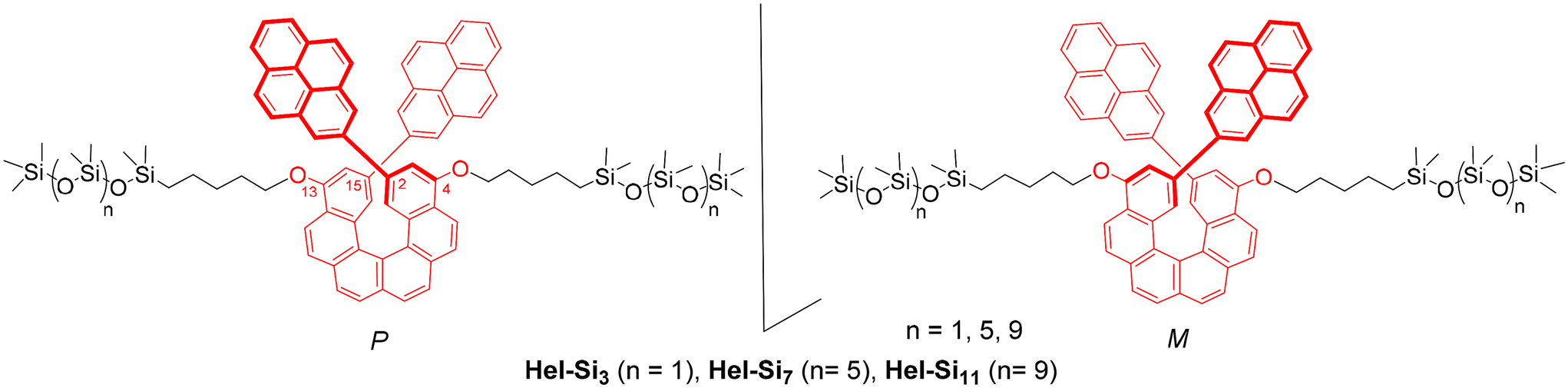DOI:
10.1039/D4NR03389B
(Paper)
Nanoscale, 2024,
16, 21351-21359
Engineering circularly polarized light emission in nanostructured oligodimethylsiloxane-helicene chiral materials†
Received
18th August 2024
, Accepted 14th October 2024
First published on 18th October 2024
Abstract
Chiroptical properties in the bulk state can be tuned by controlling the formation of chiral ordered nanostructures. Here, we present a series of discrete oligodimethylsiloxane-helicene-pyrene block molecules with varying oDMS lengths and study the nanostructures formed in both bulk and solution, including their chiroptical properties. In bulk, ordered 2D nanostructures self-assemble, driven by phase segregation induced by the siloxane oligomers, with clear differences in the properties of the racemic and enantiopure versions. Moreover, intermolecular pyrene interactions lead to excimer emission. As a result, up to a 5-fold increase in circularly polarized luminescence is observed in the solid state as compared to the solution, accompanied by a clear influence of the pyrene excimer emission on the overall emission process. Interestingly, in the ordered lamellar packing achieved from long oDMS units, the excimer emission shows very little net circular polarization, while in the disordered state achieved from shorter oDMS units, this excimer emission displays a significant degree of circular polarization. These results demonstrate that functionalizing chiroptical building blocks with discrete oligodimethylsiloxane chains is a versatile strategy to control photophysical properties and modulate chiroptical emission in bulk. This approach advances the integration of chiroptical materials into devices, enabling diverse applications ranging from optoelectronics to communication technologies.
Introduction
Helicenes are helical semiconducting molecules formed from ortho-fused aromatic rings that exhibit attractive properties such as strong chiroptical activity (intense electronic circular dichroism – ECD, and circularly polarized luminescence – CPL). Helicenes therefore hold great promise for applications in diverse domains such as optoelectronics, spintronics and bioimaging.1 While their photophysical and chiroptical properties have been well studied and examined in solution, studies in the condensed phase, as in ordered gels, mesophases or the solid state, remain scarce. Indeed, the assembly of helicene derivatives is particularly challenging as it requires a rational design to balance the complementary recognition units connecting the helicene monomers into an ordered structure and the compatibility between the various peripheral functional groups.2 For instance, the formation of fibers through columnar organization has been reported by Katz and coworkers for a carbo[6]helicene bearing terminal para-quinones and four dodecyloxy chains.2a,b These supramolecular stacks result in gigantic optical rotation values, strong circular dichroic responses and linearly/circularly polarized emission. Similarly, supramolecular trimeric aggregates displaying a fibrous structure with improved CPL responses compared to monomeric phthalhydrazide helical units have been described by Takeuchi and coworkers.2c Following a self-assembly strategy developed by Sánchez and coworkers on carbo[5]helicenes,2e our group has recently evidenced a CPL inversion effect upon self-assembly of carbo[6]helicenes decorated with bis-amide units.2f In addition, the group of Yamaguchi has thoroughly examined the self-assembly processes of 1,14-dimethyl-tetrahelicenic oligomers (see ref. 2h and references therein). These reports highlight the ongoing progress in the field and serve as inspiration for the development of new methods to control photophysical properties in bulk, to integrate helicene materials into devices, as well as to understand the mechanisms at play.3
Recently, the functionalization of crystalline moieties with discrete oligodimethylsiloxanes (oDMS) has emerged as a versatile strategy to achieve long-range order with sub-5 nm domain spacings.4 These block co-oligomers (BCOs) or block molecules consist of a “hard” molecular core and a “soft” amorphous oDMS chain that induce phase separation into different morphologies.5 In such systems, synergistic effects between directional interactions and phase separation promote the assembly of complex nanostructures in a highly controlled manner with unique properties. For example, oDMS chains have been functionalized with azobenzenes resulting in photo-switchable nanomaterials,5a dinitrophenylhydrazones for the construction of highly ordered thermosensitive nanomaterials,5b as well as diphenylanthracenes for the emission of upconverted, linearly polarized light.5c
Inspired by this work, we intended to combine carbo[6]helicene as a π-conjugated helical core with oDMS side chains and generate self-assembled ordered nanostructures. We hypothesized that the amorphous liquid-like siloxane matrix should influence the chiroptical response of helicenes. Herein, we report the synthesis of oDMS end-functionalized carbo[6]helicenes and their resulting physicochemical and morphological properties in bulk. A series of enantiopure P- and M-carbo[6]helicenes coupled with siloxane oligomers of different chain lengths at both the 4- and 13-positions were synthesized (Hel-Si3, Hel-Si7 and Hel-Si11 in Fig. 1) and their photophysical and chiroptical properties were studied, both in solution and in bulk. This was performed with the aim of achieving optimized CPL activity through lamellar morphology. The helical core was decorated with pyrene units at its 2,15-positions and the typical pyrene-based monomeric/excimeric emission response was analysed. The formation of 2D layered bulk assemblies was characterized in solution and in the solid state by (chir)optical spectroscopy (UV/Vis, ECD, emission and CPL), polarized optical microscopy (POM), differential scanning calorimetry (DSC), and medium/wide-angle X-ray scattering (MAXS/WAXS) to identify the formed nanostructures and to elucidate the molecular features leading to the emerging macroscopic photophysical and chiroptical properties. Pleasingly, up to a 5-fold increase in CPL activity was observed in the lamellar ordered phase as compared to the solution, accompanied by a clear influence of the pyrene excimer emission on the overall emission process.
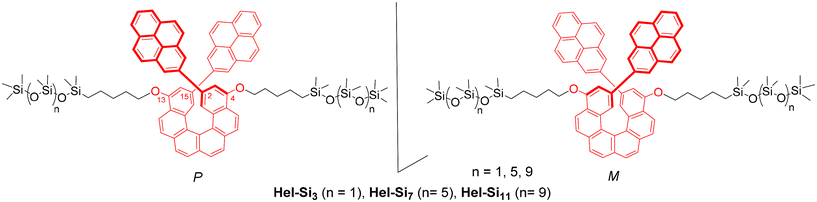 |
| | Fig. 1 Chemical structures of the carbo[6]helicene derivatives studied, bearing 2-pyrenyl chromophores at its 2,15-positions and two oDMS chains of diverse lengths at the 4,13-positions. | |
Results and discussion
Design and synthesis of oligodimethylsiloxane-helicenes
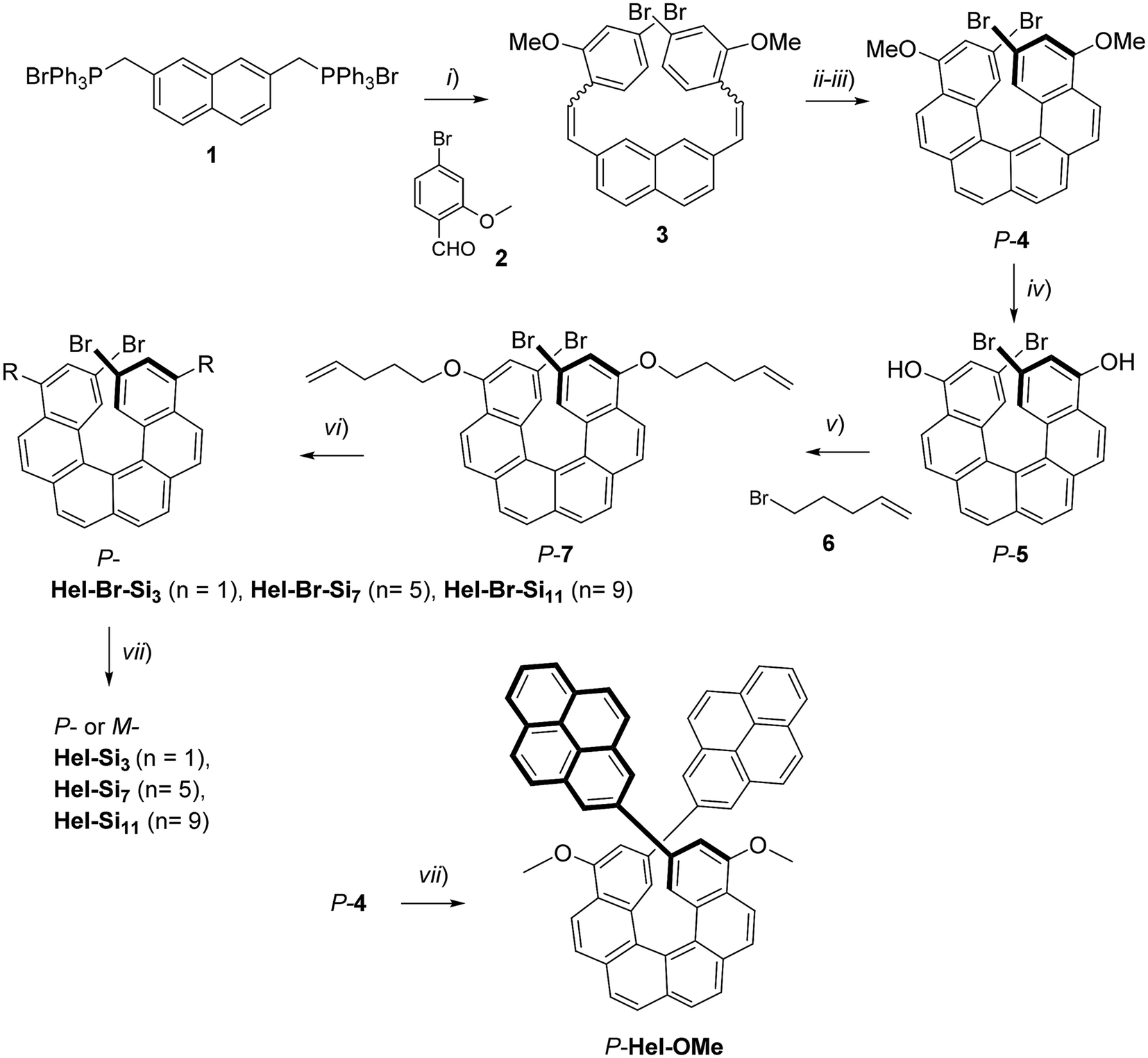
The synthesis of enantiopure P-carbo[6]helicene derivatives Hel-Si3, Hel-Si7 and Hel-Si11 as helical BCOs is depicted in Scheme 1. The same procedure is followed for the M-derivative. First, enantiopure P- and M-7 derivatives were prepared, bearing two bromides and two pent-1-enyl-oxy groups at the 2,15- and 4,13-positions, respectively (see the ESI† for detailed procedures). A classical Wittig reaction between (2,7-naphthalenedimethylene)-bis[triphenylphosphonium bromide] 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and 4-bromo-2-methoxy-benzaldehyde 2 was followed by an oxidative photocyclization (Mallory reaction)7 to yield 2,15-dibromo-4,13-dimethoxy-carbo[6]helicene 4, as a novel helicene derivative which was subsequently resolved into its pure enantiomers by using HPLC separations over a chiral stationary phase (see conditions in the ESI†). Demethylation using BBr3 yielded P- and M-2,15-dibromo-4,13-dimethoxy-carbo[6]helicene 5. Since 5 appeared to be unstable, it was thus directly engaged in a nucleophilic substitution using bromopent-1-ene. The obtained enantiopure helical derivatives P- and M-7, bearing two terminal olefins, were ready to react with siloxanes of different lengths through a hydrosilylation reaction using Karstedt's catalyst. The helicenes were coupled with three different lengths of oDMS chains, respectively, with 3, 7 and 11 silicon atoms yielding P- and M-Hel-Br-Si3, Hel-Br-Si7, and Hel-Br-Si11 in 59–73% yields. Finally, a classical Suzuki coupling with pyrene-2-boronic acid gave access to enantiopure helical compounds P- and M-Hel-Si3, Hel-Si7, and Hel-Si11 as waxy solids (Fig. 1 and Scheme 1), which were fully characterized by NMR spectroscopy and mass spectrometry (see Fig. S1–S38†).
6 and 4-bromo-2-methoxy-benzaldehyde 2 was followed by an oxidative photocyclization (Mallory reaction)7 to yield 2,15-dibromo-4,13-dimethoxy-carbo[6]helicene 4, as a novel helicene derivative which was subsequently resolved into its pure enantiomers by using HPLC separations over a chiral stationary phase (see conditions in the ESI†). Demethylation using BBr3 yielded P- and M-2,15-dibromo-4,13-dimethoxy-carbo[6]helicene 5. Since 5 appeared to be unstable, it was thus directly engaged in a nucleophilic substitution using bromopent-1-ene. The obtained enantiopure helical derivatives P- and M-7, bearing two terminal olefins, were ready to react with siloxanes of different lengths through a hydrosilylation reaction using Karstedt's catalyst. The helicenes were coupled with three different lengths of oDMS chains, respectively, with 3, 7 and 11 silicon atoms yielding P- and M-Hel-Br-Si3, Hel-Br-Si7, and Hel-Br-Si11 in 59–73% yields. Finally, a classical Suzuki coupling with pyrene-2-boronic acid gave access to enantiopure helical compounds P- and M-Hel-Si3, Hel-Si7, and Hel-Si11 as waxy solids (Fig. 1 and Scheme 1), which were fully characterized by NMR spectroscopy and mass spectrometry (see Fig. S1–S38†).
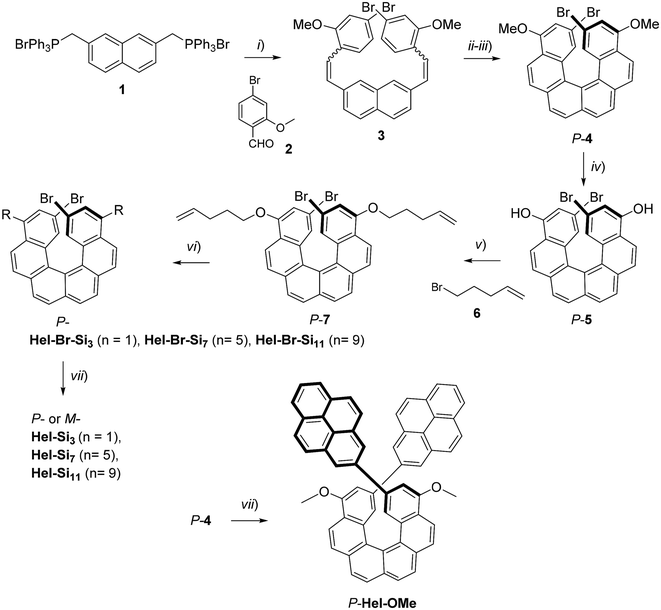 |
| | Scheme 1 Synthetic route to enantiopure oDMS end-functionalized carbo[6]helicenes P-Hel-Si3,7,11 and Hel-OMe. (i) n-BuLi, rt, Ar, 3 h, 94%; (ii) I2, toluene, Ar, propylene oxide, hν, 7 h, 61%; (iii) chiral HPLC resolution (see the ESI†); (iv) BBr3, CH2Cl2, rt, Ar, 12 h; (v) K2CO3, DMF, Ar, 65 °C, overnight, 67%; (vi) Karstedt's catalyst, Si(2+n)H, CH2Cl2, rt, Ar, 12 h, 73% (n = 1), 59% (n = 5, 9); (vii) pyrene-2-boronic acid, Pd(PPh3)4, Na2CO3, DMF, 80 °C, Ar, 12 h, 43–55%. The same strategy was used for the M enantiomers. The detailed R substituents can be seen in Fig. 1. | |
UV-vis absorption and ECD responses of Hel-Sin in solution and the solid state
UV-vis and ECD spectroscopies are dependent on the absolute configuration of a molecule and on its overall conformation, making them powerful tools for analysing aggregation in solution. The UV-vis absorption responses of Hel-Sin in methylcyclohexane (MCH) solutions are shown in Fig. 2a–c. All enantiomers show identical UV and mirrored ECD spectra, as expected for enantiomerically pure compounds. Under these conditions, the three systems clearly show very similar responses, with strong absorption bands at 240, 280, 325, 345 and 375 nm, and ε values up to 7 × 104 M−1 cm−1. As anticipated, all the absorption bands are strongly ECD active (see Fig. 2a–c, upper curves). For instance, the P-Hel-Si3 enantiomer exhibits respective Δε values of −206, −130, −57, −82 and +250 M−1 cm−1, when going from higher to lower energies. Due to their strong similarities, we conclude that the ECD response originates from the helical backbone without the oDMS units, since they are similar to those measured in CH2Cl2 and to the Hel-OMe enantiomers (Fig. S43–S55†), thus confirming the absence of assembly at 10−5 M concentrations in MCH. It should be noted that all the samples were also analysed in solvent/antisolvent THF/MCH mixtures to induce the aggregation in solution (Fig. S56–S61†). No significant changes in the ECD spectra were observed, indicating that the optical properties in the ground state do not reflect an aggregation process in solution. This observation also indicates that the emission in solution comes from the molecularly dissolved state (vide infra).
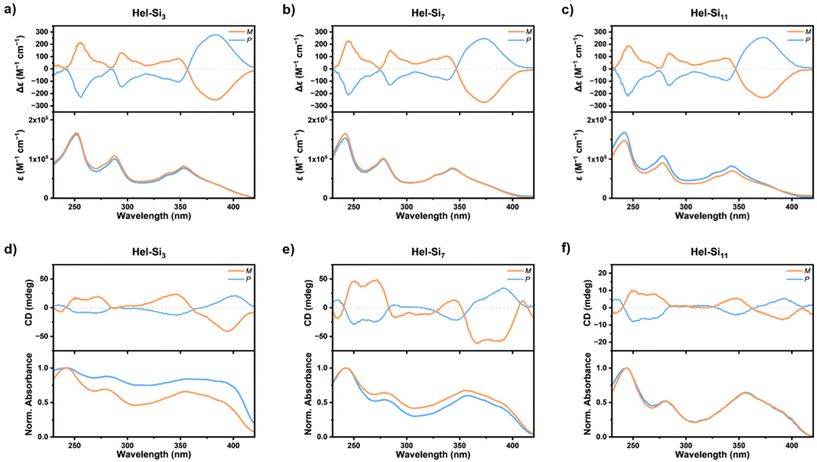 |
| | Fig. 2 (a–c) Comparison of UV-vis absorption and ECD responses of Hel-Sin in MCH solutions (C ∼ 1–2 × 10−5 M) and (d–f) in the solid state (solutions dropcast onto quartz substrates and annealed). | |
The molecules also exhibit interesting chiroptical properties in the solid state (Fig. 2d–f). Solid-state samples were prepared by drop casting onto quartz substrates, and the films were then annealed (see the ESI† for more details). In the solid state, the enantiomers show different UV spectra and non-perfectly mirrored ECD spectra. These differences are rationalized to be due to sample preparation, which tends to create inhomogeneities in the thickness and arrangement of the samples. Indeed, due to the low quantities of material available, it was difficult to adjust the film thickness for obtaining the mirror-image relationship. In addition, the UV-vis and ECD responses differed from those previously observed in solution. While at higher energies (i.e. between 200 and 330 nm) the UV-vis responses were similar with the presence of two bands, and the bands above 350 nm were found to be broader, with higher intensities around 400 nm and with an onset starting around 430 nm, thus highlighting a bathochromic shift of these low-energy bands when going from solution to the solid state. Interestingly, one can notice that there is a systematic extinction of the ECD bands corresponding to the absorption around 280 nm. This phenomenon is most probably related to the supramolecular alignment of the helical species, as already highlighted by Wade et al. in helicene systems well-oriented on surfaces.8 It should be noted that although only slight anisotropy effects were observed when the samples were rotated or the back and front surfaces were switched (see Fig. S59–S65†), the averaged ECD spectra were still considered.
Nanostructures of Hel-Sin in bulk
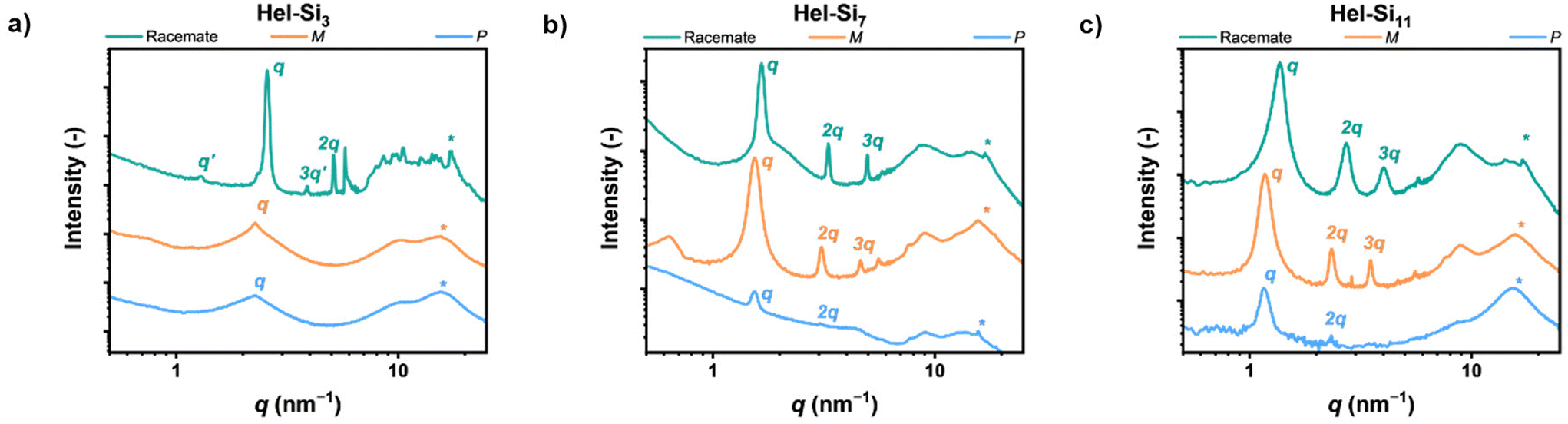
To elucidate the thermal stability of Hel-Sin, the materials were analysed by DSC (Fig. S39,†Table 1). Functionalization of the helicenes with a discrete oDMS chain resulted in a reversible transition at elevated temperatures. Hel-Si3 showed this transition at the highest temperature (Tm = 187–196 °C), which was expected since the volume fraction of the crystalline unit is highest in this material. Moreover, both enantiomers as well as the racemic mixture of Hel-Si3 showed cold crystallization between 108–116 °C. Enantiomerically pure Hel-Si7 and Hel-Si11 exhibited regular crystallization and melting transitions, with temperatures decreasing almost linearly with increasing siloxane content. Besides, similar to the solid-state UV-vis and ECD data (Fig. 2d–f), the M- and P-Hel-Sin enantiomers show differences in Tm, Tc, and ΔHfus due to sample inhomogeneity. Strikingly, the racemic mixtures of Hel-Si7 and Hel-Si11 exhibited lower Tc and these exothermic transitions were observed only upon heating. We hypothesize that this effect originates from slower crystallization in the racemic mixture compared to the enantiomerically pure substrates. In these racemic mixtures, we also observed a decreasing Tc with increasing siloxane content, similar to the enantiomerically pure materials. This trend indicates that the oDMS side chains have a large influence on the thermal stability of the crystalline phase of Hel-Sin. The presence of an ordered structure below Tm was confirmed by the observation of birefringent structures in all enantiomerically pure Hel-Sin under the POM (Fig. S40 and S41†). In this part, we thus emphasize the fact that these 2D ordered structures follow the typical racemate properties observed in the crystalline bulk solid state,9 highlighting a clear difference in behaviour and organization between racemic and enantiopure mixtures. Such different organization is usually not studied in chiral siloxane-based systems10 although it has direct implications for the material's properties such as semi-conducting properties and charge carrier mobility.11
Table 1 Bulk characterization of Hel-Sin
| Name |
Enantiomer |
T
m [°C] |
T
c [°C] |
ΔHfus [kJ mol−1] |
Phasea |
d
[nm] |
|
Morphology of the nanostructure determined with MAXS/WAXS at room temperature. DIS = disordered, LAM = lamellar phase. Domain spacing (d) was calculated using d = 2π/q.
Observed as cold crystallization.
|
|
Hel-Si3
|
M
|
196 |
108b |
11.8 |
DIS |
— |
|
P
|
186 |
115b |
7.0 |
DIS |
— |
|
Racemate
|
188 |
116b |
27.8 |
LAM |
2.4 |
|
Hel-Si7
|
M
|
151 |
136 |
10.4 |
LAM |
4.1 |
|
P
|
145 |
125 |
8.3 |
LAM |
4.1 |
|
Racemate
|
108 |
76b |
9.4 |
LAM |
3.8 |
|
Hel-Si11
|
M
|
116 |
90 |
10.3 |
LAM |
5.3 |
|
P
|
114 |
88 |
8.7 |
LAM |
5.3 |
|
Racemate
|
110 |
49b |
35.6 |
LAM |
4.6 |
The origin of the phase-separated structures was then investigated using MAXS/WAXS (Fig. 3). Pure enantiomers of Hel-Si3 showed a primary scattering peak at 2.3 nm−1, but no subsequent scattering peaks were present, indicating the absence of long-range order. In contrast, in the racemate rac-Hel-Si3, we observed several scattering peaks positioned at integers from the primary peak, indicating a lamellar morphology (q, 2q, 3q, …). Moreover, a low-intensity reflection was positioned at ½q (q′). We attribute this to a packing defect, similar to those previously reported in the literature.5a The resolved lamellar structure has a domain spacing of 2.4 nm. In contrast to the pure enantiomers of Hel-Si3 and similar to rac-Hel-Si3, Hel-Si7 and Hel-Si11 displayed lamellar morphologies in the racemic and enantiopure forms. For P- and M-Hel-Si7, the lamellar morphology was observed with a domain spacing of 4.1 nm. Assuming the side chains are fully stretched, the calculated theoretical length of Hel-Si7 was approximately 4.0 nm, suggesting that the side chains are indeed present in a stretched conformation. In rac-Hel-Si7, however, we found the lamellar spacing to be slightly smaller (3.8 nm), indicating partial intercalation of the siloxane side chains. We rationalize that this less efficient packing is also the cause of the slower crystallization observed in the DSC experiments. Moreover, in P- and M-Hel-Si11 the dlam increased by 1.2 nm compared to P- and M-Hel-Si7 to reach 5.3 nm, which is in good agreement with the length of four additional oDMS units on both side chains. Hence, we conclude that the phase-separated structures that are formed by P- and M-Hel-Si7 and P- and M-Hel-Si11 consist of stretched molecules. For M-Hel-Si7, a reflection at q′ was observed, similar to that previously described for rac-Hel-Si3. Moreover, the intensity of the peaks for P-HelSi7 is limited compared to the other spectra of Hel-Si7. Due to the waxy nature of this material and the limited amount available, its spectrum was recorded as a thin film on Kapton tape. A more zoomed-in version of the spectrum is shown in Fig. S42.† Interestingly, each Hel-Sin showed a distinct peak at 15.6 nm−1, indicated with *, corresponding to a distance of 0.4 nm. We rationalize that this peak originates from the intermolecular distance in the helicene, since it is persistent in all materials. Indeed, the distance between the benzene rings in a [6]helicene has been previously reported to be about 0.4 nm.12
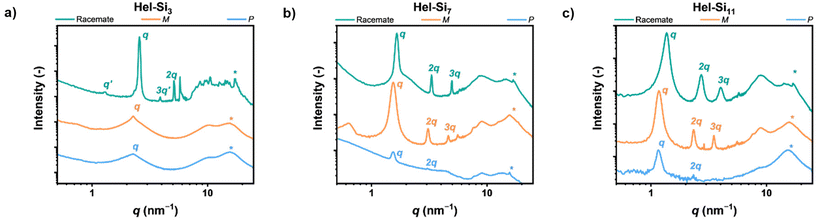 |
| | Fig. 3 MAXS/WAXS spectra of the rac-, P-, and M-forms of Hel-Si3 (a), Hel-Si7 (b) and Hel-Si11 (c). It should be noted that the curves of P-Hel-Sin are recorded on Kapton tape due to the small amount synthesized and therefore differences with the enantiomer M-Hel-Sin are observed due to the different processing conditions and the lower sensitivity of the measurement. | |
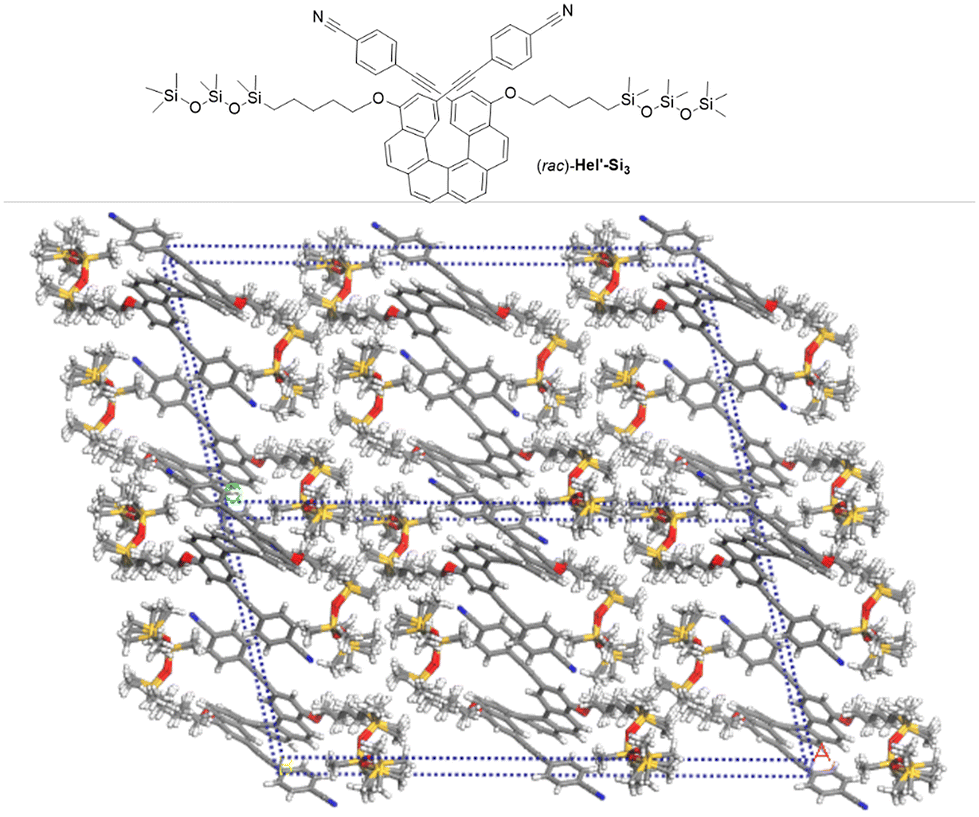
To further confirm the validity of our strategy to efficiently and systematically generate lamellar structures in oDMS-helicene BCOs, we synthesized Hel′-Si3 bearing cyanophenyl moieties at the 2,15-positions, which crystallizes readily and provides new insights into the molecular packing (see the ESI†). The X-ray crystallographic structure of Hel′-Si3 has been resolved (C2/c centrosymmetric space group) and shows that order is present along both the b- and c-axes, indicating a tendency to assemble into 2D layered materials (Fig. 4). The widths of both the helicene layers and the oDMS layers were found to be 12 Å, which together corresponds well to the lamellar dimension of rac-Hel-Si3 (2.4 nm, vide supra). It should be noted that in Hel′-Si3 no clear π–π stacking is observed because the centroid–centroid distance of 6.4 Å between the intermolecular p-CN-phenyl rings is large, and the silicon atoms in the siloxane chains adopt a classical tetrahedral geometry, while the pentyl chains are in a linear zigzag manner, resulting in overall elongated chains. The reported helicity (dihedral angle between the terminal rings of 47.3°) was in agreement with previously reported values for carbo[6]helicene.1 For comparison, single crystals of rac-4 were grown and the X-ray analysis revealed that in the absence of siloxane chains, a columnar alignment along the c-axis is observed between heterochiral species (Pbcn centrosymmetric space group), with π–π stacking between the terminal rings (centroid–centroid distances of 3.9 Å, see Fig. S43 and S44†), confirming that substitution with siloxane chains leads to the formation of a 2D lamellar morphology.
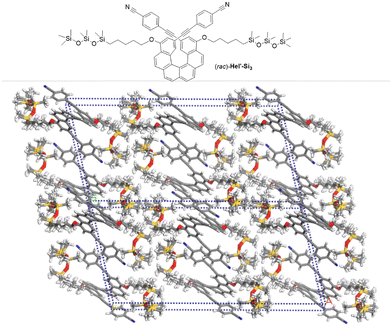 |
| | Fig. 4 Chemical structure and supramolecular assembly of 2,15-bis-cyanophenyl-carbo[6]helicene-siloxane adduct rac-Hel′-Si3 obtained from X-ray data analysis. | |
Consequences of self-assembly on the emissive properties of Hel-Sin in solution and the solid state
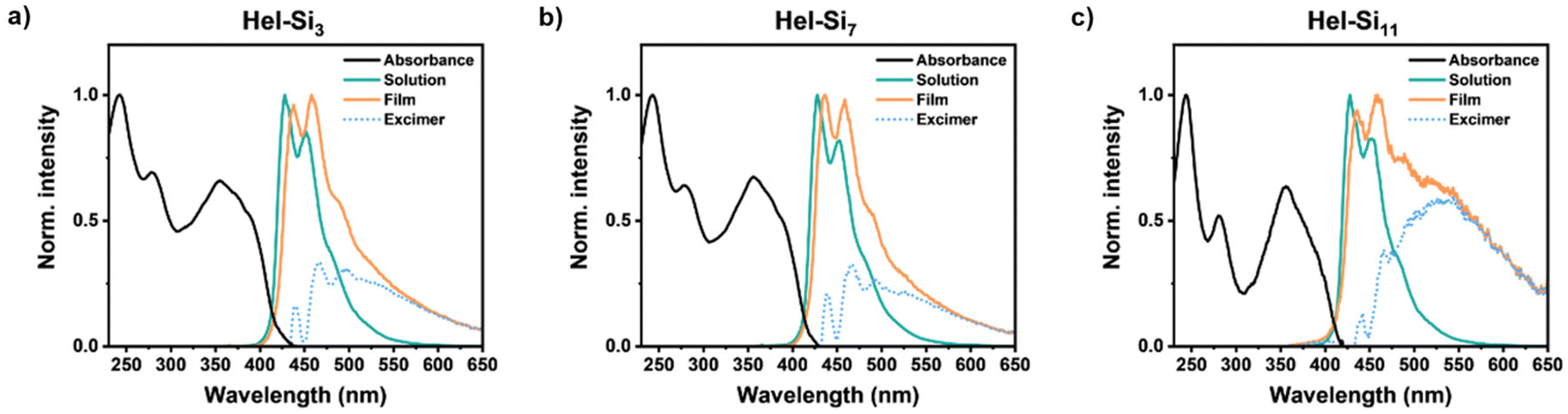
The emissive properties of Hel-Sin in bulk were then studied to compare with the emissive properties in dilute solution and to understand the role of excimers in the observations. Overall, each Hel-Sin shows a similar trend in the solid state and in solution, with deep blue emission in solution and light blue emission in the solid state (Fig. 5, Fig. S70†). The spectrum in solution has the typical signature of helicene compounds and consists mainly of two peaks, one at 427 nm, the most intense, and one at 450 nm. The solid-state emission shows a bathochromic shift of the two peaks of 7 nm and 5 nm, respectively, with the same relative intensity and a broad band appearing around 550 to 650 nm. This band is attributed to the formation of an excimer between the pyrene moieties at positions 2 and 15 of the helicene, as can be seen by plotting the difference between emissions of enantiomeric Hel-Sin in the solid state and monomeric Hel-Sin in solution. In contrast, no excimer was observed in dilute solution (10−5 M). This lack of visible excimer formation in the monomer indicates that excimer formation in the solid state likely involves two pyrene moieties from different molecular points, supporting the intermolecular nature of the excimer.
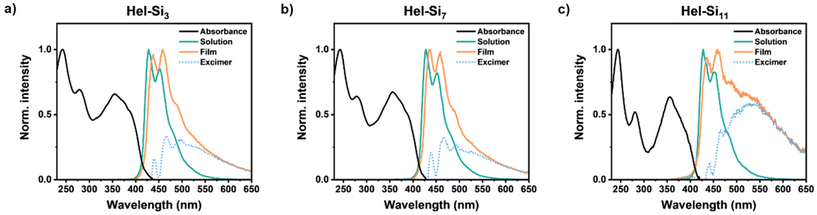 |
| | Fig. 5 Absorption and emission spectra of M-Hel-Sin (a, b, and c for n = 3, 7 and 11, respectively) in 10−5 M THF solution and in the solid state recorded as a thin film. The dotted blue trace is the difference between the emission spectra in the solid state and solution to show the excimer. The spectra have all been normalized to unit intensity. The spectra for P enantiomers are similar (Fig. S70†). | |
Consequences in the CPL activity of Hel-Sin in solution and in the solid state
The chiroptical properties of helicenes prompted us to investigate the consequences of the excimer state on the CPL activity of Hel-Sin. Fig. 6 shows the solution CPL spectra of the three enantiomer pairs of the Hel-Sin molecules, which generally have the same shapes and CPL dissymmetry factors (glum values) on the order of ±7 × 10−3. Interestingly, these values are similar in both magnitude and sign to the gabs at lower energy (of positive sign for the P enantiomers, see Table 2), suggesting that the same excited state is involved upon excitation and emission, with very low electronic and geometric reorganization. We rationalize that this similarity originates from a behaviour closely related to that of the classical helicene derivatives in the solution.1
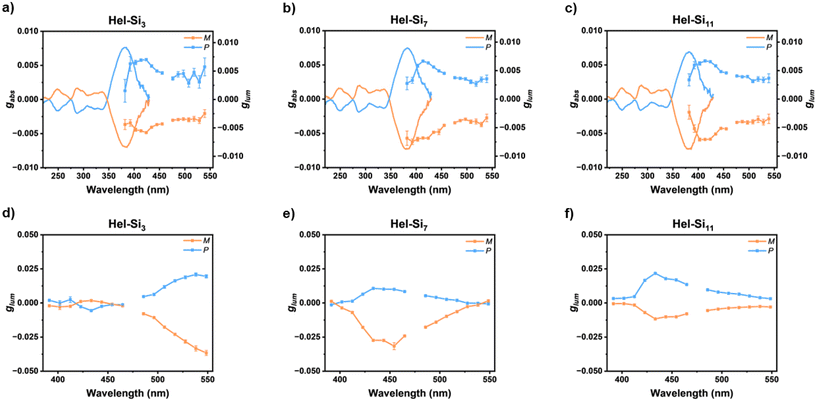 |
| | Fig. 6 (a–c) Absorption and emission dissymmetry factors gabs and glum of Hel-Sin in solution (C = 10−5 M in THF) and (d–f) emission dissymmetry factors glum of Hel-Sin in the solid state. | |
Table 2 Absorption and emission dissymmetry factors gabs and glum found for Hel-Sin (a) in 10−5 M THF solutions and (b) in the lamellar solid state (see the ESI† for excitation wavelengths)
| |
|
M-Hel-Si3 |
P-Hel-Si3 |
M-Hel-Si7 |
P-Hel-Si7 |
M-Hel-Si11 |
P-Hel-Si11 |
P-Hel-OMe |
M-Hel-OMe |
|
Measured for the racemic form (see Table S4†).
Measured for the racemic form in CH2Cl2 (see Table S5†).
Calculated ratio of glum values between the lamellar solid state and the solution.
|
| Solution (THF) |
g
abs (λabs) |
−7.0 × 10−3 (382 nm) |
+7.6 × 10−3 (382 nm) |
−7.2 × 10−3 (382 nm) |
+7.4 × 10−3 (382 nm) |
−7.2 × 10−3 (382 nm) |
+6.8 × 10−3 (382 nm) |
|
|
|
g
lum (λlum) |
−5.5 × 10−3 (422 nm) ± 1.3 × 10−4 |
+6.9 × 10−3 (422 nm) ± 2.3 × 10−4 |
−6.8 × 10−3 (422 nm) ± 1.0 × 10−4 |
+6.3 × 10−3 (422 nm) ± 1.2 × 10−4 |
−7.0 × 10−3 (422 nm) ± 1.1 × 10−4 |
+6.7 × 10−3 (422 nm) ± 1.3 × 10−4 |
−5 × 10−3 (440 nm) |
+6 × 10−3 (440 nm) |
|
Φ
lum (%) |
13.4a |
|
12.3a |
|
13.2a |
|
10b |
|
| Lamellar solid state |
g
lum (λlum) |
−1.9 × 10−2 (538 nm) ± 1.9 × 10−3 |
+3.6 × 10−2 (538 nm) ± 1.1 × 10−3 |
−2.7 × 10−2 (440 nm) ± 2.2 × 10−4 |
1.0 × 10−2 (440 nm) ± 2.6 × 10−4 |
−1.2 × 10−2 (433 nm) ± 3.0 × 10−4 |
2.1 × 10−2 (433 nm) ± 2.7 × 10−4 |
|
|
| X-fold increasec |
3.4 |
5.2 |
3.9 |
1.6 |
1.7 |
3.1 |
|
|
|
Φ
lum (%) |
5.9a |
|
6.6a |
|
8.1a |
|
|
|
The situation is different for the ordered nanostructures in bulk where higher glum values were observed (up to 5 times higher, see Table 2 and Fig. 6). Furthermore, while the CPL bands for Hel-Si7 and Hel-Si11 are centred at monomer emission wavelengths similar to those in the solution state, this is not the case for Hel-Si3. In fact, Hel-Si3 showed high CPL activity around 550 nm, i.e. in the excimer emission region, but showed almost no signal in the monomer emission region, indicating intermolecular interactions between pyrenes in a chiral arrangement. In contrast, as the length of the siloxane chain is increased, the lamellar packing prevents the chiral intermolecular arrangement of pyrenes for CPL emission of excimers. This nicely illustrates how the solid-state supramolecular organization affects the overall chiroptical properties when going from Hel-Si3, which exhibits excimer of chiral nature, to Hel-Si7 and Hel-Si11, which show chiroptical properties similar to the monomer. It seems that the disordered nature of Hel-Si3 gives more degrees of freedom to the system, enabling it to achieve chiral excimeric nature through the pyrene units.
Conclusion
We have presented the assembly and chiroptical properties of oligodimethylsiloxane-helicene with varying lengths of discrete siloxane oligomers in solution and bulk. In apolar and polar solutions, these BCOs do not aggregate and exhibit the same chiroptical response as the helical backbone without the oDMS units. Different properties were found between the racemic and enantiopure systems. In bulk, pure enantiomers of Hel-Sin with short oDMS chains (3 repeating units) form a disordered phase, while with longer oDMS chains (7 and 11 repeating units), they assemble into an ordered 2D lamellar morphology. As a result of these different assembled structures, different chiroptical properties are obtained. While the less-ordered state of Hel-Si3 exhibits CPL emission from an excimer of chiral nature, the lamellar structures of Hel-Si7 and Hel-Si11 show CPL emission from the monomer but hardly any circular polarization in the emission of the excimer. Overall, up to a 5-fold increase in CPL was observed in the solid state as compared to the solution, with a clear influence of the pyrene excimer emission. These results demonstrate that control of the bulk assembly by molecular design allows for the modulation of chiroptical emission. The transition from single molecules in solution to functional supramolecular structures in soft matter is enabled by the functionalization of helicenes with discrete oDMS chains. Hereby, a step forward is made in the construction of bulk supramolecular systems with tuneable chiroptical properties.13
Author contributions
S. Cadeddu: synthesis and spectroscopic characterization; B. W. L. van den Bersselaar: POM, DSC, and MAXS/WAXS measurements; Bas de Waal: synthesis of discrete oDMS; S. Cadeddu and B. W. L. van den Bersselaar: data curation, formal analysis, visualization, and writing; S. Meskers: chiroptical and emissive properties and writing; N. Vanthuyne: isolation of enantiomers; M. Cordier: X-ray analysis; G. Vantomme and J. Crassous: conceptualization, supervision, validation, writing – review & editing and funding.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the Ministère de l'Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS). The European Commission Research Executive Agency (Grant Agreement number: 859752 – HEL4CHIROLED – H2020-MSCA-ITN-2019), the Dutch Research Council (OCENW.M20.256), and the Dutch Ministry of Education, Culture and Science (Gravity program 024.005.020) are thanked for financial support. Prof. E. W. Meijer is warmly thanked for insightful discussions.
References
-
(a) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed
 ;
(b) M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC ;
(c) M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC ;
(d)
C.-F. Chen and Y. Shen, Helicene Chemistry: From Synthesis to Applications, Springer, Berlin, Heidelberg, 2017 CrossRef ;
(e) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed ;
(f)
Helicenes – Synthesis, Properties and Applications, ed. J. Crassous, I. G. Stará and I. Starý, Wiley, 2022 Search PubMed .
;
(b) M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC ;
(c) M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC ;
(d)
C.-F. Chen and Y. Shen, Helicene Chemistry: From Synthesis to Applications, Springer, Berlin, Heidelberg, 2017 CrossRef ;
(e) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed ;
(f)
Helicenes – Synthesis, Properties and Applications, ed. J. Crassous, I. G. Stará and I. Starý, Wiley, 2022 Search PubMed .
-
(a) A. J. Lovinger, C. Nuckolls and T. J. Katz, J. Am. Chem. Soc., 1998, 120, 264–268 CrossRef CAS ;
(b) K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899–11907 CrossRef CAS PubMed ;
(c) T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684–3687 CrossRef CAS PubMed ;
(d) T. Hirose, N. Ito, H. Kubo, T. Sato and K. Matsuda, J. Mater. Chem. C, 2016, 4, 2811–2819 RSC ;
(e) J. S. Valera, R. Gómez and L. Sánchez, Org. Lett., 2018, 20, 2020–2023 CrossRef CAS PubMed ;
(f) R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T.-K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709–7719 CrossRef PubMed ;
(g) R. Rodríguez, C. Naranjo, A. Kumar, K. Dhbaibi, P. Matozzo, F. Camerel, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, Chem. – Eur. J., 2023, 29, e202302254 CrossRef PubMed ;
(h) N. Saito and M. Yamaguchi, Molecules, 2018, 23, 277 CrossRef PubMed .
- F. Wang, F. Gan, C. Shen and H. Qiu, J. Am. Chem. Soc., 2020, 142, 16167–16172 CrossRef CAS PubMed .
-
(a) B. Oschmann, J. Lawrence, M. W. Schulze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pester, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6, 668–673 CrossRef CAS PubMed ;
(b) W. Zhang, W. Yang, H. Pan, X. Lyu, A. Xiao, D. Liu, Y. Liu, Z. Shen, H. Yang and X.-H. Fan, Soft Matter, 2022, 18, 3430–3436 RSC ;
(c) T. Nishimura, S. Katsuhara, C. Lee, B. J. Ree, R. Borsali, T. Yamamoto, K. Tajima, T. Satoh and T. Isono, Nanomaterials, 2022, 12, 1653 CrossRef CAS PubMed .
-
(a) R. H. Zha, G. Vantomme, J. A. Berrocal, R. Gosens, B. de Waal, S. Meskers and E. W. Meijer, Adv. Funct. Mater., 2018, 28, 1703952 CrossRef ;
(b) B. A. G. Lamers, R. Graf, B. F. M. De Waal, G. Vantomme, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 15456–15463 CrossRef CAS PubMed ;
(c) W. Yang, W. Zhang, L. Luo, X. Lyu, A. Xiao, Z. Shen and X.-H. Fan, Chem. Commun., 2020, 56, 10341–10344 RSC ;
(d) M. H. C. van Son, A. M. Berghuis, F. Eisenreich, B. de Waal, G. Vantomme, J. Gómez Rivas and E. W. Meijer, Adv. Mater., 2020, 2004775 CrossRef CAS PubMed .
- A. Terfort, H. Gorls and H. Brunner, Synthesis, 1997, 79–86 CrossRef CAS .
-
(a) M. Jakubec, T. Beranek, P. Jakubik, J. Sykora, J. Zadny, V. Cirkva and J. Storch, J. Org. Chem., 2018, 83, 3607–3616 CrossRef CAS PubMed ;
(b)
F. B. Mallory and C. W. Mallory, Organic Reactions, J. Wiley &, Sons, Inc., 1984, vol. 30, pp. 3 Search PubMed .
- J. Wade, F. Salerno, R. C. Kilbride, D. K. Kim, J. A. Schmidt, J. A. Smith, L. M. LeBlanc, E. H. Wolpert, A. A. Adeleke, E. R. Johnson, J. Nelson, T. Mori, K. E. Jelfs, S. Heutz and M. J. Fuchter, Nat. Chem., 2022, 14, 1383–1389 CrossRef CAS PubMed .
-
J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates and Resolutions, John Wiley & Sons, 1981 Search PubMed .
-
(a) B. A. G. Lamers, B. van Genabeek, J. Hennissen, B. F. M. de Waal, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2019, 52, 1200–1209 CrossRef CAS PubMed ;
(b)
B. A. G. Lamers, A. Herdlitschka, T. Schnitzer, M. F. J. Mabesoone, S. M. C. Schoenmakers, B. F. M. de Waal, A. R. A. Palmans, H. Wennemers and E. W. Meijer.
-
(a) F. Pop, P. Auban-Senzier, A. Frąckowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed ;
(b) F. Pop, N. Zigon and N. Avarvari, Chem. Rev., 2019, 119, 8435–8478 CrossRef CAS PubMed ;
(c) B. Rice, L. M. LeBlanc, A. Otero-de-la-Roza, M. J. Fuchter, E. R. Johnson, J. Nelson and K. E. Jelfs, Nanoscale, 2018, 10, 1865–1876 RSC ;
(d) L. Favereau, C. Quinton, C. Poriel, T. Roisnel, D. Jacquemin and J. Crassous, J. Phys. Chem. Lett., 2020, 11, 6426–6434 CrossRef CAS PubMed .
- Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS PubMed .
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfold and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Bas
de Waal
b,
Marie
Cordier
b,
Bas
de Waal
b,
Marie
Cordier