
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon-supported Au25 cluster catalysts partially decorated with dendron thiolates: enhanced loading weight and durability for hydrogen evolution reaction†
Kosuke
Sakamoto
,
Shinya
Masuda
 ,
Shinjiro
Takano
and
Tatsuya
Tsukuda
*
,
Shinjiro
Takano
and
Tatsuya
Tsukuda
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tsukuda@chem.s.u-tokyo.ac.jp
First published on 17th October 2024
Abstract
In order to establish a design principle for efficient Au electrocatalysis, it is desirable to synthesize a highly loaded, robust, and atomically precise Au cluster catalyst on a conductive carbon support. In this work, heterogeneous Au25 catalysts were prepared by calcining 5.0 wt% of mixed ligated [Au25(D2S)x(PET)18−x]0 (D2S = second generation Fréchet-type dendron thiolate, PET = 2-phenylethanethiolate) on a carbon support. X-ray absorption fine structure analysis, powder X-ray diffraction, and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) revealed the successful synthesis of carbon-supported partially thiolated Au25 clusters by calcining [Au25(D2S)10.7(PET)7.3]0 at 425 °C for ≥12 h, whereas calcination of [Au25(PET)18]0 under the same conditions resulted in thermally induced aggregation into larger Au nanoparticles. The D2S-modified Au25 catalyst showed better durability than PET-modified Au25 in electrocatalytic hydrogen evolution reaction. The higher durability was attributed to the suppression of aggregation of Au25 clusters during the reaction, as confirmed by AC-HAADF-STEM. These results indicate that the residual D2S ligands on Au25 enhance the stability against aggregation more than the residual PET due to stronger non-covalent interactions with carbon supports and/or greater steric hindrance of dendritic structure. This work demonstrates that the stability of Au catalysts can be improved by partial decoration with designed ligands.
Introduction
Electrochemical catalysis has long been considered a promising environmentally friendly approach to producing clean energy sources and valuable fine chemicals.1–10 For example, carbon-supported platinum (Pt/C) and copper (Cu/C) catalysts are benchmark catalysts for electrocatalytic hydrogen evolution reaction (HER) and carbon dioxide reduction reaction (CO2RR), respectively, which can be used to synthesize valuable chemicals without toxic waste.11–15 The fabrication method and the catalytic mechanism have been extensively studied for the further development of efficient and novel catalysts.Among a variety of metal nanoparticles (NPs) used for electrocatalysis, gold NPs have recently attracted interest due to their high selectivity toward CO in CO2RR16–19 and less poisoning of the active sites by the formed CO.18,20 For the development of highly efficient Au electrocatalysts, it is desirable to increase the Au surface area by loading surface-rich Au nanoclusters with high density onto a conductive carbon support.21,22 However, it is challenging to synthesize Au cluster catalysts by the conventional methods such as deposition–precipitation and impregnation, due to the intrinsic instability of the clusters against aggregation, especially on the carbon supports due to their weak interaction.23 Improving the stability of the clusters during the electrocatalytic reaction is another challenge as the clusters often undergo aggregation during catalysis.
A promising approach to synthesizing the heterogeneous Au cluster catalysts is to use atomically precise, ligand-protected Au clusters as starting materials.24–35 Since the protecting ligands usually block the active sites for catalysis, they are conventionally removed completely from the Au surface by calcination on the solid support,36–40 which often leads to aggregation.23,41 Recently, we found that controlled calcination of the ligand-protected Au clusters on the support allows partial removal of the thiolate ligands and preferentially remains the ligands that interact non-covalently with the solid support.42,43 For example, when [Au25(PET)18]0 (PET: 2-phenylethanethiolate) was calcined on a carbon support under vacuum, the PET ligands exposed to the vacuum were first removed, yielding partially thiolated Au25(PET)∼12/C as an intermediate. The remaining PET ligands were probably located at the cluster/carbon support interface, as more severe calcinations were required to remove them due to multiple non-covalent interactions such as van der Waals (vdW), CH/π, and π/π interactions between the ligands and the carbon support. The partially thiolated Au25(PET)∼12/C showed higher activity and durability than the ligand-free Au25/C in benzyl alcohol oxidation at 80 °C, and retained the original cluster size after the reaction.42
Although the loading amount in Au25(PET)∼12/C could be improved up to 3 wt%,42 which is much higher than those of ligand-free Au25/C (e.g., 0.2 wt%), the stability of high-loading catalyst was not sufficient and underwent aggregation upon longer calcination. To further increase the loading amount and to improve the durability of the catalysts, we focused on the design of the residual anchoring ligands. In particular, we believed that this task could be accomplished by allowing bulky, aryl-containing ligands to remain on the cluster surface for the following reasons: (1) the aryl-containing ligands enhance the interaction with the carbon supports through the CH/π and π/π interactions; and (2) the bulky ligands prevent the physical contact of the Au cores even after thermally induced diffusion.
To test the above hypothesis, we first prepared starting materials by introducing a second-generation Fréchet-type dendron thiol (D2S-H) into [Au25(PET)18]0,44 based on the successful introduction of this ligand without the core deformation into [Au23(SCy)16]− (SCy: cyclohexanethiolate) and [Au25(PET)18]−.45 Multiple structural characterization revealed that the aggregation was suppressed by calcining 5.0 wt% of [Au25(D2S)10.7(PET)7.3]0 on carbon at 425 °C for ≥12 h, whereas calcination of [Au25(PET)18]0 under the same conditions resulted in thermally induced aggregation into larger Au NPs. Furthermore, the resulting catalyst exhibited higher durability than that without D2S introduction in the electrocatalytic HER, due to the suppression of aggregation during the reaction. This work demonstrates that the loading amount and durability of partially thiolated Au cluster catalysts can be improved by introducing appropriate ligands into the precursor clusters.
Results and discussion
Synthesis of catalysts
In the previous report, the D2S-H ligand was introduced by ligand exchange into an anionic cluster [Au25(PET)18]− with tetraoctylammonium (TOA+) as the counter cation.45 To avoid the involvement of the counter cation in the calcination process and the catalysis, we used a neutral cluster [Au25(PET)18]0 to obtain [Au25(D2S)x(PET)18−x]0. Two samples of [Au25(D2S)x(PET)18−x]0 were synthesized by adding 5 or 20 equivalents of D2S-H to [Au25(PET)18]0 (Fig. 1A), followed by purification by gel permeation chromatography (GPC) (Fig. S1†). The ultraviolet-visible (UV-vis) absorption spectra of the purified products (Fig. S2†) showed peaks at comparable positions to those of [Au25(PET)18]0 except for an additional peak at ∼280 nm due to the D2S-H ligand (Fig. S2†). These results indicate that the ligand exchange on [Au25(PET)18]0 proceeded while maintaining the compositions and structures.45 The matrix-assisted laser desorption/ionization (MALDI) mass spectra in the negative mode of the purified products (Fig. 1B) showed a series of mass peaks assigned to [Au25(D2S)x(PET)18−x]−. The average number of x was calculated to be 2.7 and 10.7 for the samples prepared by 5 and 20 equivalents of D2S-H, respectively, by fitting the ion intensity distribution with a Gaussian function.45 The corresponding full width half maximum value of the distribution was 0.8 and 1.0, respectively. The ion intensity distribution remained almost unchanged as long as the laser power was below the critical values (Fig. S3†). These results indicate that the observed distribution is not affected by laser-induced fragmentation, but reflects the abundance in the sample. Thermogravimetric (TG) analysis (Fig. 1C) of [Au25(PET)18]0 under air flow showed that the ligand desorption started at ∼200 °C, while those of [Au25(D2S)x(PET)18−x]0 showed a two-step desorption at ∼200 and ∼400 °C, which most likely corresponds to the loss of PET and D2S, respectively. The weight loss after complete desorption from the sample prepared by 0, 5, and 20 equivalents of D2S-H were 32.3, 47.1, and 64.0%, respectively. These values were in good agreement with those theoretically calculated for [Au25(D2S)x(PET)18−x]0 (33.4, 45.8, and 65.0% for x = 0, 2.7, and 10.7, respectively). These results confirmed that the desired starting materials were successfully synthesized: the samples prepared with 0, 5, and 20 equivalents of D2S-H are hereafter referred to as [Au25(D2S)x(PET)18−x]0 with x = 0, 2.7, and 10.7, respectively.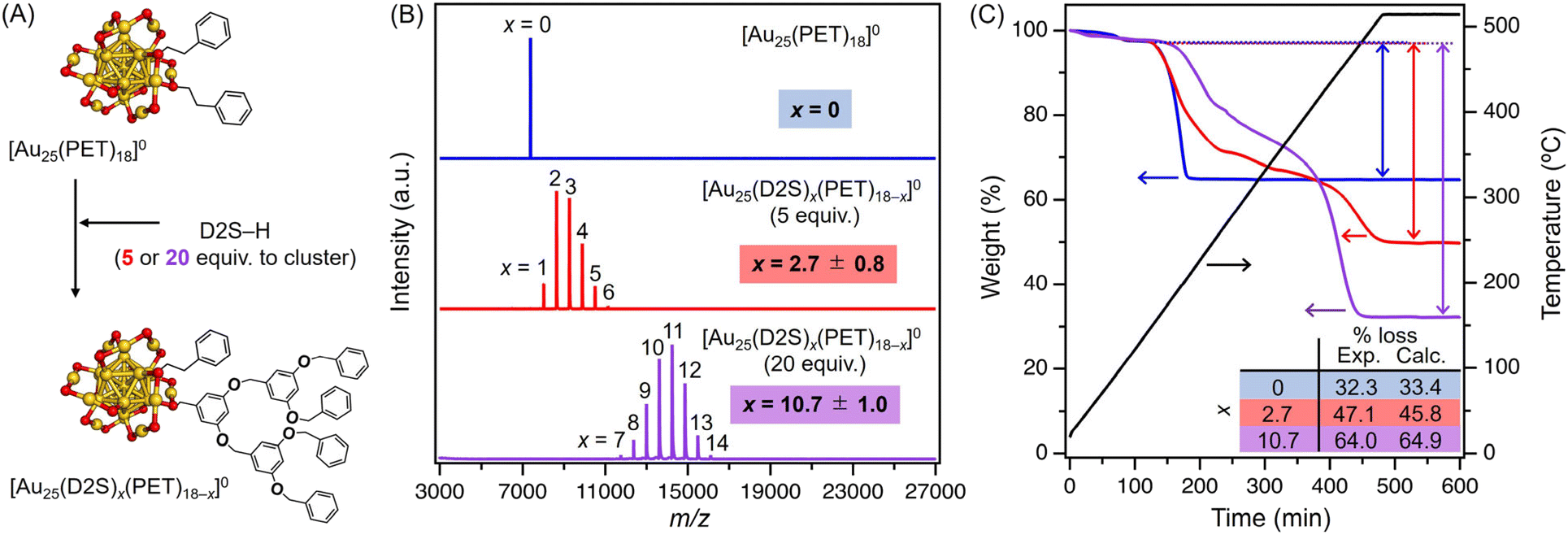 | ||
| Fig. 1 (A) Schematic representation of the ligand exchange from [Au25(PET)18]0 to [Au25(D2S)x(PET)18−x]0. (B) Negative ion MALDI mass spectra of purified products prepared with 0 (blue), 5 (red), and 20 (purple) equivalents of D2S-H. (C) TG analyses of purified products prepared with 0 (blue), 5 (red), and 20 (purple) equivalents of D2S-H in air. A black line represents the temperature change during the measurement. The inset table shows the experimental (Exp.) and theoretically calculated (Calc.) weight loss in percent. | ||
Subsequently, 1.0 or 5.0 wt% of [Au25(D2S)x(PET)18−x]0 (based on Au) was mixed with commercially available porous carbon material (CNovel) in toluene at 0 °C. The filtrate showed no absorption of [Au25(D2S)x(PET)18−x]0 in the UV-vis absorption spectra (Fig. S4†), indicating successful adsorption of the expected amounts of [Au25(D2S)x(PET)18−x]0. We believe that multiple non-covalent interactions such as vdW, CH/π, and π/π interactions between the ligands and the carbon support play a key role in the adsorption of the Au25 clusters on carbon. The obtained composites are hereafter referred to as w-Au25(D2S)x/C, where w represents the loading amount (1.0 or 5.0 wt%).
Finally, the composites w-Au25(D2S)x/C were calcined in vacuo to obtain catalysts. First, the calcination temperature was optimized using 1.0-Au25(D2S)x/C (x = 0, 2.7, and 10.7). The catalytic activity of benzyl alcohol oxidation was compared between the catalysts prepared by calcining at 400 or 425 °C (Fig. S5†). The catalytic activity of the catalyst obtained by calcining at 400 °C for 12 h was poor regardless of x (Fig. S5A†). This poor activity indicates that most of the ligands were still present and passivated the Au cluster surface after calcination at 400 °C for 12 h.42 In contrast, all catalysts obtained by calcining at 425 °C for 12 h showed high catalytic activity. The main products were benzoic acid and benzyl benzoate, suggesting the presence of exposed Au cluster surfaces as active sites. Based on these results, the calcination temperature was fixed at 425 °C and the catalysts are hereafter denoted as w-Au25(D2S)x/C-t, where t is the calcination period in hours. The optimal calcination temperature was slightly different from that derived from TG analyses due to the calcination atmosphere: TG analyses were performed in air, while the calcination was done in a vacuum to prevent the oxidation of carbon support. Then, the calcination time t was optimized using 1.0-Au25(D2S)x/C-t as a model to maximize the exposed area while suppressing the aggregation (Fig. S5B†). Regardless of x, the catalytic activity increased between t = 8 and 12 and remained comparable for t ≥ 15. This indicates that the exposed surface area was formed by calcining for 12 h, but did not increase significantly for t > 12 h. The powder X-ray diffraction (PXRD) patterns of 1.0-Au25(D2S)x/C-t did not show the diffraction peak due to the Au(111) plane at ∼38° regardless of t, indicating that the clusters did not aggregate during the calcination at 425 °C (Fig. S6†). The broad peak at ∼44° is assigned to diffraction from the (10l) (l = –1, 0, and 1) planes of the turbostratic carbon.46,47
Structural characterization of catalysts
A more detailed structural characterization of the catalysts was performed by X-ray absorption fine structure (XAFS) measurement at the Au L3 edge at room temperature.48 Fig. S7† shows the X-ray absorption near edge structure (XANES) spectra of 1.0-Au25(D2S)x/C-t with x = 0, 2.7, and 10.7 and t = 0, 8, 12, 15, 18, 21, and 24. Regardless of x, the catalysts with t = 0 showed a featureless peak, while those with t ≥ 8 showed the peaks assigned to metallic Au. The extended X-ray absorption fine structure (EXAFS) oscillations and their Fourier transformations (FT-EXAFS) of 1.0-Au25(D2S)x/C-t are presented in Fig. S8 and S9,† respectively. The coordination numbers for the Au–Au and Au–S bonds (CNsAu–Au and CNsAu–S, respectively) determined from the curve fitting analyses are summarized in Table S1† and plotted as blue and red blank squares against t in Fig. 2A. The following behaviors are generally observed regardless of x. At t = 0, the CNsAu–S values were in the range of 1.5–1.6, which are comparable to those calculated from the single crystal structure of [Au25(PET)18]0. Meanwhile, the CNsAu–Au values were 0.6–1.3, which are significantly smaller than those calculated from the crystal structure (3.3), due to the thermal fluctuation of the Au–Au bonds in the Au2(SR)3 units and ultrasmall Au13 core.49 At t = 8, the CNsAu–S values decreased to 1.0–1.2 while the CNsAu–Au increased to 4.1–4.5, indicating the partial removal of the thiolates and the growth of the Au core. This increase of CNsAu–Au is not due to the aggregation of Au25 clusters induced by thermal diffusion, but to the attachment of Au atoms originating from the Au2(SR)3 units to the Au13 core. At t ≥ 12, the CNsAu–S and CNsAu–Au values of the catalysts remained almost constant, suggesting that the removal of the remaining thiolates from the partially thiolated Au25 cluster became more difficult due to the non-covalent interactions between the thiolates and the carbon support.42 The CNsAu–Au values for t ≥ 12 were in the range of 4.3–5.6, which are smaller than those estimated from hemispherical face-centered cubic (fcc) model structures for Au25 (6.0–6.5). Aggregation of the Au25 clusters was negligible in 1.0-Au25(D2S)x/C-t regardless of x.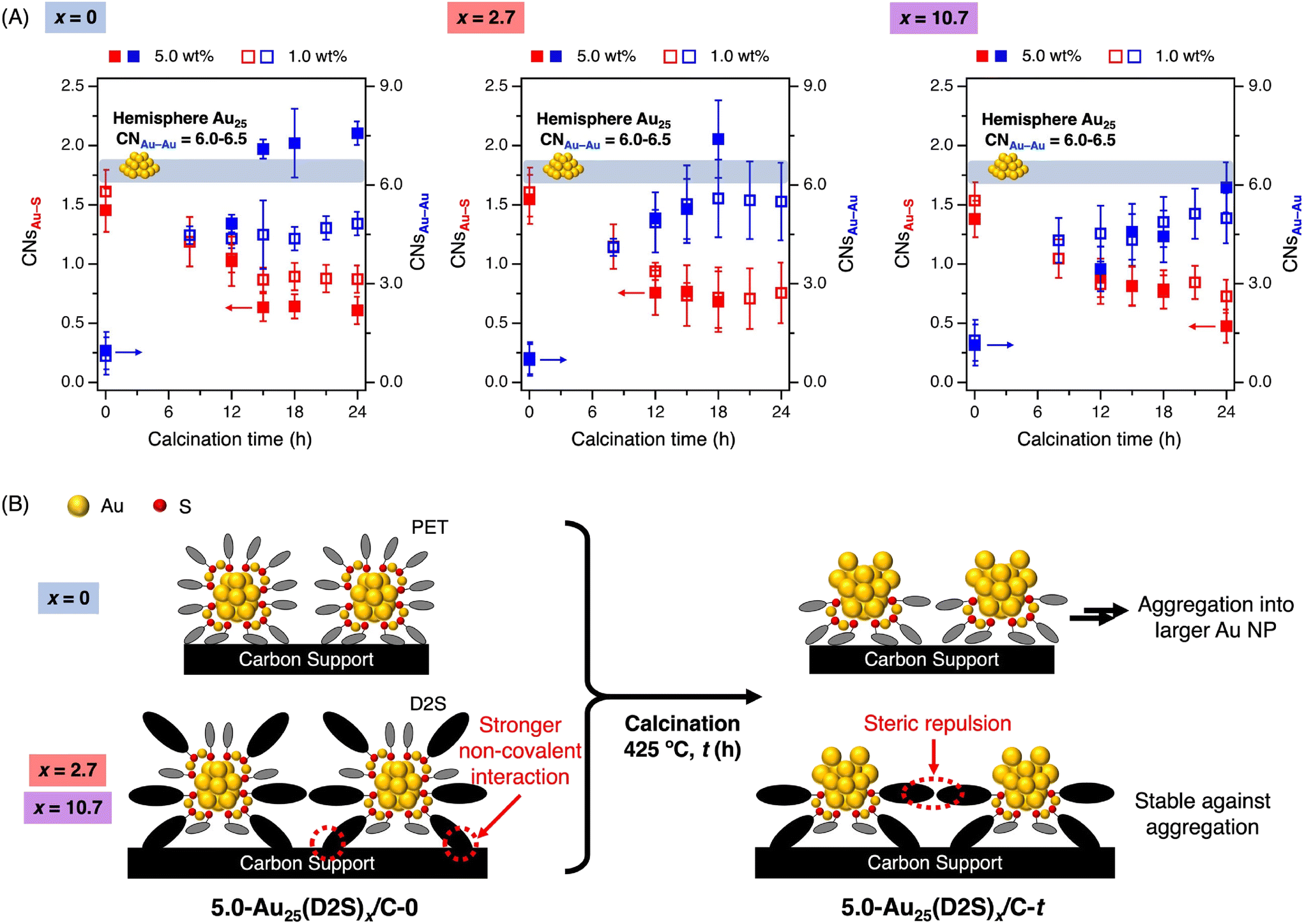 | ||
| Fig. 2 (A) Time course of CNsAu–S (red) and CNsAu–Au (blue) of w-Au25(D2S)x/C-t with x = 0, 2.7, and 10.7 estimated from curve fitting analyses of Au L3-edge EXAFS oscillations. Blank and filled squares correspond to the data for w = 1.0 and 5.0, respectively. (B) Schematic representation of the ligand desorption from 5.0-Au25(D2S)x/C and the model structures of 5.0-Au25(D2S)x/C-t. | ||
On the other hand, the structures of higher loaded 5.0-Au25(D2S)x/C-t prepared by calcination at 425 °C differed depending on whether D2S was introduced (x = 2.7, 10.7) or not (x = 0). The PXRD patterns for 5.0-Au25(D2S)0/C-t exhibited the diffraction peak due to Au(111) planes at t ≥ 15 (Fig. S10A†), indicating the aggregation of the Au25 clusters during the calcination. In contrast, the diffraction peak for Au(111) was greatly suppressed for x = 2.7, 10.7 (Fig. S10B and C†), indicating that D2S suppressed the thermal-induced aggregation of the clusters. Au L3-edge XANES spectra, EXAFS oscillations, and FT-EXAFS of 5.0-Au25(D2S)x/C-t are presented in Fig. S11–S13,† respectively. The results of the curve fitting analyses are summarized in Table S2† and the CNsAu–Au and CNsAu–S are plotted as blue and red filled squares in Fig. 2A. The CNAu–Au value of 5.0-Au25(D2S)0/C-t was 4.8 at t = 12, but increased to 7.1–7.6 at t ≥ 15, which are larger than the theoretical values of hemispherical Au25 (6.0–6.5). This change indicates that D2S-free Au25(PET)18 clusters with the loading of 5.0 wt% undergo aggregation by calcination for t ≥ 15. In contrast, the CNsAu–Au value for 5.0-Au25(D2S)2.7/C-t was smaller than the theoretical values of hemispherical Au25 (6.0–6.5) at t = 12 and 15, but increased to ∼7.4 at t = 18. Notably, the CNsAu–Au values of 5.0-Au25(D2S)10.7/C-t remained smaller than the theoretical values of hemispherical Au25 (6.0–6.5) up to t = 24. These results indicated that the D2S ligands played a key role in enhancing the stability of the partially thiolated Au25 cluster against thermally induced aggregation. In conclusion, 5.0-Au25(D2S)10.7/C-t (t ≥ 12) consists of the partially thiolated Au25 clusters individually immobilized on the carbon support by non-covalent interactions with D2S ligands (Fig. 2B).
The gradual removal of thiolate ligands in 5.0-Au25(D2S)x/C-t upon calcination was also supported by the Au 4f X-ray photoelectron (XP) spectra in Fig. S14.† The Au 4f7/2 electron binding energies of 5.0-Au25(D2S)x/C-t were ∼84.5 eV before calcination (t = 0) regardless of x due to the electron withdrawal by the thiolates.50,51 The binding energies then gradually decreased to 84.1–84.2 eV with t, suggesting the gradual removal of thiolate ligands upon calcination. In addition, the S 2p XP signal was detected even at t = 24 for all samples, supporting the residual thiolates (Fig. S14†).
The morphologies of the Au clusters were investigated by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) for 5.0-Au25(D2S)0/C-t and Au25(D2S)10.7/C-t with t = 0, 12, and 18. Typical images and the size distributions are shown in Fig. 3. The average diameter Dave and standard deviation σ of 5.0-Au25(D2S)0/C-t with t = 0, 12, and 18 was 1.1 ± 0.2, 1.2 ± 0.4, and 1.5 ± 0.7 nm, respectively. The increase in Dave and σ with t indicated that the Au clusters became polydisperse due to the aggregation, in agreement with the results of PXRD (Fig. S10A†) and XAFS (Fig. 2A). In contrast, the Dave and σ of 5.0-Au25(D2S)10.7/C-t with t = 0, 12, and 18 was 1.1 ± 0.2, 1.1 ± 0.3, and 1.2 ± 0.3 nm, respectively. Almost no change in Dave and σ with t indicates the suppression of aggregation during calcination. The results of AC-HAADF-STEM, together with those of PXRD, XAFS, and XP spectroscopy, demonstrate that the partially thiolated Au25 cluster catalyst with the loading amount of 5.0 wt% was successfully synthesized by using [Au25(D2S)10.7(PET)7.3]0 as the precursors.
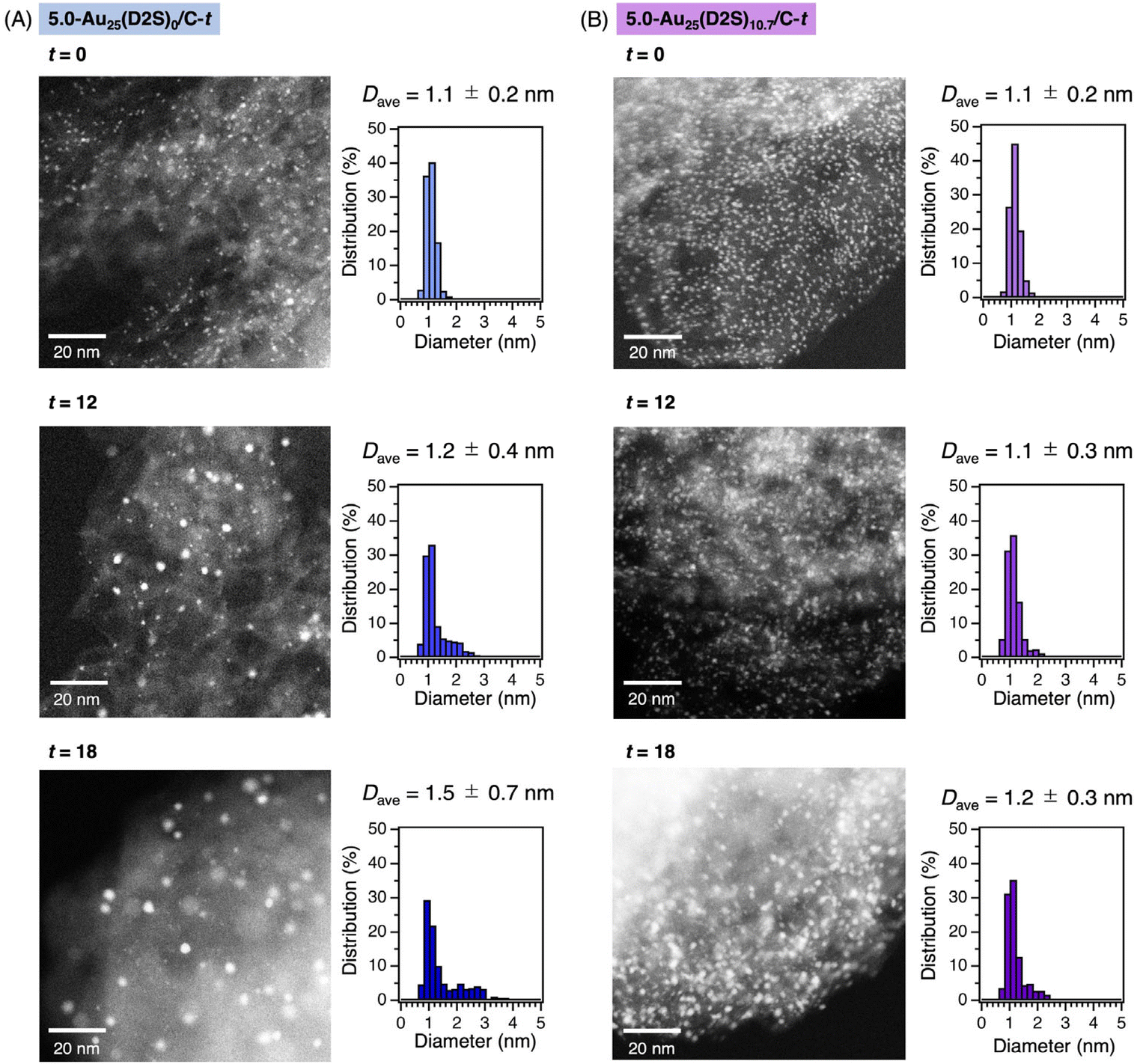 | ||
| Fig. 3 Typical AC-HAADF-STEM images and particle size distributions of (A) 5.0-Au25(D2S)0/C-t and (B) 5.0-Au25(D2S)10.7/C-t with t = 0, 12, and 18. The average diameter of the particles Dave was determined by measuring >300 particles. | ||
A carbon-supported Au NP catalyst, 5.0-AuNP/C, was also prepared as a reference catalyst by the deposition-reduction method at 5.0 wt%. The XANES spectra (Fig. S15A†) showed peaks corresponding to metallic Au, indicating the successful reduction of Au. The CNAu–Au determined by EXAFS analysis (Fig. S15B–D and Table S2†) was 10.1, which was much larger than those of 5.0-Au25(D2S)x/C-t. In addition, the sharp diffraction peak of Au was observed in the PXRD pattern (Fig. S16A†), supporting the formation of Au NPs. The typical AC-HAADF-STEM images are shown in Fig. S17.† The particle size was polydisperse, ranging from ∼1 to ∼300 nm. The electronic state was metallic, as suggested by the Au 4f XP spectrum (Fig. S16B†).
Electrocatalysis for hydrogen evolution reaction (HER)
First, the effect of the residual thiolates on electrocatalytic HER was investigated using 5.0-Au25(D2S)0/C-t, 5.0-Au25(D2S)10.7/C-t with t = 0, 12, and 18 (Fig. 4 and Table S3†). HER activity was evaluated by the overpotential (η), at which the current density (j) reaches 10 mA cm−2, determined by linear sweep voltammetry (LSV): the catalyst with less negative η is more active. The LSV measurement was performed in 0.5M H2SO4 using a standard three-electrode setup with reference Ag/AgCl electrode. Hereafter, η is referred to the reversible hydrogen electrode (RHE), which was calibrated by measuring potential of Ag/AgCl electrode against RHE under the reaction condition. The η value for Au25(D2S)0/C-t became less negative with t: −0.53, −0.34 and −0.27 V for t = 0, 12 and 18, respectively (Fig. 4A). A similar trend was observed for 5.0-Au25(D2S)10.7/C-t: the η value became less negative from −0.59 V at t = 0 to −0.34 and −0.29 V for t = 12 and 18, respectively (Fig. 4B). In terms of the η value and current density, 5.0-Au25(D2S)x-t with t = 12 and 18 showed much higher activity than non-calcined, thiolate (SR)-protected clusters, Au25(SR)18, reported previously (Table S3†). These data clearly indicated that the electrocatalytic HER takes place on the exposed Au cluster surface.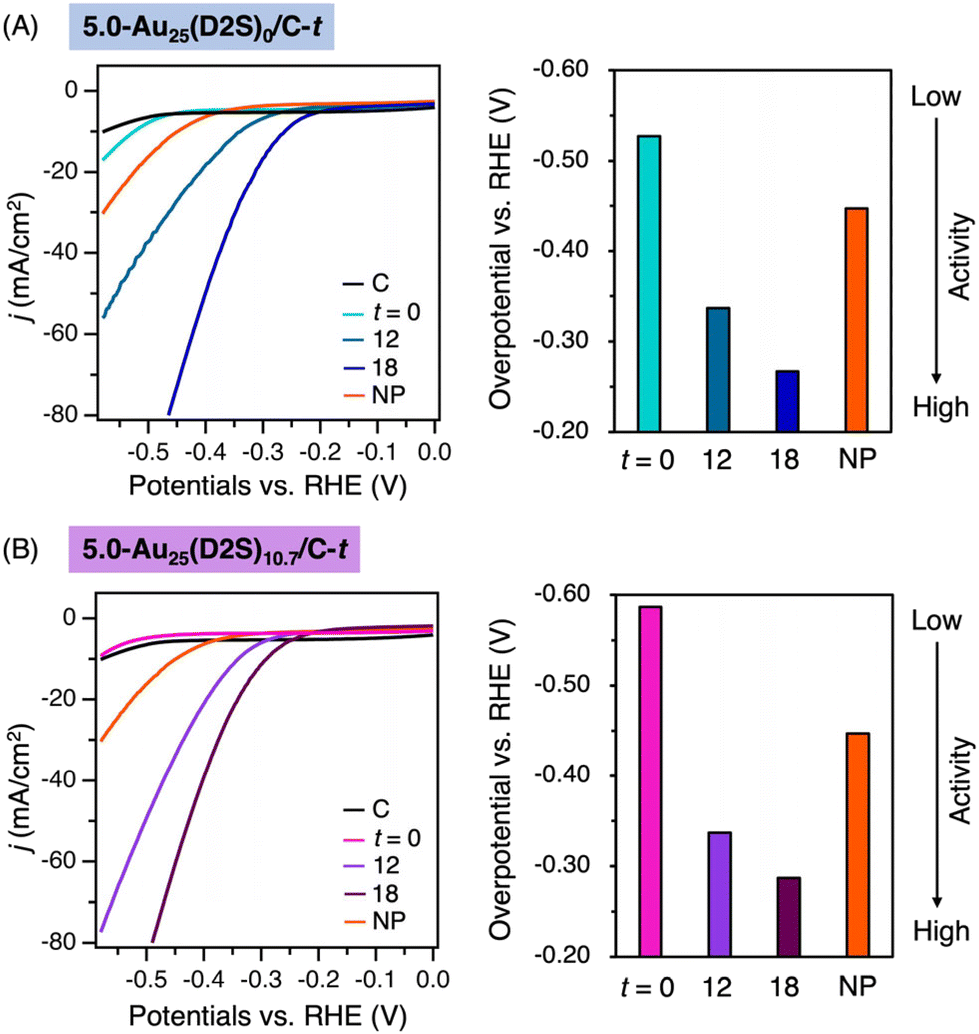 | ||
| Fig. 4 LSV curves of HER and comparison of overpotentials catalyzed by (A) 5.0-Au25(D2S)0/C-t and (B) 5.0-Au25(D2S)10.7/C-t with t = 0, 12, and 18. “C” and “NP” represent pristine carbon and 5.0-AuNP/C, respectively. | ||
Second, the cluster size effect on electrocatalytic HER was investigated using 5.0-AuNP/C as a reference. The catalytic activities of 5.0-Au25(D2S)0/C-18 and 5.0-Au25(D2S)10.7/C-18 were comparable, but significantly higher than 5.0-AuNP/C in terms of the η value (Fig. 4), demonstrating that the smaller sized Au clusters with exposed surface are more active.52 As expected, the HER activity was significantly reduced when using the same weight of the catalysts with 1.0 wt% loading (Fig. S18†). This indicates that the catalysts with 5.0 wt% loading are practically superior in terms of activity per gram (gcat).
Finally, the durability of 5.0-Au25(D2S)0/C-t and 5.0-Au25(D2S)10.7/C-t with t = 12 and 18 in electrocatalytic HER was compared. LSV measurements were performed after sweeping the potentials in the range of −0.1 to −0.8 V (vs. Ag/AgCl) by cyclic voltammetry (Fig. S19†). The j values using 5.0-Au25(D2S)0/C-t (t = 12 and 18) gradually decreased with increasing the cycle numbers (Fig. S19A†). As a result, the η value became more negative from −0.34 to −0.40 V for t = 12 after 2000 cycles and from −0.26 to −0.36 V for t = 18 after 4000 cycles, respectively (Fig. S19C†). The shift in η value after the first 100 voltammetric cycles was significantly greater for t = 18 than for t = 12, due to the poorer stability of the former clusters with fewer residual PETs (Table S2†). In contrast, 5.0-Au25(D2S)10.7/C-12 retained the η value even after 2000 cycles, while 5.0-Au25(D2S)10.7/C-18 showed a greater shift in η value in the first 100 cycles (Fig. S19B†): the η value changed from −0.34 to −0.36 V for t = 12 and from −0.29 to −0.34 V for t = 18 (Fig. S19C†). The durability of 5.0-Au25(D2S)0/C-t and 5.0-Au25(D2S)10.7/C-t was further investigated by using controlled potential electrolysis (chronopotentiometry). The time course of j under the constant potential at −0.6 V (vs. Ag/AgCl) is shown for 5.0-Au25(D2S)0/C-t and 5.0-Au25(D2S)10.7/C-t in Fig. 5A and B, respectively. Although the j values decreased at the early stage for 5.0-Au25(D2S)0/C-t regardless of t (Fig. 5A), 5.0-Au25(D2S)10.7/C-t especially with t = 18 retained larger j values than 5.0-Au25(D2S)0/C-t (Fig. 5B). These results clearly demonstrate that the Au25 clusters with residual D2S, especially 5.0-Au25(D2S)10.7/C-t, exhibited the higher robustness in the electrocatalytic HER.
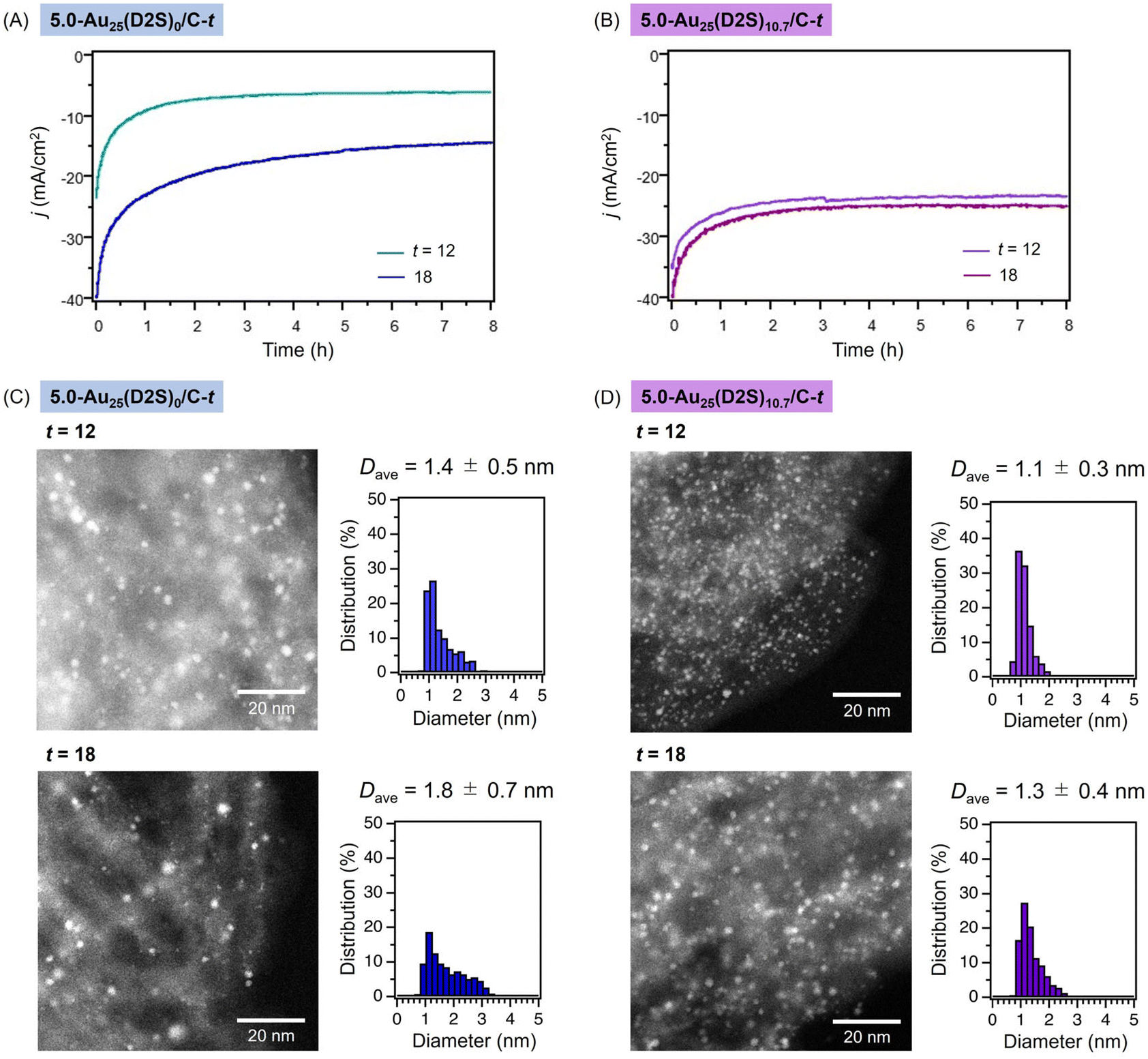 | ||
| Fig. 5 Time course of the electrocatalytic current density j of HER under the constant voltage of −0.6 V (vs. Ag/AgCl) in 0.5 M H2SO4 catalyzed by (A) 5.0-Au25(D2S)0/C-t and (B) 5.0-Au25(D2S)10.7/C-t with t = 12 and 18. Typical AC-HAADF-STEM images and particle size distributions of (C) 5.0-Au25(D2S)0/C-t and (D) 5.0-Au25(D2S)10.7/C-t with t = 12 and 18 collected after reaction under constant voltage of −0.6 V (vs. Ag/AgCl) in 0.5 M H2SO4 for 8 h. | ||
To gain insight into the reason for the higher durability and catalytic activity of the D2S-introduced catalysts, the catalysts were collected after the reaction under the conditions of Fig. 5A and B and characterized by AC-HAADF-STEM. Typical AC-HAADF-STEM images and the particle size distributions of the used 5.0-Au25(D2S)0/C-t and 5.0-Au25(D2S)10.7/C-t are shown in Fig. 5C and D, respectively. The Dave value of the Au clusters of 5.0-Au25(D2S)0/C-t apparently increased from 1.2 ± 0.4 to 1.4 ± 0.5 nm for t = 12 and from 1.5 ± 0.7 nm to 1.8 ± 0.7 nm for t = 18 (Fig. 5C). On the other hand, the Dave values for 5.0-Au25(D2S)10.7/C-12 before and after usage of 1.1 ± 0.3 and 1.1 ± 0.3 nm for t = 12 and 1.2 ± 0.3 nm and 1.3 ± 0.4 nm for t = 18, respectively. The cluster aggregation during the reaction was successfully suppressed especially in 5.0-Au25(D2S)10.7/C-12. Taken together, the introduction of D2S ligands enabled the immobilization of Au25 clusters with a high loading amount on the carbon support and remarkably improved the robustness against the aggregation during electrocatalytic HER.
Conclusions
In summary, we successfully synthesized the neutral Au25 cluster protected by D2S and PET, [Au25(D2S)x(PET)18−x]0 by ligand exchange and synthesized carbon-supported partially thiolated Au25 cluster catalyst co-stabilized by D2S and PET. Although the catalyst stabilized only by PET underwent aggregation during the removal of ligands by calcination when the loading amount was 5.0 wt%, the introduction of D2S ligands suppressed aggregation, enabling the stabilization of exposed Au surface with high density. The ligand removal increased the catalytic activity of electrocatalytic HER by enabling protons to access the Au surface more easily. Furthermore, D2S stabilized partially thiolated catalysts showed higher durability in HER than those without D2S. Characterization of used catalysts suggested the aggregation during the reaction could be suppressed by the D2S introduction, achieving both high activity and durability. This work demonstrates that the catalytic performances and durability of partially thiolated Au cluster catalysts can be further improved by introducing rationally designed ligands to the precursor Au clusters.Experimental
All methods are summarized in the ESI file.†Author contributions
K. S. conducted most of the experiments and analyses, and wrote the manuscript. S. M. synthesized Au nanoparticle catalyst (5.0-AuNP/C), performed AC-HAADF-STEM and XP spectroscopy measurements, and advised in some experiments. S. T. carried out the determination of the potential of Ag/AgCl vs. RHE in electrocatalysis and advised in some experiments. T. T. supervised the project and wrote the manuscript. The manuscript was written through discussions among all the authors, and all approved the final version.Data availability
The data supporting this article have been included as part of the ESI.† Additional data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Haru Hirai (The University of Tokyo) for assistance in electrocatalysis. The authors appreciate Prof. Kazuya Yamaguchi (The University of Tokyo) and Dr Takafumi Yatabe (The University of Tokyo) for fruitful discussions. This research was financially supported by JST, CREST (Grant No. JPMJCR20B2), “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Grant No. JPMXP1223UT0082) and a Grant-in-Aid for Scientific Research (A) (Grant No. 20H00370, and 23H00284), a Grant-in-Aid for Scientific Research (B) (23H01917), a Grant-in-Aid for Early-Career Scientists (Grant No. 21K14476 and 23K13617), and a Grant-in-Aid for JSPS Fellows (Grant No. 24KJ0676) from the Japan Society for the Promotion of Science (JSPS), and the World-leading Innovative Graduate Study Program for Materials Research, Information, and Technology (MERIT-WINGS) from the University of Tokyo. The synchrotron radiation experiments were performed under the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2023A1635 and 2023B2004).References
- A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2021, 11, 2003323 CrossRef CAS.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- A. Hameed, M. Batool, Z. Liu, M. A. Nadeem and R. Jin, ACS Energy Lett., 2022, 7, 3311–3328 CrossRef CAS.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, e2304021 CrossRef PubMed.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- C. Li and J.-B. Baek, ACS Omega, 2020, 5, 31–40 CrossRef CAS PubMed.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. M. Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS PubMed.
- P. Rodriguez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2014, 16, 13583–13594 RSC.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- C. Li, O. J. H. Chai, Q. Yao, Z. Liu, L. Wang, H. Wang and J. Xie, Mater. Horiz., 2021, 8, 1657–1682 RSC.
- L.-W. Chen, Y.-C. Hao, J. Li, L. Hu, Y. Guo, S. Li, D. Liu, Z. Zhu, S.-Q. Wu, H.-Z. Huang, A.-X. Yin, B. Wang and Y.-W. Zhang, Sci. China: Chem., 2022, 65, 2188–2196 CrossRef CAS.
- T. Szumełda, A. Drelinkiewicz, E. Lalik, R. Kosydar, D. Duraczyńska and J. Gurgul, Appl. Catal., B, 2018, 221, 393–405 CrossRef.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- J. Luo, G. I. N. Waterhouse, L. Peng and Q. Chen, Ind. Chem. Mater., 2023, 1, 486–500 RSC.
- J. Fang, J. Li, B. Zhang, X. Yuan, H. Asakura, T. Tanaka, K. Teramura, J. Xie and N. Yan, Nanoscale, 2015, 7, 6325–6333 RSC.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- M. H. Naveen, R. Khan and J. H. Bang, Chem. Mater., 2021, 33, 7595–7612 CrossRef CAS.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- R. H. Adnan, J. M. L. Madridejos, A. S. Alotabi, G. F. Metha and G. G. Andersson, Adv. Sci., 2022, 9, 2105692 CrossRef CAS PubMed.
- S. Masuda, K. Sakamoto and T. Tsukuda, Nanoscale, 2024, 16, 4514–4528 RSC.
- S. Yamazoe, T. Yoskamtorn, S. Takano, S. Yadnum, J. Limtrakul and T. Tsukuda, Chem. Rec., 2016, 16, 2338–2348 CrossRef CAS PubMed.
- V. Sudheeshkumar, K. O. Sulaiman and R. W. J. Scott, Nanoscale Adv., 2020, 2, 55–69 RSC.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Akinaga, R. Takahata, K. Wakamatsu, Y. Fujiki, M. Kataoka, S. Kikkawa, A. S. Alotabi, S. Hossain, D. J. Osborn, T. Teranishi, G. G. Andersson, G. F. Metha, S. Yamazoe and Y. Negishi, Angew. Chem., Int. Ed., 2021, 60, 21340–21350 CrossRef CAS PubMed.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Iwamatsu, S. Hossain and Y. Negishi, Nanoscale Horiz., 2021, 6, 409–448 RSC.
- S. Masuda, S. Takano, S. Yamazoe and T. Tsukuda, Nanoscale, 2022, 14, 3031–3039 RSC.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- K. Sakamoto, S. Masuda, S. Takano and T. Tsukuda, ACS Catal., 2023, 13, 3263–3271 CrossRef CAS.
- S. Masuda and T. Tsukuda, ACS Catal., 2023, 13, 16179–16187 CrossRef CAS.
- J. A. Trindell, J. Clausmeyer and R. M. Crooks, J. Am. Chem. Soc., 2017, 139, 16161–16167 CrossRef CAS PubMed.
- T. Omoda, S. Takano, S. Masuda and T. Tsukuda, Chem. Commun., 2021, 57, 12159–12162 RSC.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- N. S. Saenko, Phys. Proc., 2012, 23, 102–105 CrossRef CAS.
- S. Yamazoe and T. Tsukuda, Bull. Chem. Soc. Jpn., 2019, 92, 193–204 CrossRef CAS.
- S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- P. Zhang, J. Phys. Chem. C, 2014, 118, 25291–25299 CrossRef CAS.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03385j |
| This journal is © The Royal Society of Chemistry 2024 |