
Novel nitrogen-doped carbon-coated SnSe2 based on a post-synthetically modified MOF as a high-performance anode material for LIBs and SIBs†
Zhiyuan
Chen
a,
Zhe
Zhang
a,
Longzhen
Wang
a,
Yifei
Li
a,
Yiting
Wang
a,
Yichuan
Rui
 a,
Ailing
Song
*b,
Min
Li
c,
Yinyu
Xiang
d,
Kaibin
Chu
e,
Lei
Jiang
*f,
Bohejin
Tang
*a,
Ning
Han
g,
Guoxiu
Wang
h and
Hao
Tian
*h
a,
Ailing
Song
*b,
Min
Li
c,
Yinyu
Xiang
d,
Kaibin
Chu
e,
Lei
Jiang
*f,
Bohejin
Tang
*a,
Ning
Han
g,
Guoxiu
Wang
h and
Hao
Tian
*h
aCollege of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, Shanghai 201620, People's Republic of China. E-mail: tangbohejin@sues.edu.cn
bCollege of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao, China. E-mail: ailing.song@ysu.edu.cn
cDepartment of Industrial Chemistry, University of Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
dSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, China
eSchool of Materials Science and Engineering, Linyi University, Linyi, 276000, P. R. China
fDepartment of Chemical Engineering, KU Leuven, Celestijnenlaan 200F, B-3001 Heverlee, Belgium. E-mail: lei.jiang@kuleuven.be
gDepartment of Materials Engineering, KU Leuven, Leuven 3001, Belgium
hCentre for Clean Energy Technology, School of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Broadway, Sydney, NSW 2007, Australia. E-mail: Hao.tian@uts.edu.au
First published on 11th July 2024
Abstract
SnSe2 with high theoretical capacity has been identified as an emerging anode candidate for lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs). However, the rate performance and cycling performance of this material in practical applications are still limited by unavoidable volume expansion and low conductivity. In this work, we designed and synthesized nitrogen-doped carbon-coated SnSe2/C–N composites using 2-aminoterephthalic acid (C8H7NO4) as a nitrogen-containing compound for modification by hydrothermal and vacuum calcination methods to achieve efficient utilization of active sites and optimization of the electronic structure. The carbon skeleton inherited from the Sn-MOF precursor can effectively improve the electronic conduction properties of SnSe2. N-doping in the Sn-MOF can increase the positive and negative electrostatic potential energy regions on the molecular surface to further improve the electrical conductivity, and effectively reduce the binding energy with Li+/Na+ which was determined by Density Functional Theory (DFT) methods. In addition, the N-doped carbon skeleton also introduces a larger space for Li+/Na+ intercalation and enhances the mechanical properties. In particular, the post-synthetically modified MOF-derived SnSe2/C–N materials exhibit excellent cyclability, with a reversible capacity of 695 mA h g−1 for LIBs and 259 mA h g−1 for SIBs after 100 cycles at 100 mA g−1.
Introduction
The emergence of the energy crisis and the escalating climate change situation have led to the consideration of large-scale application of secondary energy resources, such as solar and wind energy.1 This is seen as a feasible solution to reduce the overuse of petroleum fuels and alleviate ecological pollution.2–6 Therefore, there is an urgent need to develop and explore a green and recyclable energy storage and conversion system.7 As an energy storage device that meets the above requirements, lithium-ion batteries (LIBs) are widely used in various academic and industrial fields such as mobile power, new energy vehicles, and aerospace due to their high quality/volumetric energy density, and no memory effect.8–17 Although LIBs have developed into a mature technology, the fact that there are limited Li resources has prompted researchers not to focus solely on Li+, thus stimulating research on sodium-ion batteries (SIBs), which are based on the more abundant Na metal element.18 Moreover, the intercalation reaction chemistry of LIBs and SIBs exhibits striking similarities, thereby making SIBs function in a manner akin to LIBs.19,20 Due to their excellent electrochemical and safety properties, LIBs and SIBs have been receiving continuing attention from researchers around the world.21,22The composition and structure of the anode material have a decisive influence on the electrochemical performance of LIBs and SIBs.23,24 Transition metal compounds have been widely investigated because they have sufficient reactive active sites and short Li+ and Na+ diffusion paths, which are conducive to the electrochemical performance of anode materials.25–28 Metal selenides (MSex) with a layered structure offer superior performance over metal chalcogenides in many aspects in typical conversion anode materials for secondary batteries.29,30 Compared with metal oxides, the weaker M–Se bond compared to the M–O bond facilitates the smooth progress of the conversion reaction, and the alkali metal ions may migrate rapidly with lower diffusion energy barriers supported by the larger layer spacing and weaker van der Waals force interactions.31 In addition, compared with metal sulfides, selenium atoms have larger atomic radii and stronger metallicity than sulfur atoms, resulting in metal selenides with larger layer spacing and higher conductivity.32
As the representatives of reactive MSex, SnSe/SnSe2 materials have high theoretical activity through successive intercalation and transformation reactions producing reactive metals that will further form alloys with Li and Na.33 For example, with 1 mol e− involved in the reaction calculations, the theoretical sodium storage capacities of SnSe and SnSe2 in SIB anodes are 780 mA h g−1 and 756 mA h g−1, respectively, which are very impressive for the field of stored Na+ research.34,35 These compounds have a large layer spacing due to their typical lamellar structure, thus facilitating easy intercalation of bulkier active metal ions.36 However, tin selenide active materials undergo unavoidable volume expansion and particle agglomeration during the alloying/de-alloying process, resulting in a significant decrease in reversible capacity.37,38 However, the inherently poor electrical conductivity of SnSe/SnSe2 and the high cost of industrialized synthesis methods severely hinder the utilization of their excellent theoretical capacity.39 To overcome the above shortcomings of tin selenides, studies have shown that carbon coatings are effective in reducing the occurrence of side reactions, enhancing the electrical conductivity of the material, and mitigating drastic volume changes in the material due to stress residues, maintaining the stability of the main structure.40–44 Combining carbon skeletons with transition metal compounds to make composites can capitalize on the powerful synergistic effects brought about by heterogeneous structural engineering, mitigating the volumetric expansion of the materials that occurs during cyclic intercalation while significantly improving electrical conductivity.45–49 What's more, by incorporating N atoms into the carbon skeleton, the electronic property modulation effect of N atoms can significantly change the charge and spin densities of carbon atoms, and the carbon material becomes non-electronic and neutral, which is favorable for the oxygen reduction reaction on carbon.50–52 That is to say, compounding with conductive carbon skeletons based on nitrogen doping may be one of the effective ways to overcome the disadvantages of existing SnSe/SnSe2 anode materials.53–56
Metal–organic frameworks (MOFs) are organic–inorganic hybrid crystalline materials with intramolecular pores that result from the self-assembly of metal ions or clusters in organic ligands and coordination bonds.57–59 They have attracted much attention from researchers because of their tuneable porosity, abundant active centres and controllable morphology.60–62 It has been reported that using MOFs as sacrificial templates can produce a specific porous structural morphology, and most of the functional materials obtained inherit the morphological structure of the MOF precursors.63,64 The use of organic ligands containing N for the pretreatment of MOF precursors and the synergistic doping effect of trace heteroatoms on the carbon skeleton can also effectively enhance the activity and stability of the carbon skeleton.65–68
In this work, we synthesized an Sn-MOF using a low-temperature hydrothermal method, and then used 2-aminoterephthalic acid (C8H7NO4) to replace the solvent sites in the MOF clusters, and further heat-treated it with selenium under vacuum to synthesize nitrogen-loaded carbon-capped SnSe2/C–N. According to the previous reports of our group, the Sn-MOF can effectively replace the solvent sites in MOF clusters by injecting an organic nitrogen source after heat treatment under vacuum, and the organic ligand (C8H7NO4) is converted to an impurity-free N-loaded carbon source during the subsequent selenization process.65 The N-loaded carbon skeleton derived from the Sn-MOF precursor can limit the volume expansion of the material during the cycling process. The modification of the Sn-MOF, employing organic ligands to replace solvent sites in MOF clusters, is not only carried out to enhance the mechanical properties of the carbon framework, but also to bring a wider layer spacing and more chemically reactive sites to the carbon skeleton and further improve the conductivity of this material.69,70 In addition, N atoms are incorporated into the carbon skeleton as dopants to further enhance the redox activity based on the synergistic effect.71 In particular, the post-synthetically modified MOF-derived SnSe2/C–N material exhibits excellent cyclability with a reversible capacity of 695 mA h g−1 for LIBs and a reversible capacity of 259 mA h g−1 for SIBs after 100 cycles at a current density of 100 mA g−1, respectively.
Experimental section
Synthesis of the Sn-MOF
The raw materials used in the experiments were commercially available, analytically pure chemicals without pretreatment. The Sn-MOF was synthesised using a simple one-step low-temperature hydrothermal method as reported in previous work. First, 1.073 g of SnSO4 was dissolved in a beaker with 25 ml of deionized (DI) water and sonicated for 10 minutes to form a homogeneous SnSO4 solution. Subsequently, 1.007 g of lithium hydroxide (LiOH) and 1.993 g of terephthalic acid (C8H6O4) were ultrasonically dissolved in 100 ml of a mixture of N,N-dimethylformamide (DMF) and DI water (VDMF/Vwater, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the same manner. The SnSO4 solution was then slowly added dropwise to the mixed LiOH + C8H6O4 solution and magnetically stirred at 50 °C for 2 h. Finally, the precipitate was washed by centrifugation with DMF and deionised water in turn to obtain a white product, which was dried under vacuum at 70 °C for one day to obtain the Sn-MOF.
:1) in the same manner. The SnSO4 solution was then slowly added dropwise to the mixed LiOH + C8H6O4 solution and magnetically stirred at 50 °C for 2 h. Finally, the precipitate was washed by centrifugation with DMF and deionised water in turn to obtain a white product, which was dried under vacuum at 70 °C for one day to obtain the Sn-MOF.
Synthesis of Sn/C and SnSe2/C
The Sn-MOF was then placed in a porcelain boat and gradually heated to 450 °C at a rate of 4 °C per minute under an Ar/H2 (10%) atmosphere and held for 6 hours to obtain Sn/C. The selenization process may lead to impurity in the final products and insufficient reaction when a protective atmosphere of inert gas is used. Based on the successful preparation of V3S4/CNF by vacuum vulcanization,72 and our previous work,65 the selenization process can be carried out under vacuum conditions, achieving complete reaction and pure products. For our work, a mixture of Sn/C and selenium powder (mass ratio, MSn/C/MSe, 1:3) was placed in a vacuum quartz tube, and then gradually heated to 600 °C and kept in a muffle furnace for 6 h. The product obtained by centrifugation was washed 3 times with ethanol and carbon disulfide to remove unreacted selenium powder, and dried under vacuum at 80 °C for 24 h to obtain SnSe2/C.
Synthesis of SnSe2/C–N
Prior to the formal synthesis of SnSe2/C–N, the Sn-MOF sacrificial template precursor required additional nitrogen doping treatment. The Sn-MOF was placed in a glass flask and dried under vacuum at 180 °C for 4 h to remove water molecules from the cluster cores. Then 2-aminoterephthalic acid (mass ratio, MSn-MOF:M2-aminoterephthalic acid = 1:1) was dissolved in DMF and quickly poured into the flask, which was heated at 200 °C for 2 h to allow the nitrogen-containing structural units to be fully absorbed by the MOF clusters. The subsequent preparation of SnSe2/C–N was carried out in the same way as that of SnSe2/C.
Characterization
First, the microscopic morphologies of the Sn-MOF, SnSe2/C and SnSe2/C–N were observed using an FE-SEM (Field Emission Scanning Electron Microscope, Japan-Hitachi S-4800) and a TEM (Transmission Electron Microscope, Hitachi S-4800, Japan). After that, the main phase compositions and crystal structures of SnSe2/C and SnSe2/C–N were determined using powder XRD (X-ray diffraction: Netherlands X'pert TROMPD (Philips), Cu-Kα radiation). The elemental compositions and the valence of each element in SnSe2/C–N were detected by XPS (X-ray photoelectron spectroscopy, USA-Escalab 250, Al-Kα source). Then to further identify the chemical bonds present in the composites to probe their structure, Fourier Transform Infrared (FT-IR) spectra in the range of 500–3000 cm−1 were measured on a Nicolet AVATAR 370 infrared spectrometer. Subsequently, an ICP (Inductively Coupled Plasma, PerkinElmer ICP 2100) test was performed in order to characterize the Sn and Se content in the composites. Afterwards, in order to investigate the proportion of carbon and reactive materials in the composites, the materials were subjected to a temperature increase test by TGA (thermogravimetric analysis, Germany-STA2500, temperature increase rate: 10 °C min−1, temperature range: 25–800 °C). Finally, the adsorption–desorption curves of N2 were tested with a Micromeritics analyzer (USA-ASAP 2460, exhaust temperature: 473 k). The specific surface area (m2 g−1) and pore size (nm) distribution of the samples were calculated utilizing the BET equation and BJH method.Electrochemical measurements
The electrochemical properties of the samples have been investigated with a semi-cell. The working electrode is composed of an active material (weight ratio: 70%), acetylene black (weight ratio: 20%) and polyvinylidene fluoride (weight ratio: 10%, PVDF). The above substances were proportionally mixed in N-methyl-pyrrolidone (NMP) solution and uniformly ground for 5 h. Subsequently, the mixture was coated onto copper foil (1.1 cm2) and dried under vacuum for 12 h (70 °C) to obtain the working electrode in which the active material (e.g. SnSe2/C–N) loading was 1.0–1.5 mg cm−1. Lithium foil and a polypropylene film were used as the reference electrode material and diaphragm for the CR2032 LIB half-cell, respectively. The electrolyte consisted of dimethyl carbonate (DMC) and ethylene carbonate (EC) dissolved (V/V, 1:1) in 1 M LiPF6. The sodium-ion button cell used for the charge/discharge tests had a sodium metal foil as the standard comparison electrode, and the electrolyte consisted of 1 M NaClO4 in ethylene carbonate (EC) and diethyl ethyl carbonate (DEC) (V/V, 1:1) with a 2% additive of fluoroethyl bicarbonate (FEC). The batteries were constructed in a glove box under standard conditions (H2O, O2 < 0.1 ppm). The electrochemical properties of the material were tested on the CT-3008 system (China-Neware, voltage window: 0.01–3 V) and they included charge/discharge curves, cycling performance, and rate performance. Cyclic voltammetry (CV, scan rate: 0.1 mV s−1) and electrochemical impedance spectroscopy (EIS, parameter setting, frequency: 0.01–100 kHz; voltage disturbance: 5 mV) were performed in electrochemical workstations (China-CHI660D). All the above activity data were calculated from the total quality of the electrodes. In addition, the electrostatic energies and binding energies at the active sites during the binding process of SnSe2/C and SnSe2/C–N with Li/Na were simulated using Gaussian 16. The reagents and their products were further optimized at the B3LYP-D3BJ/Def2-SVP level, using Density Functional theory (DFT) methods.73
Results and discussion
Synthesis and characterization
As shown in Fig. 1a, the Sn-MOF precursor and two electrode samples required for the experiments were prepared by hydrothermal and calcination methods, respectively, using SnSe2/C–N as an example. The morphologies and microstructures of Sn-MOF, SnSe2/C, and SnSe2/C–N were characterized by SEM as shown in Fig. 1b–d. The Sn-MOF clearly exhibits a relatively homogeneous hexahedral structure (Fig. 1b), with a diameter of about 1.5–1.8 μm. The SEM images of SnSe2/C after vacuum selenization (Fig. 1c and d) show that it inherits the hexahedral structure of the Sn-MOF and forms a number of thin layers of SnSe2 nanosheets with irregular thickness clusters on the hexahedral surface. The SEM images of SnSe2/C–N (Fig. 1d) confirm that the Sn-MOF doped with nitrogen by the implantation method formed a structure similar to that of SnSe2/C after vacuum vulcanization, which indicates that the doping of C8H7NO4 does not affect the formation of the SnSe2 composite structure. Unlike SnSe2/C, the size and morphology of the nanosheets on the surface of the material are different. From the comparison, it can be clearly seen that the thickness of nanosheets relatively increases after doping with C8H7NO4 and slight agglomeration occurs. Fig. S1† shows the SEM image of SnSe2/C–N after 100 cycles of charge/discharge (LIBs), which indicates that the skeleton structure of the material can still be preserved intact even after several reactive ion (de-)/intercalations, confirming the reason for its long cycling stability. The deeper microstructure and crystalline morphology of the SnSe2/C–N composites were characterised by TEM, as shown in Fig. 1e–h. The clearly visible lattice stripes from the HRTEM images (Fig. 1f–h) proved that the SnSe2 nanocomplexes have excellent crystal properties with spacings of 0.29 and 0.22 nm, which correspond to the (101) and (102) planes of the standard SnSe2 as determined by XRD. In addition, Fig. 1i–l and Fig. S2† reflect the elemental mapping analysis of the composites. The results confirm that the composites contain Sn, Se, N and C elements, and the distribution of each element is uniform and clearly visible, which indicates that the carbon coating in the composite structure is well preserved and not damaged during calcination at 600 °C. Comparing the images with different colours, the Sn and Se elements are uniformly dispersed in the selected areas without agglomeration, further proving the dispersion of SnSe2 nanoparticles. | ||
| Fig. 1 (a) The schematic representation of the SnSe2/C–N process; SEM images: (b) Sn-MOF, (c) SnSe2/C, and (d) SnSe2/C–N; (e) TEM image of SnSe2/C–N; (f), (g) and (h) HRTEM images of SnSe2/C–N; and EDS mapping images for SnSe2/C–N: (i) Sn, (j) Se, (k) C and (l) N. | ||
The crystallinities of the samples were characterized using XRD. Fig. 2a shows the XRD patterns of the Sn-MOF, SnSe2/C and SnSe2/C–N. Sharp peaks of SnSe2 can be clearly seen in the test data plot lines, while the carbon skeleton does not show any distinct peaks, indicating that the C and C–N skeletons in the sample are amorphous. In addition, all the sharp diffraction peaks of SnSe2/C and SnSe2/C–N are consistent with those of the original SnSe2 standard materials (JCPDS #23-0602). It can be seen that the diffraction peaks of the Sn-MOF are accurate and distinct,74 and the diffraction peaks of SnSe2/C and SnSe2/C–N samples are not consistent with that of the Sn-MOF, which suggests that the composite samples are well-crystallized and free of impurity components.
 | ||
| Fig. 2 (a) XRD patterns of the Sn-MOF, SnSe2/C and SnSe2/C–N composites; XPS analysis of (b) Sn 3d, (c) Se 2p, (d) C 1s, and (e) N 1s in SnSe2/C–N; and N2 adsorption (desorption) isotherms and pore size distributions for (f) SnSe2/C and (g) SnSe2/C–N. | ||
To investigate the structural composition of the amorphous part of the composites, SnSe2/C and SnSe2/C–N composites were characterized using FTIR (Fig. S3†). The characteristic peak at 1558 cm−1 common to both composites is attributed to the C–C stretching vibration.75,76 In addition, the characteristic peaks of SnSe2/C–N observed in the spectral bands 1408 cm−1 and 1252 cm−1 are then related to the C–N stretching vibration, confirming the presence of nitrogen-doped carbon, which promotes the electron mobility.77–79 The obtained results indicate that the prepared SnSe2/C and SnSe2/C–N composites exhibit characteristic vibrations with the expected structures, which further proves the successful synthesis of nitrogen-doped carbon skeletons.80 Regarding the ICP test, we tested and analyzed the Sn and Se elements in the SnSe2/C–N samples, and the test results showed that the mass percentage of the Sn element was 29.62%, while that of the Se element was 39.11%, which corresponds to an atomic number ratio of Sn:Se of about 1:2.
Fig. 2b–e and Fig. S4† show the XPS spectra of the SnSe2/C–N composite, mainly to analyze the chemical state of the sample surface in more depth. The presence of C, N, Sn and Se in the composites can be confirmed from Fig. S4.† Quantitative analysis of the elements C, N, Sn, and Se in the samples based on XPS peak area calculations showed that the surface atomic concentrations were 81.55%, 0.08%, 12.14%, and 6.23%, respectively, indicating that the atomic ratios of Sn to Se were around 1:2, and it proves the conclusion of nitrogen doping. In addition, the elemental binding behavior in the SnSe2/C–N samples can be evaluated by high-resolution XPS of the N 1s, Se 3d and Sn 3d regions. As can be seen from the high-resolution XPS results of Sn 3d in Fig. 2b, the two major peaks at 486.2 eV and 494.6 eV correspond to 3d5/2 and 3d3/2, respectively, which confirms the presence of Sn–Se.81,82 The two additional peaks in Fig. 2b at 487.2 and 495.6 eV can be assigned to Sn–C bonds,83,84 and the peaks in Fig. 2b at 486.7 and 495.1 eV can be attributed to Sn–N bonds,85,86 respectively. The peak in the Se 3d spectrum (Fig. 2c) can recoil into two smaller peaks, which are formed mainly by the higher peaks at 55.3 eV and 56.2 eV corresponding to the 3d5/2 and 3d3/2 binding energies of Se.82,87 In the XPS spectra of C1s (Fig. 2d), the peaks at 283.5 eV, 284.5 eV and 285.3 eV correspond to Sn–C, C–C and C–N, respectively.88,89 Finally, the peaks of 398.2 eV, 400.0 eV and 402.0 eV in the N1s spectrum (Fig. 2e) correspond to pyridine-N, pyrrole-N and quaternary-N, respectively.30,52,90 In addition, previous studies have demonstrated that the introduction of pyrrole nitrogen in carbon layers improves the electrical conductivity and creates structural defects to form active sites for Li+ intercalation.65 To further determine the content of SnSe2 and C in the composites, TGA tests were performed on SnSe2/C and SnSe2/C–N materials under an air atmosphere at a heating rate of 10 °C min−1, and the results are shown in Fig. S5.† In the case of SnSe2/C, for example, the decomposition and oxidation of SnSe2 and sublimation of SeO2 occur at temperatures above 300 °C, resulting in a sharp weight loss. The weight loss between 650 °C and 800 °C is due to carbon combustion.91 Therefore, the content of C and SnSe2 in the SnSe2/C composite can be calculated to be approximately 35.7% and 64.3%, respectively. Following the same calculation method, the contents of C and SnSe2 in SnSe2/C–N can be calculated to be about 36.2% and 63.8%, respectively. This further confirms that the injected organic ligands have been successfully introduced into the MOF cluster structure and completely dehydrogenated into N-containing carbon skeletons during the subsequent hydrogen calcination and vacuum selenization processes. By analyzing the nitrogen adsorption–desorption curves, the specific surface area and pore size assignments of SnSe2/C–N and SnSe2/C were obtained as shown in Fig. 2f and g. The BET specific surface areas of SnSe2/C and SnSe2/C–N were 12.85 m2 g−1 (average pore size: 11.3 nm) and 6.76 m2 g−1 (average pore size: 8.8 nm), respectively. The isothermal curves follow the characteristics of mesoporous materials.92 According to the above data, the metal ions are connected to the lone pair of electrons of N through ligand bonds, and N generates a carbon coating during vacuum carbonization. This carbon partially clogs or closes the Sn-MOF pores, resulting in a decrease in the specific surface area.
Electrochemical performance
| SnSe2 + 4Li+ + 4e− → Sn + 2Li2Se | (1) |
| Sn + xLi+ + xe− ↔ LixSn(0 < x ≪ 4.4) | (2) |
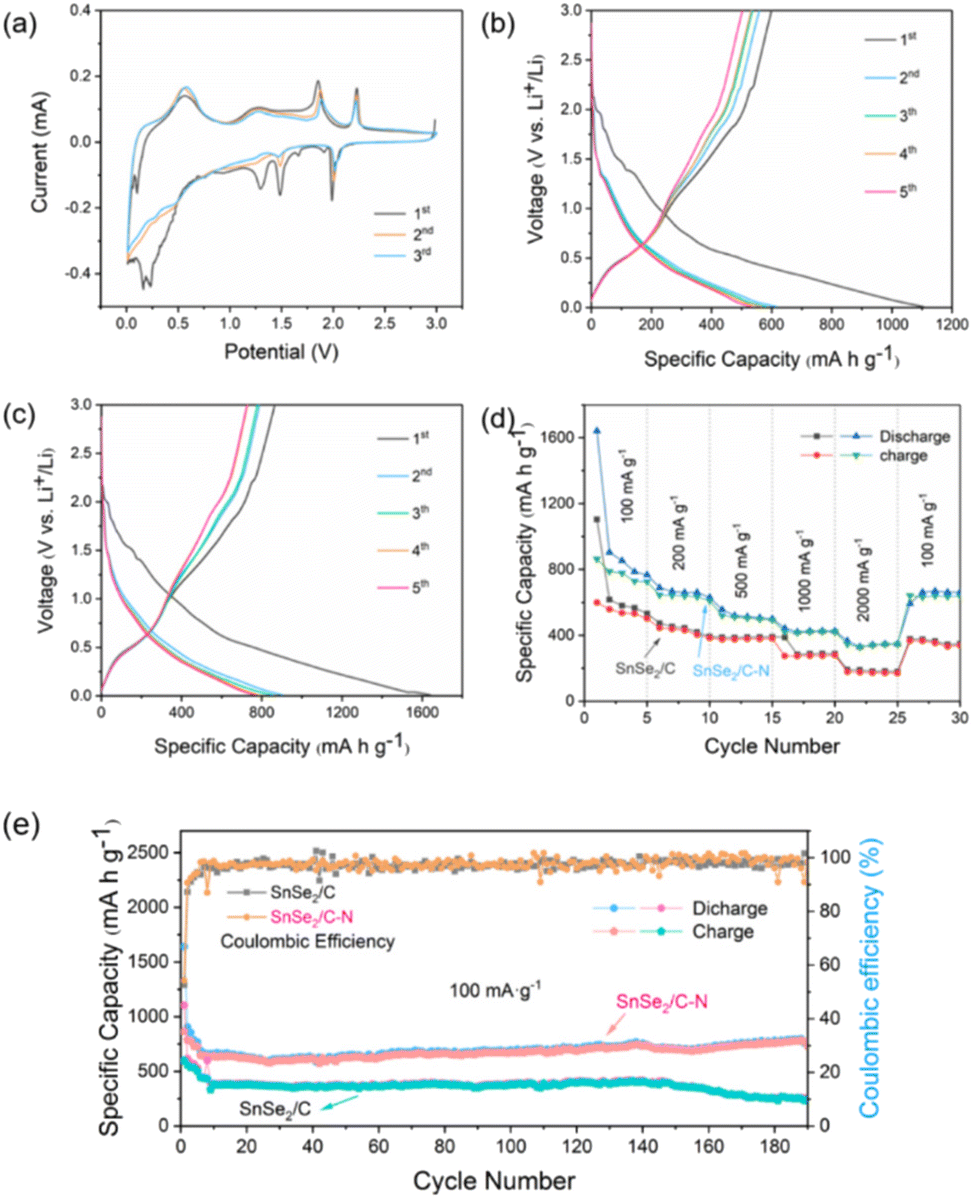 | ||
| Fig. 3 (a) CV curves of SnSe2/C–N at 100 mV s−1. Constant current charge (discharge) curves at 100 mA g−1 of (b) SnSe2/C and (c) SnSe2/C–N. (d) Rate performance and charge (discharge) profiles of SnSe2/C-NB at different rates. (e) Performance of all electrodes for cycling at 100 mA h g−1. | ||
During the first cathodic scan, lithiation begins with a reduction peak at ∼1.98 V, marking the initial insertion of Li+. The sharp reduction peak at ∼1.48 V is attributed to the formation of Sn and Li2Se from SnSe2via a conversion reaction (eqn (1)). The sharp reduction peak at ∼1.29 V is irreversible due to the formation of a solid–electrolyte interface (SEI) film. In addition, the reduction peak between 0.3 V and 0.01 V is attributed to the alloying reaction of Sn with Li+ (eqn (2)).91 For the first anodic scan, the oxidation peaks between 0.5 V and 0.6 V were attributed to the dealloying process of LixSn and the oxidation peaks at ∼1.85 and ∼2.22 V corresponded to the decomposition of Li2Se and the oxidation of Sn to SnSe2, respectively (eqn (1)). In subsequent cycles, the redox pairs at ∼0.56/0.25 V are assigned to the dealloying/alloying between Li and Sn, whereas the redox pairs at ∼1.87/1.49 V and ∼2.22/1.99 V come from the reversible conversion of SnSe2 and Li2Se. The intensity of these peaks remains constant, implying highly reversible Se alloying/dealloying and conversion reactions.93
The initial and reversible specific capacity obtained from charging/discharging tests is one of the important indexes for evaluating the energy storage characteristics of batteries. The specific capacity obtained under the same current density is a very intuitive evaluation standard, and the performance of the battery is positively correlated with this data. The constant current charge and discharge curves when SnSe2/C and SnSe2/C–N are used as anode materials for LIBs are shown in Fig. 3b and c. The initial discharge capacities of SnSe2/C and SnSe2/C–N materials are 1110.67 mA h g−1 and 1621.94 mA h g−1, and it can be clearly seen that the latter has a higher initial specific capacity. The charging capacity values are 600.1 and 849.7 mA h g−1, and the initial coulombic efficiency (ICE) values are 54.5% and 53.1% respectively. As shown in Fig. 3d, the average specific capacity of discharge at different current densities exhibited 793.0, 655.5, 503.2, 347.7, and 166.0 mA h g−1 for the cycling tests at 100, 200, 500, 1000, and 2000 mA g−1, respectively. Back to 100 mA g−1, SnSe2/C–N still show an impressive reversible specific capacity (662.1 mA h g−1), and the capacity retention of 83.5% indicates its good rate performance. In terms of long cycle life testing (Fig. 3e), it can maintain a reversible capacity of 695 mA h g−1 after 100 cycles. It is worth mentioning that its reversible specific capacity has the tendency to continuously increase and subsequently level off during the subsequent cycling process, even rising to 791.4 mA h g−1 after 200 cycles. This may be caused by the gradual activation of the composite nanoparticles during the cycling process which improves the Li+ diffusion rate.95
The SnSe2/C–N anode material can exhibit extremely high reversible capacity and excellent rate performance for the following reasons: firstly, the MOF-derived carbon skeleton has good electrical conductivity, which leads to a greatly enhanced electrochemical reaction rate of the electrode material. In addition, the pretreatment of the MOF with the organic nitrogen source resulted in the introduction of nitrogen on the surface of the final sample nanosheets, which not only increased the stability of the intrinsic carbon skeleton of the MOF, but also brought more active sites for chemical reactions. In order to reflect the dynamics of the SnSe2/C and SnSe2/C–N charge transfer processes at room temperature, the composites were subjected to Electrochemical Impedance Spectroscopy (EIS) tests. The Nyquist equivalent circuit diagrams fitted to the curves are shown in Fig. S6† and each diagram consists of four parts. Initially, the SEI surface resistance (Rs) corresponds to a compressed semicircle in the high frequency region. In addition, the semicircle in both the mid-frequency and low-frequency domains corresponds to the charge transfer resistance (Rct) of the unit system.96 Equivalent circuit fitting results show that SnSe2/C–N (27.1 Ω) has a lower Rct value compared to SnSe2/C (57.3 Ω). The lower Rct value of the SnSe2/C–N anode suggests that the nitrogen-doped skeleton further enhances the solid-state diffusion rate of Li+ inside the electrode as compared to that of the pristine carbon skeleton.97
In addition, the pseudocapacitive contribution to the lithium storage capacity was also dynamically analyzed, since the charge storage mechanism of the energy storage system is divided into diffusion control and capacitance control. The quantitative analysis was performed based on i (peak current) and v (scan rate) in eqn (3), where a and b are random parameters. When b approaches 0.5, the electrochemical response tends to be generated by diffusion control contribution, whereas when b is closer to 1, it tends to be generated more by capacitive control contribution.15,98–100 The b-values corresponding to the five peaks were calculated by linearly fitting the log(i) versus log(v) curves (eqn (4)). The b-values for each redox peak are 0.90, 0.80, 0.81, 0.99, and 0.91, respectively (Fig. 4b), which suggests that capacitance control dominates the charge storage mechanism of the material.
| i = a·vb | (3) |
| log(i) = b·log(v) + log(a) | (4) |
| i = k1v + k2v1/2 | (5) |
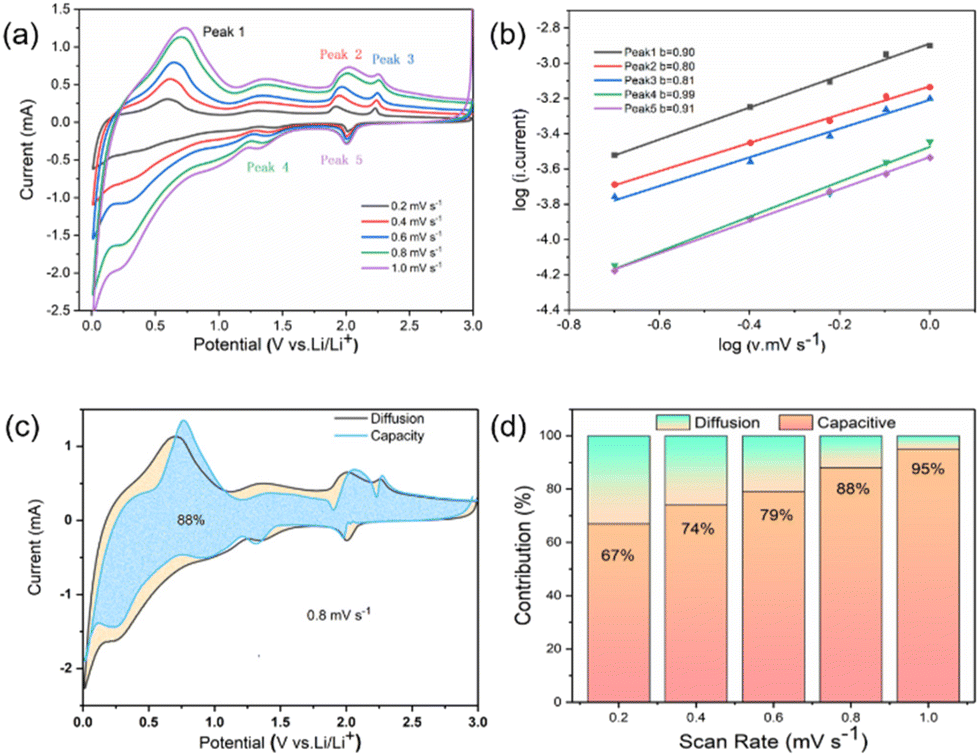 | ||
| Fig. 4 (a) CV curves of SnSe2/C–N for different scan rates (0.2–1.0 mV s−1), (b) log(scan rate)/log(current) plots, (c) pseudocapacitive contribution (orange area) at 0.8 mV s−1, and (d) histogram of the pseudocapacitive contributions at different scan rates. | ||
The specific contributions of capacitive control (k1v) and diffusion control (k2v1/2) values were calculated by graphical fitting of eqn (5). For example, the capacitive control of SnSe2/C–N contributes 88% at 0.8 mV s−1 (Fig. 4c) and as the scan rate increases from 0.2 to 1.0 mV s−1, its capacitance contribution increases sequentially from 67% to 95%. As a comparison, the capacitance contribution of the undoped SnSe2/C composite is much lower than that of SnSe2/C as can be seen in Fig. S7,† which indicates that the high mass transfer conductivity of the nitrogen-doped carbon skeleton composites is further verified.101,102
| SnSe2 + 4Na+ + 4e− → Sn + 2Na2Se | (6) |
| Sn + 3.75Na+ + 3.75e− ↔ Na3.75Sn | (7) |
 | ||
| Fig. 5 (a) CV curves of SnSe2/C–N at 0.1 mV s−1; constant current charge/discharge curves of (b) SnSe2/C and (c) SnSe2/C–N at 0.1 A g−1; (d) rate performance of SnSe2/C, and SnSe2/C–N electrodes at different rates; and (e) cycling performance at 0.1 A g−1. | ||
A reversible reduction peak around 1.0–1.2 V was detected in the first cathodic scan, which can be attributed to the generation of Sn with Na2Se (eqn (6)). The peaks around 0.5–0.7 V are due to the alloying reaction to produce Na3.75Sn (eqn (7)). The two anodic peaks around 1.61 V and 2.12 V are clearly visible in the subsequent anodic scans, and they correspond to phase transitions during the dealloying process and the reoxidation of Sn. Fig. 5b and c exhibit the charge–discharge curves of the two composites in the voltage range of 0.01–0.3 V and 100 mA g−1 for investigating the electrochemical performance of the materials in SIB anode applications. The initial discharge/charge capacities of SnSe2/C–N are 1092.1 mA h g−1 and 421.4 mA h g−1, while the corresponding initial capacities of SnSe2/C are only 570.2 mA h g−1 and 200.1 mA h g−1. The SIBs were tested for rate and cycling performance at the same current density settings as the LIBs in the previous subsection (Fig. 5d and e). As shown in Fig. 5e, the SnSe2/C–N electrode achieves a good reversible specific capacity (271 mA h g−1) after 100 cycles at 100 mA g−1. The temperature difference between day and night during the testing of SnSe2/C–N materials may lead to capacity fluctuations. In addition, the Coulombic Efficiency (CE) for SnSe2/C–N materials was stable at around 100%, indicating that the interface between the SnSe2/C–N electrodes and the electrolyte can be stabilized for a long time. Therefore, it can be proved that the electrochemical reaction of SnSe2/C–N is highly stable and reversible.73
The dynamic testing and subsequent analysis of the SIBs were performed with the same parameter settings as for the LIBs (Fig. 6). According to the calculation results in Fig. 6b, the ionic contribution in SIBs is dominated by diffusion. For example, at a scan rate of 0.4 mV s−1, the capacitance control contribution is 50.9% based on the fitted plot line (Fig. 6c). However, it can be seen from Fig. 6d that the capacitance contribution increases from 43.3% to 72.6% as the scan rate increases (0.2 to 1.0 mV s−1), indicating that the effect of nitrogen doping on the carbon skeleton in the SIB application has a similar gain to that in the LIBs.
 | ||
| Fig. 6 (a) CV of SnSe2/C–N curves for different scan rates (0.2–1.0 mV s−1), (b) log(scan rate)/log(current) plots, (c) pseudocapacitive contribution (orange area) at 1.0 mV s−1, and (d) histogram of the pseudocapacitive contributions at different scan rates. | ||
 | ||
| Fig. 7 Molecular surface electrostatic energy of (a) SnSe2/C and (b) SnSe2/C–N using Gaussian 16. | ||
The binding energies of SnSe2/C and SnSe2/C–N after the attack of the active site by Li in SnSe2/C–N were also specifically calculated using Gaussian 16, and the results are shown in Table 1. Using Li with SnSe2/C as an example, the adsorption capacity of SnSe2/C and SnSe2/C–N for Li+/Na+ was explored according to the following equation: Ebind = ELi-SnSe2/C − ESnSe2/C − ELi. In the comprehensive analysis, the larger nucleophilic/electrophilic region of the electrostatic potential energy of the molecular surface of SnSe2/C–N enhances its ability to attract more Li+/Na+ and electrons, which promotes the electrochemical process. Furthermore, the smaller binding energy of SnSe2/C–N with Li+/Na+ also indicates the higher reversible specific capacity during long cycling.
| Different category | E (Hatree) | E bind (Hatree) |
|---|---|---|
| Li | −7.4909023 | — |
| Na | −162.2866299 | — |
| SnSe2/C | −5414.9401514 | — |
| SnSe2/C–N | −5524.4772155 | — |
| Li with SnSe2/C | −5421.2881263 | 1.1429274 |
| Li with SnSe2/C–N | −5531.8693801 | 0.0987377 |
| Na with SnSe2/C | −5576.1884092 | 1.0383721 |
| Na with SnSe2/C–N | −5686.5860736 | 0.1777718 |
Conclusions
In this work, the Sn-MOF-derived SnSe2/C–N anode materials synthesized after nitrogen doping modification showed excellent rate and cycling performance for both LIB and SIB applications, with reversible capacities of 695 mA h g−1 and 259 mA h g−1 after 100 cycles at 100 mA g−1, respectively. This work has the following breakthroughs compared to other studies: firstly, the innovative concept of using MOFs as precursors was utilized to form SnSe2/C–N composites by hydrogen reduction, resulting in a stable carbon framework. Secondly, the carbon skeleton inherited from the Sn-MOF precursor can further improve the electrical conductivity of the SnSe2/C–N materials, effectively improving the multiplicity and cycling performance of the electrode material. Thirdly, the modification of the Sn-MOF by using nitrogen-containing organic ligands in the MOF clusters enhances the mechanical properties of the carbon skeleton, which further improves the stability of the Li+/Na+ storage skeleton in the SnSe2/C–N materials. Finally, the successful application of SnSe2/C–N materials modified by N-doping only by a simple process in SIBs demonstrates that there is still much room for exploring the potential of Sn-based anode materials in SIBs, and our work will provide new ideas for the application of other N-doped materials in SIBs.Author contributions
L. Jiang, A. L. Song, B. H. J. Tang, and H. Tian designed and supervised the study; Z. Y. Chen and Z. Zhang synthesized the samples, analysed the data and wrote the manuscript; L. Z. Wang, Y. F. Li, and Y. T. Wang performed the DFT calculations; Y. C. Rui characterized the structure; A. L. Song, M. Li, Y. Y. Xiang and K. B. Chou recorded the XRD and XPS spectra; N. Han and G. X. Wang proofread this manuscript. All authors discussed and commented on the manuscript.Data availability
The data that support the findings of this study are available on request from the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52202178), Natural Science Foundation of Shanghai (22ZR1426300), and Class III Peak Discipline of Shanghai-Materials Science and Engineering (High-Energy Beam Intelligent Processing and Green Manufacturing). The authors extend their gratitude to Mr Yang Junjie from Shiyanjia Lab (https://www.shiyanjia.com) for providing invaluable assistance with the BET analysis.References
- D. J. Wuebbles and A. K. Jain, Fuel Process. Technol., 2001, 71, 99–119 CrossRef CAS.
- X. B. Cheng, H. Liu, H. Yuan, H. J. Peng, C. Tang, J. Q. Huang and Q. Zhang, SusMat, 2021, 1, 38–50 CrossRef CAS.
- K. B. Chin, E. J. Brandon, R. V. Bugga, M. C. Smart, S. C. Jones, F. C. Krause, W. C. West and G. G. Bolotin, Proc. IEEE, 2018, 106, 419–428 CAS.
- B. Crew, Nature, 2022, 609, S1–S1 CrossRef CAS.
- W. A. Braff, J. M. Mueller and J. E. Trancik, Nat. Clim. Change, 2016, 6, 964–969 CrossRef.
- M. S. Ziegler, Joule, 2021, 5, 1925–1927 CrossRef.
- J. M. Amanor-Boadu, M. A. Abouzied and E. Sánchez-Sinencio, IEEE Trans. Ind. Electron., 2018, 65, 7383–7394 Search PubMed.
- J. Li, Z. Du, R. E. Ruther, S. J. An, L. A. David, K. Hays, M. Wood, N. D. Phillip, Y. Sheng and C. Mao, JOM, 2017, 69, 1484–1496 CrossRef CAS.
- T. S. Pathan, M. Rashid, M. Walker, W. D. Widanage and E. Kendrick, JPhys Energy, 2019, 1, 044003 CrossRef CAS.
- J. Wang, Q. Zheng, M. Fang, S. Ko, Y. Yamada and A. Yamada, Adv. Sci., 2021, 8, 2101646 CrossRef CAS.
- M. Fan, X. Chang, Q. Meng, L. J. Wan and Y. G. Guo, SusMat, 2021, 1, 241–254 CrossRef CAS.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters and K.-J. Zhou, Nature, 2020, 577, 502–508 CrossRef CAS.
- C. Wang, G. Zhang, S. Ge, T. Xu, Y. Ji, X. Yang and Y. Leng, Nature, 2016, 529, 515–518 CrossRef CAS.
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS.
- J. He, C. Lu, H. Jiang, F. Han, X. Shi, J. Wu, L. Wang, T. Chen, J. Wang and Y. Zhang, Nature, 2021, 597, 57–63 CrossRef CAS PubMed.
- K. J. Griffith, K. M. Wiaderek, G. Cibin, L. E. Marbella and C. P. Grey, Nature, 2018, 559, 556–563 CrossRef CAS.
- Z. Liao, S. Zhang, K. Li, G. Zhang and T. G. Habetler, J. Power Sources, 2019, 436, 226879 CrossRef CAS.
- J. Darga, J. Lamb and A. Manthiram, Energy Technol., 2020, 8, 2000723 CrossRef CAS.
- F. Xie, L. Zhang, C. Ye, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2019, 31, 1800492 CrossRef PubMed.
- Q. Bai, L. Yang, H. Chen and Y. Mo, Adv. Energy Mater., 2018, 8, 1702998 CrossRef.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- H. Tian, A. Song, H. Tian, J. Liu, G. Shao, H. Liu and G. Wang, Chem. Sci., 2021, 12, 7656–7676 RSC.
- J. Chai, N. Han, S. Feng, X. Huang, B. Tang and W. Zhang, Chem. Eng. J., 2023, 453, 139768 CrossRef CAS.
- J. Liu, D. Xie, W. Shi and P. Cheng, Chem. Soc. Rev., 2020, 49, 1624–1642 RSC.
- Y. Ma, Y. Ma, G. T. Kim, T. Diemant, R. J. Behm, D. Geiger, U. Kaiser, A. Varzi and S. Passerini, Adv. Energy Mater., 2019, 9, 1902077 CrossRef CAS.
- P. Lou, Z. Cui, Z. Jia, J. Sun, Y. Tan and X. Guo, ACS Nano, 2017, 11, 3705–3715 CrossRef CAS PubMed.
- L. Wang, Q. Li, Z. Chen, Y. Wang, Y. Li, J. Chai, N. Han, B. Tang, Y. Rui and L. Jiang, Small, 2024, 2310426 CrossRef CAS.
- J. Du, Q. Li, J. Chai, L. Jiang, Q. Zhang, N. Han, W. Zhang and B. Tang, Dalton Trans., 2022, 51, 9584–9590 RSC.
- Y. Gong, Y. Li, Y. Li, M. Liu, Y. Bai and C. Wu, Small, 2023, 19, 2206194 CrossRef CAS.
- H. Tian, H. Tian, S. Wang, S. Chen, F. Zhang, L. Song, H. Liu, J. Liu and G. Wang, Nat. Commun., 2020, 11, 5025 CrossRef CAS PubMed.
- H. Tan, Y. Feng, X. Rui, Y. Yu and S. Huang, Small Methods, 2020, 4, 1900563 CrossRef CAS.
- R. Nkum, A. Adimado and H. Totoe, Mater. Sci. Eng., B, 1998, 55, 102–108 CrossRef.
- W. Yang, Y. Chen, X. Yin, X. Lai, J. Wang and J. Jian, ACS Appl. Mater. Interfaces, 2023, 15, 42811–42822 CrossRef CAS.
- Z. Zhang, X. Zhao and J. Li, Electrochim. Acta, 2015, 176, 1296–1301 CrossRef CAS.
- Y. Kim, Y. Kim, Y. Park, Y. N. Jo, Y.-J. Kim, N.-S. Choi and K. T. Lee, Chem. Commun., 2015, 51, 50–53 RSC.
- Z. Wei, L. Wang, M. Zhuo, W. Ni, H. Wang and J. Ma, J. Mater. Chem. A, 2018, 6, 12185–12214 RSC.
- X. Zhao, W. Wang, Z. Hou, X. Fan, G. Wei, Y. Yu, Q. Di, Y. Liu, Z. Quan and J. Zhang, Inorg. Chem. Front., 2019, 6, 562–565 RSC.
- J. Xia, Y. Yuan, H. Yan, J. Liu, Y. Zhang, L. Liu, S. Zhang, W. Li, X. Yang and H. Shu, J. Power Sources, 2020, 449, 227559 CrossRef CAS.
- Q. Yu, B. Wang, J. Wang, S. Hu, J. Hu and Y. Li, Front. Chem., 2020, 8, 590 CrossRef CAS PubMed.
- K. Wang, W. Ye, W. Yin, W. Chai, Y. Rui and B. Tang, Electrochim. Acta, 2019, 322, 134790 CrossRef CAS.
- X. Li, J. Zheng, C. He, K. Wang, W. Chai, Y. Duan, B. Tang and Y. Rui, Electrochim. Acta, 2019, 326, 134960 CrossRef CAS.
- G. Chang, X. Yin, S. Shi, Y. Zhao and J. Zhang, J. Electrochem. Soc., 2020, 167, 140531 CrossRef CAS.
- L. Jiang, N. Han, T. Luo, Z. Zhang, F. Liang, D. Wu, X. Li, F. Liu, Y. Rui and W. Zhang, Chem. Eng. J., 2023, 458, 141451 CrossRef CAS.
- L. Jiang, W. Yin, C. He, T. Luo, Y. Rui and B. Tang, Colloids Surf., A, 2022, 644, 128843 CrossRef CAS.
- H. Tian, J. Liang and J. Liu, Adv. Mater., 2019, 31, 1903886 CrossRef CAS PubMed.
- H. Tian, F. Huang, Y. Zhu, S. Liu, Y. Han, M. Jaroniec, Q. Yang, H. Liu, G. Q. M. Lu and J. Liu, Adv. Funct. Mater., 2018, 28, 1801737 CrossRef.
- H. Tian, H. Tian, W. Yang, F. Zhang, W. Yang, Q. Zhang, Y. Wang, J. Liu, S. R. P. Silva and H. Liu, Adv. Funct. Mater., 2021, 31, 2101796 CrossRef CAS.
- Z. Ma, A. Song, Z. Liu, Y. Guo, X. Yang, Q. Li, Y. Fan, L. Dai, H. Tian and X. Qin, Adv. Funct. Mater., 2023, 33, 2301112 CrossRef CAS.
- H. Tian, J. Zhao, X. Wang, L. Wang, H. Liu, G. Wang, J. Huang, J. Liu and G. Lu, Natl. Sci. Rev., 2020, 7, 1647–1655 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2012, 124, 11664–11668 CrossRef.
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen and H. Liu, Adv. Mater., 2023, 35, 2210714 CrossRef CAS.
- H. Tian, X. Liu, L. Dong, X. Ren, H. Liu, C. A. H. Price, Y. Li, G. Wang, Q. Yang and J. Liu, Adv. Sci., 2019, 6, 1900807 CrossRef CAS PubMed.
- J. Zheng, X. Li, C. He, C. Zhou, H. Zhang, B. Tang and Y. Rui, ChemElectroChem, 2020, 7, 782–791 CrossRef CAS.
- Z. Li, W. Zhang, Y. Tan, J. Hu, S. He, A. Stein and B. Tang, Electrochim. Acta, 2016, 214, 103–109 CrossRef CAS.
- X. Li, C. He, J. Zheng, W. Ye, W. Yin, B. Tang and Y. Rui, J. Alloys Compd., 2020, 842, 155605 CrossRef CAS.
- Y. Xu, T. Yuan, Z. Bian, J. Yang and S. Zheng, J. Power Sources, 2020, 453, 227467 CrossRef CAS.
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- L. Zhang, H. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2019, 388, 293–309 CrossRef CAS.
- L. Chen and Q. Xu, Matter, 2019, 1, 57–89 CrossRef.
- Q. Li, N. Han, J. Chai, W. Zhang, J. Du, H. Tian, H. Liu, G. Wang and B. Tang, Energy, 2023, 128378, DOI:10.1016/j.energy.2023.128378.
- J. H. Na, H. G. Oh, S. Lee and S.-K. Park, J. Mater. Chem. A, 2024, 12, 2848–2855 RSC.
- Z. Bian, A. Li, R. He, H. Song, X. Chen, J. Zhou and Z. Ma, Electrochim. Acta, 2018, 289, 389–396 CrossRef CAS.
- H. G. Oh and S. K. Park, Int. J. Energy Res., 2022, 46, 10677–10688 CrossRef CAS.
- Z. Zhang, L. Jiang, D. Wu, F. Liang, X. Li, Y. Rui and B. Tang, J. Alloys Compd., 2021, 883, 160834 CrossRef CAS.
- L. Jiang, F. Liang, Z. Zhang, D. Wu, J. Chai, T. Luo, N. Han, W. Zhang, Y. Rui and B. Tang, Chem. Eng. J., 2022, 433, 133568 CrossRef CAS.
- H. G. Oh, S. H. Yang, Y. C. Kang and S. K. Park, Int. J. Energy Res., 2021, 45, 17738–17748 CrossRef CAS.
- J. H. Na, Y. C. Kang and S.-K. Park, Chem. Eng. J., 2021, 425, 131651 CrossRef.
- L. Wang, Y. Zheng, X. Wang, S. Chen, F. Xu, L. Zuo, J. Wu, L. Sun, Z. Li and H. Hou, ACS Appl. Mater. Interfaces, 2014, 6, 7117–7125 CrossRef CAS.
- S. Guo, J. Liu, Q. Zhang, Y. Jin and H. Wang, J. Alloys Compd., 2021, 855, 157538 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS.
- L. Chen, Z. X. Rao, Z. Chen, Z. H. Wu, S. Yang, Z. J. Feng, H. Y. Yang, F. Y. Zeng, X. B. Li, J. Y. Hu and J. T. Huang, Rare Met., 2024, 43, 1836–1844 CrossRef CAS.
- Y. Li, Y. Wang, Z. Chen, Q. Li, J. Du, J. Chai, L. Wang, Y. Rui, L. Jiang and B. Tang, Chem. Eng. J., 2023, 476, 146577 CrossRef CAS.
- N. Wu, T. Jia, Y. R. Shi, Y. J. Yang, T. H. Li, F. Li and Z. Wang, Ionics, 2020, 26, 1547–1553 CrossRef CAS.
- H. S. Kim, D. H. Park, Y. B. Lee, D. C. Kim, H. J. Kim, J. Kim and J. Joo, Synth. Met., 2007, 157, 910–913 CrossRef CAS.
- B. Tian and G. Zerbi, J. Chem. Phys., 1990, 92, 3886–3891 CrossRef CAS.
- Y. Zou, C. Cai, C. Xiang, P. Huang, H. Chu, Z. She, F. Xu, L. Sun and H. B. Kraatz, Electrochim. Acta, 2018, 261, 537–547 CrossRef CAS.
- B. Cong, S. Sun, B. Wang, C. Lv, J. Zhao, F. Jin, J. Jia and G. Chen, Chem. Eng. J., 2022, 435, 135185 CrossRef CAS.
- B. Guo, Y. Yang, Z. Hu, Y. An, Q. Zhang, X. Yang, X. Wang and H. Wu, Electrochim. Acta, 2017, 223, 74–84 CrossRef CAS.
- W. Xie, X. Yun, D. Lu, W. Song, H. Gao, Q. Guo, Y. Han, H. Wang, P. Xiao and Y. Chen, ACS Sustainable Chem. Eng., 2023, 12, 375–385 CrossRef.
- G. Xi, X. Jiao and X. Hu, Ionics, 2023, 29, 119–128 CrossRef CAS.
- F. Zhang, Y. Shen, M. Shao, Y. Zhang, B. Zheng, J. Wu, W. Zhang, A. Zhu, F. Huo and S. Li, ACS Appl. Mater. Interfaces, 2019, 12, 2346–2353 CrossRef.
- Y. Chen, Q. Yang, F. Zhou, J. Wu, J. Wang and Y. Li, Ionics, 2021, 27, 3801–3809 CrossRef CAS.
- J. Tian, Y. Yao, L. Yang, L. Zha, G. Xu, S. Huang, T. Wei, J. Cao and X. Wei, New J. Chem., 2022, 46, 5848–5860 RSC.
- E. Lewin and J. Patscheider, J. Alloys Compd., 2016, 682, 42–51 CrossRef CAS.
- Y. Wu, F. Li, J. Xue and Z. Lv, Turk. J. Chem., 2020, 44, 393–408 CrossRef CAS PubMed.
- K. Yang, X. Zhang, K. Song, J. Zhang, C. Liu, L. Mi, Y. Wang and W. Chen, Electrochim. Acta, 2020, 337, 135783 CrossRef CAS.
- D. Gao, Y. Wang, Y. Liu, H. Sun, M. Wu and H. Zhang, J. Colloid Interface Sci., 2019, 538, 116–124 CrossRef CAS.
- Y. Cheng, J. Huang, H. Qi, L. Cao, X. Luo, J. Li, Z. Xu and J. Yang, Nanoscale, 2017, 9, 18681–18689 RSC.
- X. Li, J. Li, R. N. Ali, Z. Wang, G. Hu and B. Xiang, Chem. Eng. J., 2019, 368, 764–771 CrossRef CAS.
- W. Yao, C. Cai, J. Xiang, L. He, J. Shi, X. Chang, L. Ruan, Z. Chen and Z. Yao, J. Electron. Mater., 2022, 51, 6654–6662 CrossRef CAS.
- Y. Zhuang, J. G. Seong, Y. S. Do, H. J. Jo, Z. Cui, J. Lee, Y. M. Lee and M. D. Guiver, Macromolecules, 2014, 47, 3254–3262 CrossRef CAS.
- Y. Cheng, J. Huang, L. Cao, H. Xie, Y. Wang, Y. Wang, H. Wu, S. Xi and J. Li, Ionics, 2020, 26, 2855–2862 CrossRef CAS.
- H. Chen, R. Liu, Y. Wu, J. Cao, J. Chen, Y. Hou, Y. Guo, R. Khatoon, L. Chen and Q. Zhang, Chem. Eng. J., 2021, 407, 126973 CrossRef CAS.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed.
- B. Sun, Q. Zhang, H. Xiang, F. Han, W. Tang, G. Yuan, Y. Cong, C. Fan, A. Westwood and X. Li, Energy Storage Mater., 2020, 24, 450–457 CrossRef.
- W. Chai, F. Yang, W. Yin, S. You, K. Wang, W. Ye, Y. Rui and B. Tang, Dalton Trans., 2019, 48, 1906–1914 RSC.
- T. Hou, B. Liu, X. Sun, A. Fan, Z. Xu, S. Cai, C. Zheng, G. Yu and A. Tricoli, ACS Nano, 2021, 15, 6735–6746 CrossRef CAS.
- T. Li, L. Wang and J. Li, Electrochim. Acta, 2022, 408, 139947 CrossRef CAS.
- L. Möller, E. Thauer, A. Ottmann, L. Deeg, R. Ghunaim, S. Hampel and R. Klingeler, J. Alloys Compd., 2020, 834, 155018 CrossRef.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- X. Wu, Z. Yang, L. Xu, J. Wang, L. Fan, F. Kong, Q. Shi, Y. Piao, G. Diao and M. Chen, J. Energy Chem., 2022, 74, 8–17 CrossRef CAS.
- A. Marrouche, B. Bouargane, A. Mabrouk, M. G. Biyoune, A. Bachar, A. Atbir, L. Boukbir, S. Mançour-Billah, R. Bellajrou and M. El Hadek, Mediterr. J. Chem., 2019, 8, 10–16 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1−S4. See DOI: https://doi.org/10.1039/d4nr02418d |
| This journal is © The Royal Society of Chemistry 2024 |