
Imaging of intracellular protein aggregates through plasmon-assisted clusteroluminescence†
Ashish Kumar
Dhillon
 a,
Pranay Eknath
Dudhe
b,
Shubhangi
Majumdar
a,
Sanmitra
Barman
c,
Dibyajyoti
Ghosh
*ad,
Karthigeyan
Dhanasekaran
*b and
Soumik
Siddhanta
*a
a,
Pranay Eknath
Dudhe
b,
Shubhangi
Majumdar
a,
Sanmitra
Barman
c,
Dibyajyoti
Ghosh
*ad,
Karthigeyan
Dhanasekaran
*b and
Soumik
Siddhanta
*a
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi-110016, India. E-mail: soumik@iitd.ac.in
bCentrosome and Cilia Laboratory, Regional Centre for Biotechnology, NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurugram Expressway, Faridabad, Haryana (NCR Delhi) 121001, India. E-mail: karthigeyan@rcb.res.in
cCenter for Advanced Materials and Devices (CAMD), BML Munjal University, Haryana, India
dDepartment of Materials Science and Engineering, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: dibyajyoti@iitd.ac.in
First published on 24th May 2024
Abstract
The formation of clusters in non-aromatic molecules can give rise to unconventional luminescence or clusteroluminescence. Typically containing heteroatoms without extended conjugation or aromatic rings, these molecules have drawn much attention owing to the prospects of label-free biological imaging. However, their applications have been limited due to the lack of knowledge of the underlying mechanism. Herein, we have elucidated the mechanism of clusteroluminescence from proteins, which were explicitly aggregated using plasmonic silver nanoparticles. The nanoparticles promoted protein aggregation and induced nitrile formation on the surface, which, along with other lone-pair-containing heteroatoms, contributed to enhanced emission in the visible range. Remarkably, this makes imaging of proteins possible with visible excitations, as co-factor-lacking proteins generally undergo electronic transitions only in the ultraviolet range. Furthermore, the inherent protein-aggregating behaviour of plasmonic nanoparticles was harnessed for imaging of intracellular Huntingtin protein aggregates overexpressed in HeLa cells through clusteroluminescence. Significant plasmon-enhanced and red-shifted fluorescence emission was observed, which helped in the imaging and localization of the intracellular aggregates. Density functional theory calculations and transient absorbance spectroscopy were used to probe the molecular interactions at the protein–nanoparticle interface and the charge transfer states, further elucidating the role of nanoparticles and the emission mechanism. This technique thus opens alternate avenues for label-free fluorescence bioimaging.
1. Introduction
Imaging of biological entities is crucial for understanding critical and complex diseases, and optical and spectroscopic modalities have played a central role. Fluorescence emission, light absorbance, vibrational scattering, and nuclear magnetic resonance provide a wealth of molecular information about biological systems.1–3 This molecular information helps in elucidating the composition, structure, and kinetics of biomolecules, which sheds light on the pathogenesis of diseases, specifically neurodegenerative ones. Optical techniques such as fluorescence or vibrational spectroscopy provide excellent spatial and temporal resolution of biological entities, even in their native conditions in the cells. However, exogenous labels or fluorescent tags and dye molecules often used in these techniques can perturb the aggregation kinetics and cause toxicity and aggregation-induced quenching or photobleaching.4–7 Due to these complexities in detecting and probing protein aggregates, there is an interest in label-free detection and imaging, which can be performed in real-time and under physiological conditions.3,8–10Intrinsic fluorescence or autofluorescence has been used for biological imaging, as reported in the literature. The intrinsic fluorescence from protein molecules arising from phenylalanine, tyrosine, and tryptophan has enabled the detection of conformational changes and ligand binding events.11,12 The recent demonstration that fluorescence can arise even from non-aromatic entities, specifically heteroatom-containing molecules, has opened up the area of unconventional fluorescence.13,14 This emission occurs in systems in an aggregated state, indicating through-space interactions. Being classified as clusteroluminogens, through-space n–n interactions and through-space n–π-interactions are found to be responsible for the emissions in non-pi conjugated systems.15 This unconventional fluorescence has been observed in macromolecules such as cellulose and proteins, which possess ‘n’ or separated non-conjugated ‘π’ electrons, respectively.16 A host of natural biological systems also have similar heteroatoms and exhibit unconventional emission properties.17 Therefore, cluster-triggered emission has the potential to be used in applications such as the imaging of biological systems. However, its utility appears to be severely affected by the requirement of a high concentration of molecules to form clusters and the low quantum yield of these entities.18 In this light, we hypothesize that such non-aromatic intrinsic emission can be coupled with metal-enhanced fluorescence for signal enhancement, as such strategies have already been adopted for fluorescence enhancement.11,19,20 The localized surface plasmon resonance of noble metal nanoparticles gives rise to intense electromagnetic intensity. It can enhance the low quantum yield and remove the requirement for higher analyte concentrations. Such a technique is harnessed by several analytical tools such as surface-enhanced Raman scattering (SERS),21–24 surface-enhanced infrared absorption, and metal-enhanced fluorescence.25 Through plasmon–protein conjugation, we have achieved the following targets in this work: (i) generating clusteroluminescence from protein–nanoparticle aggregates, (ii) red-shifting the fluorescence to separate the target molecule's fluorescence from the autofluorescence background, often seen in biological systems, (iii) enhancing the clusteroluminescence of biological samples through plasmonic attachment, and (iv) applying clusteroluminescence to directly observe protein aggregation inside complex biological systems such as cells.
Fluorescence enhancement was achieved by harnessing through-space interactions from the inherently present heteroatoms within the aggregated protein–nanoparticle conjugates. The catalytic formation of nitrile on the nanoparticle surface and the restricted intramolecular motion due to the self-assembly-driven aggregation on the surface of the plasmonics contribute to the improved emission quantum yield of the biomolecules.26 The fluorescence of the biomolecules was red-shifted due to the enhanced intra- and inter-molecular through-space interactions. The Stark effect type of charged interaction within the aggregates also contributes to this shift. First-principles-based calculations showed the dominant interaction of nitrogen-containing groups of proteins with the nanoparticle surface. The relatively weak but evident charge accumulation on the nitrogen atom highlights the through-space intermolecular electronic coupling, which results in fluorescence emission. The charge transfer state in such a scenario with longer lifetimes has been demonstrated through transient absorbance spectroscopy studies. The overall mechanism of clusteroluminescence is illustrated in Fig. 1.
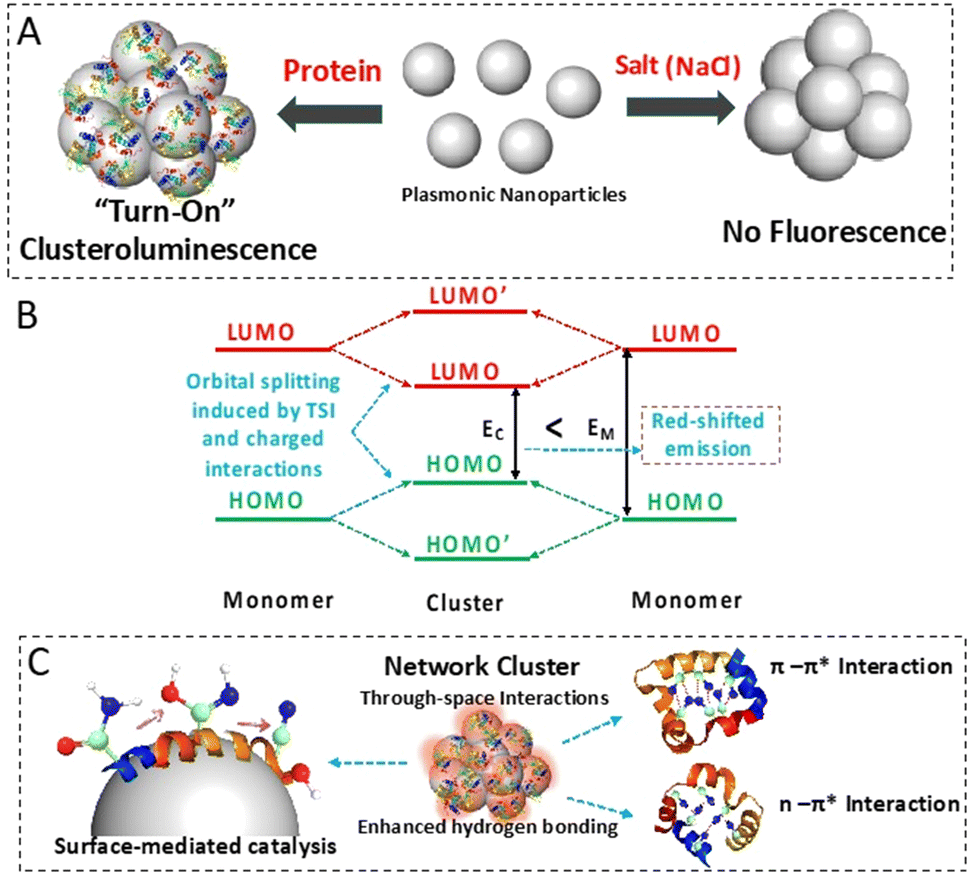 | ||
| Fig. 1 Schematic diagram showing the overall workflow. (A) “Turn-on” clusteroluminescence was observed when protein molecules were aggregated using colloidal AgNPs. Similar fluorescence emission was absent when the AgNPs were aggregated using a salt solution. (B) The electronic transitions responsible for red-shifted emission due to through-space and charged interactions. (C) The formation of Ag surface species such as nitrile groups (carbon, nitrogen and oxygen atoms in the ball and stick model are colored in green, blue and red, respectively) results in through-space intermolecular electronic coupling (TSI) leading to the clusteroluminescence phenomenon. The enhanced hydrogen bonding in the protein–AgNP aggregate and the TSI through the π–π* and n–π* interactions also contribute to this phenomenon. | ||
Herein, for the first time, we utilize such metal-nanoparticle-enhanced clusteroluminescence to image protein aggregates inside the cells without using any exogenous fluorophores. This phenomenon is shown by silver nanoparticles (AgNPs) which were able to induce visible or red emission from aggregation-prone Huntingtin proteins in cells in a label-free manner.27 This study highlights the role of non-functionalized nanoparticles as they have been found to promote protein aggregation in aqueous solution and, when exposed to live cells, can do the same. Such behaviour has been observed in the case of the aggregation of α-synuclein.28 Gold nanoparticles decorated with polyglutamine-binding amphiphilic peptides have been used to target polyQ-rich mutant Huntingtin proteins.29 Interestingly, we observed a similar trend when the AgNPs were incubated with HeLa cells overexpressing the Huntingtin protein. The flip from a significantly quenched fluorescence state to a highly enhanced state in the event of aggregation provides an excellent contrast ratio. It also acts as an indicator of the aggregation itself. These properties make these aggregation-induced fluorescence techniques ideal for sensing and tracing applications.26,30
2. Experimental section
2.1. Chemicals
Silver nitrate (99.99% purity), lysozyme from chicken egg white (crystalline powder, 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg), sodium chloride (>99.0% purity), Hoechst dye, sodium citrate dihydrate (≥99.5% purity), protamine sulfate, 4-nitrothiophenol (4-NTP), thiophenol (TP), and paraformaldehyde (PFA), all with 99% purity, were purchased from Sigma Aldrich. Fetal bovine serum (RM9955) was purchased from Himedia, rabbit polyclonal anti-HA antibody (1:500; BB-AB0050) was sourced from Bio Bharati Life Sciences, goat anti-rabbit Alexa Fluor 647 antibody (1:2000; A21245) was sourced from Invitrogen, and Dulbecco's Modified Eagle Medium (DMEM) and penicillin–streptomycin were purchased from Gibco. Plasmid (pHM6-Q74) was received as a gift from David Rubinsztein (Addgene plasmid # 40264). Other reagents and solvents were of analytical grade and used as such without purification. MilliQ water was used for all the experiments.
000 units per mg), sodium chloride (>99.0% purity), Hoechst dye, sodium citrate dihydrate (≥99.5% purity), protamine sulfate, 4-nitrothiophenol (4-NTP), thiophenol (TP), and paraformaldehyde (PFA), all with 99% purity, were purchased from Sigma Aldrich. Fetal bovine serum (RM9955) was purchased from Himedia, rabbit polyclonal anti-HA antibody (1:500; BB-AB0050) was sourced from Bio Bharati Life Sciences, goat anti-rabbit Alexa Fluor 647 antibody (1:2000; A21245) was sourced from Invitrogen, and Dulbecco's Modified Eagle Medium (DMEM) and penicillin–streptomycin were purchased from Gibco. Plasmid (pHM6-Q74) was received as a gift from David Rubinsztein (Addgene plasmid # 40264). Other reagents and solvents were of analytical grade and used as such without purification. MilliQ water was used for all the experiments.
2.2. Silver nanoparticle synthesis
The Lee–Meisel method was used to synthesize colloidal AgNPs.31 In 100 mL of MilliQ water, 18.0 mg of silver nitrate was dissolved. 1% sodium citrate (2 mL) was added to the boiling solution. The mixture was heated with stirring for 1 h and then cooled to room temperature while stirring. The product obtained was a greenish-yellow colloidal fluid. Then, the obtained AgNPs were characterized using several techniques. A Shimadzu UV-2450 spectrometer was used to perform the UV-visible spectroscopic measurement to study the lysozyme–AgNP interactions using a quartz cuvette with a 1 cm path length. Photoluminescence (PL) spectra were obtained from the same solution using a QuantaMaster 8450-22 spectrofluorometer (Horiba) with excitation and emission slit widths of 3 nm and 3 nm, respectively, while the attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra were recorded on an Agilent Cary 600 Series (Cary 660) FTIR spectrometer equipped with cryogenically cooled InSb detectors. A Tecnai TF20 electron microscope with an accelerating voltage of 200 kV was used for high-resolution transmission electron microscopy (HRTEM) imaging of the prepared AgNPs and the size distribution was investigated by dynamic light scattering (DLS) using a Zetasizer from Malvern Panalytical.2.3. Surface-enhanced Raman studies
The SERS spectra were recorded using an Xplora Raman spectrometer from Horiba Jobin Yvon containing two excitation lasers of wavelengths 532 nm and 785 nm and a Peltier-cooled CCD. The SERS spectra were recorded with 1200 grooves per mm of holographic grating with a 100 μm slit width and a 10× objective. A typical spectral acquisition time was 20 s at room temperature. The Raman spectra were recorded at least three times to check the reproducibility, and the Raman peak at 520.7 cm−1 of the silicon wafer was used to calibrate the instrument.32 The post-processing of the spectra was performed using OriginLab Pro 2020b, including background subtraction and smoothening using a 5-point FFT algorithm.2.4. Computational methods
We used density functional theory (DFT)-based simulations as implemented in the Vienna ab initio simulation package for all the computational simulations.33,34 All the calculations employed the projected augmented wave method and a plane-wave basis set with a cut-off of 400 eV.35 The generalized gradient approximation with the Perdew–Burke–Ernzerhof functional form was used for approximating the exchange and correlation interactions.36 The geometry optimization ensured that the interatomic forces are less than 0.01 eV Å−1. For these structural relaxation simulations, we employed a 4 × 4 × 1 Γ-centered k-point mesh for Brillouin zone sampling. To model the noncovalent interactions, we considered the dispersion correction as prescribed by Grimme et al. in their DFT-D3 scheme.37 For self-consistent field (SCF) calculations, a finer Γ-centered k-point grid of 6 × 6 × 1 was used. The Gaussian smearing of 0.01 eV was considered for all SCF calculations. To model AgNP–molecule interactions, we constructed an AgNP slab with an exposed (111) surface. The AgNP slab contained four layers of metal (64 atoms) with a 4 × 4 supercell in the ab-plane. To introduce vacuum in the nonperiodic c direction, we increased the cell parameter by 20 Å in that direction. Such a model suppresses the spurious interaction between periodic images. The molecules were adsorbed on top of the (111) plane of the AgNPs to achieve AgNP layer–molecule interactions. A similar computational model has been recently employed by some of us to model the AgNP–alloy surface.38 To model the optimized geometry and total electronic energy of isolated molecules, we kept those in the middle of the three-dimensional simulation box used for AgNP–molecule simulations. For these simulations, we used a single 1 × 1 × 1 Γ-point and retained all other parameters the same as before.2.5. Transient absorption spectroscopy
Ultrafast transient absorption measurements were conducted using a commercial Ti:sapphire amplified laser system (Astrella 1K-F, 100 fs, 5 mJ per pulse, and 1 kHz repitition rate, Coherent Inc.). The amplified pulses with 5 mJ per pulse energy were obtained after seeding with 20 fs and 100 nm bandwidth laser pulses obtained from an oscillator (Vitara-S) operating at an 80 MHz repetition rate. The amplified output of the central wavelength of 800 nm was divided into two parts. One part was used to produce the femtosecond probe pulse by focusing it on a CaF2 and 2 mm thick sapphire crystal by generating a white light continuum (310–800 nm), and the other part was used to generate a tunable femtosecond pump pulse using an optical parametric amplifier (Coherent OPerA Solo ultrafast optical parametric amplifier system). After the sample, the probe beam was collimated and then focused into a fiber-coupled spectrometer with CCD sensors and detected at a frequency of 1 kHz. The power of the pump pulse used in the experiment was controlled by a variable neutral-density filter wheel and was kept at 1 mJ per pulse. The delay between the pump and probe pulses was controlled by a motorized delay stage. The pump pulses were chopped by a synchronized chopper at 500 Hz and the absorbance change was calculated with two time-adjacent probe pulses (pump-blocked and pump-unblocked).The samples soluble in solvent were taken in a 2 mm quartz cuvette and stirred constantly using a magnetic stirrer during the measurements. For transient measurements, 400 nm was used as the pump wavelength, and a sapphire crystal was used for visible range white light as the probe. Concentrations of the freshly prepared samples were adjusted to maintain an optical density in the range of 0.02–0.04 in a 2 mm quartz cuvette. To check for charring or any damage in the sample during transient absorption measurements, ground state absorption spectra were recorded before and after laser exposure. Surface Xplorer version 4.5 was used to analyze the transient absorption spectra.
2.6. In cellulo AgNP labelling for cellular imaging
HeLa cells were maintained in DMEM supplemented with 10% fetal bovine serum and 1× penicillin–streptomycin in a humidified incubator at 37 °C with 5% CO2. For the imaging experiments, 2 × 104 cells were seeded 12 h prior to transfection onto 35 mm dishes containing 10 mm coverslips. Upon achieving 30% confluency, these cells were transfected with 2 μg of the plasmid (pHM6-Q74) and allowed to express the aggregation prone hemagglutinin (HA)-tagged Huntingtin (HA-HTT) protein for the next 48 h before harvesting. The transfected population without the AgNPs was used as a control while the un-transfected cells were used as a negative control. The cells were treated with 60 μL of 0.5 nM AgNP suspension made in 0.25 mM trehalose 3 h prior to collection.39 In the end, cells on coverslips were fixed with 4% PFA at room temperature for 15 min and then subsequently washed thrice with 1× PBS. These fixed cells were further labelled to visualize the HTT aggregates by immunofluorescence staining and fluorescence microscopy using a Zeiss LSM880 confocal microscope. Z-Stack images (0.2 μm step size) were acquired under a 63× magnification oil immersion objective lens with a numerical aperture of 1.4. The AgNPs were excited with a 488 nm laser and the emission between 550 and 700 nm was acquired using HyD detectors. Similarly, 647 and 405 nm lasers were used for HA-HTT and DNA visualization, respectively, in a sequential manner. The acquired images were visualized and analyzed using ImageJ. Furthermore, Pearson's coefficient representing the colocalization status for the AgNP clusters and HTT aggregates was estimated using 30 cells showing HTT aggregation. The mean and standard deviation (SD) were calculated for the same from three independent biological repeats and represented using the GraphPad Prism software.2.7. Immunofluorescence assay
Briefly, cells grown on glass coverslips were fixed with 2% PFA for 10 min, permeabilized with 0.3% Tween20 for 20 min and then blocked with 5% bovine serum albumin for 1 h at room temperature. The coverslips were then incubated with a rabbit polyclonal anti-HA antibody for 1 h, followed by incubation with goat anti-rabbit Alexa Fluor 647 antibody. DNA was stained using Hoechst dye for 5 min and mounted using 70% glycerol.2.8. Cell viability assay
HeLa cells were seeded in 35 mm dishes at a density of 0.3 × 106 and cultured overnight. At 70–80% confluency, each dish was treated with different concentrations of AgNPs, i.e. 0, 5, 10, 30, 50 and 100 μL with equal volumes of 0.5 M trehalose solution to achieve the final concentrations of 0, 0.08, 0.16, 0.5, 1.6, and 3.2 nM, respectively, and incubated for 24 h. After which the cells were trypsinized and resuspended in serum-free media. One part of the cell suspension, i.e. 10 μL, was mixed with one part of 0.4% trypan blue and allowed to incubate for ∼1 min at room temperature and the cells were counted, unstained (viable) and stained (nonviable) separately in a haemocytometer.3. Results and discussion
The main aim of this study was to comprehensively probe the mechanism behind the intensified, red-shifted fluorescence observed in protein–nanoparticle conjugates. This distinctive optical trait was targeted for its potential in the imaging of a neurodegenerative disease. The underlying mechanism responsible for the emergence of “turn-on” far-red fluorescence within non-fluorescent protein–nanoparticle aggregates was investigated by combining spectroscopic techniques and quantum theoretical calculations.3.1. Protein–nanoparticle interactions and the characterization of the aggregates
The size of the AgNPs employed in the experiments was 60 nm, with a localized surface plasmon resonance (LSPR) peak at 410 nm. The size of the nanoparticles was confirmed by TEM (Fig. S1†). For an in-depth exploration of surface interactions of proteins at the molecular level, lysozyme was used as an appropriate model protein due to its well-established characterization and several biologically significant applications. Of significance, lysozyme, a globular protein, has demonstrated interactions with nanomaterials that lead to amyloid-type aggregates.40 The LSPR was obtained through UV-visible spectroscopy both in the presence and absence of the protein. The positive charge on the lysozyme surface at physiological pH is due to its isoelectric point around 11, which facilitates attachment with the negatively charged AgNPs. It has been shown previously that a dynamic equilibrium exists between the adsorbed protein molecules on the nanoparticle surface, and the point of interaction is not random but through specific domains on the protein.41 When lysozyme was added to the AgNPs, a slight change in the location of the LSPR band was observed, which was attributed to the change in the refractive index surrounding the nanoparticles (Fig. 2(A and B)). Interestingly, when the nanoparticles were incubated with different protein concentrations, a drastic change in the longitudinal surface plasmon resonance peak in the UV-visible spectrum was observed (Fig. 2(A)). The transverse plasmon band of the AgNPs was significantly diminished, accompanied by an increased longitudinal (tail) band intensity. Such observation is typical of the formation of protein–nanoparticle aggregates as reported by Halas et al.42 Since the protein molecules undergo partial denaturation on the surface of the nanoparticles, the favourably interacting domains come together and form amyloid-type aggregates, thus pulling the nanoparticles together. This results in the loss of the dipolar plasmon peak as the particles are no longer spherical and aggregated together to form differently sized nanoparticle clusters. This gives rise to the increasing intensity of multipolar LSPR modes at a higher wavelength (around 800 nm), and the heterogeneity of the system makes the peak much broader.43 The change in the LSPR profile indicates that the maximum aggregation occurs at a concentration of 10−5 M of protein incubated with 0.5 nM of AgNPs. Furthermore, the change in the protein structure due to surface-induced aggregation was observed from the changes in the intensity of the characteristic peaks of lysozyme in the UV-visible spectrum at around 260–280 nm (Fig. 2(A)). These peaks emerge due to the lateral chains of aromatic residues like Tyr, Trp, Phe, and Cys. The changes in the protein structure could also be observed through the deconvolution of the amide I band of the SERS spectra of the protein–nanoparticle conjugates (Fig. 2(C) and (D)). The protein exhibits alterations in its conformational state, specifically in the proportion of its helical secondary structure following the aggregation process. Consequently, a reduction of 6% in helical conformation was observed as compared to the native protein.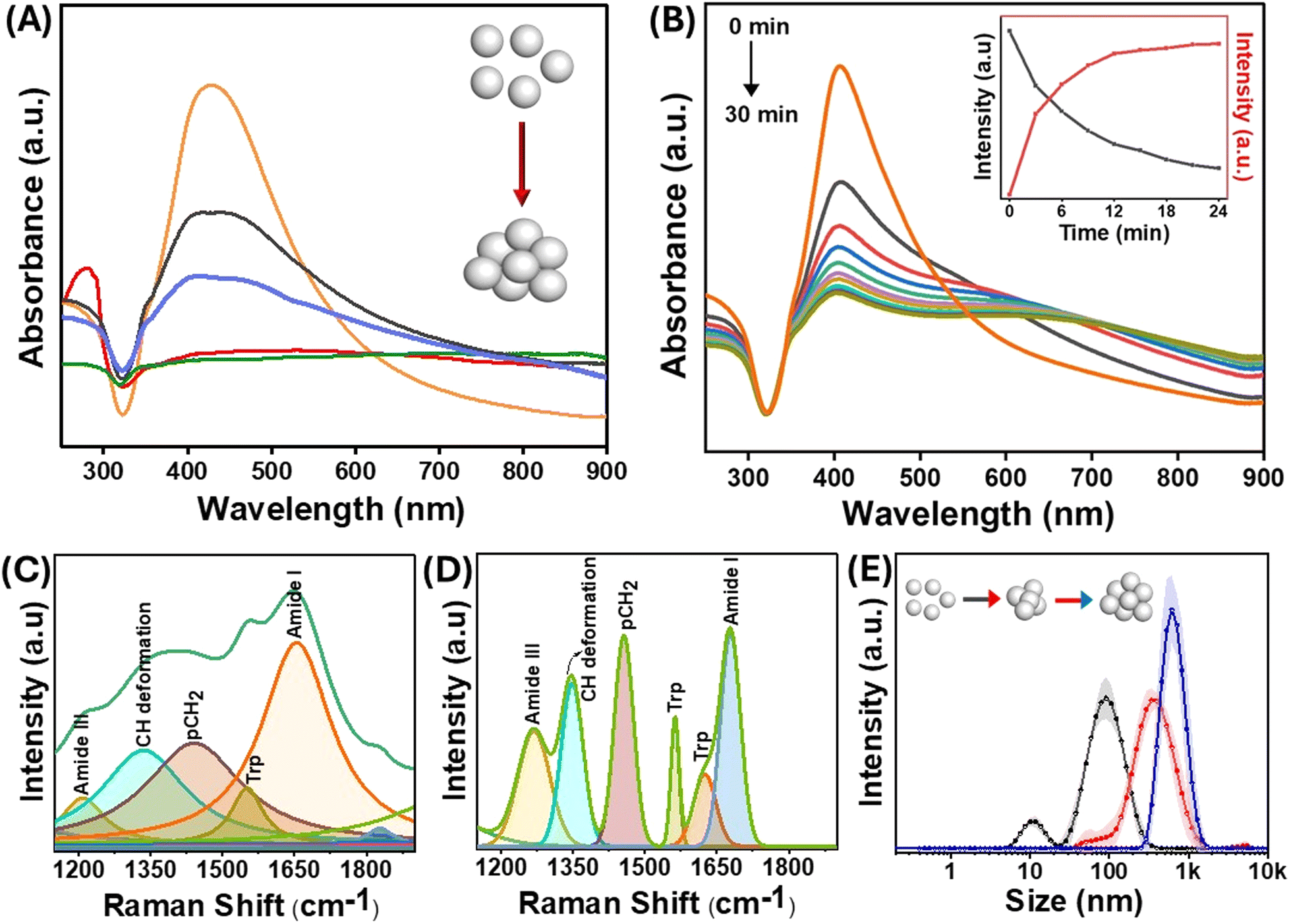 | ||
| Fig. 2 (A) UV-visible spectra of the lysozyme–AgNP complex with different concentrations of protein [10−3 M (red), 10−5 M (green), 10−7 M (purple), 10−9 M (black), and AgNP (orange)] incubated for 30 min. (B) Time-dependent UV-visible spectra from 0 to 30 min. The inset shows the variation of dipolar plasmonic peak intensity (black) and longitudinal tail intensity (red) with time. Deconvoluted SERS spectra of (C) the lysozyme–AgNP complex and (D) pure lysozyme showing a change in the secondary structure of proteins. (E) Dynamic light scattering spectra of AgNPs (black), salt aggregated AgNPs (red), and protein aggregates (blue) showing the different size distributions of the aggregates, while the shaded part of the line corresponds to the standard deviation. A protein concentration of 10−5 M was used in the lysozyme–AgNP conjugates used in (B–E). | ||
In addition to the appearance of multipolar surface plasmon modes, the time-dependent UV-visible spectrum in Fig. 2(B) and Fig. S2† shows a blue shift in the transverse surface plasmon mode of the AgNPs with the increase in the incubation time. This blue shift in the absorption spectrum of the AgNPs can be due to events such as leaching or charge transfer at the protein–nanoparticle interface.44 The addition of lysozyme resulted in a slight increase in the hydrodynamic diameter of bare nanoparticles, which was directly observed by the DLS technique. The formation of large aggregates of the AgNPs as a function of time was evident from the evolving hydrodynamic radius of nanoparticles incubated with lysozyme (Fig. 2(E)).
Since charge-based interactions are critical for aggregate formation, we have also investigated the effect of ionic species such as salts. Fig. 2(E) compares the sizes of salt- and protein-induced nanoparticle aggregates. The size distribution plots from the DLS data revealed that the lysozyme-catalyzed aggregation produced tightly packed and well-ordered aggregates. The analysis of the full-width at half-maximum of these peaks showed that the polydispersity of the salt-aggregated AgNPs was more pronounced than that formed using lysozyme. These observations indicate the possible existence of an additional interaction mechanism between the protein and the nanoparticles, specifically at the interface, which is discussed in the subsequent sections.
3.2. The emergence of far-red fluorescence from the protein–nanoparticle aggregates
SERS, a predominantly surface-sensitive technique, was used to explore the precise mechanism of interactions between proteins and nanoparticles. SERS has been recently used to probe minute structural changes in the binding of proteins to the surface of nanoparticles.45–47 The vibrational features of aromatic amino acids Phe, Tyr, and Trp, as well as amide and protein secondary structural characteristics, dominate the SERS spectrum of lysozyme, which is consistent with previous research.48,49 It is worth noting that the position and amplitude of the protein's modes are influenced by its orientation to the nanoparticle surface, which is determined by surface selection criteria.50 Because the electromagnetic field augmentation decays with the third power of the circumferential distance from the surface, vibrational modes near the surface are enhanced more than those further away.51 Interestingly, as the aggregation of the protein–nanoparticle conjugate increases with time, the fluorescence background seen from SERS also increases, indicating the emergence of “turn-on” fluorescence in the system (Fig. 3(A) and S3†). | ||
| Fig. 3 SERS spectra (A) lysozyme aggregated AgNPs at different time points and (B) salt aggregated AgNPs with 4-NTP at different time points. The SERS spectra were collected using a 532 nm laser source. (C) Confocal fluorescence images of (I) AgNPs, (II) lysozyme, and (III) AgNPs incubated with lysozyme (10−5 M) obtained with a laser excitation source of 543 nm. (D) Time-dependent fluorescence intensity variation calculated from the confocal fluorescence images. The clusteroluminescence intensity of the AgNPs is quantified and represented as a histogram plot showing the mean fluorescence intensity in arbitrary units (au). Error bars represent standard deviations of 6 individual data sets under the same conditions. | ||
The evolution of protein-induced nanoparticle aggregation with time and the corresponding fluorescence emission were monitored using confocal fluorescence microscopy [Fig. 3]. An increase in the fluorescence intensity with incubation time was observed, as aggregation is a time-dependent phenomenon.42 The extent of aggregation could be directly correlated with the increase in fluorescence emission intensity. Confocal microscopy also showed this red-shifted fluorescence, aligning with the substantial fluorescence background noted earlier in the SERS of the same samples. In Fig. 3(C)-(i), it could be seen that the confocal fluorescence microscopy image of the pure and fresh lysozyme solution obtained with 543 nm laser excitation had no fluorescence emission. Similarly, no red fluorescence was observed in the case of bare AgNPs [Fig. 3(C)-(ii)]. However, when the nanoparticles were aggregated using lysozyme, they exhibited strong red fluorescence [Fig. 3(C)-(iii)]. The red emission was observed in the range of 645–680 nm. Contrasting results were obtained when the same AgNPs were aggregated with sodium chloride, and no fluorescence emission could be observed.
Interestingly, small molecules such as 4-NTP also displayed fluorescence emission, as confirmed by the Raman and fluorescence data. However, the confocal fluorescence image of the aggregated nanoparticles in the presence of TP, where nitrogen and oxygen atoms are absent, showed no fluorescence emission (Fig. S4†), which essentially highlights the role of functional groups containing these atoms in fluorescence emission in the red region. This unique emission has been observed previously in a few reports and also in the case of clusteroluminescence.52,53 Even non-conjugated systems possessing lone pair-containing atoms also have enhanced red-shifted emissions.54,55 Therefore, biomolecules containing a high percentage of nitrogen-containing functional groups show clusteroluminescence and cluster-dependent biological activities.18,56 In the subsequent sections, we investigated this unique emission through various spectroscopic techniques.
3.3. Spectroscopic analysis of the origin of red emission from the nanoparticle–protein conjugate
Absorbance and emission spectroscopy provided significant insights into the photophysics of the system. Protein–nanoparticle aggregates of different sizes were created using different nanoparticle-to-protein ratios, and their average hydrodynamic radius was obtained by DLS. The emission spectra of these different aggregates were found to be red-shifted with the increase in size (Fig. 4(A)). Unlike conventional fluorescence, the emission wavelengths varied when the same aggregate was excited by different excitation wavelengths (Fig. 4(B)). This excitation-dependent luminescence is a unique feature of clusteroluminescence. According to Kasha's rule, luminescence happens from the lowest excited state, and the emission wavelength is constant for a particular molecule with a definite structure, due to which the change in excitation will only result in a change in the emission intensity but not the emission wavelength.57 However, in the case of clusteroluminescence, the electron delocalization differs in the clustered state, resulting from the different energy gaps of the emitters. These results confirm the emergence of clusteroluminescence from the protein–nanoparticle aggregates. | ||
| Fig. 4 (A) PL spectra of the AgNP–lysozyme complex with different concentrations of lysozyme. (B) Laser excitation-dependent PL spectra of AgNP aggregates in the presence of lysozyme. | ||
Since the fluorescence in proteins emerges from aromatic amino acids such as Trp, Tyr, His, and Phe, the initial hypothesis was that proteins bereft of these amino acids would not show such red-shifted fluorescence. However, a small positively charged protein molecule, protamine sulfate, which lacks amino acids, also produced considerable fluorescence background on aggregation (Fig. S5(A)†). The SERS background fluorescence from protamine was higher than that from 4-NTP (Fig. 3(B)) and TP (Fig. S5(B)†), where the nanoparticle–molecule conjugates were aggregated using salt solutions. These observations point toward specific surface chemistry of nanoparticles, giving rise to this “turn-on” fluorescence. The interaction of the protein with the nanoparticles was further studied through ATR–FTIR, which provided information on molecular-scale site-specific interactions. The ATR spectrum from the nanoparticle–protein conjugate showed new peaks in the 2200–2400 cm−1 range. Fig. 5(A) shows the infrared spectra of lysozyme incubated with AgNPs at different durations. The newly emergent peak at 2330 cm−1 can be assigned to the nitrile (CN) triple bond stretching mode, as reported earlier.58 As the concentration of the nitrile group increases with the incubation time, the intensity of this peak increases. Therefore, it can be concluded that some of the amine or amide bonds were successfully converted to the nitrile group. The nitrile group absorbs at 2250–2270 cm−1 and is well-separated from naturally occurring vibrational modes in proteins. The local environment might vary dramatically within a protein (the bond in question might be in proximity to or buried in the protein's hydrophobic core), which causes the absorption peak of the nitrile group to shift to around 2330 cm−1. The amine or amide group can be converted to the nitrile group through various mechanisms, such as the silver-catalyzed dehydration of the amide bond or the silver-catalyzed oxidation to the nitrile group.59 As the aggregation increases, the reactive sites captured between the aggregates are locked at a specific position, making the conversion more probable. Therefore, the silver atom catalyzes the formation of the nitrile group by acting as the electron acceptor from the nitrogen atom in the peptide chain, as confirmed by the charge transfer quantum calculation between the Ag surface and the binding moieties. As reported in earlier studies, nitrile group clusters have been strongly linked to the hyperbranched-enhanced-emission effect due to the through-space conjugation.60 These moieties or sub-fluorophores, such as –NH–, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and nitrile, are generally composed of electron-rich heteroatoms and exhibit unusual fluorescence due to the formation of a pendent group cluster. This clusterization-mediated restricted movement of the nitrile groups triggers the emission caused by non-radiative relaxation and electron delocalization by through-space conjugation. We envisage that the oxygen-rich moieties in the protein molecules, specifically the formation of a silver-catalyzed nitrile group on the surface of the AgNPs, give rise to the clusteroluminescence in the nanoparticle aggregates.
O, and nitrile, are generally composed of electron-rich heteroatoms and exhibit unusual fluorescence due to the formation of a pendent group cluster. This clusterization-mediated restricted movement of the nitrile groups triggers the emission caused by non-radiative relaxation and electron delocalization by through-space conjugation. We envisage that the oxygen-rich moieties in the protein molecules, specifically the formation of a silver-catalyzed nitrile group on the surface of the AgNPs, give rise to the clusteroluminescence in the nanoparticle aggregates.
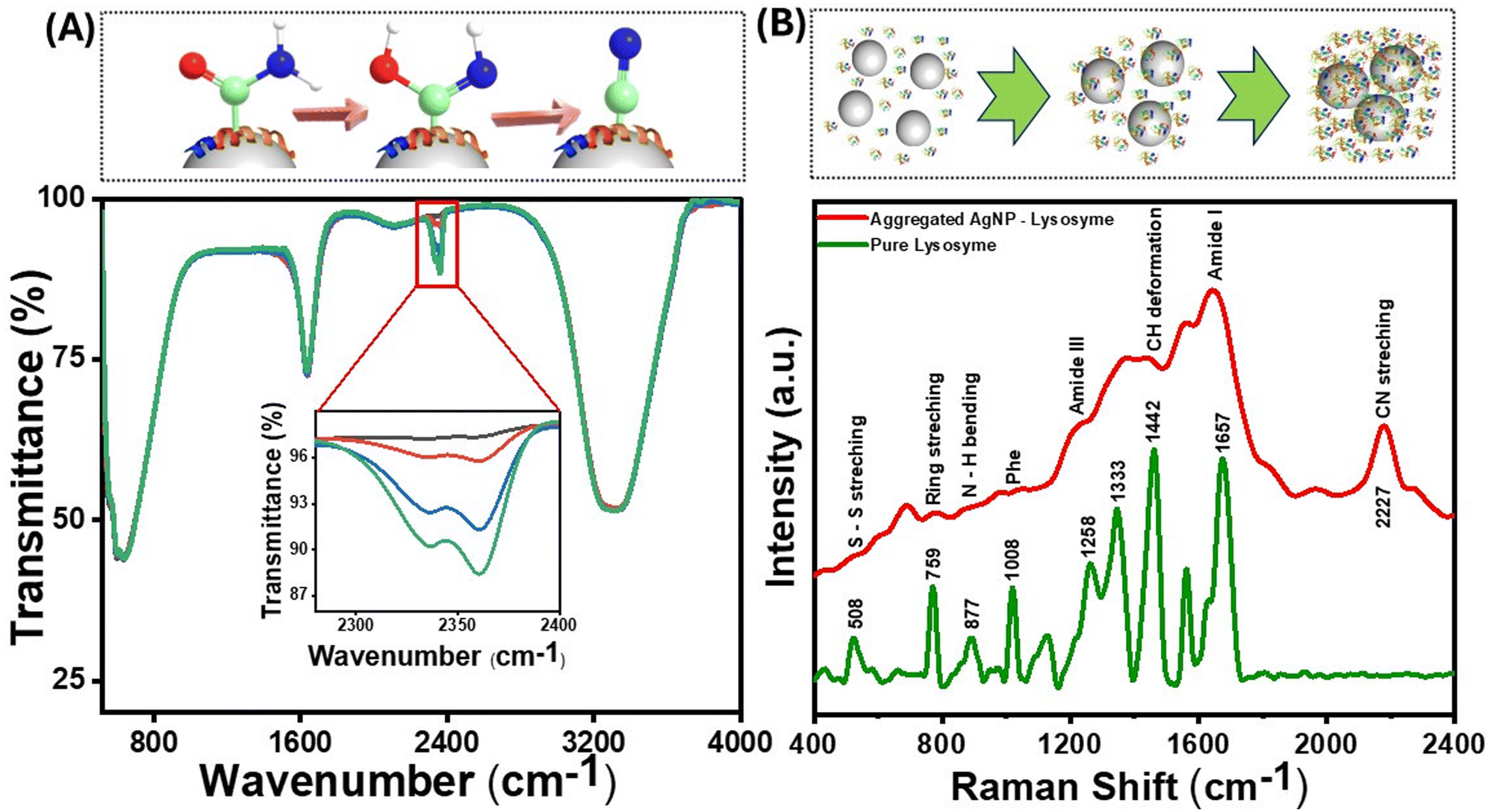 | ||
| Fig. 5 (A) (Top) Pictorial representation of the conversion of amide to nitrile. The carbon, nitrogen and oxygen atoms are shown in light green, blue and red respectively. The ribbon representation depicts the adsorption of the protein and the AgNP-mediated catalysis happening on the AgNP surface (shown as grey spheres). (Bottom) ATR-FTIR spectra of the lysozyme–AgNP complex containing 10−3 M (green), 10−5 M (blue), 10−7 M (red), and 10−9 M (black) lysozyme concentrations (the inset shows the zoomed view from 2250 to 2400 cm−1). (B) (Top) Schematic of the nanoparticle-induced formation of protein aggregates. (Bottom) Raman spectra of lysozyme (green) and the lysozyme–AgNP complex (red) obtained using a 532 nm laser source. | ||
The emergence of a nitrile group can also be seen from the SERS spectra of lysozyme (Fig. 5(B)) and protamine sulfate (Fig. S5(A)†), along with a significant fluorescence background when excited with a 532 nm laser source. The SERS band at around 1442 cm−1 is due to the methylene group, and the band corresponding to 877 cm−1 is assigned to Trp. This band belongs explicitly to the N–H site of the indole ring on aggregation inside the Trp moiety and is sensitive to environmental changes.51 The peak at 233 cm−1 is assigned to the Ag–N bond, which explains the binding site to the AgNPs. The nitrile peak at 2227 cm−1 increased with the nanoparticle incubation time and the consequent increase in aggregation. However, this peak was absent in the SERS spectra of TP, further highlighting the surface-catalyzed conversion of the nitrogen-containing functional groups to the nitrile group and its role in inducing red emission due to the clusteroluminescence.61
The significant red shift of the emission can be attributed to the interaction of the dipole moment of the luminogens with the electrostatic charges within the protein–nanoparticle aggregate.62 This interaction can be compared to the Stark effect, where the dipole moment is renormalized. The altered dipole can change the energy gap between the highest occupied molecular orbitals and the lowest unoccupied molecular orbitals, causing the observed red shift during emission.20 The red emission can also be attributed to the coexistence of intrachain and interchain through-space interactions of the luminogens in the aggregated protein molecules. The aggregated state also promotes abundant intramolecular and intermolecular hydrogen-bond interactions, which impart stability to n⋯n (N⋯O) and n⋯π (N⋯CO) through-space type of interaction. Electrons from peptides can be delocalized through intramolecular or intermolecular hydrogen bond formation and have been previously found to have electronic transitions in longer wavelength regions, resulting in a red shift in the emission spectrum.63
3.4. Protein–nanoparticle charge transfer probed through quantum calculations and ultrafast spectroscopy
To understand the atomistic details of interactions between the AgNP surface and different building units of protein, we performed thorough first-principles-based computational investigations. We initially optimized the ground state of the adsorbed molecules on the AgNP surface at 0 K. The (111) surface of AgNPs has been considered for exploring molecular interactions with these metal nanoparticles. Fig. 6(A) and Table 1 include the optimized geometries and detailed structural parameters.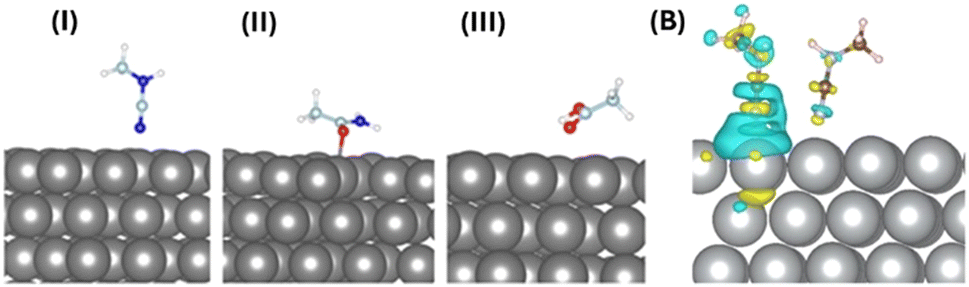 | ||
| Fig. 6 (A) The optimized structures of molecular moieties on the AgNP (111) surface. Surfaces with (i) NCNHCH3, (ii) OC(NH2)CH3, and (iii) OHC(O)CH3 are presented. The non-covalent interactions between Ag atoms and anchoring atoms (C/N) give rise to physiosorbed molecules of the surface. Key: AgNPs (gray), C (cyan), N (blue), O (red), and H (white). (B) The charge transfer between the single CH3NCN and CH3NCN–AgNP (111) surfaces. The yellow and blue iso-surfaces indicate charge depletion and accumulation, respectively. The charge transfer plot shows intermolecular as well as molecule–AgNP (111) surface electronic coupling. | ||
| Molecules | Binding energy (meV per atom) | Charge transfer (e−) | Ag–N/O distance (Å) |
|---|---|---|---|
| NCNHCH3 | −28.3 | +0.05 (molecules to AgNPs) | 2.29 |
| OHCOCH3 | −27.0 | +0.05 (AgNPs to molecules) | 2.86 |
| NHCHCH3 | −31.1 | +0.05 (molecules to AgNPs) | 2.57 |
The molecular moieties in the protein are expected to interact with the (111) surface of AgNPs while forming aggregates. The anchoring atoms (O and N) of the molecules remain placed on top of the AgNP surface. The non-aromatic molecular moieties with different anchoring groups exhibit physisorption and shorter AgNP-X (X = anchoring atom) distances (2.29–2.86 Å), as shown in Table 1. The calculated binding energies of the different protein moieties narrowly range from −27 to −31 meV per atom, indicating weak but stable nanoparticle–protein interactions under ambient conditions.
The intermolecular interactions between adjacent fluorescent CH3NCN molecules on the (111) surface of AgNPs were studied computationally. It should be noted that we are only considering nitrogen-containing fluorescent moieties for all further studies. The modelled supercell of the AgNP surface contains two CH3NCN molecules that are spatially close to each other (Fig. 6(B)). The binding energy calculations in the presence and absence of the (111) surface of AgNPs depict that these molecules remain stacked only on the metallic surface. The positive binding energy of 0.11 eV between pairs of molecules in the gas phase indicates that AgNPs play a crucial role in aggregating these molecules by initiating intermolecular electronic coupling. The aggregation of molecules on the (111) surface of AgNPs further suggests their limited thermal motion, which can enhance the emission properties of the overall system by suppressing non-radiative recombination processes. As tabulated in Table 1, the Bader charge calculation depicts the direction of charge transfer between the binding entities that closely depend on the anchoring groups’ chemical nature. The interacting nitrogen atoms donate electrons to the AgNP surface, whereas moieties with oxygen as the binding site accept electrons from the metal surface.
The charge density difference was also studied to understand the electronic coupling between protein moieties and AgNP surfaces. The plotted charge density difference reveals that the non-covalent interaction between the AgNP (111) surface and the nitrogen of CH3NCN is the dominant kind of electronic coupling present in these systems, as shown in Fig. 6(B). Moreover, relatively weak but evident charge accumulation on the nitrogen atom of the NCN group away from the AgNP surface in one of the molecules depicts the through-space intermolecular electronic coupling.15 The lone pair of anchoring nitrogen in CH3NCN donates the electron cloud to the AgNP surface atoms, depicting metal surface–molecule electronic coupling. Thus, the overall charge transfer and electronic coupling between protein and AgNPs can be tuned by modifying anchoring group chemistry.
The charge transfer processes that AgNPs experience when interacting with proteins were probed through transient absorption spectroscopy. AgNPs (Fig. 7(A)) exhibited prominent excited state absorption with a maximum at about 470 nm, showing no probe dependency on the decay kinetics. A faster interband transition to the ground state occurred within ∼6.5 ps of the same probe pulse at 470 nm in the presence of AgNPs (Fig. 7(B)). On the other hand, AgNPs exhibit intriguing probe-dependent dynamics in the presence of protein (∼10 μM) (Fig. 7(C)), which depicts the formation of a photoproduct state (525–650 nm) at the compensation of AgNP decay followed by its progressive ground state transition. Proteins are found to delay the electron–hole recombination time in AgNPs up to ∼237 ps (at 470 nm), which is the probable signature of charge transfer from the nanoparticle to the protein (Fig. 7(D)). The growth component of 33 ps further confirms the formation of a new photoproduct with an absorption at around 600 nm, which decays down within 305 ps (Fig. 7(E)). Therefore, these ultrafast measurements further confirm the electron transfer between the nitrogen and oxygen atoms of the proteins and the AgNP surface.
 | ||
| Fig. 7 Transient absorption spectra of (A) AgNPs and (B) AgNPs with protein and the principal wavelength decay kinetics of (C) AgNPs at 470 nm and AgNPs with protein probing at (D) 470 nm and (E) 600 nm. | ||
3.5. Utilizing clusteroluminescence for imaging Huntingtin protein aggregates in cells
Clusteroluminescence-based cellular imaging of Huntingtin protein aggregates overexpressed in HeLa cells was performed during AgNP incubation. Upon overexpressing the human influenza HA-HTT partial exon (1-Q74) in HeLa cells for 48 h, the protein aggregates were labelled in the presence or absence of AgNP (0.5 nM) suspension for the final 3 h prior to harvesting (Fig. 8A). Untransfected cells were used as a negative control, and similarly, the overexpressing population without the AgNP labelling was used as an additional negative control to trace the protein aggregates. Interestingly, our observations revealed that only the cells marked with AgNPs exhibited positive clusteroluminescence. Moreover, the cells expressing the aggregation-prone species, which were colocalized with the HTT clusters, were detected using an anti-HA antibody tagged with Alexa Fluor under excitations at 488 and 647 nm respectively (Fig. 8B). As the untransfected population failed to form these protein aggregates, no clusteroluminescence was observed in these negative control cells as expected, and no signal was seen in either of the control cells. The colocalization was also confirmed through the visualization of the aggregates along with the nucleus through Hoechst dye staining. The Z-stack image of the HTT aggregates with AgNPs showing clusteroluminescence clearly shows their localization within the cells (as shown in Movies M1 and M2†). Thus, the surface of plasmonic AgNPs enables a complex interplay of various forces between the nanoparticles and proteins and the chemical reactions catalyzed by the constituent materials, as shown in the earlier sections. This study is also consistent with earlier reports of metallic gold nanoparticles, which have been found to interact with proteins and interfere with amyloid formation. Proteins have a high affinity to adsorb onto nanosurfaces, and the interaction is driven by entropic and enthalpic factors such as redistribution of amino acids, dehydration at the interface, and conformational changes in the protein molecule itself. Notably, foreign entities in cells or physiological systems often trigger nucleation on the surface, promoting accelerated aggregation and amyloid formation.64 The nanoparticles also attach to oligomeric and aggregated species, particularly soft and flexible proteins and aggregates, which have longer residence times on the nanoparticle surface. Furthermore, trypan blue viability assays demonstrated that concentrations exceeding 0.5 nM exhibited heightened toxicity, as shown in Fig. S6(B).† Furthermore, extending the duration of AgNP labelling up to 24 h did not yield a significant increase in clusteroluminescent signals, as evidenced by Fig. S6(A) and S6(C).† The observed mean Pearson's coefficient (R = 0.5802) between AgNP and HA-HTT signals in green and magenta underscores a robust positive correlation between clusteroluminescence emanating from AgNPs and their specific binding to HTT protein aggregate clusters within cells. The direct observation of AgNP aggregation with protein molecules again corroborates the specificity of the AgNPs to exhibit “turn-on” clusteroluminescence when they are strictly in a bound form with the protein aggregates, which is the Huntingtin protein in this case. The plasmonic nanoparticles and the protein–nanoparticle aggregate ensure (1) higher emission intensity from the clusteroluminogens on the nanoparticle surface and (2) red-shifting of the fluorescence to differentiate from autofluorescence and increase the detection selectivity. Since the emission depends on the excitation wavelength, interfering optical windows can be selectively avoided. We anticipate that “turn-on” clusteroluminescence would provide a simple, robust, and functional method for molecular imaging and increase the accuracy of diagnosing debilitating diseases.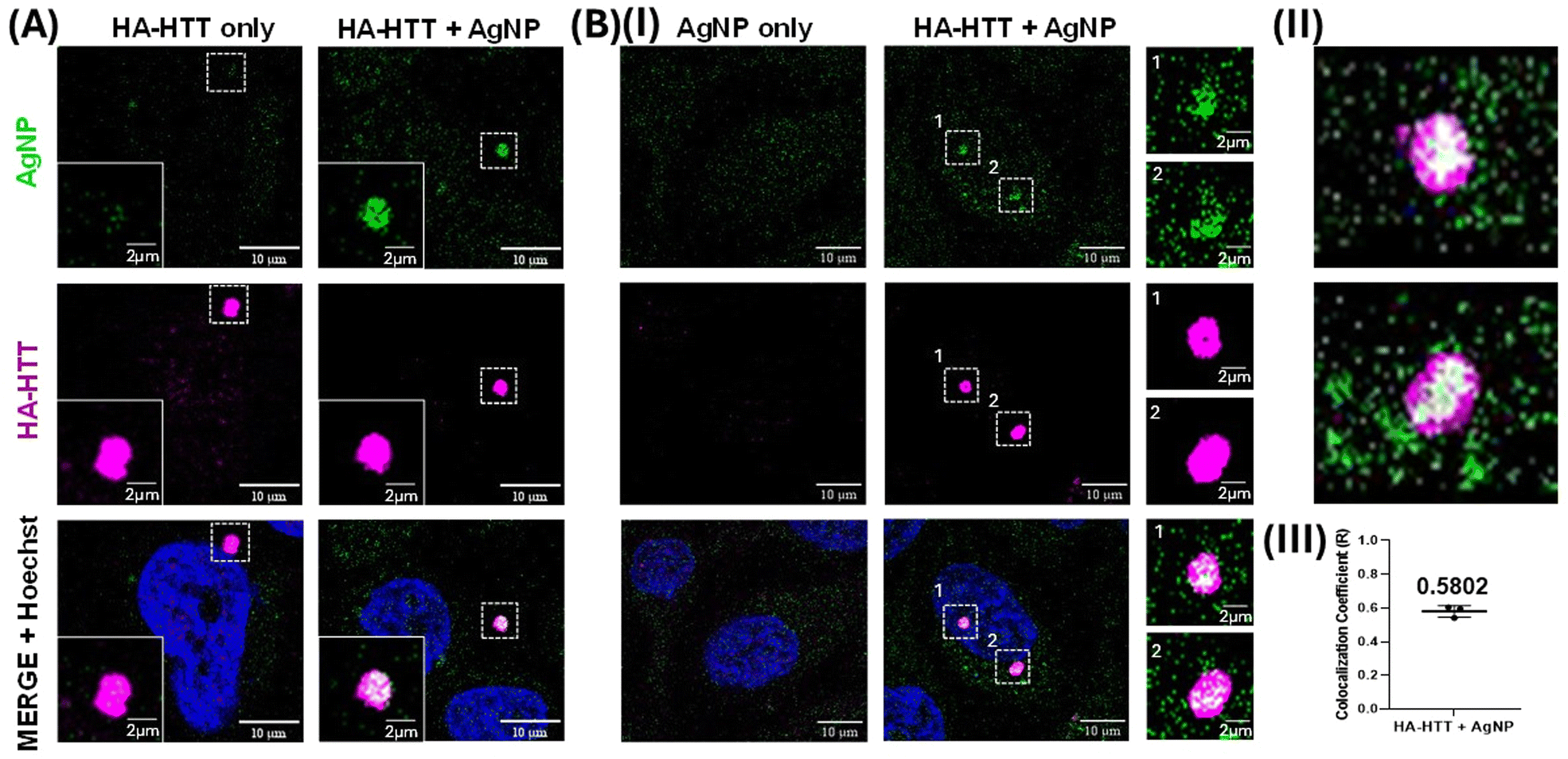 | ||
| Fig. 8 Clusteroluminescence labelling of Huntingtin protein aggregates using AgNPs in HeLa cells: (A) Cells expressing the aggregation prone HA tagged Huntingtin protein (HA-HTT) were allowed to form the aggregates and the luminescence (green) was traced in the 488 nm excitation range with and without the AgNPs. (B) (I) Similarly, HeLa cells with or without the expression of HA-HTT were assessed for luminescence in the presence of AgNPs. The 3D projection videos of the insets are shown in the XZ rotational axis in panel (II), while Pearson's coefficient (R) for colocalizing green and magenta signals is quantified with 30 individual images across three independent biological replicates and represented as a dot plot indicating that the mean 0.5802 with a p value of 0.0012 is shown in panel (III). An error bar represents SD. Magenta represents anti-HA staining with Alexa 647, while green represents the clusteroluminescent signal from AgNPs. DNA is visualized with Hoechst dye shown in cyan. | ||
4. Conclusions
An alternative method of label-free imaging of protein aggregates in cells is demonstrated using clusteroluminescence. Plasmonic AgNPs undergo aggregation with protein molecules, leading to unconventional “turn-on” fluorescence in an aggregated state that is significantly red-shifted. This clusteroluminescence results from the interaction of the electron-rich nitrogen heteroatoms at the nanoparticle interface, specifically the plasmon-catalyzed nitrile groups, which contribute to the through-space intermolecular electron coupling, leading to fluorescence emission. The red shift results from the hydrogen-bonded solid network of the aggregated protein molecules, which has higher electron delocalization and the Stark effect type of phenomenon where the charged aggregates enable the renormalization of dipoles and change of the HOMO–LUMO energy gap. This combined effect of plasmonic enhancement and clusteroluminescence in the protein–nanoparticle aggregates was harnessed to visualize intracellular accumulations of Huntingtin proteins. This unconventional, red-shifted fluorescence can potentially facilitate fluorescence imaging of biological systems with minimum interference from the physiological environment.Author contributions
Ashish Kumar Dhillon: investigation and analysis, writing – original draft, and writing – review and editing. Pranay Eknath Dudhe: cellular investigation and analysis, and writing – review and editing. Shubhangi Majumdar: transient absorbance spectroscopy investigation and analysis, and writing – original draft. Sanmitra Barman: writing – review and editing. Dibyajyoti Ghosh: conceptualization, investigation, first-principles calculations and analysis, and writing – review and editing. Karthigeyan Dhanasekaran: conceptualization, funding acquisition, cellular investigation and analysis, and writing – review and editing. Soumik Siddhanta: conceptualization, funding acquisition, investigation and analysis, project administration, supervision, writing – original draft, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the IIT Delhi SEED grant (IITD/Plg/Budget/2020-2021/204758), the DST SERB (CRG/2023/006527), and the CSIR Research grant (01(3042)/21/EMR-II). Ashish Kumar Dhillon thanks UGC, Government of India, for the student fellowship. AKD and SS acknowledge the central research facility, IIT Delhi, for the use of TEM, PL, and departmental instrument facility for characterization. SM thanks MHRD (Govt. of India) for the Prime Minister’s Research Fellowship (PMRF ID: 1401240). SM and SS acknowledge the Department of Science and Technology, DST-FIST (SR/FST/CS-II027/2014), New Delhi, India, for providing funds for the ultrafast TA facility. DG acknowledges the IIT Delhi SEED grant (PLN12/04MS), the Science and Engineering Research Board (SERB), the Department of Science and Technology (DST), India for Start-up Research Grant SRG/2022/00l234 and the IIT Delhi HPC facility for computational resources. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by the Los Alamos National Laboratory (contract 89233218CNA000001) and Sandia National Laboratories (contract DE-NA-0003525). KD acknowledges RCB Faridabad core funding and instrument facilities for supporting this work. The research activities of KD, being a Ramalingaswami fellow (BT/RLF/Re-entry/47/2020), are partly funded by the Department of Biotechnology (DBT), Ministry of Science and Technology, India and partly by the Start-up Research Grant (SRG/2021/001466) from the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India.References
- J. D. Ciubuc, K. E. Bennet, C. Qiu, M. Alonzo, W. G. Durrer and F. S. Manciu, Biosensors, 2017, 7, 43 CrossRef.
- S. Ashrafpour, T. T. Moghadam and B. Ranjbar, Int. J. Chem. Mol. Eng., 2014, 8, 985–988 Search PubMed.
- W. R. Silva, C. T. Graefe and R. R. Frontiera, ACS Photonics, 2016, 3, 79–86 CrossRef CAS.
- A. Vertegel, R. Siegel and J. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- X. Wu and G. Narsimhan, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1694–1701 CrossRef CAS PubMed.
- G. Vecchio, A. Galeone, V. Brunetti, G. Maiorano, L. Rizzello, S. Sabella, R. Cingolani and P. Pompa, Nanomedicine, 2012, 8, 1–7 CrossRef CAS PubMed.
- J. Karlo, A. K. Dhillon, S. Siddhanta and S. P. Singh, J. Biophotonics, 2023, e202300341–e202300341 Search PubMed.
- A. J. Schain, R. A. Hill and J. Grutzendler, Nat. Med., 2014, 20, 443–449 CrossRef CAS.
- A. K. Dhillon, A. Sharma, V. Yadav, R. Singh, T. Ahuja, S. Barman and S. Siddhanta, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2023, e1917 Search PubMed.
- J. Karlo, A. K. Dhillon, S. Siddhanta and S. P. Singh, J. Biophotonics, 2023, 16, e202200341 CrossRef CAS PubMed.
- H. Szmacinski, K. Ray and J. R. Lakowicz, Anal. Biochem., 2009, 385, 358–364 CrossRef CAS PubMed.
- T. Imoto, L. S. Forster, J. A. Rupley and F. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 1151–1155 CrossRef CAS.
- B. He, J. Zhang, J. Zhang, H. Zhang, X. Wu, X. Chen, K. Kei, A. Qin, H. Sung, J. Lam and B. Tang, Adv. Sci., 2021, 8, 2004299 CrossRef CAS PubMed.
- A. D. Stephens, M. N. Qaisrani, M. T. Ruggiero, G. Díaz Mirón, U. N. Morzan, M. C. González Lebrero, S. T. E. Jones, E. Poli, A. D. Bond and P. J. Woodhams, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020389118 CrossRef CAS PubMed.
- H. Zhang and B. Z. Tang, JACS Au, 2021, 1, 1805–1814 CrossRef CAS PubMed.
- Q. Wang, X. Dou, X. Chen, Z. Zhao, S. Wang, Y. Wang, K. Sui, Y. Tan, Y. Gong, Y. Zhang and W. Yuan, Angew. Chem., Int. Ed., 2019, 58, 12667–12673 CrossRef CAS PubMed.
- M. Mittal, S. Gautam, P. K. Chowdhury, S. Deep and S. Sapra, Z. Phys. Chem., 2019, 233, 41–54 CrossRef CAS.
- H. Zhang, Z. Zhao, P. McGonigal, R. Ye, S. Liu, J. Lam, R. Kwok, W. Yuan, J. Xie, A. Rogach and B. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- B. Lochocki, B. D. C. Boon, S. R. Verheul, L. Zada, J. J. M. Hoozemans, F. Ariese and J. F. de Boer, Commun. Biol., 2021, 4, 474 CrossRef CAS PubMed.
- S. Sarkar, B. Kanchibotla, J. D. Nelson, J. D. Edwards, J. Anderson, G. C. Tepper and S. Bandyopadhyay, Nano Lett., 2014, 14, 5973–5978 CrossRef CAS PubMed.
- N. Abu Hatab, J. Oran and M. Sepaniak, ACS Nano, 2008, 2, 377–385 CrossRef CAS PubMed.
- A. Campion and P. Kambhampati, Chem. Soc. Rev., 1998, 27, 241–250 RSC.
- K. Ikeda, S. Suzuki and K. Uosaki, J. Am. Chem. Soc., 2013, 135, 17387–17392 CrossRef CAS PubMed.
- R. Livingstone, X. Zhou, M. Tamargo, J. Lombardi, L. Quagliano and F. Jean-Mary, J. Phys. Chem. C, 2010, 114, 17460–17464 CrossRef CAS.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- J. Qian and B. Tang, Chem, 2017, 3, 56–91 CAS.
- K. Miao and L. Wei, ACS Cent. Sci., 2020, 6, 478–486 CrossRef CAS PubMed.
- P. Jiang, M. Gan, S.-H. Yen and D. W. Dickson, Front. Mol. Neurosci., 2021, 14, 738535 CrossRef CAS.
- D. Wahyuningtyas, W. Chen, R. He, Y. Huang, C. Tsao, Y. He, C. Yu, P. Lu, Y. Chen, S. Wang, K. Ng, B. Chen, P. Wei, J. Shie, C. Kuo, Y. Sun and J. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 60894–60906 CrossRef CAS.
- S. Li, J. He and Q.-H. Xu, ACS Omega, 2019, 5, 41–48 CrossRef PubMed.
- P. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- P. Borowicz, M. Latek, W. Rzodkiewicz, A. Łaszcz, A. Czerwinski and J. Ratajczak, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 045003 CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. Mishra, D. Ghosh and B. P. Tripathi, J. Catal., 2022, 411, 1–14 CrossRef CAS.
- S. Siddhanta, C. Zheng, C. Narayana and I. Barman, Chem. Sci., 2016, 7, 3730–3736 RSC.
- A. Sharma, D. Kesamsetty, J. Debnath and K. S. Ghosh, J. Mol. Liq., 2023, 372, 121156 CrossRef CAS.
- L. Calzolai, F. Franchini, D. Gilliland and F. Rossi, Nano Lett., 2010, 10, 3101–3105 CrossRef CAS PubMed.
- D. Zhang, O. Neumann, H. Wang, V. M. Yuwono, A. Barhoumi, M. Perham, J. D. Hartgerink, P. Wittung-Stafshede and N. J. Halas, Nano Lett., 2009, 9, 666–671 CrossRef CAS PubMed.
- A. Agrawal, I. Kriegel and D. Milliron, J. Phys. Chem. C, 2015, 119, 6227–6238 CrossRef CAS.
- R. Han, W. Song, X. Wang, Z. Mao, X. Han and B. Zhao, Phys. Chem. Chem. Phys., 2018, 20, 5666–5673 RSC.
- D. Karthigeyan, S. Siddhanta, A. Kishore, S. Perumal, H. Agren, S. Sudevan, A. Bhat, K. Balasubramanyam, R. Subbegowda, T. Kundu and C. Narayana, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10416–10421 CrossRef CAS PubMed.
- S. Siddhanta and C. Narayana, Nanomater. Nanotechnol., 2012, 2, 1 CrossRef.
- S. Siddhanta, D. Karthigeyan, P. P. Kundu, T. K. Kundu and C. Narayana, RSC Adv., 2013, 3, 4221–4230 RSC.
- J. Hu, R. S. Sheng and Z. San Xu, Spectrochim. Acta, Part A, 1995, 51, 1087–1096 CrossRef.
- G. Chandra, K. Ghosh, S. Dasgupta and A. Roy, Int. J. Biol. Macromol., 2010, 47, 361–365 CrossRef CAS PubMed.
- S. Schlucker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- S. Siddhanta, I. Barman and C. Narayana, Soft Matter, 2015, 11, 7241–7249 RSC.
- L. Fang, C. Huang, G. Shabir, J. Liang, Z. Liu and H. Zhang, ACS Macro Lett., 2019, 8, 1605–1610 CrossRef CAS PubMed.
- H. Wang, B. Aydiner, Z. Seferoglu, F. Bures and J. Liu, Dyes Pigm., 2022, 205, 110354 CrossRef CAS.
- E. Zhao, J. W. Y. Lam, L. Meng, Y. Hong, H. Deng, G. Bai, X. Huang, J. Hao and B. Z. Tang, Macromolecules, 2015, 48, 64–71 CrossRef CAS.
- C. Xing, J. W. Y. Lam, A. Qin, Y. Dong, M. Haeussler, W. Yang and B. Z. Tang.
- S. C. Lee and R. H. Holm, Chem. Rev., 2004, 104, 1135–1158 CrossRef CAS PubMed.
- Y. Shen, Z. An, H. Liu, B. Yang and Y. Zhang, Angew. Chem., 2023, 135, e202214483 CrossRef.
- L. Xue, F. Zou, Y. Zhao, X. Huang and Y. Qu, Spectrochim. Acta, Part A, 2012, 97, 858–863 CrossRef CAS PubMed.
- M. Ganesan and P. Nagaraaj, Org. Chem. Front., 2020, 7, 3792–3814 RSC.
- L. Fang, C. Huang, G. Shabir, J. Liang, Z. Liu and H. Zhang, ACS Macro Lett., 2019, 8, 1605–1610 CrossRef CAS PubMed.
- X. Wei, Y. Sun, C. Liu, Z. Li, X. Zou, D. Zhang, W. Zhang, J. Shi, X. Huang and Y. Li, Sens. Actuators, B, 2021, 329, 129075 CrossRef CAS.
- R. Bhattacharya, P. Mukherjee, Z. Xiong, A. Atala, S. Soker and D. Mukhopadhyay, Nano Lett., 2004, 4, 2479–2481 CrossRef CAS.
- A. Shukla, S. Mukherjee, S. Sharma, V. Agrawal, K. V. R. Kishan and P. Guptasarma, Arch. Biochem. Biophys., 2004, 428, 144–153 CrossRef CAS PubMed.
- E. Lipiec, D. Perez-Guaita, J. Kaderli, B. Wood and R. Zenobi, Angew. Chem., Int. Ed., 2018, 57, 8519–8524 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRTEM images, Raman spectroscopic analyses, cell viability studies, cell images and movies. See DOI: https://doi.org/10.1039/d4nr01803f |
| This journal is © The Royal Society of Chemistry 2024 |