 Electrochemical studies on Zn-ion intercalation into Mo6S8 electrode in 2 M ZnSO4. (a) Cyclic voltammetry profiles of the 1st and 2nd cycles recorded at 1 mV s−1, and (b) CV recoded at different scan rates from 0.5 to 5 mV s−1. (c) The log(I) versus log(v) plots of the main peak located at ∼0.25 V vs. Zn2+/Zn in CV curves. (d) The GCD (voltage–capacity) profiles recorded at different current density. Here, the 1 C rate was calculated as 128 mA g−1 based on the theoretical capacity of the four-electron reaction from Mo6S8 to Zn2Mo6S8. | ||
The Zn2+ insertion process was found to follow a multistep intercalation mechanism whereby ZnxMo6S8 is initially formed at a potential of approximately ∼0.5 V vs. Zn2+/Zn (eqn (1)) with its subsequent transformation to Zn2Mo6S8 at around ∼0.3 V vs. Zn2+/Zn as Zn2+ are further inserted into the Mo6S8 cavities (eqn (1)). This observation is consistent with previous work by Cheng et al.,35 as presented in their supplementary data (Fig. S2† of ref. 35). A noteworthy feature of Fig. 1a is that during the second cycle, the position of the cathodic insertion peak shifts from ∼0.38 V to approximately ∼0.5 V vs. Zn2+/Zn, and the peak current is significantly reduced. Following that, as the number of cycles proceeds, the peak position and current remained roughly unchanged. On first sight, this might be related to zinc ion trapping during the Znx formation: this process was reported by Aurbach et al.36 for CPs prepared with microsized structure, although they did not thoroughly investigate the composition or properties of the phase that formed as a result of this process.
The intercalation of zinc into Mo6S8, was confirmed by testing bare and modified Mo6S8 GC electrodes in 2 M ZnSO4 (Fig. S4†). It is clear that the redox peaks shown in Fig. S4b,† and Fig. 1a are due to the zinc intercalation/de-intercalation process and not due to the zinc plating/striping process. In order to further investigate the electrochemical kinetics of Mo6S8 towards zinc ions, a series of cyclic voltammetry tests were recorded at different scan rates. Fig. 1b shows that the cathodic and anodic peaks slightly shift with increase of scan rate from 0.5 to 5 mV s−1, due to increased polarization with higher sweep rates, a phenomenon that is associated with battery-type electrode materials.7,20 To study the zinc ion energy storage mechanism in detail, the relationship between the peak current (I) and scan rate (v) was investigated based on the power-law formula I = a × vb, where the value of the exponent b (the slope of log(I) vs. log(v) plot), provides information about the charge storage mechanism. For a diffusion-controlled process, I is proportional to the square root of v (b = 0.5). However, for processes that involve surface ion adsorption/desorption, the current response is proportional to the scan rate (b = 1).20 As seen in Fig. 1c, the results from log(I) and log(v) show that the calculated b values are ca. 0.50, and 0.58 for zinc intercalation and de-intercalation, respectively, suggesting that the zinc storage mechanism into nanosized Mo6S8 is controlled by diffusion. The charge/discharge behaviour of zinc ions in the electrode was evaluated in a two-electrode coin cell system comprised of Mo6S8 as working electrode, 2 M ZnSO4 as electrolyte, and Zn foil as counter and reference electrode. The observations of the galvanostatic charge–discharge shown in Fig. 1d were consistent with those of the CV, in which two voltage plateaus are clearly observed. In particular, an ill-defined shoulder is recorded in the higher voltage region at ca. ∼0.53 V vs. Zn2+/Zn followed by a clear plateau at ∼0.34 V vs. Zn2+/Zn. The electrode shows a capacity of 66 mA h g−1 at current density of 0.1 A g−1 (∼0.8 C), which decreases to 35 mA h g−1, as the current density increases to 0.3 A g−1 (2.4C).
In order to provide insights into the rather uncharted phenomenon of zinc trapping, we investigate the effect of (electro)chemical parameters, i.e., electrolyte identity and applied potential bias, on the overall process. Fig. S5† shows the CVs derived from the experimental protocol adopted for the data presented in Fig. 2(a and b), using two other zinc electrolytes, namely 2 M ZnCl2 and saturated zinc triflate (Zn(OTF)2). From the observed response, it is evident that zinc trapping is practically unaffected by the anion identity. The influence of the cathodic potential limit is shown in Fig. S6.† It was found that even when the latter is confined to potentials more positive than the second step in the previously proposed reaction scheme (eqn (1)), the position and magnitude of the peak attributed to zinc trapping remains unchanged. Both findings strongly suggest that trapping of Zn ions in the Mo6S8 cavities is related to the intrinsic properties of the host material.
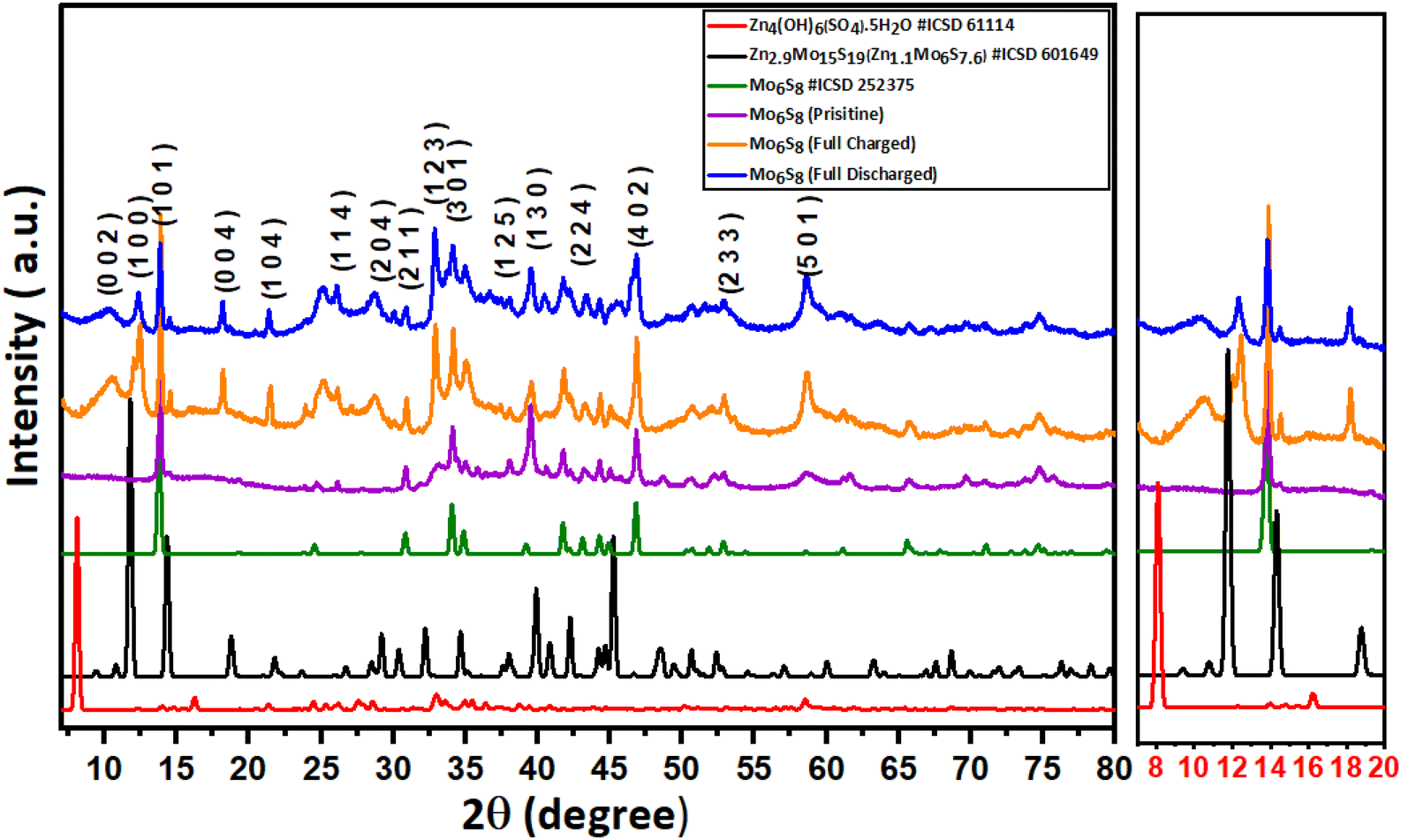 | ||
| Fig. 2 Post-mortem powder-XRD pattern recorded on the pristine, fully charged to 0.15 V vs. Zn2+/Zn, and fully discharged to 1 V vs. Zn2+/Zn Mo6S8 electrode, along with the standard pattern of Mo6S8 (ICSD#252376), Zn4(OH)6(SO4)·5H2O (ICSD#61114), and Zn2.9Mo15S19 (ICSD#601649). | ||
The intercalation of Zn2+ ions into Mo6S8 nanocubes was investigated with ex situ post-mortem X-ray diffraction (XRD) on cycled electrodes. Initially, the Mo6S8 electrode was charged and discharged at a rate of 0.1 A g−1. In the second step, the electrodes were removed from the Zn/Mo6S8 half-cell, washed thoroughly with de-ionized water, and dried before the XRD measurements were recorded. In the XRD pattern of the charged Mo6S8 presented in Fig. 2, four peaks of high intensity can be seen at 2θ = 10.7°, 12.4°, 18.2°, and 21.4°: these closely match the standard XRD pattern of the sulfur-deficient phase of Zn2.9Mo15S19 (Zn1.16Mo6S7.6, ICSD#601649). These peaks are distinct from the ZnMo6S8 (ICSD#252375) and Zn2Mo6S8 (ICSD#252374) phases, indicating the formation of the Zn1.16Mo6S7.6 phase upon Zn intercalation.35 Although Rietveld refinement was not possible on the data shown in Fig. 2 due to the broad nature of the peaks, we were able to perform Rietveld refinement of the diffraction data from another coin cell sample with stronger and sharper X-ray reflections (Fig. S7†). The results indicate that one of the crystalline products is Zn1.16Mo6S7.6, which is produced as a mixture with a small side-product of Zn4(OH)6(SO4).5H2O.
In the fully discharged state, the sharp crystalline peaks at 2θ = 10.7°, 12.4°, 18.2°, and 21.4° resulting from Zn2+ insertion are still evident, albeit with reduced intensity. This finding demonstrates that zinc is trapped within the Mo6S8 structure, and the Mo6S8 structure is not fully recovered.
It is worth noting that the Zn1.16Mo6S7.6 phase we observe is distinct from the zinc hydroxide sulfate phases e.g. Zn4(OH)6SO4·0.5H2O and Zn4(OH)6(SO4)·5H2O, which are observed when zinc metal is used as the electrode material in Zn–MnO2 batteries or Zinc hybrid supercapacitors. This hypothesis is supported by comparing the experimental XRD data of both the charged and discharged Mo6S8 samples with the standard XRD diffraction pattern of the zinc basic sulfate pattern, and the Rietveld refinement on the highly crystalline sample. Typically, the formation of the zinc hydroxide sulfate phase is a result of localized pH changes, caused by H+ insertion in the MnO2 during the discharge process, which leads to an increase in OH− concentration and thus, the formation of flakelike product of zinc hydroxide sulfate hydrate (Zn4(OH)6(SO4)·5H2O) on the surface of MnO2.37 It is also known that the zinc hydroxide sulfate phase undergoes a reversible precipitation/dissolution process during the charge/discharge process. On the contrary, this reversibility is not observed in the Zn1.16Mo6S7.6 phase. This is evident as the peaks corresponding to Zn1.16Mo6S7.6 are consistently identifiable in both the fully charged and discharged samples within the XRD patterns, albeit with different intensity.
The surface and chemical composition of Mo6S8 was examined with ex situ hard X-ray photoelectron spectroscopy (HAXPES, 9252 eV) in combination with conventional XPS (1486 eV) during the zinc ions’ intercalation and (de)intercalation processes (Fig. 3). The higher sampling depth of HAXPES, being ca. 43 nm based on the Mo 2p core level in the Mo6S8 sample, enables it to probe the structure of the material more deeply than conventional XPS (with a sampling depth of ca. 5 nm). Consequently, HAXPES can provide information about the physicochemical characteristics of the material at deeper sampling depth, in addition to the outermost surface layers. By comparing the data recorded using the two photon energies we are able to monitor any changes occurring on the surface and sub-surface of Mo6S8 during the zinc ions’ (de)intercalation process. In addition, HAXPES allows the use of deeper core levels at relatively higher binding energies, such as S 1s (∼2469 eV BE). Fig. 3(a and b) displays the narrow-window Mo 3d/S 2s core level scans of the blank CP sample (the electrode where no (de)intercalation process occurred), recorded using XPS and HAXPES techniques. Mo 3d5/2 photoelectron peaks are present at 228 eV corresponding to the Mo2+ oxidation state, and 229.1 eV corresponding to the Mo3+ oxidation state in Mo6S8, which agrees with earlier reports.34 Furthermore, the XPS and HAXPES data shows the presence of an additional peak ∼232.5 eV indicating the presence of an oxide surface film corresponding to the Mo6+ oxide state. These results suggest that the oxides are located on the surface of the sample, which could be attributed to the exposure of the sample to ambient air prior to the measurements. However, upon zinc insertion into the Mo6S8 the narrow window spectrum of the Mo 3d/S 2s core levels tend to change significantly as a function of the insertion potential (0.5 V and 0.3 V vs. Zn2+/Zn), indicating the successful insertion of the ions into the Mo6S8 structure. Furthermore, upon the insertion of Zn2+ ions, the binding energy experiences a shift towards higher energy levels in comparison to the pristine Mo6S8. This shift in both XPS and HAXPES data indicates that the [Mo6]m cluster, where m represents the number of electrons involved in the formation of metallic bonds within the pristine Mo6 cluster, is oxidized to [Mo6]m−n, where n with values between 0 and 1 denotes the number of electrons lost. This abnormal oxidation of Mo differs from the typical reduction of transition metals observed in conventional cathode materials upon cation insertion.38 This finding is also consistent with the observation made when Al3+ was inserted into Mo6S8 using an ionic liquid electrolyte.39 The XPS spectra also show another species with Mo 3d5/2 at ∼234 eV, which we assign as a higher oxide state that could include MoOx.40 Upon full discharge the spectrum almost returns to its original state, with however, a particularly notable decrease in the concentration of the S 2s peak as inferred from both photon energies. This finding may be attributed to the formation of a sulfur-deficient phase of the type Zn1.16Mo6S7.6, as shown by XRD data (see Fig. 2 and the discussion, vide supra). As for S 2p and S 1s, we can notice that the intensity of sulphide (at 226.0 eV and 2469.0 eV) changes significantly while two different oxide forms emerge (at 235.0 eV and ∼2478 eV) upon zinc insertion/de-insertion (Fig. 3(c and d)), respectively. Such changes in the subsurface S 1s manifest electron redistribution between Mo and S and therefore cannot be attributed to surface oxidation by air as initially speculated. Most importantly, the fact that Zn still exists on the subsurface following full discharge (Fig. 3e) further demonstrates that Zn ions are trapped in the Mo6S8 in line with the electrochemistry data (see above).
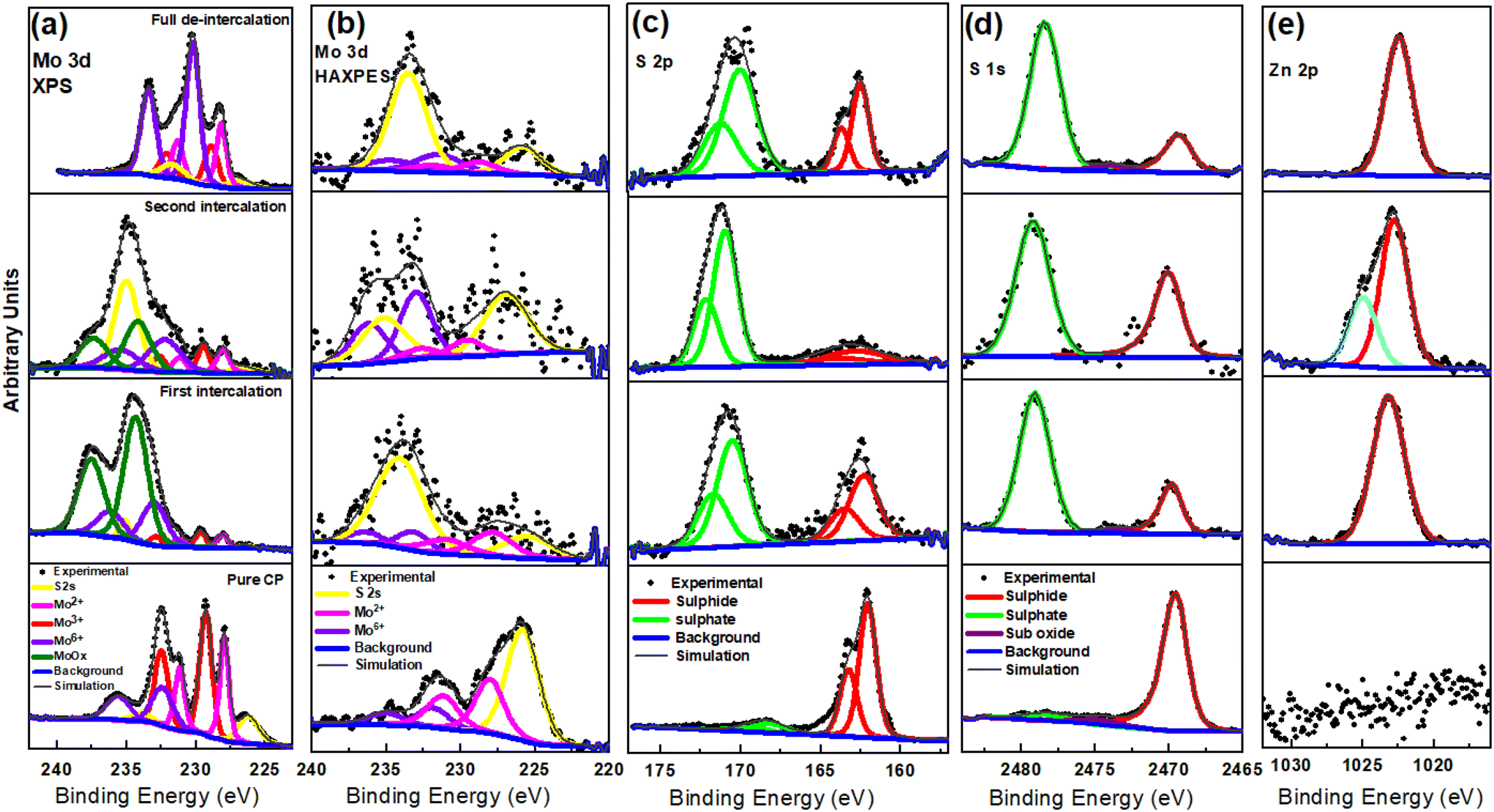 | ||
| Fig. 3 High resolution HAXPES and XPS spectra results of the fully/partially charged and fully discharged Mo6S8 electrode in 2 M ZnSO4 half-cell in the region of (a) Mo 3d/S 2s (XPS), (b) Mo 3d/S 2s (HAXPES), (c) S 2p, (d) S 1s (HAXPES), and (e) Zn 2p (HAXPES). Data panels start from bottom to up as follow: pure CP, first intercalation, second intercalation, and full-deintercalation. | ||
The zinc elemental composition during Zn2+ intercalation staging was further examined by means of EDX (Fig. S8†). From the derived data it is seen that the Zn atomic % increased from 8.42 during the first stage of intercalation (Fig. 1b), to 17.53 at complete intercalation (Fig. S8c†). Interestingly, the fully deintercalated sample contains almost the same percentage of Zn ions as the first intercalation sample, which further confirms zinc trapping. Furthermore, it is worth noting that the theoretical capacity of the sulfur deficient phase Zn1.16Mo6S7.6 is ∼65.69 mA h g−1, a value that aligns closely with both our experimental results (Fig. 1d) and the derived discharge capacity recorded following the initial cycles in previous works at nearly equal discharge current.18,19
To further confirm the zinc trapping during the intercalation process, we employed in situ Raman spectroscopy experiments. Fig. 4 shows the fully intercalated/de-intercalated Raman spectra for Mo6S8 in 2 M ZnSO4 during the first and second cycle. The free-standing Mo6S8 at open-circuit potential (OCP) showed characteristic peaks at 133, 227, 254, 301, 322, 383 and 404 cm−1 corresponding to various vibrational modes in Mo6S8.21,41 During the zinc intercalation process, the A1g characteristic peak at 404 cm−1 becomes broader and the relative intensity ratio of A1g/E12g decreases from 2.67 to 1.32, corresponding to the insertion of Zn2+ into the cavities of Mo6S8 (Fig. 4b). Furthermore, upon Zn intercalation from +0.7 V to +0.17 V vs. Zn2+/Zn, the bands soften and the peaks at lower wavenumber almost disappear. Such softening suggests that this mode primarily involves motion of the sulfur atoms, since the force constants between the sulfur atoms are likely to decrease as the sulfur cubes expand, whereas the force constants between the molybdenum atoms should increase as the octahedra shrink.41 On the other hand, during the discharge process (zinc de-intercalation), the A1g characteristic peak gradually enhances and the relative A1g/E12g intensity ratio increases from 1.32 to 2.32 (Fig. 5c). The relative decrease in A1g/E12g intensity after full zinc de-intercalation (2.32) compared to that at the OCP (no zinc intercalation) (2.67), suggests the zinc is trapped inside the Chevrel phase cavities during the first charging process. This could be associated with the formation of the new, sulfur deficient Zn2.9Mo15S19 (Zn1.16Mo6S7.6) phase because the force constant between the cubic sulfur atoms decreased with Zn intercalation as previously discussed. Following the first cycle we noted a reversible change of the Raman spectrum, where the ratio between A1g/E12g after the first cycle is almost the same as that of the second cycle, Fig. 4(d and e). These results are consistent with the CV data presented in Fig. 4a in which the CV plots are reversible following the first cycle. Overall, the analysis of Raman spectra further supports the hypothesis of a quasi-reversible Zn2+ ion intercalation/deintercalation mechanism during the first cycle. This results in a decrease in the specific capacity for the second discharge cycle due to the trapping of zinc during the first cycle. It is worth mentioning that, to the best of our knowledge, no previous studies on zinc ion intercalation into Mo6S8 using in situ Raman spectroscopy have been reported in the literature.
 | ||
| Fig. 4 In situ Raman spectral of Mo6S8 electrode at different charge and discharge potential in 2 M ZnSO4. (a) Cyclic voltammograms of Zn/Mo6S8 cell at 1 mV s−1 during 1st, 2nd, 3rd, and 4th cycle: (b, c), and (d, e) Raman spectra of the first and second cycle during the intercalation/de-intercalation process, respectively. | ||
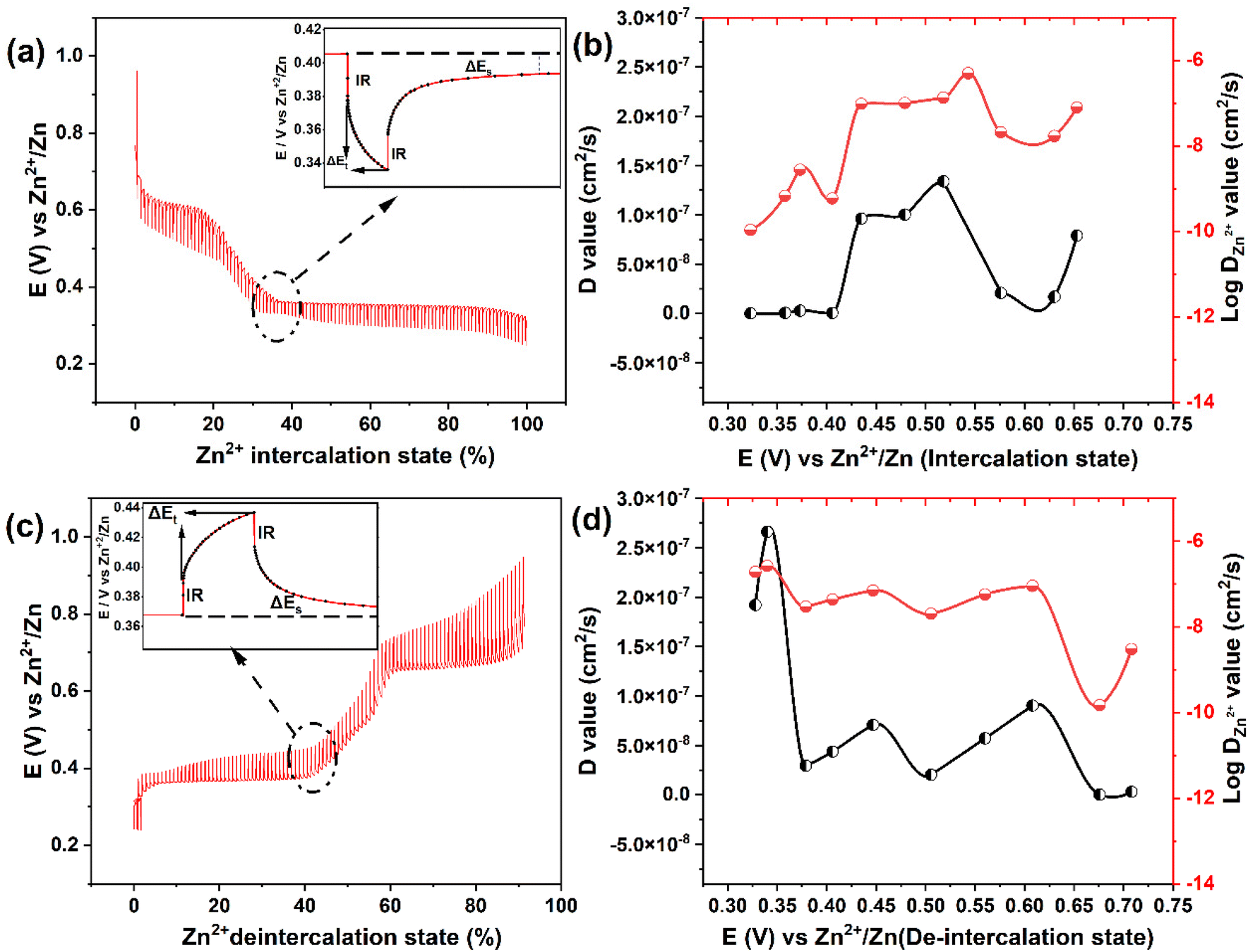 | ||
| Fig. 5 Galvanostatic intermittent titration technique (GITT) curves of in 2 M ZnSO4 at 0.1 A g−1 recorded during the Zn2+ intercalation (a) and deintercalation process (c) as a function of state of (de)intercalation process. The insets in (a and c) scheme for a single step of a GITT experiment. The corresponding Zn2+ diffusion coefficients values among the selected steps during intercalation (b) and deintercalation (d) process. | ||
To further investigate the potential cause of Zn trapping within the first cycle, we conducted periodic density functional theory (DFT) calculations. As a first possibility we checked for agglomeration within the Mo6S8 structure by constructing a (2 × 2 × 3) supercell consisting of 12 CP units. This supercell was filled with either two Zn or two Mg atoms at varying distances of one, two, or three unit cells apart (equivalent to approximately 6, 12, and 18 Å, respectively), and the system energies were compared in order to check whether there is any mutual interaction between the two inserted metal atoms. The energetic differences for both Zn and Mg at all considered distances were in the meV range, which is within the typical DFT accuracy for such systems. Therefore, the DFT calculations suggest that there is no direct interaction between the charge carriers within the Mo6S8 structure itself.
We also investigated the possibility of Chevrel phase decomposition into MoS2 in the form of layered sulfides and compared the formation energies to determine whether the process is thermodynamically feasible. Both zinc and magnesium were considered as potential charge carriers as zinc ions are trapped in the anode, while this behavior is not observed for magnesium. Specifically, we looked at the formation energy per atom of MgMo2S4, Mg2Mo2S4, ZnMo2S4, and Zn2Mo2S4, as well as their corresponding Chevrel phase counterparts such as MgMo6S8etc. The trends were similar for both charge carriers at one charge carrier per supercell, with a small difference of 0.01 eV per p.a., and the Mg2Mo6S8 being slightly more stable with a difference of 0.05 eV per p.a. These differences are negligible and unlikely to cause significant Zn trapping.
Due to the large computational effort associated with the identification of diffusion barriers and the evaluation of their energetic height in such large supercells, we did not determine these barriers. However, in a previous work,42 we found a linear dependence of the diffusion barrier on the lattice parameter for the CP. Using the results of this study, we estimate that the diffusion barrier in this system with a lattice constant of 6.43 Å should be of the order of 0.6 eV.
The XRD measurements of Fig. 2 suggest the emergence of a Zn2.9Mo15S19 phase during cell cycling. This phase has a different structure from the Chevrel cluster structure, with 4 Zn positions at 100% occupancy (A positions) and 6 positions at 30% occupancy (B positions) which are split into two layers, for a visual representation of the different intercalation positions, refer to Fig. S9.† With a Zn content of 5.8 (2 × 2.9) spread over 10 possible sites, we model the phase by incrementally filling 1 to 10 Zn atoms per cell. Note that our unit cell contains two formula units (30 Mo atoms and 38 S atoms), so that increasing the Zn content by one atom at a time results in stoichiometries from Zn0.5Mo15S19 to Zn5.0Mo15S19. The Zn3.0Mo15S19 phase is closest to the experimentally measured Zn content of 2.9 (refer to the XRD data above and the relevant discussion therein). The formation energies of the phase were then examined by filling it with Zn atoms and comparing each phase with n + 1 Zn atoms to its previous most stable phase with n Zn atoms. When we fill the empty phase by putting the Zn atoms in the A positions, the formation energies with respect to adding new Zn are positive, which means that it is energetically unfavourable to insert the Zn atom at these sites. This suggests that the B positions are filled first in an empty phase. Here the formation energy is −1.15 eV for the first Zn atom, regardless of which B position is occupied. For the second Zn the formation energy is −1.18 eV if it is located in a different layer than the first Zn and −0.67 eV if they lie in the same layer. As Fig. 1 shows the working window of the battery extends up to ca. 0.8 eV, hence we can surmise that the first two Zn atoms are too strongly bound to be fully deintercalated within the operational window of the battery, which likely results in the observed trapping. For the third Zn atom the formation energy for three Zn atoms in the B positions is only slightly more stable (−0.72 eV) than putting the third Zn atom in one of the 4 A positions (−0.67 eV). For 4 Zn atoms the occupations of 2 Zn in A and 2 in B, 3 in B and 1 in A and all 4 Zn in B are respectively −0.55, −0.68 and −0.78 eV, making it most favourable for the Zn atoms to be spread equally over the two layers of the B positions (see Fig. S9†). The configuration with 4 Zn in the B positions and any additional Zn atoms in the A positions is consistently the most stable for Zn content ranging from 5 to 8. The formation energy of this phase always falls within the range of −0.66 to −0.68 eV. The positive formation energies obtained upon attempting to fill the phase beyond an occupancy of 8 Zn atoms suggest that this is the maximum capacity of the phase. The optimal occupancy of Zn atoms in the phase was thus found to be between 1.0 and 4.0 Zn atoms per formula unit, with an equal spread of Zn atoms over the two layers of B positions being the most favourable. The Zn atoms in this range bind to the anode with an energy that enables them to be easily deintercalated within the battery's operating range. In conclusion, our theoretical analysis reveals that during the initial cycle, the Zn2.9Mo15S19 phase we examined forms alongside the charged Chevrel phase. However, the phase cannot compete with the CP in terms of reversibility, as it traps 1 Zn atom per formula unit in its structure, while still performing reasonably as an anode by loosely binding 3 additional Zn atoms.
To investigate the diffusion coefficient of zinc ions (DZn2+) into the Mo6S8 cavities, the galvanostatic intermittent titration technique (GITT) was employed (Fig. 5), and the DZn2+ values were calculated according to the following equation:43
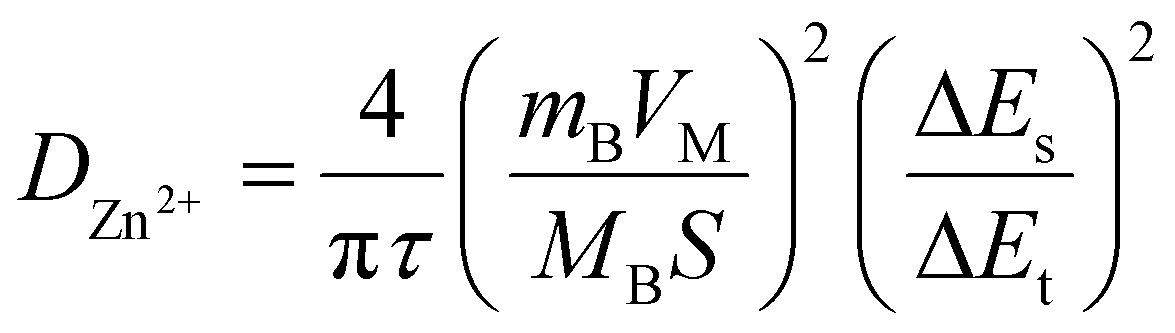 | (2) |
Based on the data derived using eqn (2), the profiles of DZn2+vs. voltage during the intercalation/deintercalation process are displayed in Fig. 5(b and d). DZn2+ values range from 4.99 × 10−7 to 1.07 × 10−10 cm2 s−1 (Zn2+ intercalation) and 1.92 × 10−7 to 1.48 × 10−10 cm2 s−1 (Zn2+ de-intercalation). The diffusion coefficient data can be divided into two characteristics regions: (i) zinc ion insertion/de-insertion processes and (ii) non-insertion/de-insertion stages. Such phenomena are typical for an intercalation-type battery electrode material.44 Initially, from the open circuit voltage (+0.95 V vs. Zn2+/Zn) to the start of the first insertion plateau (+0.70 V vs. Zn2+/Zn), the values of DZn2+ are in the range of 7.89 × 10−8 to 2.07 × 10−8 cm2 s−1. As the intercalation potential increases to +0.47 V vs. Zn2+/Zn, DZn2+ increases to 1.0 × 10−7 cm2 s−1 due to the formation of the ZnxMo6S8 phase. In the next stage, the ZnxMo6S8 phase is gradually being transformed to Zn2Mo6S8 as Zn2+ intercalation advances (+0.43 to +0.32 V vs. Zn2+/Zn) and the determined DZn2+ values lie within the range of 9.6 × 10−8 to 1.07 × 10−10 cm2 s−1. The decrease in diffusion coefficient upon transformation from ZnxMo6S8 to Zn2Mo6S8 could be correlated to the structural differences between the two phases. In particular, the larger average interatomic distance of Mo–Mo in ZnxMo6S8 (2.71 Å) compared to Zn2Mo6S8 (2.61 Å) may facilitate Zn2+ insertion kinetics. Interestingly in the reverse de-intercalation scan, Zn2+ diffuses back quickly from the Zn2Mo6S8 phase (1.9 × 10−7 cm2 s−1) to ZnxMo6S8, which could be due to the reversibility of this process (Zn2Mo6S8) as inferred from the peak separation in cyclic voltammetry data and the increased stability of ZnxMo6S8 relative to Zn2Mo6S8.19,45 Following this, nearly identical evolution of diffusion coefficients of Zn2+ can be observed as compared to the intercalation process. A detailed list of the calculated diffusion coefficients values at different intercalation/de-intercalation potentials are presented in Table S1† (representative experimental data is presented in Fig. S10†). It is worth mentioning that the calculated diffusion coefficient values of the nanoscale Mo6S8 during charge/discharge process are higher than those of most electrode materials previously reported for aqueous Zn-storage e.g., TiS2,13 1.33 × 10−11–5.71 × 10−9 cm2 s−1; TiSe2,7 10−10–10−9 cm2 s−1; layered MnO2,46 10−14–10−12 cm2 s−1; V2O5·6H2O47 10−11–10−10 cm2 s−1 as well as microsized Mo6S8-based materials 4.5 × 10−10–1.5 × 10−9 cm2 s−1.48 These findings demonstrate the fast kinetics of the prepared Mo6S8 driven by the nanosized structure.
3.3. Characterization of the YEC-8A cathode material in a half-cell
The structural characterisation and the electrochemical performance of activated carbon (YEC-8A) was examined in 2 M ZnSO4. The XRD pattern (Fig. 6a) shows a broad diffraction peak at 43.39° associated with the (101) plane reflection.49 The broad peak feature of the XRD pattern is indicative of the amorphous structure of the YEC-8A sample. The surface morphology of YEC-8A was examined by SEM (Fig. 6b). The activated carbon particles have an irregular shape with a relatively rough surface and particle size within the range of 5 μm to 20 μm. Fig. 6(c and d) shows the cyclic voltammograms within a potential window of 1–1.8 V vs. Zn2+/Zn at various scan rates from 2 to 100 mV s−1. The CVs display a quasi-rectangular shape, reflecting the capacitive behaviour of the YEC-8A electrode. The GCD curves (Fig. 6e) at different current densities are triangular with negligible IR-drop, implying favourable reversibility and good electrical conductivity of YEC-8A electrode. The specific capacitance was calculated from the GCD data and presented in Fig. 6f. We note that with increasing current density, from 0.2 A g−1 to 2 A g−1, the specific capacitance decreases from ∼153 F g−1 to 113 F g−1, respectively (for specific capacitance calculations, refer to the ESI†). | ||
| Fig. 6 Structural characterisation and electrochemical performance of YEC-8A. (a) XRD pattern; (b) SEM image; (c and d) Cyclic voltammograms as a function of scan rate; (e) GCD curves at current densities from 0.2 to 2 A g−1; (f) Specific capacitance as a function of current density. | ||
3.4. Full cell Zn-ion hybrid supercapacitor (ZIHSC)
In view of the high performance and fast Zn2+ diffusion into Mo6S8, as well as to demonstrate the potential application of the nanoscale synthesised material, we assembled a ZIHSC device. Fig. 7a shows the schematic diagram of the assembled ZIHSC, in which Mo6S8, YEC-8A, and 2.0 M ZnSO4 serve as the anode, cathode, and electrolyte, respectively. During the charge/discharge process, zinc ions intercalate/de-intercalate into the anode and reversible ion adsorption/desorption on the YEC-8A cathode surface enables the ZIHSC to repeatedly store/deliver electrical energy. For an ideal symmetric capacitor, the voltage is assumed to be equally divided between both electrodes, since both electrodes have the same mass and amount of charge. In this ZIHSC, however, we have shown that each electrode has a stable potential region and stores a different amount of charge. It is therefore necessary to assemble an optimised, mass-balanced cell in order to maximise the overall performance of the electrochemical ZIHSC.50,51 The optimum mass ratio of YEC-8A to Mo6S8 electrodes was calculated so as to balance the amount of charge. The charge (Qsp) stored is related to the specific capacity (Csp), the potential evolution at each electrode (ΔV) and the active mass (m) of the corresponding electrode as follows: Qsp = m·Csp·ΔV. Thus, the mass ratio in theory can be further expressed . As discussed above, the calculated specific capacities of Mo6S8 and YEC-8A electrodes are 55 and 31.6 mA h g−1 at 0.2 A g−1; thus, the best mass ratio of cathode to anode was ∼1.76. Subsequently, the electrochemical performance of Mo6S8//YEC-8A ZIHSC was evaluated, and the relevant measurements are presented in Fig. 8.
. As discussed above, the calculated specific capacities of Mo6S8 and YEC-8A electrodes are 55 and 31.6 mA h g−1 at 0.2 A g−1; thus, the best mass ratio of cathode to anode was ∼1.76. Subsequently, the electrochemical performance of Mo6S8//YEC-8A ZIHSC was evaluated, and the relevant measurements are presented in Fig. 8.
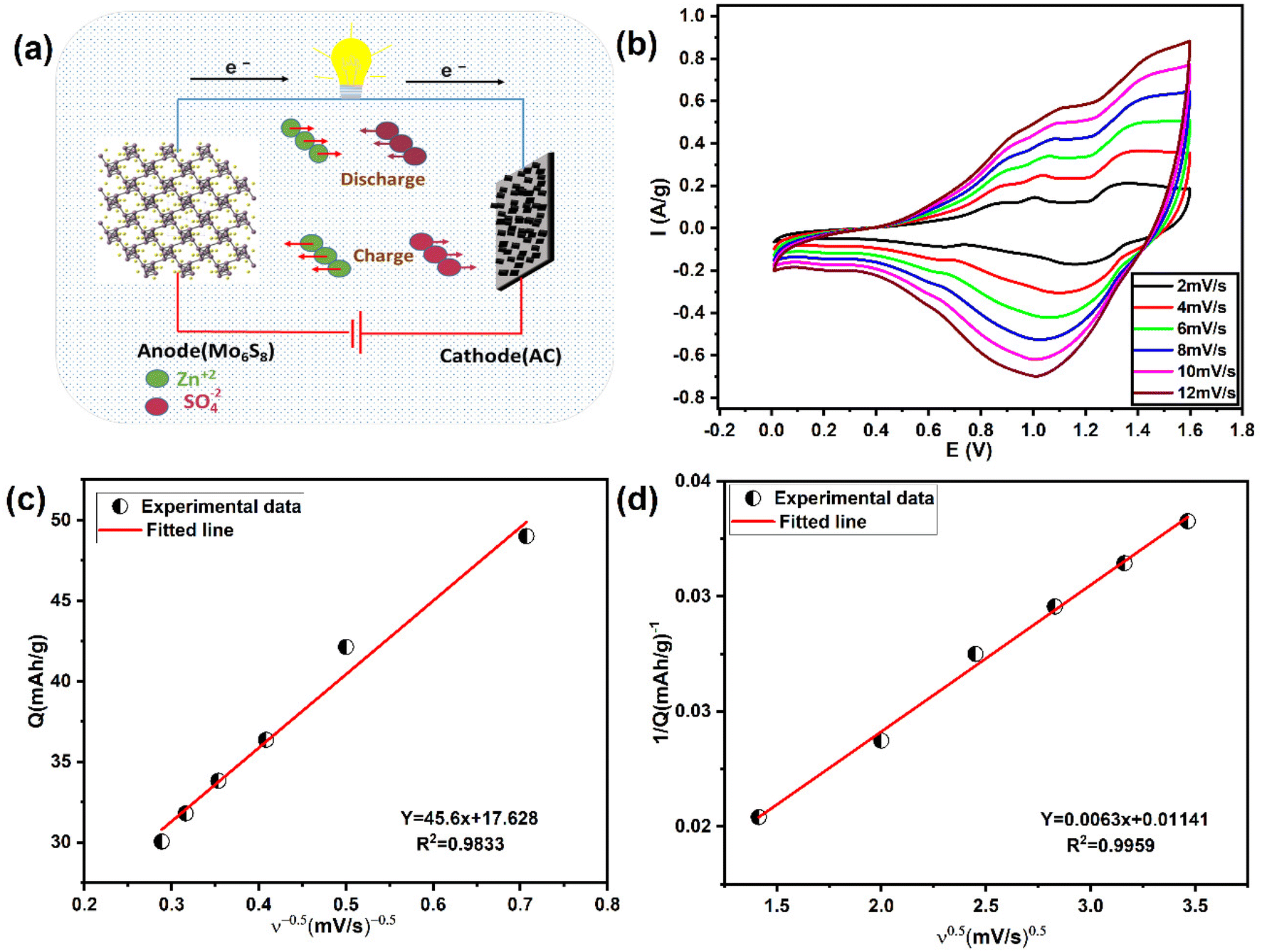 | ||
| Fig. 7 Schematic and charge storage mechanism of the fabricated energy storage system. (a) Schematic diagram of Mo6S8/YEC-8A ZIHSC. (b) CVs of Mo6S8/YEC-8A at different scan rates from 2 to 12 mV s−1. Voltammetric charge (Q) calculated as a function of scan rate according to Trasatti's method: (c) Q vs. ν−1/2 plot and (d) 1/Q vs. ν1/2 plot. | ||
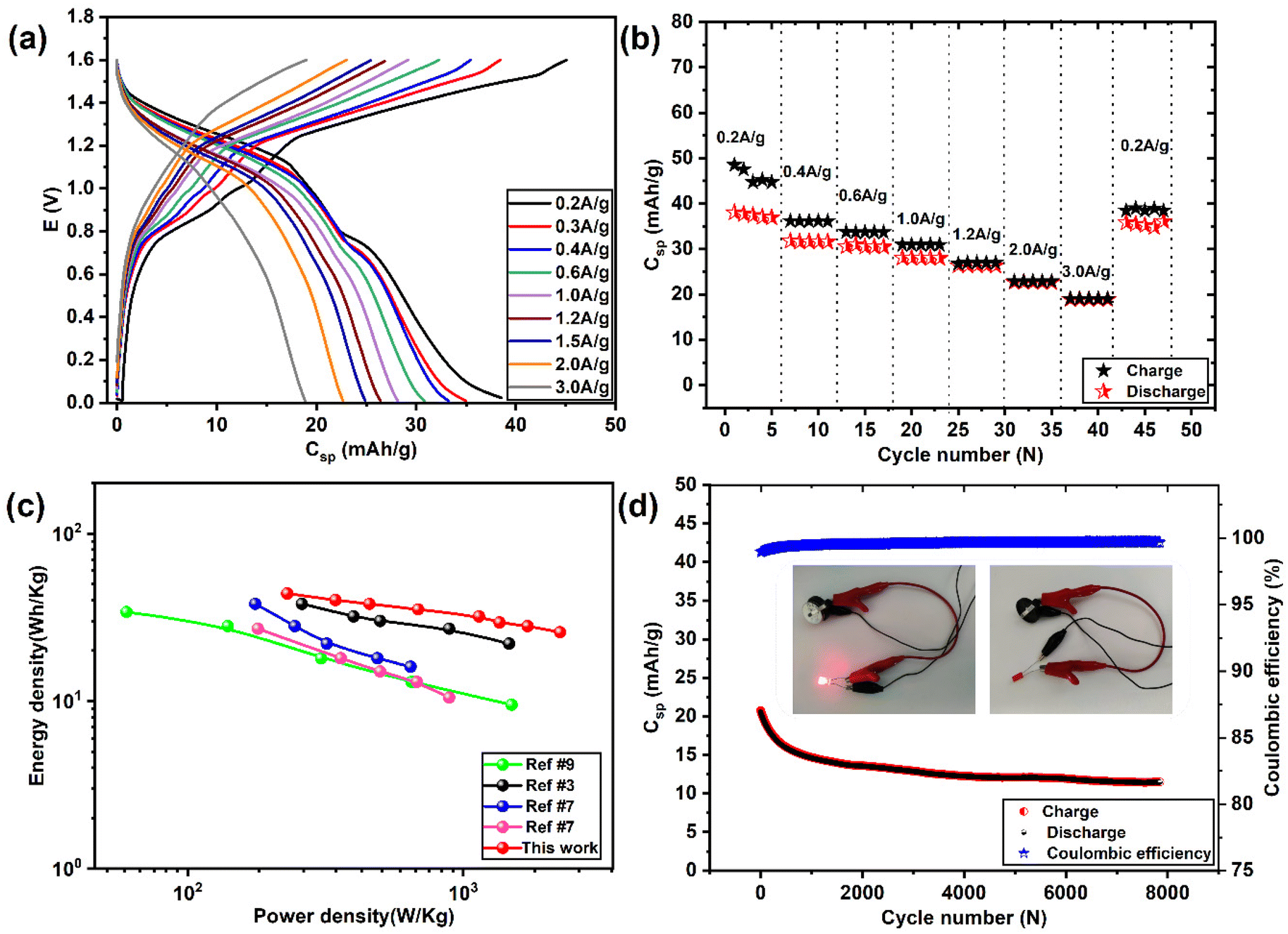 | ||
| Fig. 8 Electrochemical performance of the Mo6S8/YEC-8A device in 2 M ZnSO4 aqueous electrolyte. (a) GCD profiles (E vs. capacity format) recorded at different current densities, (b) corresponding rate capability curves, (c) Ragone's plot comparing Mo6S8/YEC-8A device with other recently published ZIHSCs, and (d) long-term cycling stability of Mo6S8/YEC-8A full cell recorded at a current density of 3A g−1 (inset shows an LED illuminated by a Mo6S8/YEC-8A device). | ||
The CVs of Mo6S8//YEC-8A within the potential range 0.0–1.6 V at various scan rates from 2 to 12 mV s−1, deviate from the ideal rectangular shape (Fig. 7b), suggesting that there are both capacitive and diffusion-controlled energy storage mechanisms.
Trasatti's method52 was used to distinguish the surface capacitive and diffusional contribution from the cyclic voltammetric charge plotted as a function of scan rate according to the following equations:1,52
| Q = Qcapacitive + Qdiffusion = Qcapacitive + Kν−0.5 | (3) |
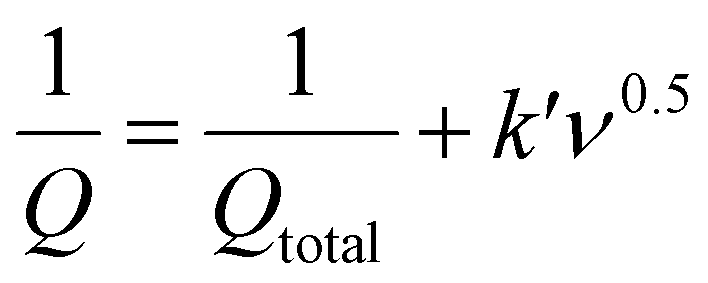 | (4) |
| Qdiffusion = Qtotal − Qcapacitive | (5) |
Fig. 8(a and b) shows the GCD plots and rate performance curves of Mo6S8//YEC-8A cell, respectively, recorded over a high voltage window of 1.6 V. We can see that the GCD profiles deviate from the capacitive triangular shape, which further demonstrates the simultaneous ionic intercalation/deintercalation and adsorption/desorption energy storage mechanisms. The cell delivered an average voltage of ∼1.1 V with an overall capacity of ∼38 mA h g−1, and 19 mA h g−1 measured at 0.2 and 2 A g−1, respectively. This means that the cell maintained 60% of its capacity when the current density was increased by a factor of 10 from 1 to 10 A g−1. The decrease in capacity at high current is mainly due to the slow Zn ion interaction into Mo6S8, a behavior related to the battery-type electrode material where more time is needed for the intercalation/deintercalation process. Notably, when the cycling rate returns to 0.2 A g−1, the recorded capacity is ca. 35.4 mA h g−1 which corresponds to a capacity retention of ca. 90% compared to the first cycle. This observation shows the excellent high-rate performance of the fabricated device.
Fig. 8c compares Ragone's plot of the Mo6S8/YEC-8A device with other recently published fabricated aqueous hybrid supercapacitors. It can be seen that the maximum specific energy density delivered from our fabricated cell is as high as 43.8 W h kg−1 with a power density of 229.9 W kg−1 at a specific current density of 0.2 A g−1. At high current density, of 2 A g−1, the device still delivers an energy density of 25.7 W h kg−1 with a high-power density of 2250 W kg−1. The relatively high specific energy delivered by our device (Mo6S8/YEC-8A) compared to TiS2/AC53 or TiSe2/AC,7 could be due to the high operating voltage of 1.6 V, lower intercalation potential of Zn2+ into (ca. 0.3 V vs. Zn2+/Zn), and hence the high operating voltage of the fabricated device of approximately 1.1 V.
The cycling stability of Mo6S8/YEC-8A was tested at 3 A g−1 for 8000 cycles as shown in Fig. 8d. The rapid decrease of capacity during the first cycles may be attributed to the aforementioned trapping of zinc inside the Mo6S8 electrode during the initial intercalation process. Following this, the device exhibits high cycling stability with a coulombic efficiency of nearly 100%. More importantly, two assembled Mo6S8/YEC-8A coin cells connected in series were able to illuminate the red light-emitting diode as shown in the inset of Fig. 8d, demonstrating its practical application. Besides, the EIS spectra of Mo6S8/YEC-8A is presented in Fig. S11.† The intercept of the abscissae (real axis) corresponds to the equivalent series resistance (Rs). This resistance is related to the internal resistance of the electrolyte, electrode material, and contact resistance at the electrode–electrolyte interface. The semicircle in the high to mid-frequency region relates to the charge-transfer process, which represents the zinc ion diffusion resistance during the electrochemical process. The Rs and Rct of the Mo6S8/YEC-8A recorded at 1.2 V are, respectively, 1.2 Ω and 6.5 Ω, which again reflects the high conductivity and rapid kinetics of the assembled device.
Post-mortem SEM physical characterisation was conducted to investigate the zinc dendrite formation and surface morphology of the cycled Mo6S8, YEC-8A, and zinc electrodes. All investigated electrodes were recovered from the fabricated coin cells, washed thoroughly with water several times, and dried before SEM measurements. Fig. 9 shows the surface morphology of YEC-8A, Mo6S8, and zinc electrodes after cycling at 3 A g−1 in 2 M ZnSO4. We can see that the YEC-8A surface and zinc metal electrodes in Zn/YEC-8A device are covered with zinc dendrites (Fig. 9(a and b)). In contrast, when Mo6S8 is used as the anode in the Mo6S8/YEC-8A device, no zinc dendrites are observed on either the anode or cathode surfaces even after 8000 cycles (Fig. 9(c and d)). This is attributed to use of a framework material, specifically the Chevrel phase, which provides a “scaffold” into which the reduced form of the metal is held. This selection also depends on the transport of Zn ions into the Chevrel phase being efficient, and the diffusion coefficient being relatively high for divalent ions as concluded from the GITT experiments (as discussed above).
 | ||
| Fig. 9 Post-mortem SEM characterization of the electrodes examined after cycling in 2 M ZnSO4 at a gravimetric current of 3 A g−1. SEM images of (a) zinc foil, and (b) YEC-8A electrode in Zn/YEC-8A system after 100 cycles. SEM images of (c) Mo6S8 electrode, and (d) YEC-8A electrode in Mo6S8/YEC-8A coin cell after 8000 cycles. | ||
4. Conclusion
We have demonstrated that Mo6S8 is a promising dendrite-free zinc anode for ZIHSC devices. A range of electrochemical measurements show that the Mo6S8 Chevrel phase follows a multi-step zinc ion intercalation/de-intercalation process, with a lower intercalation potential of approximately 0.3 V vs. Zn2+/Zn. Additionally, the nanosized CP offers fast zinc ion kinetics, as inferred by the GITT data, compared to other available anode materials such as TiSe2, and TiS2. Post-mortem (P-XRD) diffraction shows the formation of a new sulfur deficient Zn1.16Mo6S7.6 phase that tends to be the most stable one upon cycling, as also confirmed theoretically by DFT calculations. Furthermore, in situ Raman spectroscopy has revealed the change in the phonon structure of the Mo6S8 upon intercalation/de-intercalation, indicating zinc trapping upon cycling. In addition, the potential application of nanosized Mo6S8 was also investigated. A ZIHSC device, with a M6S8 anode and YEC-8A cathode, delivered a capacity of 38 mA h g−1, with a superior energy density of 43.8Wh kg−1 at a specific current density of 0.2 A g−1. In addition, the ZIHSC shows excellent cycling response, up to 8000 cycles (3 A g−1) with ∼100% coulombic efficiency. Interestingly, the post-mortem SEM analysis show no dendrite formation over the Mo6S8 anode compared to the usage of zinc metal. This work provides a proof-of-concept of using Mo6S8 compound as zinc-dendrite free anode material for zinc hybrid capacitors. This success could motivate further developments in the adoption of new materials, with lower zinc intercalation potential than the Mo6S8 materials demonstrated in this work, for practical application in next-generation ZIHSC energy storage systems.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work presented in this paper.Acknowledgements
Amr Abdelkader Ahmed Sadek (also known as Amr Elgendy) expresses his gratitude to the Newton-Mosharafa Fund, a partnership between the British Council and the Ministry of Higher Education- Missions Sector in Egypt- for providing him with a full PhD scholarship. Further support from EPSRC is gratefully acknowledged (EP/T01816X/1). Further support for this work was provided via the Henry Royce Institute, funded through EPSRC grants EP/R00661X/1, EP/P025021/1 and EP/P025498/1. Support by the German Research Foundation (DFG) under Germany's Excellence Strategy - EXC 2154 - Project number 390874152 (POLiS Cluster of Excellence) is gratefully acknowledged. Computational resources have been provided by the state of Baden-Wuerttemberg through bwHPC and the German Research Foundation (DFG) through Grant No. INST 40/575-1 FUGG (JUSTUS 2 cluster).References
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- L. Dong, X. Ma, Y. Li, L. Zhao, W. Liu, J. Cheng, C. Xu, B. Li, Q.-H. Yang and F. Kang, Energy Storage Mater., 2018, 13, 96–102 CrossRef.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- P. He, Q. Chen, M. Yan, X. Xu, L. Zhou, L. Mai and C.-W. Nan, EnergyChem, 2019, 1, 100022 CrossRef.
- L. Wen, Y. Wu, S. Wang, J. Shi, Q. Zhang, B. Zhao, Q. Wang, C. Zhu, Z. Liu and Y. Zheng, Nano Energy, 2021, 106896 Search PubMed.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- X. Ma, J. Cheng, L. Dong, W. Liu, J. Mou, L. Zhao, J. Wang, D. Ren, J. Wu and C. Xu, Energy Storage Mater., 2019, 20, 335–342 CrossRef.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- B. Liu, S. Wang, Z. Wang, H. Lei, Z. Chen and W. Mai, Small, 2020, 16, 2001323 CrossRef CAS PubMed.
- Y. Tian, Y. An, C. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, Adv. Energy Mater., 2021, 11, 2002529 CrossRef CAS.
- L. Wang, J. Zou, S. Chen, G. Zhou, J. Bai, P. Gao, Y. Wang, X. Yu, J. Li, Y.-S. Hu and H. Li, Energy Storage Mater., 2018, 12, 216–222 CrossRef.
- T. Xiong, Y. Zhang, Y. Wang, W. S. V. Lee and J. Xue, J. Mater. Chem. A, 2020, 8, 9006–9012 RSC.
- Z. Xu, M. Li, W. Sun, T. Tang, J. Lu and X. Wang, Adv. Mater., 2022, 2200077 CrossRef CAS PubMed.
- R. Schöllhorn, M. Kümpers and J. O. Besenhard, Mater. Res. Bull., 1977, 12, 781–788 CrossRef.
- L. Mei, J. Xu, Z. Wei, H. Liu, Y. Li, J. Ma and S. Dou, Small, 2017, 13, 1701441 CrossRef PubMed.
- M. S. Chae and S.-T. Hong, Batteries, 2019, 5, 3 CrossRef CAS.
- M. S. Chae, J. W. Heo, S.-C. Lim and S.-T. Hong, Inorg. Chem., 2016, 55, 3294–3301 CrossRef CAS PubMed.
- A. Elgendy, A. A. Papaderakis, R. Cai, K. Polus, S. J. Haigh, A. S. Walton, D. J. Lewis and R. A. W. Dryfe, Nanoscale, 2022, 14, 10125–10135 RSC.
- A. Elgendy, A. A. Papaderakis, C. Byrne, Z. Sun, J. V. Lauritsen, E. P. C. Higgins, A. Ejigu, R. Cernik, A. S. Walton, D. J. Lewis and R. A. W. Dryfe, ACS Appl. Energy Mater., 2021, 4, 13015–13026 CrossRef CAS.
- A. Regoutz, M. Mascheck, T. Wiell, S. K. Eriksson, C. Liljenberg, K. Tetzner, B. A. D. Williamson, D. O. Scanlon and P. Palmgren, Rev. Sci. Instrum., 2018, 89, 73105 CrossRef PubMed.
- B. F. Spencer, S. Maniyarasu, B. P. Reed, D. J. H. Cant, R. Ahumada-Lazo, A. G. Thomas, C. A. Muryn, M. Maschek, S. K. Eriksson and T. Wiell, Appl. Surf. Sci., 2021, 541, 148635 CrossRef CAS.
- N. Fairley, AES, SIMS More, Casa Softw. Ltd, 2017. See: https://www.casaxps.com Search PubMed.
- D. J. H. Cant, B. F. Spencer, W. R. Flavell and A. G. Shard, Surf. Interface Anal., 2022, 54, 442–454 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- H. Euchner and A. Groß, Phys. Rev. Mater., 2022, 6, 40302 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- M. Mao, Z. Lin, Y. Tong, J. Yue, C. Zhao, J. Lu, Q. Zhang, L. Gu, L. Suo, Y.-S. Hu, H. Li, X. Huang and L. Chen, ACS Nano, 2020, 14, 1102–1110 CrossRef CAS PubMed.
- Y. Cheng, L. Luo, L. Zhong, J. Chen, B. Li, W. Wang, S. X. Mao, C. Wang, V. L. Sprenkle and G. Li, ACS Appl. Mater. Interfaces, 2016, 8, 13673–13677 CrossRef CAS PubMed.
- E. Levi, G. Gershinsky, D. Aurbach and O. Isnard, Inorg. Chem., 2009, 48, 8751–8758 CrossRef CAS PubMed.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- P. Yan, J. Zheng, J. Liu, B. Wang, X. Cheng, Y. Zhang, X. Sun, C. Wang and J.-G. Zhang, Nat. Energy, 2018, 3, 600–605 CrossRef CAS.
- Y. Tong, A. Gao, Q. Zhang, T. Gao, J. Yue, F. Meng, Y. Gong, S. Xi, Z. Lin and M. Mao, Energy Storage Mater., 2021, 37, 87–93 CrossRef.
- J. Zou and G. L. Schrader, J. Catal., 1996, 161, 667–686 CrossRef CAS.
- D. J. Holmgren, R. T. Demers, M. V. Klein and D. M. Ginsberg, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 1952–1955 CrossRef CAS PubMed.
- K. Helmbrecht, H. Euchner and A. Groß, Batteries Supercaps, 2022, 5, e202200002 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569 CrossRef CAS.
- P. Li, X. Zheng, H. Yu, G. Zhao, J. Shu, X. Xu, W. Sun and S. X. Dou, Energy Storage Mater., 2019, 16, 512–518 CrossRef.
- E. Gocke, W. Schramm, P. Dolscheid and R. Scho, J. Solid State Chem., 1987, 70, 71–81 CrossRef CAS.
- B. Lee, H. R. Lee, H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, Chem. Commun., 2015, 51, 9265–9268 RSC.
- N. Zhang, M. Jia, Y. Dong, Y. Wang, J. Xu, Y. Liu, L. Jiao and F. Cheng, Adv. Funct. Mater., 2019, 29, 1807331 CrossRef.
- E. Levi, G. Gershinsky, D. Aurbach, O. Isnard and G. Ceder, Chem. Mater., 2009, 21, 1390–1399 CrossRef CAS.
- S. Ghosh, R. Santhosh, S. Jeniffer, V. Raghavan, G. Jacob, K. Nanaji, P. Kollu, S. K. Jeong and A. N. Grace, Sci. Rep., 2019, 9, 1–15 CrossRef PubMed.
- E. Redondo, L. W. Le Fevre, R. Fields, R. Todd, A. J. Forsyth and R. A. W. Dryfe, Electrochim. Acta, 2020, 360, 136957 CrossRef CAS.
- A. Elgendy, N. M. El Basiony, F. E.-T. Heakal and A. E. Elkholy, J. Power Sources, 2020, 466, 228294 CrossRef CAS.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- Q. Wang, S. Wang, J. Li, L. Ruan, N. Wei, L. Huang, Z. Dong, Q. Cheng, Y. Xiong and W. Zeng, Adv. Electron. Mater., 2020, 6, 2000388 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01238k |
| This journal is © The Royal Society of Chemistry 2024 |