
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interlaboratory comparison of endotoxin contamination assessment of nanomaterials†
Gary
Hannon
 *ab,
Bethany J.
Heaton
cd,
Alexander
Plant-Hately
cd,
Christopher
David
cd,
Neill J.
Liptrott
cd,
Ainhoa
Egizabal
e,
Ana
Ayerdi-Izquierdo
e,
Noelia
Alvarez
e,
Oihane
Ibarrola
f,
Andres Arbona
Celaya
f,
Angel
Del Pozo Perez
f,
Nerea
Lazcanoiturburu
f,
Iris
Luzuriaga
f,
Fikirte Debebe
Zegeye
g,
Shanbeh
Zienolddiny-Narui
g,
An
Jacobs
h,
Alexandra
Van Driessche
h,
Inge
Nelissen
h,
Ibane
Abasolo
ijk,
Fernanda
Andrade
ij,
Nora
Ventosa
jl,
Elisabet
González-Mira
jl,
Aida
Carreño
jl and
Adriele
Prina-Mello
*abm
*ab,
Bethany J.
Heaton
cd,
Alexander
Plant-Hately
cd,
Christopher
David
cd,
Neill J.
Liptrott
cd,
Ainhoa
Egizabal
e,
Ana
Ayerdi-Izquierdo
e,
Noelia
Alvarez
e,
Oihane
Ibarrola
f,
Andres Arbona
Celaya
f,
Angel
Del Pozo Perez
f,
Nerea
Lazcanoiturburu
f,
Iris
Luzuriaga
f,
Fikirte Debebe
Zegeye
g,
Shanbeh
Zienolddiny-Narui
g,
An
Jacobs
h,
Alexandra
Van Driessche
h,
Inge
Nelissen
h,
Ibane
Abasolo
ijk,
Fernanda
Andrade
ij,
Nora
Ventosa
jl,
Elisabet
González-Mira
jl,
Aida
Carreño
jl and
Adriele
Prina-Mello
*abm
aNanomedicine and Molecular Imaging Group, Department of Clinical Medicine, Trinity Translational Medicine Institute, Trinity College Dublin, Dublin, Ireland. E-mail: prinamea@tcd.ie
bLaboratory of Biological Characterization of Advanced Materials (LBCAM), Trinity Translational Medicine Institute, Trinity College Dublin, Dublin, Ireland
cImmunocompatibility Group, Department of Pharmacology and Therapeutics, Institute of Systems, Molecular, and Integrative Biology, University of Liverpool, Liverpool, UK
dCentre of Excellence for Long-Acting Therapeutics, Department of Pharmacology and Therapeutics, Institute of Systems, Molecular, and Integrative Biology, University of Liverpool, Liverpool, UK
eTECNALIA, Basque Research and Technology Alliance (BRTA), Mikeletegi Pasealekua 2, 20009 Donostia-San Sebastián, Spain
fBiokeralty Research Institute AIE, Parque Tecnológico de Álava, Vitoria- Gasteiz, Álava, Spain
gNational Institute of Occupational Health, Oslo, Norway
hHealth department, Flemish Institute for Technological Research (VITO), Mol, Belgium
iDrug Delivery & Targeting, Vall d'Hebron Institute de Recerca (VHIR), Universitat Autònoma de Barcelona (UAB), 08035, Barcelona, Spain
jNetworking Research Centre for Bioengineering, Biomaterials, and nanomedicine (CIBER-BBN), Instituto de Salud Carlos III, 28029, Madrid, Spain
kFunctional Validation & Preclinical Research (FVPR)/U20 ICTS Nanobiosis, Vall d'Hebron Institut de Recerca (VHIR), Universitat Autònoma de Barcelona (UAB), 08035, Barcelona, Spain
lInstitut de Ciència de Materials de Barcelona, ICMAB-CSIC, Campus UAB, 08193 Bellaterra, Spain
mAdvanced Materials and Bioengineering Research (AMBER) Centre, CRANN Institute, Trinity College Dublin, Dublin, Ireland
First published on 24th October 2024
Abstract
Endotoxin contamination is a significant hurdle to the translation of nanomaterials for biomedical applications. Multiple reports now describe that more than one-third of nanomaterials fail early pre-clinical assessment due to levels of endotoxin above regulatory requirements. Additionally, most immunological studies or in vivo studies testing nanomaterials in the literature lack inclusion of this assessment, which may lead to false-positive or false-negative results if high levels of the contaminant are present. The currently approved methods for endotoxin contamination assessment rely on enzymatic activity and wavelength absorbance as their endpoint, and many nanomaterials can interfere with such assays. For this reason, we devised an interlaboratory comparison of endotoxin contamination assessment for a range of nanomaterials to challenge the current international organization for standardization and pharmacopeia standards. Herein, we show that detected endotoxin levels could vary considerably between groups, and, in some instances, nanomaterials could both pass and fail regulatory endotoxin limits for medical devices depending on the group undertaking the assessment, all while passing all quality criteria standards. This work emphasises the requirement for multiple assays to fully assess the endotoxin levels in a nanomaterial and highlights the need for additional assays to be developed in this space.
Introduction
Nanomaterials (NM) possess unique physicochemical properties that make them attractive tools for many biomedical applications.1 These novel characteristics also come with caveats, however, as their translation comes with many added challenges compared to conventional drugs.2 One such safety hurdle for NM is endotoxin contamination. The large surface-to-volume ratios and multi-component design associated with NM can accommodate high levels of endotoxin. Additionally, endotoxin's structure facilitates binding to both hydrophobic and cationic structures and its high thermal stability makes it incredibly difficult to remove once bound.3 Intravenous injections of 2–4 ng kg−1 of endotoxin have consistently induced systemic inflammation in humans,4,5 while lower doses (0.3 ng kg−1) have been associated with low-grade inflammation.6 Therefore, endotoxin represents a significant early safety consideration for NM.In 2013, the National Cancer Institute's Nanotechnology Characterization Laboratory (NCI-NCL) reported that more than one-third of the nanomedicines they tested during a one-year period failed endotoxin contamination assessment.7 More recently, another group published similar findings.3 In addition to its high prevalence on NM, there is also evidence to suggest that the vast majority of publications detailing immunological assessments and in vivo studies with NM lack inclusion of endotoxin contamination assessment, potentially resulting in misleading findings.8 Hence, endotoxin is a commonly overlooked, early hurdle to the translation of NM for biomedical applications.9–11
Testing for endotoxin contamination on NM is not a trivial endeavour, with the unique physicochemical properties of NM commonly leading to issues with assay interference.12,13 The surface chemistry of NM can interfere with the constituents of the assay, while their optical properties commonly overlap with absorbance endpoints in these tests.3,14 Accordingly, current international and Pharmacopeia standards for endotoxin testing require the inclusion of an inhibition-enhancement control (IEC) on top of common acceptance criteria to account for this potential interference.15,16 The IEC is a spike-recovery control consisting of a known concentration of endotoxin spiked into a NM test sample that is subsequently detected to determine its propensity to introduce an inhibition or enhancement into the assay. An inhibition is considered a spike-recovery <50% of the concentration introduced, whereas an enhancement is defined as >200%.13 However, despite this additional interference control, different results between assays have been reported.12,17
Here we present the results from an interlaboratory comparison (ILC) by the SAFE-N-MEDTECH (safety testing in the life cycle of nanotechnology-enabled medical technologies for health) consortium for the endotoxin contamination assessment of three NM and a medical implant coated with a nanosurface. These NM were analysed for their physicochemical characteristics (PCC) and tested for endotoxin contamination by eight different groups -including a Good Laboratory Practice (GLP) certified laboratory- using either the chromogenic, turbidimetric or gel clot Limulus amoebocyte lysate (LAL) assays or the recombinant factor C assay. Results showed that while no changes in PCC occurred during shipment, detected endotoxin levels could differ considerably between groups. Importantly, some of these differences could result in a NM both passing and failing regulatory requirements for a medical device, even when the same LAL assay is used. This study highlights potential issues with the currently accepted standards for endotoxin contamination of NM.
Materials and methods
Materials
The four chosen NM used for this study were (I) iron oxide nanoparticles (IONP) purchased from Chemicell GmbH (Germany) and consisted of a maghemite core with dextran coating and dispersed in water; (II) gold nanoparticles (GNP) purchased from NanoComposix, USA and were coated in polyethylene glycol and dispersed in water; (III) polystyrene nanoparticles (PsNP) purchased from Sigma Aldrich, Ireland and were also dispersed in water; and (IV) titanium-coated femoral hip stem medical device implant with a hydroxyapatite nanosurface purchased from Stryker Orthopaedics, Ireland. These four materials were utilised for the ILC as they all possessed a NM-component in their design and they varied in their structural, optical and chemical properties which enabled a robust comparison of the different endotoxin detection methods. The suppliers for all endotoxin detection kits are outlined in ESI Table 1.† Additionally, LAL reagent water (<0.001 EU ml−1 and <1.56 pg ml−1 (1,3)-β-D-glucan) used for aliquoting the solutions was supplied by Associates of Cape Cod Incorporated, United Kingdom.Methods
The IONP were supplied at 25 mg ml−1 stock concentration and then diluted to 10 mg ml−1 in LAL reagent water to ensure sufficient sample volume for the consortium. The GNP were supplied as a stock solution of 0.053 mg ml−1 concentration and were not diluted any further as sufficient volume was available as stock. The PsNP were also diluted from 100 mg ml−1 to 50 mg ml−1 concentration using LAL reagent water to provide enough sample volume for the consortium. The implant was completely submerged in 100 ml LAL reagent water for 24 h. After this time, the ‘implant fluid’ (or IF) was extracted and used to indirectly test the levels of endotoxin on the structure. Each sample was aliquoted into 5 ml pyrogen-free Eppendorf ™ tubes (Fisher Scientific, Ireland) for subsequent testing and sealed with Parafilm® M (Sigma Aldrich, Ireland) to avoid environmental exposure around the screw cap. The samples were then ready to be shipped to the consortium.
For shipment, samples were placed in sealed polystyrene boxes containing cold packs at 2–6 °C and arrived at their respective destinations within 24 hours. Upon arrival, samples were immediately stored at 2–6 °C until testing by the various partners.
 | ||
| Fig. 1 Schematic workflow for the interlaboratory study. Stocks of IONP, GNP, PsNP and implant fluid are aliquoted into tubes and the samples tested by the study coordinator for size distribution by nanoparticle tracking analysis (NTA) and dynamic light scattering (DLS). The 4 samples were then shipped to 7 additional groups within the consortium who repeated the size distribution analysis of the samples with either NTA or DLS and also undertook endotoxin contamination assessment with a least one assay along with the coordinator. The size distribution and endotoxin contamination levels were statistically compared across all groups. [Note: size distribution analysis was not undertaken by the GLP laboratory prior to endotoxin testing]. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in Milli-Q® Type 1 ultrapure water (Wasserlab, QA03DP30GR model) and measured for surface zeta potential using a minimum of 3 measurements of 3 runs. Cuvettes used were Malvern's disposable folded capillary tubes (DTS1070). Equilibrium times lasted 120 seconds and 20-second delays were employed between measurements. Attenuation and voltage selection were automatic and analysis was also undertaken in automatic mode. Each measurement met Malvern's quality criteria for average count rate, phase plot and frequency plot.
:100 in Milli-Q® Type 1 ultrapure water (Wasserlab, QA03DP30GR model) and measured for surface zeta potential using a minimum of 3 measurements of 3 runs. Cuvettes used were Malvern's disposable folded capillary tubes (DTS1070). Equilibrium times lasted 120 seconds and 20-second delays were employed between measurements. Attenuation and voltage selection were automatic and analysis was also undertaken in automatic mode. Each measurement met Malvern's quality criteria for average count rate, phase plot and frequency plot.
Following this confirmation, a spike-recovery control was performed. Here, the samples were tested at a concentration that does not clot the gel in replicates of four. At this concentration, the samples were spiked with endotoxin at a concentration of 2X, X, 0.5X and 0.25X the sensitivity of the lysate, along with samples alone. In parallel to the spike-recovery control, a sensitivity test was also run again in duplicate as a quality control. For this test to be deemed successful, the samples alone must not clot the gel and the lysate sensitivity must be confirmed again in the parallel test. Additionally, if the lysate retained its sensitivity when endotoxin was added, they were deemed to not interfere with the assay; however, if the sensitivity is outside the 50–200% range then the samples did interfere. If the samples pass the spike-recovery control, they were tested for the quantity of endotoxin they possess in a final test.
For the limit test, the samples were diluted continuously from their stock in LAL reagent water (not exceeding the MVD) and tested in duplicate until no clotting of the gel was observed. At this concentration, an additional test was run with samples alone, samples spiked with 2X the sensitivity of the lysate, an additional 2X control in water and water alone in duplicate. If the spiked sample and water-spiked control were positive, and samples alone and water alone were negative, the limit test was deemed successful and the endotoxin levels were reported.
Statistical analysis
GraphPad version 7 was used to determine statistical distribution (min, mean, max, range and coefficient of variance) of size and contamination results between the consortium.Results
Physicochemical characterization of nanomaterials
The PCC of the NM used for this interlaboratory study are summarized in Table 1 below and data graphs are provided in ESI Fig. 1–3.† Notably, the IONP had a mean hydrodynamic size range of 79–89 nm, GNP ranged from 128–151 nm and PsNP ranged from 98–116 nm, depending on whether NTA or DLS was used for the analysis. Additionally, each material was monodispersed, with respective polydispersity indexes of 0.111 for IONP, 0.008 for GNP and 0.028 for PsNP, based on DLS measurements. The GNP had a lower stock concentration (0.053 mg ml−1) and so was expected to have a lower particle count (2 × 109 NP ml−1) compared to the IONP (6.46 × 1013 NP ml−1) and PsNP (8.78 × 1013 NP ml−1). All NM were negatively charged at pH 7.5 for IONP and pH 7 for GNP and PsNP. As the IF consisted solely of LAL reagent water that had been exposed to the implant, it was excluded from PCC analysis.| Sample | NTA mean size (nm) | NTA (NP ml−1) | DLS mean size (nm) | DLS PDI | Zeta potential (mV) |
|---|---|---|---|---|---|
| NTA and DLS were used to measure the size distribution of the NM. Dynamic light scattering was also used to measure zeta potential at pH 7.5 for IONP and pH 7 for GNP and PsNP. Abbreviations: NP, nanoparticle; PDI, polydispersity index. Values indicate mean ± standard deviation. | |||||
| IONP | 79 ± 0.9 | 6.46 × 1013 ± 1.43 × 1012 | 89 | 0.111 | −13.0 ± 1.2 |
| GNP | 128 ± 1.6 | 2.08 × 109 ± 1.61 × 108 | 151 | 0.008 | −21.3 ± 2.4 |
| PsNP | 98 ± 0.4 | 8.78 × 1013 ± 6.10 × 1011 | 116 | 0.028 | −48.4 ± 1.8 |
Beta-glucan, microbial and mycoplasma contamination of samples
All samples were assessed for contamination with beta-glucans, microorganisms and mycoplasma. Detectable levels of beta-glucans were noted in the IONP, GNP and PsNP, whereas the levels in the IF were below the lowest value in the standard curve (5 pg ml−1). None of the samples had detectable CFU, indicating sterility (confirming the data provided by the suppliers in each case). Importantly, in both of these cases, the spike-recovery controls for beta-glucan and microbial contamination were within 50–200%, indicating no interference in the tests. Mycoplasma was also considered negative for each sample tested as they produced a reading ratio <0.9, while the positive and negative controls in this test showed positive (34.57) and negative (0.16) ratios, respectively. Importantly, the viability of the cells used for this assay was >98% after incubation with the samples; hence, the samples did not interfere in this regard (Table 2).| Sample | Beta-glucan contamination (pg ml−1) | Spike recovery (%) | Microbial contamination (CFU ml−1) | Spike recovery (%) | Mycoplasma reading ratio |
|---|---|---|---|---|---|
| Both beta-glucan and microbial contamination levels reflect stock concentrations for each sample. Spike-recovery controls indicate the percentage of the contaminant retrieved following spiking into the sample. Mycoplasma reading ratio refers to the ratio of readings before and after the detection substrate was added to each sample. Abbreviations: CFU, colony forming units. | |||||
| IONP | 1629.1 | 103 | 0 | 102 | 0.4011 ± 0.0423 |
| GNP | 7.5 | 96 | 0 | 67 | 0.5134 ± 0.2377 |
| PsNP | 4239.3 | 78 | 0 | 108 | 0.5885 ± 0.1822 |
| IF | <5.0 | 91 | 0 | 71 | 0.5371 ± 0.0655 |
Size comparison by the consortium
Hydrodynamic size results from NTA and DLS ensured the NM were not compromised during shipment (Fig. 2). For NTA, IONP had a mean size of 85.9 nm and a standard deviation of 6.7 nm (7.8% CV); GNP had a mean size 127.1 nm and a standard deviation of 1.4 nm (1.1% CV); PsNP had a mean size of 104.3 nm and standard deviation of 5.2 nm (5.0% CV). For DLS, IONP had a mean size of 91.7 nm and a standard deviation of 1.6 nm (3.8% CV); GNP had a mean size of 153.1 nm and a standard deviation of 2.0 nm (1.3% CV); PsNP had a mean size of 117.5 nm and a standard deviation of 3.0 nm (2.5% CV). | ||
| Fig. 2 Size comparison between groups. Bars depict mean hydrodynamic size (nm) ± standard deviation and shapes represent individual results for both NTA and DLS measurements. For NTA, the results are measurements from 3 different groups; for DLS, the results are measurements from 5 different groups. | ||
Endotoxin contamination
The statistical variability of endotoxin results is summarized in Fig. 3 and Table 3. As beta-glucan could be detected for the 3 NM tested, assays for detecting both endotoxin and beta-glucan and endotoxin-specifically were used. All results obtained and reported herein abided by ISO and Pharmacopeia standards for endotoxin assessment of NM (summarized in ESI Table 2†).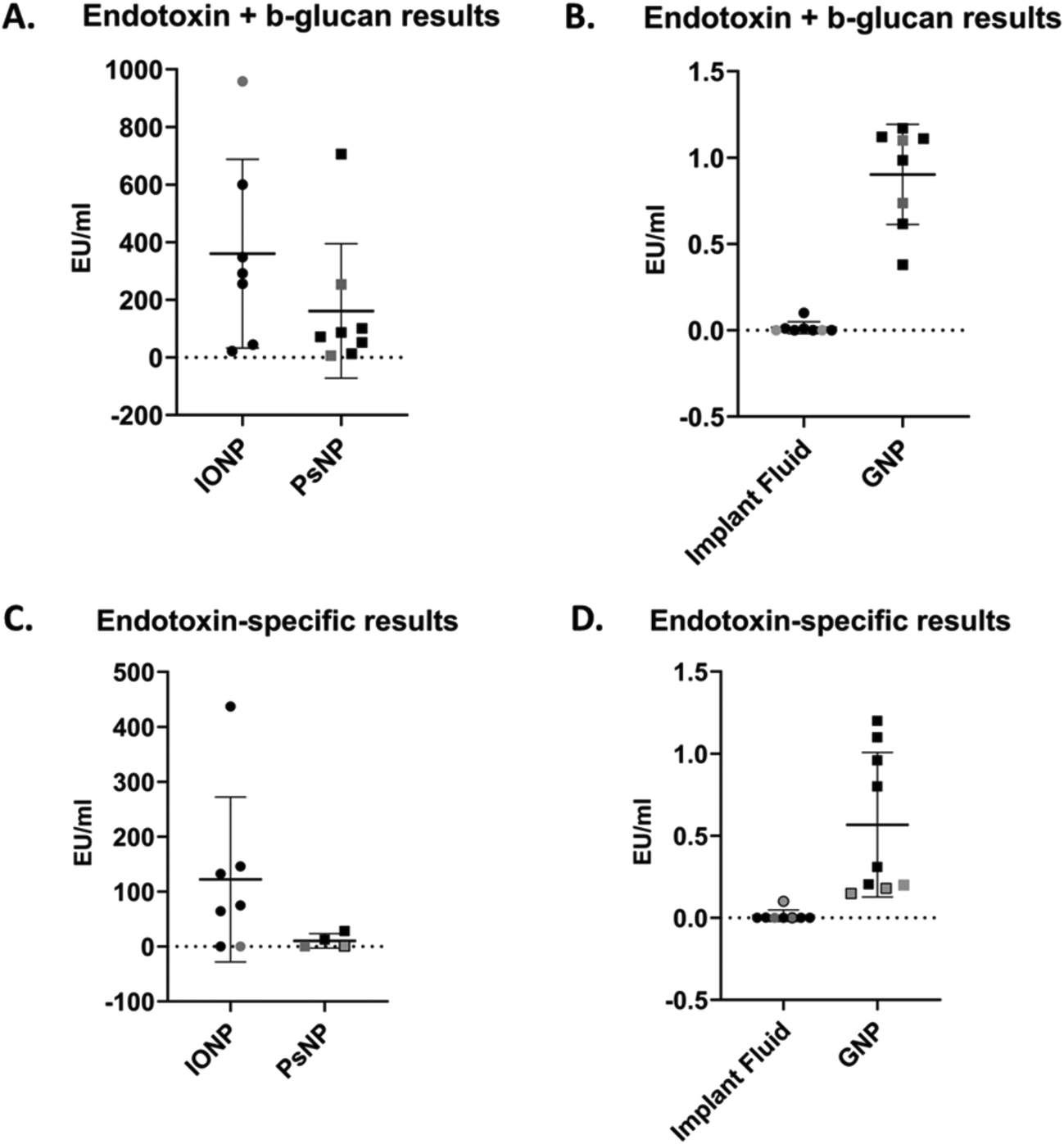 | ||
| Fig. 3 Summary of results from endotoxin and beta-glucan contamination assessment of samples. (A) Endotoxin and beta-glucan results for IONP (n = 7) and PsNP (n = 8). (B) Endotoxin and beta-glucan results for IF (n = 8) and GNP (n = 8). (C) Endotoxin- specific results for IONP (n = 7) and PsNP (n = 4). (D) Endotoxin- specific results for IF (n = 8) and GNP (n = 9). Lines reflect mean values ± standard deviation and shapes indicate individual results. Black shapes indicate chromogenic assay results, grey shapes indicate turbidimetric assay results and grey shapes with black borders indicate recombinant factor C assay results. Note: for PsNP endotoxin-specific measurements, three values were semi-quantitative and were excluded from this data. These values are specified in ESI Table 2.† | ||
| Parameter | IONP | PsNP | Implant fluid | GNP |
|---|---|---|---|---|
| Abbreviations: SD, standard deviation; CV, coefficient of variation. | ||||
| Endotoxin and beta-glucan results (EU ml −1 ) | ||||
| Mean | 360.10 | 161.30 | 0.02 | 0.90 |
| SD | 327.80 | 233.10 | 0.04 | 0.29 |
| Min | 22.18 | 6.07 | 0.00 | 0.38 |
| Max | 958.00 | 706.10 | 0.10 | 1.17 |
| Range | 935.80 | 700.00 | 0.10 | 0.79 |
| CV | 91% | 145% | 231% | 32% |
| Endotoxin-specific results (EU ml −1 ) | ||||
| Mean | 122.10 | 10.36 | 0.00 | 0.57 |
| SD | 150.10 | 13.17 | 0.00 | 0.44 |
| Min | 0.005 | 0.059 | 0.00 | 0.15 |
| Max | 437.00 | 28.16 | 0.1 | 1.20 |
| Range | 432.00 | 28.16 | 0.1 | 1.05 |
| CV | 123% | 127% | 277% | 78% |
The results of endotoxin and beta-glucan quantification reflected seven groups’ data for the chromogenic (n = 6) and turbidimetric (n = 2) assays are presented in Fig. 3(A and B) and Table 3. The gel clot assay was also performed by two groups, however, the MVD was reached for the IONP, GNP and PsNP with these assays and values could not be reported. Additionally, the IF levels were below the sensitivity of the assays (0.03 EU ml−1). For the more sensitive LAL assays (chromogenic and turbidimetric), when the endotoxin levels of IF were below the limit of detection they were reported 0.00 EU ml−1 in the results. For the NM, specific concentrations could be determined for both the turbidimetric and chromogenic assays in all but one case, where IONP reached the MVD for one turbidimetric assay and could not be reported. IONP had a mean value of 360.1 EU ml−1, but the min and max values of 22.18 EU ml−1 and 958.00 EU ml−1 respectively noted a large range in the results. This range corresponded to standard deviation of 327.8 EU ml−1. Similar results could also be seen with PsNP, as the mean concentration measured 161.3 EU ml−1 and the min and max values were 6.07 EU ml−1 and 706.10 EU ml−1 respectively. These values equated to a standard deviation of 233.10 EU ml−1. GNP and IF contained considerably less endotoxin (0.90 EU ml−1 and 0.02 EU ml−1 respectively) and therefore a smaller range in results (0.38–1.17 EU ml−1 for GNP and 0–0.1 EU ml−1 for IF). Accordingly, these materials also exhibited considerably less standard deviation in their data (0.29 EU ml−1 for GNP and 0.04 EU ml−1 for IF).
The endotoxin-specific results for eight groups’ data using the chromogenic (n = 6), turbidimetric (n = 1) and recombinant factor C (n = 2) assays are reported in Fig. 3(C and D) and Table 3. Once again, the gel clot assay was also performed by one group but the MVD was reached for the IONP, GNP and PsNP, and the sensitivity was reached for the IF (0.06 EU ml−1). The IF endotoxin levels were below the level of detection for some groups with the chromogenic, turbidimetric or recombinant factor C assays, and were therefore also reported as 0.00 EU ml−1 in the results. Similar findings were observed with PsNP at its MVD for three chromogenic assays, and only ranges for endotoxin could be obtained in these cases. These ranges are provided in ESI Table 2.† Additionally, the recombinant factor C assay was unable to overcome the interference of the IONP in both cases. For both the chromogenic and turbidimetric assays, however, the specific endotoxin concentrations of IONP and GNP could be reported in all cases. IONP had a mean value of 122.10 EU ml−1 with a min and max value of 0.005 EU ml−1 and 437.00 EU ml−1 respectively; PsNP had a mean value of 10.36 EU ml−1 with a min and max value of 0.059 EU ml−1 and 28.16 EU ml−1 respectively. This resulted in standard deviations of 150.10 EU ml−1 for IONP and 13.17 EU ml−1 for PsNP. As before, GNP and IF contained considerably less endotoxin than the other NM, on average, which resulted in a smaller range of results. GNP had a mean endotoxin centration of 0.57 EU ml−1, with a range of 0.15–1.2 EU ml−1 and a standard deviation of 0.44 EU ml−1. The endotoxin levels of IF were undetectable except for two groups, where the concentrations were quantified as 0.002 and 0.1 EU ml−1.
Discussion
Three NM were aliquoted into pyrogen-free tubes and characterized for their PCC by the ILC coordinator prior to batch-shipment to each partner. The fourth material -a medical implant coated with a nanosurface- was exposed to endotoxin-free water and this liquid was also aliquoted for subsequent testing. NTA and DLS were utilised (prior and after shipment) to measure the hydrodynamic size distribution of the NM and the low CV values (<10% in all cases) ensured each sample was not altered during shipment. The zeta potential of each NM -determined by DLS- was negative at pH 7–7.5 and so interference with endotoxin detection assays via electrostatic interaction through the negatively-charged phosphate groups on endotoxin could be avoided, as positively charged NM can evoke false negatives in these assays through direct binding to endotoxin.3Next, each sample was tested for microbial and mycoplasma contamination along with presence of beta-glucans. No microbial or mycoplasma contamination was detected for each sample, and confirming this sterility was essential to ensure endotoxin or beta-glucan-harbouring microorganisms were not present in each of the formulations. In IONP, GNP and PsNP, however, beta-glucans could be detected. Notably, IONP and PsNP possessed high levels of beta-glucan within the nanogram range. Beta-glucans are not as potent as endotoxin, but do possess immune-stimulatory properties and are common contaminants in NM.23,24 Moreover, LAL assays are responsive to both endotoxin and beta-glucans through factor G activity (downstream of endotoxin-specific factor C signalling), and so this contaminant could lead to an overestimation of the endotoxin present in each sample.23 Because of this, the ILC was expanded to include LAL assays specific to endotoxin along with assays to detect a combination of endotoxin and beta-glucans to compare results between each.
The results for both endotoxin-specific and endotoxin and beta-glucan results varied considerably between groups, particularly for IONP and PsNP. This can be noted with the reported range values, which increased to several hundred EU when the levels of contamination were higher. Importantly, this variation existed even within the same assays, and so this is not a result of inter-assay differences but exists regardless of the assay used. While this large variability range is concerning, it is expected that higher concentrations of endotoxin will lead to greater variations in results as larger dilutions of the NM are required. Therefore, in these cases, if the goal of these assays is to specifically ascertain whether the NM is within endotoxin limits, this large range at higher concentrations is not a major concern.
For the samples with lower endotoxin and beta-glucan contamination -GNP and IF- the range was expectedly lower, however, at this level, small differences were the difference between a pass or fail with regards to regulatory endotoxin limits for medical devices (0.5 EU ml−1). With the endotoxin-specific results, in particular, the GNP and PsNP could pass or fail between assays all while abiding by ISO and Pharmacopeia standards for endotoxin testing of NM. Once again, these differences were not a result of inter-assay variability in the case of GNP, as the same assays were also providing large ranges in results. This is a concerning finding as NM could potentially harbour greater levels of endotoxin than what has been reported, and this may lead to unexpected immune stimulation at a later point in their preclinical or even clinical evaluation. Notably, mice are 10000–1000000 times less sensitive to pure lipopolysaccharide treatment than humans;25,26 therefore, the immune-stimulatory effects of a NM containing undetected, excessive levels of endotoxin may go unnoticed until late preclinical or even clinical evaluation.
This variation in results is likely due to the propensity of NM to interfere with LAL assays. While none of these NM were cationic, they all have the potential to influence absorbance at the wavelengths used for the chromogenic, turbidimetric and recombinant factor C assays.3,12 Moreover, the gel clot assay -that does not use optical absorbance as an endpoint- was an unsuitable alternative for the NM tested here as the MVD was reached in most cases before the endotoxin levels could be quantified. Hence, while current controls are in place in ISO and Pharmacopeia standards to account for the potential of NM to interfere with LAL assays, limitations remain in these tests that may result in misleading findings. The IEC appears not enough to sufficiently determine the extent of NM interference with the LAL assays, and the combination of multiple assay types is advised. This recommendation has also been voiced by the NCI-NCL previously,13 and its importance is emphasised here as the use of two assays was more likely to pick up a positive result.
The monocyte activation test is the only other regulatory approved, non-animal alternative to the LAL assays.27 This test also comes with limitations, however, as it also relies on optical absorbance as an endpoint (although NM are mostly washed away prior to detection), the test is not endotoxin-specific and NM with inherent cytotoxicity or immune modulating properties can also interfere with it.3 It is therefore essential for more work to be carried out in the future to advance the current options for endotoxin assessment of NM.
Conclusion
This study summarized results from an ILC on the endotoxin contamination assessment of four samples containing NM: IONP, GNP, PsNP and a nanosurface- based implant. It was hypothesised that the interference widely reported in the literature with NM and endotoxin assessment would result in variable data between groups, even though ISO and Pharmacopeia quality control standards were met in all cases. While it was ensured that these samples were sterile, and PCC analysis was performed to ensure that none of the samples were altered during batch-preparation and shipment to the various partners, large variations in results were observed, even within the same assays. Notably, for 2 of the 4 samples, individual results showed that each could both pass and fail endotoxin assessment depending on the group performing the assessment. Therefore, this study highlights the limitations of LAL assays to the endotoxin contamination assessment of NM and the need for more work in this space to improve current testing options in the future.Authors contribution
GH and APM conceptualised and coordinated the study. GH generated data for the study, wrote the initial draft of the manuscript, reviewed subsequent drafts and finalised it for submission. APM reviewed the initial draft, reviewed subsequent drafts, finalised it for submission. BJH, APH, CD, NJL, AE, AAI, NA, OI, AAC, ADPP, NL, IL, FDZ, SZN, AJ, AVD, IN, IA, FA, NV, EGM and AC all contributed equally to the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work was funded by the European Commission's Horizon 2020 research and innovation programme (grant agreement: 814607). The authors would also like to acknowledge BioRender software used for the creation of Fig. 1 and the Graphical Abstract.References
- A. C. Anselmo and S. Mitragotri, Nanoparticles in the clinic: An update post COVID-19 vaccines, Bioeng. Transl. Med., 2021, 6(3), e10246 CrossRef CAS PubMed.
- T. J. MacCormack, et al., Commentary: Revisiting nanoparticle-assay interference: There's plenty of room at the bottom for misinterpretation, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2021, 255, 110601 CrossRef CAS PubMed.
- G. Hannon and A. Prina-Mello, Endotoxin contamination of engineered nanomaterials: Overcoming the hurdles associated with endotoxin testing, WIREs Nanomed. Nanobiotechnol., 2021, 13(6), e1738 CrossRef CAS PubMed.
- S. E. Calvano and S. M. Coyle, Experimental Human Endotoxemia: A Model of the Systemic Inflammatory Response Syndrome?, Surg. Infect., 2012, 13(5), 293–299 CrossRef.
- J. N. Fullerton, et al., Intravenous Endotoxin Challenge in Healthy Humans: An Experimental Platform to Investigate and Modulate Systemic Inflammation, J. Visualized Exp., 2016,(111), e53913 Search PubMed.
- S. Taudorf, et al., Human Models of Low-Grade Inflammation: Bolus versus Continuous Infusion of Endotoxin, Clin. Vaccine Immunol., 2007, 14(3), 250–255 Search PubMed.
- R. M. Crist, et al., Common pitfalls in nanotechnology: lessons learned from NCI's Nanotechnology Characterization Laboratory, Integr. Biol., 2013, 5(1), 66–73 CrossRef CAS PubMed.
- G. Hannon, et al., Immunotoxicity Considerations for Next Generation Cancer Nanomedicines, Adv. Sci., 2019, 6(19), 1900133 CrossRef CAS PubMed.
- Y. Li and D. Boraschi, Endotoxin contamination: a key element in the interpretation of nanosafety studies, Nanomedicine, 2016, 11(3), 269–287 CrossRef CAS PubMed.
- Y. Li, M. Fujita and D. Boraschi, Endotoxin Contamination in Nanomaterials Leads to the Misinterpretation of Immunosafety Results, Front. Immunol., 2017, 8, 472 CrossRef.
- Y. Li, et al., Bacterial endotoxin (lipopolysaccharide) binds to the surface of gold nanoparticles, interferes with biocorona formation and induces human monocyte inflammatory activation, Nanotoxicology, 2017, 11(9–10), 1157–1175 CrossRef CAS.
- Y. Li, et al., Optimising the use of commercial LAL assays for the analysis of endotoxin contamination in metal colloids and metal oxide nanoparticles, Nanotoxicology, 2015, 9(4), 462–473 Search PubMed.
- M. A. Dobrovolskaia, Pre-clinical immunotoxicity studies of nanotechnology-formulated drugs: Challenges, considerations and strategy, J. Controlled Release, 2015, 220(Pt B), 571–583 CrossRef CAS.
- M. Mangini, et al., Interaction of nanoparticles with endotoxin Importance in nanosafety testing and exploitation for endotoxin binding, Nanotoxicology, 2021, 15(4), 558–576 CrossRef CAS.
- ISO, Nanotechnologies:Endotoxin test on nanomaterial samples for in vitro systems: Limulus amebocyte lysate (LAL) test. 2010.
- USP, <85> Bacterial Endotoxin Test. 2012.
- M. A. Dobrovolskaia, D. R. Germolec and J. L. Weaver, Evaluation of nanoparticle immunotoxicity, Nat. Nanotechnol., 2009, 4(7), 411–414 CrossRef CAS PubMed.
- EUNCL. Measuring Batch Mode DLS., 2016[cited 2023 January]; Available from: https://www.euncl.org/about-us/assay-cascade/PDFs/Prescreening/EUNCL-PCC-001.pdf?m=1468937875&.
- EUNCL. Particle Tracking Analysis. 2018[cited 2023 January]; Available from: https://www.euncl.org/about-us/assay-cascade/PDFs/PCC/EUNCL_PCC_023.pdf?m=1526712237&.
- EUNCL. Detection of Bacterial Contamination by Agar Plate Test. 2016[cited 2023 January]; Available from: https://www.euncl.org/about-us/assay-cascade/PDFs/Prescreening/EUNCL-STE-002_2.pdf?m=1476164583&.
- USP. USP provides guidelines for Recombinant Factor C (rFC) a non-animal-derived reagent critical to development of vaccines and other sterile pharmaceutical products. 2020[cited 2023 January]; Available from: https://www.usp.org/news/rfc-horseshoe-crabs-statement.
- H. Grallert, et al., EndoLISA®: a novel and reliable method for endotoxin detection, Nat. Methods, 2011, 8(10), iii–v CrossRef.
- B. W. Neun, et al., Detection of Beta-Glucan Contamination in Nanotechnology-Based Formulations, Molecules, 2020, 25(15), 3367 CrossRef CAS.
- B. Han, et al., Structure-Functional Activity Relationship of β-Glucans From the Perspective of Immunomodulation: A Mini-Review, Front. Immunol., 2020, 11, 658 CrossRef CAS PubMed.
- J. Lasselin, et al., Comparison of bacterial lipopolysaccharide-induced sickness behavior in rodents and humans: Relevance for symptoms of anxiety and depression, Neurosci. Biobehav. Rev., 2020, 115, 15–24 CrossRef CAS.
- H. S. Warren, et al., Resilience to bacterial infection: difference between species could be due to proteins in serum, J. Infect. Dis., 2010, 201(2), 223–232 Search PubMed.
- T. Hartung, Pyrogen testing revisited on occasion of the 25th anniversary of the whole blood monocyte activation test, ALTEX - Alternatives to animal experimentation, 2021, 38(1), 3–19 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02821j |
| This journal is © The Royal Society of Chemistry 2024 |