
Orotic acid-capped Tb(III)-doped calcium sulphate nanorods for the selective detection of tryptophan†
Jaydeep
Kumar
 a,
Neha
Yadav
b,
Viplove
Mishra
a,
Heramba V. S. R. M.
Koppisetti
a,
Avishek
Roy
a,
Antarip
Mitra
a and
Venkataramanan
Mahalingam
*a
a,
Neha
Yadav
b,
Viplove
Mishra
a,
Heramba V. S. R. M.
Koppisetti
a,
Avishek
Roy
a,
Antarip
Mitra
a and
Venkataramanan
Mahalingam
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, 741246, West Bengal, India. E-mail: mvenkataramanan@yahoo.com
bDepartment of Biological Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, 741246, West Bengal, India
First published on 21st October 2024
Abstract
Lanthanide-based luminescent materials have gained huge attention due to their applications in optoelectronic devices, sensing, bio-imaging, anti-counterfeiting, and more. In this work, we report a luminescence-based sensor for the detection of tryptophan using orotic acid-capped Tb3+-doped CaSO4 nanorods (NRs). Orotic acid (OA) was found to play a dual role as a capping agent to control the growth of the nanorods and as a sensitizer for Tb3+ ions. The resulting nanorods exhibited excellent dispersibility and strong photoluminescence signals characteristic of Tb3+ ions in the visible region. Nearly 10-fold enhancement in the emission intensity was noted through OA sensitization compared to direct excitation of Tb3+ ions (acceptors). Interestingly, the strong emission intensity of the NRs reduced significantly with the addition of tryptophan. In contrast, hardly any change was noted with the addition of other amino acids and metal ions, suggesting greater selectivity for tryptophan. Moreover, there is barely any notable interference from other amino acids toward the detection of tryptophan. The limit of detection is found to be ∼0.61 μM. Finally, the sensing study was extended to biological samples to detect tryptophan present in blood plasma, urine, and saliva samples. The nanorods demonstrated high detection abilities, indicating the potential of the developed materials for biomedical applications.
Introduction
Tryptophan is one of the essential aromatic amino acids that has numerous applications in different fields.1 Its detection can provide crucial insights into the structural and functional properties of biomolecules and also be a suitable biomarker for different diseases, especially in medicine.2 For instance, tryptophan has been shown to be a precursor for serotonin, a neurotransmitter that regulates mood and sleep patterns.3 Besides, tryptophan has been used to diagnose and monitor cancer.4 In certain types of cancer, tryptophan metabolism is altered, leading to increased levels of specific metabolites, like in the case of gastric cancer, where an increased level of tryptophan in gastric juice can be a marker of potential gastric cancer.5 Hence, the detection of tryptophan can help in the early diagnosis of the disease, which is quite essential in the field of medicine. The free tryptophan level in plasma has been found to be linked with major depressive disorders (MDD), type 2 diabetes, tryptophan metabolism, etc.6–10 Also, the tryptophan level in saliva was found to be correlated with levels of serotonin, salivary cortisol, etc.11–13 Additionally, tryptophan levels in urine were also reported to be correlated to vitamin B6 deficiency, breast cancer, and autistic patients, just to mention a few.14–16 The detection of tryptophan is also essential in environmental applications such as monitoring water quality.17 Since tryptophan is also present in natural organic matter, its presence in organic pollutants can also serve as a marker for microbial activities.18,19 Adding these up, analyzing tryptophan can provide valuable information in various applications.Traditional methods for tryptophan detection include mass spectrometry, inductively coupled plasma mass spectrometry (ICP-MS), infrared spectroscopy, gas chromatography, electrochemical methods, AAS, voltammetry, high-performance liquid chromatography (HPLC), and inductively coupled plasma atomic emission spectroscopy (ICP-AES).20–23 Despite their high sensitivity, they are unsuitable for real-time and on-site detection. Fluorescence-based methods are advantageous as they are relatively simpler and provide high sensitivity, easy sample preparation methods, and quick response times.24–27 Furthermore, hand-held optical spectrometers are available for on-site detection. There are few reports on fluorescence sensors for the detection of tryptophan. For example, Zhang et al. reported a hydrothermally prepared carbon spheres-based fluorescence sensor for detecting L-tryptophan.28 The carbon spheres have limited capacity in terms of sensing due to challenges associated with achieving precise control over their emission properties. Additionally, the stability and photostability (i.e., resistance to photobleaching) of carbon spheres are not promising, nor do the photophysical properties of the carbon spheres allow them to show low detection limits and sensitivity.29 In another work, Pang and co-workers have synthesized terbium hybrids with a silica framework to detect tryptophan.30 However, the obtained detection limit was low, and the synthesis protocol involves multiple complex steps with long reaction times. Alternatively, lanthanide (Ln3+)-doped nanomaterials are interesting for the following reasons. For example, the 4f electrons of lanthanides are well-shielded from the 5s and 5p orbitals, making them unperturbed by the surrounding crystal fields.31,32 This results in quite narrow intra-4f–4f transitions.33,34 They possess long luminescence lifetimes, allowing efficient excited-state processes like luminescence resonance energy transfer (LRET) to occur.35,36 LRET can occur over a larger distance in Ln3+-doped materials or complexes (>10 nm).37 This allows efficient sensitization of the Ln3+ emission, thereby minimizing the problem of low absorbance of Ln3+ ions. Additionally, Ln3+ ions show a large Stokes shift, which minimizes optical interference from other foreign molecules.38,39
There are quite a few reports on the use of Ln3+-doped nanomaterials for the detection of toxic metal ions and other small molecules.40–50 Nevertheless, there are few reports on using these nanomaterials for amino acid detection, particularly tryptophan. For instance, Hazra et al. used Tb3+-doped nanocrystals for the detection of aromatic amino acids at nanomolar concentrations.51 However, the obtained nanocrystals were not selective towards the detection of only tryptophan but rather for all three aromatic amino acids, thus urging the need to develop specific and selective sensor materials. Our objective is to choose a multifunctional ligand that not only controls the nanoparticle size but acts as both a sensitizer to Ln3+ ions and a linker to tryptophan. The direct binding of tryptophan to the sensitizer is expected to alter the energy transfer efficiency of the latter to the Ln3+ ions. We have synthesized orotic acid (OA)-capped Tb3+-doped CaSO4 nanorods (NRs) using a simple microwave method. A 3% doping concentration of Tb3+ ions was chosen based on optimization results (Fig. S1, ESI†).
The use of microwave synthesis is advantageous due to its fast reaction times, high product yield, and minimal by-products.52 We have chosen OA as a capping molecule as it possesses a pyrimidinedione unit and the carboxyl group, which can bind to the nanoparticles. The pyrimidinedione unit is expected to have hydrogen bonding interaction with tryptophan. In addition, OA has strong absorbance in the UV region and can thus sensitize the Tb3+ ions possessing multiple excited energy levels in the UV and visible regions. Interestingly, DMSO plays a dual role as a suitable solvent and sulphate source.53 CaSO4 is chosen as the host matrix as there is a close match of ionic radii of Ca2+ (1.0 Å) with Tb3+ (0.92 Å), which is expected to facilitate the doping of Tb3+ ions.54 The close match in size is essential for preparing Tb3+-doped NRs with fewer crystal defects. The successful incorporation of Tb3+ ions into the CaSO4 host matrix resulted in strong green emission upon excitation at 290 nm. The strong intensity of the green emission is attributed to the efficient energy transfer from OA to the Tb3+ ions.
The addition of tryptophan selectively reduces the emission intensity of the Tb3+ ions. Furthermore, there is barely any interference in the detection of tryptophan from other amino acids or metal ions. Finally, we have extended the detection of tryptophan using OA-capped Tb3+-doped CaSO4 NRs in various biological samples.
Results and discussion
Phase analysis
Powder X-ray diffraction measurements (PXRD) were performed to check the phase purity of as-prepared Tb3+-doped CaSO4 nanorods. Fig. 1 shows the PXRD pattern of OA-capped CaSO4:Tb3+ NRs, along with the standard pattern of bulk CaSO4. The PXRD pattern of the NRs matched well with the standard pattern of orthorhombic phase bulk CaSO4 (ICDD: 01-072-0916), confirming the formation of orthorhombic phase materials. The observed sharp diffraction peaks indicate good crystallinity in the synthesized NRs. | ||
| Fig. 1 PXRD pattern of OA@CaSO4:Tb3+ NRs compared with the standard pattern of pure CaSO4. | ||
Morphology analysis
The formation of the rod-shaped morphology of the OA-capped NRs is confirmed by the field-emission scanning electron microscopy (FESEM) images presented in Fig. 2(a, b, e & f). The formation of rod-shaped nanostructures is further supported by transmission electron microscopy (TEM) analysis (Fig. 2(c, d, g & h)). The high-resolution TEM image of the OA–CaSO4 NRs reveals the presence of lattice fringes, suggesting high crystallinity of the NRs. The d-spacing between the planes is 2.84 Å, indicating that the observed lattice fringes correspond to the (102) planes. | ||
| Fig. 2 SEM images (a, b, e, and f) and TEM images (c and d) of OA@CaSO4:Tb3+ NRs at different magnifications. (g and h) HRTEM image and SAED pattern of the NC NRs showing diffraction planes corresponding to CaSO4. | ||
Surface functionalization
FTIR analysis confirmed the binding of OA molecules to the NRs, presumably at the surface. Fig. 3(a) shows the FTIR spectra of pure OA and OA-capped CaSO4 NRs. The shift in the “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” stretching frequency of the –COOH group from 1716 cm−1 in pure OA to 1623 cm−1 in OA-capped CaSO4 indicates the successful binding of OA to the surface of the nanorods.55 This shift is attributed to the binding of the carboxylate group to the NRs through the oxygen atom, leading to a decrease in the strength of the “CO” bond and a subsequent reduction in the corresponding stretching frequency. The other peaks corresponding to different functional groups remained unaffected, confirming that binding has occurred through the –COOH group.
O” stretching frequency of the –COOH group from 1716 cm−1 in pure OA to 1623 cm−1 in OA-capped CaSO4 indicates the successful binding of OA to the surface of the nanorods.55 This shift is attributed to the binding of the carboxylate group to the NRs through the oxygen atom, leading to a decrease in the strength of the “CO” bond and a subsequent reduction in the corresponding stretching frequency. The other peaks corresponding to different functional groups remained unaffected, confirming that binding has occurred through the –COOH group.
 | ||
| Fig. 3 (a) FT-IR spectra of pure OA and the same after being attached to the Tb3+-doped CaSO4 NRs. (b) Excitation and photoluminescence spectra of OA@CaSO4:Tb3+ NRs. The concentration of OA@CaSO4:Tb3+ nanorods in DMSO used was 1 mg per 10 mL. | ||
UV-vis and photoluminescence analyses
UV-vis absorption spectra of pure OA and OA-capped CaSO4:Tb3+ nanorods are presented in Fig. S2 (ESI†). The absorption spectrum of pure OA exhibited a broad peak ranging from 271 to 330 nm, with a maximum at 295 nm. In the case of OA@CaSO4:Tb3+ NRs, a broad hump with peak maxima at 291 nm is observed, closely resembling the absorption spectrum of pure OA. This confirms the attachment of OA molecules to the nanorods. Fig. 3(b) represents the excitation and emission spectra of OA-capped Tb3+-doped CaSO4 NRs. All PL studies were carried out on the NRs dispersed in DMSO. The PL spectrum shows peaks at approximately 490 nm, 542 nm, 586 nm, and 622 nm, attributed to the 5D4 excited level to 7F6, 7F5, 7F4, and 7F3 ground state transitions, respectively. A strong band with a maximum at 291 nm is observed in the excitation spectrum (λem = 542 nm, 7F5 ← 5D4 transition of Tb3+ ions). The excitation peak position matches with the maxima in the UV-vis absorbance peak of OA, indicating the possible sensitization of the Tb3+ emission by OA. In comparison with direct excitation (i.e., λex = 378 nm) of Tb3+ ions, excitation via OA resulted in almost ∼10 times enhancement in the emission intensity of Tb3+ ions.56 To confirm the occurrence of energy transfer from OA to the Tb3+ ions inside the NRs, a control experiment was performed by synthesizing citric acid (CA)-capped CaSO4:Tb3+ NRs. CA was chosen as it lacks any characteristic absorption or emission in the UV region and is expected to show hardly any energy transfer process between citric acid and Tb3+ ions. Fig. S3 (ESI†) shows the comparison of the excitation and PL spectra of Tb3+-doped CaSO4 NRs capped with OA and CA. As expected, barely any enhancement in the characteristic Tb3+ emission peaks is observed when excited at 291 nm for the CA@CaSO4:Tb3+ nanorods compared to the peaks obtained by exciting at 378 nm, which is the direct excitation of the Tb3+ ions (Fig. S3(c), ESI†).A schematic of the proposed energy transfer process between OA and Tb3+ ions in OA@CaSO4:Tb3+ nanorods is shown in Fig. S4 (ESI†). Briefly, upon excitation of the NRs at 291 nm, OA becomes excited from the ground state (S0) and reaches the excited singlet state (S1). This is followed by a non-radiative intersystem crossing (ISC), which results in the long-lived triplet (T1) state.57 The 5D4 level of the Tb3+ ion lies below the triplet state of OA, facilitating energy transfer from the higher triplet state to the low-lying energy levels of Tb3+ ions. This leads to the population of the excited states of Tb3+ ions and subsequent radiative transitions from 5D4 to 7FJ (J = 3–6) ground state energy levels. The photoluminescence quantum yield was determined using tyrosine as a reference, as its absorbance matches well with that of orotic acid. The quantum yield of OA-capped CaSO4:Tb3+ nanorods is calculated using the following equation (eqn (1)):58
| Qsample = Qref(A/Aref)(Iref/I)(n2/nref2) | (1) |
To investigate the detection capabilities of the OA@CaSO4:Tb3+ nanorods, a 1 mM concentration of tryptophan was added to the NR dispersion, and the PL characteristics were measured. A strong reduction in the emission intensity of the Tb3+ was observed. To understand the selectivity of the detection, a range of different amino acid solutions (at a concentration of 1 mM) were tested.
The results are shown as a bar diagram in Fig. 4a. There is barely any change in the emission intensity with the addition of other amino acids. Additionally, various other analytes are tested, such as EDTA, Cu2+, Hg2+, Ni2+, Mn4+, Cd2+, Fe3+, Zn2+, Co2+, Pb2+, and other relevant cations and anions. Fig. 4b shows a reduction in the emission intensity to approximately 90%, noted only with the addition of tryptophan. The above results confirm the good selectivity of the OA@CaSO4:Tb3+ nanorods toward detecting tryptophan.
 | ||
| Fig. 4 Bar diagram showing (a) the effect of different amino acids on the PL intensity of OA@CaSO4:Tb3+ NRs, (b) the effect of different metal ions on the PL intensity of OA@CaSO4:Tb3+ NRs, and (c) the interference of other amino acids on the selective detection of tryptophan using OA@CaSO4:Tb3+ nanorods, and (d) the interference of metal ions on the selective detection of tryptophan using OA@CaSO4:Tb3+ nanorods. The concentration of OA@CaSO4:Tb3+ nanorods in DMSO used was 1 mg per 10 mL. | ||
To understand the reliability of the assay, we have studied the colloidal stability of the nanoprobe. The photoluminescence of the colloidal dispersion was measured up to one hour at 10-minute intervals (Fig. S5, ESI†). The intensity of the OA@CaSO4:Tb3+ NRs showed minimal change, with a maximum decrease of approximately 16% compared to the fresh samples. Please note that all tryptophan detection analyses were done within a minute of addition. This indicates that the nanoprobe exhibits good stability during the assay.
To examine any interference from other amino acids, ions, and molecules towards quenching of the Tb3+ emission intensity by tryptophan, we conducted PL measurements of OA@CaSO4:Tb3+ NRs in a mixture containing equimolar (1 mM) tryptophan and other analytes. The results shown in Fig. 4(c & d) suggest that the quenching of the PL emission intensity by tryptophan is hardly influenced by the presence of other interfering ions and molecules. Furthermore, to check if any change in the pH and temperature of the medium with the addition of different amino acids is responsible for the observed differences in the PL intensity, the pH and temperature of the dispersion containing NRs are measured for the corresponding mixtures. There is barely any noticeable change in the pH and temperature of the NR dispersion in DMSO after adding different amino acids (Fig. S6, ESI†).
To study the photoluminescence response of OA@CaSO4:Tb3+ NRs towards tryptophan, different concentrations of tryptophan were added to the NR dispersion. As noted in Fig. 5(b), a gradual decrease in the intensity of Tb3+ emission is observed with an increase in the concentration of tryptophan in the dispersion. Almost a 90% reduction in the emission intensity of Tb3+ is observed with the addition of 5 mM tryptophan. In the excitation spectra of OA@CaSO4:Tb3+, a shift towards the higher wavelength (redshift) is observed for the characteristic tryptophan peak at higher concentrations of tryptophan (Fig. 5(a)). The redshift observed in the excitation spectra of OA@CaSO4:Tb3+ NRs can be attributed to the tryptophan binding to the OA present on the surface of the NRs. This likely alters the energy level of the OA, thus affecting the energy transfer efficiency between OA and Tb3+ ions present in the nanorods.60–62
 | ||
| Fig. 5 Excitation (a) and PL (b) spectra of OA@CaSO4:Tb3+ NRs collected with the gradual addition of tryptophan solutions of increasing concentration. The concentration of OA@CaSO4:Tb3+ nanorods in DMSO used was 1 mg per 10 mL. | ||
The sensitivity of OA@CaSO4:Tb3+ NRs towards the detection of tryptophan ions was evaluated using the Stern–Volmer analysis, which relates the luminescence intensity of the nanorods in the presence of an analyte to its intensity in its absence. In the following Stern–Volmer equation (eqn (2)):
| I0/I = 1 + KSV[C] | (2) |
 | ||
| Fig. 6 The Stern–Volmer plot of OA@CaSO4:Tb3+ NRs (3%) with the addition of increasing concentrations of tryptophan in DMSO. The concentration of OA@CaSO4:Tb3+ nanorods in DMSO used is 1 mg per 10 mL. | ||
Tryptophan detection in biological samples
The detection of tryptophan using OA@CaSO4:Tb3+ NRs is extended to biological samples like human blood plasma, saliva, and urine specimens. The determination of tryptophan in clinical specimens is essential, though challenging, given the presence of heterogeneous organic (proteins) and inorganic (for example, metal ions) components.64 To assess the tryptophan level in blood plasma, we have taken a 2 mL dispersion of OA@CaSO4:Tb3+ NRs and added 100 μl of pure plasma. The PL analysis of the mixture shows quenching of the luminescence intensity of Tb3+ ions in OA@CaSO4:Tb3+ NRs (Fig. 6). The limit of detection (LOD) is determined using the equation 3σ/K, where σ is the standard deviation of the blank and K is the slope of the calibration plot. The calculated LOD is 0.61 μM.To check the effectiveness of our approach, a mixed solution of amino acids (2 mmol each), except tryptophan, was added to the above mixture containing NRs and blood plasma. The corresponding dispersion shows no further quenching. Next, the NR dispersion is mixed with pure plasma containing a mixture of amino acids, including tryptophan (2 mmol). The corresponding PL spectrum shows a further reduction in the emission intensity of Tb3+ ions (Fig. 7(a)). The above results suggest that the reduction in the emission intensity from the NRs is caused primarily by the presence of tryptophan in the dispersion.
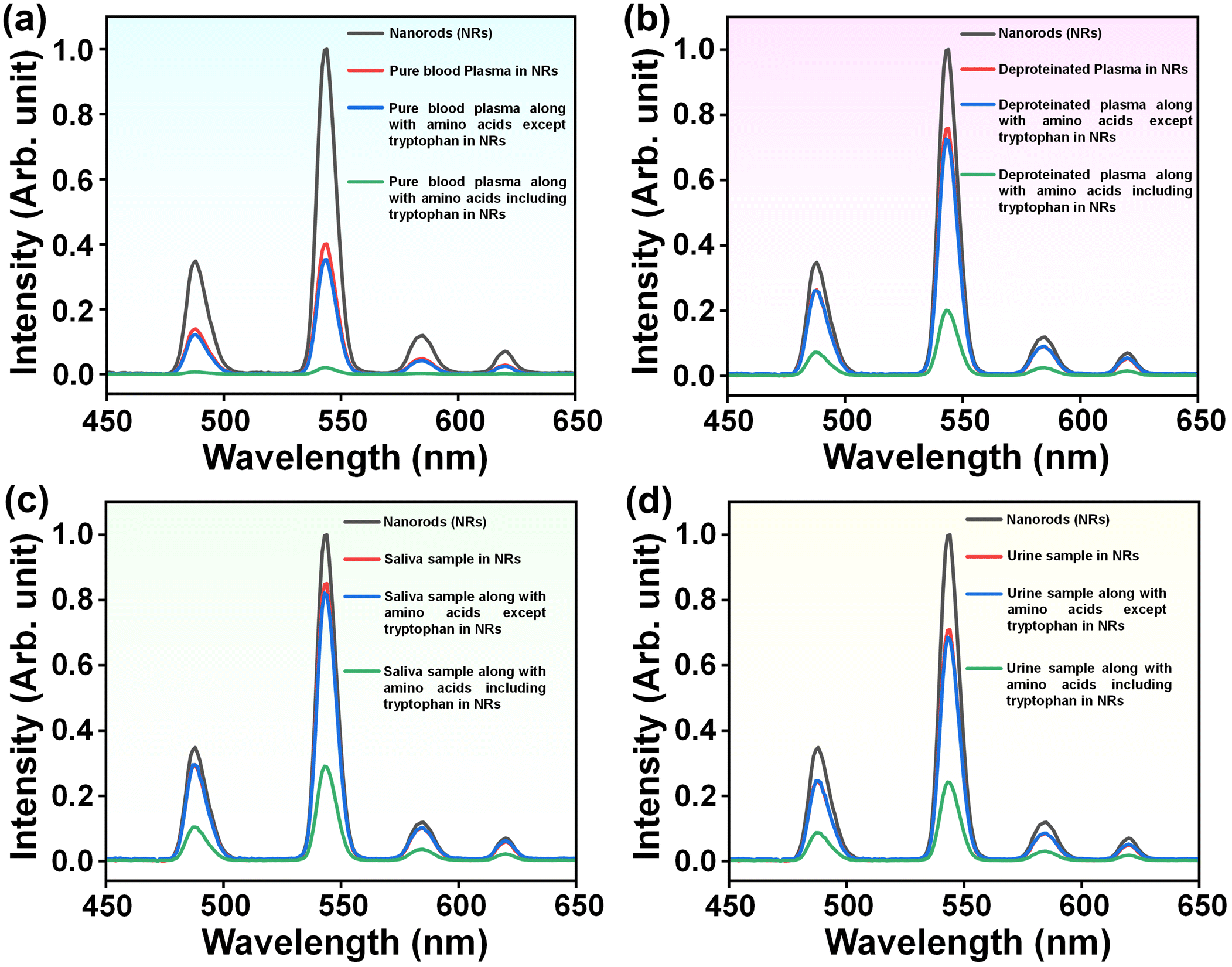 | ||
| Fig. 7 PL analysis: (a) emission spectra of pure OA@CaSO4:Tb3+ NRs, after addition of pure blood plasma sample, pure blood plasma sample with a mixture of amino acids without tryptophan, and pure sample with a mixture of amino acids including tryptophan to OA@CaSO4:Tb3+ NRs. (b) emission spectra of pure OA@CaSO4:Tb3+ NRs, after addition of deproteinated blood plasma sample, deproteinated sample with a mixture of amino acids without tryptophan, and deproteinated sample with a mixture of amino acids including tryptophan to OA@CaSO4:Tb3+ NRs; (c) emission spectra of pure OA@CaSO4:Tb3+ NRs, after addition of saliva, a saliva sample with a mixture of amino acids without tryptophan, and saliva with a mixture of amino acids including tryptophan to OA@CaSO4:Tb3+ NRs; and (d) emission spectra of pure OA@CaSO4:Tb3+ NRs, after addition of a urine sample, a urine sample with a mixture of amino acids without tryptophan, and a urine sample with a mixture of amino acids including tryptophan to OA@CaSO4:Tb3+ NRs. The concentration of OA@CaSO4:Tb3+ nanorods in DMSO is 1 mg per 10 mL. | ||
Furthermore, a detection analysis was performed on the plasma sample after removing the proteins to detect the presence of free tryptophan in the blood plasma. A 100 μL deproteinated plasma sample was added to a 2 mL dispersion of NRs. The PL spectrum of the resulting sample shows a reduction in the luminescence intensity of Tb3+ ions in OA@CaSO4:Tb3+. However, the quenching observed was relatively lower than that observed for pure plasma samples, primarily due to the absence of tryptophan bound to plasma proteins (Fig. 7(b)). Again, a sample of deproteinated blood plasma and the mixture of amino acids (2 mmol each), except tryptophan, was added to the NR dispersion. Again, no further quenching of the emission intensity is observed. Finally, the NR dispersion was treated with a sample of deproteinated blood plasma with a mixture of amino acids, including tryptophan (2 mmol each). In this case, further quenching was observed, confirming the efficiency of our method in detecting tryptophan in human blood plasma.
The above results motivated us to extend the study to detect tryptophan in human saliva and urine. We followed a procedure similar to that performed with the blood plasma study. In this case, saliva and urine samples were used directly without further processing. In both studies, the PL analysis showed considerable quenching of the luminescence intensity of Tb3+ ions in OA@CaSO4:Tb3+ NRs. Similarly, the NR dispersion with the saliva and urine samples, with a mixture of amino acids (2 mmol each), except tryptophan, shows hardly any quenching (Fig. 7(c & d)). The addition of a mixture containing all the amino acids, including tryptophan (2 mmol), to the saliva and urine samples in the NR solution resulted in further quenching of the Tb3+ emission intensity. These results suggest that OA@CaSO4:Tb3+ NRs could be an efficient optical probe for detecting tryptophan in biological samples without much interference from other analytes. In addition to the emission spectra, we also tried to study the excitation spectra at λem = 542 nm after the addition of pure plasma to the OA@CaSO4:Tb3+ NRs, and the same in the presence of an additional 2 mmol concentration of tryptophan (Fig. S7, ESI†). It is noted that the excitation peaks showed a similar shift, as noted in the case of the addition of different concentrations of tryptophan in NRs (vide supra). Additionally, the results of the recovery test of tryptophan in the bio-related samples are shown in Table 1. The results showed that recoveries of tryptophan in these samples are within the range of 99–102.5%.
Proposed detection mechanism
To investigate the cause of the quenching of Tb3+ ion emission intensity upon the addition of tryptophan, some additional experiments and analyses were performed. The attachment or interaction of tryptophan with OA present on the surface of OA@CaSO4:Tb3+ nanorods was validated through FTIR analysis. The FTIR spectrum shows a shift in the N–H bond stretching to a lower wavenumber (from 3400 cm−1 to 3130 cm−1) for the OA@CaSO4:Tb3+ NRs after the addition of tryptophan (Fig. S8, ESI†). This indicates a probable tryptophan binding to the OA on the NR surface through the pyrimidine N–H-group. Furthermore, to confirm any possible interaction between the amino acid tryptophan and OA, NMR analysis was performed. First, the 1H NMR spectra of tryptophan and OA were recorded in DMSO-d6. The 1H NMR spectrum of tryptophan consists of 3 peaks within the 3 to 3.5 ppm range, which belongs to the aliphatic C–H bonds (Fig. S9, ESI†). Similarly, OA shows a peak at 6 ppm corresponding to the sp2 C–H unit. The peaks at 11.33 and 10.89 ppm represent the two N–H bonds of OA (Fig. S10, ESI†). In a separate experiment, a few drops of tryptophan were mixed with a DMSO-d6 solution containing OA. The NMR spectrum of the mixture shows a shift in the peak position in the aliphatic region of tryptophan (Fig. S11, ESI†). We believe that this shift is because of the interaction of tryptophan's amino and –COOH groups with OA. This interaction likely resulted in a change in the electron density around the carbon of the tryptophan molecules, causing a shift in the chemical shift positions of these hydrogen atoms of the aliphatic regions of tryptophan. The concentration of OA is much greater than that of tryptophan in the above experiment. As a result, only the shift in tryptophan protons is observed. To acquire more information, a 1H NMR spectrum was recorded for a solution containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of tryptophan and OA in DMSO-d6 (Fig. S12, ESI†). Interestingly, a shift in the peak positions of both OA and tryptophan is noted, suggesting a chemical interaction between both molecules. This interaction is likely to cause changes in the triplet state of the OA, thereby decreasing the probability of energy transfer from OA to Tb3+ ions present inside the NRs.
:1 molar ratio of tryptophan and OA in DMSO-d6 (Fig. S12, ESI†). Interestingly, a shift in the peak positions of both OA and tryptophan is noted, suggesting a chemical interaction between both molecules. This interaction is likely to cause changes in the triplet state of the OA, thereby decreasing the probability of energy transfer from OA to Tb3+ ions present inside the NRs.
The decrease in the energy transfer probability from OA to Tb3+ inside the NRs reduces the excited state population of Tb3+ ions, decreasing the emission intensity. This is validated by the reduction in the lifetime of the 5D0 level of Tb3+ ions in OA@CaSO4:Tb3+ NRs, shifting from 2.00 ms to 0.01 ms after the addition of tryptophan. In contrast, minimal change was observed in the case of serine addition (2.00 ms to 1.99 ms). These dynamics are illustrated in Fig. 8. The decrease in lifetimes indicates the presence of another relaxation pathway that can depopulate the excited state. The potential role of π–π interactions, whereby tryptophan engages with the aromatic systems in the nanorods, is also noteworthy and could contribute to luminescence quenching by perturbing electronic transitions responsible for the emission.65–70
 | ||
| Fig. 8 The lifetime decay curves of OA@CaSO4:Tb3+ NRs before (blue) and after the addition of serine (red) and tryptophan (green). The concentration of OA@CaSO4:Tb3+ nanorods in DMSO was 1 mg per 10 mL. | ||
To further check any competitive absorption occurring between OA and tryptophan for the excitation energy, UV-vis absorption analysis was performed. The UV-vis spectra shown in Fig. S13† show a clear overlap between the absorption peaks of OA and tryptophan (between 250 nm to 300 nm). However, near 290 nm, the absorbance is higher for OA compared to tryptophan. This suggests that there is competitive absorption to some extent between OA and tryptophan for the excitation light. Thus, part of the reduction in the Tb3+ emission intensity in the presence of tryptophan is likely due to the competitive absorption. Therefore, one of the reasons for the observed reduction in the emission intensity of Tb3+ ions upon the addition of tryptophan could be the combination of competitive absorption of excitation energy between OA@CaSO4:Tb3+ and tryptophan. In addition, intermolecular interactions between the OA capping agent and tryptophan alter the energy of the triplet state, thereby reducing the probability of energy transfer efficiency between OA and Tb3+ present inside the nanorods.
Conclusions
In summary, we utilized a microwave synthesis method to prepare OA-capped Tb3+-doped CaSO4 colloidal nanorods. When excited at the OA absorption wavelength of 291 nm, these colloidal nanorods exhibited strong emissions at 542 nm along with weak emissions at 488, 585, and 620 nm, which are characteristic emission peaks of Tb3+ ions. The addition of tryptophan selectively quenches the emission intensities of the Tb3+ ions due to a reduction in the energy transfer efficiency between OA and Tb3+ ions, presumably caused by the overlapping of tryptophan emission with that of OA along with the chemical interaction between OA and tryptophan altering the energy transfer efficiency between OA and Tb3+ ions. This quenching effect is specific to tryptophan, as adding other amino acids and analytes had little impact on the Tb3+ ion emission intensity. In addition, the developed nanorods efficiently detect tryptophan in human blood plasma, saliva, and urine, indicating its potential use in biomedical applications.Author contributions
J. Kumar and H. V. S. R. M. Koppisetti conducted a critical analysis of experimental data. J. Kumar contributed to the writing of the manuscript. N. Yadav helped with biological experiments. V. Mishra assisted with the synthesis. A. Roy and A. Mitra assisted with the physical characterizations and NMR analyses, respectively. V. Mahalingam planned the study, providing motivation and oversight throughout its execution.Data availability
The data that support the findings of this study are available from the corresponding author (Venkataramanan Mahalingam) upon reasonable request.Conflicts of interest
The authors have no conflicts to declare neither in the financial interests nor in the personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
VM thanks SERB, CSIR, and IISER Kolkata for providing the instrumental facilities and funding. The authors thank DST-FIST for the TEM facility at IISER Kolkata through the project SR/FST/CS11-029/2014. JK thanks his family for their encouragement and support.References
- A. Agazzi, F. De Ponti, R. De Giorgio, S. M. Candura, L. Anselmi, E. Cervio, A. Di Nucci and M. Tonini, Dig. Liver Dis., 2003, 35, 590–595 CrossRef CAS PubMed
.
- L. Palego, L. Betti, A. Rossi and G. Giannaccini, J. Amino Acids, 2016, 2016, 1–13 CrossRef
- D. M. Richard, M. A. Dawes, C. W. Mathias, A. Acheson, N. Hill-Kapturczak and D. M. Dougherty, Int. J. Tryptophan Res., 2009, 2, 45 CAS
- A. A. B. Badawy, Biosci. Rep., 2022, 42, 20221682 CrossRef
- Y. Chen, J. Chen, D. Guo, P. Yang, S. Chen, C. Zhao, C. Xu, Q. Zhang, C. Lin, S. Zhong and S. Zhang, Front. Oncol., 2022, 12, 800291 CrossRef CAS PubMed
- D. L. Head and C. G. McCarty, Tetrahedron Lett., 1973, 14, 1405–1408 CrossRef
- J. D. Fernstrom and R. J. Wurtman, Science, 1971, 173, 149–152 CrossRef CAS PubMed
- T. Chen, X. Zheng, X. Ma, Y. Bao, Y. Ni, C. Hu, C. Rajani, F. Huang, A. Zhao, W. Jiia and W. Jia, PLoS One, 2016, 11, e0162192 CrossRef
- A. Coppen, E. G. Eccleston and M. Peet, Lancet, 1973, 302, 60–63 CrossRef
- A. B. Badawy, J. Psychopharmacol., 2010, 24, 809–815 CrossRef CAS PubMed
- S. Ogawa, T. Fujii, N. Koga, H. Hori, T. Teraishi, K. Hattori, T. Noda, T. Higuchi, N. Motohashi and H. Kunugi, J. Clin. Psychiatry, 2014, 75, e906–e915 CrossRef CAS PubMed
- R. J. Porter, P. Gallagher and J. T. O'Brien, J. Psychopharmacol., 2007, 21, 71–75 CrossRef CAS
- K. Vielhaber, D. Riemann, B. Feige, A. Kuelz, C. Kirschbaum and U. Voderholzer, Pharmacopsychiatry, 2005, 38, 87–94 CrossRef CAS PubMed
- G. Lindseth, B. Helland and J. Caspers, Arch. Psychiatr. Nurs., 2015, 29, 102 CrossRef PubMed
- J. K. Yeh and R. R. Brown, J. Nutr., 1977, 107, 261–271 CrossRef CAS PubMed
- H. L. Davis Jr, R. R. Brown, J. Leklem and I. H. Carlson, Cancer, 1973, 31, 1061–1064 CrossRef CAS
- J. S. Ward, D. J. Lapworth, D. S. Read, S. Pedley, S. T. Banda, M. Monjerezi and A. M. MacDonald, Sci. Total Environ., 2021, 750, 141284 CrossRef CAS PubMed
- C. E. Garner, S. Smith, B. de Lacy Costello, P. White, R. Spencer, C. S. J. Probert and N. M. Ratcliffem, FASEB J., 2007, 21, 1675–1688 CrossRef CAS
- I. A. Ratiu, V. Bocos-Bintintan, F. Monedeiro, M. Milanowski, T. Ligor and B. Buszewski, Crit. Rev. Anal. Chem., 2020, 50, 501–512 CrossRef CAS
- J. W. Sam, X. J. Tang and J. Peisach, J. Am. Chem. Soc., 1994, 116, 5250–5256 CrossRef CAS
- S. E. Jackson, N. J. Pearson, W. L. Griffin and E. A. Belousova, Chem. Geol., 2004, 211, 47–69 CrossRef CAS
-
J. Dedina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, Wiley, 1995, vol. 130, p. 526
- P. W. J. M. Boumans, Fresenius’ Z. Anal. Chem., 1979, 299, 337–361 CrossRef CAS
- G. Dantelle, M. Matulionyte, D. Testemale, A. Cantarano, A. Ibanez and F. Vetrone, Phys. Chem. Chem. Phys., 2019, 21, 11132–11141 Search PubMed
- K. Saidi, M. Yangui, C. Hernández-Álvarez, M. Dammak, I. Rafael Martín Benenzuela and M. Runowski, ACS Appl. Mater. Interfaces, 2024, 16, 19137–19149 CrossRef CAS
- B. B. S. Ramin, W. G. Santos, Y. Messaddeq, E. Deffune, M. L. Moraes and S. J. L. Ribeiro, J. Lumin., 2024, 271, 120590 Search PubMed
- J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417 RSC
- R. Zhang, L. X. Wang, Y. Di Zhang, C. H. Ge, J. P. Wang, Y. Zhang and X. D. Zhang, J. Fluoresc., 2018, 28, 439–444 CrossRef CAS PubMed
- B. S. Naidu, B. Vishwanadh, V. Sudarsan and R. K. Vatsa, Dalton Trans., 2012, 41, 3194–3203 RSC
- S. Pang, Z. Zhou and Q. Wang, J. Nanopart. Res., 2013, 15, 1–9 CrossRef
- U. Hömmerich, E. E. Nyein, D. S. Lee, A. J. Steckl and J. M. Zavada, Appl. Phys. Lett., 2003, 83, 4556–4558 CrossRef
- B. Joo Han Kim, P. H. Holloway, P. H. Holloway and P. Holloway, Adv. Mater., 2005, 17, 91–96 CrossRef
- D. A. Hirsh, N. J. J. Johnson, F. C. J. M. Van Veggel and R. W. Schurko, Chem. Mater., 2015, 27, 6495–6507 CrossRef CAS
- J. M. Gonçalves, A. R. N. Bastos, S. J. L. Ribeiro, L. D. Carlos, R. L. Longo, J. M. A. Caiut and R. A. S. Ferreira, Nanoscale Adv., 2024, 6, 1486 RSC
- J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC
- J. G. Jesu Raj, M. Quintanilla, K. A. Mahmoud, A. Ng, F. Vetrone and M. Zourob, ACS Appl. Mater. Interfaces, 2015, 7, 18257–18265 CrossRef CAS
- D. Sarkar, S. Ganguli, T. Samanta and V. Mahalingam, Langmuir, 2019, 35, 6211–6230 CrossRef CAS
- R. Z. Wu, X. Yang, L. W. Zhang and P. P. Zhou, Dalton Trans., 2017, 46, 9859–9867 RSC
-
Luminescence of lanthanide ions in coordination compounds and nanomaterials, ed. A. de Bettencourt-Dias, John Wiley & Sons, 2014 Search PubMed
- Q. Li, C. Wang, H. Tan, G. Tang, J. Gao and C. H. Chen, RSC Adv., 2016, 6, 17811–17817 RSC
- R. Lv, M. Raab, Y. Wang, J. Tian, J. Lin and P. N. Prasad, Coord. Chem. Rev., 2022, 460, 214486 CrossRef CAS
- M. Chatti, S. Sarkar and V. Mahalingam, Microchim. Acta, 2016, 183, 133–140 CrossRef CAS
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS
- Z. Liu, Z. Ju, H. Liu, Z. Wang and R. Lv, J. Innovative Opt. Health Sci., 2024, 2441002 CrossRef
- H. Shao, D. Xu, Y. Ding, X. Hong and Y. Liu, Microchim. Acta, 2018, 185, 1–8 CrossRef CAS
- B. Li, A. A. Ansari, A. K. Parchur and R. Lv, Coord. Chem. Rev., 2024, 514, 215922 CrossRef CAS
- S. Sarkar, M. Chatti and V. Mahalingam, Chem. – Eur. J., 2014, 20, 3311–3316 CrossRef CAS
- S. Sarkar, M. Chatti, V. N. K. B. Adusumalli and V. Mahalingam, ACS Appl. Mater. Interfaces, 2015, 7, 25702–25708 CrossRef CAS PubMed
- C. Lv, W. Di, Z. Liu, K. Zheng and W. Qin, Analyst, 2014, 139, 4547–4555 RSC
- B. Meesaragandla, A. Verma, V. Bheemireddy and V. Mahalingam, ChemistrySelect, 2016, 1, 4927–4934 CrossRef
- C. Hazra, T. Samanta and V. Mahalingam, J. Mater. Chem. C, 2014, 2, 10157–10163 RSC
- R. Krishnan, S. N. Shibu, D. Poelman, A. K. Badyal, A. K. Kunti, H. C. Swart and S. G. Menon, Mater. Today Commun., 2022, 32, 103890 CrossRef CAS
- Y. Deguchi, M. Kono, Y. Koizumi, Y. I. Izato and A. Miyake, Org. Process Res. Dev., 2020, 24, 1614–1620 CrossRef CAS
- T. Fujii, H. Ohfuji and T. Inoue, Phys. Chem. Miner., 2016, 43, 353–361 CrossRef CAS
- H. J. K. Kim, K. E. Kaplan, P. Schindler, S. Xu, M. M. Winterkorn, D. B. Heinz, T. S. English, J. Provine, F. B. Prinz and T. W. Kenny, ACS Appl. Mater. Interfaces, 2019, 11, 9594–9599 CrossRef CAS
- F. Schiffmann, J. Vandevondele, J. Hutter, R. Wirz, A. Urakawa and A. Baiker, J. Phys. Chem. C, 2010, 114, 8398–8404 CrossRef CAS
- A. P. Bassett, S. W. Magennis, P. B. Glover, D. J. Lewis, N. Spencer, S. Parsons, R. M. Williams, L. De Cola and Z. Pikramenou, J. Am. Chem. Soc., 2004, 126, 9413–9424 CrossRef CAS PubMed
- C. Hazra, T. Samanta, A. V. Asaithambi and V. Mahalingam, Dalton Trans., 2014, 43, 6623–6630 RSC
- R. F. Chen, Anal. Lett., 1967, 1, 35–42 CrossRef CAS
- C. P. Pan, P. L. Muino, M. D. Barkley and P. R. Callis, J. Phys. Chem. B, 2011, 115, 3245–3253 CrossRef CAS PubMed
- D. M. Uriza-Prias, A. Méndez-Blas and J. F. Rivas-Silva, Open J. Phys. Chem., 2021, 11, 87–105 CrossRef CAS
- J. T. Vivian and P. R. Callis, Biophys. J., 2001, 80, 2093–2109 CrossRef CAS
- R. Krishnan, S. N. Shibu, D. Poelman, A. K. Badyal, A. K. Kunti, H. C. Swart and S. G. Menon, Mater. Today Commun., 2022, 32, 103890 CrossRef CAS
- R. W. Yip, W. D. Riddell and A. G. Szabo, Can. J. Chem., 1970, 48, 987–999 CrossRef CAS
- C. Q. Jiao, M. Sun, F. Liu, Y. N. Zhou, Y. Y. Zhu, Z. G. Sun and J. Li, ACS Omega, 2018, 3, 16735–16742 CrossRef CAS PubMed
- Y. Zhou and J. Yoon, Chem. Soc., 2012, 41, 52–67 CAS
- C. He, J. Wang, P. Wu, L. Jia, Y. Bai, Z. Zhang and C. Duan, Chem. Commun., 2012, 48, 11880–11882 RSC
- Z. Zhao, D. Yang, B. Xing, C. Ma, Z. G. Sun, Y. Y. Zhu, H. Y. Li and J. Li, RSC Adv., 2016, 6, 92175–92185 RSC
- H. Tsukube, M. Wada, S. Shinoda and H. Tamiaki, Chem. Commun., 1999, 11, 1007–1008 RSC
- C. Q. Jiao, M. Sun, F. Liu, Y. N. Zhou, Y. Y. Zhu, Z. G. Sun and J. Li, ACS Omega, 2018, 3, 16735–16742 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02774d |
| This journal is © The Royal Society of Chemistry 2024 |