
A silver cluster-assembled material as a matrix for enzyme immobilization towards a highly efficient biocatalyst†
Jin
Sakai
a,
Kohki
Sasaki
a,
Riki
Nakatani
a,
Saikat
Das
 *b and
Yuichi
Negishi
*ab
*b and
Yuichi
Negishi
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.tus.ac.jp
bResearch Institute for Science & Technology, Tokyo University of Science, Tokyo 162-8601, Japan. E-mail: saikatdas@rs.tus.ac.jp
First published on 16th September 2024
Abstract
Silver cluster-assembled materials (SCAMs) epitomize well-defined extended crystalline frameworks that combine the ingenious designability at the atomic/molecular level and high structural robustness. They have captivated the interest of the scientific fraternity because of their modular construction which enables to systematically tailor their functions, and their capacity to not only inherit the characteristics of component building units but also introduce their uniqueness in endowing the final material with extraordinary properties. Herein, we demonstrate the synthesis of a novel (3,6)-connected two-dimensional (2D) SCAM [Ag12(StBu)6(CF3COO)6(THIT)6]n (described as TUS 5, THIT = 2,4,6-tri(1H-imidazol-1-yl)-1,3,5-triazine) composed of Ag12 cluster nodes and tritopic imidazolyl linkers. We have leveraged, for the first time, this precisely architected extended SCAM structure as a support matrix for enzyme immobilization. The electrostatic attraction between the negatively charged amano lipase PS and positively charged TUS 5 as well as the surface hydrophobicity of TUS 5 catered to great binding of lipase onto the TUS 5 matrix, in addition to boosting the activity of lipase via interfacial activation. Capitalizing on the cooperative benefits of organic and inorganic support matrices wherein organic supports impart with cost-efficiency, biocompatibility, and improved enzyme stability and reusability and inorganic supports confer high thermal, mechanical and microbial resistance, we have utilized the immobilized lipase on TUS 5 SCAM (lipase@TUS 5) for the kinetic resolution of (R,S)-1-phenylethanol by transesterification reaction. Importantly, lipase@TUS 5 could attain appreciably higher conversion into (R)-1-phenylethyl acetate, besides featuring superior thermal stability, solvent tolerance and recyclability, over the native lipase.
Introduction
The emergence of atomically precise metal nanoclusters (NCs) has radically transformed the nanomaterials research landscape and has imparted a wide range of applications serving materials science, chemistry, biological sciences and healthcare.1–9 Characterized with a core size ranging from sub-nanometre to 3 nm and acting as a bridge between metal atoms and nanoparticles, NCs comprise peripheral ligands that fundamentally perform as stabilizers in addition to regulating the shapes and physical properties like solubility, luminescence etc. of NCs.10–14 Silver (Ag) NCs have been a constant source of interest owing to their elegant structures and strong luminescence produced by electronic transitions between the energy levels.15–20 Despite showing its mettle, silver has greater tendency to oxidize as compared to gold because the standard electrode potential of silver (E°(Ag+/Ag) = 0.79 V) is less than that of gold (E°(Au+/Au) = 1.83 V), which adds to the concern over Ag NCs synthesis and its widespread application.21,22 To alleviate the stability issue, substantial research effort has been directed toward the assembly of Ag NCs.23–25 Inter-cluster self-assembly aids in augmenting the stability; however, the crystal structure of Ag NCs cannot be modulated with self-assembly.26,27 On the other hand, organic linkers were leveraged as struts to bridge the metal ion/cluster nodes in the construction of customizable crystal structures of metal–organic frameworks (MOFs).28 Such research accomplishments triggered the incorporation of organic linkers in the assembly of Ag NC nodes.29 Stitching discrete polynuclear NCs by organic linkers furnishes aesthetically-appealing extended network structures that not merely feature the distinct attributes of the NCs but as well demonstrate superior features attributed to their synergistic cooperation.The fundamental function of organic molecular building units is to establish strong directional coordinate bonding interactions with Ag NC nodes that drive the molecular assembly to harvest silver cluster-assembled materials (SCAMs).30,31 Secondly, the customizability of organic linkers at the atomic/molecular level enables precise control over the crystal structure, network topology, surface property and pore environment of SCAMs. Thirdly, the capacity to accommodate various functional groups in SCAM structures through the utilization of organic linkers imparts with copious active sites and enhances host–guest interactions, thereby increasing the efficacy of SCAMs for distinct applications. Systematic choice of linkers coupled with strategic control over the organization of molecular building blocks to single-crystalline SCAMs can help regulate the behaviour of the assembled NC structures as well as furnish a critical knowledge of the key drivers contributing to the assembly processes.
A groundbreaking study by Zang and colleagues involved the substitution of coordinated acetonitrile ligands of Ag12 NCs with 4,4′-bipyridine (bpy) linkers, generating a rigid two-dimensional (2D) framework.32 The structural rigidity imparted upon coordination network formation gave Ag12bpy SCAM an edge over discrete Ag NCs in stability under different conditions, including long-term exposure to air and light irradiation, as well as in photoluminescence quantum yield owing to suppressed nonradiative decay rates. The library of SCAMs thereafter has enriched significantly upon the adoption of varied linkers to connect Ag(I) cluster nodes and the application extended to diverse sectors,33–51 with a substantial proportion of these structures featuring dodecanuclear Ag NC nodes.
In the quest for new synthesis approaches to the sustainable production of fine chemicals, pharmaceuticals and materials, catalysts serve as an invaluable tool enabling improved energy efficiency and reduced environmental footprint. Attributed to the high reaction specificity, substrate specificity and efficiency of enzymes that far outweigh those of traditional chemical catalysts, enzymatic catalysis has aroused considerable interest for contributing to sustainable catalytic processes.52,53 However, the potential benefits of natural enzymes are undermined by their instability and sensitivity to the working environment (elevated temperatures, organic/non-aqueous media etc.) for which it is crucial to implement a strategic solution of how to immobilize enzymes onto solid host matrices, which could not only enhance the enzymatic activity and stability over a broader range of operating conditions but could also be productive for recycling use.54,55 The earliest instance of industrial implementation of immobilized enzyme can be traced back to 1969 when aminoacylases was physically adsorbed onto Diethylethanolamine–Sephadex and used for the industrial production of L-methionine by Tanabe Seiyaku Co., Ltd, Japan.56,57 Choice of suitable support material as carrier for effectively arresting the enzymes has become a significant research thrust recently. Sol–gel matrices have garnered immense attention for the immobilization of enzymes inasmuch as they improve the mechanical and solvent resistance of enzymes and stabilize them against denaturation.58,59 Nevertheless, their potential is impaired by the limited diffusion of the substrate to the enzyme.60 The competency to absorb water provides hydrogels an upper hand for immobilizing enzymes in non-aqueous media; however, low mass transfer is a significant setback.61 The appeal of inorganic supports in enzyme immobilization derives from their thermal, mechanical and microbial resistance, but fall short of biocompatibility and affinity to enzymes.62,63 On the other hand, organic supports hold considerable promise in enzyme immobilization for their cost-efficiency and high biocompatibility, in addition to boosting the enzyme's operational stability and recyclability.64 The merits of crystalline ordered architectures, structural designability and diversity, and notable stabilities notwithstanding, SCAMs built from the periodic arrangement of inorganic and organic building blocks have not yet been applied as support matrices for enzyme immobilization.
In this contribution, by carefully controlling the synthesis conditions, we accomplished the coordination-driven crystallization of a novel two-dimensionally connected SCAM [Ag12(StBu)6(CF3COO)6(THIT)6]n, designated as TUS 5 (THIT = 2,4,6-tri(1H-imidazol-1-yl)-1,3,5-triazine). This intricate extended structure reinforces the structural stability and leads to intriguing properties, unleashing the potential for a broad scope of applications. We have leveraged this precisely constructed crystalline network as a support for enzyme immobilization. Lipases, conventionally used for the hydrolysis of triglycerides, are now highly sought-after catalysts for a broad assortment of reactions and are in great demand across the pharmaceutical, food and biodiesel industries for their remarkable regio-, enantio- and chemoselectivity as well as specificity.65 Unfortunately, free lipase fails to attain sufficiently good activity, stability and reusability. We herein utilized the immobilized lipase on TUS 5 SCAM for the kinetic resolution of (R,S)-1-phenylethanol via transesterification reaction. Significantly, the immobilized lipase could reach substantially higher conversion into (R)-1-phenylethyl acetate, in addition to showing greater thermal stability, solvent tolerance and recycling performance, over its free form. This work paves the way for the development of SCAM-enzyme composites as prolific biocatalysts at the frontier of synthetic organic chemistry and chemical biology.
Results and discussion
The synthesis of TUS 5 was achieved through a convenient two-step route, commencing with the synthesis of Ag12 NC nodes followed by the incorporation of organic moieties. The first reaction proceeded by mixing [AgStBu]n and CF3COOAg in N,N-Dimethylacetamide (DMAc). At the outset, the mixture of [AgStBu]n in DMAc became cloudy, which afterwards turned clear upon the addition of CF3COOAg that substantiated the development of Ag12 cluster vertices. At the same time, the linker THIT was dissolved in a methanol/acetonitrile (MeOH/MeCN) binary solvent mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Following this, the linker solution was slowly added into the Ag12 NC node solution. The generated mixture was allowed to stand still at room temperature under dark conditions. In the course of 1 day, colorless hexagonal-shaped crystals suitable for single-crystal X-ray diffraction (SCXRD) characterization were obtained from the bottom of the vial in 64.73% yield (based on Ag). Optical microscopy attested to the three-dimensional (3D) growth of the single crystal, besides revealing its hexagonal-shaped morphology (Fig. 1).
:1, v/v). Following this, the linker solution was slowly added into the Ag12 NC node solution. The generated mixture was allowed to stand still at room temperature under dark conditions. In the course of 1 day, colorless hexagonal-shaped crystals suitable for single-crystal X-ray diffraction (SCXRD) characterization were obtained from the bottom of the vial in 64.73% yield (based on Ag). Optical microscopy attested to the three-dimensional (3D) growth of the single crystal, besides revealing its hexagonal-shaped morphology (Fig. 1).
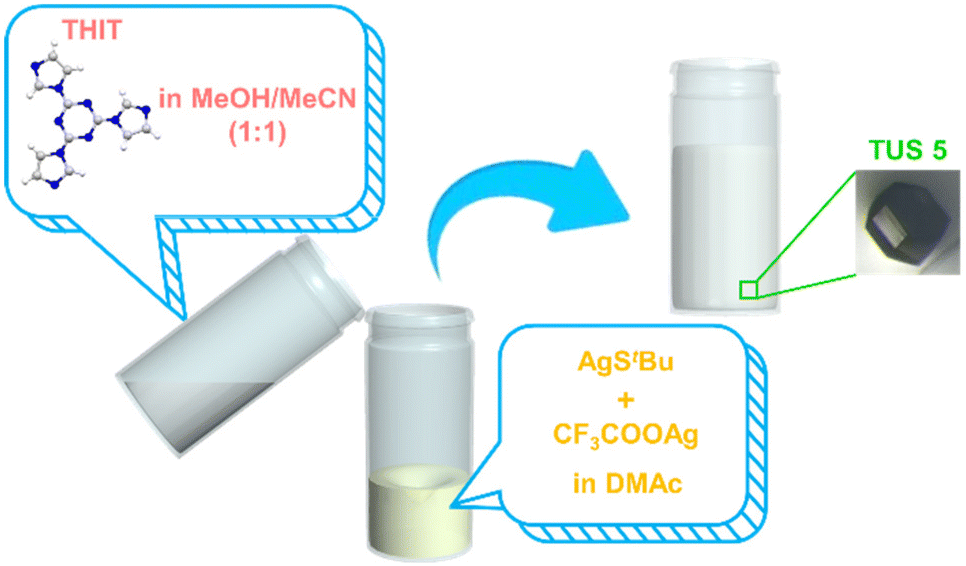 | ||
| Fig. 1 Schematic diagram portraying the synthesis procedure of TUS 5 single crystals. The magnified view of the bottom surface of the product vial displays the optical microscopy image of the single crystal. Color scheme: N, blue; C, grey; and H, white. | ||
SCXRD was accomplished to procure in-depth information and knowledge of the elaborate crystal structure of the SCAM. As reflected from SCXRD analysis, TUS 5 crystallizes in a trigonal crystal system and adopts the P3 (no. 143) space group (Table S2†). Each NC node comprises a Ag12 core co-protected by six StBu ligands and six CF3COO− ligands, and coordinated to six linker molecules (Fig. S2†). The cluster core carrying twelve Ag(I) atoms can be represented as an empty cuboctahedron scaffold (Fig. 2a). This geometric form exemplifies an Archimedean solid characterized by fourteen faces: eight triangles and six trapezoids. To delve deeper into the structure, the Ag12 core can be dissected into three layers: a middle hexagonal layer, bearing six Ag atoms, with two equilateral triangular layers above and beneath, each accommodating three Ag atoms (Fig. S1†). The Ag–Ag edges of the upper and lower triangular layers measure 3.017 to 3.055 Å in length. On the other hand, the Ag–Ag bonding between the hexagonal layer and equilateral triangular layers that help hold the structure together, range from 3.031 to 3.207 Å (Table S3†). Since both these bond lengths lie below the sum of the van der Waals radii of two Ag atoms (3.44 Å),66 the cuboctahedral architecture is regulated by pronounced argentophilic interactions.67 The trapezoidal faces are passivated by six bulky thiolate (–StBu) ligands that project out from the Ag12 NC nodes and function as surface capping agents (Fig. 2b). Each thiolate ligand features μ4-η1,η1,η1,η1 ligation behavior to connect four Ag atoms: two Ag atoms from the upper or lower triangular layer and two Ag atoms from the middle hexagonal layer via Ag–S bonds with an average bond length of 2.506 Å (Table S4†). As depicted in Fig. 2c, the metal skeleton is further protected by six trifluoroacetate (–CF3COO−) ligands. Specifically, each CF3COO− ligand taking the μ2-η1,η1 ligation mode bridges two different Ag atoms, one from the top or bottom triangular layer and another from the middle hexagonal layer, through O atoms with an average Ag–O bond length of 2.529 Å (Table S5†). Moreover, six coordination sites at the middle hexagonal layer holding six Ag atoms in each Ag12 cluster node are bound to six N donors from the imidazolyl groups of THIT linkers (Fig. 2d), showcasing an average Ag–N bond length of 2.219 Å (Table S6†).
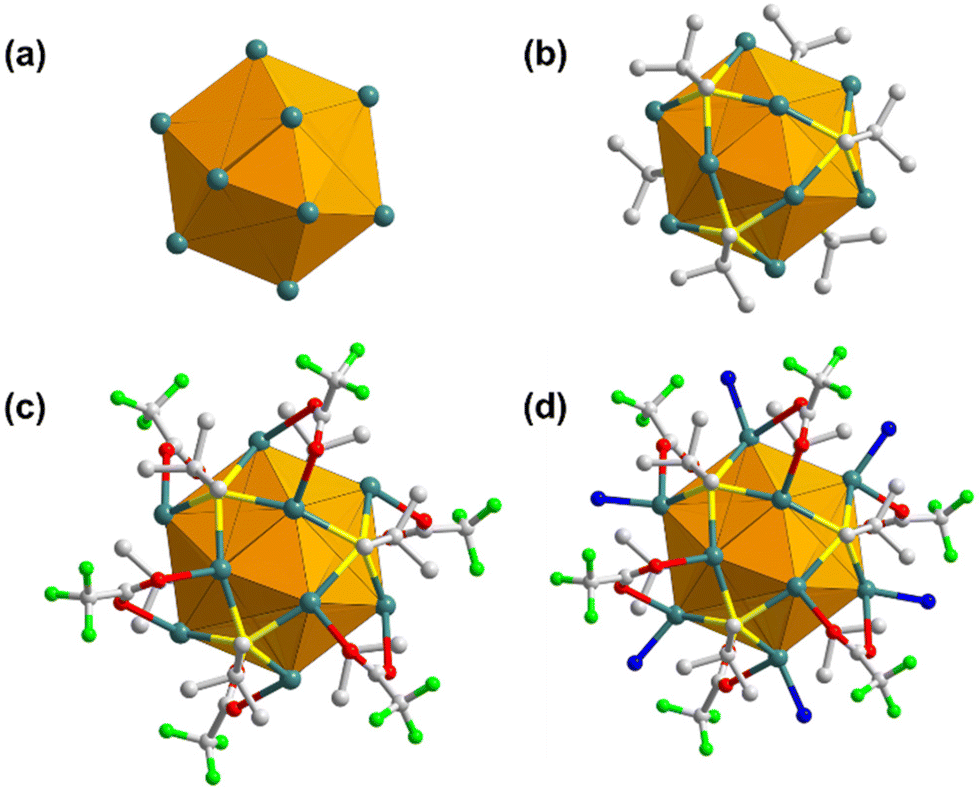 | ||
| Fig. 2 Stepwise construction of each NC node: (a) empty cuboctahedron scaffold representing the Ag12 cluster core; attachment of (b) StBu ligands, (c) CF3COO− ligands, and (d) organic linkers, on the Ag12 NC node. H atoms and linker parts have been excluded for clarity. | ||
In more detail, each THIT linker assumes the role of a 3-c organic node linking with three neighboring Ag NC nodes. At the same time, each Ag NC node acts as a 6-c inorganic node coordinated to six linker molecules to generate a periodically extending structure along the ab-plane (Fig. 3a). Satisfyingly, the cluster–linker connectivities resulted in the exquisite construction of 2D (3,6)-connected layers, that are further vertically stacked to form 3D array via interlayer noncovalent interactions (Fig. 3b). An interlayer spacing of 11.1 Å between adjacent 2D layers was obtained from the crystal structure (Fig. S7†). This stacking geometry afforded open pore nanochannels orthogonal to the ab-plane. The porosity of TUS 5 was examined through N2 uptake experiments at 77 K. Type-I adsorption–desorption isotherms hallmarked by a steep rise under low relative pressures (P/P0 < 0.01) was observed, illustrative of the microporous characteristics of the SCAM (Fig. 4a). The Brunauer–Emmett–Teller (BET) surface area was determined to be 270 m2 g−1 (Fig. S8†). Implementation of the nonlocal density functional theory (NLDFT) yielded a pore-size distribution centered at 0.6 nm (Fig. S9†).
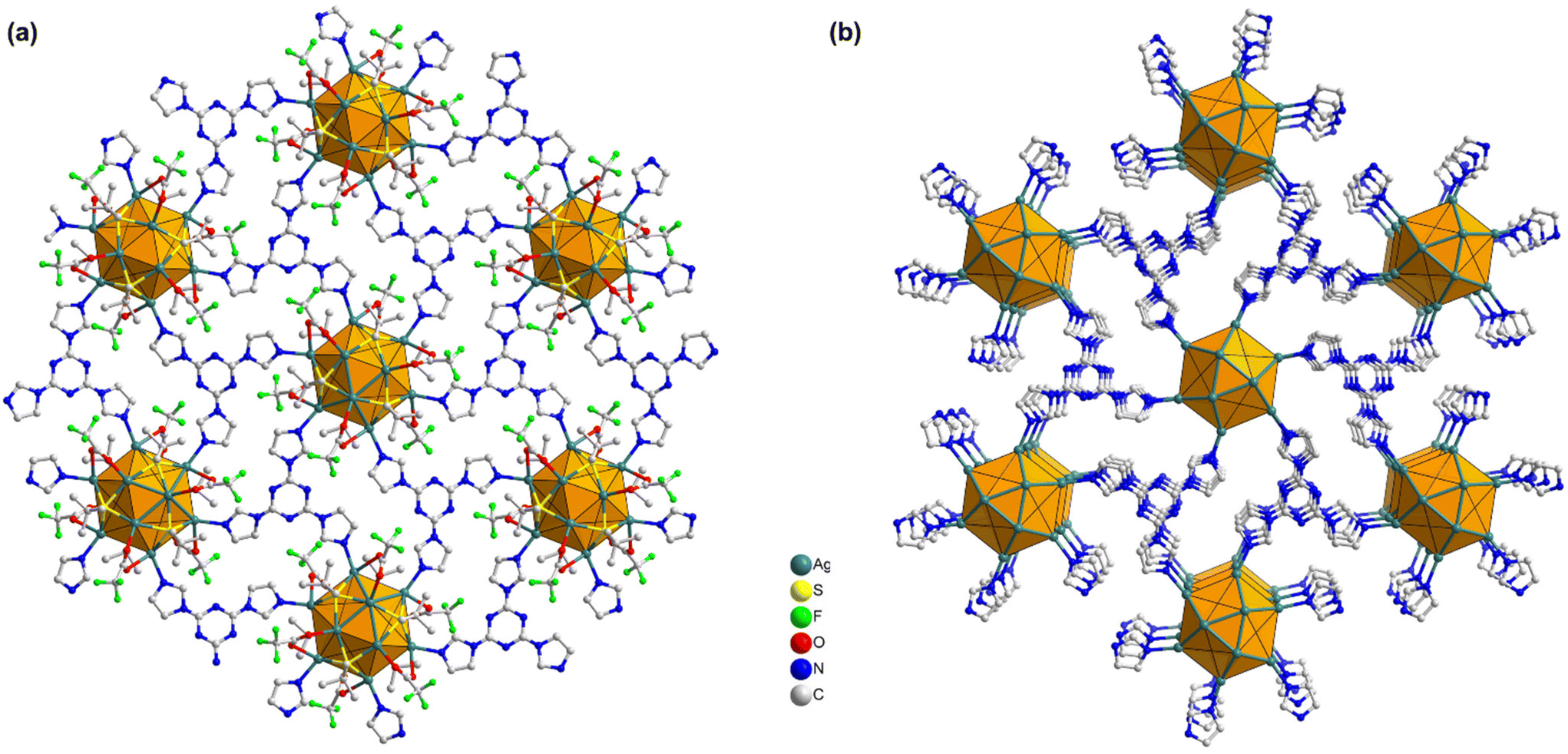 | ||
| Fig. 3 (a) 2D (3,6)-connected layer of TUS 5 constructed by Ag12 cluster vertices and organic linkers through coordination interactions, (b) 3D array arising out of the stacked layers of TUS 5. All H atoms and occasionally ligands have been excluded for clarity. | ||
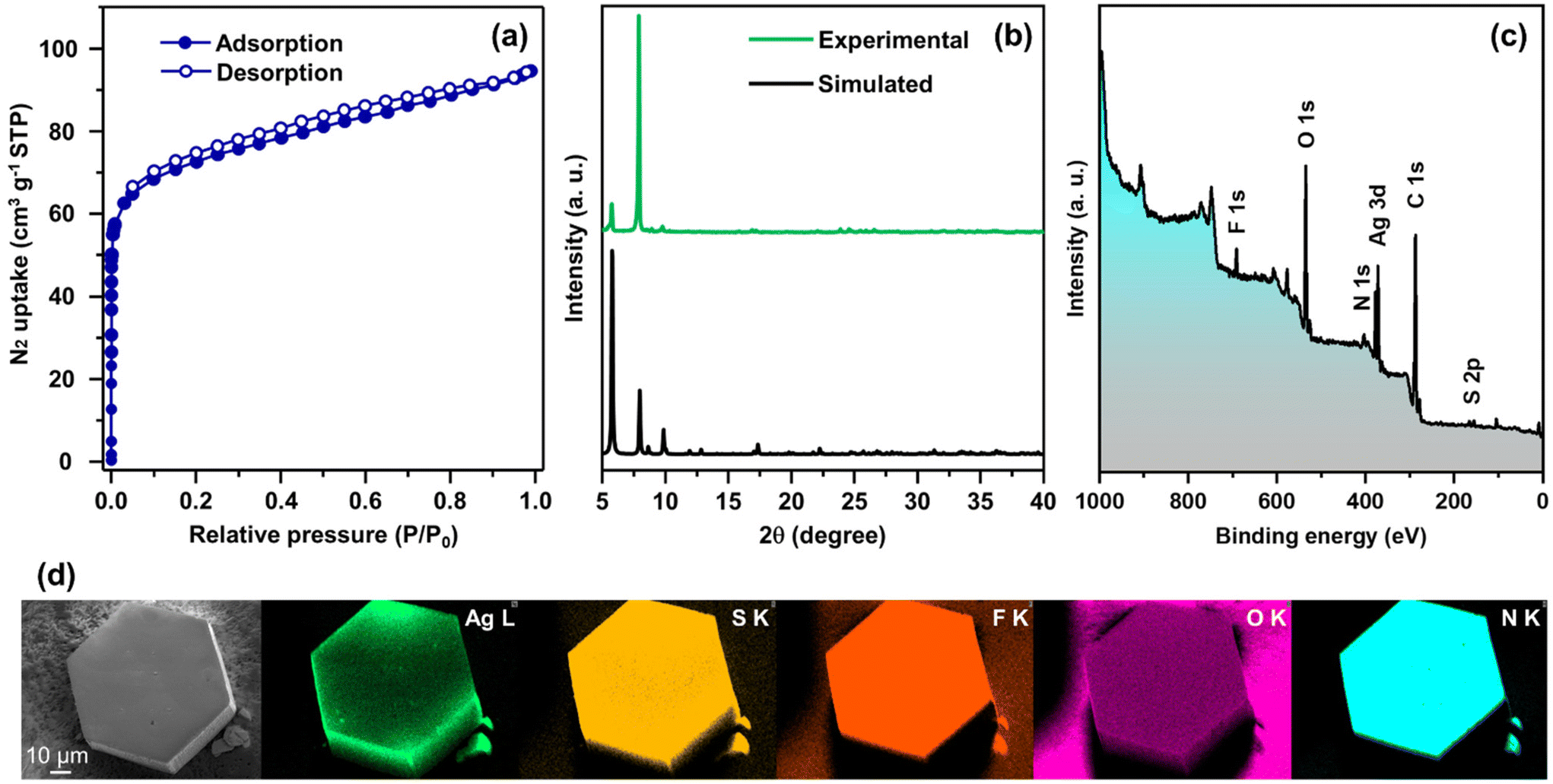 | ||
| Fig. 4 (a) N2 physisorption isotherms, (b) comparison between experimental and simulated PXRD patterns, (c) wide-scan XPS spectrum, and (d) SEM image and corresponding EDX elemental maps, of TUS 5. | ||
With a focus on ascertaining the crystalline phase purity of TUS 5 bulk crystal samples, we accomplished powder X-ray diffraction (PXRD) scans on the outgassed samples. The powder X-ray diffractogram bore a close resemblance to the simulated diffraction pattern from single-crystal data, providing testimony that the crystals were in pure phase (Fig. 4b). Next, we sought to explore the chemical composition and electronic structure of the SCAM through X-ray photoelectron spectroscopy (XPS) upon irradiating the sample in vacuo with soft X-rays (Mg Kα). The occurrence of Ag, S, N, F, O, and C could be evidenced by the wide-scan survey spectrum in Fig. 4c. High-resolution narrow scans of these constituent elements were collected to gather information about their oxidation states (Fig. S10†). As manifested in Fig. S10a,† the Ag 3d spectrum features two conspicuous peaks at 373.4 and 367.4 eV, corresponding to the spin–orbit coupling of Ag 3d3/2 and Ag 3d5/2 with splitting of 6.0 eV, which signifies the existence of Ag+. On account of the S 2p binding energies of 162.3 and 161.1 eV, it is assessed that the XPS signals arise from S 2p1/2 and S 2p3/2, respectively that imply the occurrence of S− (Fig. S10d†). The acquired O 1s binding energy spectrum is characterized by a single peak positioned at 530.8 eV that can refer to the Ag–O–C bonds (Fig. S10e†). When it comes to the N 1s spectrum, the peak at 398.5 eV traces its roots to the Ag–N bonds (Fig. S10f†). While a hexagonal-shaped morphology was furthered through scanning electron microscopy (SEM) inspection of TUS 5, the energy dispersive X-ray spectroscopy (EDX) elemental maps concurred with the surface elemental compositions obtained through XPS measurements (Fig. 4d). As can be noted from the thermogravimetric analysis (TGA) trace in Fig. S11,† TUS 5 displayed modestly high thermal stability up to ca. 135 °C.
After this, we sought to immobilize amano lipase PS on TUS 5 in the interest of augmenting the lipase activity and stability by treating the SCAM with 5 mL of phosphate buffer solution (pH 7.0) containing 120 mg of lipase. The loading capacity of lipase by TUS 5 was calculated to be 0.65 mg mg−1 from the standard curve and by measuring the absorbances of different concentrations of lipase by UV-Vis spectroscopy at 561 nm using bicinchoninic acid (BCA) assay (Fig. S12†). Considering that the size of lipase (30 Å × 32 Å × 60 Å)68 exceeds the pore size of TUS 5 (6 Å), it is logical to conclude that the enzyme is only attached to the surface of TUS 5. As evident from the consistency of the peaks in the XRD diffractogram of lipase-loaded TUS 5 (expressed as lipase@TUS 5) with that of pristine TUS 5, the crystallinity of TUS 5 was not affected upon the attachment of the enzyme (Fig. 5b). Besides, SEM investigation divulged that the hexagonal-shaped morphology of TUS 5 crystals (marked in yellow, Fig. S13†) was also maintained after lipase immobilization. While a few hexagonal-shaped TUS-5 crystals can be prominently observed in the SEM image, there are a number of TUS 5 crystals covered under the enzyme surface, as indicated by the spatial distribution of Ag, S, F, O, and N elements in the EDX elemental maps of lipase@TUS 5 (Fig. 5d). The surface constituent elements and their oxidation states of lipase@TUS 5 were not significantly affected after enzyme attachment, as reflected from XPS analysis (Fig. 5c and S14†).
 | ||
| Fig. 5 (a) Zeta potential of TUS 5, lipase and lipase@TUS 5 at pH 7.0, (b) comparison between the experimental PXRD patterns of lipase@TUS 5 and TUS 5, (c) wide-scan XPS spectrum of lipase@TUS 5, and (d) SEM image and the corresponding EDX elemental maps of lipase@TUS 5. | ||
To probe the interactions between lipase and TUS 5, we carried out zeta potential tests at the immobilization conditions (pH 7.0) on TUS 5, lipase and lipase@TUS 5 samples. The zeta potential values observed for TUS 5, lipase and lipase@TUS 5 were 6.09, −3.46 and −6.16 mV, respectively (Fig. 5a). The electrostatic attraction between negatively charged lipase and positively charged TUS 5 led to the greater binding of lipase onto the TUS 5 matrix. Besides, a salient characteristic of lipases is their interfacial activation upon adsorption on hydrophobic supports.69–71 In its closed structure, lipases are characterized by a lid that shrouds the active sites and renders them inaccessible to substrates. Hydrophobic supports enable the lids of lipases to open, thereby facilitating the substrates to gain access to the active sites. The surface hydrophobicity of TUS 5 was ascertained by water contact-angle measurement, which revealed a high contact angle of 145.84° (Fig. S15†). Consequently, the strong hydrophobic interactions between TUS 5 and lipase translated to the enhanced activity of lipase.
Enantiopure 1-phenylethanol is a potential synthetic intermediate for pharmaceutical and chemical industries, and enzyme-catalyzed kinetic resolution of (R,S)-1-phenylethanol represents a sustainable and economical route to its preparation compared to asymmetric synthesis.72,73 As shown in Fig. 6a, the transesterification reaction was carried out with (R,S)-1-phenylethanol as the substrate, vinyl acetate as the acyl donor, n-hexane as the reaction solvent, and immobilized lipase on TUS 5 or free lipase as the catalyst. Notably, all experiments regarding the kinetic resolution of (R,S)-1-phenylethanol were carried out in triplicate, and the conversion data depict the mean ± standard deviation. High-performance liquid chromatography (HPLC) analysis of the solution obtained after the kinetic resolution of (R,S)-1-phenylethanol attested to the acylation of (R)-1-phenylethanol only, and no transformation of (S)-1-phenylethanol could be detected (Fig. S16†). When free lipase was used as the catalyst, we found a mere 4% conversion of (R,S)-1-phenylethanol after 20 min which increased to 14% after extending the reaction time to 100 min. Strikingly different, the immobilized lipase on TUS 5 furnished far higher 24% and 46% conversions of (R,S)-1-phenylethanol after 20 and 100 min, respectively (Fig. 6b). Note that the kinetic resolution of racemic mixtures can give a maximum theoretical yield of 50%.74 Inspired, we next sought to survey the effect of enzyme immobilization on the thermal stability of lipase@TUS 5. While an 8% conversion of (R,S)-1-phenylethanol was observed after the incubation of native lipase at 60 °C for 8 h, the conversion reached 34% for the immobilized counterpart after incubated under identical conditions. Besides, lipase@TUS 5 could maintain its activity with 34% conversion of (R,S)-1-phenylethanol even when the incubation time was extended to 16 h. Contrarily, the conversion reduced to only 4% with the native enzyme after incubation at 60 °C for 16 h. Upon increasing the incubation temperature of lipase@TUS 5 to 100 °C and for 3 h, we observed a drop in (R,S)-1-phenylethanol conversion to 18%; however, it is still substantially ahead of a 3% conversion of (R,S)-1-phenylethanol for an identical incubation condition of free lipase (Fig. 6c). Consequently, attributed to the reduction of the conformational flexibility of lipase after immobilization, lipase@TUS 5 displayed markedly superior stability under high temperatures when evaluated against free lipase. After this, we appraised the significance of immobilization of lipase in preserving its catalytic activity upon exposure to organic solvents for 24 h. In case of free lipase after treatment by different solvents, the conversion of (R,S)-1-phenylethanol was appreciably low (Fig. S17†). As distinct from free lipase, lipase@TUS 5 maintained 99%, 92%, 83%, 45% and 39% of its initial activity after treatment by isopropyl ether, toluene, THF, ethyl acetate and acetone, respectively, for 24 h, which implies a considerable improvement in the chemical stability (Fig. 6d). Catalyst recyclability is a prime determinant of its cost-efficiency and industrial importance. The recyclability of lipase@TUS 5 was investigated for five consecutive cycles, each reaction cycle lasting 24 h. While free lipase could not be collected in the fresh state due to its solubility in the phosphate buffer solution after the first catalytic cycle, lipase@TUS 5 demonstrated 94% residual activity after the third run which decreased to 42% after the fifth run. Hence, recyclability studies substantiated that the immobilized lipase on TUS 5 can be reused over three cycles (Fig. 6e).
 | ||
| Fig. 6 Kinetic resolution of (R,S)-1-phenylethanol utilizing vinyl acetate as the acyl donor and n-hexane as the reaction solvent: (a) transesterification reaction, (b) comparison of conversion rates when catalyzed by lipase@TUS 5 and free lipase, (c) retention of the catalytic activities of immobilized and free lipases after incubation at different temperatures, (d) retention of the catalytic activities of the immobilized lipase upon treatment with different organic solvents for 24 h, (e) recyclability of lipase@TUS 5 and free lipase. The error bars represent the standard deviation from three measurements. | ||
Conclusions
In summary, we report the designed construction of a new 2D SCAM from tritopic imidazolyl linkers and dodecanuclear Ag(I) NC nodes via a [3 + 6] approach. The node–linker coordination bonding generated a 2D layered network, and the layers are further vertically stacked to develop 3D array through noncovalent interlayer interactions furnishing unidirectional pore channels. Relying on the unique crystalline structure, noteworthy stability and advantageous characteristics from both organic and inorganic components, we used the SCAM as a support for lipase immobilization. In the kinetic resolution of (R,S)-1-phenylethanol, the immobilized lipase on SCAM showed considerably greater conversion into (R)-1-phenylethyl acetate, as well as better thermal stability, stronger solvent resistance and recyclability, in comparison with its free counterpart. This contribution showcases the overwhelming prospects that SCAMs hold to expanding its application domain and carves exciting avenues where nanochemistry meets biological catalysis.Author contributions
S. D. and Y. N. conceived the idea for this study and supervised the project. J. S. conducted the synthesis and characterization. J. S. and K. S. performed the application. R. N. carried out the XPS tests. S. D. and R. N. wrote the manuscript. All authors discussed the results and approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The crystallographic data for [Ag12(StBu)6(CF3COO)6(THIT)6]n has been deposited at the CCDC under CCDC number 2352291.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Zhan Li (Kobe University) for assisting with the water contact angle test. This study was enabled by support from the JSPS KAKENHI (grant no. 20H02698, 20H02552), Ogasawara Foundation for the Promotion of Science and Engineering, Yazaki Memorial Foundation for Science and Technology, and “Aquatic Functional Materials” (grant no. 22H04562).References
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- K. L. D. M. Weerawardene, H. Häkkinen and C. M. Aikens, Annu. Rev. Phys. Chem., 2018, 69, 205–229 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- Z. Wang, R. K. Gupta, F. Alkan, B.-L. Han, L. Feng, X.-Q. Huang, Z.-Y. Gao, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2023, 145, 19523–19532 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- B.-L. Han, Z. Liu, L. Feng, Z. Wang, R. K. Gupta, C. M. Aikens, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2020, 142, 5834–5841 CrossRef CAS PubMed.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- Z. Gan, J. Chen, J. Wang, C. Wang, M.-B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS PubMed.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- Y.-P. Xie, Y.-L. Shen, G.-X. Duan, J. Han, L.-P. Zhang and X. Lu, Mater. Chem. Front., 2020, 4, 2205–2222 RSC.
- I. Díez and R. H. A. Ras, Nanoscale, 2011, 3, 1963–1970 RSC.
- W.-M. He, Z. Zhou, Z. Han, S. Li, Z. Zhou, L.-F. Ma and S.-Q. Zang, Angew. Chem., Int. Ed., 2021, 60, 8505–8509 CrossRef CAS PubMed.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- P. Vanýsek, Electrochemical Series, in Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, 93rd edn, 2012, pp. 5–80 Search PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, CRC Press, 1st edn, 1985 Search PubMed.
- S. Biswas, P. Sun, X. Xin, S. Mandal and D. Sun, Chapter 15 – Atom-Precise Cluster-Assembled Materials, in Atomically Precise Nanochemistry, ed. R. Jin and D. Jiang, John Wiley & Sons Ltd, Chichester, 2023, pp. 453–478. DOI:10.1002/9781119788676.ch15.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- Y.-P. Xie, J.-L. Jin, G.-X. Duan, X. Lu and T. C. W. Mak, Coord. Chem. Rev., 2017, 331, 54–72 CrossRef CAS.
- Z. Wang, X.-Y. Li, L.-W. Liu, S.-Q. Yu, Z.-Y. Feng, C.-H. Tung and D. Sun, Chem. – Eur. J., 2016, 22, 6830–6836 CrossRef CAS PubMed.
- S. A. Claridge, A. W. Castleman Jr., S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. Nakatani, S. Das and Y. Negishi, Nanoscale, 2024, 16, 9642–9658 RSC.
- Z.-Y. Wang and S.-Q. Zang, Chapter 16 – Coinage Metal Cluster-Assembled Materials, in Atomically Precise Nanochemistry, ed. R. Jin and D. Jiang, John Wiley & Sons Ltd, Chichester, 2023, pp. 479–501. DOI:10.1002/9781119788676.ch16.
- S. Biswas, S. Das and Y. Negishi, Coord. Chem. Rev., 2023, 492, 215255 CrossRef CAS.
- R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- M. Cao, R. Pang, Q.-Y. Wang, Z. Han, Z.-Y. Wang, X.-Y. Dong, S.-F. Li, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2019, 141, 14505–14509 CrossRef CAS PubMed.
- Y.-M. Wang, J.-W. Zhang, Q.-Y. Wang, H.-Y. Li, X.-Y. Dong, S. Wang and S.-Q. Zang, Chem. Commun., 2019, 55, 14677–14680 RSC.
- J.-Y. Wang, Y. Si, X.-M. Luo, Z.-Y. Wang, X.-Y. Dong, P. Luo, C. Zhang, C. Duan and S.-Q. Zang, Adv. Sci., 2023, 10, 2207660 CrossRef CAS PubMed.
- X.-S. Du, B.-J. Yan, J.-Y. Wang, X.-J. Xi, Z.-Y. Wang and S.-Q. Zang, Chem. Commun., 2018, 54, 5361–5364 RSC.
- Z. Wei, X.-H. Wu, P. Luo, J.-Y. Wang, K. Li and S.-Q. Zang, Chem. – Eur. J., 2019, 25, 2750–2756 CrossRef CAS PubMed.
- X.-H. Wu, P. Luo, Z. Wei, Y.-Y. Li, R.-W. Huang, X.-Y. Dong, K. Li, S.-Q. Zang and B. Z. Tang, Adv. Sci., 2019, 6, 1801304 CrossRef PubMed.
- M. Cao, S. Wang, J.-H. Hu, B.-H. Lu, Q.-Y. Wang and S.-Q. Zang, Adv. Sci., 2022, 9, 2103721 CrossRef CAS PubMed.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed.
- A. K. Das, S. Biswas, A. Thomas, S. Paul, A. S. Nair, B. Pathak, M. S. Singh and S. Mandal, Mater. Chem. Front., 2021, 5, 8380–8386 RSC.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Inorg. Chem., 2021, 60, 18234–18241 CrossRef CAS PubMed.
- S. Biswas, A. K. Das, A. Nath, S. Paul, M. S. Singh and S. Mandal, Nanoscale, 2021, 13, 17325–17330 RSC.
- Z. Wang, Y.-J. Zhu, Y.-Z. Li, G.-L. Zhuang, K.-P. Song, Z.-Y. Gao, J.-M. Dou, M. Kurmoo, C.-H. Tung and D. Sun, Nat. Commun., 2022, 13, 1802 CrossRef CAS PubMed.
- M. Zhao, S. Huang, Q. Fu, W. Li, R. Guo, Q. Yao, F. Wang, P. Cui, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2020, 59, 20031–20036 CrossRef CAS PubMed.
- W. A. Dar, A. Jana, K. S. Sugi, G. Paramasivam, M. Bodiuzzaman, E. Khatun, A. Som, A. Mahendranath, A. Chakraborty and T. Pradeep, Chem. Mater., 2022, 34, 4703–4711 CrossRef CAS.
- T. Sekine, J. Sakai, Y. Horita, H. Mabuchi, T. Irie, S. Hossain, T. Kawawaki, S. Das, S. Takahashi, S. Das and Y. Negishi, Chem. – Eur. J., 2023, 29, e202300706 CrossRef CAS PubMed.
- R. Nakatani, S. Biswas, T. Irie, J. Sakai, D. Hirayama, T. Kawawaki, Y. Niihori, S. Das and Y. Negishi, Nanoscale, 2023, 15, 16299–16306 RSC.
- J. Sakai, S. Biswas, T. Irie, H. Mabuchi, T. Sekine, Y. Niihori, S. Das and Y. Negishi, Nanoscale, 2023, 15, 12227–12234 RSC.
- S. Das, T. Sekine, H. Mabuchi, S. Hossain, S. Das, S. Aoki, S. Takahashi and Y. Negishi, Chem. Commun., 2023, 59, 4000–4003 RSC.
- R. Nakatani, S. Biswas, T. Irie, Y. Niihori, S. Das and Y. Negishi, ACS Mater. Lett., 2024, 6, 438–445 CrossRef CAS.
- V. L. Schramm, Chem. Rev., 2006, 106, 3029–3030 CrossRef CAS.
- L. Hedstrom, Enzyme Specificity and Selectivity, in Encyclopedia of Life Sciences (ELS), John Wiley & Sons, Ltd, Chichester, 2010. DOI:10.1002/9780470015902.a0000716.pub2.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- J. M. Guisan, G. Fernandez-Lorente, J. Rocha-Martin and D. Moreno-Gamero, Curr. Opin. Green Sustainable Chem., 2022, 35, 100593 CrossRef CAS.
- D. Vasic-Racki, Chapter 1 – History of Industrial Biotransformations – Dreams and Realities, in Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH, Weinhiem, 2006, pp. 1–36. DOI:10.1002/9783527608188.ch1.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277–6289 RSC.
- I. Gill and A. Ballesteros, Ann. N. Y. Acad. Sci., 1996, 799, 697–700 CrossRef CAS PubMed.
- D. Weiser, F. Nagy, G. Bánóczi, M. Oláh, A. Farkas, A. Szilágyi, K. László, Á. Gellért, G. Marosi, S. Kemény and L. Poppe, Green Chem., 2017, 19, 3927–3937 RSC.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185–205 CrossRef PubMed.
- J. Meyer, L.-E. Meyer and S. Kara, Eng. Life Sci., 2022, 22, 165–177 CrossRef CAS PubMed.
- V. L. Sirisha, A. Jain and A. Jain, Adv. Food Nutr. Res., 2016, 79, 179–211 CAS.
- J. Zdarta, A. S. Meyer, T. Jesionowski and M. Pinelo, Catalysts, 2018, 8, 92 CrossRef.
- S. Zahirinejad, R. Hemmati, A. Homaei, A. Dinari, S. Hosseinkhani, S. Mohammadi and F. Vianello, Colloids Surf., B, 2021, 204, 111774 CrossRef CAS PubMed.
- B. R. Facin, M. S. Melchiors, A. Valério, J. V. Oliveira and D. de Oliveira, Ind. Eng. Chem. Res., 2019, 58, 5358–5378 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- I. Akpinar, X. Wang, K. Fahy, F. Sha, S. Yang, T.-w. Kwon, P. J. Das, T. Islamoglu, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2024, 146, 5108–5117 CrossRef CAS PubMed.
- J. M. Palomo, G. Muñoz, G. Fernández-Lorente, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, J. Mol. Catal. B: Enzym., 2002, 19–20, 279–286 CrossRef CAS.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisán, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS PubMed.
- F. I. Khan, D. Lan, R. Durrani, W. Huan, Z. Zhao and Y. Wang, Front. Bioeng. Biotechnol., 2017, 5, 16, DOI:10.3389/fbioe.2017.00016.
- A. Bozan, R. Songür and Ü. Mehmetoğlu, Turk. J. Chem., 2020, 44, 1352–1365 CrossRef CAS PubMed.
- Q. Sun, C.-W. Fu, B. Aguila, J. Perman, S. Wang, H.-Y. Huang, F.-S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef CAS PubMed.
- F. Golombek, M. Haumann, M. S. G. Knoll, A. P. Fröba and K. Castiglione, ACS Omega, 2021, 6, 29192–29200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2352291. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr02506g |
| This journal is © The Royal Society of Chemistry 2024 |