
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The interaction of glycogen nanoparticles with human blood†
Nadiia
Davydiuk‡
,
Vaidehi
Londhe‡
,
Manfred F.
Maitz
 ,
Carsten
Werner
,
Andreas
Fery
and
Quinn A.
Besford
*
,
Carsten
Werner
,
Andreas
Fery
and
Quinn A.
Besford
*
Leibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: besford@ipfdd.de
First published on 4th November 2024
Abstract
Glycogen, a naturally sourced highly branched polysaccharide nanoparticle, has been receiving attention in the field of nanomedicine due to its inherent non-toxicity and biodegradability. However, often in the literature glycogen nanoparticles (NPs) are used that come from different commercial sources (animals and tissues), which have significantly different sizes, molecular weights, and protein content, meaning a comprehensive overview of the interactions of these differently-sourced NPs with the human immune system is missing. Herein, we investigated coagulation, immune cell association and inflammation responses triggered by source-dependent interactions of glycogen NPs in human blood, utilising four types of commercially available glycogen: phytoglycogen (PG) isolated from sweet corn kernels, oyster glycogen (OG), rabbit liver glycogen (RLG), and bovine liver glycogen (BLG). Our results reveal that glycogen NPs exhibit minimal immune cell association, low complement factor, granulocyte, and platelet activation, and have no impact on blood clotting. This is despite the significant physico-chemical differences between the NP types, and when studied at exceptionally high particle concentrations (orders of magnitude higher than other typically studied synthetic systems). Given the similarities in the interactions with blood, either of the commercial glycogens can be leveraged in nanomedicine with respect to immuno-interactions, though PG provides a sustainable and ethically sourced form of NPs from plants. Together, our results highlight a key benefit of using glycogen NPs as injectable biomaterials for therapeutic applications.
Introduction
Within the context of nanomedicine, some of the most important features of polymer nanoparticles include tissue targeting abilities, controllable conjugate-drug release rates, low-fouling or stealthy behaviour, high water solubility, biocompatibility (e.g., cell toxicity, inflammation responses, etc.), and biodegradability in the long-term.1–4 In line with this, significant work has gone into developing polymer nanoparticle systems that meet most, if not all of these criteria. These include systems based on zwitterionic poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC),5 polyethylene glycol (PEG),6 and poly(2-oxazoline),7 in a variety of architectures, including as polymer replica particles,8–10 bottle-brush polymers,11–13 or as layers on secondary substrates,14,15 as some examples amongst many. While these systems have proved very useful, there is also a strong-case to move towards minimally synthetic polymer nanoparticles sourced directly from nature, which may have more predictable biological outcomes.16 Furthermore, this can potentially allow for greener production methods with fewer synthetic steps. With this in mind, biomaterial glycogen nanoparticles have been gaining significant traction for use in nanomedicine.17,18Glycogen is a naturally occurring polysaccharide nanoparticle that functions as a primary storage form of cellular glucose,2,17,19 where it acts as a vital energy source for cells in the human body by storing glucose and releasing it when needed.20,21 Glycogen is already present in most human cells, meaning our cells already possess the cellular machinery to degrade the nanoparticles in the long-term, yielding only glucose and a small amount of protein.22 Furthermore, it also makes these nanoparticles inherently biocompatible23–25 within our cells, with a very high-water solubility.26 Glycogen is composed of linear chains of approximately 8–14 glucosyl residues joined in α-1,4-glucosidic linkages, which are branched by α(1 → 6) linkages, to form randomly hyperbranched polysaccharide nanoparticles that are approximately 20–90 nm in diameter27 (Fig. 1). The particles contain a small amount of associated protein that participates in the synthesis pathway.28
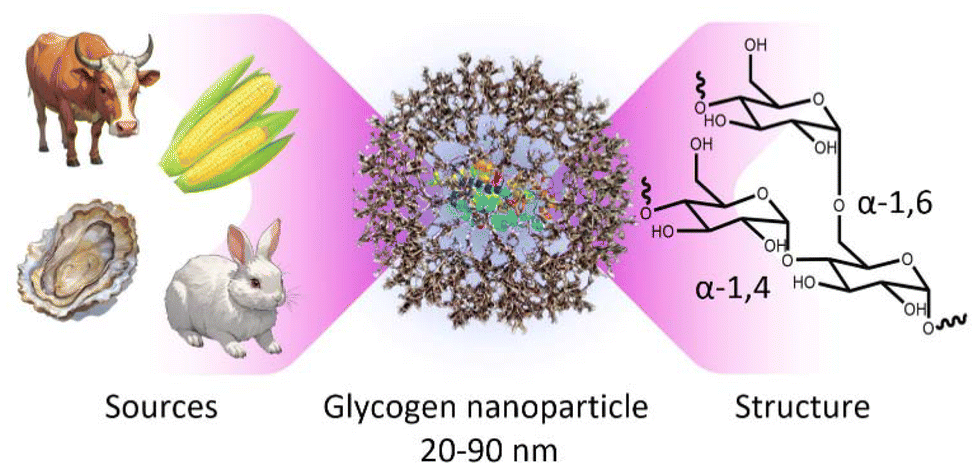 | ||
| Fig. 1 Schematic of a glycogen nanoparticle sourced from currently available commercial animal, shellfish, and plant sources, along with the internal chain structure. | ||
Glycogen can be readily isolated from liver, muscle, brain, and heart tissues of various animals.29 The liver is a particularly common source because it is highly concentrated.20 Additionally, certain shellfish, such as oysters,30 and the kernels of sweet corn31 are also rich sources of glycogen and can be extracted on an industrial scale.29,31 Importantly, glycogen nanoparticles are commercially available as a relatively low-cost off-the-shelf nanoparticle from a variety of sources,29 supplied in a stable powder form, available on the gram-to-tonne scale.
Glycogen has therefore been utilised in numerous studies for treating and diagnosing diseases,32–34 and as functional macroscopic hydrogel materials.23,24,35–37 Recently, more studies have used glycogen as a circulating nanoparticle in mice (for tumour accumulation), and as circulating glucose-responsive insulin-conjugates for treating diabetes in mice.38 However, there is no clear understanding of what are the biological interactions of glycogen obtained from different sources (animals or tissues) once they are integrated into human blood with a therapeutic purpose. It is known that glycogen from different sources differ in protein content, as well as other physicochemical properties such as size, density and charge, which may lead to different cell association and inflammation responses in human blood. Herein, we look to full characterise the physicochemical properties and in-blood interactions of glycogens obtained from four common commercial sources. We systematically compare rabbit liver glycogen (RLG), bovine liver glycogen (BLG), oyster glycogen (OG), and phytoglycogen (PG) (isolated from the kernels of sweet corn). Our results provide insight into source-dependent effects of glycogen interactions, which can better inform of injectable nanoparticle choices for therapeutic applications.
Experimental section
Materials
Glycogen from bovine liver (BLG), rabbit liver (RLG), and oysters (OG) were purchased from Sigma Aldrich. Phytoglycogen (PG) sourced from the kernels of sweet corn was purchased from Mirexus (Guelph, Canada). Alexa Fluor™ 488 NHS dye (AF488-NHS), dimethyl sulfoxide (DMSO), α-amylase from Aspergillus oryzae (activity ∼30 U mg−1), trifluoroacetic acid (TFA, >99%), phenol (≥99%), sulfuric acid (>95%), sodium bicarbonate (≥99.7%), sodium carbonate (≥99.5%), ethylenediaminetetraacetic acid (EDTA), formaldehyde solution (wt 37%) and PBS buffer tablets were purchased from Sigma Aldrich. A Micro BCA™ Protein assay Kit (product number 23235), Amicon Ultra Centrifugal Filters (molecular weight cut-off of 10 and 100 kDa), and 96-well microplates (black walled, clear bottom, non-sterile; product number M33089) were purchased from Thermo Fisher Scientific. Lysis of red blood cells (RBCs) was performed using Pharm Lyse buffer from BD Biosciences (Heidelberg, Germany). For cell phenotyping, antibodies against CD3 AF700 (SP34-2), CD11b Pacific Blue (ICRF44), CD14 APC-H7 (MΦP9), CD66b BV421 (G10F5), CD45 V500 (HI30), CD56 PE (B159), lineage-1 cocktail APC, HLADR PerCP-Cy5.5 (G46-6), and CD19 BV650 (HIB19) were used, all purchased from BD Biosciences, except for lineage-1 cocktail, CD15, and CD19, which were obtained from BioLegend (CA, USA). C5a, prothrombin fragment F1 + 2 and platelet factor 4 (PF4) concentration were determined using an enzyme-linked immunosorbent assay (C5a ELISA from DRG Diagnostics, Marburg, Germany; Enzygnost F1 + 2, Siemens Healthineers, Erlangen, Germany; Zymotest PF4, CoaChrom, Vienna, Austria). Positive controls for the blood assays, namely Kaolin and Zymosan A (Saccharomyces cerevisiae) were purchased from Sigma Aldrich while silica particles (1000 μm) were synthesized in-house. The pH of the solutions was measured by using a Mettler-Toledo FiveEasy FE20 pH meter. Milli-Q water with a resistivity greater than 18.2 MΩ cm, obtained from a three-stage Millipore Milli-Q Plus 185 purification system (Millipore Corporation, USA), was used in all experiments unless otherwise stated. All materials were used as received or passed purification.Fluorescence labelling of glycogen NPs
The fluorescent labelling of the particles was carried out according to the previously published protocol.39 The particles (10 mg) were dissolved in 0.2 mL of 0.1 M sodium bicarbonate buffer at pH 8. AF488-NHS (1 mg) previously dissolved in 1 mL of anhydrous DMSO, was added to the glycogen types as follows: PG-0.69 μL, OG-3.01 μL, RG-4.76 μL, BG-100 μL. These ratios were chosen in order to provide each nanoparticle the same number of fluorophores (considering the variations in nanoparticle Mn), which is important for later biological studies. The mixture was then incubated for 24 hours at 24 °C with continuous shaking. Subsequently, any unbound dye was removed using centrifugal filters with a pore size of 10 kDa. The particles were centrifuged (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 5 min), washed with Milli-Q water (2 times), freeze-dried, and stored in the freezer (−20 °C) in the dark prior to use.
000 rpm, 5 min), washed with Milli-Q water (2 times), freeze-dried, and stored in the freezer (−20 °C) in the dark prior to use.
Gel permeation chromatography
Molecular weight and molar mass determination were performed using an AF4 system Eclipse DUALTEC (Wyatt Technologies Europe) with Agilent pump system (1260er Serie) with detection at various wavelengths (λ = 254, 280, 400, 500 nm). A solvent PBS-buffer (10 mM, pH = 7.4) was used as the eluent with a flow rate of 0.8 mL min−1.Protein content of NPs by bicinchoninic acid (BCA) assay
Colorimetric protein detection was performed following a standard protocol provided by the supplier. Glycogen NP solutions (1 mg mL−1) and diluted albumin standards (0.5–0.0 mg mL−1) were prepared in Milli-Q water. To prepare working reagents (WR), mixed 25 parts of Reagent MA (1.725 mL, mixture of sodium carbonate monohydrate 8% w/v, sodium hydroxide 1.6% w/v, sodium tartrate 1.6% w/v, and sodium bicarbonate) with 24 parts of Reagent MB (1.656 mL of bicinchoninic acid disodium salt hydrate, 4% w/v) and 1 part of Reagent MC (0.069 mL of copper(II) sulphate pentahydrate, 4% w/v). A 150 μL of each diluted albumin standard and glycogen sample replicate was pipetted into a 96-well microplate. Then, 150 µL of the WR was then added to each well followed by thorough mixing by a plate shaker for 30 seconds. The plate was covered and incubated at 37 °C for 2 h, then cooled and the absorbance was measured at λ = 562 nm on a Tecan SPARK 10 M Luminescence Multi Mode Microplate Reader.Biodegradation of NPs by α-amylase
The enzymatic degradation of PG, OG, RG and BG were assessed using a phenol-sulfuric acid assay.39 Glycogen NP solutions (2 mg mL−1) were prepared in Milli-Q water, and α-amylase (1.0 U mL−1) solutions in PBS buffer (pH 7.4) To begin the assay, glycogen solutions (70 μL) were added to the enzyme solution (200 μL), and the resulting mixtures were incubated at 23 °C with gentle agitation (150 rpm) for 3 h. Then, the undigested glycogen and enzyme were separated from the free glucose using a centrifugal filter (100 kDa). The filtrate was collected and divided into sample aliquots of 50 μL, to which concentrated sulfuric acid (150 μL) was added, followed by the addition of 5% phenol solution (30 μL, previously prepared in Milli-Q water). The samples were subsequently incubated at 90 °C with agitation (700 rpm) for 15 min. The UV–vis absorbance of the digested solution was then measured at 480 nm with a Tecan SPARK 10M Luminescence Multi Mode Microplate Reader. To measure the glucose content in each type of particle, the initial glycogen solutions (70 μL) were treated with 0.2 M TFA (200 μL) and subjected to agitation (150 rpm) at 80 °C for 3 hours. Sample aliquots of 50 μL was added to concentrated sulfuric acid (150 μL) and 5% phenol solution (30 μL). Then samples were incubated at 90 °C with agitation (700 rpm) for 15 min. The mixtures were then analysed by UV–vis absorbance reading at λ = 480 nm on the microplate reader.Dynamic light scattering (DLS) and zeta-potential (ZP)
Size distribution and ZP of the glycogen NPs were measured with a Malvern Zetasizer Nano (ZS) instrument. Samples were analysed at 298 K with disposable plastic sizing cuvettes and clear disposable zeta cells, containing 1 mg of glycogen NPs (dissolved in 1 mL of Milli-Q water, additively sonicated for 5 min in an ultrasonic bath). The reported data show means and standard deviations of three measurements.Atomic force microscopy
Measurements were performed on a MFP-3D Asylum research instrument, with Bruker OTESPA-R3 (force constant = 26 N m−1, resonance frequency = 300 kHz, tip radius – 25 nm) cantilevers in tapping mode. The 0.2 mL of NPs with a concentration of 0.001 mg mL−1 were dispersed onto freshly cleaved mica sheets (15 mm diameter) and left unperturbed for 1 h to adsorb, then the mica surfaces were dried under a stream of nitrogen. AFM topography images were recorded in dry air, using the NanoScope v9.40 software package.Blood assay to measure association of glycogen NPs with human immune cells
Blood assays were performed following a recently published protocol.5 The study was approved by the ethics committee of the Sächsische Landesärztekammer under EK-BR-95/20-1. Fresh blood was collected from healthy human volunteers after obtaining informed consent. The blood was drawn into sodium heparin vacuettes (Grenier Bio-one). The glycogen nanoparticles, at concentrations of 1 × 1010 and 5 × 106 per μL blood were incubated in whole blood (100 μL) for 1 h at 37 °C. 1 × 106 silica NPs served as the positive control. RBCs were lysed by adding Pharm Lyse buffer at 40× blood volume and washed with phosphate-buffered saline (pH 7.2, 4 mL, 2×) (500g, 5 min, 4 °C). Cells were phenotyped on ice for 1 h using titrated concentrations of antibodies against CD3 AF700 (SP34-2), CD14 APC-H7 (MΦP9), CD66b BV421 (G10F5), CD45 V500 (HI30), CD56 PE (B159), lineage-1 cocktail APC, HLA-DR PerCP-Cy5.5 (G46-6), and CD19 BV650 (HIB19). Unbound antibodies were removed by washing and centrifugation (500g, 5 min, 4 °C) in a PBS buffer containing 0.5% w/v BSA and 2 mM EDTA at pH 8. Cells were fixed in 1% w/v formaldehyde in PBS. The samples were directly used for cell association analysis by flow cytometry (LSRFortessa, BD Biosciences, USA) and analyzed using BD FACSDiva software.Confocal laser scanning microscopy of glycogen NPs with immune cells
Glycogen NPs (500 µg) were incubated in whole human blood for 1 h, with incubation conditions and processes similar to the association assay, though with a large excess of NPs. RBCs were lysed by adding Pharm Lyse buffer at 40× blood volume and washed with phosphate-buffered saline (pH 7.2, 4 mL, 2×) (500g, 5 min, 4 °C) leaving only the leukocyte population. The leukocytes were then fixed with 1% formaldehyde at room temperature for 30 min. After fixation, the samples were permeabilised using Triton X-100 (0.1% in PBS) for 10 min followed by staining with AF633-Phalloidin (Atto-TEC, 1:200) conjugate. Between each step, the cells were washed (2×, 500g, 5 min). For imaging, the cells were transferred onto poly-L-lysine coated coverslips and imaged using Dragonfly Spinning Disc confocal microscope (Andor Technology Ltd, Belfast, UK).
Hemocompatibility of glycogen NPs
Hemocompatibility assessments were carried out as previously described.28,40 In brief, fresh venous blood was collected from 2 ABO compatible healthy human volunteers after obtaining informed consent. Blood was anticoagulated with 0.5 U mL−1 heparin. A point-of-care assay for C-reactive protein (Diagnostik-Nord, Schwerin, Germany) and a differential blood cell count (DxH500, Beckman Coulter, Germany) were determined before performing subsequent experiments. All experiments were done in triplicates and started within 15 min of blood collection. The nanoparticles, or PBS (vehicle control), were mixed with 2 mL of whole blood to obtain a final concentration of 5 × 106 NPs per μL of blood and 1 × 1010 NPs per μL of blood in accordance to the concentration used in the association assay. The blood-nanoparticle suspension was incubated in polypropylene tubes for 2 h at 37 °C under constant overhead rotation. For positive controls, 10 µg mL−1 of kaolin. 10 µg mL−1 zymosan, and 1 × 107 silica NPs were also incubated in 2 mL of blood. After 2 h incubation, the blood was analyzed with regards to CD11b expression on CD15-gated neutrophils, CD14-gated monocytes, and CD62P expression on platelets by flow cytometry (LSRFortessa, BD Biosciences, USA) and analyzed using BD FACSDiva software. Additionally, blood samples were split and mixed with recommended stabilizers for subsequent ELISA analysis of prothrombin fragment F1 + 2 (F1 + 2 Enzygnost, Siemens Engineers, Germany), and complement factor fragment C5a (DRG Instruments GmbH, Marburg, Germany). The ELISAs were carried out according to the manufacturer's instructions.Results and discussion
Physicochemical properties
An investigation on the morphology of glycogen nanoparticles from different sources was conducted via atomic force microscopy (AFM, Fig. 2) and dynamic light scattering (DLS, Fig. 3A). From AFM, glycogen NPs exhibited a roughly spherical-like morphology in the dry (dehydrated) state. From DLS it was found that PG exhibited the largest hydrodynamic diameter (72 ± 0.3 nm), compared with OG (48 ± 0.2 nm), and RLG (43 ± 0.3 nm) (Fig. 3A), while the smallest size distribution was observed for BLG (21 ± 0.4 nm). The differences in size values between AFM and DLS attribute to the fact that AFM examines particles in a dry state, where particles can become depressed (“pancake” structure), leading to larger apparent diameters compared to DLS measurements in the solvated state. | ||
| Fig. 2 AFM dry-state topographical images of (A) PG, (B) OG, (C) RLG and (D) BLG nanoparticles dispersed on freshly cleaved mica. Scale bars are 200 nm, with independent z-scales (in panels). | ||
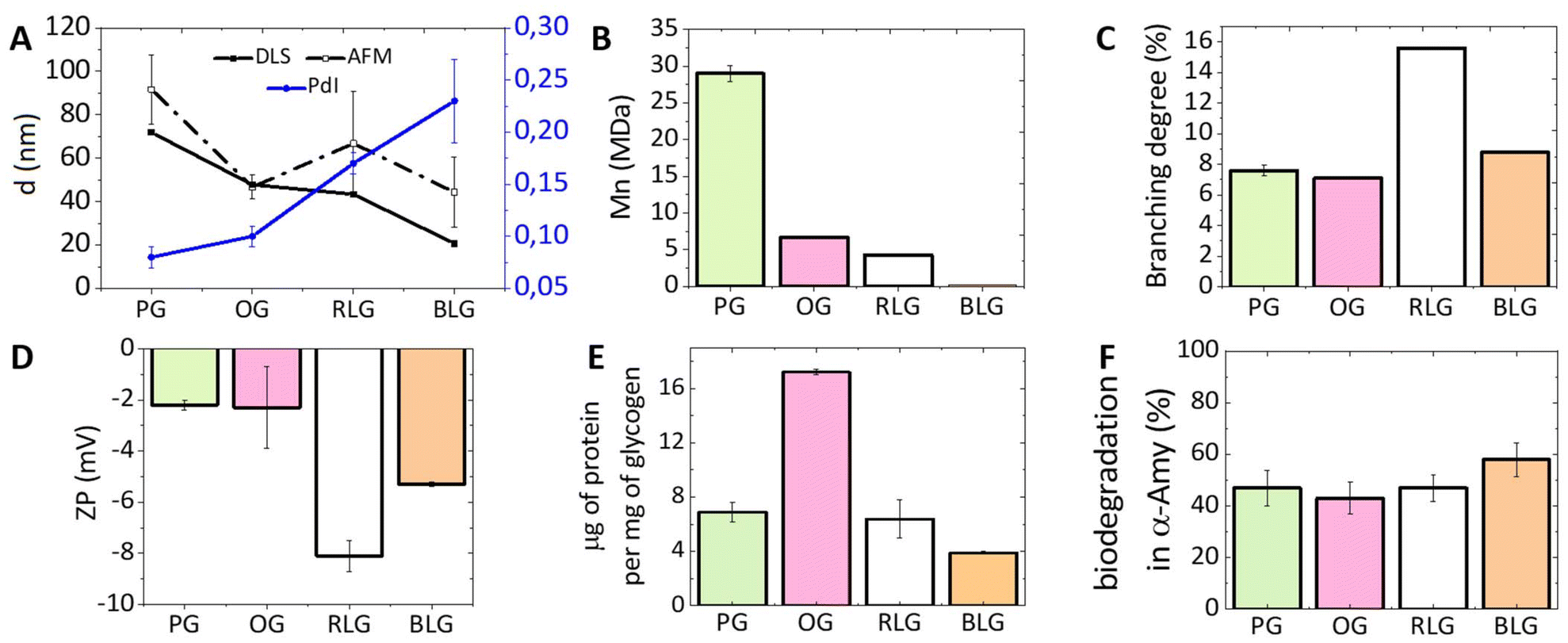 | ||
| Fig. 3 The physicochemical properties of differently-sourced glycogen NPs. (A) The NP sizes from DLS and AFM, along with PdI, (B) the Mn measured by GPC, (C) the branching degree (data taken from published papers (PG,41 OG,42 RLG,39 BLG39)), (D) the zeta-potential (ZP), (E) the protein content, and (F) the biodegradability towards α-amylase for the different glycogen NPs. Additional data (size and molar mass distributions, PdI, ZP) are provided in the ESI (Tables S1 and S2, Fig. S1 and S2†). Error bars indicate standard deviations on at least three replicate measurements. | ||
The polydispersity index (PdI) indicates the degree of heterogeneity in the size distribution of nanoparticles (Fig. 3A). A small PdI value (<0.1) suggests a homogeneous population of particles, while a value of >0.3 indicates a higher degree of heterogeneity.43 The PdIs of PG and OG nanoparticles are 0.08 and 0.10, respectively, indicating a relatively homogeneous population. RLG nanoparticles have a PdI of 0.17 ± 0.01, suggesting a slightly higher degree of heterogeneity but still within an acceptable range. BLG nanoparticles have the highest PdI of 0.23 ± 0.04, indicating a more heterogeneous population compared to the others.
The zeta-potential (ZP) of most commercial glycogen particles are slightly negative, depending on the source (Fig. 3D). RLG nanoparticles have the most negative ZP (−8 ± 0.6 mV), followed by BLG nanoparticles (−5 ± 0.1 mV). PG and OG nanoparticles have relatively less negative ZPs (−2 ± 0.2 mV and −2 ± 1.6 mV, respectively). This charge suggests the presence of a minor quantity of leftover phosphate resulting from biological synthesis and degradation processes.25,44
The number-average molecular weight (Mn) was determined for all glycogens by gel-permeation chromatography (GPC), which showed that the Mn increases with increasing particle diameter (Fig. 3B). PG nanoparticles, with the largest size, have the highest Mn of 29.1 ± 1.61 MDa, followed by OG, RLG, and BLG nanoparticles with progressively lower Mn values and smaller sizes. The degree of branching in NPs was obtained by comparing previous values reported in the literature measured using 1H NMR.39,42,45 This was achieved by assessing the ratio between the H1-6 proton signal at δ = 4.93 ppm, associated with glucose residues linked by α-1,6-glycosidic bonds branching, and the H1-4 proton signal at δ = 5.32 ppm, indicative of the total glucose units within the glycogen structure.39 As shown in Fig. 3C, the percentage of α-1,6 linkages of RLG, BLG, PG and OG were 15.6%, 8.8%, 7.6% and 7.1%, respectively, which indicated that the following branching order of RLG > BLG > PG > OG, therefore showing no clear correlation between particle size and branching density.
Protein content in glycogen NPs
During the initial stages of glycogen synthesis, a glycogenin protein dimer serves as the anchoring core and initiates glycogen formation.46 Other enzymes, including glycogen synthase and glycogen branching enzyme, then work in tandem to synthesise the glycogen NPs. These proteins likely contributes to the protein content found in glycogen NPs, perhaps also containing an intracellular protein corona, which is important to understand from the standpoint of differently sourced glycogens. From Fig. 3E, it is evident that OG shows the highest protein content at about 17.2 μg of protein per mg of NPs, implying a richer presence of associated proteins. Conversely, BLG exhibits the lowest protein content at 3.9 μg mg−1, suggesting a comparatively lower protein proportion among the tested glycogen sources. The exact nature of the protein content has not been investigated further.Biodegradability by α-amylase
The breakdown of glycogen involves enzymatic steps where the α-1,6-glycosidic bond branch points and the α-1,4-glycosidic bonds of the straight chain are cleaved by glycogen-debranching enzyme and glycogen phosphorylase.22,47 However, α-amylase, a glucosidase enzyme, primarily catalyses the hydrolysis of α-1,4-glycosidic bonds in glycogen,48 specifically targeting these bonds to break them down into smaller glucose units.25 Treating glycogen with trifluoroacetic acid (TFA) can fully hydrolysed glycogen into glucose units.49 The biodegradability is therefore determined by the ratio of the glucose yielded from incubation with α-amylase to that achieved through complete hydrolysis with TFA. It was found that all glycogens exhibit a similar biodegradability towards α-amylase (PG: 47%, OG: 43%, RLG: 47% and BLG: 58%, Fig. 3F). A degradation percentage falling within the 40–60% range is considered efficient, especially given that the assay employs only a single enzyme. In vivo, multiple glucosidase enzymes participate in the degradation of glycogen, further contributing to its biological breakdown.22Nanoparticle interactions with immune cells in human blood
Nanoparticles can interact with immune cells, such as phagocytic cells (neutrophils, monocytes, dendritic cells) and nonphagocytic cells (T cells, B cells, NK cells).50 These interactions impact biocompatibility and alter the therapeutic efficacy of nanoparticles designed for drug delivery and diagnostics.51 Human blood assays provide a unique environment which allows examination of the nanoparticle-immune system interactions across all immune cells.The interactions of PG, OG, RLG, and BLG with human immune cells was examined at two different particle concentrations. Identification of different white blood cell populations and association with glycogen NPs was performed by flow cytometry, according to a published gating strategy5 (ESI Fig. S3†). The systems were tested with nanoparticle concentrations ranging from 5 × 106 to 1 × 1010 NPs per μL to assess concentration-dependent effects on cell association. PG and BLG showed minimal association with B cells and dendritic cells, with values not exceeding 6% (Fig. 4A). There was negligible association with neutrophils or NK cells, and less than 3% association with T cells and monocytes. A decreasing association was noted with lower nanoparticle concentrations (from 1 × 1010 to 5 × 106 NPs per μL). Overall, the nanoparticles, even at high concentration in blood exhibited no significant association with immune cells, indicating a stealthy character. For example, non-stealthy materials, such as mesoporous silica nanoparticles, exhibit immune cell associations between 20% to 85%.5 While the magnitude of nanoparticle-immune cell interactions had slight differences across donors (ESI Fig. S4†) and immune cell subsets, the overall level of NP-immune cell association remained below 10%. Furthermore, 106 silica NPs, used as positive control, exhibited significantly higher immune cell associations (ESI Fig. S5†). Furthermore, we note that the cell associations observed for all glycogen NPs are significantly lower than that for other stealthy PMPC NP systems previously studied (associations between 5% to 35%),5 where the PMPC-based systems were analysed at a concentration 6-orders of magnitude lower (1 × 104 particles per μL) than what has been studied here for concentrated glycogen. The mechanism that leads to this stealthiness towards immune cells is not clear, as it seems independent of the physicochemical parameters of the glycogens (Fig. 3), but is likely related to the low-fouling nature of the NPs, and due to the presence of glycogen within cells to begin with.
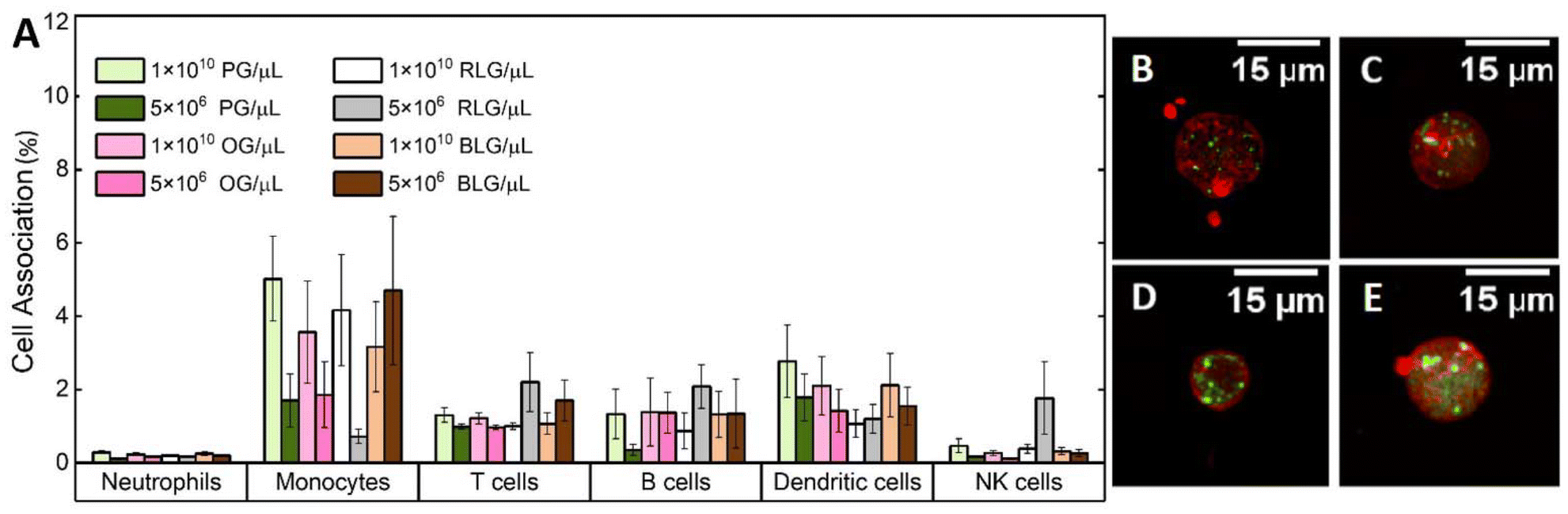 | ||
| Fig. 4 (A) Immune cell association to glycogen NPs (PG, OG, RLG and BLG) at different concentrations. Data are shown as mean ± standard errors (n = 6). Confocal laser scanning microscopy images of leukocytes following (B) PG, (C) OG, (D) RLG and (E) BLG NPs after 1 h incubation at 0.5 mg mL−1 concentration. Cells were stained with a phalloidin-AF633 conjugate (red) and the nanoparticles were labelled with AF488 (green). Scale bars are 15 µm. Statistical significance is determined by ANOVA followed by Tukey's test and is represented as *p < 0.05, **p < 0.005. | ||
To validate the stability and interaction of the NPs with leukocytes, a larger excess of glycogen NPs were incubated with the immune cells, making it possible to visualise leukocyte cell uptake by CLSM. Bright spots could be seen localised intracellularly for all particle types (Fig. 4B–E). It was also possible to visualise dispersed dye (cloudy regions), suggesting intracellular degradation of the NPs once internalised and released into the cytosol. Note that the type of leukocytes could not be confirmed for the CLSM imaging.
Hemocompatibility of glycogen NPs
Hemocompatibility is a crucial property to determine for nanomaterials that contact with blood components, where preventing coagulation and inflammation are key challenges.52 During hemocompatibility testing, aspects including activation of platelets, coagulation, and complement system are assessed. We investigated the hemocompatibility of all glycogen NPs by assessing platelet activation, complement activation and initiation of the coagulation cascade. Additionally, cellular activation of neutrophils and monocytes as a marker of inflammation has been investigated. Due to the absence of dramatic dose-dependent trends in the association assay, the highest (1 × 1010 NPs per μL) and a representative lower NP concentration (5 × 106 NPs per μL) were selected for the subsequent haemostasis and inflammation investigations. The coagulation pathway involves the activation of Factor XII by contact with negatively charged foreign surfaces (bacteria, viruses, yeast, glass, medical apparatus).53 This drives thrombin generation and fibrin formation through the intrinsic pathway.54 Fibrin forms a fibrous mesh that traps red blood cells.55 Thrombin promotes platelet activation and stabilizes the clot by binding to fibrin.56Coagulation activation is detected by a prothrombin fragment 1 + 2 (F1 + 2)57 biomarker, which is released during thrombin formation.52 We found that at a concentration of 1 × 1010 NPs per μL the BLG and OG NPs induced higher F1 + 2 levels in comparison to PG and RLG. The observed elevation consequently reduced to the control level at the lower NP concentrations. Notably, PG and RLG, even at the high concentration exhibited levels similar or slightly lower than the blank control (Fig. 5A). Overall, the F1 + 2 levels in response to the NPs remained close to baseline suggesting negligible influence of glycogen NPs on coagulation activation, though some differences between particle types were evident. In order to validate the observed glycogen responses; kaolin, zymosan, and silica NPs were used as positive controls in our coagulation assays (F1 + 2) (ESI Fig. S6†).
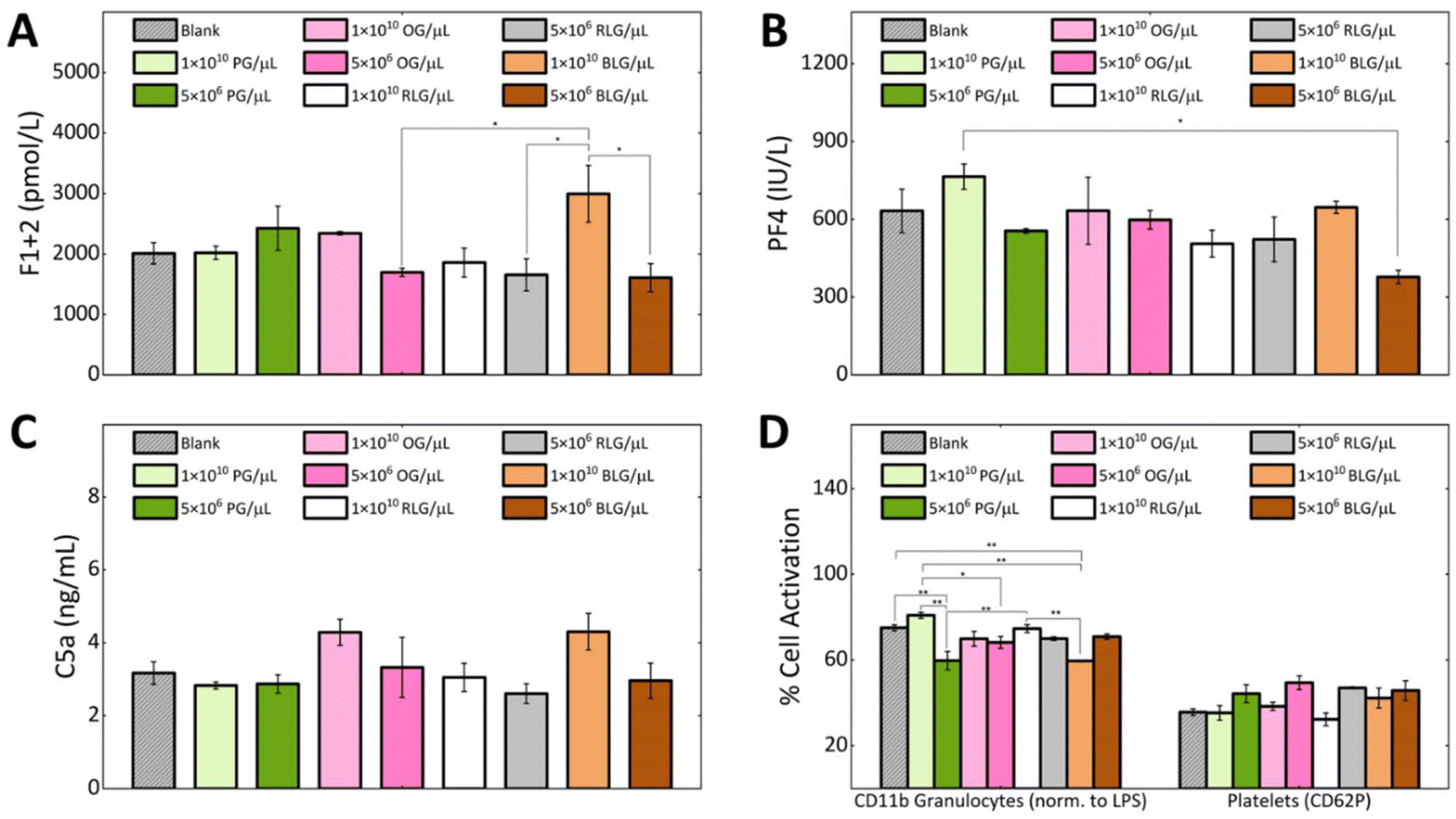 | ||
| Fig. 5 Activation of coagulation and inflammation parameters in response to PG, OG, RLG and BLG in human whole blood: (A) prothrombin fragment 1 + 2 (F1 + 2); (B) platelet factor 4 (PF4); (C) complement fragment C5a; (D) CD11b expression of granulocytes (% normalised to lipopolysaccharide (LPS)), and platelets P-selectin CD62P expression. Data are shown as mean ± standard error of n = 3. Statistical significance is determined by ANOVA followed by Tukey's test and is represented as *p < 0.05, **p < 0.005. | ||
To study platelet activation, we used the P-selectin CD62P marker. The percentage of platelets expressing CD62P was the same for all types of glycogen at a concentration of 1 × 1010 NPs per μL, as the blank control (Fig. 5D). While, a slightly higher activation was observed for all glycogen types at the lower concentration of 5 × 106 NPs per μL (Fig. 5D), though the levels remained comparable to blank. PF4 is a specific protein found in platelet alpha-granules that is released during platelet activation58 and degranulation.59 PF4 electrostatically binds and neutralizes heparin, enhances blood coagulation, inhibits local antithrombin activity, attracting granulocytes and fibroblasts through chemotaxis.59 In Fig. 5B we analysed the platelet factor 4 (PF4). The lowest PF4 activation was observed at a concentration of 5 × 106 BLG per μL. The PG system showed the highest PF4 activation at 1 × 1010 NPs per μL, but stabilizes at 5 × 106 NPs per μL as compared to the control. OG, RLG did not significantly change at 1 × 1010 NPs per μL or 5 × 106 NPs per μL concentrations.
If glycogen nanoparticles were to be recognised, the complement system would be sequentially activated, with the pathway subsequently generating anaphylatoxins C3a and C5a, promoting inflammation.60,61 The C5a fragment therefore serves as a marker of inflammation mediated by total complement activation.57 At a concentration of 1 × 1010 NPs per μL, the complement factor C5a levels were higher in response to the OG and BLG systems. For OG, this possibly comes from the higher protein content. The PG and RLG at either concentration remained comparable to the blank control (Fig. 5C). Although differences in C5a activation were observed between the NP systems, likely owing to their physicochemical variations. However, none of the NPs induced a substantial C5a-mediated humoral inflammatory response.
The CD11b expression was used as a marker of granulocyte activation (cellular inflammation) in response to glycogen NPs (Fig. 5D). Overall, the activation of granulocytes induced by glycogen nanoparticles was less than that induced by LPS, indicating their non-inflammatory nature. At the higher concentration of 1 × 1010 NPs per μL, CD11b expression was elevated in response to PG as compared to OG, RLG and BLG. The increased CD11b levels at the higher concentration could be attributed to slightly larger particle size of PG. The increased levels dropped at the lower 5 × 106 PG per μL concentration. In contrast, OG and RLG systems exhibited concentration-independent granulocyte activation across both NP concentrations. BLG displayed significantly lower granulocyte activation at the higher NP concentration as compared to all other NPs and control. While the activation level increased slightly at the lower concentration, it was not a significant change. Overall, the OG, RLG and BLG NP systems did not elicit granulocyte activation, while PG elicited a dose-dependent response.
The minimal complement activation, negligible coagulation activation, and lack of platelet and granulocyte activation highlights the low immunogenicity of the glycogen nanoparticles. This indicates a high degree of hemocompatibility as well as reduced likelihood of recognition as foreign agents despite being derived from non-human sources. This further serves to highlight the benefits of biomaterial glycogen NPs in nanomedicine as injectable biomaterials. An interesting aspect to be pursued in future investigations would be to clarify if glycogen nanoparticles induce anti-glycogen antibodies, between the different glycogen types. This has become evident as a hurdle for PEGylated vaccine technologies,62 and given that glycogen is already within us, perhaps this effect can be avoided.
Conclusions
We have investigated the physicochemical and biological interactions with human blood of various commercial glycogen nanoparticles (NPs) derived from different sources: phytoglycogen (PG) from sweet corn kernels, oyster glycogen (OG), rabbit liver glycogen (RLG), and bovine liver glycogen (BLG). These NPs have sizes ranging from 20 to 90 nm, with significant differences in molecular weights. Our evaluation revealed no significant differences in degradability among the NPs, with an average degradability of 49 ± 9%. OG had the highest protein content at 17.2 μg mg−1, but this did not appear to significantly affect inflammation or coagulation processes in human blood. At a concentration of 1 × 1010 NPs per μL of blood, all types of glycogen showed no significant recognition by the immune system or impact on the coagulation pathway. PG and RLG could be used at higher concentrations of 1 × 1010 NPs per μL, although OG and BLG at this concentration exhibited increased activation of clotting and immune cells. These properties suggests glycogen NPs to be highly suitable for drug delivery, at particle concentrations several orders of magnitude greater than other reported synthetic systems. Our data suggest that glycogen can circulate in the bloodstream without causing clotting or inflammation, allowing for the efficient and safe transport of therapeutic agents can reach their targets without being intercepted or neutralized by the body's defence mechanisms. Additionally, glycogen holds significant promise for imaging and diagnostic applications due to this biocompatibility and also ease of functionalisation. Our results further clarify the benefits of glycogen going ahead in theranostic applications.Author contributions
N. D., V. L., and Q. A. B. conceived and planned the experiments. Q. A. B., A. F., and C. W. supervised the work. N. D. synthesised the materials and performed all physicochemical characterisation. N. D. and V. L. performed cell association assays. M. F. M. performed hemocompatibility assays. N. D. wrote the manuscript, with contributions from all authors. All authors have discussed the results and commented on the final manuscript.Data availability
Data available within the article. The authors confirm that the data supporting the findings of this study are available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Deutsche Forschungsgemeinschaft (DFG) for funding (451339898), and Dr Alessia C. G. Weiss, A/Prof. Francesca Cavalieri, and Dr Shayan Vazirieh Lenjani for discussions, and Dr Susanne Boye for assistance with measurements. This work received funding from the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement 872233 (“PEPSA-MATE”).References
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- L. P. Wu, D. Wang and Z. Li, Mater. Sci. Eng., C, 2020, 106, 110302 CrossRef CAS PubMed.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Front. Chem., 2018, 6, 360 CrossRef PubMed.
- S. Su and P. M. Kang, Nanomaterials, 2020, 10, 656 CrossRef CAS.
- A. C. G. Weiss, H. G. Kelly, M. Faria, Q. A. Besford, A. K. Wheatley, C.-S. Ang, E. J. Crampin, F. Caruso and S. J. Kent, ACS Nano, 2019, 13, 4980–4991 CrossRef CAS PubMed.
- J. Cui, M. Björnmalm, Y. Ju and F. Caruso, Langmuir, 2018, 34, 10817–10827 CrossRef CAS.
- P. Wilson, P. C. Ke, T. P. Davis and K. Kempe, Eur. Polym. J., 2017, 88, 486–515 CrossRef CAS.
- D. Song, J. Cui, H. Sun, T.-H. Nguyen, S. Alcantara, R. De Rose, S. J. Kent, C. J. H. Porter and F. Caruso, ACS Appl. Mater. Interfaces, 2017, 9, 33683–33694 CrossRef CAS.
- M. Müllner, J. Cui, K. F. Noi, S. T. Gunawan and F. Caruso, Langmuir, 2014, 30, 6286–6293 CrossRef.
- S. Lück, R. Schubel, J. Rüb, D. Hahn, E. Mathieu, H. Zimmermann, D. Scharnweber, C. Werner, S. Pautot and R. Jordan, Biomaterials, 2016, 79, 1–14 CrossRef PubMed.
- N. Zhang, M. Steenackers, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 5345–5351 CrossRef CAS.
- D. Wang, J. Lin, F. Jia, X. Tan, Y. Wang, X. Sun, X. Cao, F. Che, H. Lu, X. Gao, J. C. Shimkonis, Z. Nyoni, X. Lu and K. Zhang, Sci. Adv., 2019, 5, eaav9322 CrossRef CAS PubMed.
- G. Charmi, M. Rahimi, K. Socha, D. A. Pham, L. Séguy, Q. T. Phan, F. Moldovan, M. Kozanecki, K. Matyjaszewski, X. Banquy and J. Pietrasik, Polym. Chem., 2023, 14, 3827–3833 RSC.
- J. Svoboda, O. Sedláček, T. Riedel, M. Hrubý and O. Pop-Georgievski, Biomacromolecules, 2019, 20, 3453–3463 CrossRef CAS PubMed.
- D. Xiong, Y. Deng, N. Wang and Y. Yang, Appl. Surf. Sci., 2014, 298, 56–61 CrossRef CAS.
- R. E. Baier, J. Mater. Sci.: Mater. Med., 2006, 17, 1057–1062 CrossRef CAS PubMed.
- Q. A. Besford, Soft Matter, 2024, 20, 3577–3584 RSC.
- V. Gopinath, S. Saravanan, A. R. Al-Maleki, M. Ramesh and J. Vadivelu, Biomed. Pharmacother., 2018, 107, 96–108 CrossRef CAS.
- J. L. Ivy, Sports Med., 1991, 11, 6–19 CrossRef CAS PubMed.
- S. Ghosh, G. G. Thakurta and K. L. Mukherjee, Indian J. Med. Res., 1989, 90, 147–153 CAS.
- P. Mergenthaler, U. Lindauer, G. A. Dienel and A. Meisel, Trends Neurosci., 2013, 36, 587–597 CrossRef CAS PubMed.
- M. M. Adeva-Andany, M. Gonzalez-Lucan, C. Donapetry-Garcia, C. Fernandez-Fernandez and E. Ameneiros-Rodriguez, BBA Clin., 2016, 5, 85–100 CrossRef PubMed.
- P. Patra, A. P. Rameshbabu, D. Das, S. Dhara, A. B. Panda and S. Pal, Polym. Chem., 2016, 7, 5426–5435 RSC.
- M. Jiratova, A. Pospisilova, M. Rabyk, M. Parizek, J. Kovar, A. Galisova, M. Hruby and D. Jirak, Drug Delivery Transl. Res., 2018, 8, 73–82 CrossRef CAS.
- Q. A. Besford, F. Cavalieri and F. Caruso, Adv. Mater., 2020, 32, e1904625 CrossRef PubMed.
- C. E. Zois and A. L. Harris, J. Mol. Med., 2016, 94, 137–154 CrossRef CAS PubMed.
- C. Li and Z. Hu, FASEB J., 2020, 34, 3–15 CrossRef CAS PubMed.
- Q. A. Besford, A. C. G. Weiss, J. Schubert, T. M. Ryan, M. F. Maitz, P. P. Tomanin, M. Savioli, C. Werner, A. Fery, F. Caruso and F. Cavalieri, ACS Appl. Mater. Interfaces, 2020, 12, 38976–38988 CrossRef CAS.
- Z. Wang, Q. Liu, L. Wang, R. G. Gilbert and M. A. Sullivan, Carbohydr. Polym., 2021, 261, 117887 CrossRef CAS PubMed.
- K. Hata, M. Hata, M. Hata and K. Matsuda, J. Jpn. Soc. Starch Sci., 1983, 30, 88–94 CrossRef CAS.
- R. Liu, S. K. Boehlein, W. F. Tracy, M. F. R. Resende Jr. and G. A. Hudalla, Molecules, 2020, 25, 637 CrossRef CAS.
- D. K. Božanić, S. Dimitrijević-Branković, N. Bibić, A. S. Luyt and V. Djoković, Carbohydr. Polym., 2011, 83, 883–890 CrossRef.
- T. M. Abdelghany, A. M. H. Al-Rajhi, M. S. Almuhayawi, E. Abada, M. A. Al Abboud, H. Moawad, R. Yahya and S. Selim, Biomass Convers. Biorefin., 2022, 13, 431–443 CrossRef.
- X. Li, X.-X. Chen, Y. Xu, X.-B. Xu, W.-F. Wu, Q. Zhao and J.-N. Hu, J. Biol. Macromol., 2022, 23, 409–423 CAS.
- L. Qiu, J. Wang, M. Conceição, S. Liu, M. Yang, W. Chen, M. Long, X. Cheng, M. J. A. Wood and J. Chen, Int. J. Biol. Macromol., 2023, 239, 124363 CrossRef CAS PubMed.
- X. Zhang, J. Zhou, H. Ying, Y. Zhou, J. Lai and J. Chen, ACS Sustainable Chem. Eng., 2020, 8, 2106–2114 CrossRef CAS.
- M. Rabyk, M. Hruby, M. Vetrik, J. Kucka, V. Proks, M. Parizek, R. Konefal, P. Krist, D. Chvatil, L. Bacakova, M. Slouf and P. Stepanek, Carbohydr. Polym., 2016, 152, 271–279 CrossRef CAS PubMed.
- R. Xu, S. K. Bhangu, K. C. Sourris, D. Vanni, M. A. Sani, J. A. Karas, K. Alt, B. Niego, A. Ale, Q. A. Besford, B. Dyett, J. Patrick, I. Carmichael, J. E. Shaw, F. Caruso, M. E. Cooper, C. E. Hagemeyer and F. Cavalieri, Adv. Mater., 2023, 35, e2210392 CrossRef PubMed.
- M. Wojnilowicz, Q. A. Besford, Y. L. Wu, X. J. Loh, J. A. Braunger, A. Glab, C. Cortez-Jugo, F. Caruso and F. Cavalieri, Biomaterials, 2018, 176, 34–49 CrossRef CAS.
- M. F. Maitz, C. Sperling, T. Wongpinyochit, M. Herklotz, C. Werner and F. P. Seib, Nanomedicine, 2017, 13, 2633–2642 CrossRef CAS.
- X. Jiang, P. Zhang, S. Li, X. Tan, Z. Hu, B. Deng, K. Wang, C. Li, M. A. Sullivan, E. Li and R. G. Gilbert, Eur. Polym. J., 2016, 82, 175–180 CrossRef CAS.
- M. Martinez-Garcia, M. C. Stuart and M. J. van der Maarel, Int. J. Biol. Macromol., 2016, 89, 12–18 CrossRef CAS PubMed.
- S. Mahmood, U. K. Mandal, B. Chatterjee and M. Taher, Nanotechnol. Rev., 2017, 6, 355–372 CAS.
- P. J. Roach, Mol. Aspects Med., 2015, 46, 78–84 CrossRef CAS PubMed.
- B. Pan, N. Zhao, Q. Xie, Y. Li, B. R. Hamaker and M. Miao, npj Sci. Food, 2023, 7, 27 CrossRef.
- J. Lomako, W. M. Lomako and W. J. Whelan, Biochim. Biophys. Acta, 2004, 1673, 45–55 CrossRef CAS PubMed.
- H. Xu, D. Stapleton and R. M. Murphy, J. Physiol. Biochem., 2015, 71, 267–280 CrossRef CAS.
- M. van der Maarel, B. Veen, J. Uitdehaag, H. Leemhuis and L. Dijkhuizen, J. Biotechnol., 2002, 94, 137–155 CrossRef CAS.
- D. Fengel and G. Wegener, ed. R. Brown, Advances in Chemistry, Washington, 1979 Search PubMed.
- Y. Ju, H. G. Kelly, L. F. Dagley, A. Reynaldi, T. E. Schlub, S. K. Spall, C. A. Bell, J. Cui, A. J. Mitchell, Z. Lin, A. K. Wheatley, K. J. Thurecht, M. P. Davenport, A. I. Webb, F. Caruso and S. J. Kent, ACS Nano, 2020, 14, 15723–15737 CrossRef CAS.
- S. Naahidi, M. Jafari, F. Edalat, K. Raymond, A. Khademhosseini and P. Chen, J. Controlled Release, 2013, 166, 182–194 CrossRef CAS PubMed.
- M. Weber, H. Steinle, S. Golombek, L. Hann, C. Schlensak, H. P. Wendel and M. Avci-Adali, Front. Bioeng. Biotechnol., 2018, 6, 99 CrossRef PubMed.
- A. H. Schmaier and E. X. Stavrou, Res. Pract. Thromb. Haemostasis, 2019, 3, 599–606 CrossRef CAS.
- Q. Cheng, E. I. Tucker, M. S. Pine, I. Sisler, A. Matafonov, M. F. Sun, T. C. White-Adams, S. A. Smith, S. R. Hanson, O. J. McCarty, T. Renne, A. Gruber and D. Gailani, Blood, 2010, 116, 3981–3989 CrossRef CAS PubMed.
- S. Kattula, J. R. Byrnes and A. S. Wolberg, Arterioscler. Thromb. Vasc. Biol., 2017, 37, e13–e21 CrossRef CAS PubMed.
- F. Swieringa, H. M. H. Spronk, J. W. M. Heemskerk and P. E. J. van der Meijden, Res. Pract. Thromb. Haemostasis, 2018, 2, 450–460 CrossRef CAS.
- M. F. Maitz, C. Sperling, T. Wongpinyochit, M. Herklotz, C. Werner and F. P. Seib, Nanomedicine, 2017, 13, 2633–2642 CrossRef CAS PubMed.
- M. M. White, L. K. Jennings and M. P. Condry, in Platelet Protocols, ed. M. M. White, L. K. Jennings and M. P. Condry, Academic Press, San Diego, 1999 Search PubMed.
- C. B. Schimper, P. Pachschwoll, M. F. Maitz, C. Werner, T. Rosenau and F. Liebner, Front. Bioeng. Biotechnol., 2023, 11, 1152577 CrossRef PubMed.
- I. Kourtzelis, M. M. Markiewski, M. Doumas, S. Rafail, K. Kambas, I. Mitroulis, S. Panagoutsos, P. Passadakis, V. Vargemezis, P. Magotti, H. Qu, T. E. Mollnes, K. Ritis and J. D. Lambris, Blood, 2010, 116, 631–639 CrossRef CAS.
- M. F. Maitz, C. Sperling, T. Wongpinyochit, M. Herklotz, C. Werner and F. P. Seib, Nanomedicine, 2017, 13, 2633–2642 CrossRef CAS.
- Y. Ju, J. M. Carreno, V. Simon, K. Dawson, F. Krammer and S. J. Kent, Nat. Rev. Immunol., 2023, 23, 135–136 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03034f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |