
One-pot synthesis of heterostructured CsPbBr3/PdSe nanowires with excellent humidity stability†
Shuai
Ye
 ab,
Mingyi
Huang
a,
Jun
Song
*ab and
Junle
Qu
*ab
ab,
Mingyi
Huang
a,
Jun
Song
*ab and
Junle
Qu
*ab
aState Key Laboratory of Radio Frequency Heterogeneous Integration (Shenzhen University); College of Physics and Optoelectronic Engineering, Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, Shenzhen University, Shenzhen 518060, P. R. China. E-mail: songjun@szu.edu.cn
bMedical Engineering and Technology College, Xinjiang Medical University, Urumqi 830011, P. R. China. E-mail: jlqu@szu.edu.cn
First published on 21st October 2024
Abstract
Lead halide perovskite nanowires (NWs) have garnered increasing attention owing to their unique properties, including axial carrier transportation, low pumping thresholds, and polarized emission. However, their intrinsic instability significantly limits their applications. In this study, heterostructured CsPbBr3/PbSe NWs with a diameter of 10 nm and lengths ranging from several to tens of microns were successfully synthesized via a one-pot solution-phase process. Photoluminescence measurements revealed that the CsPbBr3/PbSe NWs emitted green fluorescence with a narrow width of 16 nm and a high photoluminescence quantum yield (PLQY) of 37.6%. The obtained CsPbBr3/PbSe NWs underwent anion exchange with I− ion precursors to transform into CsPbBr3−xIx/PbSe, but not with Cl− ion precursors. Remarkably, these heterostructured CsPbBr3/PbSe NWs remained stable when immersed in water or exposed to air for several months, demonstrating excellent humidity stability. This study offers an effective approach for preparing stable CsPbBr3 NWs and fosters their future applications in optoelectronic devices.
Introduction
Lead halide perovskite nanocrystals (NCs) represent one of the most promising materials for optoelectronic applications due to their exceptional properties.1–5 Significant advancements have been achieved in synthesizing perovskite NCs with diverse morphologies, including 0D quantum dots (QDs),6,7 1D nanowires (NWs),8 and 2D nanoplates (NPs).9 Among these, 1D NWs have garnered considerable attention owing to their unique characteristics such as axial carrier transport, low pumping thresholds, and polarized emission, demonstrating substantial potential in applications such as photodetectors, lasers, and X-ray scintillators.10–13 However, perovskite NCs face challenges in maintaining stability over extended periods in polar solvents, moisture, or high temperatures due to their ionic nature.14,15 The intrinsic instability of lead-halide perovskites severely limits their practical applications, necessitating urgent solutions.Numerous studies have been conducted in the past decade to enhance the stability of perovskite NCs. For instance, all-inorganic perovskite QDs (CsPbX3) have been extensively explored. Common strategies for enhancing the stability of CsPbX3 QDs involve their encapsulation within a hydrophobic matrix or coating with protective shells, isolating them from the surrounding environment.16,17 More recently, a variety of heterostructured CsPbX3 QDs have been developed, significantly improving their stability.18–20 These heterostructured CsPbX3 QDs are typically synthesized using straightforward methods. For instance, Zhang et al. employed an effective sol–gel process to prepare CsPbX3-based heterostructures with ZrO2 or SiO2.21,22 Wang et al. sulfureted the surface of CsPbX3 QDs with hexamethyldisilathiane at room temperature to produce heterostructured CsPbX3–PbS (X = Cl, Br, I) QDs.23 Manna et al. reported a simple hot-injection method for fabricating CsPbX3–Pb4S3Br2 QDs heterostructures.24
These successful strategies for fabricating CsPbX3 QDs can also enhance the stability of CsPbX3 NWs. However, relatively few related studies have been conducted to date. Core–shell CsPbBr3/MoO3 NWs were synthesized by a vapor–liquid–solid (VLS) process.25 The MoO3 shell effectively passivated the surface of the CsPbBr3 NW cores, thereby reducing the diffusion of harmful water and oxygen molecules and enhancing air stability. Additionally, core–shell PbSe@CsPbBr3 wire heterostructures were prepared through conformal epitaxy of a CsPbBr3 shell on a PbSe wire core using chemical vapor deposition (CVD).26 Compared to the VLS and CVD processes, the solution-phase process is simpler, more economical, and widely used for preparing perovskite NWs. Here, we propose a one-pot strategy for synthesizing heterostructured CsPbBr3/PbSe NWs using a solution-phase process. The resulting CsPbBr3/PbSe NWs had a diameter of approximately 10 nm and lengths ranging from several to tens of microns. They remained stable even after immersion in water or exposure to air for several months, exhibiting excellent humidity stability.
Experimental
Materials
PbCl2 (99.999%), PbBr2 (99.999% pure), PbI2 (99.999% pure), Cs2CO3 (99.9%), 1-octadecene (ODE, 90%), oleic acid (OA, 90%), oleylamine (OLA, 70% pure), 1-dodecanethiol (DDT, 99.9%) and selenium powders (Se, 99.999%) were purchased from Sigma-Aldrich (St Louis, MO, USA). Analytical hexane was purchased from Macklin Biochemical Co., Ltd (Shanghai, China). All chemicals were used without further purification.Preparation of Pb precursor solution
0.3 mmol of PbBr2 powders was added to a 50 ml stand-up flask containing 5 mL of ODE, 0.25 mL of OA, and 0.5 mL of OLA. The flask was heated to 160 °C in a flowing argon atmosphere under vigorous stirring until the PbBr2 powders were dissolved completely. Then, the obtained Pb/Br precursor solution was cooled to room temperature and stored for subsequent use. The Pb/Cl precursor solution and Pb/I precursor solution were prepared by a similar process.Preparation of Cs precursor solution
0.1 mmol of Cs2CO3 powders was added to a 50 ml stand-up flask containing 5 mL of ODE and 0.25 mL of OA. The flask was heated to 160 °C in a flowing argon atmosphere under vigorous stirring until the Cs2CO3 powders were dissolved completely. Then, the obtained solution was cooled to room temperature and stored for subsequent use.Preparation of Se solution
The Se solution was prepared by a previous approach developed by Yang et al. 1.5 mmol of Se powders were added to a brown vial containing 0.75 ml of DDT and 0.75 ml of OLA. The mixture in the vial was stirred vigorously using a magnetic bar at room temperature for more than 4 hours. The Se powders were dissolved completely and the Se solution was obtained.Preparation of CsPbBr3/PbSe nanowires (NWs)
5.75 ml of Pb/Br precursor solution, 5.25 ml of Cs precursor solution and 30 μl of Se solution were simultaneously added to a 50 ml stand-up flask at room temperature. Note that an additional 0.5 ml of OA and 0.5 ml of OLA must be added to prepare the CsPbBr3/PbSe NWs. The mixed solutions were stirred under a flowing Ar atmosphere and then heated quickly to 160 °C (∼5 min). After maintaining the temperature for 20 min at 160 °C, the reacted flask was allowed to cool naturally to room temperature. The crude solutions were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min to precipitate the products. The supernatants were removed and the precipitates were dispersed in 10 mL of hexane. The dispersed solution was centrifuged at 1000 rpm for 10 min to precipitate the CsPbBr3/PbSe NWs. Finally, the collected CsPbBr3/PbSe NWs were dispersed in 10 mL of hexane again and stored for subsequent use.
000 rpm for 10 min to precipitate the products. The supernatants were removed and the precipitates were dispersed in 10 mL of hexane. The dispersed solution was centrifuged at 1000 rpm for 10 min to precipitate the CsPbBr3/PbSe NWs. Finally, the collected CsPbBr3/PbSe NWs were dispersed in 10 mL of hexane again and stored for subsequent use.
Characterization
An X-ray diffractometer (XRD, X'Pert PRO, Netherlands) with Cu Kα radiation was used to analyze the microstructures of the NWs. Transmission electron microscopy (TEM, JEM-T7700, Japan) was used to observe the samples’ morphologies. Ultraviolet/visible (UV/vis) absorption spectra were recorded using a spectrophotometer (U-2800, Hitachi, Japan). Photoluminescence (PL) spectra were recorded and time-resolved fluorescence measurements were taken with a Fluorolog®-3 spectrofluorometer (HORIBA Scientific, Japan). The photoluminescent quantum yield (PLQY) was also measured with the Fluorolog®-3 spectrofluorometer by direct methods. All spectra were measured at room temperature in a quartz cuvette.Results and discussion
To synthesize the CsPbBr3/PbSe NWs, PbBr2 and Cs2CO3 powders were pre-dissolved in ODE/OA/OLA and ODE/OA, respectively, to prepare the precursor solutions. The Pb precursor, Cs precursor, and OLA/DDT solutions with dissolved Se were then mixed simultaneously at room temperature; subsequently, the mixture was rapidly heated to 160 °C and the reaction was maintained for 20 min. Notably, the amounts of OA and OLA needed to be adjusted to control the morphologies of the products. The detailed procedure is described in the ESI.† Three crucial factors must be considered when preparing the CsPbBr3/PbSe NWs. The first is the reaction precursor: CsPbBr3/PbSe NWs are only produced when the pre-dissolved Pb precursor, Cs precursor, and OLA/DDT solutions with dissolved Se are simultaneously mixed at room temperature. CsPbBr3 nanocubes and nanorods are produced when the precursors are PbBr2 and Cs2CO3 powders, respectively (Fig. S1†). A similar process was used in the solvothermal process for preparation of CsPbX3 NCs.27 The second factor is the stoichiometric ratio of OA to OLA: products with a high content of CsPbBr3/PbSe NWs are obtained when 1 ml of OA and 1 ml of OLA are used (Fig. S2, S3 and Table S1†). The last factor is the addition of DDT and Se: synthesizing CsPbBr3/PbSe NWs in the absence of DDT or Se in the initial solution is challenging (Fig. S4†).The formation mechanism of CsPbBr3/PbSe NWs was investigated initially. Samples were quenched at various stages during the preparation process, and their morphologies and absorption spectra were analyzed (see Fig. 1 and S5, and Table S2†). During the heating stage from room temperature to 160 °C (Fig. 1a–e), the products exhibited diverse morphologies at different temperatures. Products quenched at 80 °C (Fig. 1a) displayed complex morphologies and non-uniform sizes, with many nanocrystals smaller than 5 nm, resulting in weak cyan fluorescence (Fig. S5a†). When the reaction temperature was increased to 100 °C (Fig. 1b), the products predominantly consisted of hexagonal nanocrystals with uniform sizes and irregularly shaped nanosheets. Analysis of the absorption spectrum (Fig. S5b†) showed a characteristic peak centered at 311 nm, suggesting that the hexagonal nanocrystals were Cs4PbBr6. Further raising the temperature to 120 °C (Fig. 1c) led to the nanosheets becoming regular squares with widths of approximately 40 nm, resulting in a change in the emission color of the products to green. Additionally, hexagonal Cs4PbBr6 nanocrystals were present. NWs several hundred nanometers in length were initially observed in products quenched at 140 °C (Fig. 1d), with their lengths increasing as the temperature was raised to 160 °C (Fig. 1e). During the heat-holding stage at 160 °C (Fig. 1f), more NWs nucleated and grew, while the hexagonal nanocrystals and square nanosheets disappeared over time. However, the NWs gradually transformed into larger crystals as the time increased beyond 20 min (Fig. 1g–i). Consequently, intermediates with various morphologies were obtained and gradually transformed under the given conditions during the formation of CsPbBr3/PbSe NWs. The formation mechanism of CsPbBr3/PbSe NWs closely resembled that of pure CsPbBr3 NWs reported by Yang et al., wherein the NWs were nucleated and grown through a surfactant-directed 1D mode, rather than by the oriented attachment of NCs or seed-mediated anisotropic growth.28 Notably, CsPbBr3/PbSe NWs were formed within 20 min, considerably faster than the time required for pure CsPbBr3 NWs (90 min).
 | ||
| Fig. 1 TEM images of samples quenched at different times during the transformation from the precursor solution into NWs: (a) 80 °C, (b) 100 °C, (c) 120 °C, (d) 140 °C, (e) 160 °C, 0 min, (f) 160 °C, 20 min, (g) 160 °C, 30 min, (h) 160 °C, 60 min, and (i) 160 °C, 90 min. | ||
The microstructure of the colloidal CsPbBr3/PbSe NWs was characterized using X-ray diffraction (XRD). The XRD pattern in Fig. 2a indicates that the synthesized NW samples exclusively comprised the CsPbBr3 and PbSe phases. As is known, CsPbBr3 exhibits three different phases, namely cubic, tetragonal, and orthorhombic, with phase transitions occurring at 130 °C from cubic to tetragonal and at 88 °C from tetragonal to orthorhombic.29 The distinct double peaks at approximately 30° corresponding to the (002) and (200) lattice planes (JCPDS 18-0364) confirmed the orthorhombic CsPbBr3 phase in the CsPbBr3/PbSe NWs. Additionally, diffraction peaks at 25.1° and 28.5° corresponded to the (111) and (200) lattice planes, respectively, of the cubic PbSe phase (JCPDS 06-0354), indicating the presence of the PbSe phase. The morphologies of the CsPbBr3/PbSe NWs were examined using transmission electron microscopy (TEM) (Fig. 2b and c). The synthesized CsPbBr3/PbSe NWs formed bundles, with each bundle consisting of separate NWs with a uniform size of approximately 10 nm. The CsPbBr3/PbSe NWs ranged from several microns to tens of microns in length. The high-resolution high-angle annular dark-field scanning TEM (HAADF-STEM) image in Fig. 2d depicted single crystal CsPbBr3/PbSe NWs in the [100] direction, with an interplanar distance of ∼0.58 nm corresponding to the (100) plane of CsPbBr3. Furthermore, tiny PbSe nanoparticles (1–3 nm in size) were embedded in the CsPbBr3 matrix. The XPS spectrum (Fig. S6†) also confirmed the coexistence of Cs+, Pb2+, Br−, and the peak at 59.2 eV in the XPS spectrum was assigned to the Se 3d signals of Se2−, indicating that Se2− had been incorporated into the NWs. EDS analysis (Fig. S7†) further confirmed the coexistence of Cs, Pb, Br, and Se in the CsPbBr3/PbSe NWs. Another experiment involved reacting the PbBr2 precursor solution solely with the Se precursor solution at 160 °C for 5 min, resulting in cubic PbSe nanoparticles with sizes in the range of 5–10 nm (Fig. S8†). This experiment confirmed the formation of PbSe nanoparticles under the reaction conditions used to prepare the CsPbBr3/PbSe NWs. Combined with the aforementioned formation mechanism, it can be inferred that the nucleation and growth of CsPbBr3/PbSe NWs occurred. In the early stages, CsPbBr3 and PbSe seeds formed simultaneously. The number of CsPbBr3 seeds was considerably higher than that of PbSe seeds due to the higher Br concentration compared to the Se concentration in the precursor solution. During the growth process of the NWs, the PbSe seeds grew into PbSe NCs and were gradually surrounded by growing CsPbBr3 seeds, forming CsPbBr3/PbSe heterostructures. These heterostructures inhibited the growth of PbSe NCs, resulting in a smaller size of the PbSe NCs in the CsPbBr3/PbSe NWs than in the PbSe NCs (Fig. S8†).
 | ||
| Fig. 2 Structural characterization of colloidal CsPbBr3/PbSe NWs: (a) XRD pattern, (b–c) TEM images, and (d) HAADF-STEM image. | ||
The optical properties of the synthesized CsPbBr3/PbSe NWs were characterized (Fig. 3). The absorption spectrum of CsPbBr3/PbSe NWs (Fig. 3a) revealed an absorption peak at 510 nm. In contrast to the sharp edges of pure CsPbBr3 QDs or NWs, an absorption tail in the long-wavelength direction was observed for CsPbBr3/PbSe NWs, attributed to the growth of PbSe nanoparticles within the CsPbBr3/PbSe NWs. The photoluminescence (PL) spectrum of CsPbBr3/PbSe NWs displayed a single narrow emission peak at 516 nm with a width of 16 nm, similar to that of CsPbBr3 QDs. The PL quantum yield of the CsPbBr3/PbSe NWs was measured at 37.6% (Fig. 3b), which closely approximates the value reported for pure 10.0 nm thick CsPbBr3 NWs (∼38%) by Manna et al.30 Time-resolved PL measurements of the CsPbBr3/PbSe NWs (Fig. 3c) revealed bi-exponential decay traces, with an average lifetime of only 6.88 ns, 42% lower than that of pure 10.0 nm thick CsPbBr3 NWs (16.4 ns).30 To further investigate the optical properties, the CsPbBr3/PbSe NWs were deposited onto a glass slide and imaged using confocal microscopy under excitation by a 488 nm continuous-wave laser (Fig. 3d–f). The 10 nm thick CsPbBr3/PbSe NWs were sufficiently long to be observed in bright field, with strong fluorescent signals detected in the fluorescence field. The merged images in Fig. 3d and e indicate fluorescence emitted by the CsPbBr3/PbSe NWs.
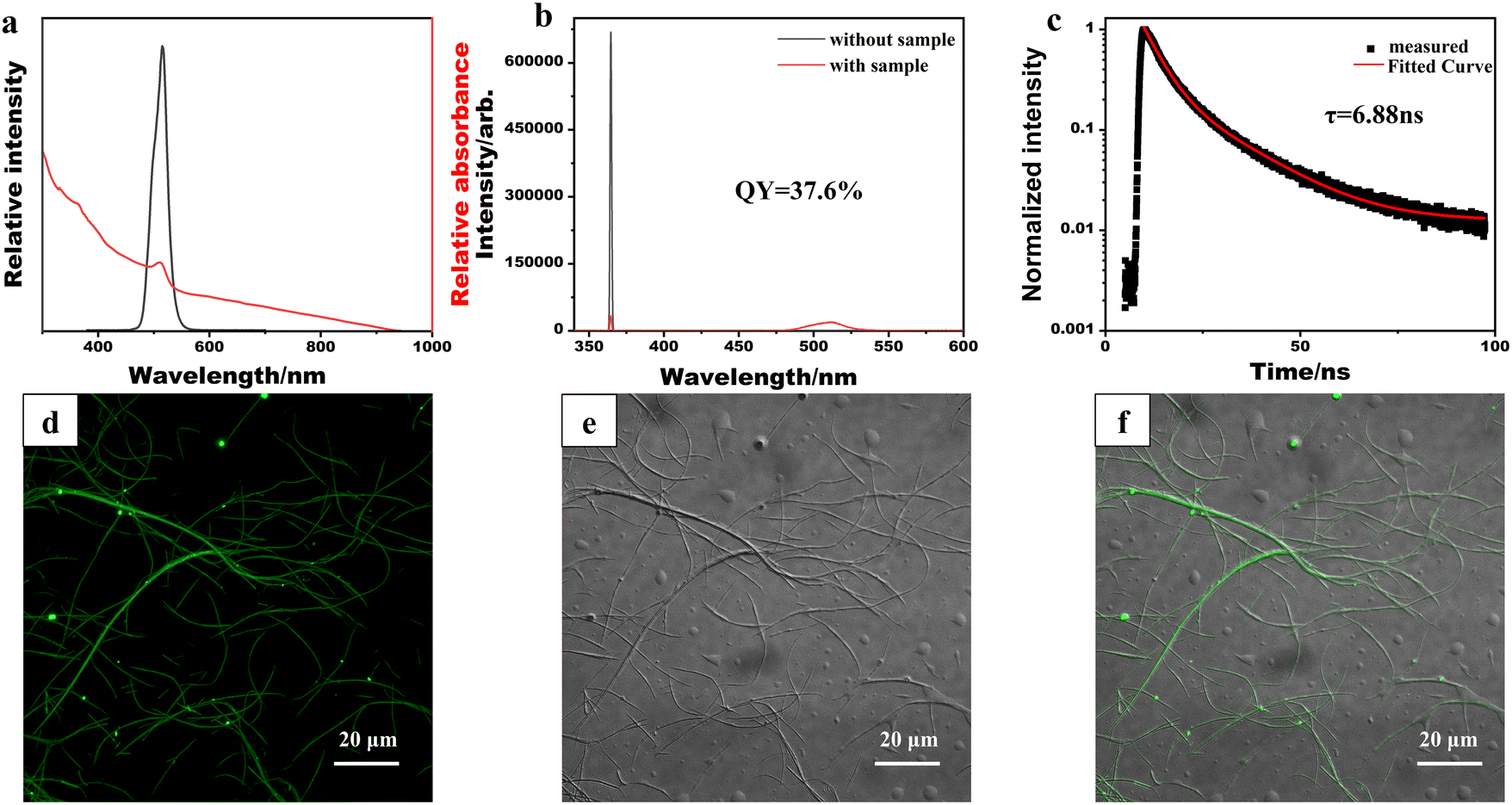 | ||
| Fig. 3 Optical characterization of CsPbBr3/PbSe nanowires: (a) UV/vis absorption and photoluminescence spectra, (b) PLQY, (c) lifetime, (d) confocal fluorescence image, (e) bright field image and (f) merged image of (d) and (e). | ||
In prior studies,31 anion exchange was utilized to adjust the emission band of halide perovskite nanocrystals. Similarly, the emission band of the synthesized CsPbBr3/PbSe NWs could be tuned via anion exchange. Fig. 4 illustrates the emission color changing from green to yellow to dark red as I− precursors were gradually introduced into the dispersion of CsPbBr3/PbSe NWs in hexane, providing a series of tunable PL signals from 516 to 660 nm with a narrow band (FWHM in the range of 15–40 nm). Anion exchange of CsPbBr3/PbSe NWs with I− ions occurred rapidly at room temperature, whereas exchange with Cl− ions was hindered. The emission color remained unchanged when Cl− precursors were added to the dispersion of CsPbBr3/PbSe NWs at room temperature (Fig. S9†). Similarly, the emission color of the CsPbBr3 QD dispersion quickly shifted from green to blue. This unusual phenomenon may be linked to internal strain within the CsPbBr3/PbSe NWs. Incorporation of PbSe nanoparticles into the CsPbBr3 matrix resulted in a 4.8% lattice mismatch, producing internal strain. Gradual release of this strain occurred upon anion exchange with I− ions due to a decreased mismatch (2.9%) with CsPbI3, making the reaction easily achievable. Conversely, the 8.8% lattice mismatch between PbSe and the CsPbCl3 lattice generated strong internal strain, hindering the anion exchange of CsPbBr3/PbSe NWs with Cl− ions.
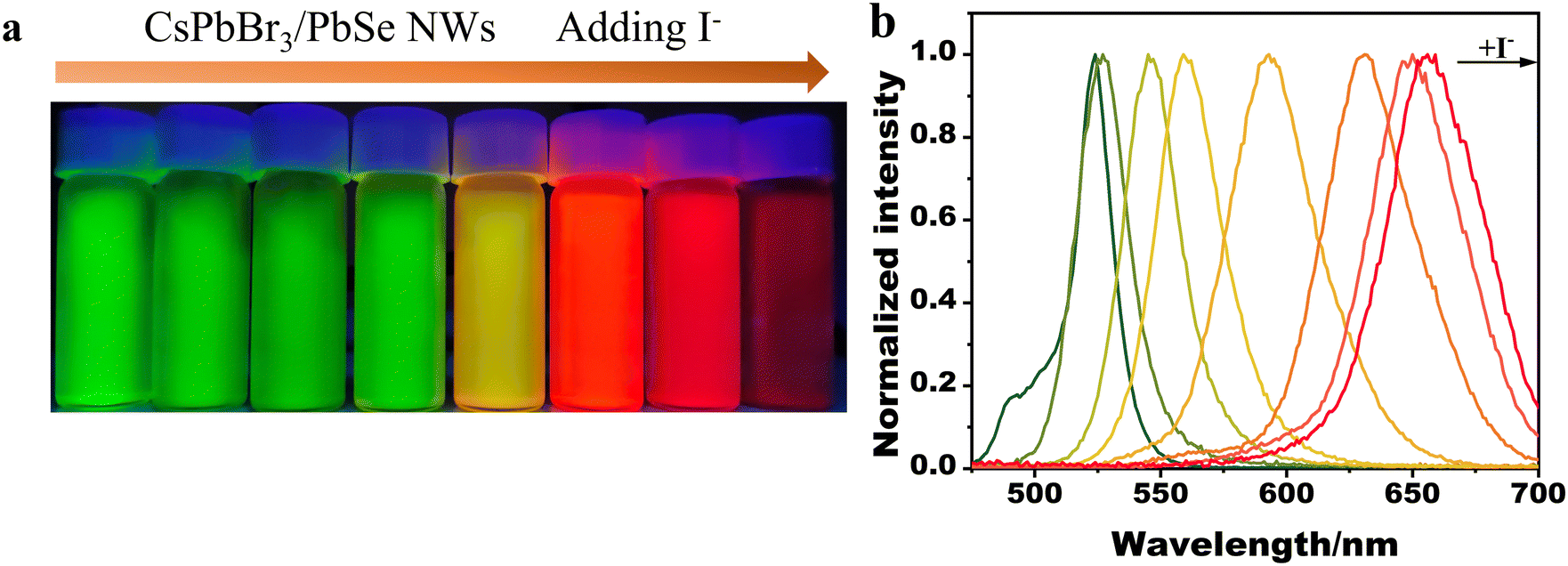 | ||
| Fig. 4 (a) Photograph of colloidal CsPbBr3/PbSe NWs reacting with different concentrations of I− ions under a 365 nm UV lamp; (b) corresponding photoluminescence spectra of colloidal CsPbX3/PbSe perovskite NWs. | ||
The integration of PbSe nanoparticles into the CsPbBr3 NWs significantly enhanced their humidity stability. Fig. 5 illustrates the stability of two CsPbBr3/PbSe NW samples. The sample labeled “in air” consisted of a CsPbBr3/PbSe NW film coated on a glass slide and exposed to air, whereas the other sample labeled “in water” comprised centrifuged CsPbBr3/PbSe NW powders immersed in a tube filled with deionized water. The CsPbBr3/PbSe NWs demonstrated robust stability in both air and water (Fig. 5a). The PL intensity of the CsPbBr3/PbSe NW film exposed to air retained approximately 95% of its original intensity after 30 days, while the fluorescence intensity of the CsPbBr3/PbSe NW film powders immersed in deionized water retained approximately 80% of its original PL intensity after the same duration. Fig. 5b presents photographs of the two CsPbBr3/PbSe NW samples under excitation by 365 nm lamps. The CsPbBr3/PbSe NW sample retained its original shape when exposed to air or immersed in water for 30 days, exhibiting strong green fluorescence under the 365 nm lamp. Notably, the PL of perovskite nanocrystals is typically completely quenched within several hours of immersion in water. The results depicted in Fig. 5 indicate the excellent humidity stability of the CsPbBr3/PbSe NWs. In previous research, core–shell CsPbBr3/MoO3 NWs have been developed to enhance the stability of CsPbBr3 NWs.25 The mobility of the CsPbBr3/MnO3 NWs was maintained at 80% after 7 days’ storage at 65% relative humidity. Another strategy to enhance the stability of CsPbBr3 NWs was to passivate their surfaces with thiourea. The passivated CsPbBr3 NWs retained approximately 80% of their original performance after 2 hours of water treatment.32 Comparatively, the CsPbBr3/PbSe NWs in this work demonstrated superior stability. However, a direct comparison of the stability between CsPbBr3/PbSe NWs and pure CsPbBr3 NWs is not feasible due to the current process's inability to produce pure CsPbBr3 NWs. To shed light on this, we refer to CsPbX3 QDs. Previous studies have highlighted that the stability of CsPbX3 QDs can be enhanced by incorporating metal chalcogenides such as ZnS,33 PbS,34 and PbTe.35 Density functional theory (DFT) calculations36 have revealed that the augmented stability of CsPbX3 QDs primarily stems from the strong interaction and high binding energy at the interface between the perovskite and the metal chalcogenide. Additionally, in the CsPbBr3 NWs passivated by thiourea,32 the enhanced stability was attributed to the surface passivation effect of thiourea, which forms a relatively stable Pb2+–thiourea complex with Pb2+. In the CsPbBr3/PbSe NWs, the incorporation of PbSe nanoparticles into the CsPbBr3 matrix led to the formation of a stable complex and high binding energy at the interface, thus improving the stability of the CsPbBr3 NWs.
 | ||
| Fig. 5 (a) PL intensity change of colloidal CsPbBr3/PbSe NWs in air and in water, (b) photographs of CsPbBr3/PbSe NW samples under a 365 nm lamp: top is the CsPbBr3/PbSe NWs film on a glass slide exposed to air, and bottom is the centrifuged CsPbBr3/PbSe NWs in a tube full of deionized wate. | ||
Conclusions
This study proposed a one-pot solution-phase process to synthesize heterostructured CsPbBr3/PbSe NWs and successfully obtained CsPbBr3/PbSe NWs with a diameter of 10 nm and lengths ranging from several to tens of microns. The CsPbBr3/PbSe NWs emitted green fluorescence with a narrow width of 16 nm and a high photoluminescence quantum yield (PLQY) of 37.6%. In contrast to pure CsPbBr3 NWs, the heterostructured CsPbBr3/PbSe NWs only underwent anion exchange with the I− ion precursor, transforming into CsPbBr3−xIx/PbSe, and did not react with the Cl− ion precursor. These heterostructured CsPbBr3/PbSe NWs demonstrated excellent humidity stability owing to the strong binding energy resulting from the incorporation of PbSe nanoparticles into the CsPbBr3 matrix; they maintained their strong emission even when immersed in water or exposed to air for several months.Author contributions
J. S. conceived the overall project and supervised the effort. S. Y. and M. H. designed the experiments, ran the synthetic protocols, and characterized the samples. J. S. and J. Q. helped in data curation. S. Y. wrote the original draft with help from all the authors. J. S. and J. Q. reviewed and revised the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study received partial support from the National Key R&D Program of China (2021YFF0502900), the National Natural Science Foundation of China (62175161/62127819), the Shenzhen Basic Research Program (JCYJ20210324095810028), and the Shenzhen Key Laboratory of Photonics and Biophotonics (ZDSYS20210623092006020). We thank the Instrument Analysis Center of Shenzhen University for their assistance with the TEM analysis.References
- Q. Zhang, Q. Shang, R. Su, T. T. H. Do and Q. Xiong, Nano Lett., 2021, 21, 1903–1914 CrossRef CAS PubMed.
- D. Yang, B. Zhao, T. Yang, R. Lai, D. Lan, R. H. Friend and D. Di, Adv. Funct. Mater., 2022, 32, 2109495 CrossRef CAS.
- H.-Y. Hou, S. Tian, H.-R. Ge, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Adv. Funct. Mater., 2022, 32, 2209324 CrossRef CAS.
- S. Ye, W. Yan, M. Zhao, X. Peng, J. Song and J. Qu, Adv. Mater., 2018, 30, 180016 Search PubMed.
- F. Zhao, A. Ren, P. Li, Y. Li, J. Wu and Z. M. Wang, ACS Nano, 2022, 16, 7116–7143 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- D. Zhang, Q. Zhang, Y. Zhu, S. Poddar, Y. Zhang, L. Gu, H. Zeng and Z. Fan, Adv. Energy Mater., 2023, 13, 2201735 CrossRef CAS.
- J. Shamsi, Z. Dang, P. Bianchini, C. Canale, F. D. Stasio and R. Brescia, J. Am. Chem. Soc., 2016, 138, 7240–7243 CrossRef CAS PubMed.
- Y. Fu, H. Zhu, C. C. Stoumpos, Q. Ding, J. Wang, M. G. Kanatzidis, X. Zhu and S. Jin, ACS Nano, 2016, 10, 7963–7972 CrossRef CAS PubMed.
- G. S. Kumar, R. R. Sumukam, R. K. Rajaboina, R. N. Savu, M. Srinivas and M. Banavoth, ACS Appl. Energy Mater., 2022, 5, 1342–1377 CrossRef CAS.
- Y. Meng, C. Lan, F. Li, S. Yip, R. Wei, X. Kang, X. Bu, R. Dong, H. Zhang and J. C. Ho, ACS Nano, 2019, 13, 6060–6070 CrossRef CAS PubMed.
- H. Dierks, Z. Zhang, N. Lamers and J. Wallentin, Nano Res., 2023, 16(1), 1084–1089 CrossRef CAS.
- Y. Zhou and Y. Zhao, Energy Environ. Sci., 2019, 12, 1495–1511 RSC.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- D. N. Dirin, L. Protesescu, D. Trummer, I. V. Kochetygov, S. Yakunin, F. Krumeich, N. P. Stadie and M. V. Kovalenko, Nano Lett., 2016, 16, 5866–5874 CrossRef CAS PubMed.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 1703535 CrossRef.
- Y. Wei, Y. Zhao, C. Liu, Z. Wang, F. Jiang, Y. Liu, Q. Zhao, D. Yu and M. Hong, Adv. Funct. Mater., 2021, 31, 2106386 CrossRef CAS.
- P. V. Kamat, N. Pradhan, K. Schanze, P. S. Weiss, J. Buriak, P. Stang, T. W. Odom and G. Hartland, ACS Energy Lett., 2020, 5, 2253–2255 CrossRef CAS.
- X. Tang, J. Yang, S. Li, Z. Liu, Z. Hu, J. Hao, J. Du, Y. Leng, H. Qin, X. Lin, Y. Lin, Y. Tian, M. Zhou and Q. Xiong, Adv. Sci., 2019, 6, 1900412 CrossRef CAS PubMed.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- H. Liu, Y. Tan, M. Cao, H. Hu, L. Wu, X. Yu, L. Wang, B. Sun and Q. Zhang, ACS Nano, 2019, 13, 5366–5374 CrossRef CAS PubMed.
- X. Zhang, X. Wu, X. Liu, G. Chen, Y. Wang, J. Bao, X. Xu, X. Liu, Q. Zhang, K. Yu, W. Wei, J. Liu, J. Xu, H. Jiang, P. Wang and X. Wang, J. Am. Chem. Soc., 2020, 142, 4464–4471 CrossRef CAS PubMed.
- M. Imran, L. Peng, A. Pianetti, V. Pinchetti, J. Ramade, J. Zito, F. D. Stasio, J. Buha, S. Toso, J. Song, I. Infante, S. Bals, S. Brovelli and L. Manna, J. Am. Chem. Soc., 2021, 143, 1435–1446 CrossRef CAS PubMed.
- Y. Meng, Z. Lai, F. Li, W. Wang, S. Yip, Q. Quan, X. Bu, F. Wang, Y. Bao, T. Hosomi, T. Takahashi, K. Nagashima, T. Yanagida, J. Lu and J. C. Ho, ACS Nano, 2020, 14, 12749–12760 CrossRef CAS PubMed.
- C. Fan, X. Xu, K. Yang, F. Jiang, S. Wang and Q. Zhang, Adv. Mater., 2018, 30, 1804707 CrossRef PubMed.
- M. Chen, Y. Zou, L. Wu, Q. Pan, D. Yang, H. Hu, Y. Tan, Q. Zhong, Y. Xu, H. Liu, B. Sun and Q. Zhang, Adv. Funct. Mater., 2017, 1701121 CrossRef.
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230–9233 CrossRef CAS PubMed.
- C. K. Moeller, Nature, 1958, 182, 1436 CrossRef.
- M. Imran, F. D. Stasio, Z. Dang, C. Canale, A. H. Khan, J. Shamsi, R. Brescia, M. Prato and L. Manna, Chem. Mater., 2016, 28, 6450–6454 CrossRef CAS PubMed.
- S. Ye, M. Zhao, J. Song and J. Qu, Nano Res., 2018, 11(9), 4654–4663 CrossRef CAS.
- P. Li, D. Yang, Y. Tan, M. Cao, Q. Zhong, M. Chen, H. Hu, B. Sun, Q. Zhong, M. Chen, H. Hu and B. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 3351–3359 CrossRef CAS PubMed.
- V. K. Ravi, S. Saikia, S. Yadav, V. V. Nawale and A. Nag, ACS Energy Lett., 2020, 5, 1794–1796 CrossRef CAS.
- M. Jagadeeswararao, P. Vashishtha, T. J. N. Hooper, A. Kanwat, J. W. M. Lim, S. K. Vishwanath, N. Yantara, T. Park, T. C. Sum, D. S. Chung, S. G. Mhaisalkar and N. Mathews, J. Phys. Chem. Lett., 2021, 12, 9569–9578 CrossRef CAS PubMed.
- L. Zhang, M. Zhu, Y. Sun, J. Zhang, M. Zhang, H. Zhang, F. Zhou, J. Qu and J. Song, Nano Energy, 2021, 90, 106506 CrossRef CAS.
- W. Chen, J. Hao, W. Hu, Z. Zang, X. Tang, L. Fang, T. Niu and M. Zhou, Small, 2017, 13, 1604085 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03554b |
| This journal is © The Royal Society of Chemistry 2024 |