
Alternating vs. random amphiphilic polydisulfides: aggregation, enzyme activity inhibition and redox-responsive guest release†
Sukanya
Bera
and
Suhrit
Ghosh
 *
*
School of Applied and Interdisciplinary Sciences, Indian Association for the Cultivation of Science, 2A and 2B Raja S. C. Mullick Road, Kolkata, India 700032. E-mail: psusg2@iacs.res.in
First published on 26th August 2024
Abstract
Herein, we report the synthesis of an alternating copolymer (ACP) with a bio-reducible amphiphilic polydisulfide backbone and highlight the impact of the alternating monomer connectivity on the self-assembly, morphology, chain-exchange dynamics, drug-release kinetics, and enzyme activity inhibition. Condensation polymerization between hydrophobic 1,10-bis(pyridin-2-yldisulfaneyl)decane and hydrophilic 2,3-mercaptosuccinic acid (1.04![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.00 ratio) generated amphiphilic ACP P1 (Mw = 8450 g mol−1, Đ = 1.3), which exhibited self-assembly in water, leading to the formation of an ultra-thin (height <5.0 nm) entangled fibrillar network. In contrast, structurally similar amphiphilic random copolymer P2 exhibited a truncated irregular disc-like morphology under the same conditions. It is postulated that due to the perfect alternating sequence of the hydrophobic and hydrophilic segments in P1, its immiscibility-driven aggregation in water leads to a pleated structure, which further assembles and forms the observed long fibrillar structures, similar to crystallization-driven self-assembly. In fact, wide-angle X-ray diffraction (WXRD) analysis of a lyophilized P1 sample showed sharp peaks, indicating its crystalline nature (approximately 37% crystallinity), and these were completely missing for P2. The effect of such distinct self-assembly on the chain-exchange dynamics was probed by fluorescence resonance energy transfer (FRET) using 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) as the FRET-donor and -acceptor, respectively. For DiI- and DiO-entrapped solutions of P1, when mixed, no prominent FRET appeared even after 24 h. In sharp contrast, for P2, intense FRET emission occurred, and the FRET ratio (approximately 0.9) reached saturation in approximately 15 h, indicating the greatly enhanced kinetic stability of P1 aggregates. Glutathione-induced release of encapsulated Nile red showed much slower kinetics for P1 compared to that of P2, which was corroborated by the observed slow chain-exchange dynamics of the highly stable alternating copolymer assembly. Furthermore, the well-ordered assembly of P1 exhibited an excellent surface-functional group display (zeta potential of −32 mV compared to −14 mV for P2), which resulted in the effective recognition of the α-chymotrypsin (Cht) protein surface by electrostatic interaction. Consequently, P1 significantly (>70%) suppressed the enzymatic activity of Cht, while in the presence of P2, the enzyme was still active with >70% efficacy.
:1.00 ratio) generated amphiphilic ACP P1 (Mw = 8450 g mol−1, Đ = 1.3), which exhibited self-assembly in water, leading to the formation of an ultra-thin (height <5.0 nm) entangled fibrillar network. In contrast, structurally similar amphiphilic random copolymer P2 exhibited a truncated irregular disc-like morphology under the same conditions. It is postulated that due to the perfect alternating sequence of the hydrophobic and hydrophilic segments in P1, its immiscibility-driven aggregation in water leads to a pleated structure, which further assembles and forms the observed long fibrillar structures, similar to crystallization-driven self-assembly. In fact, wide-angle X-ray diffraction (WXRD) analysis of a lyophilized P1 sample showed sharp peaks, indicating its crystalline nature (approximately 37% crystallinity), and these were completely missing for P2. The effect of such distinct self-assembly on the chain-exchange dynamics was probed by fluorescence resonance energy transfer (FRET) using 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) as the FRET-donor and -acceptor, respectively. For DiI- and DiO-entrapped solutions of P1, when mixed, no prominent FRET appeared even after 24 h. In sharp contrast, for P2, intense FRET emission occurred, and the FRET ratio (approximately 0.9) reached saturation in approximately 15 h, indicating the greatly enhanced kinetic stability of P1 aggregates. Glutathione-induced release of encapsulated Nile red showed much slower kinetics for P1 compared to that of P2, which was corroborated by the observed slow chain-exchange dynamics of the highly stable alternating copolymer assembly. Furthermore, the well-ordered assembly of P1 exhibited an excellent surface-functional group display (zeta potential of −32 mV compared to −14 mV for P2), which resulted in the effective recognition of the α-chymotrypsin (Cht) protein surface by electrostatic interaction. Consequently, P1 significantly (>70%) suppressed the enzymatic activity of Cht, while in the presence of P2, the enzyme was still active with >70% efficacy.
Introduction
Amphiphilic polymers exhibit immiscibility-driven aggregation in water, leading to different nanostructures.1 They are of interest for different applications, including drug delivery, antibacterial materials, tissue engineering, protein delivery, and others.2 Most of these systems are block copolymers,1,2 with only a relatively small number of reports on amphiphilic homopolymers,3 or random4 or alternating copolymers (ACPs).5 ACPs are unique in the sense that there is a defined sequence of the two different monomers6 in this structure. Because of this unique sequence, alternating copolymers exhibit distinct self-assembly behaviour7 or functional properties.8The first report on the synthesis of an ACP of stilbene with maleic anhydride was published in 1930.9 However, it continues to be a challenge to prepare structurally diverse copolymers with an alternating sequence through chain-growth mechanisms. Steric hindrance, pseudoconnectivity, increased difference in electron density, or a pre-defined sequence between the two comonomers are strategies that have resulted in limited success for synthesis of ACPs by chain polymerization.5 However, in step-growth polymerization between AA- and BB-type monomers, the resulting polymer intrinsically contains the alternating sequence.
Hence, if these monomers can be designed in such a way that one is hydrophobic and the other is hydrophilic, it will produce amphiphilic ACPs without any ambiguity in their sequence. Similar strategies have been used for the synthesis of different amphiphilic ACPs that exhibit chain-folding regulated self-assembly or crystallization.10,11 We envisaged that such well-defined self-assembled systems may be highly relevant for biological applications, for which it is imperative to achieve control over several important parameters such as functional group display, chain-exchange dynamics, or stability.
We recently developed a synthetic methodology for producing polydisulfides (PDSs) through a condensation polymerization pathway.12 In a sense, it is a unique polymerization technique, in which polymerization of any dithiol (AA) can proceed with commercially available 2,2′-dipyridyl disulfide (BB) by thiol-disulfide exchange reaction. Although it is an AA + BB-type polymerization, the same polydisulfides are produced that would have been achieved by oxidative polymerization of the dithiol monomer.
We are particularly interested in disulfide-containing polymers because they present a bio-reducible linker,13 which can be degraded in the presence of glutathione (GSH), especially in an intracellular location due to its significantly higher concentration.14 We realized that this methodology can be easily extended to prepare amphiphilic ACPs with a degradable PDS backbone, which would enable testing of the relevance of such perfectly sequenced polymers for aqueous self-assembly and biological applications. With this objective, in the current study, we synthesized an amphiphilic ACP with a PDS backbone (P1, Scheme 1) and compared its self-assembly, chain-exchange dynamics, and enzyme activity inhibition with a structurally similar random copolymer (P2, Scheme 1).
 | ||
| Scheme 1 Synthesis of P1 and P2. | ||
Results and discussion
Scheme 1 shows the synthesis of P1 and P2. Monomer M1 was synthesized from commercially available 1,10-decanedithiol by reacting it with excess 2,2′-dipyridyl disulfide and isolating a pale yellow oil in 58% yield. The condensation polymerization reaction at rt between 1,10-bis(pyridin-2-yldisulfaneyl)decane (M1, hydrophobic) and commercially available 2,3-dimercaptosuccinic acid (M2, hydrophilic) produced the desired poly(disulfide)-based amphiphilic ACP P1. It was isolated as a colourless solid in 42% yield. During the polymerization, M1 was intentionally used in slight excess (M1:M2 = 1.04:1.00) to avoid the presence of reactive thiol groups at the chain terminal.
P1 was characterized by 1H NMR (Fig. 1) in which all the peaks were unambiguously assigned. An enlarged view of the selected region (δ = 7.0–8.0 ppm) showed the presence of characteristic peaks for the aromatic protons of the pyridyl-disulfide groups, present in the chain-ends. Also visible was the peak at δ = 13 ppm for the carboxylic acid protons. By comparing the intensity of the end-group protons (He) and backbone proton (Ha), the average degree of polymerization could be calculated to be 17. The UV/Vis spectrum (Fig. S1†) showed a band (λ = 250–300 nm) corresponding to the end-groups, from which the molecular weight of P1 was estimated to be 8560 g mol−1. This was a very good match with that estimated from size-exclusion chromatography (SEC) (Fig. S2†) or end-group analysis using the NMR spectrum (Table 1).
 | ||
| Fig. 1 1H NMR of P1 (solvent DMSO-d6); the peaks marked with an ‘X’ were derived from the solvent. | ||
Likewise, the amphiphilic random copolymer P2 was synthesized by condensation polymerization between 1,10-decanedithiol, 2,3-dimercaptosuccinic acid (M2), and 2,2′-dipyridyl disulfide, in which the reactive pyridyl-disulfide and the thiol groups were taken in a 1.04:1.00 ratio, while the two dithiols were taken in a 1:1 ratio. It produced an amphiphilic random copolymer, which was characterized by SEC studies (Fig. S2†), 1H NMR (Fig. S3†), and UV/Vis (Fig. S1†).
The molecular weight of P2, estimated from SEC or end-group analysis, was comparable to that of P1 (Table 1). From the end-group analysis by 1H NMR, the average number of hydrophobic and hydrophilic units was 9 and 10, respectively. It is imperative to note that the molecular weight and dispersity values for both polymers are nearly identical, and therefore, their comparison is free from any intrinsic structural effects other than the monomer connectivity sequence. Also, the dispersity values in the range of approximately 1.2 for polymers synthesized by the step-growth route are appreciable and indicate well-defined polymerization.
The aggregation of P1 was tested in aqueous medium at a basic pH to ensure that the carboxylic acid groups remain as carboxylates. An atomic force microscopy (AFM) image (Fig. 2a) shows long cylindrical structures with an interconnected network formation.15 Interestingly, the height of these worm-like structures is <5.0 nm, indicating that these are extremely thin flat structures. Contrary to P1, the random copolymer P2 shows an irregular disc-like morphology (Fig. 2b) with a height <2 nm, indicating significant difference in the aggregation behaviour of the alternating and random amphiphilic copolymers.
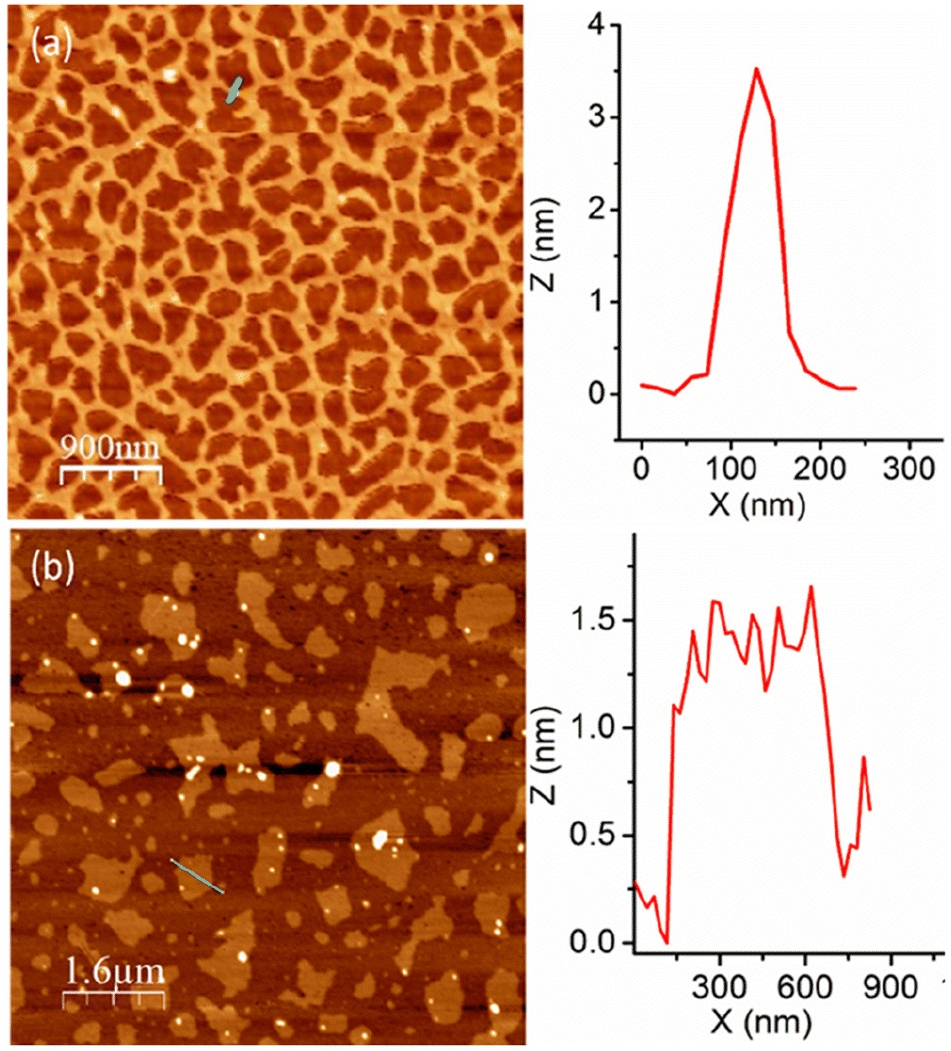 | ||
| Fig. 2 AFM images of (a) P1 and (b) P2 aggregates. Height profile along the green line is shown at the right. | ||
It was proposed that due to the strictly alternating sequences of the hydrophilic and hydrophobic segments in P1, immiscibility-driven aggregation in water leads to a pleated structure (Fig. 3a), which further assembles to the observed long fibrillar structures,15 similar to those observed for the crystallization-driven assembly.16 In fact, wide-angle X-ray diffraction (WXRD) analysis of the sample, obtained by lyophilization of the aqueous solution of P1, showed (Fig. 3b) sharp peaks at 2θ = 12.8, 25.6, 32.2, and 39.0°, indicating the crystalline nature of the aggregate.17 In sharp contrast, no such peaks were noted for random copolymer P2 (Fig. 3b), which highlights the importance of the alternating sequence for the observed well-defined aggregation.
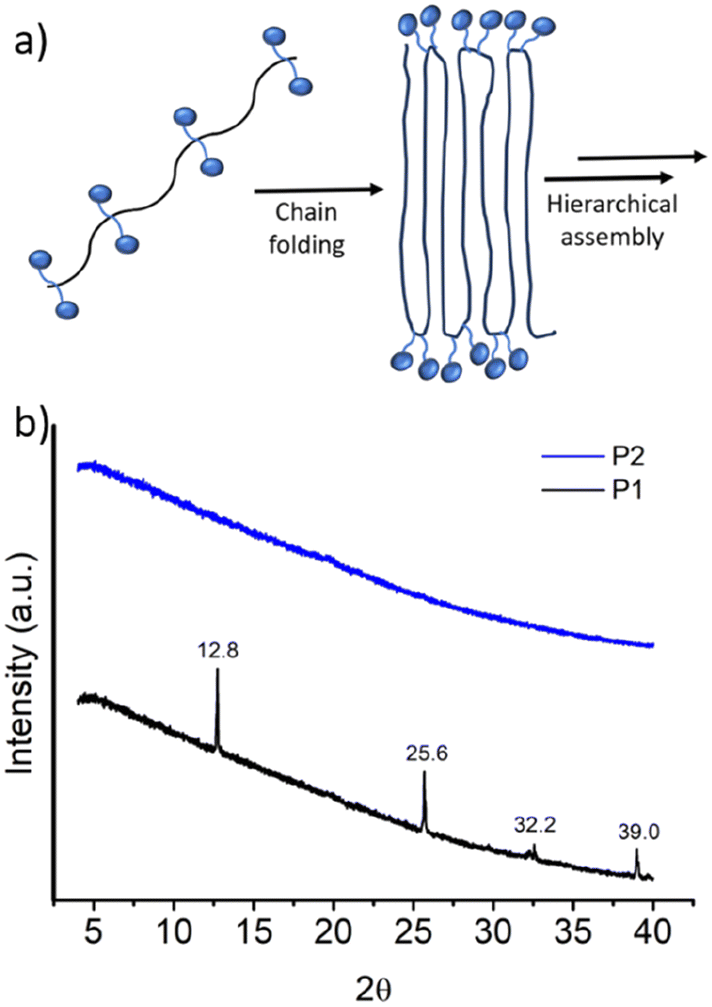 | ||
| Fig. 3 (a) Schematic showing the immiscibility-driven assembly of P1, with its crystalline nature. (b) Wide angle X-ray diffraction pattern of P1 and P2. Experiments were carried out with solid samples obtained after lyophilization of aqueous aggregates (c = 3.0 mg mL−1) of P1 and P2. | ||
The container property of these polymer aggregates was examined by encapsulation of the hydrophobic dye Nile red (NR). In both cases, NR-treated polymer solution showed an intense red colour, indicating successful dye encapsulation in the hydrophobic pocket of these polymer aggregates. The fluorescence spectra showed a typical emission band for NR. By concentration-dependent fluorescence experiments (Fig. S4†), the critical aggregation concentration (CAC) of P1 and P2 was estimated to be roughly 15 μg mL−1 and 27 μg mL−1, respectively.
Next, we examined the chain-exchange dynamics of the aggregates of P1 and P2 by fluorescence resonance energy transfer (FRET) using 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) as the FRET- donor and -acceptor, respectively (Fig. S5†).18 DiI and DiO were separately encapsulated in the P1 aggregate, and then, the two different dye-encapsulated solutions were mixed together, and the FRET emission was monitored as a function of time (Fig. 4).
 | ||
| Fig. 4 (a) Emission spectra (λex = 485 nm) of the mixed solution of DiO- and DiI-encapsulated P1 (black line) and P2 (blue line) just after mixing (dotted line) and after 24 h (solid line). For P1 and P2, peak intensities were normalized to 1.0 at 520 nm and 575 nm, respectively. The concentration of polymer and each dye = 20 μM and 10 μM, respectively. (b) Variation of the FRET ratio as a function of time. | ||
Excitation was set at 485 nm, which corresponded to the absorption of the donor (DiO), and resulted in intense emission at 508 nm corresponding to DiO. However, no such prominent emission band was noted for the acceptor DiI at 570 nm, indicating negligible FRET. Emission spectra were recorded up to 24 h, when no increase in the FRET efficiency was noted. Rather, it remained constant at a very low value of approximately 0.3, which can also be due to the contribution from the direct excitation of the acceptor chromophore. This indicates remarkably slow exchange dynamics in the P1 aggregate. In sharp contrast, for the P2 aggregate, after mixing the DiO and DiI encapsulated solutions, the FRET efficiency spontaneously reached a very high value of approximately 0.7 (Fig. 4), which gradually increased and reached a value of >0.9 in 24 h, suggesting a much faster chain exchange in this case.
The contrast, as shown in Fig. 4b, is truly noteworthy because it clearly indicates that the alternating sequence leads to a stable assembly with a negligible chain exchange, which can be attributed to the crystalline nature of the assembly. The irregular sequence in P2 leads to relatively ineffective chain-packing and consequently loosely bonded aggregates and fast chain exchange. It is noteworthy that for applications such as drug delivery and others, it is imperative to have systems with slow exchange dynamics to minimize leakage of the encapsulated therapeutic molecules. This has generally been achieved by crosslinking and other techniques.19 In that sense, the present system is unique because it demonstrates that crystalline assembly intrinsically can lead to a situation where the chain exchange is extremely slow.
Polydisulfides are of interest because the disulfide linkage can be cleaved in the presence of GSH, which is of biological relevance due to its significantly higher intra-cellular concentration compared to its extracellular domain. GSH is a highly polar tripeptide, while the disulfide linkage in the aggregates is located in the hydrophobic domain. Hence, cleavage of the disulfide bond by diffusion of the GSH to the hydrophobic pocket of the aggregates may not be very effective. Instead, earlier studies with disulfide-containing small molecule surfactants predicted that cleavage may occur in the unimer state of the surfactant, which is always in dynamic equilibrium with the aggregates.20 In that case, the sharp contrast in the chain-exchange dynamics between P1 and P2 may significantly influence the kinetics of GSH-triggered disulfide cleavage and disassembly.
To test such possibilities, the Nile red-encapsulated P1 and P1 aggregates were treated with GSH (10 mM), and the emission spectra of Nile red were monitored as a function of time (Fig. S6,†Fig. 5). The Nile red emission intensity was comparable in both samples.
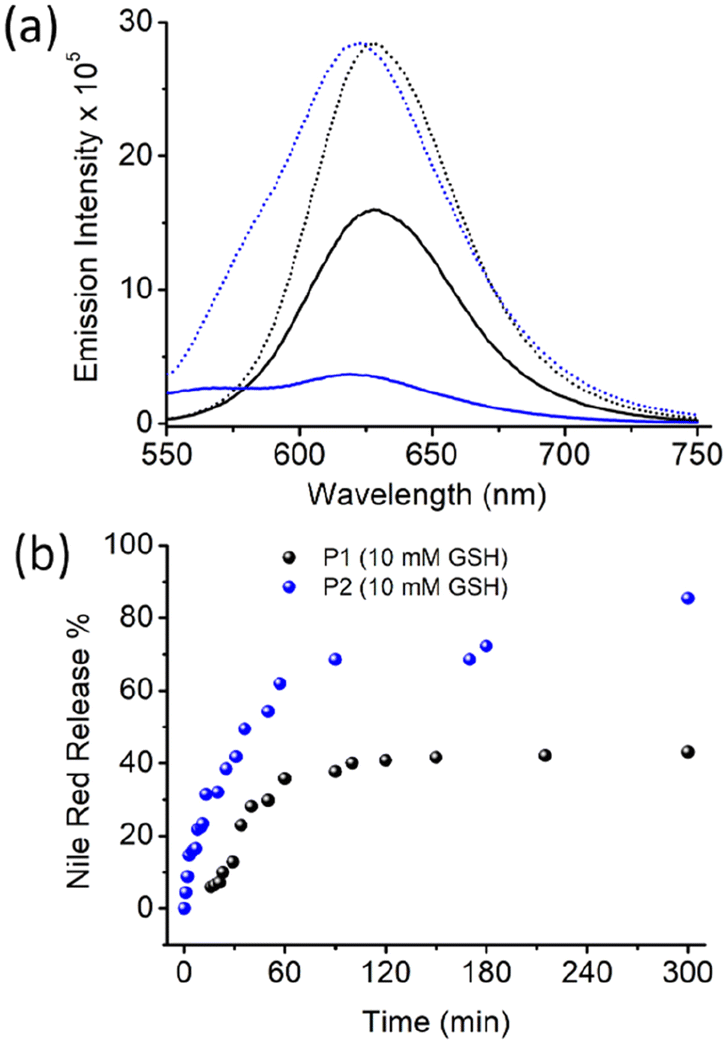 | ||
| Fig. 5 (a) The emission spectra of Nile red encapsulated in P1 (black line) or P2 (blue line) aggregates before (dotted line) and after GSH treatment for 5 h (solid line). The concentrations of Nile red, P1/P2, and GSH were 10−5 M, 0.5 mg mL−1, and 10 mM, respectively. λex = 530 nm. (b) Cumulative % release of Nile red as a function of time after GSH addition. The release % was calculated using an emission intensity of 622 nm. | ||
However, with time, the emission intensity decreased at a much faster rate for P2, and eventually it became negligible after approximately 5 h. For P1, the intensity decreased at a much slower rate, and after the same time interval, significant band intensity remained. The % of dye release was estimated using the band intensity at 622 nm, and was plotted in Fig. 5b. A clear difference was noted, with a much faster release rate for P2, where approximately 90% of the dye was released after 5 h. In the case of P1, a much slower release rate was noted, with only approximately 50% dye release after 5 h. A similar trend was observed when dye release experiments were performed using UV/Vis absorption spectroscopy (Fig. S7†).
This clearly indicates a remarkable influence of the chain-exchange dynamics on the rate of disassembly of these polydisulfide aggregates, which can be clearly corroborated by earlier reports suggesting that such disulfide cleavage occurs in the unimer state of the surfactant rather than in the aggregated state. It is imperative to note that depending on the specific need, both fast release and sustained release are important. Hence, the ability to tune the release rate to such an extent by controlling the polymer sequence is not only of fundamental interest, but also could be of importance for practical applications.
Finally, we examined the effect of the well-ordered assembly of P1 on the surface-functional group display and attempted to correlate it with recognition of the protein surface by electrostatic interaction. For this study, α-chymotrypsin (Cht) with a cationic surface charge was used as the model protein.
The polymers are negatively charged due to the presence of the carboxylate groups, and therefore, it is anticipated that they will bind to the protein surface by electrostatic interaction, which in turn will hamper the enzymatic activity of Cht, depending on the effectiveness of a given polymer aggregate to bind to the protein surface.21 To test this, activity assays were carried out with a chromogenic substance, namely N-succinyl-L-phenylalanine-p-nitroanilide (SPNA) (Fig. 6).
 | ||
| Fig. 6 (a) Scheme showing the Cht-induced product formation of the SPNA substrate. (b) Time versus concentration plot of p-nitroaniline generated from the hydrolysis of SPNA (2 mM) in the presence of Cht (3.2 μm) after incubation with P1 or P2 (0.1 mM) or without addition of polymer (control) at pH 9.0. (c) Relative Cht activity in the presence of P1 and P2. | ||
It is known that Cht, in its active form, can hydrolyse SPNA, which produces p-nitroaniline with an absorption band in the visible region (λmax = 405 nm). The activity of Cht in the absence of any polymer was initially confirmed, and a sharp increase in the absorption intensity at 405 nm with time was observed, which indicated that the protein was in its active form. Cht, preincubated with P1, showed negligible production of p-nitroaniline, indicating a prominent decrease in the enzymatic activity by approximately 70%. However, in the presence of P2, the decrease in enzymatic activity was only 30%, which was significantly less than that of P1.
Therefore, it is evident that the strict alternating sequence in P1 not only leads to stable self-assembly, but also assists in well-defined chain packing, which leads to a more optimal functional group display. This was also evident from a significantly higher negative zeta potential (−32 mV) for P1 compared to −14 mV for P2. Finally, HeLa cells were subjected to the MTT assay (Fig. S8†) with P1 and P2 (concentration up to 400 μg mL−1), which showed that >85% of cells were alive even after 48 h. This indicated excellent compatibility of cells with the polymers and the possibility of their future biomedical application.
Conclusions
Herein, we described an amphiphilic polydisulfide with an alternating sequence of hydrophobic and hydrophilic monomers, and demonstrated its facile synthesis by condensation polymerization involving the thiol-activated disulfide exchange reaction. Self-assembly studies showed that such an alternating sequence is highly useful to achieve distinct morphology and negligible chain-exchange dynamics, which can be attributed to the immiscibility-driven crystalline chain-packing that is missing in structurally similar random copolymers.Kinetic stability of the aggregate assists in slow degradation of the backbone disulfide linker in the presence of GHS, which is highly useful for sustained drug release. Furthermore, well-ordered chain packing leads to a distinct ultrathin fibrillar morphology with excellent functional group (carboxylate) display that enables electrostatic interaction-mediated surface recognition of the enzyme chymotrypsin (Cht), with approximately 70% inhibition of its enzymatic activity in contrast to the random copolymer, which exhibited negligible enzyme activity inhibition.
Despite significant research progress in the self-assembly of amphiphilic polymers and their functional utility, alternating copolymers are relatively uncommon, perhaps due to the difficulty in producing such polymers with a perfect alternating sequence. However, easy access to such polymers can be attained by a condensation polymerization approach, and the results reported in this manuscript should be highly inspiring to explore such systems for diverse functional materials.
Author contributions
SB performed all the experimental work and collected the data. The data analysis and manuscript writing were jointly performed by SB and SG. SG conceptualized the work, supervised the project, and raised research funding.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
SB thanks IACS for a research fellowship. SG thanks the Technical Research Centre, IACS for funding.References
- (a) Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC; (b) P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7055 RSC; (c) T. H. Epps III and R. K. O'Reilly, Chem. Sci., 2016, 7, 1674 RSC; (d) H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61 CrossRef CAS; (e) F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468 RSC.
- (a) N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602 CrossRef CAS PubMed; (b) H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844 CrossRef CAS PubMed; (c) E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866 CrossRef CAS; (d) Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542 RSC; (e) R. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836 CrossRef CAS; (f) B. S. Bolu, R. Sanyal and A. Sanyal, Molecules, 2018, 23, 1570 CrossRef PubMed; (g) S. R. Mane, A. Sathyan and R. Shunmugam, ACS Appl. Nano Mater., 2020, 3, 2104 CrossRef CAS; (h) R. Aluri, S. Saxena, D. Joshi and M. Jayakannan, Biomacromolecules, 2018, 19, 2166 CrossRef CAS PubMed; (i) S. Saxena and M. Jayakannan, Biomacromolecules, 2020, 21, 171 CrossRef CAS PubMed.
- T. S. Kale, A. Klaikherd, B. Popere and S. Thayumanavan, Langmuir, 2009, 25, 9660 CrossRef CAS.
- (a) L. Li, K. Raghupathi, C. Song, P. Prasad and S. Thayumanavan, Chem. Commun., 2014, 50, 13417 RSC; (b) Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 5084 CrossRef CAS.
- K. Nishimori and M. Ouchi, Chem. Commun., 2020, 56, 3473 RSC.
- N. Badia and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383 RSC.
- (a) S. G. Fenimore, L. Abezgauz, D. Danino, C. C. Ho and C. C. Co, Macromolecules, 2009, 42, 2702 CrossRef CAS; (b) M. Ueda, A. Hashidzume and T. Sato, Macromolecules, 2011, 44, 2970 CrossRef CAS; (c) K. Nishimori, E. Cazares-Cortes, J. M. Guigner, F. Tournilhac and M. Ouchi, Polym. Chem., 2019, 10, 2327 RSC; (d) B. Saha, N. Choudhury, S. Seal, B. Ruidas and P. De, Biomacromolecules, 2019, 20, 546 CrossRef CAS.
- (a) E. Zhao, J. W. Y. Lam, L. M. Meng, Y. Hong, H. Q. Deng, G. X. Bai, X. H. Huang, J. H. Hao and B. Z. Tang, Macromolecules, 2015, 48, 64 CrossRef CAS; (b) B. Saha, K. Bauri, A. Bag, P. K. Ghorai and P. De, Polym. Chem., 2016, 7, 6895 RSC; (c) J. J. Yan, Z. K. Wang, X. S. Lin, C. Y. Hong, H. J. Liang, C. Y. Pan and Y. Z. You, Adv. Mater., 2012, 24, 5617 CrossRef CAS; (d) B. Saha, N. Choudhury, A. Bhadran, K. Bauri and P. De, Polym. Chem., 2019, 10, 3306 RSC.
- T. Wagner-Jauregg, Ber. Dtsch. Chem. Ges. B, 1930, 63, 3213 CrossRef.
- R. Barman, A. Mukherjee, A. Nag, P. Rajdev and S. Ghosh, Chem. Commun., 2023, 59, 13951 RSC.
- V. Damodara, H. Sardana and S. Ramakrishnan, Eur. Polym. J., 2024, 112818 CrossRef CAS.
- D. Basak, R. Kumar and S. Ghosh, Macromol. Rapid. Commun., 2014, 35, 1340 CrossRef CAS.
- (a) R. Bej, P. Dey and S. Ghosh, Soft Matter, 2020, 16, 11 RSC; (b) I. Altinbasak, M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 7603 RSC; (c) J. Zhuang, M. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421 RSC; (d) J. Quinn, M. Whittaker and T. Davis, Polym. Chem., 2017, 8, 97 RSC; (e) H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef; (f) J.-H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246 CrossRef CAS; (g) I. Altinbasak, S. Kocak, R. Sanyal and A. Sanyal, Biomacromolecules, 2022, 23, 3525 CrossRef CAS PubMed.
- (a) G. K. Such, Y. Yan, A. P. R. Johnston, S. T. Gunawan and F. Carus, Adv. Mater., 2015, 27, 2278 CrossRef CAS PubMed; (b) A. Russo, W. DeGraff, N. Friedman and J. B. Mitchell, Cancer Res., 1986, 46, 2845 CAS; (c) F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191 CrossRef CAS.
- (a) J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, J. Am. Chem. Soc., 2019, 141, 2742 CrossRef CAS; (b) A. Sikder, S. Chakraborty, P. Rajdev, P. Dey and S. Ghosh, Acc. Chem. Res., 2021, 54, 2670 CrossRef CAS.
- (a) X. He, Y. He, M.-S. Hsiao, R. L. Harniman, S. Pearce, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2017, 139, 9221 CrossRef CAS; (b) X. He, M. S. Hsiao, C. E. Boott, R. L. Harniman, A. Nazemi, X. Li, M. A. Winnik and I. Manners, Nat. Mater., 2017, 16, 481 CrossRef CAS; (c) M. Inam, G. Cambridge, A. Pitto-Barry, Z. P. L. Laker, N. R. Wilson, R. T. Mathers, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2017, 8, 4223 RSC; (d) P. J. Hurst, A. M. J. Rakowski and P. Patterson, Nat. Commun., 2020, 11, 4690 CrossRef CAS; (e) A. Rajak and A. Das, Angew. Chem., Int. Ed., 2022, 61, e202116572 CrossRef CAS.
- C. Zhang, H. Pan and Y. Zhou, Macromolecules, 2023, 56, 7870 CrossRef CAS.
- P. Rajdev and S. Ghosh, J. Phys. Chem. B, 2019, 123, 327 CrossRef CAS.
- (a) J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227 CrossRef CAS; (b) P. Rajdev, D. Basak and S. Ghosh, Macromolecules, 2015, 48, 3360 CrossRef CAS.
- S. Ghosh, K. Irvine and S. Thayumanavan, Langmuir, 2007, 23, 7916 CrossRef CAS PubMed.
- (a) H. S. Park, Q. Lin and A. D. Hamilton, J. Am. Chem. Soc., 1999, 121, 8 CrossRef CAS; (b) B. S. Sandanaraj, D. R. Vutukuri, J. M. Simard, A. Klaikherd, R. Hong, V. M. Rotello and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 10693 CrossRef CAS PubMed; (c) M. De, S. S. Chou and V. P. Dravid, J. Am. Chem. Soc., 2011, 133, 17524 CrossRef CAS; (d) A. Sikder, A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2015, 54, 6755 CrossRef CAS; (e) P. Khanra, P. Rajdev and A. Das, Angew. Chem., Int. Ed., 2024, 63, e202400486 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the polymers, materials and methods, additional self-assembly data and experimental detail. See DOI: https://doi.org/10.1039/d4nr02494j |
| This journal is © The Royal Society of Chemistry 2024 |