DOI:
10.1039/D4NR03249G
(Paper)
Nanoscale, 2024,
16, 19756-19762
pH-Responsive self-assembled polymer-photosensitizer conjugate for activable photodynamic therapy†
Received
7th August 2024
, Accepted 22nd September 2024
First published on 23rd September 2024
Abstract
This paper reports synthesis, aqueous self-assembly and relevance of the pH-triggered activable photodynamic therapy of amphiphilic polyurethane (P1S) functionalized with a heavy-atom free organic photosensitizer. Condensation polymerization between 1,4-diisocyanatobutane and two different dihydroxy monomers (one having a pendant hydrophilic wedge and the other having 1,3-dihydroxypropan-2-one with a reactive carbonyl group) in the presence of a measured amount of (S)-2-methylbutan-1-ol (chain-stopper) and DABCO catalyst produces a reactive pre-polymer P1. Hydrazide-functionalized thionated-naphthalenemonoimide (NMIS), which acts as a photosensitizer, reacted with the carbonyl-functionality of P1 to obtain the desired polymer-photosensitizer conjugate P1S in which the dye was attached to the polymer backbone via an acid-labile hydrazone linker. In water, P1S adopted an intra-chain H-bonding stabilized folded structure, which further assembled to produce a polymersome structure (Dh ≈ 200 nm), in which the hydrophobic membrane consists of aggregated NMIS and trialkoxy-benzene chromophores, as evident from UV/vis, CD and small-angle X-ray diffraction studies. In the aggregated state, NMIS loses its reactive oxygen species (ROS) generation ability and remains in a dormant state. However, under acidic conditions (pH 5.5), the photosensitizer is detached (presumably because of the cleavage of the hydrazone linker) and regains its full ROS-generation activity under photoirradiation, as evidenced from the standard DCFH assay. To test the possibility of such pH-activable intra-cellular ROS generation, P1S was treated with HeLa cells, as it is known that cancer cells are more acidic than normal cells. Indeed, photoirradiation-induced intra-cellular ROS generation was evident by the DCFH assay, resulting in efficient cell killing.
Introduction
The therapeutic efficiency of light has been known for thousands of years. Modern photodynamic therapy (PDT) has been clinically tested and used for the treatment of various types of cancers, such as skin, brain, lung, and breast cancer.1 In photodynamic therapy (PDT), a non-toxic photosensitizer2 gets activated by absorbing visible or near infrared light and transfers its energy or electron to molecular oxygen to produce reactive oxygen species, such as superoxide anions, hydrogen peroxide, oxygen radicals, or hypochlorous acid, which cause cellular death.3 Traditionally, porphyrin-based photosensitizers4 have been used as they exhibit an excellent singlet oxygen quantum yield. Despite being used clinically, they suffer from multistep synthesis, low solubility and dark cytotoxicity beyond a certain concentration within cells. To circumvent these issues, research on heavy-atom-free photosensitizers has been gaining interest recently.5 It has been reported that in organic π-conjugated chromophores, substitution of oxygen atoms with sulfur can cause complete fluorescence quenching and leads to an excellent singlet oxygen quantum yield,6 even in the tumor hypoxic environment.7 Although these systems are promising for future biomedical applications, suitable strategies for making activable-photosensitizers8 may be highly beneficial because they provide secondary shielding if the photosensitizer becomes active by a specific biological stimulus, rather than just light activation. Such systems may be useful for tissue specific on-demand delivery of photosensitizers and for reducing phototoxicity in healthy tissue. Recently, this concept was tested with a few organic small-molecule photosensitizers by our group and others.9 Generally, a photosensitizer is non-covalently sequestered in an aggregated system, causing its inactivation. With a specific external stimulus, disassembly of the aggregate causes the release of the photosensitizer in its active form. However, non-covalent encapsulation or aggregation of small molecules may not be adequately stable under biological conditions and may cause undesired leakage similar to the problems encountered for non-covalent encapsulation-based drug delivery systems.10 Instead, we envisaged polymer-photosensitizer conjugate (similar to polymer–drug conjugates)11 might be a better approach if events such as (i) easy covalent conjugation, (ii) aggregation of the polymer-photosensitizer conjugate and inactivation of the photosensitizer and (iii) stimuli-responsive release and re-activation of the photosensitizer can be achieved using a single system.
Polymeric-aggregated systems are also attractive for target specific cellular uptake due to the well-known EPR effect.12 To test these possibilities, we develop polymer-photosensitizer conjugate P1S (Scheme 1), consisting of a hydrophobic polyurethane (PU) backbone and pendant hydrophilic oligo-oxyethylene wedge and the thionated derivative of naphthalene-monoimide (NMIS), which is linked by an acid labile hydrazone linker.13 NMIS is known to be a good photosensitizer but becomes dormant in the aggregated state.9a Recently, we showed that similar PU derivatives exhibit chain-folding regulated hierarchical assembly,14 producing the polymersome structure.15 Similarly, we envisaged that P1S would self-assemble (Scheme 1), leading to the segregation of the NMIS chromophores in the hydrophobic aromatic domain of the aggregate, which in turn would take away its ability to produce ROS. However, in acidic pH, cleavage of the labile hydrazone linker should release the photosensitizer in its active form (Scheme 1). This may be particularly useful for target specific delivery as cancer tissue has a lower pH compared to that of healthy cells.16 With this proposition, herein, we describe the synthesis of polymer-photosensitizer conjugate P1S, self-assembly leading to polymersome structure and its utility as a pH-responsive activable photosensitizer system.
 |
| | Scheme 1 Synthesis of activable polymer-photosensitizer conjugate (P1S) and the control polymer (P1O); its chain-folding regulated polymersome assembly and pH-triggered release of the active photosensitizer and ROS generation upon photoirradiation. Reagents and conditions: (a) DABCO, THF, 65 °C, 12 h, 79%. (b) NMIS-NHNH2 or NMIO-NHNH2, DMF, 70 °C, 12 h, quantitative. | |
Results and discussion
Synthesis
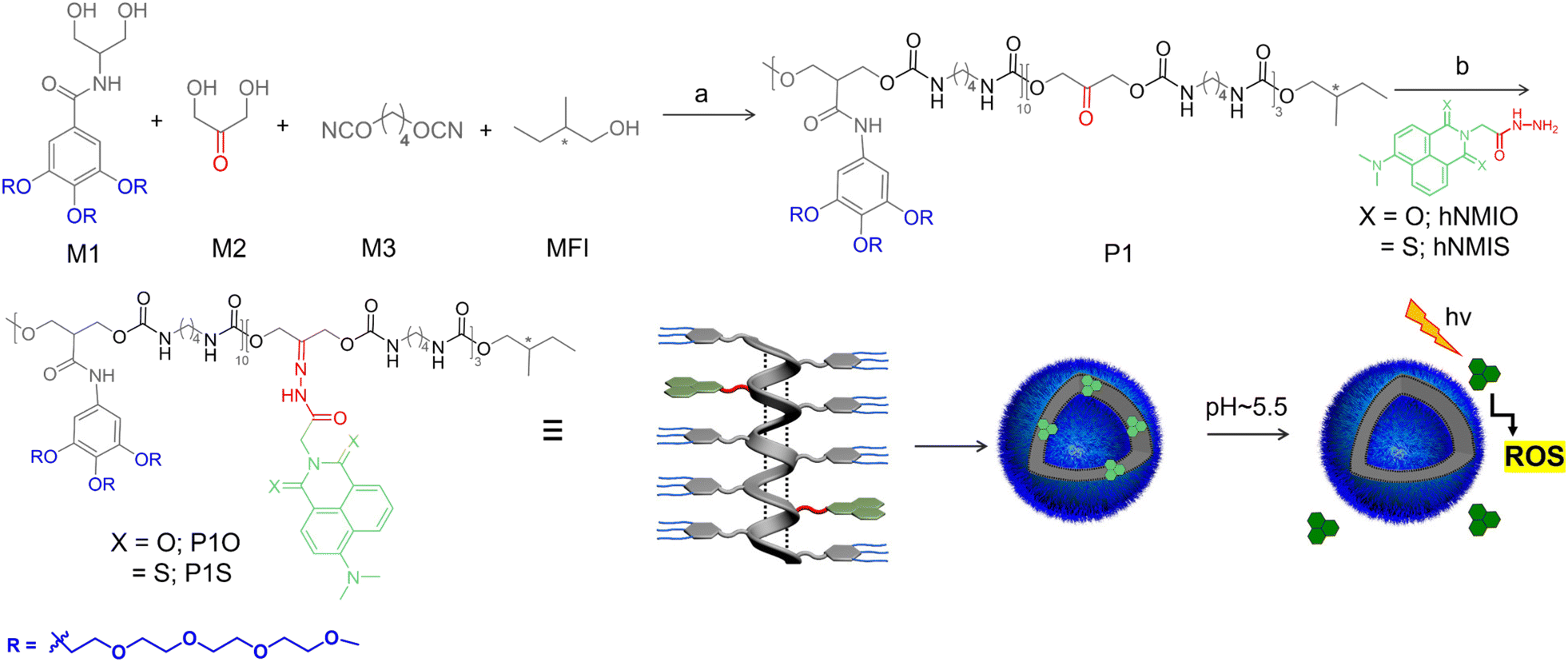
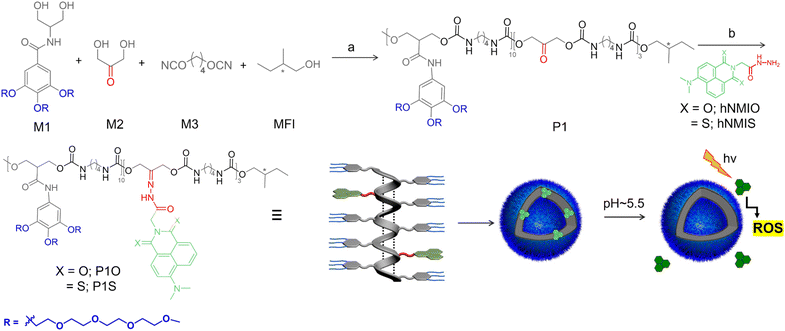
The multi-step synthesis of P1S is shown in Scheme 1. To synthesize the parent polymer P1, a commercially available hydrophobic di-isocyanate (M3) was reacted with a mixture of two di-hydroxy compounds (M1 and M2) in the presence of catalyst 1,4-diazabicyclo [2.2.2] octane (DABCO) and a monofunctional impurity (MFI) (−) S-2-methyl butanol at 65 °C in DMAc. The ratio of M1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M2:M3:MFI = 0.7:0.3:1.1:0.02 was maintained to ensure that (i) about 30% (with respect to the total di-hydroxy compounds) of the reactive monomer is incorporated in the polymer chain and (ii) all chains are terminated with the chiral chain-stopper to achieve a predictable degree of polymerization.17 The chiral chain-stopper also helps in probing the chain-folding by CD spectroscopy.18 P1 was isolated as a white sticky solid in 79% yield and could be characterized unambiguously by 1H NMR spectroscopy (Fig. S1†). Size exclusion chromatography (Fig. S2†) revealed Mn = 9000 g mol−1 (Đ = 1.2), which corroborated with the theoretically predicted value (∼10000 g mol−1). Hydrazide-functionalized thionated-naphthalene derivative hNMIS (Scheme S1†) was synthesized in a few steps (see ESI† for detail) and reacted with P1 to obtain the desired polymer-photosensitizer conjugate (P1S, Scheme 1). As a control system, hNMIO (Scheme S1†), lacking any sulfur substitution, was also prepared and attached with P1 in a similar way to produce P1O (Scheme 1). Post-polymer conjugation19 was confirmed by 1H NMR spectra of the polymers (Fig. S3 and S4†), clearly showing the presence of aromatic peaks for the attached chromophores. UV/vis spectra of P1S and P1O are shown in Fig. 1 along with those of the free dye.
:M2:M3:MFI = 0.7:0.3:1.1:0.02 was maintained to ensure that (i) about 30% (with respect to the total di-hydroxy compounds) of the reactive monomer is incorporated in the polymer chain and (ii) all chains are terminated with the chiral chain-stopper to achieve a predictable degree of polymerization.17 The chiral chain-stopper also helps in probing the chain-folding by CD spectroscopy.18 P1 was isolated as a white sticky solid in 79% yield and could be characterized unambiguously by 1H NMR spectroscopy (Fig. S1†). Size exclusion chromatography (Fig. S2†) revealed Mn = 9000 g mol−1 (Đ = 1.2), which corroborated with the theoretically predicted value (∼10000 g mol−1). Hydrazide-functionalized thionated-naphthalene derivative hNMIS (Scheme S1†) was synthesized in a few steps (see ESI† for detail) and reacted with P1 to obtain the desired polymer-photosensitizer conjugate (P1S, Scheme 1). As a control system, hNMIO (Scheme S1†), lacking any sulfur substitution, was also prepared and attached with P1 in a similar way to produce P1O (Scheme 1). Post-polymer conjugation19 was confirmed by 1H NMR spectra of the polymers (Fig. S3 and S4†), clearly showing the presence of aromatic peaks for the attached chromophores. UV/vis spectra of P1S and P1O are shown in Fig. 1 along with those of the free dye.
 |
| | Fig. 1 (a) UV-vis and (b) fluorescence spectra of hNMIS, hNMIO, P1S and P1O in THF (concentration of P1S/P1O = 0.5 mg ml−1; hNMIO/hNMIS = 0.15 mM; l = 1 cm). | |
For hNMIO, a clear absorption band is visible (350–450 nm), which exhibits significant bathochromic shift and tails up to the near IR region in hNMIS as a consequence of the sulfur-substitution.6a Similarly, in the P1S spectra (Fig. 1a), a prominent absorption band is observed with λmax = 470 nm, which confirms the successful attachment of the photosensitizer to the polymer backbone. From the intensity of the band λmax = 450 nm and using the extinction coefficient of hNMIS (Fig. S5†), the amount of attached dye was estimated to be ∼30 wt% in P1S. Emission spectra of hNMIO or the P1O showed a strong emission band (450–600 nm), which quenched completely in hNMIS or P1S due to the excited state electron transfer to the triplet state,9a as previously established in the literature for such thionated photosensitizers.20
pH-Responsive self-assembly and ROS generation
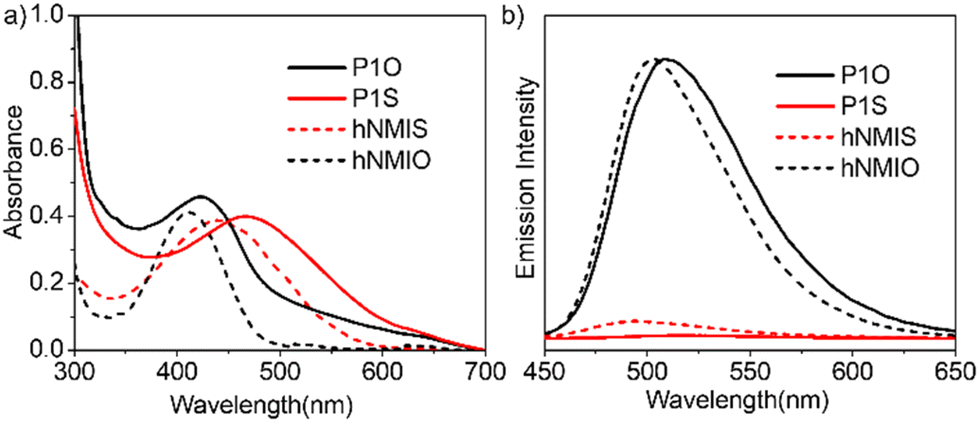
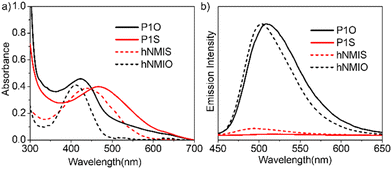
Self-assembly of P1S was tested in water. The transmission electron microscopy image (Fig. 2a) showed the presence of hollow spherical structures (diameter about 200 nm), indicating polymersome morphology. Dynamic light scattering studies (Fig. 2b) showed a single sharp peak, revealing a hydrodynamic diameter of 230 nm, which approximately matches the size obtained from the TEM. The presence of a hydrophilic lacuna, as should be the case for the polymersome structure, was examined by encapsulating the water-soluble dye calcein in P1S, which exhibits self-quenching properties. UV/vis spectra of calcein-treated P1S, after removing any free dye by dialysis, showed a prominent absorption band in the range of 400–550 nm (Fig. S6†), confirming successful dye encapsulation. The absorption-matched fluorescence spectra (Fig. 2c) showed significant quenching of the emission intensity compared to the free dye solution. When encapsulated in the confined water inside the vesicle, its actual concentration increases compared to that in a free aqueous solution. Hence, the absorption-normalized emission intensity shows a significant reduction for the encapsulated dye, which in turn is taken as indirect proof of its location in the confined water inside the polymersome.21
 |
| | Fig. 2 (a) TEM image (c = 0.1 mg mL−1) and (b) DLS of P1S in water (c = 0.1 mg mL−1). (c) Absorption normalized emission spectra (λex = 450 nm) of calcein encapsulated in P1S and free calcein in water. (d) Fluorescence microscopy image of the calcein-encapsulated P1S polymersome. | |
It is postulated that the intra-chain H-bonded folding (Scheme 1) leads to a pleated structure that assembles to produce the polymersome, similar to what has been reported by us for related amphiphilic polyurethane derivatives before.14 Small-angle powder-XRD of the drop-cast sample, prepared from an aqueous solution of P1S, showed a prominent peak at 2θ = 1.6° (d = 54.4 Å), which roughly matches the width of the folded chain from its energy minimized structure (Fig. S7†), and thus supports the model.
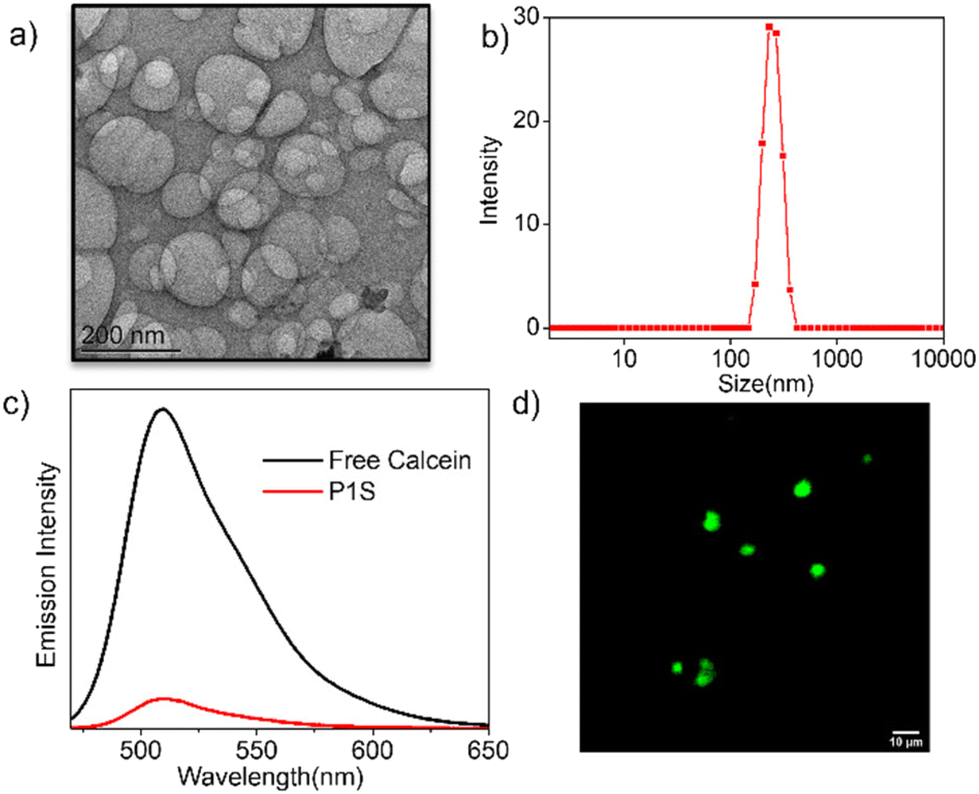
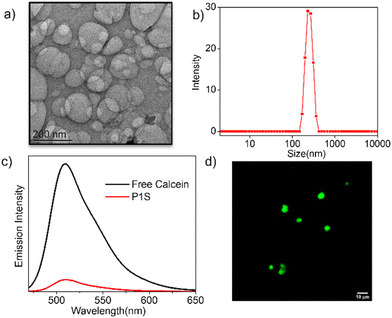
Furthermore, the CD spectra of P1S in water showed (Fig. 3a) an intense band in the region of 200–325 nm and a bisignated CD signal in the range of 400–550 nm corresponding to the absorption of the phenyl-ring and the NMIS chromophores, respectively. These results further support the proposed chain-folding, which brings the tri-alkoxy-benzene rings and the NMIS chromophores within the stacking distance, and the chirality is induced from the chiral chain-stopper. The aggregation of the NMIS chromophore is also evident from the UV/vis spectra (Fig. 3b), showing significant bathochromic shift with concomitant intensity-reduction and broadening of the absorption band going from THF to water. We further tested the pH-responsive release of the NMIS dye by UV/vis spectroscopy (Fig. 3c and d). The P1S polymersome sample was placed in a dialysis bag and treated with a pH 5 buffer solution. Aliquot was taken periodically from the dialysis bag and analyzed using UV/vis spectroscopy. A sharp decrease in the band intensity clearly indicates the removal of the dye. % of release reached up to ∼70% in about 1 h and then saturated. In a control experiment performed at pH 7.0, no such decrease in the band intensity was observed (Fig. S8† and Fig. 3d), confirming the pH-selective release of the NMIS dye, which is expected considering the acid-labile nature of the hydrazone-linker. Now, to check the impact of aggregation and pH responsive release of the NMIS dye on the ROS generation, the DCFH-assay22 was performed.
 |
| | Fig. 3 (a) CD and (b) UV/vis spectra of P1S in water and THF. (c) Time-dependent UV/vis spectra of P1S at pH 5 buffer solution. Each spectrum was taken at 15 min time intervals. (d) %release of NMIS from aqueous solution of P1S solution at pH 5 or pH 7 buffer (concentration of P1S = 1.0 mg mL−1). | |
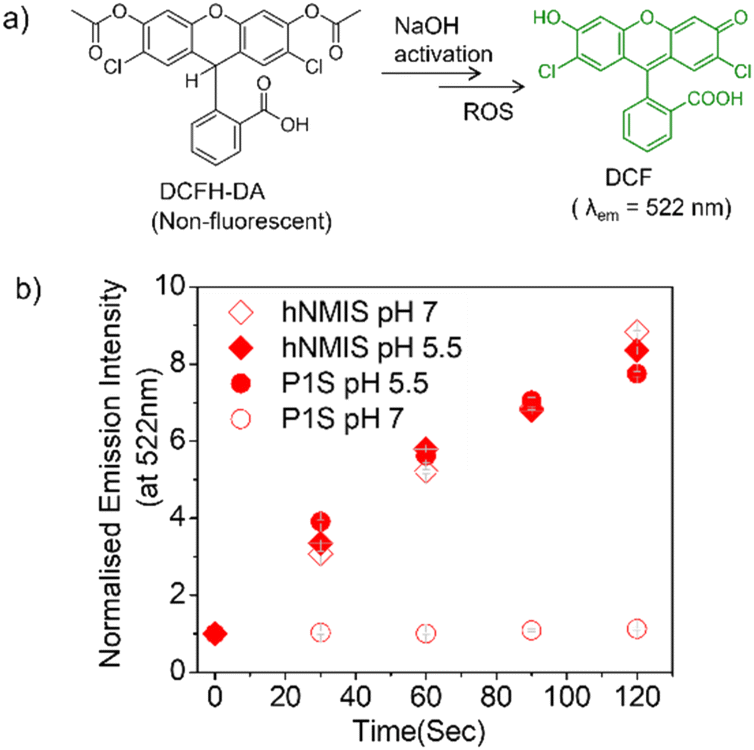
Commercially available DCFH-DA (2,7-dichlorofluorescein diacetate) (Fig. 4a) was pretreated with NaOH to produce active DCFH, a weak fluorophore. In the presence of ROS, DCFH is oxidized to DCF, which is a strongly emissive dye with an emission of around 520 nm. Therefore, for a given photosensitizer, monitoring the emission spectra of the so-produced DCF as a function of time enables probing the ROS generation. First, the experiment was conducted with the free hNMIS, which showed about a 9-fold increase in the fluorescence intensity in ∼2 min (Fig. S9† and Fig. 4b), confirming the ROS generation ability of the dye. However, in the presence of P1S, no such increase in the fluorescence intensity was observed at pH 7 (Fig. 4b and S9†), indicating that when attached to the polymer backbone and aggregated, the dye goes to the dormant state and loses its ROS-generation ability. In contrast, at pH 5, a sharp change in the fluorescent intensity was observed (Fig. 4b and S9†), and the increase in the band intensity was almost comparable to that of the free dye. This clearly reveals that under acidic pH, when the photosensitizer is detached from the polymer backbone (Fig. 3d), it regains its ROS-generation ability upon photoirradiation.
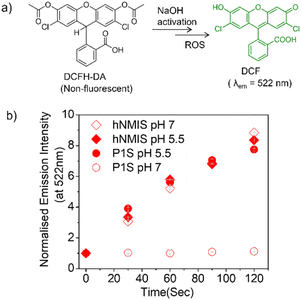 |
| | Fig. 4 (a) Reaction scheme of the DCFH assay. (b) Variation of normalized DCF emission intensity in aqueous solution of P1S (pH = 5/7), and free NMIS after light irradiation (λ = 550 nm). Concentration of P1S = 1 mg mL−1. Error bars indicate the mean of three different independent experiments (±S.E.M.). | |
Biological studies
Having achieved the pH-triggered ROS-generation ability of P1S, we sought to examine its ROS-generation efficiency in cancer cells. This is particularly relevant because it is known that the microenvironment of cancer cells is acidic compared to that of healthy cells.16
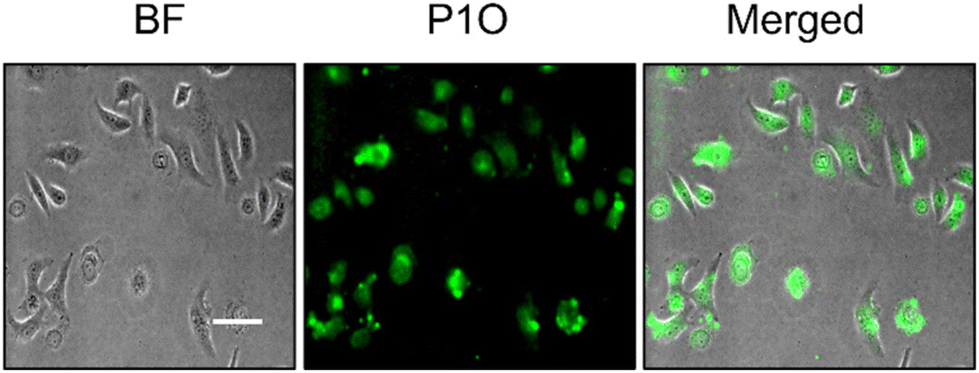
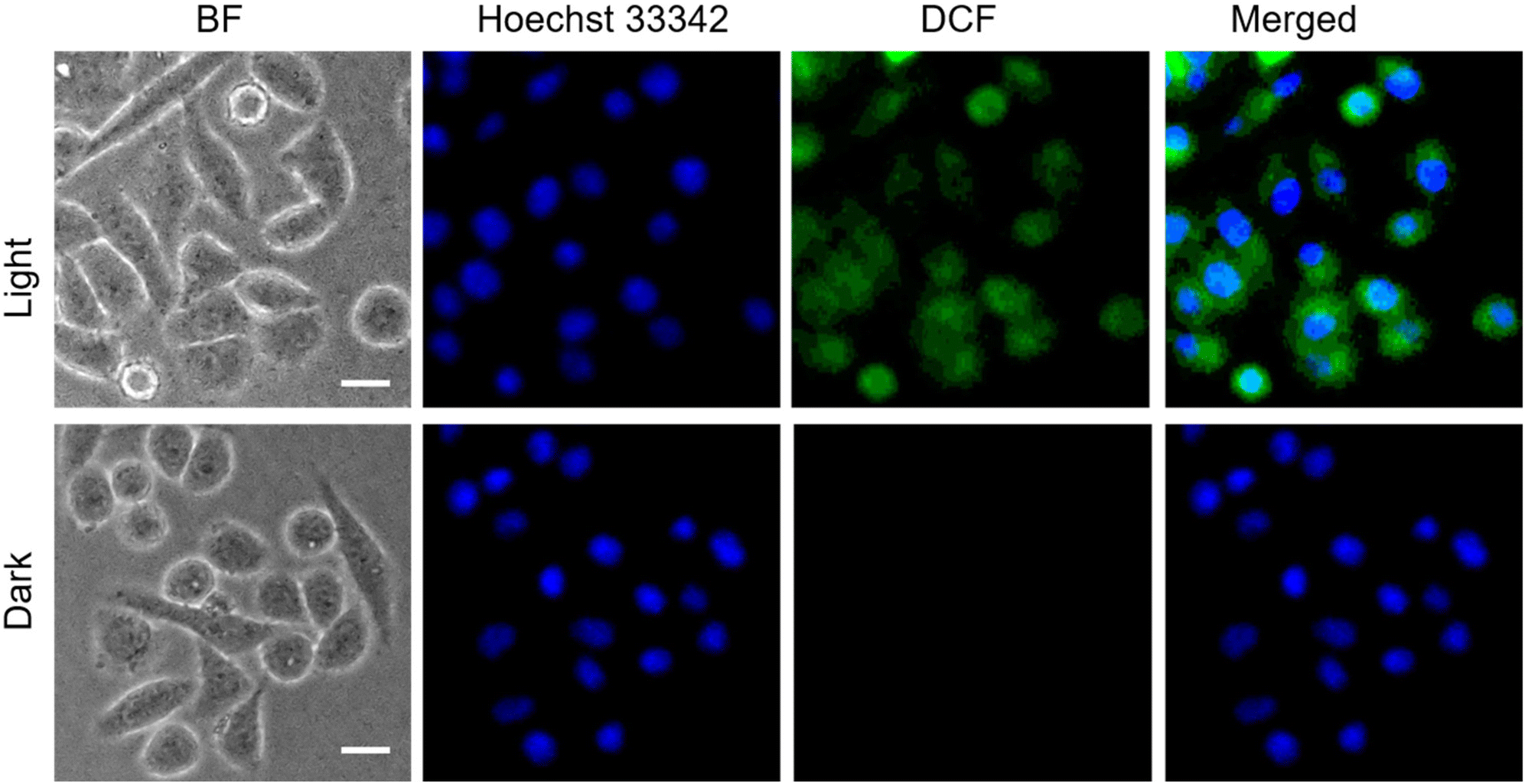
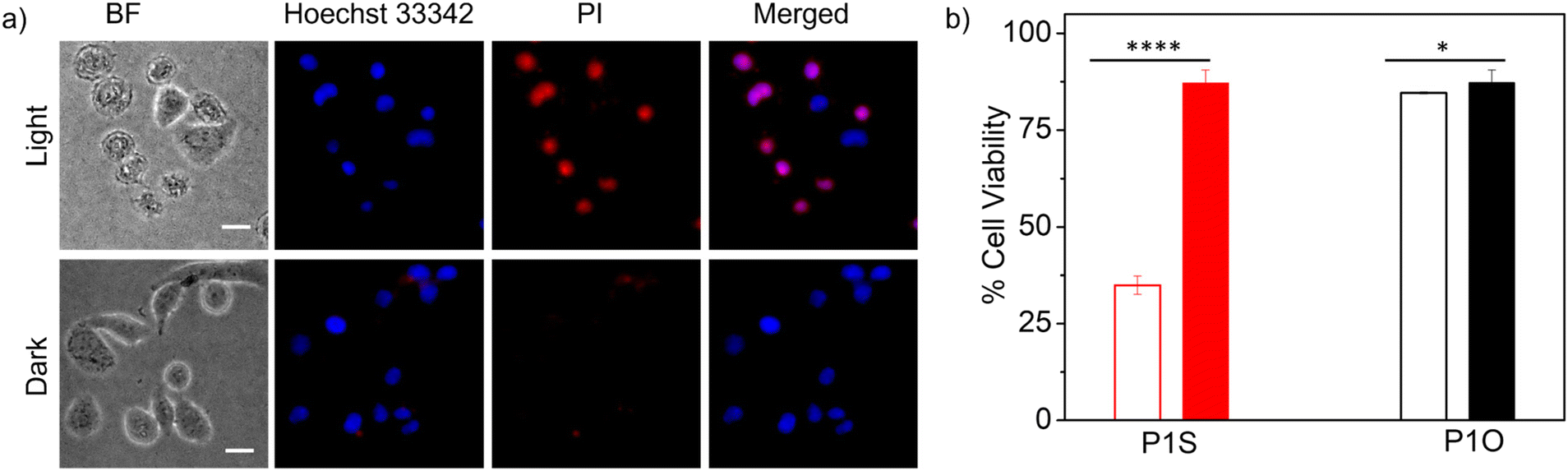
Prior to testing this, we wanted to examine the cellular uptake of P1O, which is structurally analogous to P1S but green emitting (Fig. 1). DLS studies of P1O (Fig. S10†) showed a sharp peak with a hydrodynamic diameter of 225 nm, which was comparable to that of P1S, thus ensuring that both polymers produced similar aggregates. P1O was treated with HeLa cells at different time intervals and lysed; then, the fluorescence emission intensity of the cell lysis solutions was measured. It was observed (Fig. S11†) that at 6 h, the cell solution showed the highest green emission, and after that, it remained constant for 12 h (Fig. S11†). Fluorescence microscopy images of P1O-treated cells (after 6 h) showed intense green emission (Fig. 5), further confirming its cellular uptake. Hence, for all subsequent experiments with P1S, a 6 h incubation time was selected. The intracellular ROS-generation ability of P1S was probed using the DCFH assay. For this, P1S-treated HeLa cells were added with DCFH-DA, and fluorescence microscopy experiments were conducted after photoirradiation. Fig. 6 shows intense green emission from the post-irradiated cells, whereas in the dark no such green emission emerged, confirming intra-cellular ROS generation. To examine the impact on cell killing, P1S-treated cells after photoirradiation were further assessed by the propidium iodide (PI) assay (PI).23 PI can easily penetrate through the pores of the dead-cell membrane and shows red emission after binding with the nuclear DNA. Fluorescence microscopy images (Fig. 7a) of P1S-treated HeLa cells upon photoirradiation and PI treatment show intense red-emission from the PI-labelled nucleus, which is absent for light-untreated cells. This confirms that ROS induces cell-killing by the activable P1S. Furthermore, P1S-treated (6 h) HeLa cells were irradiated with light (λ = 560–590 nm) and examined by the MTT assay (Fig. 7b), which showed merely <30% cell viability. In contrast, without light irradiation, ∼85% cell viability was noticed. Furthermore, >80% cell viability was observed (even after light irradiation) for P1O-treated cells under identical conditions. These results firmly establish that cell killing does not occur due to any intrinsic toxicity of the polymer/cleaved polymer or only by light irradiation; however, it occurs explicitly due to the light-induced intra-cellular ROS generation by P1S.
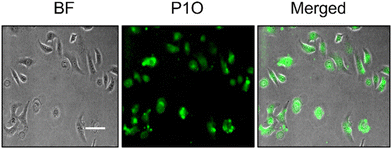 |
| | Fig. 5 Fluorescence microscopy image of HeLa cells after 6 h incubation with P1O (concentration = 1 mg mL−1). Scale bar = 10 μm. | |
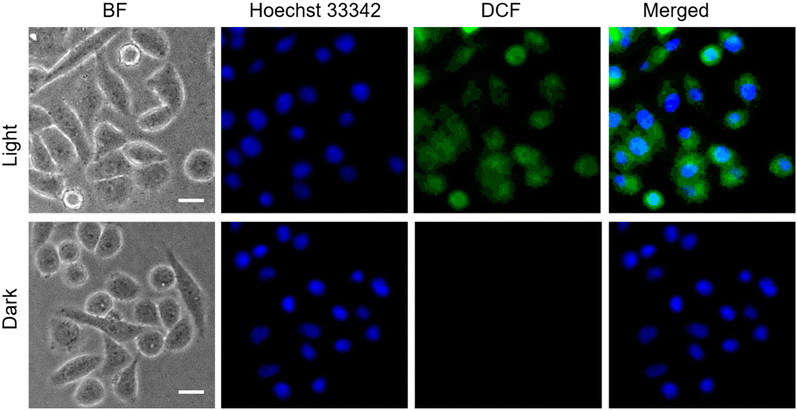 |
| | Fig. 6 ROS generation assay (DCFH assay) with HeLa cells after incubation with P1S for 6 h in the presence of light (λ = 560–590 nm), (concentration of P1S = 1.0 mg mL−1), scale bar = 10 μm. | |
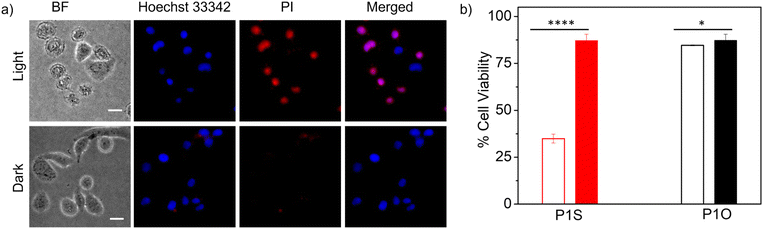 |
| | Fig. 7 (a) Propidium iodide (PI) assay in HeLa cells after incubation with P1S for 6 h in the presence of light (λ = 560–590 nm) or in the dark. Concentration of P1S = 1.0 mg mL−1, scale bar = 10 μm. (b) Cell viability (MTT assay) of HeLa cells after incubation with P1S for 6 h, followed by light irradiation for 1 h or dark (empty bar represents post photo irradiated sample and filled bar represents non-irradiated one). Each experiment was repeated three times with three replicates in each case. Data are represented as mean ± S.E.M. ****P < 0.0001, *P > 0.1 obtained using an unpaired t test. In the case of P1O, the same cleaved polymer should be formed inside cells, but it shows almost >90% cell viability both in light and the dark, suggesting lack of toxicity of the cleaved polymer in our working concentration. | |
Conclusions
In summary, we demonstrated an attractive strategy for pH-triggered activable photodynamic therapy (PDT) by conjugating a heavy-atom-free photosensitizer (thionated naphthalene-monoimide derivative) with an amphiphilic polyurethane (PU) via an acid-labile hydrazone linker. The PU backbone, with alternating sequences of hydrophobic and hydrophilic segments, exhibits H-bonding driven intra-chain folding in water, and the hierarchical assembly of such folded chains produces the polymersome structure. In such assembly, the photosensitizer remains in the aggregated form, which deactivates its ROS generation ability. However, under acidic pH, cleavage of the hydrazone linker leads to the release of the photosensitizer in its active form, which, upon light irradiation, generates reactive oxygen species, resulting in cell killing. At a neutral pH, the same photosensitizer remains in the dormant state. Recently, activable PDT has emerged as an important breakthrough in the field. In this context, the present work may be of significant importance because (i) the dormant photosensitizer-embedded polymersome can be useful for targeted delivery by taking advantage of the EPR effect; (ii) polyurethane is known to be a biodegradable and biocompatible polymer; and (iii) cancer cells have a lower pH than normal cells. Furthermore, the polymersome can also be very useful for the non-covalent encapsulation of hydrophilic or hydrophobic drug molecules to achieve simultaneous PDT and chemotherapy. Decorating the surface of such uni-lamellar polymersome with ligands for specific cell targeting is within the reach of synthetic modification of the polymer structure. Such possibilities are currently being explored in our laboratory along with testing the scope for in vivo studies.
Author contributions
T. B. carried out synthesis and self-assembly studies. K. D. performed cellular work and raised research funding. S. G. conceptualized and supervised the project and raised research funding. The manuscript was written through contributions of all authors.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
TB thanks IACS, Kolkata, for a research fellowship. KD thanks DST, India (DST/WOSA/CS-141/2021) for financial support. SG thanks TRC, IACS for financial support.
References
-
(a) D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS;
(b) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS;
(c) R. A. Akasov, N. V. Sholina, D. A. Khochenkov, A. V. Alova, P. V. Gorelkin, A. S. Erofeev, A. N. Generalova and E. V. Khaydukov, Sci. Rep., 2019, 9, 9679 CrossRef CAS;
(d) X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC;
(e) H. Zou, F. Wang, J.-J. Zhou, X. Liu, Q. He, C. Wang, Y.-W. Zheng, Y. Wen and L. Xiong, J. Gastrointest. Oncol., 2020, 11, 431–442 CrossRef PubMed;
(f) Y. Li, Y. Ding, Y. Zhang, Z. Sun, J. Liu, M. Dai, J. Feng, B. Li, C. Wang, Y. Wei and J. W. Shen, Nanoscale, 2023, 15, 10067–10078 RSC;
(g) M. Zhang, X. Qin, W. Xu, Y. Wang, Y. Song, S. Garg and Y. Luan, J. Colloid Interface Sci., 2021, 594, 493–501 CrossRef CAS PubMed.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS.
- M. Tampa, M.-I. Sarbu, C. Matei, C.-I. Mitran;, M.-I. Mitran, C. Caruntu, C. Constantin, M. Neagu and S.-R. Georgescu, Oncol. Lett., 2019, 17, 4085–4093 CAS.
-
(a) A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed;
(b) J. Kou, D. Dou and L. Yang, Oncotarget, 2017, 8, 81591–81603 CrossRef PubMed.
-
(a) V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS;
(b) V. Nguyen, Y. Yim, S. Kim, B. Ryu, K. M. K. Swamy, G. Kim, N. Kwon, C. Kim, S. Park and J. Yoon, Angew. Chem., Int. Ed., 2020, 59, 8957–8962 CrossRef CAS PubMed.
-
(a) J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701–6708 RSC;
(b) L. A. Ortiz-Rodríguez and C. E. Crespo-Hernández, Chem. Sci., 2020, 11, 11113–11123 RSC.
- M. Li, Y. Xu, X. Peng and J. S. Kim, Acc. Chem. Res., 2022, 55, 3253–3264 CrossRef CAS.
-
(a) J. Tian, B. Li, F. Zhang, Z. Yao, W. Song, Y. Tang, Y. Ping and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307288 CrossRef CAS;
(b) Z. Li, Z. Zhou, Y. Wang, J. Wang, L. Zhou, H.-B. Cheng and J. Yoon, Coord. Chem. Rev., 2023, 493, 215324–215352 CrossRef CAS;
(c) B. Yuan, H. Wang, J.-F. Xu and X. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 26982–26990 CrossRef CAS.
-
(a) T. Banerjee, K. Dan, A. K. Pal, R. Bej, A. Datta and S. Ghosh, ACS Macro Lett., 2023, 12, 928–934 CrossRef CAS PubMed;
(b) J. Yin, C. Ouyang, S. Shen, Y. Zhou, G. He, H. Zhang, K. Zhou, G. Chen and L. Ren, Mol. Pharm., 2022, 19, 2441–2455 CrossRef CAS PubMed;
(c) D. Fu, Y. Wang, K. Lin, L. Huang, J. Xu and H. Wu, RSC Adv., 2023, 13, 22367–22374 RSC;
(d) M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505–5616 CrossRef CAS.
-
(a) B. S. Bolu, R. Sanyal and A. Sanyal, Molecules, 2018, 23, 1570 CrossRef;
(b) H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS;
(c) K. Ulbrich, K. Hola, V. Subr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed;
(d) R. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS;
(e) S. Mukherjee and R. Shunmugam, J. Nanomed. Res., 2017, 5, 1–12 Search PubMed.
-
(a) Y. Liu, G. Yang, S. Jin, L. Xu and C. X. Zhao, ChemPlusChem, 2020, 85, 2143–2157 CrossRef CAS;
(b) I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed;
(c) R. Bej and S. Ghosh, Bioconjugate Chem., 2019, 30, 101–110 CrossRef CAS PubMed;
(d) S. Gong, J. Qiu and S. Thayumanavan, J. Am. Chem. Soc., 2024, 146, 33–38 CrossRef CAS PubMed;
(e) I. Altinbasak, M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 7603–7624 RSC;
(f) B. Saha, U. Haldar and P. De, Macromol. Rapid Commun., 2016, 37, 1015–1020 CrossRef CAS PubMed;
(g) S. Mukherjee, D. Patra, T. K. Dash, I. Chakraborty, R. Bhattacharyya, S. Senapati and S. Shunmugam, Polym. Chem., 2019, 10, 3066–3078 RSC.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef.
-
(a) Y. Bae, N. Nishiyama, S. Fukushima, H. Koyama, M. Yasuhiro and K. Kataoka, Bioconjugate Chem., 2005, 16, 122–130 CrossRef CAS;
(b) N. Vijaykameswara Rao, M. Shivshankar, K. Abhinoy, J. Das Sarma and R. Shunmugam, Biomacromolecules, 2012, 13, 221–230 CrossRef.
- R. Barman, A. Mukherjee, A. Nag, P. Rajdev and S. Ghosh, Chem. Commun., 2023, 59, 13951–13961 RSC.
-
(a) H. De Oliveira, J. Thevenot and S. Lecommandoux, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 525–546 CAS;
(b) F. R. Wurm, K. Landfester and P. Schwille, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
-
(a) B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS;
(b) E. Boedtkjer and S. F. Pedersen, Annu. Rev. Physiol., 2020, 82, 103–126 CrossRef CAS PubMed.
-
(a) M. L. Chabinyc and C. J. Hawker, Macromolecules, 2013, 46, 6431 CrossRef.
- A. Nag, K. Banerjee, R. Barman, J. Kar, D. P. Sarkar, S. S. Jana and S. Ghosh, J. Am. Chem. Soc., 2023, 145, 579–584 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- V. N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed.
-
(a) B. Kalyanaraman, V. Darley-Usmar, K. J. A. Davies, P. A. Dennery, H. J. Forman, M. B. Grisham, G. E. Mann, K. Moore, L. J. Roberts and H. Ischiropoulos, Free Radical Biol. Med., 2012, 52, 1–6 CrossRef CAS PubMed;
(b) M. J. Reiniers, R. F. Van Golen, S. Bonnet, M. Broekgaarden, T. M. Van Gulik, M. R. Egmond and M. Heger, Anal. Chem., 2017, 89, 3853–3857 CrossRef CAS PubMed.
-
(a) S. Kasibhatla, G. P. Amarante-Mendes, D. Finucane, T. Brunner, E. Bossy-Wetzel and D. R. Green, Cold Spring Harb. Protoc., 2006, 4495 CrossRef, pdb. Prot.;
(b) H. Zhao, J. Oczos, P. Janowski, D. Trembecka, J. Dobrucki, Z. Darzynkiewicz and D. Wlodkowic, Cytometry, Part A, 2010, 77, 399–405 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the polymers, materials and methods, additional self-assembly data and experimental detail. See DOI: https://doi.org/10.1039/d4nr03249g |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*