
Surface effects on functional amyloid formation†
Alexander J.
Dear
 ad,
Georg
Meisl
a,
Christopher G.
Taylor
a,
Umberto Capasso
Palmiero
b,
Susanne Nordby
Stubbe
c,
Qian
Liu
c,
Paolo
Arosio
b,
Sara
Linse
d,
Tuomas P. J.
Knowles
a and
Maria
Andreasen
*c
ad,
Georg
Meisl
a,
Christopher G.
Taylor
a,
Umberto Capasso
Palmiero
b,
Susanne Nordby
Stubbe
c,
Qian
Liu
c,
Paolo
Arosio
b,
Sara
Linse
d,
Tuomas P. J.
Knowles
a and
Maria
Andreasen
*c
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
bInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Zürich, Switzerland
cDepartment of Biomedicine, Aarhus University, Wilhelm Meyers Allé 3, Aarhus DK-8000, Denmark. E-mail: mariaj@biomed.au.dk
dDepartment of Biochemistry and Structural Biology, Lund University, SE-221 00 Lund, Sweden
First published on 3rd August 2024
Abstract
Functional amyloids formed by the protein FapC in Pseudomonas bacteria are key structural components of Pseudomonas biofilms, which mediate chronic infections and also contribute to antimicrobial resistance. Here, we combine kinetic experiments with mechanistic modelling to probe the role of surfaces in FapC functional amyloid formation. We find that nucleation of new fibrils is predominantly heterogeneous in vitro, being catalysed by reaction vessel walls but not by the air/water interface. Removal of such interfaces by using microdroplets greatly slows heterogeneous nucleation and reveals a hitherto undetected fibril surface-catalysed “secondary nucleation” reaction step. We tune the degree of catalysis by varying the interface chemistry of the reaction vessel and by adding nanoparticles with tailored surface properties that catalyse fibril nucleation. In so doing, we discover that the rate of nucleation is controlled predominantly by the strength with which FapC binds to the catalytic sites on the interface, and by its surface area. Surprisingly, neither primary nucleation rate nor catalytic site binding strength appear closely correlated to the charge and hydrophilicity of the interface. This indicates the importance of considering experimental design in terms of surface chemistry of the reaction container while also highlighting the notion that fibril nucleation during protein aggregation is a heterogeneous process.
Introduction
Amyloid fibrils are linear polymeric structures formed by the self-assembly of monomeric protein molecules. Although usually held together by noncovalent interactions, they are highly thermodynamically stable. Historically the formation of amyloid fibrils via protein aggregation has been associated with various diseases like Parkinson's disease and Alzheimer's disease but more recently another type of protein aggregates have been found that are utilized to gain functionality hence coining the term functional amyloids.1–3 To date functional amyloids have been found in many different organisms, from humans to fungi and bacteria.2 In bacteria, functional amyloids are found extracellularly in the biofilm where they add to the stability of biofilm. Occurrence of functional amyloids in biofilm renders the biofilm very hard to disassemble.4 Note that while these and other amyloids clearly have functional properties, and amyloids such as tau tangles are clearly disease-associated, the functional and pathological properties of the amyloid form of many other proteins remains unknown, and not necessarily mutually exclusive.The mechanisms of formation of different kinds of amyloid have been extensively investigated in recent years using chemical kinetic modelling of data from aggregation experiments in plate readers to obtain information about the molecular steps involved.5–10 Formation of the observed linear fibril structures necessarily requires a rapid fibril elongation step, and a slow initial fibril formation step. The latter is typically modelled as a coarse-grained “primary nucleation” reaction involving the direct association of multiple monomers to form the smallest species capable of rapid fibril elongation. In addition to primary nucleation and elongation, so-called “secondary processes” have been frequently demonstrated to contribute to amyloid formation. The simplest secondary process is fragmentation of existing fibrils, which increases the number of growing fibril ends. The most prevalent is “secondary nucleation”, whereby monomers bind to the surfaces of existing fibrils and react with one another to form new fibrils.11
Using chemical kinetic modelling it has been shown that the functional amyloids from Pseudomonas and E. coli, formed by FapC and CsgA respectively, aggregate via a nucleation–elongation dominated mechanism, with no significant involvement of secondary processes, such as fragmentation or secondary nucleation.12 Conversely, other functional amyloids have been shown to aggregate in a secondary nucleation dominated mechanism13 such as the phenol soluble modulins from Staphylococcus aureus. We have recently demonstrated that the lack of an observable rate for secondary processes as seen for FapC and CsgA in standard in vitro experiments is a rare and unique feature in protein aggregation since the majority of aggregating proteins that have been analyzed using chemical kinetics display a secondary process that is responsible for the formation of the majority of new aggregates.8 For the few proteins for which there is no significant contribution from secondary processes, new aggregates are formed predominantly via primary nucleation. This process does not involve any existing aggregates, although in most cases it likely does not happen homogeneously in solution, but instead on an interface which catalyzes the formation of a nucleus.
During biofilm formation, specialized nucleator proteins have evolved to trigger aggregation,14,15 but the amyloid-forming protein can encounter a multitude of different interfaces and surfaces that can potentially interfere with or contribute to the aggregation process. These interfaces range from biological surfaces such as lipid bilayers of cell membranes, to extracellular matrix components of the human body, to surfaces of medical devices or non-medical related surfaces such as air–water interfaces, plastics, metal, glass and hydrogel surfaces. In fact, several of these surfaces and interfaces have been observed to affect the aggregation of amyloid forming protein.16 α-Synuclein, a protein associated with Parkinson's disease pathology, has been shown to aggregate rapidly in the presence of lipid mono- and bilayers due to these lipid layers enabling both primary nucleation and elongation of protein–lipid co-aggregates.6,17 Its aggregation has also been shown to be accelerated by hydrophobic surfaces18 and polystyrene nanoparticles (NPs).19 Indeed, NPs with various properties have been found to accelerate aggregation of several other proteins.19–22 CsgA also aggregates rapidly in the presence of lipid bilayers.23 Furthermore, the presence of an air–water interface is essential for the aggregation of hydrophobins (functional amyloids found in fungi), and lack of an air–water interface abolishes aggregation.24 In fact, in most studies where it has been specifically tested, fibril primary nucleation has been found to be heterogeneous.24–29 This is perhaps unsurprising, given that primary nucleation in other contexts such as crystallization and bubble formation is also almost always heterogeneous.30–34
Here, we investigate the effects of various surfaces on the aggregation mechanism of FapC.
Experimental section
All chemicals were purchased from Sigma-Aldrich.Recombinant expression and purification of FapC
BL21 (DE3) E. coli cells were transformed with the pET 28a vector containing the gene for FapC (residues 25–250) from the Pseudomonas fluorescens strain UK4 without the signal sequence (residue 1–24) and with six residues His-tag at the C-terminus. Cells were grown on LB-agar plates with kanamycin at 37 °C and the colonies were transferred to LB medium with kanamycin and grown to OD600 ∼ 1. Protein expression was induced by adding IPTG to a final concentration of 1 mM followed by incubation for 3 h. Cells were harvested and resuspended in 20 mL 50 mM tris-HCl pH 8, 8 M guanidine hydrochloride pr. L culture, and lysed by sonication. Cell debris was removed by centrifugation for 30 min at 5000g. The supernatant was then loaded onto a His-trap column (Nickel NTA resin, Qiagen, Netherlands). The column was washed with increasing concentrations of imidazole (0, 30, 60 and 120 mM in 50 mM tris-HCl pH 8, 8 M guanidine hydrochloride) and eluted with 300 mM imidazole, 50 mM tris-HCl pH 8, 8 M guanidine hydrochloride. The resulting fractions were analyzed by SDS-PAGE following ethanol precipitation and the guanidine hydrochloride was removed immediately before use, using a PD10 desalting column (GE Healthcare, USA) equilibrated in 20 mM sodium phosphate pH 7 using the gravity protocol.Aggregation in 96 well plates
Samples of desalted FapC were passed through a 0.22 μm filter and diluted to the required concentrations. ThT was added to the protein solutions to a final concentration of 40 μM and the solutions were transferred to 96-well microtiter plates, sealed to prevent evaporation, and placed in a Fluostar Omega plate reader (BMG Labtech, Germany). The plates were incubated at 37 °C, quiescent conditions and the ThT fluorescence (excitation 450 nm, emission 482 nm) was measured every 10 min. The experiments were carried out in black Corning polystyrene half-area microtiter plate with a non-binding surface (Corning 3881), black Corning polystyrene non-treated microtiter plate (Corning 3661), a black Greiner COC polymer microtiter plate (Greiner 655801), transparent ELISA microtiter plate with high binding surface, black Thermo Fisher microtiter plate non-treated (265301). When no specific plate is mentioned the default black Corning polystyrene half-area microtiter plate with a non-binding surface (Corning 3881) were used for aggregation reaction in the plate reader. In samples where the air–water interface was removed 30 μL mineral oil was carefully pipetted on top of the reaction mixture in the 96 well plate prior to incubation in the plate reader. To verify that ThT is an accurate reporter on fibril mass concentration, the ThT fluorescence signal after the plateau at the end of the aggregation reaction was plotted against the initial FapC monomer concentration for multiple conditions (Fig. S3†). It is well-established that the final fibril mass concentration must always be linear in the initial monomer concentration.35,36 Fig. S3† also shows a linear relationship, implying that the ThT fluorescence increase is proportional to fibril mass concentration. Therefore, normalized, baseline-subtracted ThT fluorescence kinetic curves are accurate representations of the kinetic curves for fibril mass concentration. Note this was also previously confirmed in an earlier FapC study.12 The high reproducibility of the kinetics when followed by ThT is observable throughout the study, with low variability between triplicate repeats used for each condition or initial protein concentration. We also found repeats of entire concentration-series kinetic experiments to be highly consistent with one another (Fig. S2†).Aggregation with nanoparticles
Nanoparticles (NPs) with hydrophobic, positively charged and negatively charged surface chemistries were synthesized as described previously.28 In NP experiments, various amounts of NPs were added to the reaction mixture to obtain different surface ratios compared to the FapC molecule. ThT and freshly desalted monomeric FapC was added to the 96 well plate along with the NPs in different ratios of surface area compared to the surface area of monomeric FapC protein. The plates were incubated at 37 °C, quiescent conditions and the ThT fluorescence (excitation 450 nm, emission 482 nm) was measured every 10 min.Microdroplet device fabrication
Microfluidic flowfocusing devices (Fig. S1†) were fabricated using standard soft-lithography techniques.37 Briefly, the device design was patterned on a silicon wafer using SU-8 negative photoresist (MicroChem) to produce a negative mold. Microfluidic channels were cast into poly(dimethylsiloxane) (PDMS; Dow Corning Sylgard 184 Silicone Elastomer) on the silicon mold, and the PDMS stamp was cured at 65 °C for 150 min before peeling off the mold. Channel inlets and outlets were punched into the PDMS stamp before oxygen plasma bonding it to a glass slide to seal the channels. To make channel walls hydrophobic, the device was flushed with the water repellent agent Aquapel (PPG Industries) for 30 min before washing with isopropanol and then blow-drying with a nitrogen stream.Aggregation in microdroplets
Aggregation reactions were encapsulated into water-in-oil-emulsions using a microfluidic device with flow focusing geometry. The sample containing monomeric desalted FapC and 40 μM ThT was injected into the middle inlet on the chip at a flow rate of 200 μL h−1. The carrier oil phase made of fluorinated FC-40 (Sigma) with 4% (w/v) triblock copolymer (ABA) surfactant (where A is a perfluorinated poly(propylene oxide) block and B a poly(ethylene oxide) block, synthesized as previously described38) was injected into the outer inlet at a flow rate of 300 μL h−1 to generate droplets. Fluid flow rates were controlled with a Cetoni neMESYS syringe pump (Cetoni GmbH). Droplets were collected at the outlet into rectangular 50 × 500 μm borosilicate capillaries (CM Scientific), and the capillaries were sealed with wax plugs to prevent sample evaporation. Before imaging the capillaries containing samples at different FapC concentration were aligned on a glass slide for incubation and imaging. The aligned capillaries were placed on the automated motorized stage of a custom-built epifluorescence laser microscope and covered with a hot plate heated to 37 °C. This setup is similar to ones used previously.39 To track the aggregation reaction within the droplets, fluorescent images were acquired every 15 min using laser excitation at 445 nm (diode laser (MLD445, Cobolt)). Individual droplets were identified manually, and the average intensity of the droplet area was extracted for all frames.Kinetic modelling
The concentrations of free protein monomers and of fibrils are denoted m(t) and P(t) respectively. The fibril mass concentration (expressed in molar units as the concentration of monomers that have aggregated into fibrillar form) is denoted M(t). Closed rate equations can be written down describing these quantities:40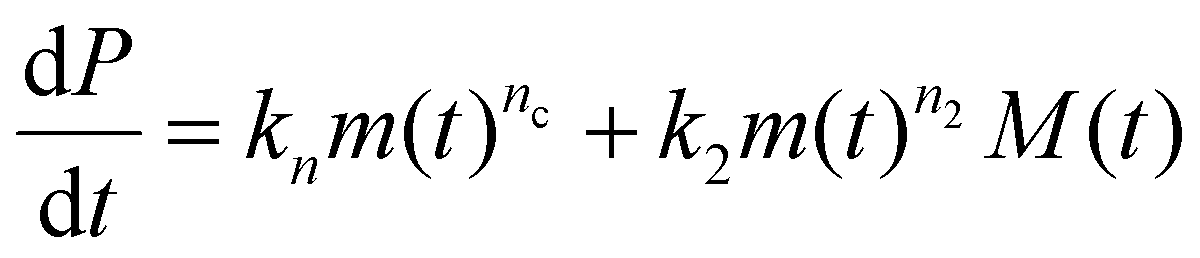 | (1) |
 | (2) |
Interpreting reaction orders
Primary nucleation in vitro is generally heterogeneous and catalyzed by interfaces. Like other catalyzed reactions, at high enough substrate concentration, the catalytic site is fully occupied at all times by substrate and the rate of product formation can no longer be increased by adding more substrate. This is referred to as “saturation”, and in the context of amyloid formation was first discovered to occur in secondary nucleation.5 The kinetics can be more completely described using the Michaelis–Menten model from enzymology: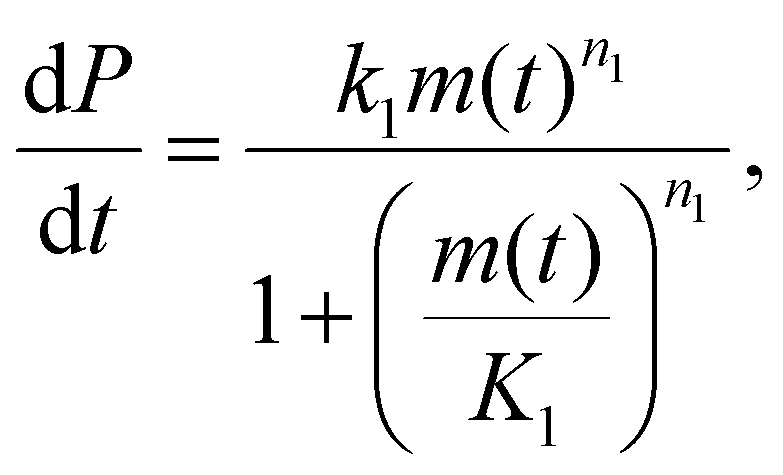 | (3) |
Transmission electron microscopy
Aggregated FapC samples were collected following the ThT fibrillation kinetics assay. 5 μL of FapC fibrils that formed with and without the presence of the different types of NPs were directly placed on carbon coated formvar grid (EM resolutions, UK), allowed to adhere for 2 min, and washed with MilliQ water followed by negative staining with 0.2% uranyl acetate for 2 min. Further, the grids were washed twice with MilliQ water and blotted dry using filter paper. The samples were examined using a Morgani 268 electron microscope (FEI Philips, USA), equipped with CCD digital camera with a resolution of 1376 × 1032, and operated at an accelerating voltage of 80 kV.Results
FapC fibril primary nucleation is heterogeneous in vitro, occurring on plate walls
The primary nucleation events occurring during in vitro aggregation of FapC can be divided into (1) homogeneous primary nucleation occurring in solution between monomeric FapC molecules and (2) heterogeneous primary nucleation occurring at an interface, Fig. 1.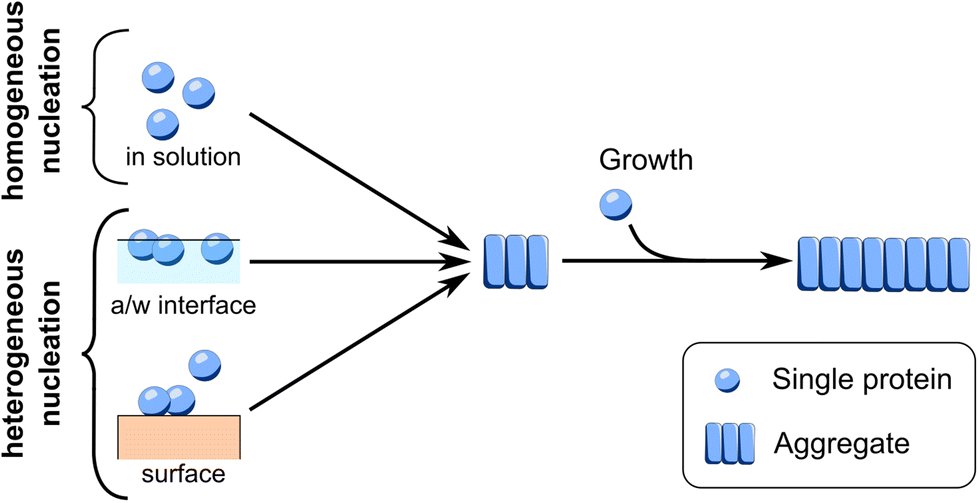 | ||
| Fig. 1 Schematic representation of possible mechanism of primary nucleation. The conversion of protein monomers into aggregation nuclei through primary nucleation can occur in different ways. Homogeneous nucleation occurs in solution. Heterogeneous nucleation occurs at an interface such as the air–water interface or the surface of the reaction container. After their initial primary nucleation, the nuclei can grow through addition of monomers at the growing ends, hence elongation. | ||
Heterogeneous primary nucleation can happen at the air–water interface or at the surface of the reaction container (or in principle on solid impurities in solution, e.g. dust particles). To identify whether the primary nucleation events that dominate FapC aggregation are homogeneous or heterogeneous, aggregation of FapC was carried out in a standard 96-well plate and compared to aggregation carried out in microdroplets. The microdroplets are produced from a surfactant-stabilized water-in-oil emulsion where the interface is effectively a surfactant monolayer, ESI Fig. 1.† The oil and surfactant used are fluorinated, which minimizes interactions with proteins.42 The very low noise level in the baseline compared to the difference between baseline and plateau in the microdroplet experiments, alongside the visible smooth increase in signal at early reaction times (Fig. 2B), indicates that Thioflavin T fluorescence is sensitive enough to accurately report on fibril aggregation kinetics at all times in microdroplets, in line with earlier studies.42
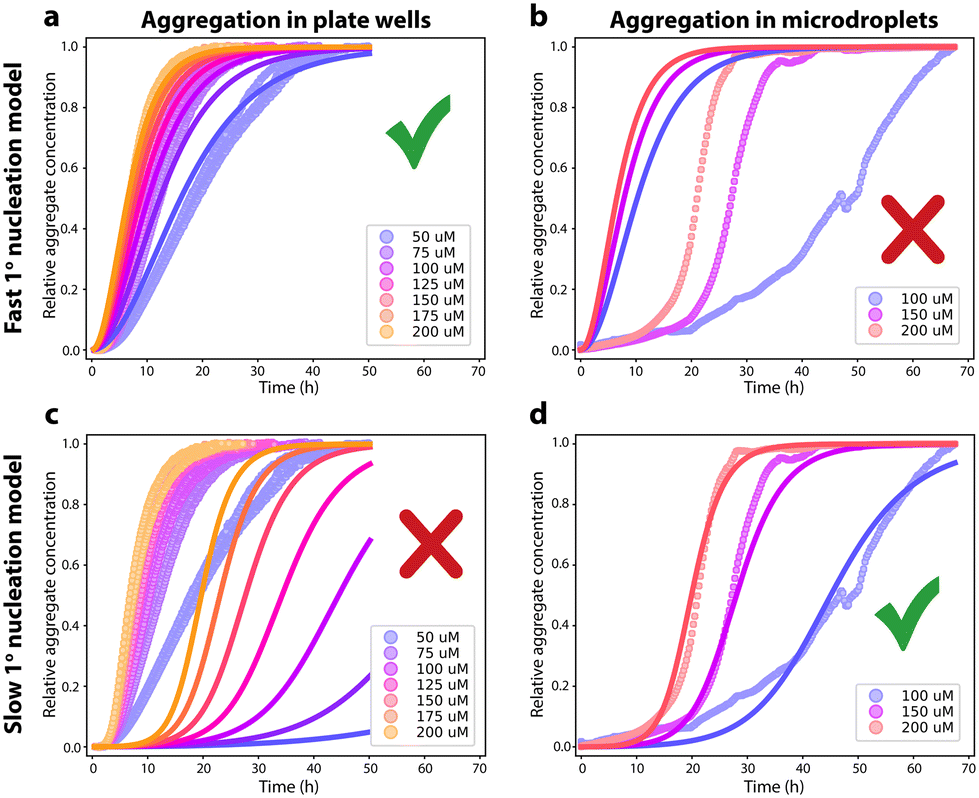 | ||
| Fig. 2 Aggregation in bulk in microtiter plate wells vs. aggregation in microdroplets without interfaces present. (a): Aggregation of FapC in microtiter plate at concentrations ranging from 50 to 200 μM at 37 °C. Triplicates are used for each concentration. The data is well-fitted by a nucleation–elongation kinetic model with nc = 1.33, knk+ = 1.34 × 103 M−nc h−2. (b): Aggregation of FapC in microdroplets in a water in oil emulsion at concentrations of 100, 150 and 200 μM at 37 °C. Kinetic traces were recorded from several thousand microdroplets. After removal of non-sigmoidal kinetic curves, this reduced to 500, 2255 and 2069 kinetic traces at 100, 150 and 200 μM; these were then averaged at each concentration. The nucleation–elongation kinetic model with rate parameters determined in (a) is overlaid, demonstrating a poor fit. (c): Same data as (a). The secondary nucleation kinetic model with rate parameters determined in (d) is shown overlaid, demonstrating a poor fit. (d): Same data as (b). The data is fitted to a secondary nucleation kinetic model with nc = 1.33, n2 = 1.72, knk+ = 4.14 M−nc h−2, k+k2 = 1.0 × 109 M−n2–1 h−2. | ||
Due also to the smoothness of a surfactant monolayer surface, we therefore expect heterogeneous primary nucleation to be greatly reduced in rate or even eliminated in these microdroplets. Indeed, the removal of the air–water and container wall interfaces with this strategy dramatically shown increased the reaction half-time (approx. 8 hours for 50 μM FapC in the plate compared to approx. 25 hours for 50 μM FapC in the microdroplets, Fig. 2). Moreover, although the data from the 96-well plate is well described by a primary nucleation and elongation model, Fig. 2A, the same kinetic model with identical kinetic parameters is not at all consistent with the data obtained in microdroplets, Fig. 2B. The kinetic data from the microdroplets can instead be fitted to a secondary nucleation dominated aggregation mechanism with n2 close to 2, Fig. 2D. In other words, elimination of the interfaces present in a 96-well plate alters the dominating aggregation mechanism of FapC. As expected, the primary nucleation rate is found to be greatly reduced (by several hundred-fold) upon removal of the air–water and plate wall interfaces, indicating that the main cause of primary nucleation in the 96-well plate is heterogeneous primary nucleation on these interfaces. Note the same secondary nucleation or fragmentation rate cannot be assumed in plate reader and in microdroplet experiments, because these rates are quite sensitive to the amount of agitation due to e.g. plate reader movements during reading.43 Such agitation also affects the primary nucleation rate,43 but this effect is small compared to the difference observed in the present study between aggregation in plate readers and in microdroplets.
Next, the identity of the specific interface causing the rapid primary nucleation of FapC in plate readers was investigated. The volume of the reaction mixture in each well in the 96 well plate was varied from 50 to 200 μL thus changing significantly the surface![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :volume ratio of air–water interface in the reaction mixture, Fig. 3A and ESI Fig. 6† (and that of the plate bottom-water interface, which is relevant if the plate base has a chemically different surface than the side walls). The geometry of the plate wells ensures that the plate wall surface:volume ratio changes relatively little by changing the fill height. No significant changes with volume were seen in the aggregation kinetics (Fig. 3A) and the kinetic data from each volume could be fitted to a nucleation–elongation model (Fig. S6†). Although the rate constants obtained from the kinetic fit to each volume data set were not identical, the knk+ values are very Similar (with a relative standard deviation of <20%) and no pattern can be seen in the variation of the knk+ rates, Fig. S6.† Next, we used light mineral oil to overlay the reaction mixture in the 96-well plate to replace the air–water interface, Fig. 3B. Other functional amyloids such as hydrophobins are known to be highly dependent on the air–water interface.24 The aggregation kinetics of FapC do not change when the air–water interface is no longer present. No changes are seen in the ThT curves in the presence and absence of mineral oil. The change in the aggregation mechanism seen in the microdroplets compared to the 96-well plates is hence not caused by removal of the air–water interface, indicating that the rapid heterogeneous primary nucleation in 96-well plates must occur at the plate walls.
:volume ratio of air–water interface in the reaction mixture, Fig. 3A and ESI Fig. 6† (and that of the plate bottom-water interface, which is relevant if the plate base has a chemically different surface than the side walls). The geometry of the plate wells ensures that the plate wall surface:volume ratio changes relatively little by changing the fill height. No significant changes with volume were seen in the aggregation kinetics (Fig. 3A) and the kinetic data from each volume could be fitted to a nucleation–elongation model (Fig. S6†). Although the rate constants obtained from the kinetic fit to each volume data set were not identical, the knk+ values are very Similar (with a relative standard deviation of <20%) and no pattern can be seen in the variation of the knk+ rates, Fig. S6.† Next, we used light mineral oil to overlay the reaction mixture in the 96-well plate to replace the air–water interface, Fig. 3B. Other functional amyloids such as hydrophobins are known to be highly dependent on the air–water interface.24 The aggregation kinetics of FapC do not change when the air–water interface is no longer present. No changes are seen in the ThT curves in the presence and absence of mineral oil. The change in the aggregation mechanism seen in the microdroplets compared to the 96-well plates is hence not caused by removal of the air–water interface, indicating that the rapid heterogeneous primary nucleation in 96-well plates must occur at the plate walls.
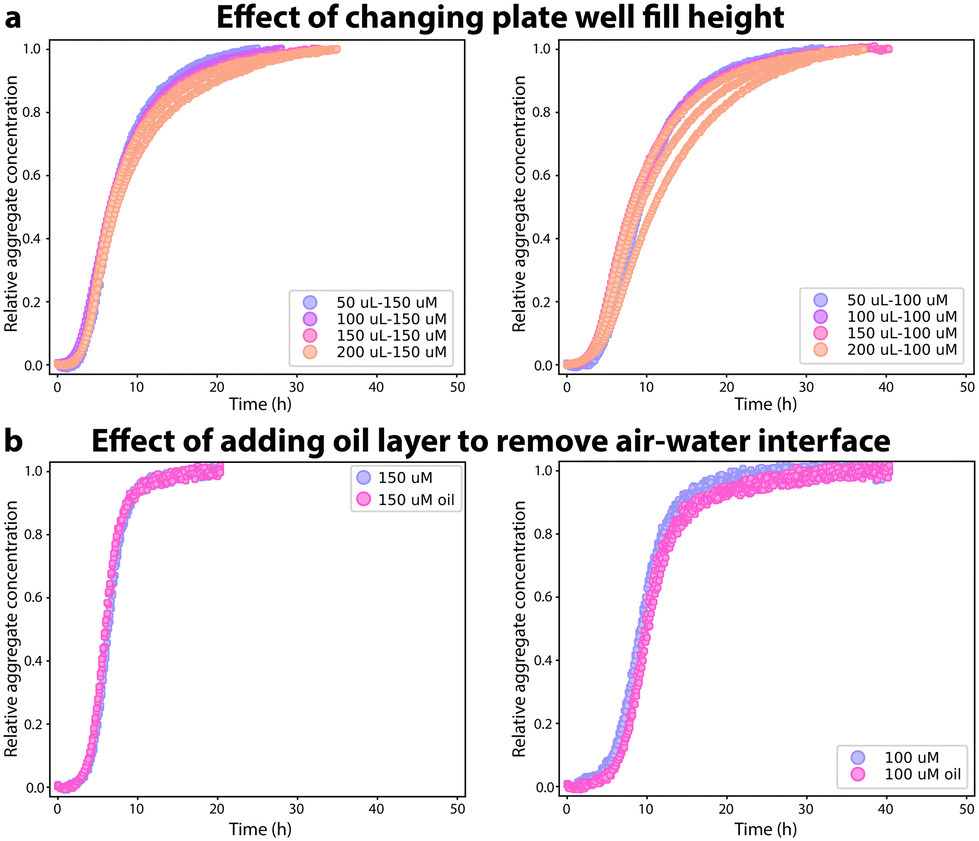 | ||
| Fig. 3 Investigating surface effects on aggregation of FapC (left column: 150 μM; right column: 100 μM). (a): Aggregation of FapC with volumes ranging from 50 μL to 200 μL loaded in the wells of a 96 well plate. (b): Aggregation of FapC in a 96-well plate with mineral oil on top to eliminate the air–water interface. | ||
Primary nucleation rate depends strongly on 96-well plate identity
We first explore the effect of the 96-well microtiter plate surface on the aggregation. The aggregation kinetics in all the different plates are faster than in microdroplets and can all be fitted to a nucleation–elongation dominated aggregation mechanism (Fig. 4), indicating that all surface chemistries tested promote primary nucleation significantly more than microdroplets. As argued in Methods, lower values of the reaction order nc indicate greater saturation of the catalytic site for primary nucleation when nc < 2, assuming the critical nucleus size does not drastically differ between different plates. This in turn implies stronger binding of monomer to the catalytic site. Since nucleation on solid surfaces occurs predominantly at topographical defects,45–47 this is not in general the same as the overall binding affinity of monomeric FapC to the plate surface, which may be affected in different ways by surface nanostructure. Relative FapC-catalytic site binding affinities in different plates can thus be inferred from comparing their nc values, but not generally from direct measurements of overall FapC-plate binding affinities.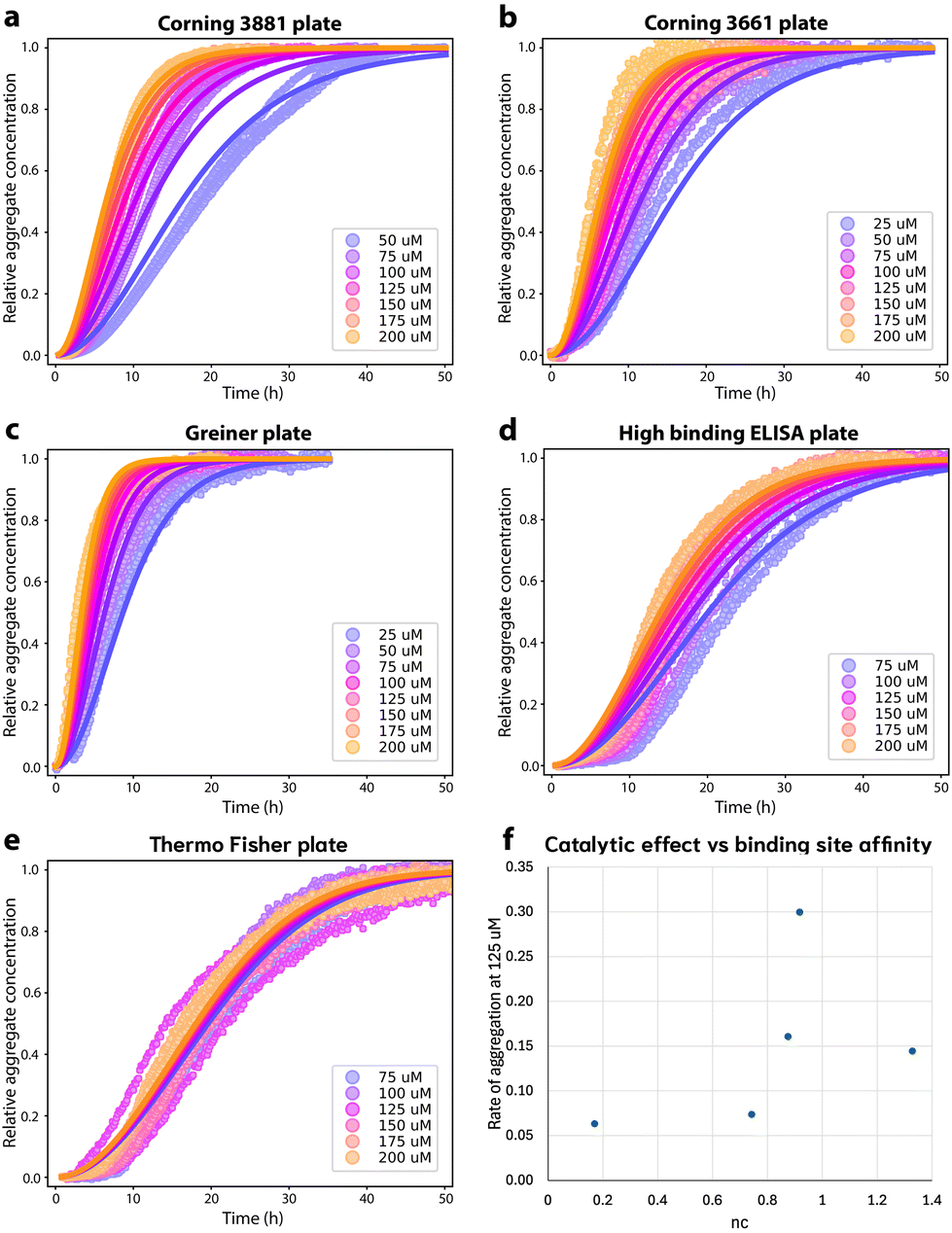 | ||
Fig. 4 Aggregation of FapC in different 96-well plates. (a): Aggregation of FapC in a standard Corning 3881 plate. The kinetic data is fitted to a nucleation–elongation model with nc = 1.33, knk+ = 1.34 × 103 M−nc h−2 and MRE = 0.00287. (b): Aggregation of FapC in a Corning 3661 plate. The kinetic data is fitted to a nucleation–elongation model with nc = 0.874, knk+ = 33.5 M−nc h−2 and MRE = 0.00254. (c): Aggregation of FapC in a Greiner plate. The kinetic data is fitted to a nucleation–elongation model with nc = 0.92, knk+ = 1.71 × 102 M−nc h−2 and MRE = 0.00133. (d): Aggregation of FapC in a high-binding ELISA plate. The kinetic data is fitted to a nucleation–elongation model with nc = 0.743, knk+ = 2.16 M−nc h−2 and MRE = 0.00349. (e): Aggregation of FapC in a Thermo Fisher 265301 plate. The kinetic data is fitted to a nucleation–elongation model with nc = 0.17, knk+ = 0.00924 M−nc h−2 and MRE = 0.00211. (f): Effective rate of aggregation 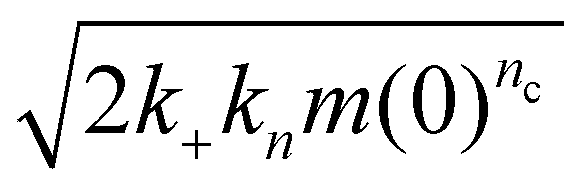 , see ref. 44 of FapC at 125 μM plotted against the nc obtained from global fitting of kinetic data from different plates. , see ref. 44 of FapC at 125 μM plotted against the nc obtained from global fitting of kinetic data from different plates. | ||
The standard plate used for FapC aggregation is the Corning 3881 polystyrene plate with surface treatment to create a nonionic hydrophilic surface to minimize molecular interactions, Fig. 4A. As might be expected, this plate has the highest nc (Table 1), indicating the lowest saturation of the catalytic site. The plate is polystyrene (PS) with a hydrophilic surface treatment (Table 2); however, the high-binding ELISA plate is also polystyrene with a hydrophilic surface treatment, and instead displays high catalytic site saturation (significantly lower nc). Presumably the hydrophilic treatments of these plates are quite different in either chemistry or nanostructure. Similarly, the 3 investigated plates with hydrophobic surfaces display a wide range of catalytic site saturations (Table 2). Overall, there is thus no apparent correlation between catalytic site binding affinity and the hydrophilic/hydrophobic nature of the plate surface alone. However, low and high binding surface treatments appear successful at modulating FapC binding strength to catalytic sites.
| Plate | Plastic type | Surface treatment | Surface nature |
|---|---|---|---|
| Corning 3881 | Polystyrene | + | Nonionic hydrophilic |
| Corning 3661 | Polystyrene | — | Hydrophobic |
| Greiner | Cyclic olefin copolymer (COC) | — | Hydrophobic |
| ELISA plate | Polystyrene | NA | Hydrophilic |
| Thermo Fisher | Polystyrene | — | Hydrophobic |
| Plate | n c | k n k + (M−nc h−2) | Representative m(0) (M) | Aggregation rate
|
|---|---|---|---|---|
| Corning 3881 | 1.33 | 1.34 × 103 | 1.25 × 10−4 | 1.4 × 10−1 |
| Corning 3661 | 0.874 | 33.5 | 1.25 × 10−4 | 1.6 × 10−1 |
| Greiner | 0.92 | 1.71 × 102 | 1.25 × 10−4 | 3.0 × 10−1 |
| ELISA plate | 0.743 | 2.16 | 1.25 × 10−4 | 7.4 × 10−2 |
| Thermo Fisher | 0.17 | 9.24 × 10−3 | 1.25 × 10−4 | 6.3 × 10−2 |
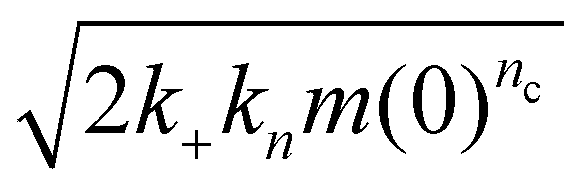
Aside from surface treatments, what correlations exist, if any, between those details of plate surface chemistry made public by the manufacturers and the degree of saturation of the catalytic site for FapC primary nucleation? Of the non-surface-treated plates, the Greiner plate, made of cyclic olefin copolymer (COC) displays lower saturation than the Corning 3661 and Thermo Fisher plates, which are made of polystyrene, suggesting that COC surfaces may interact less strongly with FapC than PS. However, despite the Corning 3661 and Thermo Fisher seemingly having identical surface chemistry to one another, the Thermo Fisher plate has higher catalytic site saturation. This implies that the Thermo Fisher plate may differ in surface nanostructure, resulting in catalytic sites whose geometry promotes stronger binding to FapC.
Surfaces catalyze the aggregation by binding reactants, intermediates and/or products, and providing an alternative pathway to nucleation of new fibrils with lower energy barriers. The strength of the catalytic effect of a surface primary nucleation site is expected to reach a peak at an intermediate binding strength of protein to this site. This peak represents a trade-off between the speed of the initial binding step of this alternative nucleation pathway, and the speed at which the products of nucleation can detach from the catalytic sites.29,48 From Table 2, we see that nucleation reaction order nc and overall reaction rate obey exactly the relationship expected if nc is indeed inversely proportional to the binding strength of FapC to catalytic sites on the plate surface. In other words, the effective aggregation rate 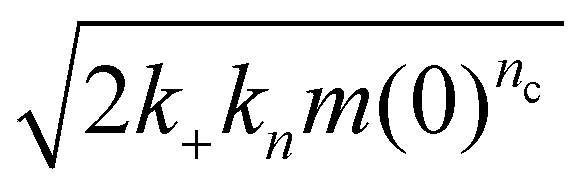 (ref. 44) initially increases to a maximum as nc decreases (Corning 3881 plate → Greiner plate), before subsequently decreasing with nc (→Corning 3661, ELISA plates → Thermo Fisher plate), Fig. 4F. This supports our earlier assumption that unsaturated reaction order n1 is not greatly different in different plates and lends strength to our argument that nc should be inversely proportional to the degree of saturation and thus the catalytic site binding affinity (see Methods).
(ref. 44) initially increases to a maximum as nc decreases (Corning 3881 plate → Greiner plate), before subsequently decreasing with nc (→Corning 3661, ELISA plates → Thermo Fisher plate), Fig. 4F. This supports our earlier assumption that unsaturated reaction order n1 is not greatly different in different plates and lends strength to our argument that nc should be inversely proportional to the degree of saturation and thus the catalytic site binding affinity (see Methods).
The high-binding ELISA plate and the Thermo Fisher plate show non-standard curve shapes especially at concentrations lower than those shown in Fig. 4, where the rate of fibril mass concentration growth changes discontinuously from zero to a large value at times >10 hours. This could conceivably be due to binding of aggregated protein to the plate surface, which could result in an initial lack of fluorescence increase, as the sides of the well are less visible to the plate reader. This would not be unexpected as these plates also display the strongest saturation (lowest nc). To test this hypothesis, we reproduce in Fig. S7† the data of Fig. 4D and E supplemented with additional lower monomer concentrations. We find that a provisional kinetic model that explicitly includes the binding of fibrils to plate surfaces gives markedly improved fits, demonstrating the plausibility of this proposed fibril sink in these plates. To positively confirm this mechanism, orthogonal experiments would in principle be needed; however, since this point is peripheral to the present study, we leave such experiments for future investigations.
Nanoparticles strongly catalyze FapC primary nucleation provided monomer binding is not too strong
The effect of differences in plate surface structure cannot be unambiguously separated from the effects of different surface chemistries by testing different types of plates. To help address this limitation, and to test additional surface chemistries, FapC was incubated in the presence of nanoparticles (NPs) with hydrophobic, hydrophilic positively charged or hydrophilic negatively charged surfaces, Fig. 5. In the presence of increasing amounts of hydrophobic NPs the aggregation of 50 μM FapC is accelerated, Fig. 5A. The aggregation data can still be well described by fitting to a nucleation–elongation model, both when varying NP concentration (Fig. 5A) and varying protein concentration, ESI Fig. 8,† further demonstrating that hydrophobic interfaces accelerate aggregation kinetics. Additionally, the parameter knk+ increases linearly with hydrophobic NP surface ratio. The constant y-intercept of this linear correlation gives the nucleation rate in the absence of NPs, i.e. on the plate interfaces. The rapid increase in nucleation rate confirms that most primary nucleation occurs on the NPs even at low NP concentrations, Fig. 5B. Note this slope is equivalent to linear proportionality; the log–log axes are chosen for convenience since the nanoparticle concentration spans several orders of magnitude.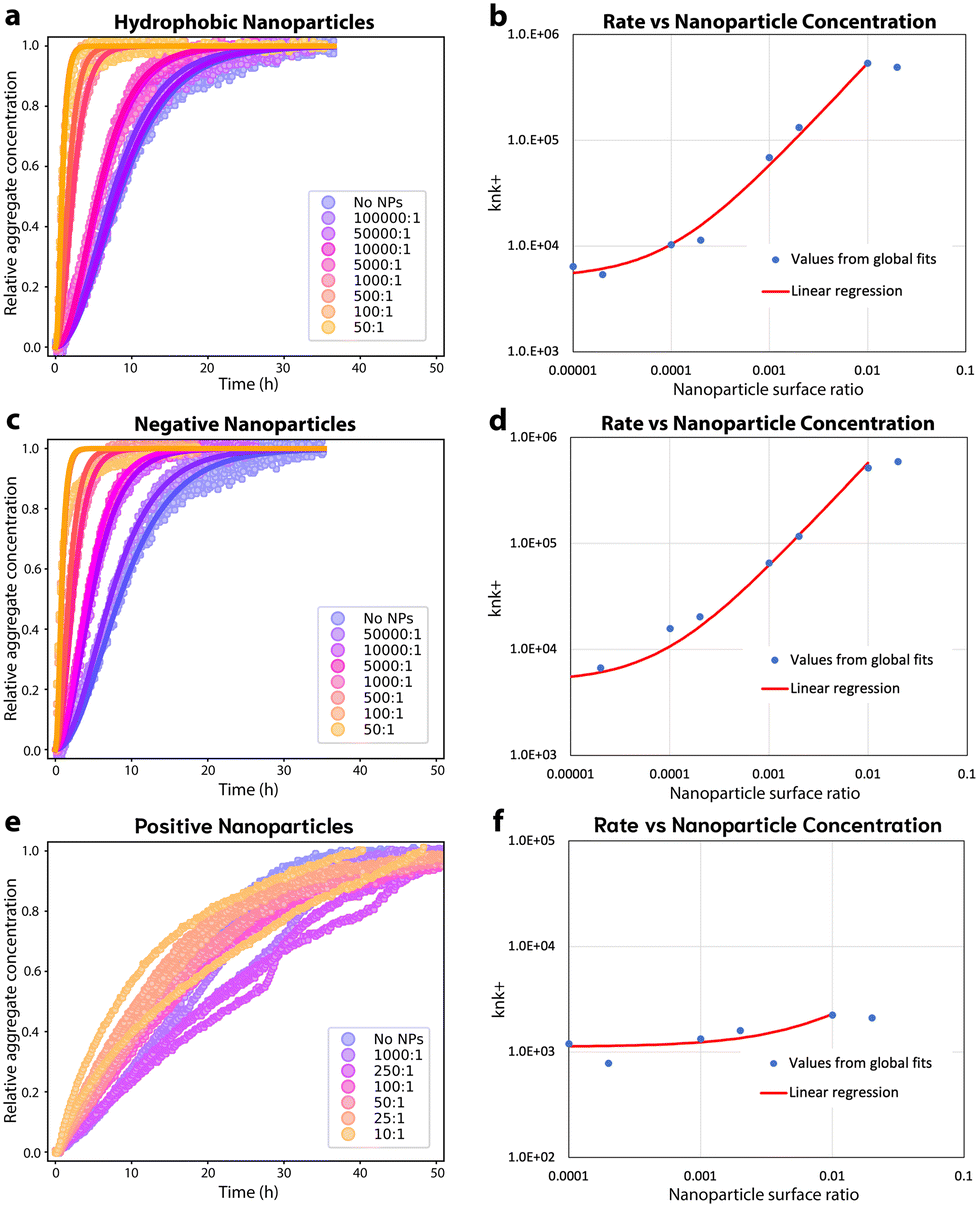 | ||
| Fig. 5 Aggregation kinetics of Fapc in the presence of NPs. (a): Aggregation of 50 μM FapC in the presence of hydrophobic NPs with different surface ratios compared to the surface ratio of FapC. The kinetic data is fitted to a nucleation–elongation model with nc = 1.33 as a global constant and knk+ varies for each concentration of NP, MRE = 0.00067. (b): The kinetic parameter knk+ from fitting 50 μM FapC in the presence of varying amounts of hydrophobic NPs plotted as a function of the surface ratio of the NPs compared to FapC on a log–log scale. The linear regression for surface ratios up to 0.01 is also plotted and exhibits curvature on logarithmic axes at lower surface ratios due to the constant term. (c): Aggregation of 50 μM FapC in the presence of negative NPs with different surface ratios compared to the surface ratio of FapC. The kinetic data is fitted to a nucleation–elongation model with nc = 1.33 as a global constant and knk+ varies for each concentration of NP, MRE = 0.00123. (d): The kinetic parameter knk+ from fitting 50 μM FapC in the presence of varying amounts of negative NPs plotted as a function of the surface ratio of the NPs compared to FapC on a log–log scale. (e): Aggregation of 50 μM FapC in the presence of positive NPs with different surface ratios compared to the surface ratio of FapC. The kinetic data is fitted to a nucleation–elongation model with nc = 1.33 as a global constant and knk+ varies for each concentration of NP, MRE = 0.00306. (f): The kinetic parameter knk+ from fitting 50 μM FapC in the presence of varying amounts of positive NPs plotted as a function of the surface ratio of the NPs compared to FapC on a log–log scale. No clear trend is seen. | ||
Similarly to hydrophobic NPs, the presence of negatively charged NPs accelerated FapC aggregation kinetics linearly with increasing NP amounts, Fig. 5C. The kinetic data in presence of the negatively charged NPs is well described by a nucleation–elongation model, both when varying NP concentration (Fig. 5C) and varying protein concentration, ESI Fig. 8.† Interestingly, the knk+ parameter displays a similar relationship to the surface ratio of negatively charged NPs as is seen for hydrophobic NPs, Fig. 5D. This implies a similar nanocatalytic effect on primary nucleation, despite the different NP surface properties. nc is reduced in the presence of both hydrophobic and negative NPs (ESI Fig. 8†), consistent with the surfaces of these NPs binding FapC more strongly than the plate walls.
The pH of 7 used in this study is close to the isoelectric point of FapC (pI = 7.25), rendering it zwitterionic, although carrying a small net positive charge. Thus, we expect it can bind to both positive and negatively charged surfaces but attain different bound conformations. Positively charged NPs clearly alter the aggregation kinetics of FapC but only at higher concentrations than used for the other NPs, Fig. 5E. At this point, the nucleation–elongation model no longer describes the data well. However, the overall rate of aggregation is not significantly accelerated or decelerated, and consequently no clear trend can be seen in the knk+ parameter observed at varying amounts of NPs, Fig. 5F. Fixed (and lower) amounts of positively charged NPs do not significantly alter the kinetics relative to no NPs, Fig. 5F and ESI Fig. 8.†
However, higher amounts of positively charged NPs appear to shift the aggregation kinetics to a first-order rate law (exponential decay of monomer concentration). This is more consistent with a coagulation or amorphous aggregation reaction than with a fibril formation reaction. Given the zwitterionic nature of FapC this could conceivably be electrostatically-driven.
We verified this hypothesis using TEM images of FapC aggregated in the presence of positive NPs, which showed the presence of amorphous aggregates surrounding and connecting the NPs, with few fibrils visible. By contrast, many fibrils are visible in TEM images of FapC aggregated in the presence of negative or hydrophobic NPs and for FapC aggregated in the absence of NPs (Fig. 6). The lack of acceleration in the kinetics even at low positive NP concentrations could be a consequence of FapC-NP binding strong enough to inhibit rather than catalyze primary nucleation, or of a bound FapC conformation not conducive to subsequent fibril formation. This is characteristic of amorphous or non-fibrillar aggregates, which can be viewed as a kinetically trapped intermediate state relative to the more thermodynamically stable fibrils.49
 | ||
| Fig. 6 TEM images of FapC in the absence and presence of NPs. Top panel: Left: 50 μM FapC aggregated in the absence of NPs. Middle: 50 μM FapC aggregated in the presence of high concentrations of negative NPs (500:1). Right: 50 μM FapC aggregated in the presence of high concentrations of hydrophobic NPs (100:1). Bottom panel: Left: 50 μM FapC aggregated in the presence of high concentrations of positive NPs (25:1). Middle: 50 μM FapC aggregated in the presence of high concentrations of positive NPs (25:1). Higher magnification. Amorphous aggregates indicated by arrows. Right: 50 μM FapC aggregated in the presence of low concentrations of positive NPs (1000:1). | ||
Discussion
The surfaces in multi-well plates where primary nucleation occurs in in vitro experiments are not usually biologically relevant. However, since the same types of plates are typically used, the relative rates of primary nucleation compared across different proteins may still retain some in vivo relevance. The primary nucleation rate of FapC is pretty typical for a known functional amyloid protein. Interestingly, at equivalent concentrations, in vitro primary nucleation rates differ little between amyloids that have been positively identified as functional or pathological, and even amyloids that do not aggregate at all in biological contexts (detailed rates across known functional and disease-associated amyloids can be found in Meisl et al.).8 This implies that primary nucleation propensity has little to do with the biological role of amyloids, or their role in disease.The rates of secondary processes have been previously compared across systems8 at in vivo-relevant concentrations tailored to each protein to identify its role in biology. Comparing rates at a single concentration (we here use 100 μM) instead gives us the innate propensity of these proteins to undergo self-replication. We find that disease-associated amyloid-forming proteins have on average a ten-fold higher rate under these circumstances than for amyloids not known to be associated with disease, implying that it is not just a matter of concentration which proteins end up associated with disease. Instead, pathological amyloids on average have an intrinsically greater propensity for secondary processes. The rate of FapC secondary nucleation is 2 orders of magnitude slower than the self-replication rate of a typical pathological amyloid-forming protein at 100 μM. However, its self-replication rate is not zero. Our results show that even functional amyloids not linked with diseases will exhibit secondary process-dominant kinetics, exactly like pathological amyloids, if the speed of other fibril formation pathways is reduced enough, for example by removing the interfaces promoting primary nucleation. Given their long, thin shape, all fibrils will fragment to some degree, and it is possible that this low intrinsic fragmentation rate is responsible for the self-replication seen when primary nucleation is slowed enough. However, the high reaction order of the secondary process observed in our data implies that the mechanism of self-replication of FapC in the droplets is in fact secondary nucleation rather than fragmentation. Remarkably, for many other proteins for which a detailed investigation has been conducted to determine the mechanism of self-replication in vitro, it has also been found to be secondary nucleation, rather than basic fragmentation.
The knowledge that surfaces are key in primary nucleation can also be used to speed up rather than slow down this process. The strong catalytic effect of NPs on primary nucleation20 may be especially useful in kickstarting aggregation in slowly nucleating systems such as tau,50,51 avoiding the need to use extreme reaction conditions. Indeed, the same NPs have been used to induce amyloid formation by human insulin under physiological conditions.28 Normally, very low pH and high temperatures are required for this protein to aggregate. By promoting primary nucleation sufficiently, the location of primary nucleation is moved from hard-to-control reaction vessel interfaces to the NPs themselves. In combination with mechanistic modelling and global fitting, this also affords the possibility of probing the mechanism of primary nucleation of other proteins in the future by tailoring the NP properties, and without the need to use multiple plate types.
It has previously been shown that for several amyloid-forming proteins that nucleate at the air–water interface or oil–water interface, the rate of nucleation first rises and then falls with the strength of binding to the interface (i.e. the adsorption free energy).29 We have shown that this also applies for FapC, that instead nucleates on the plate wall, provided the overall interface binding strength correlates strongly with the binding strength to the catalytic sites for primary nucleation that are located on the interface. We have further shown that the efficacy of nanocatalysts at promoting FapC primary nucleation is governed by the same principles, being a trade-off between their ability to promote reactant binding and their inability to release oligomeric or fibrillar products. Conversely, addition of nanoparticles has often been found to inhibit rather than promote aggregation.21,22,52,53 This can occur when a significant fraction of monomeric protein is adsorbed onto the surfaces of the nanoparticles, but the protein can aggregate only slowly on these surfaces. There is tentative evidence that this occurs in the present study with positive NPs, at concentrations low enough that flocculation does not occur. These principles must all be borne in mind when designing nanocatalysts for the primary nucleation of other fibrils or supramolecular structures from other kinds of monomeric protein. In fact, because excessively strong binding to nanoparticles can induce their flocculation, it is even more important to avoid this than with ordinary catalytic surfaces.
Author contributions
Study conception and design: A. D., G. M. and M. A.; data collection: C. G. T., S. N. S., Q. L., and M. A.; analysis and interpretation of results: A. D., G. M. and M. A.; visualization: A. D., G. M., and M. A.; resources: C. G. T., U. C. P., P. A., T. P. J. K., and M. A.; funding acquisition: P. A., T. P. J. K., and M. A.; writing – original draft: A. D., G. M., and M. A.; writing – review and editing: A. D., G. M., C. G. T., U. C. P., S. N. S., Q. L., P. A., T. P. J. K., and M. A.Data availability
The data presented in this study are openly available in FigShare. https://doi.org/10.6084/m9.figshare.26233268.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from The Danish Council for Independent Research | Natural Sciences (FNU-11-113326) (M. A.), Aarhus University Research Foundation (AUFF-E-2017-7-16) (M. A.). We would like to acknowledge funding from the European Research Council under the European Union's Horizon 2020 research and innovation program through the ERC grant DiProPhys (agreement ID 101001615) (G. M., T. P. J. K.). This work was funded by the Novo Nordisk Foundation (#NNF19OC0054635) (SL), and the Swedish Research Council, VR (2015-00143) (SL). This work was supported by the Frances and August Newman Foundation (C. G. T., T. P. J. K.).References
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef PubMed.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef PubMed.
- K. Schwartz, A. K. Syed, R. E. Stephenson, A. H. Rickard and B. R. Boles, PLoS Pathog., 2012, 8, e1002744 CrossRef.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. Cohen, C. M. Dobson, S. Linse and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef PubMed.
- C. Galvagnion, J. W. Brown, M. M. Ouberai, P. Flagmeier, M. Vendruscolo, A. K. Buell, E. Sparr and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7065–7070 CrossRef.
- P. Flagmeier, G. Meisl, M. Vendruscolo, T. P. Knowles, C. M. Dobson, A. K. Buell and C. Galvagnion, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10328–10333 CrossRef.
- G. Meisl, C. K. Xu, J. D. Taylor, T. C. T. Michaels, A. Levin, D. Otzen, D. Klenerman, S. Matthews, S. Linse, M. Andreasen and T. P. J. Knowles, Sci. Adv., 2022, 8, eabn6831 CrossRef PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef PubMed.
- S. I. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef.
- S. I. Cohen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, J. Mol. Biol., 2012, 421, 160–171 CrossRef PubMed.
- M. Andreasen, G. Meisl, J. D. Taylor, T. C. T. Michaels, A. Levin, D. E. Otzen, M. R. Chapman, C. M. Dobson, S. J. Matthews and T. P. J. Knowles, mBio, 2019, 10, 02279-18 CrossRef.
- M. Zaman and M. Andreasen, eLife, 2020, 9, e59776 CrossRef PubMed.
- M. M. Barnhart and M. R. Chapman, Annu. Rev. Microbiol., 2006, 60, 131–147 CrossRef PubMed.
- N. D. Hammer, J. C. Schmidt and M. R. Chapman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12494–12499 CrossRef.
- F. Grigolato and P. Arosio, Biophys. Chem., 2021, 270, 106533 CrossRef PubMed.
- A. J. Dear, X. Teng, S. R. Ball, J. Lewin, R. I. Horne, D. Clow, N. Harper, K. Yahya, T. C. T. Michaels, S. Linse, T. P. J. Knowles, X. Yang, S. C. Brewerton, J. Thomson, J. Habchi and G. Meisl, bioRxiv, 2023, DOI:10.1101/2023.10.20.563279.
- J. Pronchik, X. He, J. T. Giurleo and D. S. Talaga, J. Am. Chem. Soc., 2010, 132, 9797–9803 CrossRef.
- R. Vacha, S. Linse and M. Lund, J. Am. Chem. Soc., 2014, 136, 11776–11782 CrossRef PubMed.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef PubMed.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, K. A. Dawson and S. Linse, ACS Chem. Neurosci., 2010, 1, 279–287 CrossRef PubMed.
- C. Cabaleiro-Lago, O. Szczepankiewicz and S. Linse, Langmuir, 2012, 28, 1852–1857 CrossRef PubMed.
- H. M. Swasthi and S. Mukhopadhyay, J. Biol. Chem., 2017, 292, 19861–19872 CrossRef PubMed.
- C. L. Pham, A. Rey, V. Lo, M. Soules, Q. Ren, G. Meisl, T. P. Knowles, A. H. Kwan and M. Sunde, Sci. Rep., 2016, 6, 25288 CrossRef PubMed.
- S. Campioni, G. Carret, S. Jordens, L. Nicoud, R. Mezzenga and R. Riek, J. Am. Chem. Soc., 2014, 136, 2866–2875 CrossRef PubMed.
- M. A. Rubio, D. E. Schlamadinger, E. M. White and A. D. Miranker, Biochemistry, 2015, 54, 987–993 CrossRef.
- A. J. Dear, G. Meisl, T. C. T. Michaels, M. R. Zimmermann, S. Linse and T. P. J. Knowles, J. Chem. Phys., 2020, 152, 045101 CrossRef PubMed.
- F. Grigolato, C. Colombo, R. Ferrari, L. Rezabkova and P. Arosio, ACS Nano, 2017, 11, 11358–11367 CrossRef PubMed.
- Z. Toprakcioglu, A. Kamada, T. C. T. Michaels, M. Xie, J. Krausser, J. Wei, A. Saric, M. Vendruscolo and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2109718119 CrossRef.
- S. Auer and D. Frenkel, Phys. Rev. Lett., 2003, 91, 015703 CrossRef CAS PubMed.
- R. P. Sear, CrystEngComm, 2014, 16, 6506–6522 RSC.
- J. R. Espinosa, C. Vega, C. Valeriani, D. Frenkel and E. Sanz, Soft Matter, 2019, 15, 9625–9631 RSC.
- H. R. Pruppacher and J. D. Klett, Microphysics of clouds and precipitation: Reprinted 1980, Springer Science & Business Media, 2012 Search PubMed.
- V. Marghussian, Nano-glass ceramics: processing, properties and applications, William Andrew, 2015 Search PubMed.
- F. Oosawa and M. Kasai, J. Mol. Biol., 1962, 4, 10–21 CrossRef.
- F. Oosawa and S. Asakura, Thermodynamics of the Polymerization of Protein, Academic Press, London, 1975 Search PubMed.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. Hutchison, F. E. Angile, C. H. Schmitz, S. Köster, H. Duan, K. J. Humphry and R. Scanga, Lab Chip, 2008, 8, 1632–1639 RSC.
- M. Pfammatter, M. Andreasen, G. Meisl, C. G. Taylor, J. Adamcik, S. Bolisetty, A. Sánchez-Ferrer, D. Klenerman, C. M. Dobson and R. Mezzenga, Anal. Chem., 2017, 89, 12306–12313 CrossRef CAS.
- S. I. A. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Int. J. Mol. Sci., 2011, 12, 5844–5852 CrossRef CAS PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef CAS.
- T. P. Knowles, D. A. White, A. R. Abate, J. J. Agresti, S. I. Cohen, R. A. Sperling, E. J. De Genst, C. M. Dobson and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14746–14751 CrossRef CAS PubMed.
- E. Axell, J. Hu, M. Lindberg, A. J. Dear, L. Ortigosa-Pascual, E. A. Andrzejewska, G. Šneiderienė, D. Thacker, T. P. Knowles and E. Sparr, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2322572121 CrossRef.
- T. C. Michaels and T. P. Knowles, Am. J. Phys., 2014, 82, 476–483 CrossRef CAS.
- J. Dash, Phys. Rev. B: Solid State, 1977, 15, 3136 CrossRef CAS.
- J. Venables, L. Giordano and J. Harding, J. Phys.: Condens. Matter, 2006, 18, S411 CrossRef.
- J. Holbrough, J. Campbell, F. Meldrum and H. Christenson, Cryst. Growth Des., 2012, 12, 750–755 CrossRef.
- A. Šarić, A. K. Buell, G. Meisl, T. C. T. Michaels, C. M. Dobson, S. Linse, T. P. J. Knowles and D. Frenkel, Nat. Phys., 2016, 12, 874–880 Search PubMed.
- T. Miti, M. Mulaj, J. D. Schmit and M. Muschol, Biomacromolecules, 2015, 16, 326–335 CrossRef.
- D. C. Rodriguez Camargo, E. Sileikis, S. Chia, E. Axell, K. Bernfur, R. L. Cataldi, S. I. A. Cohen, G. Meisl, J. Habchi, T. P. J. Knowles, M. Vendruscolo and S. Linse, ACS Chem. Neurosci., 2021, 12, 4406–4415 CrossRef.
- S. Lövestam, D. Li, J. L. Wagstaff, A. Kotecha, D. Kimanius, S. H. McLaughlin, A. G. Murzin, S. M. V. Freund, M. Goedert and S. H. W. Scheres, Nature, 2024, 625, 119–125 CrossRef PubMed.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef PubMed.
- C. Cabaleiro-Lago, I. Lynch, K. A. Dawson and S. Linse, Langmuir, 2010, 26, 3453–3461 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01496k |
| This journal is © The Royal Society of Chemistry 2024 |