
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thermally driven rotaxane–catenane interconversion with a dynamic bis(hindered amino) disulfide†
Rikito
Takashima
a,
Daisuke
Aoki
 *b,
Akira
Takahashi
a and
Hideyuki
Otsuka
*a
*b,
Akira
Takahashi
a and
Hideyuki
Otsuka
*a
aDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: otsuka@mac.titech.ac.jp
bDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba university, 1-33 Yayoi-cho, Inage-ku, Chiba-shi, Chiba 263-8522, Japan. E-mail: daoki@chiba-u.jp
First published on 13th November 2023
Abstract
We have developed a versatile and simple synthetic method to produce a [3]catenane. Heating a rotaxane with bis(hindered amino) disulfide groups at both ends spontaneously and selectively produces the [3]catenane. The successful polymerization of the obtained [3]catenane provides a platform for the synthesis of various interlocking polymers.
Catenanes1–5 are mechanically interlocked molecular architectures that contain two or more rings connected through space, a structure which gives rise to characteristic rotational motion. Due to their unique topology and the supramolecular interactions between the two or more rings, polymeric materials that contain catenated structures6–8 are flexible and tough, respond to stimuli, and dissipate energy well. For example, Huang et al. synthesized [2]catenane crosslinked polymers9 and revealed that the elastic modulus of the gels changed significantly as the catenane switched from a rigid to a flexible state in response to pH. A variety of catenane-containing polymeric materials have already been studied, such as those that contain [2]catenane crosslinking9–11 and molecular necklace crosslinking,12 as well as poly[2]catenanes13–16 and poly[n]catenanes.17–19
Much research has been conducted on cyclization reactions for the construction of catenanes and this has enabled us to access catenanes more efficiently. For example, esterification,20 amidation,21 ring-closing olefin metathesis,4,22 and azide–alkyne cycloaddition23,24 are efficient cyclization reactions which we can use for the synthesis of catenanes. However, to synthesize polymeric materials that have characteristics that reflect those of catenanes, it remains important to develop a more universal and simple cyclization method that can create catenanes with various functional groups and molecular structures.
Recently, we have developed a method for the selective isolation of macrocycles that contain bis(2,2,6,6-tetramethylpiperidin-1-yl) disulfide (BiTEMPS25–27) units in their cyclic structure.28 The BiTEMPS unit is a dynamic moiety, whose disulfide linker can exchange with other disulfides merely upon heating to ∼100 °C. Upon heating, linear precursors that have either repeating BiTEMPS units or units at both ends of the main chain are spontaneously converted into entropically favored macrocycles (Scheme 1a). The great functional tolerance of BiTEMPS also allows the successful introduction of various structures into macrocycles. Accordingly, it should be possible to achieve rotaxane–catenane conversion, an effective way to spontaneously obtain a catenane via the dynamic nature of BiTEMPS, if a rotaxane end-capped with BiTEMPS at both ends is prepared as a precursor for the cyclization reaction (Scheme 1b). Herein, we demonstrate the applicability of this approach, which only requires heating and produces a thermally dynamic catenane that represents a unique catenane design with dynamic covalent bonds and desirable functional structures. The resulting catenanes can be used in polymerization reactions29–32 through intermolecular bond-exchange reactions of BiTEMPS-containing macrocycles.
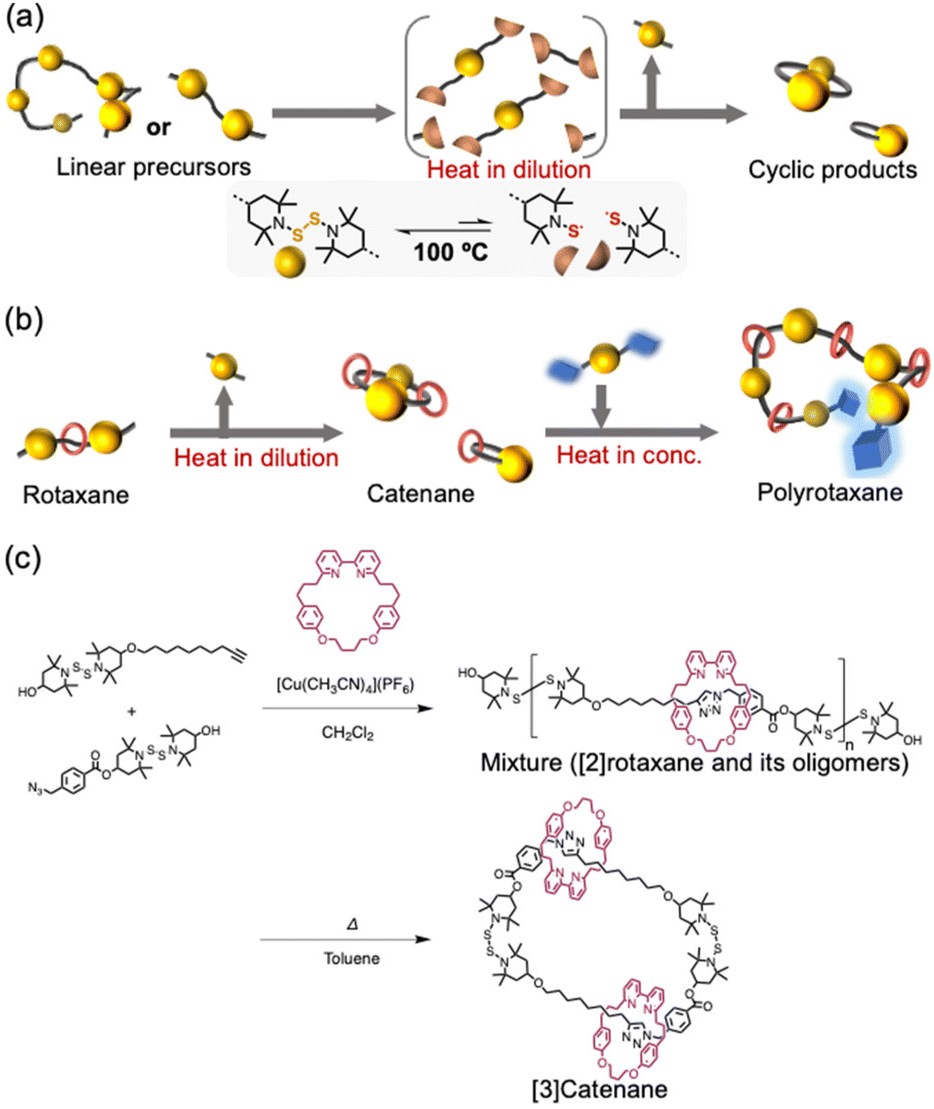 | ||
| Scheme 1 (a) Cyclization reactions based on the dynamic behavior of BiTEMPS. (b) Conceptual overview of this study. (c) Syntheses of the rotaxane precursors and the [3]catenane. | ||
To demonstrate that rotaxane–catenane interconversion is a useful tool for the creation of unique interlocking molecules and polymers, the rotaxane precursors were prepared via a bipyridine-mediated and active template Cu-mediated azide–alkyne cycloaddition (AT-CuAAC33) reaction (Scheme 1c). We chose this reaction because of its simplicity and applicability. We prepared BiTEMPS derivatives that contain azide or alkyne groups from a diol derivative of BiTEMPS (for details, see the ESI†) and conducted the AT-CuAAC according to literature procedures reported by Leigh and Goldup.33,34 The reaction was quenched with ethylenediaminetetraacetic acid (EDTA) and an NH3-saturated aqueous solution. We followed the progress of the reaction using gel-permeation chromatography (GPC) (Fig. 1a) and unexpectedly found that oligomers and a diol derivative of BiTEMPS were produced as byproducts in addition to the targeted [2]rotaxane. This result indicates that the BiTEMPS exchange reaction was caused by the AT-CuAAC reaction system without the need for heating. We isolated the [2]rotaxane from the mixture via preparative GPC (isolated yield 21%) and characterized the isolated product using electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS), nuclear magnetic resonance (NMR) spectroscopy, and Fourier-transform infrared spectroscopy (FT-IR) (Fig. S5, S12, and S16†). We observed an ionization peak in the ESI-TOF MS spectrum consistent with the theoretical value of the [2]rotaxane (Fig. S15†). A signal in the 1H NMR spectrum derived from the proton on the triazole ring was shifted to higher ppm compared to that of the reference axle which does not have a macrocycle (Fig. S18,† signal 15). This chemical shift indicates the formation of a hydrogen bond between the bipyridine and triazole moieties, a feature that has already been observed in similar rotaxanes.33,34 The disappearance of the absorption at 2100 cm−1 in the FT-IR spectrum confirmed the complete consumption of the azide precursor (Fig. S16†).
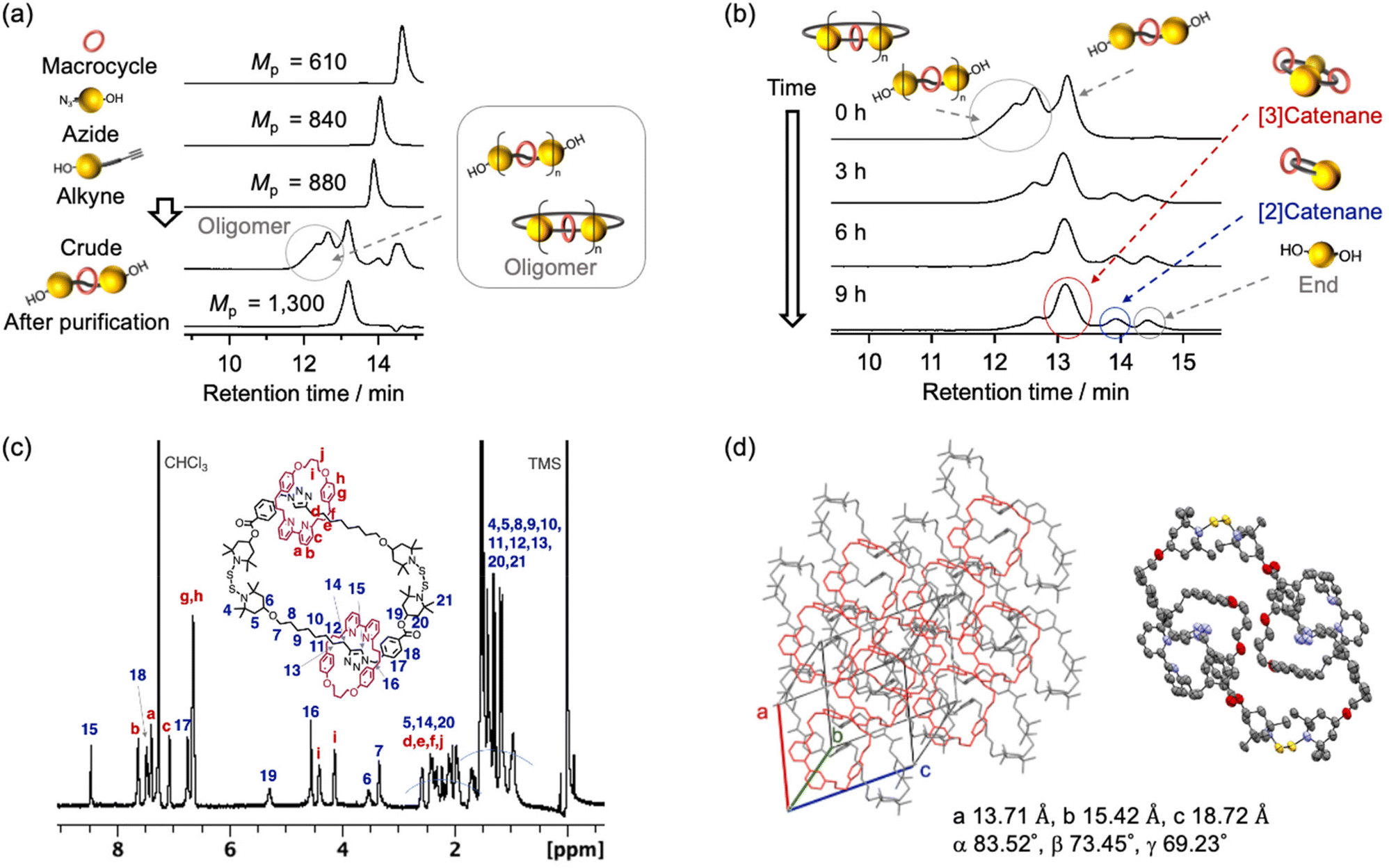 | ||
| Fig. 1 (a) GPC profiles of the [2]rotaxane after purification, its precursors, and the crude products (PS standards; eluent: THF; flow rate: 0.6 mL min−1; detected by UV). (b) Changes in the GPC profiles during the cyclization reaction of a mixture of the rotaxane and its oligomers. (c) 1H NMR spectrum of the [3]catenane (500 MHz, 25 °C, CDCl3). (d) Solid-state structure of the [3]catenane. | ||
We also investigated the stability of the rotaxane in the presence of the Cu catalyst to check for side reactions, i.e., the BiTEMPS exchange reaction that occurs during the AT-CuAAC without the need for heating. For that purpose, the isolated rotaxane was dissolved with an equivalent quantity of [Cu(CH3CN)4][PF6] in methylene chloride and stirred for 72 h at the same concentration used in the AT-CuAAC reaction. Fig. S26† shows the GPC profiles before and after the reaction and suggests that oligomerization occurred. Specifically, BiTEMPS was activated by the Cu catalyst and creates a system with a ring–chain equilibrium state during the AT-CuAAC reaction. To the best of our knowledge, this is the first example of a Cu-catalyzed BiTEMPS exchange reaction. This reactivity will be investigated in detail and developed into a stimulus-response system in a separate study in the near future.
The obtained rotaxane can be cyclized by heating it in a dilute solution to activate the dynamic behavior of the BiTEMPS structure, thus producing catenanes. Considering the efficiency of the synthesis, a mixture containing the [2]rotaxane and its oligomers (isolated yield: 68%) was used for the cyclization reaction instead of pure [2]rotaxane (isolated yield: 21%) (Scheme 1c). We characterized the mixture of the [2]rotaxane and its oligomers after we removed impurities other than the oligomers via column chromatography on silica gel (Fig. 1b: 0 h and Fig. S21a†). The peaks at earlier elution times in the GPC are due to oligomerization. In the 1H NMR spectrum of the mixture, compared to that of the isolated [2]rotaxane, the peak integrals derived from the proton adjacent to the carbon with a terminal hydroxy group are decreased (Fig. S5 and S17;† peaks 1 and 24), confirming elimination of a part of the end structure during AT-CuAAC. These results indicate that the rotaxane and its oligomers were successfully isolated without any other impurities. The cyclization reaction of the rotaxane oligomers was carried out in toluene (1 g L−1) at 100 °C under atmospheric conditions for 9 h. The reaction was monitored by GPC, as shown in Fig. 1b. The changes in peak heights and retention times revealed that the peaks are shifted to later elution times than those observed for the original oligomers and that the peaks originate from both the [3]catenane (dimer) and the end component. The existence of a peak that was attributed to the [2]catenane (monomer) was also detected, although it was present in very low quantities (<10%). In this system, a short chain length and low flexibility favor the formation of the dimer instead of the monomer. In other words, using this system, we can almost selectively produce [3]catenanes. It should also be noted that no peaks derived from the bipyridine rings appeared on the GPC curve after the reaction, demonstrating that the bipyridine rings do not undergo deslipping from the axle during the cyclization process. The steric demand of the BiTEMPS moiety is thus sufficient to act as a stopper for the rotaxane, which prevents deslipping in this system. Similar cyclization behavior was observed when we used isolated [2]rotaxane as a precursor for the [3]catenane (Fig. S27b†), showcasing the benefits of using the oligomer mixture instead of pure [2]rotaxane. We also conducted a cyclization reaction of the axle molecule as a control experiment and found that the dimer was produced as the main product (Fig. S27c†). This result indicates that the selective formation of the [3]catenane depends on the structure of the axle structure.
We subsequently isolated the [3]catenane using flash column chromatography and characterized it via GPC, MS, and NMR (Fig. 1c and Fig. S20–S23†). A unimodal GPC peak indicated the successful isolation of the [3]catenane. The ionization peak in the mass spectrum of the obtained sample is consistent with the theoretical value of the [3]catenane. The 1H NMR spectrum of the [3]catenane did not show any resonances associated with the proton adjacent to the hydroxy group, indicating the successful removal of the terminal moieties of the oligomer chain after the cyclization reaction. We also investigated the solid-state structure of the [3]catenane by means of single-crystal X-ray diffraction analysis. We successfully confirmed that the solid-state structure of the [3]catenane consists of two bipyridine rings that are penetrated and connected by a larger ring that contains two BiTEMPS units (Fig. 1d). Interestingly, we only observed a head-to-tail structure in the crystal structure, instead of a mixture of isomers (for the isomeric structure, see Scheme 2). To check for the presence of isomers in the sample prior to recrystallization, we hydrolyzed the ester group of the [3]catenane. Assuming that cleavage of the ester group occurs by hydrolysis as shown in Scheme 2, one intermediate would be formed from the head-to-tail isomer and two intermediates would be formed from the head-to-head isomer. Then, we finally observed two products (i.e., bipyridine rings and diol derivatives of BiTEMPS) from the organic layer because bipyridine rings were detached from the axle. Therefore, the ratio of isomers of the [3]catenane, i.e., head-to-tail vs. head-to-head, can be determined based on the hydrolyzed products. For that purpose, we added an excess of KOH to the [3]catenane and stirred the mixture at room temperature for 24 h in an ethanol solution. The mixture was then washed with toluene and the organic layer was collected. The 1H NMR spectrum of the obtained product revealed the isomeric ratio, i.e., head-to-tail![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :head-to-head = 2.35:1 (for details, see Fig. S28 and S29†), suggesting that the head-to-head isomer is present prior to recrystallization and that the head-to-tail isomer is produced preferentially, not statistically. At this point, we assume that only the head-to-tail isomer is observed in the X-ray structure because the head-to-tail dimer is likely more crystalline than its counterpart and therefore more likely to form single crystals. We also achieved a synthesis on the scale of hundreds of mg (665 mg of rotaxane-containing oligomers were converted to 339 mg of [3]catenane). This cyclization method only requires heating, and thus is highly efficient for a reaction that selectively furnishes a [3]catenane. This study is also groundbreaking in that a unique [3]catenane with a dynamic structure can be synthesized efficiently.
:head-to-head = 2.35:1 (for details, see Fig. S28 and S29†), suggesting that the head-to-head isomer is present prior to recrystallization and that the head-to-tail isomer is produced preferentially, not statistically. At this point, we assume that only the head-to-tail isomer is observed in the X-ray structure because the head-to-tail dimer is likely more crystalline than its counterpart and therefore more likely to form single crystals. We also achieved a synthesis on the scale of hundreds of mg (665 mg of rotaxane-containing oligomers were converted to 339 mg of [3]catenane). This cyclization method only requires heating, and thus is highly efficient for a reaction that selectively furnishes a [3]catenane. This study is also groundbreaking in that a unique [3]catenane with a dynamic structure can be synthesized efficiently.
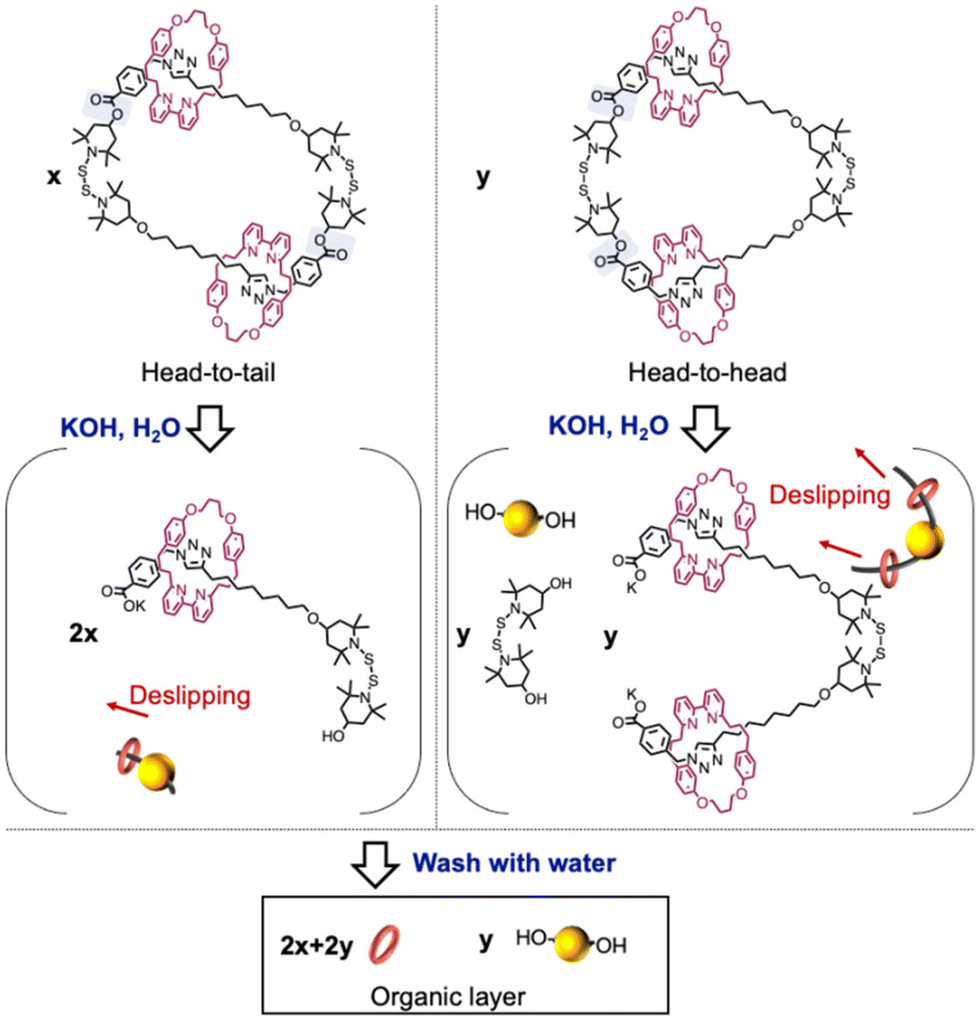 | ||
| Scheme 2 Cleavage of the ester group by hydrolysis of the [3]catenane. | ||
Since the [3]catenane has two BiTEMPS units, it can be polymerized via an intermolecular reaction in concentrated solution (Fig. 2a), as we have already demonstrated.29–31 We conducted a copolymerization reaction to demonstrate the applicability of the [3]catenane in the synthesis of various polyrotaxanes. For that purpose, we prepared Macrocycle as the comonomer and BiTEMPS-anthracene as the end source (Fig. 2a). We then mixed these components and the [3]catenane in an 18/1/2 molar ratio under conditions that resulted in a BiTEMPS concentration of 500 mM. The mixture was then heated in 1,4-dioxane to 100 °C. The GPC elution curve of the resulting polymer indicated the formation of a high-molecular-weight polymer (Mn = 9600; Mw/Mn = 1.57) (Fig. S30b†). The integral ratio of the signals derived from each reactant in the 1H NMR spectrum of the product was consistent with the theoretical value, i.e., the feed ratio, indicating the successful progress of the polymerization without any side reactions (Fig. S30a†). The signal relating to the polymer terminal and repeat units had the same diffusion coefficient in the diffusion-ordered NMR (DOSY) spectrum, thus showing that the copolymer was formed and that the anthracene groups were introduced at the polymer ends (Fig. 2b). These results demonstrate that our [3]catenane enables access to various polyrotaxanes with specific compositions via the intermolecular reaction of BiTEMPS, which follows our previous reports.29–31
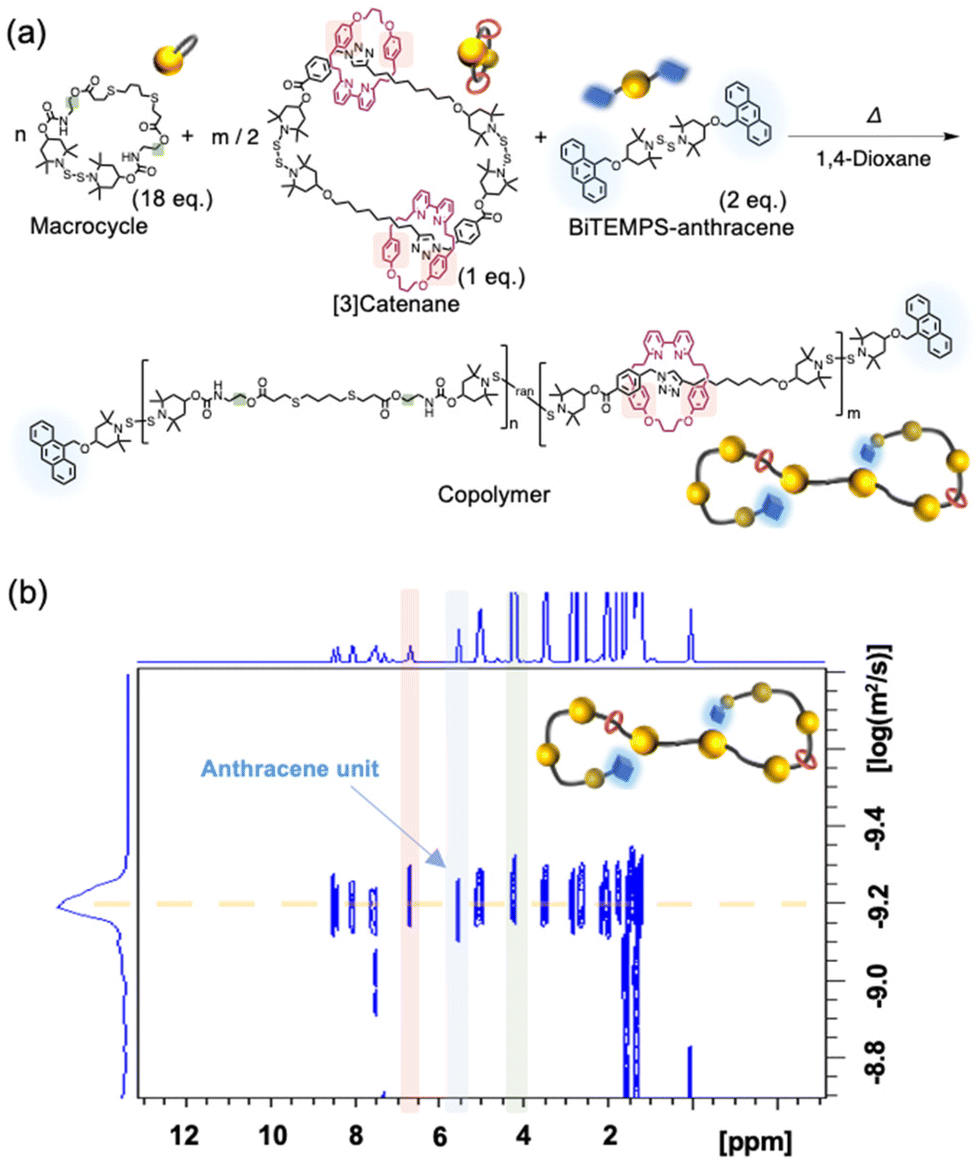 | ||
| Fig. 2 (a) Synthetic scheme for the copolymerization of the [3]catenane and (b) the 1H DOSY spectrum of the resulting copolymer. | ||
Conclusions
In summary, we have successfully developed a versatile and simple synthetic method for the creation of a catenane via a BiTEMPS exchange reaction. By merely heating an oligorotaxane with BiTEMPS units at both ends in a dilute solution, a [3]catenane was spontaneously and selectively produced. The formation of a dimer, i.e., the [3]catenane, was favored compared to the formation of the corresponding monomer, i.e., the [2]catenane, most likely because the chain connecting the two BiTEMPS units in this study was short and rigid. The obtained [3]catenane was characterized using NMR, MS, GPC, and single crystal X-ray diffraction analysis, demonstrating its successful isolation. The successful polymerization of the obtained [3]catenane significantly expands the utility of our system for the synthesis of various interlocking polymers. We believe that this study can be applied to the synthesis of thermally responsive dynamic catenanes with various structures (such as cyclodextrin, crown ether, cucurbituril, and so on), leading to the synthesis of polymers with a variety of new topologies and unique physical properties.Conflicts of interest
There are no potential conflicts of interest to declare.Acknowledgements
This work was supported by the JST in the form of a PRESTO grant JPMJPR18L1 (to D. A.), Mirai Program grant JPMJMI18A2 (to H. O.), as well as JSPS KAKENHI grants JP21J20562 (to R. T.) and JP21K18996 (to D. A.). The authors also thank the Materials Analysis Division and the Open Facility Center (OFC) at the Tokyo Institute of Technology for the MS and X-ray measurements.References
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef.
- Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed.
- R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed.
- M. Weck, B. Mohr, J. P. Sauvage and R. H. Grubbs, J. Org. Chem., 1999, 64, 5463–5471 CrossRef CAS PubMed.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS PubMed.
- Q. Wu, P. M. Rauscher, X. Lang, R. J. Wojtecki, J. J. De Pablo, M. J. A. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- G. Liu, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Chem. Soc. Rev., 2022, 51, 4928–4948 RSC.
- S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC.
- H. Xing, Z. Li, Z. L. Wu and F. Huang, Macromol. Rapid Commun., 2018, 39, 1–6 CrossRef.
- M. A. Nosiglia, N. D. Colley, M. K. Danielson, M. S. Palmquist, A. O. Delawder, S. L. Tran, G. H. Harlan and J. C. Barnes, J. Am. Chem. Soc., 2022, 144, 9990–9996 CrossRef CAS PubMed.
- W. Wang and H. Xing, Polym. Chem., 2018, 9, 2087–2091 RSC.
- Z. Zhang, J. Zhao, Z. Guo, H. Zhang, H. Pan, Q. Wu, W. You, W. Yu and X. Yan, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- D. Muscat, W. Köhler, H. J. Räder, K. Martin, S. Mullins, B. Müller, K. Müllen and Y. Geerts, Macromolecules, 1999, 32, 1737–1745 CrossRef CAS.
- R. Bai, Z. Zhang, W. Di, X. Yang, J. Zhao, H. Ouyang, G. Liu, X. Zhang, L. Cheng, Y. Cao, W. Yu and X. Yan, J. Am. Chem. Soc., 2023, 145, 9011–9020 CrossRef CAS PubMed.
- B. Lee, Z. Niu and S. L. Craig, Angew. Chem., Int. Ed., 2016, 55, 13086–13089 CrossRef CAS PubMed.
- H. Xing and B. Shi, Polym. Chem., 2016, 7, 6159–6163 RSC.
- K. Endo, T. Shiroi and N. Murata, Polym. J., 2005, 37, 512–516 CrossRef CAS.
- P. M. Rauscher, K. S. Schweizer, S. J. Rowan and J. J. De Pablo, Macromolecules, 2020, 53, 3390–3408 CrossRef CAS.
- P. Hu, J. Madsen, Q. Huang and A. L. Skov, ACS Macro Lett., 2020, 9, 1458–1463 CrossRef CAS PubMed.
- N. Kihara and T. Takata, Org. Chem., 2001, 59, 206–218 CAS.
- J. E. M. Lewis, Org. Biomol. Chem., 2019, 17, 2442–2447 RSC.
- N. D. Colley, M. A. Nosiglia, L. Li, F. Amir, C. Chang, A. F. Greene, J. M. Fisher, R. Li, X. Li and J. C. Barnes, Inorg. Chem., 2020, 59, 10450–10460 CrossRef CAS.
- K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC.
- J. E. M. Lewis, F. Modicom and S. M. Goldup, J. Am. Chem. Soc., 2018, 140, 4787–4791 CrossRef CAS PubMed.
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS.
- J. Hobich, B. Huber, P. Theato and H. Mutlu, Macromol. Rapid Commun., 2021, 2100118 CrossRef CAS.
- M. Bin Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 21–27 Search PubMed.
- N. Tsurumi, R. Takashima, D. Aoki, S. Kuwata and H. Otsuka, Angew. Chem., Int. Ed., 2020, 59, 4269–4273 CrossRef CAS PubMed.
- R. Takashima, D. Aoki and H. Otsuka, Macromolecules, 2020, 53, 4670–4677 CrossRef CAS.
- H. Yokochi, R. Takashima, D. Aoki and H. Otsuka, Polym. Chem., 2020, 11, 3557–3563 RSC.
- R. Takashima, D. Aoki and H. Otsuka, Macromolecules, 2021, 54, 8154–8163 CrossRef CAS.
- H. Yokochi, M. Ohira, M. Oka, S. Honda, X. Li, D. Aoki and H. Otsuka, Macromolecules, 2021, 54, 9992–10000 CrossRef CAS.
- V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS.
- E. A. Neal and S. M. Goldup, Chem. Sci., 2015, 6, 2398–2404 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2292612. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01693e |
| This journal is © The Royal Society of Chemistry 2024 |