
Synthesis of a metal–organic framework Cu-Mi-UiO-66-based fluorescent nanoprobe for the simultaneous sensing and intracellular imaging of GSH and ATP†
Yun
Liu‡
a,
Shuqi
Xia‡
a,
Meng
Xiao
a,
Mo
Yang
 d,
Mengsu
Yang
c and
Changqing
Yi
*ab
d,
Mengsu
Yang
c and
Changqing
Yi
*ab
aKey Laboratory of Sensing Technology and Biomedical Instruments (Guangdong Province), School of Engineering, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: yichq@mail.sysu.edu.cn
bResearch Institute of Sun Yat-Sen University in Shenzhen, Shenzhen, 518057, China
cDepartment of Biomedical Sciences, City University of Hong Kong, Hong Kong SAR, China
dDepartment of Biomedical Engineering, The Hong Kong Polytechnic University, Hong Kong SAR, China
First published on 8th July 2024
Abstract
This study reports a fluorescent nanoprobe operated in fluorescence turn-on mode for simultaneously sensing and imaging intracellular GSH and ATP. By using maleimide-derivatives as the ligand, the bimetallic nanoscale metal–organic framework (NMOF) Cu-Mi-UiO-66 has been synthesized for the first time using a straightforward one-step solvothermal approach, serving as a GSH recognition moiety. Subsequently, a Cy5-labeled ATP aptamer was assembled onto Cu-Mi-UiO-66 via strong coordination between phosphate and zirconium, π–π stacking and electrostatic adsorption to develop the dual-responsive fluorescence nanoprobe Cu-Mi-UiO-66/aptamer. Due to the photoinduced electron transfer (PET) effect between maleimide groups and the benzene ring of the ligand and the charge transfer between Cy5 and the Zr(IV)/Cu(II) bimetal center of the NMOF, the Cu-Mi-UiO-66/aptamer exhibits a fluorescence turn-off status. The Michael addition reaction between the thiol group of GSH and the maleimide on the NMOF skeleton results in turning on of the blue fluorescence of Cu-Mi-UiO-66. Meanwhile, upon specific interaction with ATP, the aptamer changes into internal loop structures and detaches from Cu-Mi-UiO-66, resulting in turning on of the red fluorescence of Cy5. The nanoprobe demonstrated an excellent sensing performance with a good linear range (GSH, 5.0–450.0 μM; ATP, 1.0–50.0 μM) and a low detection limit (GSH, 2.17 μM; ATP, 0.635 μM). More importantly, the Cu-Mi-UiO-66/aptamer exhibits good performance for tracing intracellular concentration variations of GSH and ATP in living HepG2 cells under different stimulations. This study highlights the potential of NMOFs for multiplexed analysis and provides a valuable tool for tumor microenvironment research and early cancer diagnosis.
1. Introduction
Cancer is a significant public health challenge worldwide, with almost 10 million deaths in 2020, making it a leading cause of death globally.1 Generally, the concentration of tumor markers has a good correlation with the occurrence, development, regression and recurrence of tumors.2 Therefore, monitoring the dynamic change of tumor markers can provide valuable information regarding tumor diagnosis, treatment efficacy, and prognosis. However, a tumor can produce multiple tumor markers,3 and different tumors or different tissue types of the same tumor can have the same tumor markers.4 In addition, different patients diagnosed with the same cancer usually present different quantitative profiles of tumor markers. Consequently, the sensitivity and specificity of cancer diagnosis or prognosis relying on individual tumor markers are often low and fail to meet clinical requirements. Over the past decade, the simultaneous determination of multiple tumor markers has been advocated in theory and practice to improve sensitivity and specificity in cancer diagnosis.3,5Cancer cells exhibit high proliferation rates and require substantial energy consumption. Adenosine triphosphate (ATP), as the most direct source of energy in living cells, is indispensable in the process of cancer cell growth.6,7 During ATP production in mitochondria, reactive oxygen species (ROS) are always generated as by-products. Excessive ROS can damage mitochondria, organelles, and other cellular components, leading to inflammation or cell apoptosis. To counteract the harmful effects of ROS, cancer cells produce reducing substances such as glutathione (GSH), glutathione peroxidase, and superoxide dismutase in response to oxidative stress.8 For example, the concentration of GSH in cancer cells is reported to be ∼4 times higher than that in normal cells; therefore, GSH is considered as an important tumor marker.9,10 Given the interactive correlation between ATP and GSH in tumor progression, research tools that enable simultaneous tracing of them both intracellularly and extracellularly can greatly contribute to a better understanding of their roles in tumorigenesis and progression.
Among all the developed strategies for dynamically tracing tumor markers, the fluorescence technique is particularly desirable due to its excellent sensitivity, high spatiotemporal resolution, and compatibility with other imaging techniques.11–13 Various fluorescent probes have been synthesized and successfully demonstrated for tracing intracellular ATP14,15 or GSH.16,17 However, these fluorescent probes are built on a single sensing mechanism, typically with a sole recognition site, thus limiting their ability to detect multiple analytes. According to the one-to-one principle, simultaneous detection of multiple analytes usually relies on a mixture of different probes, which is often hindered by factors such as probe uptake, localization, metabolism, and interference effects between probes.18,19 To overcome the difficulties in the simultaneous tracing of multiple analytes at an identical location, development of novel fluorescent probes which well integrate multiple recognition moieties is compelling, but still challenging.20,21
The rapidly evolving nanotechnology has demonstrated that nanomaterials are an excellent framework to be integrated with various functional modules for diverse applications such as chemo-/bio-sensing, bioimaging, theranostics, etc.22,23 Among numerous nanomaterials, NMOFs are a new type of porous material formed from self-assembled metal ions or clusters and organic ligands,24 exhibiting a high surface area, adjustable pore size and morphology, abundant functional sites, and high surface area allowing for the loading of various recognition components including organic probes, proteins, enzymes and nucleic acids.25,26 For example, the insertion of free fluorescent probes (NP-HPZ) into the PCN-224 NMOF through post-synthetic modification enabled two-photon fluorescence imaging of pH and phosphorylation in mouse models. The protonation of the N atom in the piperazine group of NP-HPZ triggered a fluorescence change of the naphthene fluorophore. Meanwhile, the specific coordination between PO43− and Zr4+ restored the fluorescence of porphyrin on the PCN-224 skeleton.25 Besides, NMOFs can serve as good nanocarriers and protect the loaded components during crossing cell membranes. In another example, taking advantage of the strong coordination between PO43− of the DNAzyme backbone and Zr4+ in UiO-66 MOFs, the Zn2+-specific DNAzyme was successfully assembled onto the surface of UiO-66 MOFs for tracing intracellular Zn2+. After being internalized by living cells, intracellular PO43− triggered DNAzyme release, while intracellular Zn2+ activated the nucleic acid cleaving function of the DNAzyme and thereafter triggered the fluorescence restoration.26
Recently, NMOF materials have also been demonstrated to exhibit remarkable fluorescence quenching capability towards various fluorophores. This feature can significantly reduce background fluorescence and herein improve the sensitivity in biosensing and bioimaging. For example, a ratiometric fluorescence nanoprobe was developed for ATP quantitation with a detection limit of 500 pM and tracing intracellular ATP by assembling a tetramethylrhodamine (TAMRA) labeled ATP-aptamer and carboxyfluorescein (FAM) labeled ATP-aptamer onto La-based MOF nanosheets via van der Waals force. Charge transfer from FAM or TAMRA to the La3+ ions resulted in their fluorescence quenching. In the presence of ATP, the conformational change of the ATP-aptamer weakened its adsorption force to MOF-La, causing the positively charged TAMRA-labeled aptamers to detach from the MOF-La surface and resulting in red fluorescence turn-on. Meanwhile, the negatively charged FAM-labeled aptamers bind to the incomplete coordination La3+ in MOF-La through electrostatic adsorption, further quenching FAM's fluorescence.27
In this study, a MOF-based fluorescent nanoprobe operating in fluorescence turn-on mode was synthesized for simultaneously sensing and imaging intracellular GSH and ATP. As depicted in Scheme 1, the bimetallic NMOF Cu-Mi-UiO-66 was facilely synthesized for the first time using a one-step solvothermal approach where ZrCl4, Cu(NO3)2 and the maleimide-containing ligand H2L1 were subtly selected as co-precursors. Then, the Cy5-labeled ATP aptamer was assembled onto the NMOF Cu-Mi-UiO-66 via strong coordination between phosphate and zirconium, π–π stacking and electrostatic adsorption to obtain the resultant nanoprobe, Cu-Mi-UiO-66/aptamer.28–30 In this design, biothiols can specifically trigger the blue fluorescence (λem = 455 nm) turn-on of maleimide-derivatives on the NMOF skeleton via the Michael addition reaction,31,32 while the specific interaction between ATP and the Cy5-labeled ATP aptamer led to the detachment of the aptamer from the NMOF surface, resulting in the red fluorescence (λem = 670 nm) turn-on of Cy5. The Cu-Mi-UiO-66/aptamer has been successfully demonstrated for quantitating GSH and ATP in aqueous solution as well as tracing them in living cells. This study reveals the great promise of the Cu-Mi-UiO-66/aptamer for developing ideal diagnostic methods for simultaneously measuring GSH and ATP in living cells, as well as studying their intrinsic link in tumorigenesis and progression.
 | ||
| Scheme 1 Schematic diagram of the synthetic route to the Cu-Mi-UiO-66/aptamer, and the dual-responsive principle of the Cu-Mi-UiO-66/aptamer towards sensing and imaging intracellular GSH and ATP. | ||
2. Experimental
2.1 Synthesis of the Cu-Mi-UiO-66/aptamer
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 40.0 mL), followed by dropwise addition of 10.0 mL NaOH solution (50 mM). Then, the mixture was kept in the dark and allowed to hydrolyze for 1 h under stirring at room temperature. After removing THF and MEOH via rotary evaporation, the resultant product was dissolved in 10.0 mL water under heating. The pH value of the solution was adjusted to pH < 2.0 using HCl to induce the formation of a yellowish precipitate. Finally, the precipitate was dried in a vacuum drying oven to obtain yellowish H2L1 (0.175 g, yield: 20%).
000 rpm and pouring off the supernatant, the resultant nanoprobe Cu-Mi-UiO-66/aptamer was collected and stored at 4 °C in a refrigerator before usage.
:1, 40.0 mL), followed by dropwise addition of 10.0 mL NaOH solution (50 mM). Then, the mixture was kept in the dark and allowed to hydrolyze for 1 h under stirring at room temperature. After removing THF and MEOH via rotary evaporation, the resultant product was dissolved in 10.0 mL water under heating. The pH value of the solution was adjusted to pH < 2.0 using HCl to induce the formation of a yellowish precipitate. Finally, the precipitate was dried in a vacuum drying oven to obtain yellowish H2L1 (0.175 g, yield: 20%).
000 rpm and pouring off the supernatant, the resultant nanoprobe Cu-Mi-UiO-66/aptamer was collected and stored at 4 °C in a refrigerator before usage.
2.2 Quantitation of GSH and ATP in cell extract
For quantitation of GSH and ATP in aqueous solution, Cu-Mi-UiO-66/aptamer was dispersed in HEPES buffer (pH = 7.4, 20 mM) at an equivalent aptamer concentration of 100 nM at first. Then, 100 μL Cu-Mi-UiO-66/aptamer solution was added with 900 μL standard solutions containing different GSH concentrations (0, 1, 5, 10, 20, 40, 80, 120, 200, 300, 450, 600, and 800 μM) or different ATP concentrations (0, 1, 5, 10, 20, 50, 100, 200, 300, 400, 800 μM). The mixture was kept in the dark and allowed to react for 80 min under shaking. Finally, 400 μL of the mixture was transferred to a quartz cuvette and subjected to an FS5 spectrofluorometer (Edinburgh, Scotland) for recording the fluorescence spectrum. The fluorescence intensity enhancement rate (ΔF = |F − F0|/F0) at 455 nm and 670 nm was plotted as functions of GSH concentration and ATP concentration to obtain calibration curves, respectively. In the equation for the fluorescence intensity enhancement rate, F0 represents the background fluorescence intensity in the absence of any analytes, while F represents the fluorescence intensity in the presence of an analyte. Of note, the excitation wavelength was set to 360 nm and 570 nm for quantitation of GSH and ATP, respectively.The quantitation capabilities of the Cu-Mi-UiO-66/aptamer in a complex biological environment were demonstrated by determining GSH and ATP in cell extracts. In this study, HepG2 cells were used for all cell experiments. To this end, HepG2 cells were seeded in 6-well plates at a density of 106 cells per well and cultured in H-DMEM medium containing 10% fetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in an incubator with 5% CO2. After 80% coverage was reached, the medium was removed, and the cells were washed twice with PBS buffer solution. Then, RIPA cell lysis buffer (250 μL per well, 1.0 mM) was added to the plates, and the lysate was agitated with a pipette to ensure complete contact between the lysate and the cells. Finally, the supernatant was collected as cell extracts for subsequent analysis by centrifugation (13000 rpm, 5 min).
To prepare spiked samples, the cell extract solution was diluted 100 times with FBS-free culture medium firstly. Then, a different amount of GSH (0, 10, 50, 100 μM) and ATP (0, 50, 100, 200 μM) was added into the diluted cell extract solution. Following the same experimental conditions and procedures as those in aqueous solution, GSH and ATP in cell extracts were determined using the Cu-Mi-UiO-66/aptamer. For validation, the standard method for GSH and ATP quantitation using commercial detection kits (BOXBIO, Beijing) was also performed following the standard protocol provided by the manufacturer (please refer to the ESI† for a detailed protocol). All the experiments were performed in triplicate, and the paired t test was used to analyze the difference between these two methods.
2.3 Fluorescence imaging of intracellular of GSH and ATP
For the assay of tracing intracellular of GSH and ATP, HepG2 cells were inoculated into 25 mm confocal glass culture dishes at a density of 1.5 × 105 cells per dish and allowed to be cultured for 24 h in H-DMEM medium. Subsequently, the cells were treated with N-ethylmaleimide (NEM) (500 μM), α-lipoic acid (ALA) (500 μM), oligomycin (3 μg mL−1), and etoposide (500 μM) for 40 min, while the cells cultured in FBS-free medium were set as the control. Then, the cells were washed with FBS-free medium three times, followed by the addition of a solution containing the Cu-Mi-UiO-66/aptamer (200 μg mL−1, 1.0 mL per dish). After incubation for another 4 h at 37 °C, the cells were washed and fixed using 4% paraformaldehyde (500 μL per dish) for 15 min. The cells were then washed again and treated with green-SYTO dyes (1.0 μM, 500 μL) for 20 min to stain the cell nucleus. After another washing step, 1.0 mL of PBS solution was added to maintain the cell morphology. Finally, a TCS SP5 confocal laser microscope (Leica, Germany) was used for the imaging of GSH and ATP in living HepG2 cells.The parameters of the microscope were set as follows: green channel (excitation wavelength: 500 nm; detection wavelength: 530 nm); red channel (excitation wavelength: 650 nm; detection wavelength: 670 nm) and blue channel (excitation wavelength: 360 nm; detection wavelength: 460 nm). Image J software was used to quantify the fluorescence intensity of fluorescence images, according to an original protocol from QBI, The University of Queensland, Australia.35
3. Results and discussion
3.1 Rationale
The principle behind this study was to design a single nanoprobe integrating two recognition moieties that can respond to GSH and ATP using two distinguishable fluorescence signals, respectively. Scheme 1 illustrates the synthetic routes for the bimetallic NMOF-based nanoprobe Cu-Mi-UiO-66/aptamer, and its dual-responsive principle towards GSH and ATP sensing. Maleimide-derivatives have been well-documented as highly specific fluorescent probes for biothiols, thanks to their features of a strong electron-withdrawing effect and fast response to thiols.31,32,36 Herein, maleimide was engineered as a key component of the ligand for the synthesis of the NMOF Cu-Mi-UiO-66/aptamer which serves as a GSH recognition moiety (Fig. 1A). Due to the PET effect between maleimide groups and the benzene ring of the ligand as well as the fluorescence quenching effect of Cu2+, the NMOF Cu-Mi-UiO-66/aptamer emitted weak blue fluorescence in the wavelength range of 400–600 nm with a peak at 455 nm (blue solid curve of Fig. 1C). The Michael addition reaction between the thiol group of GSH and maleimide on the NMOF skeleton results in the formation of a thioether, blocking the PET process and turning on the blue fluorescence of the Cu-Mi-UiO-66/aptamer (blue dotted curve of Fig. 1C). These results validated the capability of the Cu-Mi-UiO-66/aptamer as a GSH recognition moiety.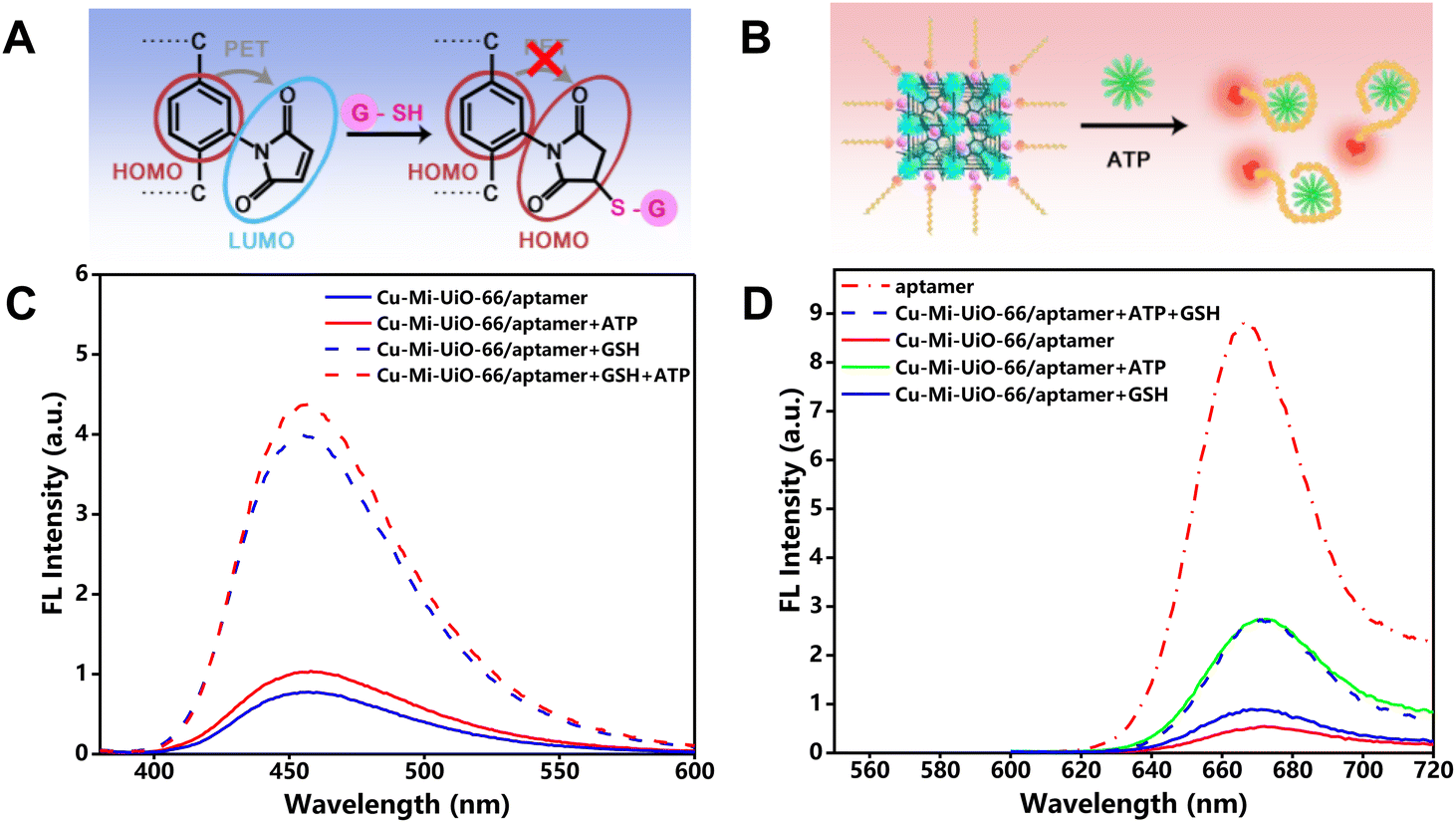 | ||
| Fig. 1 Rationale. (A and B) Schematic diagram of the mechanism of GSH (A) and ATP (B) sensing. (C and D) Fluorescence spectra of the Cu-Mi-UiO-66/aptamer in the presence of different analytes: the blue fluorescence channel (C) and the red fluorescence channel (D). | ||
To synthesize the designed dual-responsive nanoprobe Cu-Mi-UiO-66/aptamer, the Cy5-labeled ATP aptamer with an emission peak at 670 nm in the wavelength range of 620–720 nm was subtly selected as the ATP recognition moiety (red dotted curve of Fig. 1D) and assembled onto NMOF Cu-Mi-UiO-66 via strong coordination between phosphate and zirconium, π–π stacking and electrostatic adsorption.28–30 Due to the charge transfer between Cy5 and the Zr(IV)/Cu(II) bimetallic centers of the NMOF, the red fluorescence of Cy5 was significantly quenched after being assembled onto the NMOF surface (red solid curve of Fig. 1D). However, upon specific interaction with ATP, the aptamer changed into internal loop structures and detached from NMOF Cu-Mi-UiO-66 (Fig. 1B),28,37 blocking the charge transfer process and turning on the red fluorescence of Cy5-labeled ATP aptamer (green solid curve of Fig. 1D). These results validated the capability of the Cy5-labeled ATP aptamer as an ATP recognition moiety.
Noteworthily, the assembly of the Cy5-labeled ATP aptamer onto the NMOF Cu-Mi-UiO-66 did not affect the GSH recognition capability of Cu-Mi-UiO-66, as evidenced by the still weak blue fluorescence in the absence of GSH (blue solid curve of Fig. 1C) and strong blue fluorescence in the presence of GSH (blue dotted curve of Fig. 1C). The presence of ATP did lead to a slight fluorescence enhancement of the Cu-Mi-UiO-66/aptamer at 455 nm (red solid curve of Fig. 1C), which might be caused by the ATP-triggered release of the Cy5-aptamer. As shown in Fig. S1A,† the assembly of the Cy5-aptamer onto Cu-Mi-UiO-66 caused a slight decrease in the emission at 455 nm, which might be attributed to fluorescence resonance energy transfer (FRET) between Cu-Mi-UiO-66 and Cy5. The presence of ATP would reverse this phenomenon via detaching the Cy5-aptamer from Cu-Mi-UiO-66, causing a slight fluorescence enhancement at 455 nm and ∼7.0% systematic error for GSH quantitation. Thanks to the excellent specificity of the aptamer, GSH had no significant effect on the opening of the Cu-Mi-UiO-66/aptamer red fluorescence (blue solid curve of Fig. 1D and Fig. S1B†). Meanwhile, the simultaneous presence of GSH and ATP does not affect the specific response of the NMOF of the Cu-Mi-UiO-66/aptamer to GSH (red dotted curve of Fig. 1C), nor does it affect the specific response of the aptamer of the Cu-Mi-UiO-66/aptamer to ATP (blue dotted curve of Fig. 1D). These results validated the capability of the designed nanoprobe Cu-Mi-UiO-66/aptamer for the simultaneous sensing of GSH and ATP without significant cross-interference and demonstrated the feasibility of real-time monitoring GSH and ATP with separate fluorescence channels using a single probe.
3.2 Synthesis and characterization of NMOFs
To endow the NMOF with GSH sensing capability, the maleimide-containing compound H2L1 was subtly selected and synthesized as a ligand for the subsequent synthesis of NMOFs Mi-UiO-66 and Cu-Mi-UiO-66 (Scheme S1†).31 After ester hydrolysis under alkaline conditions, the disappearance of the proton peak of methyl H at 3–4 ppm and the appearance of the proton peak of carboxyl H at 11.27 ppm were observed in 1H-NMR spectra (Fig. S2†). And the shift of the characteristic molecular ion peak from 289.059 m/z to 278.033 m/z was observed in mass spectrometry (MS) spectra (Fig. S3†). These results confirmed the successful synthesis of H2L1.Then, NMOFs Mi-UiO-66 and Cu-Mi-UiO-66 were synthesized using a one-step solvothermal approach where ZrCl4 and the maleimide-containing ligand H2L1 were used as co-precursors in the absence and presence of Cu(NO3)2, respectively. X-ray photoelectron spectroscopy (XPS) analysis revealed that Mi-UiO-66 was comprised of C, N, O and Zr exhibiting the characteristic peaks corresponding to C1s (284.2 eV), O1s (531.1 eV), N1s (399.0 eV), Zr3p (333.1 eV and 346.1 eV), and Zr3d (182.1 eV) (Fig. 2A & S4†),38 while Cu-Mi-UiO-66 was comprised of C, N, O, Zr and Cu exhibiting an additional characteristic peak corresponding to Cu2p (932.7 eV) (Fig. 2A & S5†). Cu-Mi-UiO-66 and Mi-UiO-66 exhibited similar Fourier transform infrared (FTIR) spectra, both exhibiting the characteristic adsorption peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1567 cm−1), CC of the benzene ring (1386 cm−1) and Zr–O (∼767 cm−1, ∼661 cm−1, and ∼482 cm−1) (Fig. 2B).39 This result suggested that both Cu-Mi-UiO-66 and Mi-UiO-66 are typical Zr-connector-based MOFs. This conclusion was also validated by their X-ray diffraction (XRD) patterns in which both exhibited main diffraction peaks at 7.4°, 8.5°, and 25.7° (Fig. 2C).40 The nearly identical XRD patterns of Mi-UiO-66, Cu-Mi-UiO-66 and UiO-66 suggested that the introduction of maleimide groups to the ligand and further introduction of Cu to the skeleton did not affect the structural motif of UiO-type MOFs. In addition, results from N2 adsorption–desorption experiments confirmed the microporous feature of both Cu-Mi-UiO-66 and Mi-UiO-66 NMOFs (Fig. 2D). Brunauer–Emmett–Teller (BET) surface areas were determined to be 589.975 m2 g−1 and 734.553 m2 g−1 for Cu-Mi-UiO-66 and Mi-UiO-66, respectively. It seems that the introduction of maleimide groups and Cu to the frameworks of UiO-type MOFs resulted in smaller BET surface values.31,41,42
O (1567 cm−1), CC of the benzene ring (1386 cm−1) and Zr–O (∼767 cm−1, ∼661 cm−1, and ∼482 cm−1) (Fig. 2B).39 This result suggested that both Cu-Mi-UiO-66 and Mi-UiO-66 are typical Zr-connector-based MOFs. This conclusion was also validated by their X-ray diffraction (XRD) patterns in which both exhibited main diffraction peaks at 7.4°, 8.5°, and 25.7° (Fig. 2C).40 The nearly identical XRD patterns of Mi-UiO-66, Cu-Mi-UiO-66 and UiO-66 suggested that the introduction of maleimide groups to the ligand and further introduction of Cu to the skeleton did not affect the structural motif of UiO-type MOFs. In addition, results from N2 adsorption–desorption experiments confirmed the microporous feature of both Cu-Mi-UiO-66 and Mi-UiO-66 NMOFs (Fig. 2D). Brunauer–Emmett–Teller (BET) surface areas were determined to be 589.975 m2 g−1 and 734.553 m2 g−1 for Cu-Mi-UiO-66 and Mi-UiO-66, respectively. It seems that the introduction of maleimide groups and Cu to the frameworks of UiO-type MOFs resulted in smaller BET surface values.31,41,42
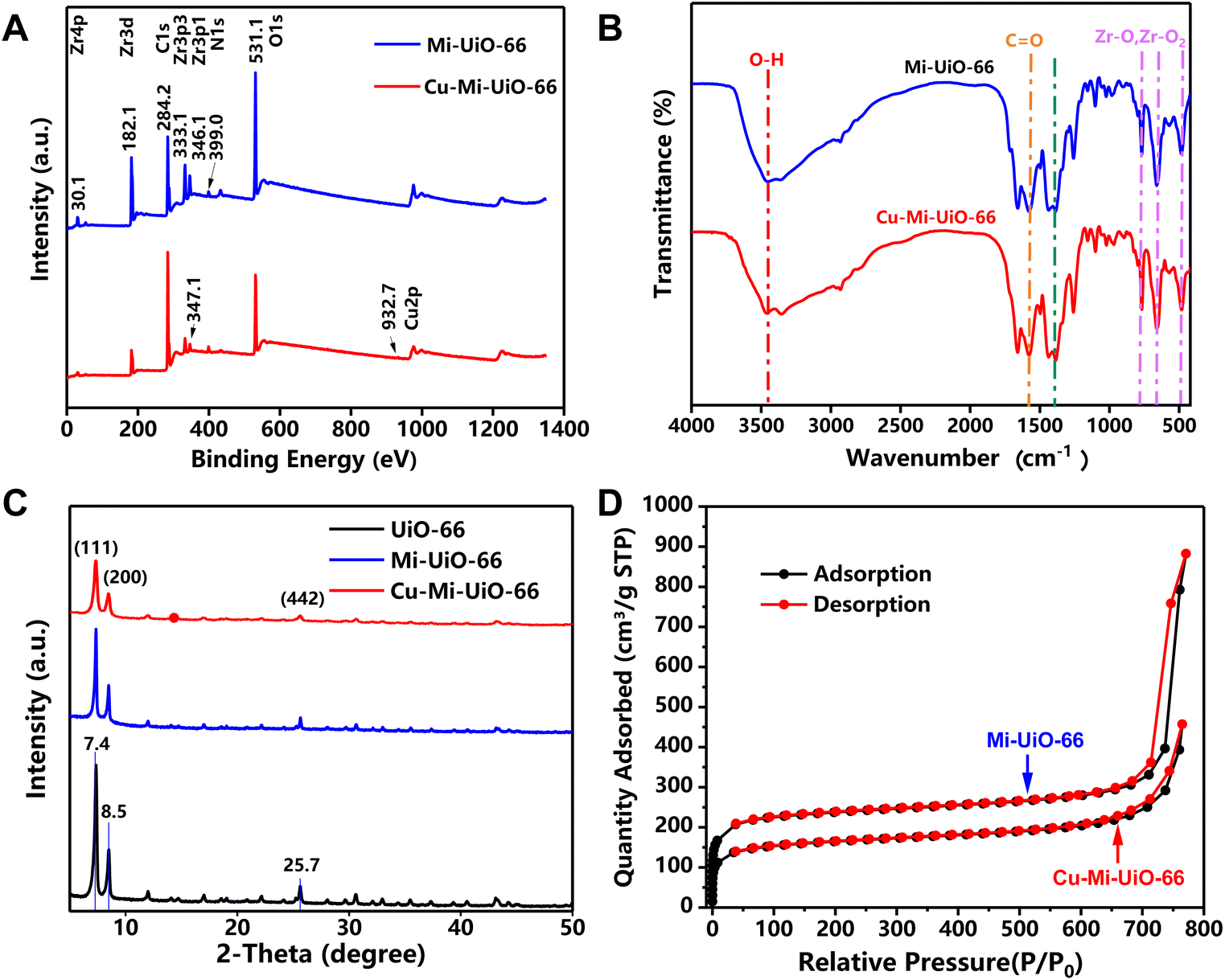 | ||
| Fig. 2 Characterization of NMOFs. (A) XPS spectra, (B) FTIR spectra, (C) XRD patterns, and (D) N2 adsorption–desorption curves of the Mi-UiO-66 NMOF and the Cu-Mi-UiO-66 NMOF. | ||
As shown in both TEM (Fig. 3A & B) and SEM (Fig. S6A† & 6B) images, the as-prepared Mi-UiO-66 and Cu-Mi-UiO-66 were cubic in shape and uniform in size, exhibiting diameters of 107.91 ± 8.30 nm and 60.65 ± 10.48 nm, respectively. The results of dynamic light scattering (DLS) measurements indicate that the hydrated particle sizes of Mi-UiO-66 and Cu-Mi-UiO-66 are 133.53 ± 25.22 nm and 64.1 ± 12.10 nm, respectively. The addition of Cu might interfere with the interactions between Zr(IV) and organic ligands and thus inhibit the growth of UiO-66 crystals, correlating well with previous findings.33,34 The larger particle size obtained by DLS than that observed by TEM provided indirect evidence for the good hydrophilicity of the synthesized NMOFs, since DLS experiment measured the hydrated diameter of the nanoparticles.
 | ||
| Fig. 3 Characterization of NMOFs. (A–C) TEM images of Mi-UiO-66 (A), Cu-Mi-UiO-66 (B), and the Cu-Mi-UiO-66/aptamer (C). The bottom right insets of each image are the corresponding particle size distribution diagrams, and the top left insets of each image are the corresponding schematic diagrams. (D–F) STEM images and the corresponding EDS mapping images of Mi-UiO-66 (D), Cu-Mi-UiO-66 (E), and the Cu-Mi-UiO-66/aptamer (F). | ||
Thanks to the maleimide-containing ligand, Mi-UiO-66 and Cu-Mi-UiO-66 were expected to have GSH sensing capability. As shown in Fig. S7,† GSH did trigger the fluorescence turn-on of NMOFs. Notably, the fluorescence intensity of Cu-Mi-UiO-66 (the red solid curve of Fig. S7A†) was substantially weaker than that of Mi-UiO-66 (blue solid curve of Fig. S7A†), possibly due to the strong fluorescence quenching capability of Cu.43 This feature is quite important because a low background signal is favourable for detection sensitivity. As anticipated, the same amounts of GSH enhanced the fluorescence intensity of Cu-Mi-UiO-66 and Mi-UiO-66 by ∼8-fold and ∼2.3-fold (Fig. S7B†), respectively. This was because, compared to Mi-UiO-66, Cu-Mi-UiO-66 had a smaller size and better water solubility (inset of Fig. S7A†), resulting in more uniform dispersion in aqueous solution. Therefore, Cu-Mi-UiO-66 reacted more thoroughly with GSH than Mi-UiO-66, leading to a more pronounced fluorescence change. All these results suggested that Cu-Mi-UiO-66 exhibited superior GSH sensing capability than Mi-UiO-66. As designed, the fluorescence intensity changed at 455 nm (ΔF455) increased linearly with the increase of GSH concentration from 1.00 to 120 μM with a correlation coefficient of 0.994 and an LOD of 0.879 μM (Fig. S7C & D†), validating the successful quantitation of GSH using Cu-Mi-UiO-66. Therefore, the NMOF Cu-Mi-UiO-66 was used in the following experiments to develop a GSH/ATP dual-responsive nanoprobe.
It has been well-established that Zr4+ exhibits strong coordination capacity to the exposed phosphoric acid groups in single-stranded nucleic acid.25,28 Cu-Mi-UiO-66 is positively charged with a zeta potential of +36.2 mV, while the Cy5-labeled ATP aptamer is negatively charged with a zeta potential of −7.44 mV (Fig. S8†). Herein, the Cy5-labeled ATP aptamer could be easily assembled onto Cu-Mi-UiO-66 via electrostatic adsorption to prepare the resultant nanoprobe Cu-Mi-UiO-66/aptamer. In addition, the ligands in Cu-Mi-UiO-66 contain abundant benzene rings, which interact via π–π stacking with the nucleobases of aptamer-Cy5, resulting in the adsorption of aptamer-Cy5 on the surface of Cu-Mi-UiO-66.28–30 Optimization experiments revealed that the nanoprobe with the optimal aptamer loading capacity was prepared by mixing 120 μg mL−1 Cu-Mi-UiO-66 with 100 nM Cy5-labeled ATP aptamer in HEPES buffer for 10 min at 37 °C (Fig. S9†).
After assembling the negatively charged aptamer onto the surface of the positively charged NMOF Cu-Mi-UiO-66, the zeta potential changed from +36.2 mV to +24.8 mV (Fig. S8†), providing indirect evidence of the successful synthesis of the Cu-Mi-UiO-66/aptamer. Although TEM observation showed almost the same size of Cu-Mi-UiO-66/aptamer (66.46 ± 11.13 nm) and Cu-Mi-UiO-66 (Fig. 3C), DLS measurement revealed an increase in hydration diameter of nearly 20 nm (85.97 ± 16.22 nm) after assembling aptamer onto Cu-Mi-UiO-66 (inset of Fig. 3C). This result provided another indirect evidence of the successful synthesis of the Cu-Mi-UiO-66/aptamer, since the hydrophilic aptamer on the surface of Cu-Mi-UiO-66 also contributed to the overall hydration diameter. Energy-dispersive spectroscopy (EDS) mapping analysis clearly showed the changes in elemental composition along with the synthesis procedures (Fig. 3D–F). This is crucial because the presence of Cu indicated the successful introduction of Cu to the skeleton of NMOF Mi-UiO-66, while the presence of P suggested the successful assembly of the aptamer onto the NMOF Cu-Mi-UiO-66. This result provided strong evidence of the successful synthesis of the resultant nanoprobe Cu-Mi-UiO-66/aptamer.
3.3 Quantitation of GSH and ATP in cell lysate
As designed, GSH triggered a gradual fluorescence increase of the Cu-Mi-UiO-66/aptamer at 455 nm (Fig. 4A), while ATP induced a gradual fluorescence increase of the Cu-Mi-UiO-66/aptamer at 670 nm (Fig. 4B). ΔF455 of the nanoprobe could be used for GSH quantitation where a linear range from 5.0 to 450.0 μM with a correlation coefficient of 0.983 and an LOD of 2.17 μM was exhibited (Fig. 4C). In parallel, ΔF670 of the nanoprobe could be used for ATP quantitation where a linear range from 1.0 to 50.0 μM with a correlation coefficient of 0.985 and an LOD of 0.635 μM was demonstrated (Fig. 4D). Noteworthily, the LODs were calculated based on the standard deviation of the response (SD) and the slope of the calibration curve (S) at levels approximating the LOD according to the formula: LOD = 3.0(SD/S). Compared to other reported methods (Tables S1 and S2†), the method designed in this study has a wider linear range, with the sensitivity for ATP detection being better than most sensors. Although the sensitivity for GSH detection is not as good as other sensors, it can still meet the detection of GSH at mM levels within cells.9 More importantly, this study can achieve the detection of both GSH and ATP biomarkers simultaneously using a kind of nano-fluorescent composite probe, simplifying experimental procedures and saving costs. It has been well documented that the amount of GSH and ATP in the cells is in the range of 1.0–10.0 mM and 1.0–10.0 mM, respectively.9,44 This result suggested that the Cu-Mi-UiO-66/aptamer could satisfy the sensitivity requirement for tracing intracellular GSH and ATP. Because of a complicated intracellular matrix, the possible interference of 12 typical amino acids to GSH detection and 5 typical triphosphates to ATP detection were examined. Thanks to the specific chemical reaction between maleimide and thiols, among all the examined substances, only Cys could slightly enhance the fluorescence of the Cu-Mi-UiO-66/aptamer at 455 nm (Fig. 4E). Considering the significantly higher concentrations of GSH (mM level) than Cys (μM level) in cells,45 the interference from Cys to intracellular monitoring GSH is nearly negligible. Owing to the high specificity of the anti-ATP aptamer to ATP, all examined substances, including GTP, CTP, UTP, AMP and ADP, only induced negligible fluorescence changes of the Cu-Mi-UiO-66/aptamer at 670 nm (Fig. 4F). All these results revealed favorable specificity of the Cu-Mi-UiO-66/aptamer for sensing GSH and ATP against possible co-existing amino acids and triphosphates simultaneously. The stability test showed that the coefficients of variation (CV) from intra-assay and inter-assay of GSH and ATP were 5.27%, 7.98%, 3.07% and 1.46% (Fig. S10,†n = 5), respectively. All of them were less than 10%, which indicated that the detection stability of this method was good.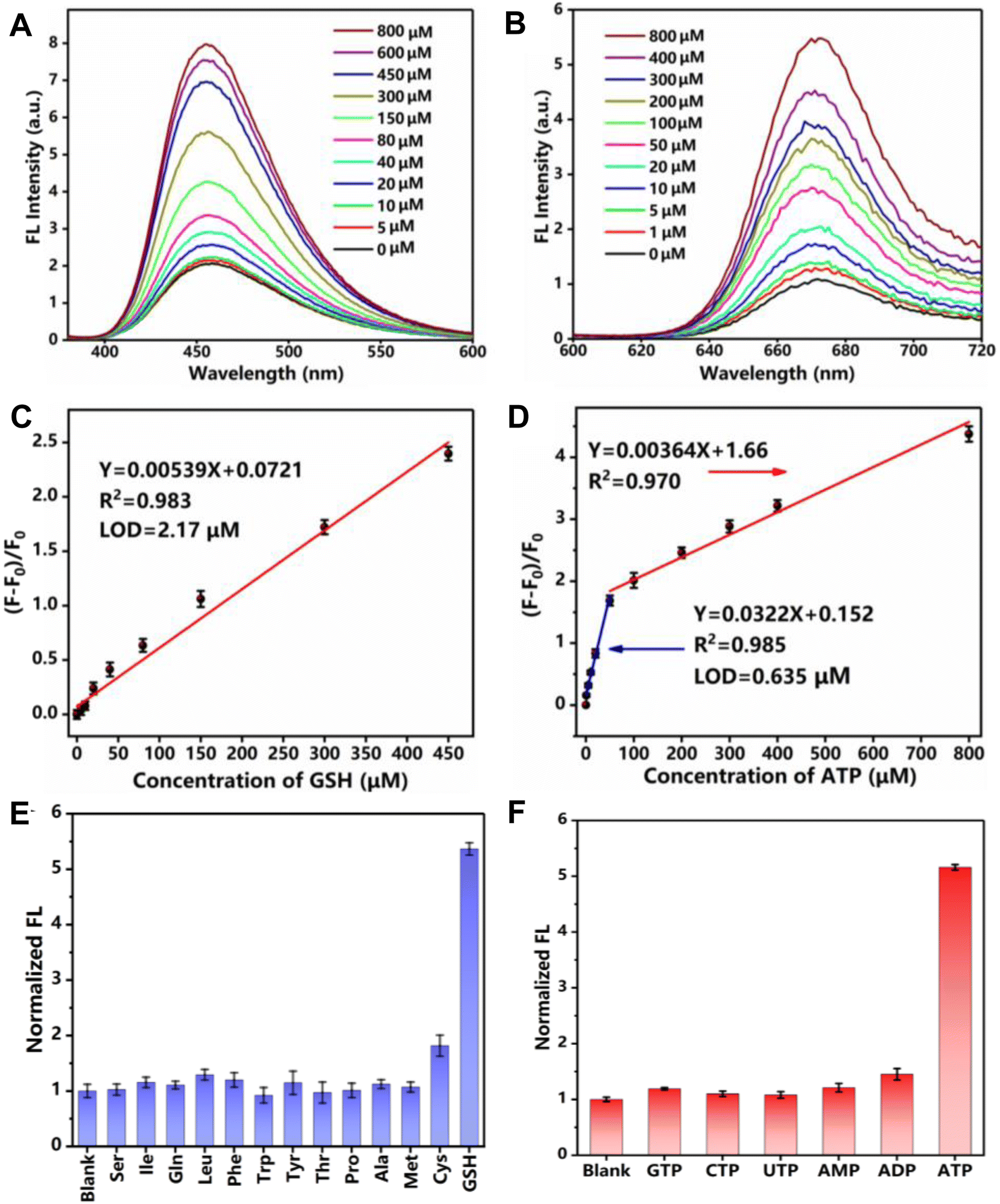 | ||
| Fig. 4 Quantitation of GSH and ATP. (A and B) Fluorescence spectra of the Cu-Mi-UiO-66/aptamer with different concentrations of GSH (0–800 μM) (A) or ATP (0–800 μM) (B) in HEPES buffer (pH 7.4). (C) Linear relationship between ΔF455 and GSH concentrations. (D) Linear relationship between ΔF670 and ATP concentrations. (E and F) The specificity of the Cu-Mi-UiO-66/aptamer to various amino acids (1.0 mM) (E) or triphosphates (1.0 mM) (F). Noteworthily, the concentration of GSH and ATP used in this assay was 0.10 mM. | ||
To demonstrate its sensing capabilities in complicated biological samples, quantitation of GSH and ATP in HepG2 cell lysate was performed using the Cu-Mi-UiO-66/aptamer and commercial assay kits. To this end, the HepG2 cell was treated with NEM (GSH inhibitor) and oligomycin (ATP inhibitor) to remove endogenous GSH and ATP. Then, the pre-treated HepG2 cells were lysed, and the lysate was used to prepare a series of GSH and ATP standard solutions with different concentrations for the purpose of minimization of the possible matrix effects. Using the calibration curves obtained in cell lysate (Fig. S11†), GSH and ATP concentrations in cell lysate were determined to be 8.52 ± 0.15 μM and 17.68 ± 0.22 μM using the Cu-Mi-UiO-66/aptamer. In parallel, following the standard protocol provided by the manufacturer, GSH and ATP concentrations in the cell lysate were determined to be 8.18 ± 0.13 μM and 16.74 ± 0.25 μM using commercial assay kits. In addition, standard addition experiments were also performed. As shown in Table 1, when using the reported assay, the recoveries were in the range of 95.6% to 103.8% with the RSD ranging from 6.50% to 13.6% for GSH quantitation, and in the range of 95.8% to 108.6% with the RSD ranging from 4.80% to 11.2% for ATP quantitation. More importantly, paired t test analysis showed no significant difference between the reported assay and commercial assay kits at the significance level of 0.05, validating the practicability and reliability of the reported assay. These results also confirmed that the detection accuracy of the reported assay was comparable with that of commercial assay kits. Noteworthily, the Cu-Mi-UiO-66/aptamer could do the work of two independent assay kits, namely the GSH content assay kit and the ATP content assay kit.
| Method | GSH spiked (μM) | GSH measured (μM) | Recovery (%) | RSD (%) (n = 5) | ATP spiked (μM) | ATP measured (μM) | Recovery (%) | RSD (%) (n = 5) |
|---|---|---|---|---|---|---|---|---|
| Measured using the Cu-Mi-UiO-66/aptamer | 0 | 8.52 ± 0.15 | — | — | 0 | 17.68 ± 0.22 | — | — |
| 10 | 17.64 ± 0.21 | 95.6 | 6.5 | 50 | 64.96 ± 0.24 | 95.8 | 4.8 | |
| 50 | 61.85 ± 0.35 | 105.7 | 5.3 | 100 | 126.35 ± 0.44 | 107.5 | 7.4 | |
| 100 | 112.27 ± 0.62 | 103.8 | 13.6 | 200 | 236.32 ± 0.58 | 108.6 | 11.2 | |
| Measured using Boxbio test kits | 0 | 8.18 ± 0.13 | — | — | 0 | 16.74 ± 0.25 | — | — |
| 10 | 18.03 ± 0.18 | 99.2 | 4.7 | 50 | 68.32 ± 0.28 | 102.4 | 5.8 | |
| 50 | 62.49 ± 0.41 | 107.5 | 8.5 | 100 | 131.75 ± 0.35 | 112.9 | 9.2 | |
| 100 | 110.26 ± 0.34 | 101.9 | 9.6 | 200 | 228.84 ± 0.46 | 105.6 | 10.8 |
3.4 Fluorescence imaging of intracellular GSH and ATP
The applicability of the Cu-Mi-UiO-66/aptamer for tracing intracellular GSH and ATP was evaluated using HepG2 cells. The results of the thiazolyl blue (MTT) assay revealed low cytotoxicity for all the materials synthesized in this study, including Mi-MOFs, Cu-Mi-MOFs and the Cu-Mi-UiO-66/aptamer (Fig. S12A†). In addition, the Cu-Mi-UiO-66/aptamer exhibited good long-term colloidal stability in ddH2O, HEPES buffer and cell lysate for up to 7 days without forming any observable aggregation (Fig. S12B†). The good bio-compatibility and long-term colloidal stability of the Cu-Mi-UiO-66/aptamer laid a solid foundation for its applicability in bioimaging.To demonstrate the capability of the Cu-Mi-UiO-66/aptamer in the intracellular imaging of GSH and ATP, HepG2 cells were incubated with 200 μg mL−1 probe for different periods of time (0, 2, 4, 6 h). To facilitate the observation, the cell nuclei was stained with SYTO to create a stable green fluorescence channel. Confocal laser scanning microscopy (CLSM) images in Fig. 5A showed that weak red emission from Cy5 and weak blue emission from Cu-Mi-UiO-66 were both observed in the cell cytoplasm. And the intensity of both red and blue fluorescence increased along with the incubation time, and peaked at 4 h. At a later incubation time point of 6 h, a decrease in both red and blue fluorescence was observed, likely due to the excessive uptake of fluorescent probes by cells via endocytosis, leading to their confinement within multivesicular bodies, preventing interaction with intracellular GSH and ATP, and subsequently expelled through exocytosis.46,47 And bright-field images revealed that HepG2 cells presented intact morphology even after exposure to the probe for up to 6 hours, demonstrating the good biocompatibility of the probe. Fluorescence intensities of CLSM images quantified using Image J software validated the time-dependent cellular uptake of the Cu-Mi-UiO-66/aptamer (Fig. 5B).
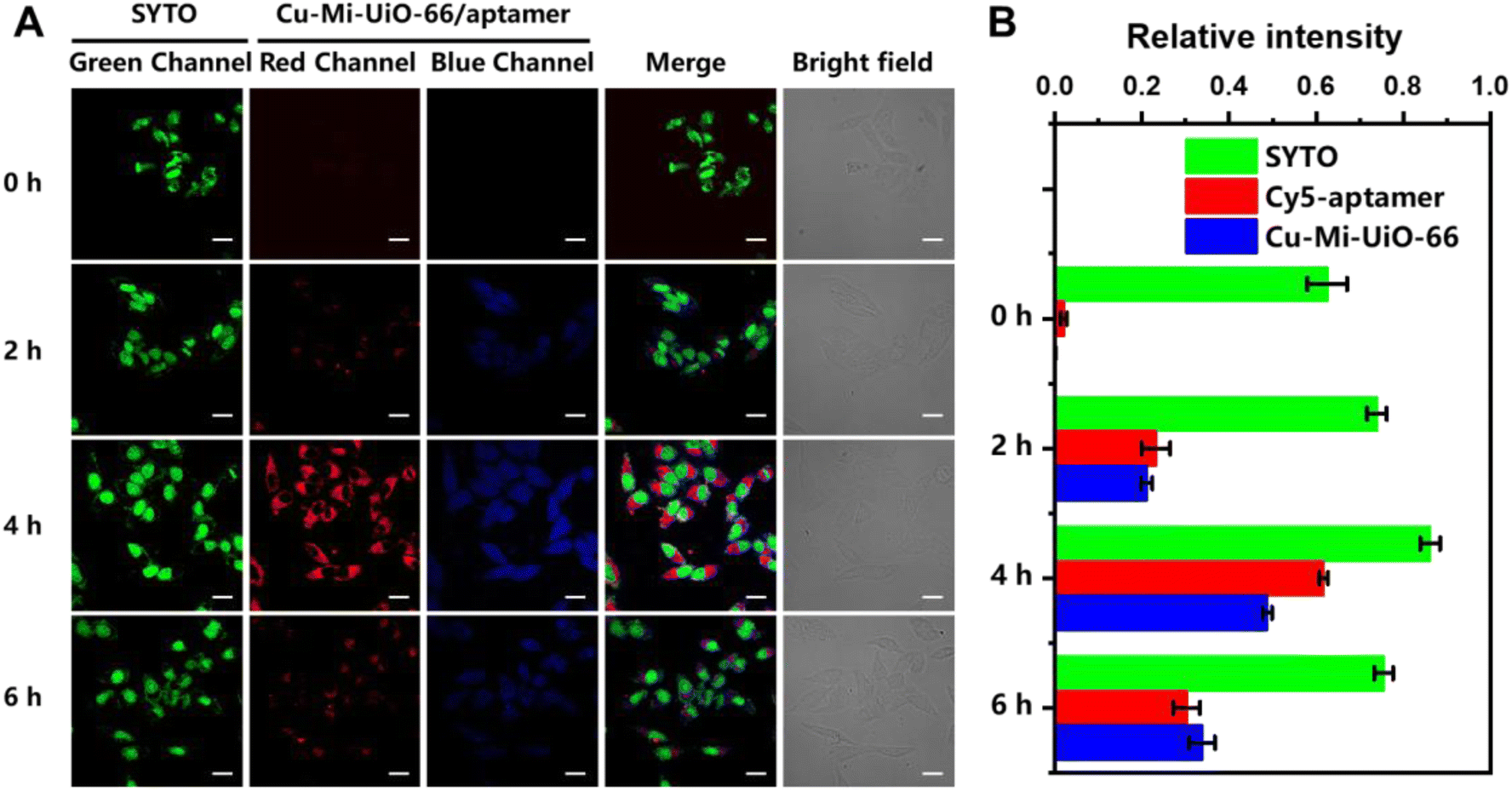 | ||
| Fig. 5 Fluorescence imaging of intracellular GSH and ATP. (A) Fluorescent, merged, and bright-field images of HepG2 cells upon exposure to Cu-Mi-UiO-66/aptamer (200 μg mL−1) for different periods of time (scale bar: 20 μm). (B) The quantitative values of different fluorescence channels quantified by Image J. | ||
In addition, z-stack images of HepG2 cells were captured from top to bottom at 1-μm “slice” intervals to show the capability of the Cu-Mi-UiO-66/aptamer in spatially tracing intracellular GSH and ATP. Video S1† illustrates that evident green (from SYTO), blue (from Cu-Mi-UiO-66) and red (from Cy5) fluorescence could be simultaneously observed, which became apparent along with scanning but faded at last. These results revealed that the Cu-Mi-UiO-66/aptamer could be efficiently internalized within the cells, evenly distributed in the cell cytoplasm, and more importantly, could be recognized by intracellular GSH and ATP to trigger its fluorescence turn-on behaviors.
To demonstrate the sensitive response of the Cu-Mi-UiO-66/aptamer to real-time changes in endogenous GSH in living cells, HepG2 cells were treated with NEM (GSH inhibitor) and ALA (GSH enhancer) to modulate the intracellular levels of GSH. As anticipated (Fig. 6A), compared to HepG2 cells without pre-treatments, blue fluorescence from Cu-Mi-UiO-66 was substantially decreased upon pre-treatment with NEM, because maleimides in NEM would clear GSH via the Michael addition reaction.48 Conversely, HepG2 cells exhibited significantly increased blue fluorescence upon pre-treatment with ALA, because ALA could induce GSH production by upregulating glutamate-cysteine ligase (γ-GCL) and glutathione reductase (GR).48,49 Quantitative analysis of fluorescence images using Image J software revealed that the fluorescence intensity of HepG2 cells treated with ALA was ∼6.0 times higher than that of HepG2 cells treated with NEM (Fig. 6B). Through paired sample t-test analysis, it was found that the GSH detection results in cells without any treatment were significantly different from those treated with the NEM inhibitor and ALA promoter, with p < 0.01.
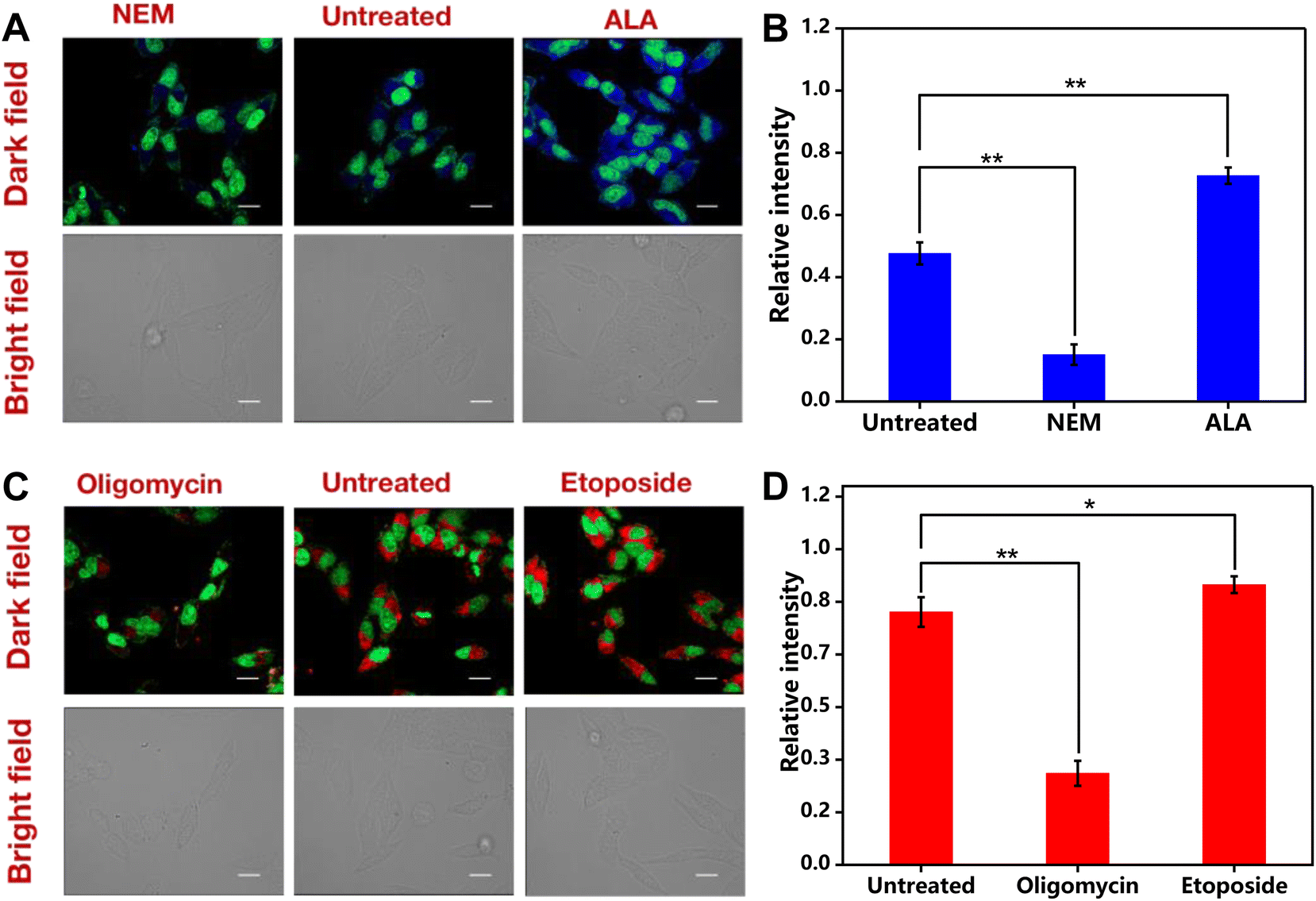 | ||
| Fig. 6 Fluorescence imaging of intracellular GSH and ATP. (A and B) Fluorescent and bright-field images (A) and the corresponding quantitative values (B) of HepG2 cells upon exposure to the Cu-Mi-UiO-66/aptamer (200 μg mL−1) alone (untreated), treated with NEM (500 μM), or ALA (500 μM). (C and D) Fluorescent and bright-field images (C) and the the corresponding quantitative values (D) of HepG2 cells upon exposure to the Cu-Mi-UiO-66/aptamer (200 μg mL−1) alone (untreated), treated with oligomycin (3 μg mL−1), or etoposide (500 μM). The statistical analysis method: the paired sample t test, *0.01 < p < 0.05 indicated a statistical difference; **p < 0.01 indicated a statistically significant difference. | ||
Similarly, treatment with oligomycin (ATP inhibitor) resulted in a substantial decrease of the red fluorescence, because ATP synthesis was inhibited by oligomycin (Fig. 6C).50 And as anticipated, HepG2 cells treated with etoposide (ATP inducer)51 resulted in an increased red fluorescence from the Cy5-labeled aptamer, because the aptamer detached from the NMOF Cu-Mi-UiO-66 by forming internal loop structures after the specific recognition of ATP.28,37 The fluorescence enhancement effect by etoposide was not prominent as revealed by quantitative analysis of fluorescence images (Fig. 6D). This is reasonable because there was only a limited amount of aptamer adsorbed onto the surface of Cu-Mi-UiO-66. High concentrations of ATP led to signal saturation. However, through paired sample t-test analysis, it was found that there was a significant difference in ATP levels between untreated cells and cells treated with the oligomycin inhibitor (p < 0.01) as well as cells treated with the etoposide promoter (0.01 < p < 0.05). All the above results validated that the Cu-Mi-UiO-66/aptamer was eligible for monitoring real-time changes of endogenous GSH and ATP simultaneously in living cells.
4. Conclusions
In summary, the NMOF Cu-Mi-UiO-66, the key components of the dual-responsive fluorescence nanoprobe Cu-Mi-UiO-66/aptamer, was facilely synthesized via a straightforward one-step solvothermal approach using ZrCl4, Cu(NO3)2 and the maleimide-containing ligand H2L1 as co-precursors. H2L1 in the skeleton of Cu-Mi-UiO-66 and the Cy5-labeled ATP aptamer assembled onto the surface of Cu-Mi-UiO-66 enables the nanoprobe Cu-Mi-UiO-66/aptamer to quantitate both GSH and ATP. Featuring high selectivity and sensitivity and good biocompatibility, the designed nanoprobe responds to GSH and ATP based on an “off-to-on” mode in both aqueous solution and living cells without an obvious crosstalk effect between the two analytes. Confocal imaging results demonstrated that the nanoprobe could be efficiently internalized within HepG2 cells and could be recognized by intracellular GSH and ATP to trace their concentration variations in HepG2 cells under different stimulations.This study offers several appealing advantages: (1) the Cu-Mi-UiO-66 NMOF presents itself as a versatile framework for developing multifunctional nanoprobes. NMOFs can integrate a variety of recognition moieties through in situ synthesis and post-synthetic functionalization. In particular, this bimetallic NMOF provides multiple sites for assembling various recognition moieties via the coordination effect, π–π stacking and electrostatic adsorption and guarantees low background thanks to the fluorescence quenching effect of the metal center. The bimetallic-center feature of NMOF can substantially simplify the probe synthesis while improving the detection sensitivity; (2) a dual-responsive nanoprobe for the simultaneous quantitation of GSH and ATP has been developed by employing the maleimide-containing ligand H2L1 and the Cy5-labeled ATP aptamer as the recognition moiety, respectively. This provides a versatile sensing platform that can be tailored to quantitate other analytes by simply changing the ligands used for NMOF's synthesis and aptamers assembled onto NMOF's surface. In addition, the tailored structures of the NMOF enable specific host–guest interactions, facilitating the identification of analytes in a highly specific manner; (3) a straightforward protocol has been established for tracing the intracellular concentration variations of GSH and ATP in living HepG2 cells under different stimulations. This provides a valuable protocol for tumor microenvironment research.
Author contributions
Yun Liu: visualization, validation, methodology, data curation, and formal analysis. Shuqi Xia: investigation, data curation, formal analysis, and writing – original draft. Meng Xiao: methodology and conceptualization. Mo Yang: supervision and resources. Mengsu Yang: supervision and funding acquisition. Changqing Yi: writing – review & editing, supervision, resources, project administration, funding acquisition, and conceptualization.Data availability
All relevant data are included in the paper and the ESI.† Data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Shenzhen Science and Technology Innovation Commission (JCYJ20210324140004013 and GJHZ20210705142200001) and the Guangdong Provincial Key Laboratory of Sensing Technology and Biomedical Instruments (2020B1212060077) is gratefully acknowledged.References
- B. J. Slotman and T. J. Eichler, Lancet Oncol., 2023, 24, 125–126 CrossRef.
- J. E. Lee and M. Y. Kim, Semin. Cancer Biol., 2022, 83, 4–14 CrossRef CAS.
- P. Johnson, Q. Zhou, D. Y. Dao and Y. Lo, Nat. Rev. Gastroenterol. Hepatol., 2022, 19, 670–681 CrossRef.
- M. Regimbeau, J. Abrey, V. Vautrot, S. Causse, J. Gobbo and C. Garrido, Semin. Cancer Biol., 2022, 86, 46–57 CrossRef CAS.
- K. Clack, N. Soda, S. Kasetsirikul, R. G. Mahmudunnabi, N. T. Nguyen and M. Shiddiky, Small, 2023, 19, e2205856 CrossRef.
- J. J. C. M. Nina and A. Mikirova, Cancer Ther. Oncol. Int. J., 2017, 3, 555623 Search PubMed.
- H. Choi, G. Park, E. Shin, S. W. Shin, B. Jana, S. Jin, S. Kim, H. Wang, S. K. Kwak, B. Xu and J. Ryu, Chem. Sci., 2022, 13, 6197–6204 RSC.
- E. C. Cheung and K. H. Vousden, Nat. Rev. Cancer, 2022, 22, 280–297 CrossRef CAS.
- Y. Xiong, C. Xiao, Z. Li and X. Yang, Chem. Soc. Rev., 2021, 50, 6013–6041 RSC.
- S. Wang, L. Zhang, J. Zhao, M. He, Y. Huang and S. Zhao, Sci. Adv., 2021, 7, eabe3588 CrossRef CAS PubMed.
- Y. Pan, Y. Shi, Z. Chen, J. Chen, M. Hou, Z. Chen, C. W. Li and C. Yi, ACS Appl. Mater. Interfaces, 2016, 8, 9472–9482 CrossRef CAS PubMed.
- N. de Souza, S. Zhao and B. Bodenmiller, Nat. Rev. Cancer, 2024, 24, 171–191 CrossRef CAS PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- X. Chai, Z. Fan, M. M. Yu, J. Zhao and L. Li, Nano Lett., 2021, 21, 10047–10053 CrossRef CAS.
- Z. Nan, H. Liu, L. Shi, H. Zhu, J. Chen, T. Ilovitsh, D. Wu, M. Wan and Y. Feng, ACS Appl. Mater. Interfaces, 2023, 15, 32732–32743 CrossRef CAS.
- Y. Shi, Y. Pan, H. Zhang, Z. Zhang, M. J. Li, C. Yi and M. Yang, Biosens. Bioelectron., 2014, 56, 39–45 CrossRef CAS.
- X. Jiang, J. Chen, A. Bajić, C. Zhang, X. Song, S. L. Carroll, Z. Cai, M. Tang, M. Xue, N. Cheng, C. P. Schaaf, F. Li, K. R. Mackenzie, A. C. M. Ferreon, F. Xia, M. C. Wang, M. Maletić-Savatić and J. Wang, Nat. Commun., 2017, 8, 16087 CrossRef CAS.
- R. Zhang, J. Zhao, G. Han, Z. Liu, C. Liu, C. Zhang, B. Liu, C. Jiang, R. Liu, T. Zhao, M. Y. Han and Z. Zhang, J. Am. Chem. Soc., 2016, 138, 3769–3778 CrossRef CAS.
- Q. Wang, Y. Shi, W. Chen, M. Yang and C. Yi, Microchim. Acta, 2021, 188, 9 CrossRef CAS PubMed.
- X. P. He, X. L. Hu, T. D. James, J. Yoon and H. Tian, Chem. Soc. Rev., 2017, 46, 6687–6696 RSC.
- C. Yu, D. Wu, L. Dai, X. He, J. Hou, J. Shen and Z. Li, Coord. Chem. Rev., 2023, 489, 215203 CrossRef CAS.
- J. Yao, M. Yang and Y. Duan, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS PubMed.
- W. Li, G. S. Kaminski Schierle, B. Lei, Y. Liu and C. F. Kaminski, Chem. Rev., 2022, 122, 12495–12543 CrossRef CAS.
- M. Nazari, A. S. Saljooghi, M. Ramezani, M. Alibolandi and M. Mirzaei, J. Mater. Chem. B, 2022, 10, 8824–8851 RSC.
- J. Li, N. Zhao, W. Zhang, P. Li, X. Yin, W. Zhang, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202215178 CrossRef CAS PubMed.
- X. Shi, H. M. Meng, X. Geng, L. Qu and Z. Li, ACS Sens., 2020, 5, 3150–3157 CrossRef CAS PubMed.
- H. Wang, J. Li, J. Li, K. Wang, Y. Ding and X. Xia, NPG Asia Mater., 2017, 9, e354 CrossRef CAS.
- L. Zhu, W. Liu, F. Tong, S. Zhang, Y. Xu, Y. Hu, M. Zheng, Y. Zhou, Z. Zhang, X. Li and Y. Liu, Spectrochim. Acta, Part A, 2024, 306, 113708 CrossRef.
- K. He, S. Dong, J. Yang, Q. Shi, L. Guan, L. Sun, Z. Chen and J. Feng, J. Environ. Chem. Eng., 2022, 10, 108178 CrossRef CAS.
- Y. Wu, X. Chen, X. Luo, M. Yang, C. Hou and D. Huo, Anal. Chim. Acta, 2021, 1183, 339000 CrossRef CAS PubMed.
- Y. Li, C. Zhao, N. Zhu, Q. Liu, G. Chen, J. Liu, X. Zhao, J. Ma, S. Zhang and Y. Dong, Chem. Commun., 2015, 51, 17672–17675 RSC.
- B. Gui, Y. Meng, Y. Xie, J. Tian, G. Yu, W. Zeng, G. Zhang, S. Gong, C. Yang, D. Zhang and C. Wang, Adv. Mater., 2018, 30, 1802329 CrossRef PubMed.
- X. Gao, P. Du, Y. Zhang, M. Wang, Y. Wang and X. Zhao, J. Environ. Chem. Eng., 2023, 11, 110934 CrossRef CAS.
- L. He, Z. Yang, X. Lu, Y. Xu, X. Yao, C. Li, C. Wu and Z. Yao, J. Environ. Chem. Eng., 2023, 11, 111419 CrossRef CAS.
- Measuring cell fluorescence using ImageJ, https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html, (accessed 2.24, 2024).
- H. S. Jung, X. Chen, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2013, 42, 6019–6031 RSC.
- L. Wu, Y. Wang, X. Xu, Y. Liu, B. Lin, M. Zhang, J. Zhang, S. Wan, C. Yang and W. Tan, Chem. Rev., 2021, 121, 12035–12105 CrossRef CAS PubMed.
- J. Lyu, H. Liu, Z. Zeng, J. Zhang, Z. Xiao, P. Bai and X. Guo, Ind. Eng. Chem. Res., 2017, 56, 2565–2572 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. Stawowy, R. Ciesielski, T. Maniecki, K. Matus, R. Bużny, J. Trawczynski, J. Silvestre-Albero and A. Bamacz, Catalysts, 2020, 10, 39 CrossRef CAS.
- Y. Pan, S. Jiang, W. Xiong, D. Liu, M. Li, B. He, X. Fan and D. Luo, Microporous Mesoporous Mater., 2020, 291, 109703 CrossRef CAS.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- A. F. Kateshali, F. Moghzi, J. Soleimannejad and J. Janczak, Inorg. Chem., 2024, 63, 3560–3571 CrossRef.
- R. Mo, T. Jiang, R. Disanto, W. Tai and Z. Gu, Nat. Commun., 2014, 5, 3364 CrossRef PubMed.
- M. Zhang, S. Wang, Y. Fu, M. Meng, H. Jin and W. Zhao, Sens. Actuators, B, 2022, 366, 132013 CAS.
- O. Muscetti, N. Blal, V. Mollo, P. A. Netti and D. Guarnieri, Nanomaterials, 2023, 13, 1999 CrossRef CAS.
- Y. Luo, M. Yin, C. Mu, X. Hu, H. Xie, J. Li, T. Cao, N. Chen, J. Wu and C. Fan, Adv. Mater., 2023, 35, 2210458 CAS.
- Z. Zhou, H. Liang, R. Yang, Y. Yang, J. Dong, Y. Di and M. Sun, Angew. Chem., Int. Ed., 2022, 61, e202202843 CrossRef CAS.
- J. Zhang, X. Zhou, W. Wu, J. Wang, H. Xie and Z. Wu, Environ. Toxicol. Pharmacol., 2017, 51, 30–37 CrossRef CAS PubMed.
- J. Symersky, D. Osowski, D. E. Walters and D. M. Mueller, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13961–13965 CrossRef CAS.
- M. Katayama, T. Kawaguchi, M. S. Berger and R. O. Pieper, Cell Death Differ., 2007, 14, 548–558 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02585g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |