
Bioinspired synthesis and biological evaluation of ent-protulactones A and B†
Changxu
Liang
ab,
Chao
Hu
a,
Pengpeng
Nie
ab,
Yuanfang
Liu
ab,
Jun
Liu
 *abc and
Yuguo
Du
abc
*abc and
Yuguo
Du
abc
aState Key Laboratory of Environmental Chemistry and Eco-toxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: junliu@rcees.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cBinzhou Institute of Technology, Weiqiao-UCAS Science and Technology Park, Binzhou 256606, Shandong Province, China
First published on 28th November 2023
Abstract
The bioinspired and stereoselective synthesis of the furo[3,2-b] furan lactone (−)-protulactone A and the dioxabicyclo[3.3.1]nonane lactone (+)-protulactone B has been achieved based on the chiron approach. The synthesis features the utilization of a number of one-pot, sequential transformations, including a cascade reaction of reductive elimination and nucleophilic addition in a one-pot process and a one-pot sequence via cross-metathesis/acetonide deprotection/O-Michael addition/lactonization to streamline the synthesis route and avoid the tedious work of product purification. Synthetic protulactones and their analogues were evaluated for their in vitro antiproliferative activity against selected tumor cell lines (MCF-7 and Capan 2) and showed minor cytotoxicity.
Introduction
As one of the ubiquitous fungi in nature, the genus Aspergillus has recently gained considerable attention owing to its impressive pharmacological properties such as antitumor, antiparasitic, immunosuppressive, and antibacterial activities.1 Protulactones A (1) and B (2) are naturally occurring bicyclic lactones that were isolated in 2010 by Sohn and Oh from the marine-derived fungus Aspergillus sp. SF-5044.2 The structures and relative configurations of protulactones A and B were determined by extensive spectroscopic analysis. However, their structures were assigned with ambiguity at most stereogenic centers and the actual structures of both bicyclic lactones remain unclear due to limited supply. Similar NMR data and chemical derivatization studies indicated that protulactones A and B were comprised of similar backbones with an extra acetate ester which attaches to C-6 and C-7, respectively. Both protulactones A and B contain five contiguous stereocenters and exhibit a clear structural similarity. Protulactone A (1) possesses a furo[3,2-b] furan backbone, whereas protulactone B (2) bears a dioxabicyclo[3.3.1]nonane ring system (Fig. 1).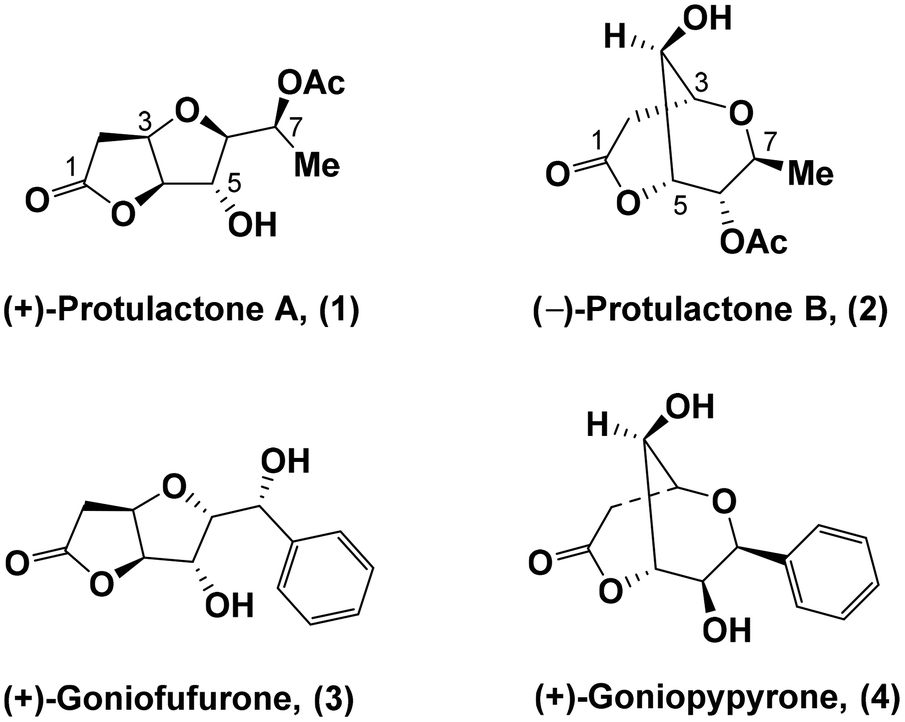 | ||
| Fig. 1 Structures of naturally occurring bicyclic lactones (1–4). | ||
The initial biological studies of protulactones have not been evaluated presumably due to their natural scarcity. Although their absolute configurations have not been fully elucidated, protulactones constitute ideal targets for total syntheses due to the high structural similarities between them and the previously isolated biologically active products goniofufurone (3) and goniopypyrone (4).3 Both goniofufurone and goniopypyrone exhibit significant cytotoxic activities against several human tumour cell lines.
To date, four syntheses of protulactone A have been disclosed, including ours.4–7 Pioneering synthetic studies of these natural bicyclic lactones were conducted by the Gracza group.4 In 2017, Gracza and co-workers reported the first total synthesis and configuration assignment of naturally occurring (+)-protulactone A (1) and its diastereomers, along with their effects on the antiproliferative activity of the NCI60 cancer cell line panel. As part of our interest in the total synthesis of biologically active natural products based on the chiral pool, we have recently accomplished the total synthesis of unnatural enantiomer (−)-protulactone A (1′) from D-mannose in nine steps and 14.7% overall yields using a cascade reaction of acetonide deprotection/lactonization/intramolecular oxa-Michael addition/acetal formation as the key step.5 Very recently, Francuz et al.6 communicated their synthesis of (+)-protulactone A (1) starting from D-galactose in seven steps with an overall yield of 4.92%. Soon after, the same group disclosed an alternative synthetic approach to synthesize (+)-protulactone A and its derivatives and evaluated their antiproliferative activity against several tumor cell lines.7 It is worth noting that, in their work, protulactone A and its structural analogues exhibited promising antiproliferative activity against a panel of tumor cell lines, especially MCF-7, HL-60, K562, and HeLa cell lines. Furthermore, none of the tested compounds exhibited toxicity toward the normal fetal lung fibroblasts (MRC-5); in contrast, doxorubicin (DOX) exhibited sub-micromolar cytotoxic activity against this cell line. Interestingly, all the structural analogues showed at least one superior cytotoxic activity against one or more tumor cell lines, comparable to that recorded for the parent compound protulactone A with IC50 values in the range of 2.2 to 38.5 μM. Moreover, a recent study by Choi et al. reported that protulactone A showed moderate activity in suppressing the growth of the plant pathogenic fungi P. infestans.8 These encouraging results suggest that protulactone A and its analogues may serve as pharmaceutical lead compounds in the synthesis of more potent and selective antitumor agents.
It was proposed by Gracza et al.4 that both protulactone A and protulactone B could be formed from intermediate lactones II and IIIvia an intramolecular O-Michael addition of hydroxyl groups at the C-6 or C-7 position relative to the double bond of the α,β-unsaturated lactone, respectively (Scheme 1). Accordingly, intramolecular additive cyclization of the hydroxyl group at C-6 followed by selective acetylation of 7-OH generated the bicyclic furo[3,2-b] furan lactone protulactone A (Route A), whereas intramolecular O-Michael addition with 7-OH and subsequent acetylation of 6-OH led to the formation of the bicyclic dioxabicyclo[3.3.1]nonane lactone protulactone B (Route B). Since these biosynthetic pathways are highly speculative, other possible mechanisms for the formation of these two compounds have been considered; for example, the O-Michael addition step precedes lactonization. Intermediate lactones II and III could be derived from the same biosynthetic precursor I, pointing to the configurational uniformity of protulactones as described above.
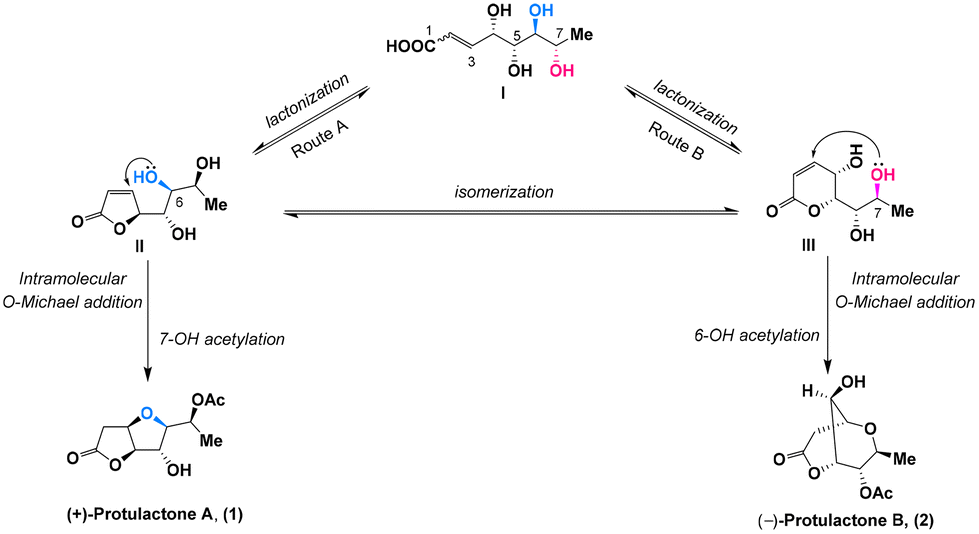 | ||
| Scheme 1 Plausible biosynthetic pathway of protulactones A and B. | ||
In view of their novel structural features and promising biological activities in conjunction with the potential medicinal application of protulactones A and B, it is therefore highly desirable to develop an efficient synthetic approach, in which a readily accessible lactone skeleton bearing a suitable functionality would serve as a key precursor for the divergent total synthesis of both natural products. In particular, to the best of our knowledge, neither the total synthesis nor the biological activity of protulactone B (2) has been reported so far. In continuation of our efforts toward the total synthesis of various complex natural products based on carbohydrate skeletons,9 herein we disclose the bioinspired and stereoselective synthesis of protulactones A and B using readily available methyl α-D-mannopyranoside as a chiral pool, along with their effects on the proliferation of selected tumor cell lines (MCF-7 and Capan 2).
Results and discussion
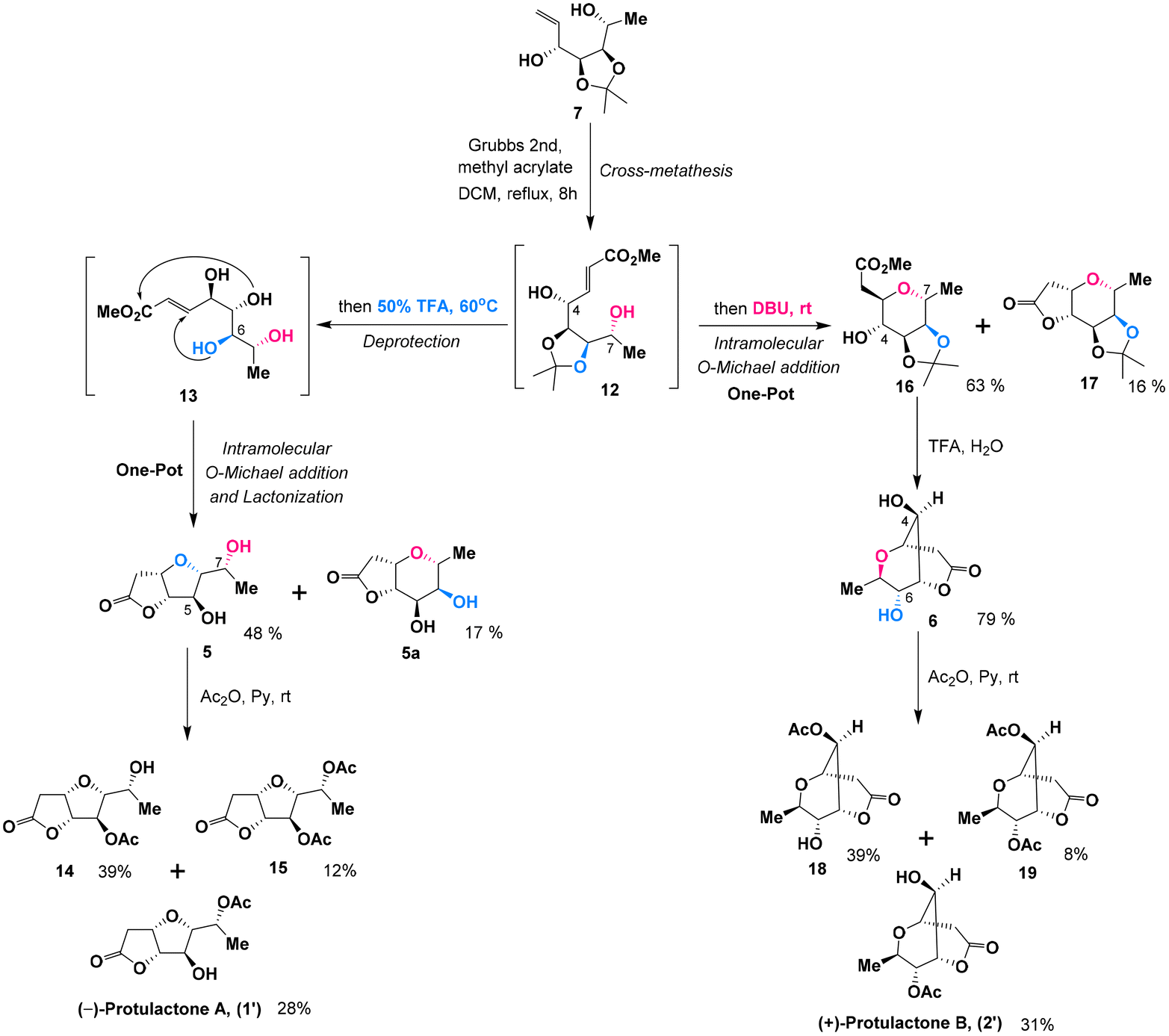
Due to the structural similarities between L-mannopyranoside and the desired stereogenic centers (C4–C7) of protulactones, we initially imagined that the natural protulactones A and B could be derived from unnatural L-mannose. Given the prohibitive cost of L-mannopyranoside and the unknown configurations of protulactone B, we were interested in synthesizing both the natural and unnatural enantiomers of protulactones for structural determination and further biological studies. Accordingly, our synthetic blueprint required swift access to the enantiomer of protulactones, based on the readily available methyl α-D-mannopyranoside.Our approach for the stereoselective synthesis of ent-protulactone A (1′) and ent-protulactone B (2′) is inspired by the above biogenetic pathway. As shown in Scheme 2, we envisioned that ent-protulactone A (1′) and ent-protulactone B (2′) could be obtained from well-functionalized bicyclic lactone 5 or 6via selective acetylation, respectively (Scheme 2). Both lactones 5 and 6, in turn, would stem from the advanced precursor 7via tandem olefin cross metathesis followed by intramolecular oxa-conjugate cyclization and lactonization. The masked tetrol 7, comprising four contiguous hydroxy groups installed with a definite configuration, could be accessible from primary iodide 8 through a cascade reaction of reductive elimination and nucleophilic addition in a one-pot process. The primary iodide 8 could be traced back to methyl α-D-mannopyranoside (9) known in the literature.
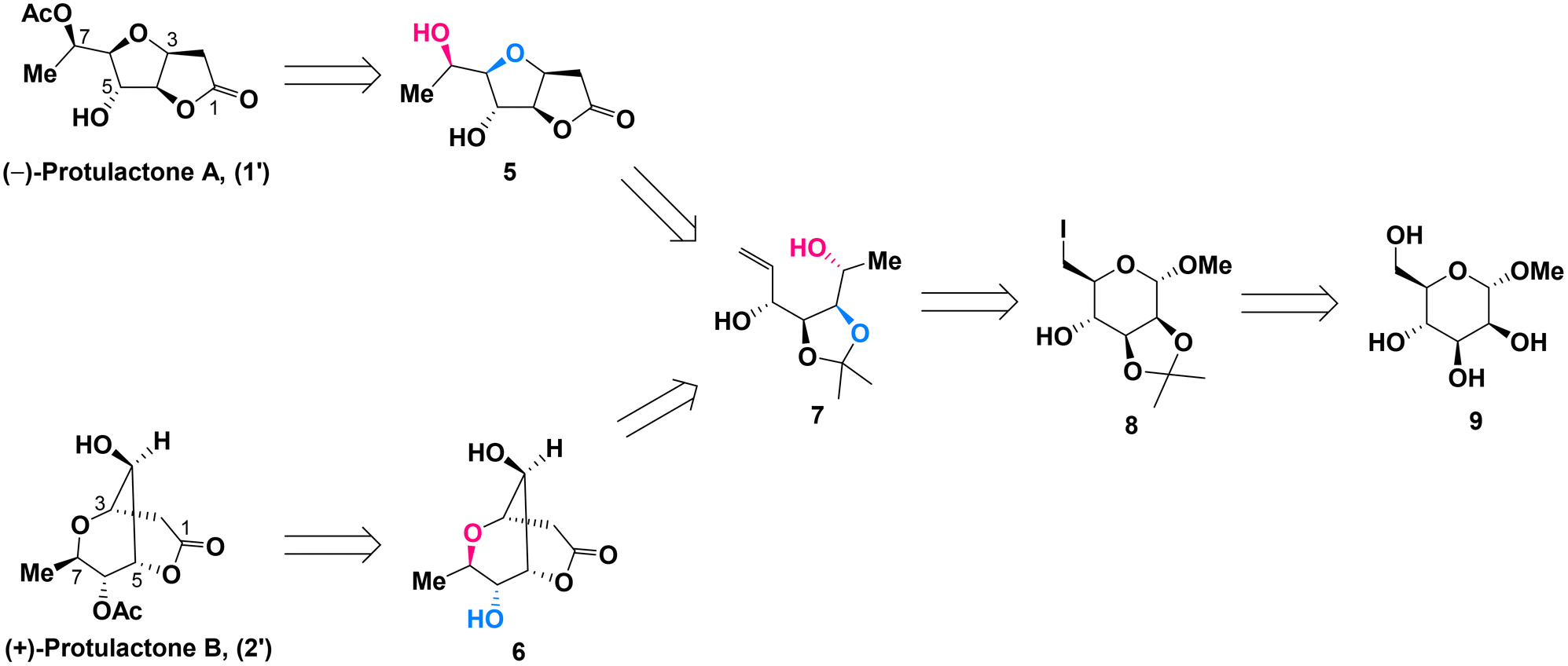 | ||
| Scheme 2 Retrosynthetic analysis of ent-protulactones A (1′) and B (2′). | ||
The synthesis of the masked tetrol 7 was accessible from commercially available methyl α-D-mannopyranoside (9) over three steps as shown in Scheme 3. Our synthesis commenced with the transformation of 9 to 6-iodo-D-mannopyranoside 10 in two steps according to a modified procedure reported by Kumamoto.10 Selective iodination of the primary hydroxy group in 9, followed by blocking the cis 2,3-diol with the isopropylidene group in the presence of a catalytic amount of camphorsulfonic acid (CSA) afforded 6-iodo-D-mannopyranoside 8 in a high yield (86%) over two steps. Subsequently, treatment of the resulting iodide 8 with excess methyllithium accomplished iodide-lithium exchange and reductive elimination followed by an immediate nucleophilic addition with the Grignard reagent in a one-pot sequence in three steps,11 affording the desired allylic alcohols 7 and 7a as an easily separable mixture of diastereomers in 83% combined yield with moderate stereoselectivity (anti-7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :syn-7a = 4.4:1, by 1H NMR analysis of the crude product). The stereochemistry of the desired major diastereomer anti-7 was further confirmed by acetalization of anti-7 with p-TSA in dry acetone to afford the corresponding diacetonide derivative 7b. The stereochemistry of 7b was confirmed by the nuclear Overhauser effect (NOE) between H5 and H6 (see the conformational diagram in Scheme 3). The complete characterization details (1H, 13C, COSY, and NOE NMR studies) of 7b can be found in the ESI.†
:syn-7a = 4.4:1, by 1H NMR analysis of the crude product). The stereochemistry of the desired major diastereomer anti-7 was further confirmed by acetalization of anti-7 with p-TSA in dry acetone to afford the corresponding diacetonide derivative 7b. The stereochemistry of 7b was confirmed by the nuclear Overhauser effect (NOE) between H5 and H6 (see the conformational diagram in Scheme 3). The complete characterization details (1H, 13C, COSY, and NOE NMR studies) of 7b can be found in the ESI.†
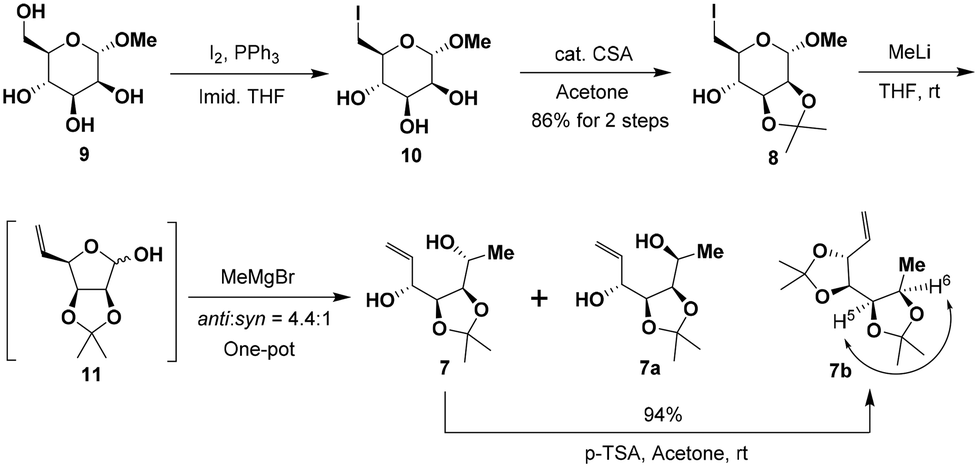 | ||
| Scheme 3 Synthesis of masked tetrol 7. | ||
To construct two entirely different bicyclic lactone systems, the development of a one-pot cross-metathesis/oxa-Michael addition/lactonization sequence under various conditions was then investigated. The olefin cross metathesis between allylic alcohol 7 and methyl acrylate was carried out smoothly using a catalytic amount of Grubbs 2nd catalyst12 generating 12 with almost full conversion. Without further purification, exposure of the resulting α,β-unsaturated ester 12 to trifluoroacetic acid (TFA, 50%) in DCM resulted in a facile cleavage of the acetonide group to afford tetrol 13, which underwent a favorable intramolecular oxa-Michael addition and spontaneous lactonization to provide the cis-disubstituted tetrahydrofuran lactone 5 in 48% yield and lactone 5a in 17% yield. Apparently, the unexpected cis-disubstituted tetrahydropyran lactone 5a was formed by a domino competitive cis-3,7-intramolecular oxa-Michael/lactonization process. Although the yield of lactone 5 was not satisfactory, it should be mentioned that this four-step domino sequence including the olefin cross metathesis of 7, hydrolysis of the acetonide protecting group of 12, intramolecular oxa-Michael addition and lactonization of 13 to give lactone 5 could take place in one pot. With the bicyclic core in place, the final task was the acetylation of 7-OH in lactone 5. The final stages of the synthesis of (−)-protulactone A (1′) are outlined in Scheme 4. Surprisingly, the desired selective acetylation of 7-OH in lactone 5 under standard acetylation conditions proved to be somewhat problematic and afforded two acetylation products in a nonselective manner. Treatment of lactone 5 with acetic anhydride in pyridine at 0 °C delivered the expected (−)-protulactone A (1′) only in a modest yield (28%) but favored the formation of the undesired 5-OH acetylated product 14 (39%) along with a small amount of bisacetylated derivative 15 (12%). Attempts to improve the selectivity of acetylation by changing solvents or reacting at lower temperatures were fruitless.
 | ||
| Scheme 4 Synthesis of ent-protulactone A (1′) and ent-protulactone B (2′). | ||
Having developed an efficient route to the furo[3,2-b] furan backbone, our focus was then shifted to the synthesis of the bicyclic dioxabicyclo[3.3.1]nonane lactone ent-protulactone B (2′). To access the trans-disubstituted tetrahydropyran skeleton, we considered base-promoted cyclization according to a stereoselectivity model proposed by Shishido.13 After investigating a number of bases and reaction conditions, it was found that a one-pot sequential CM/oxa-Michael addition process in the presence of 1,8-diazabicyclo-[5,4,0]-undec-7-ene (DBU) at ambient temperature initiated a rapid and clean intramolecular cyclization of intermediate 12 to give trans-3,7-disubstituted tetrahydropyran 16 in 63% yield, along with the cis-disubstituted tetrahydropyran lactone 17 (16%) as a side product. One-pot tandem acetonide deprotection and lactonization of 16 was accomplished with 50% TFA and afforded the desired bicyclic dioxabicyclo[3.3.1]nonane lactone 6 in a good yield of 79%. Subsequently, acetylation of 6 was carried out following a similar synthetic sequence as described for ent-protulactone A (1′) to deliver ent-protulactone B (2′) in 31% yield and favoring the formation of undesired 18 in 39% yield along with its corresponding bisacetylated derivative 19 (8%). The low selectivity of the final acetylation probably originated from a similar steric crowding between the two hydroxyl groups.
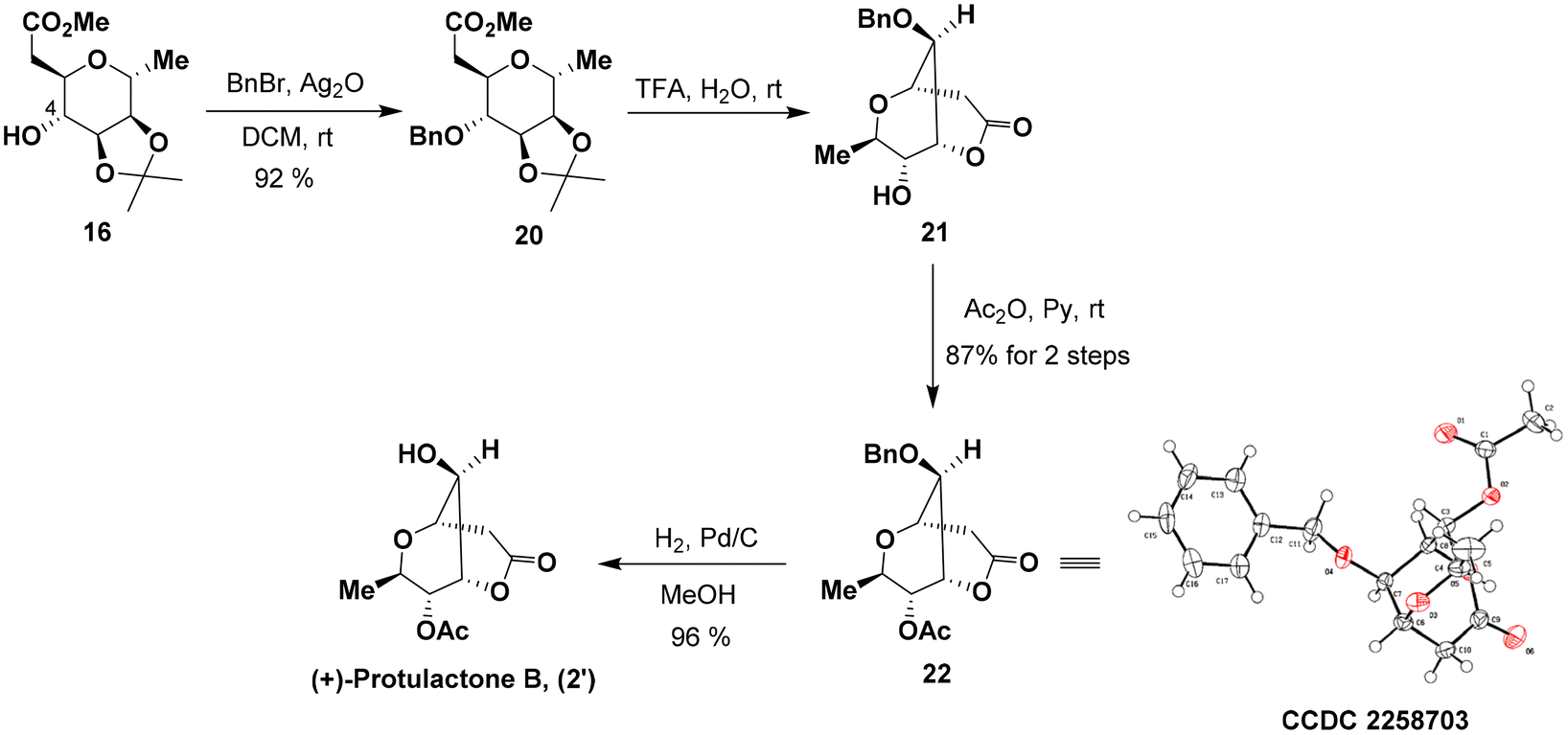
While we were unable to conduct the chemoselective acetylation of 6-OH in lactone 6, we found that the simple protection of the 4-OH group in 16 significantly increased the efficiency of this final acetylation. To this end, blocking the hydroxyl group at C-4 in 16 as the benzyl ether with freshly prepared silver oxide afforded 20 in 92% yield (Scheme 5). Subsequently, the hydrolytic removal of the acetonide group followed by lactonization with 50% aqueous trifluoroacetic acid provided the desired bicyclic dioxabicyclo[3.3.1]nonane lactone 21 with a satisfactory result over two reactions in one pot. 21 was subjected to the same acetylation conditions to furnish acetate 22 in a yield of 87% over two steps. The structure and stereochemistry of acetate 22 were confirmed by single-crystal X-ray diffraction analysis.14 Hydrogenolysis of 22 with a 10% Pd/C catalyst under a H2 atmosphere provided the target (+)-protulactone B (2′) in an essentially quantitative yield.
 | ||
| Scheme 5 Synthesis of (+)-protulactone B (2′). | ||
The 1H and 13C NMR spectra of our synthetic (−)-protulactones A (1′) and (+)-B (2′) are in complete agreement with the spectra reported for these natural products (Table 1). However, the specific rotations obtained for the synthetic samples are opposite in sign to the value reported for the natural protulactones, which shows that our synthetic protulactones A (1′) and B (2′) are the enantiomers of the natural products.15
| No. | 1H-Naturala | 1H-Syntheticb | 13C-Naturala | 13C-Syntheticb |
|---|---|---|---|---|
| a Spectra were recorded at 400 MHz (1H NMR) and 100 MHz (13C NMR) in CDCl3 with drops of CD3OD.2 b Spectra were recorded at 400 MHz (1H NMR) and 100 MHz (13C NMR) in CDCl3 with 15 μL CD3OD. | ||||
| 1 | — | — | 168.6 | 168.5 |
| 2 | 2.92 (1H, dd, 18.6, 1.1) | 2.93 (1H, dd, 19.2, 0.8) | 35.3 | 35.3 |
| 2.85 (1H, dd, 18.6, 4.7) | 2.85 (1H, dd, 19.2, 4.6) | |||
| 3 | 4.12 (1H, m) | 4.17–4.05 (2H, m) | 69.5 | 69.6 |
| 4 | 4.09 (1H, m) | 65.4 | 65.5 | |
| 5 | 4.70 (1H, ddd, 4.6, 2.3, 2.3) | 4.71 (1H, ddd, 4.6, 2.2, 2.2) | 75.0 | 75.0 |
| 6 | 4.97 (1H, dd, 10.1, 2.3) | 4.96 (1H, dd, 10.0, 2.6) | 71.1 | 71.1 |
| 7 | 3.78 (1H, dq, 6.0, 10.1) | 3.78 (1H, dq, 5.8, 10.6) | 64.5 | 64.6 |
| 8 | 1.22 (3H, d, 6.0) | 1.22 (3H, d, 6.0) | 17.8 | 17.8 |
| 1′ | — | — | 170.7 | 170.6 |
| 2′ | 2.07 (3H, s) | 2.07 (3H, s) | 21.0 | 21.0 |
As Francuz and co-workers6,7 had demonstrated previously that natural protulactone A and its analogues exhibited promising antiproliferative activity against a panel of tumor cell lines, we sought to determine whether these unnatural enantiomers have similar cytotoxic activity in human cancer cells. Therefore, synthetic ent-protulactone A (1′) and ent-protulactone B (2′) and their derivatives 5, 6, 15, and 19 were evaluated for their in vitro antiproliferative activity toward ER+ breast adenocarcinoma (MCF-7) and human pancreatic cancer Capan-2 cells. Cytotoxic activities were evaluated using the standard MTT assay after exposure of the cells to the tested compounds for 72 h. The commercial cytotoxic agent doxorubicin (DOX) was used as a reference compound in this assay. Unfortunately, none of these newly synthesized compounds showed significant activities (IC50 > 30 μM). The results are shown in Table 5 in the ESI.†
Conclusions
In summary, we have conducted a novel bioinspired synthesis of the enantiomer of natural bicyclo lactones ent-protulactone A (1′) as well as the first total synthesis of ent-protulactone B (2′). Our synthesis validated the proposed biosynthetic hypothesis from a chemical point of view and may serve as a platform for the synthesis of other natural products containing bicyclic 2,6-dioxabicyclo[3.3.1]nonan-3-one systems, such as goniochelienlactone and goniopypyrone. Further application of these cascade one-pot reactions to the synthesis of the natural protulactones A and B based on the bioinspired strategy, and their biological evaluation are currently in progress in our laboratory.Experimental
General experimental procedures
Unless noted otherwise, commercially available materials were used without further purification. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. All solvents were dried according to the established procedures before use, and the reaction process was monitored by thin-layer chromatography (TLC). The crude products were purified by flash chromatography using 100–200 mesh silica gel. The optical rotation data were obtained using a polarimeter at 25 °C. High-resolution mass spectrometry data (HRMS) were recorded by Fourier-transform mass spectrometry (FT-ICR-MS), and the solvent methanol was of chromatographic purity. 1H NMR and 13C NMR spectra were recorded on 400 MHz or 100 MHz spectrometers (NMR in CDCl3 with TMS as an internal standard). Chemical shifts (δ) are given in ppm relative to residual solvents (usually chloroform, δ 7.26 for 1H NMR or 77.2 for proton decoupled 13C NMR; methanol, δ 3.31 for 1H NMR or 49.0 for proton decoupled 13C NMR), and coupling constants (J) are expressed in Hz. s means singlet state, d means doublet state, t means triplet state, q means quartet state, and m means multiplet state.Experimental procedures
:1, 15.25 g, 86% over 2 steps) as a white amorphous solid. 1H NMR (400 MHz, chloroform-d): δ 4.93 (s, 1H), 4.17–4.09 (m, 2H), 3.59 (dd, J = 10.6, 2.6 Hz, 1H), 3.56–3.45 (m, 2H), 3.49 (s, 3H), 3.31 (dd, J = 10.6, 7.2 Hz, 1H), 1.53 (s, 3H), 1.36 (s, 3H); 13C NMR (100 MHz, chloroform-d): δ 110.0, 98.6, 78.5, 75.9, 73.3, 69.3, 55.8, 28.2, 26.3, 6.9. Data are consistent with a previously characterized compound.10
:1) gave both compound 7 (633 mg, 70%) and compound 7a (145 mg, 16%) as colorless oils. For compound 7, Rf = 0.4 (petroleum ether/EtOAc 2:1); [α]25D = −18.50 (c 0.69, MeOH); 1H NMR (400 MHz, chloroform-d): δ 6.04 (ddd, J = 17.2, 10.6, 5.6 Hz, 1H), 5.40 (d, J = 17.2 Hz, 1H), 5.26 (d, J = 10.6 Hz, 1H), 4.51 (ddt, J = 5.0, 3.2, 1.6 Hz, 1H), 4.18 (dd, J = 6.0, 3.2 Hz, 1H), 4.15–4.06 (m, 1H), 3.89 (dd, J = 8.6, 6.0 Hz, 1H), 2.53 (br s, 2H), 1.48 (s, 3H), 1.36 (s, 3H), 1.30 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, chloroform-d): δ 138.3, 116.5, 108.4, 81.6, 79.3, 70.5, 66.0, 27.5, 25.3, 21.1; HRMS (ESI): calcd for C10H18O4Na+ [M + Na]+, 225.1098; found 225.1098.
For compound 7a, Rf = 0.35 (petroleum ether/EtOAc 2:1); [α]25D = −0.92 (c 1.95, MeOH); 1H NMR (400 MHz, chloroform-d) δ 5.93 (ddd, J = 17.2, 10.4, 6.0 Hz, 1H), 5.39 (d, J = 17.2, 1H), 5.26 (d, J = 10.4 Hz, 1H), 4.35 (ddt, J = 6.0, 3.2, 1.4 Hz, 1H), 4.12 (dd, J = 7.2, 3.2 Hz, 1H), 4.07 (qd, J = 6.4, 3.2 Hz, 1H), 4.02 (dd, J = 7.2, 3.0 Hz, 1H), 2.69 (br s, 2H), 1.55 (s, 3H), 1.39 (s, 3H), 1.29 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 137.7, 117.2, 108.5, 80.8, 79.6, 70.4, 65.4, 26.8, 24.8, 21.1; HRMS (ESI): calcd for C10H18O4Na+ [M + Na]+, 225.1098; found 225.1098.
:1) to afford pure compound 7b (101 mg, 94%) as a colorless oil. Rf = 0.55 (petroleum ether/EtOAc 6:1); [α]25D = −13.64 (c 2.18, MeOH); 1H NMR (400 MHz, chloroform-d): δ 5.92 (ddd, J = 17.2, 10.6, 5.6 Hz, 1H), 5.40 (d, J = 17.2 Hz, 1H), 5.16 (d, J = 10.6 Hz, 1H), 4.48–4.30 (m, 2H), 3.97 (dd, J = 9.2, 5.6 Hz, 1H), 3.69 (dd, J = 9.2, 7.6 Hz, 1H), 1.38 (s, 2 × 3H), 1.36 (s, 3H), 1.30 (s, 3H), 1.29 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 136.6, 116.4, 109.5, 108.4, 81.3, 79.9, 78.2, 73.8, 28.3, 27.2, 27.1, 25.7, 15.1; HRMS (ESI): calcd for C13H22O4Na+ [M + Na]+, 265.1411; found 265.1410.
:1) until compound 7 disappeared. To the above mixture was added TFA (4 mL, 50% in H2O) at room temperature. After stirring at 60 °C for 8 h, the mixture was quenched with Et3N (5 mL) and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/EtOAc 1:2) to give compound 5 (43 mg, 48%) and 5a (15 mg, 17%).
For compound 5, Rf = 0.25 (petroleum ether/EtOAc 1:2); [α]25D = −49.36 (c 0.37, MeOH); 1H NMR (400 MHz, methanol-d4): δ 4.80–4.77 (m, 2H), 4.35 (d, J = 4.8 Hz, 1H), 3.77 (qd, J = 6.4, 5.2 Hz, 1H), 3.61 (t, J = 5.0 Hz, 1H), 2.83 (dd, J = 18.2, 4.8 Hz, 1H), 2.57 (d, J = 18.2 Hz, 1H), 1.18 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 177.8, 92.5, 92.0, 79.0, 76.6, 68.0, 37.0, 19.4; HRMS (ESI): calcd for C8H12O5Na+ [M + Na]+, 211.0577; found 211.0577.
For compound 5a, Rf = 0.35 (petroleum ether/EtOAc 1:2); [α]25D = −36.77 (c 0.39, MeOH); 1H NMR (400 MHz, methanol-d4) δ 4.43 (d, J = 3.2 Hz, 2H), 4.11 (t, J = 3.0 Hz, 1H), 3.73 (dq, J = 9.0, 6.4 Hz, 1H), 3.33–3.30 (m, 1H), 2.82 (dd, J = 17.2, 4.0 Hz, 1H), 2.37 (d, J = 17.2 Hz, 1H), 1.19 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 178.0, 84.1, 72.0, 71.5, 70.7, 67.6, 38.5, 18.3; HRMS (ESI): calcd for C8H12O5Na+ [M + Na]+, 211.0577; found 211.0578.
The crude α,β-unsaturated ester 12 could be used for the next step without further purification. A small sample was purified on a silica gel column (petroleum ether/EtOAc 2:1) to obtain the physical data. For compound 12, Rf = 0.5 (petroleum ether/EtOAc 1:1); [α]25D = −45.10 (c 0.33, MeOH); 1H NMR (400 MHz, chloroform-d): δ 7.10 (dd, J = 15.6, 4.0 Hz, 1H), 6.15 (dd, J = 15.6, 2.0 Hz, 1H), 4.68–4.65 (m, 1H), 4.25 (dd, J = 6.2, 3.2 Hz, 1H), 4.10 (dq, J = 8.4, 6.2 Hz, 1H), 3.92 (dd, J = 8.6, 6.2 Hz, 1H), 3.73 (s, 3H), 3.63 (br s, 1H), 3.01 (br s, 1H), 1.46 (s, 3H), 1.34 (s, 3H), 1.29 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 167.0, 148.1, 121.1, 108.6, 81.3, 78.6, 69.4, 66.1, 51.9, 27.1, 24.9, 21.4; HRMS (ESI): calcd for C12H20O6Na+ [M + Na]+, 283.1153; found 283.1153.
:1 to 2:3) to afford pure (−)-protulactone A (1′) (17 mg, 28%) as a colorless oil and compounds 14 (24 mg, 39%) and 15 (9 mg, 12%) as colorless oils.
For (−)-protulactone A (1′), Rf = 0.4 (petroleum ether/EtOAc 1:2); [α]25D = −30.38 (c 0.23, MeOH); Gracza's synthetic sample: {Lit.3 [α]25D = −33.8 (c 0.34, MeOH)}; 1H NMR (400 MHz, methanol-d4) δ 5.07 (dq, J = 4.0, 6.6 Hz, 1H), 4.82–4.74 (m, 2H), 4.33 (d, J = 4.8 Hz, 1H), 3.79 (dd, J = 4.8, 4.0 Hz, 1H), 2.84 (dd, J = 18.2, 4.8 Hz, 1H), 2.57–2.47 (m, 1H), 1.99 (s, 3H), 1.24 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 177.5, 172.1, 92.3, 90.2, 79.2, 76.6, 70.6, 37.0, 21.0, 16.3; HRMS (ESI): calcd for C10H14O6Na+ [M + Na]+, 253.0683; found 253.0683.
For compound 14, Rf = 0.5 (petroleum ether/EtOAc 1:2). [α]25D = +20.26 (c 0.08, MeOH); 1H NMR (400 MHz, methanol-d4) δ 5.29 (d, J = 4.2 Hz, 1H), 4.90 (d, J = 4.1 Hz, 1H), 4.80 (t, J = 4.7 Hz 1H), 3.85–3.75 (m, 2H), 2.85 (dd, J = 18.3, 5.3 Hz, 1H), 2.62 (d, J = 18.3 Hz, 1H), 2.10 (s, 3H), 1.16 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 177.2, 171.5, 89.6, 89.3, 79.5, 78.3, 68.0, 36.8, 20.7, 19.4; HRMS (ESI): calcd for C10H14O6Na+ [M + Na]+, 253.0683; found 253.0684.
For compound 15, Rf = 0.75 (petroleum ether/EtOAc 1:2); [α]25D = −24.68 (c 0.58, MeOH); 1H NMR (400 MHz, chloroform-d) δ 5.35 (d, J = 4.8 Hz, 1H), 5.13 (qd, J = 6.6, 3.6 Hz, 1H), 4.83–4.80 (m, 2H), 4.00 (dd, J = 4.8, 3.6 Hz, 1H), 2.71 (d, J = 1.2 Hz, 2H), 2.12 (s, 3H), 2.04 (s, 3H), 1.24 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 174.1, 170.5, 169.5, 87.6, 86.9, 78.2, 76.8, 69.1, 36.0, 21.2, 20.9, 15.7; HRMS (ESI): calcd for C12H16O7Na+ [M + Na]+, 295.0789; found 295.0788.
:1 petroleum ether–EtOAc 2:1) until compound 7 disappeared. To the above mixture was added a catalytic amount of DBU (49 μL, 0.33 mmol) at room temperature. The reaction was stirred for another 12 h. After the completion of the reaction, the solvent was evaporated off and the crude residue was purified by flash column chromatography (petroleum ether/EtOAc 2:1) to give pure compound 16 (266 mg, 63%) as a white amorphous solid and compound 17 (59 mg, 16%) as an amorphous solid.
For compound 16, Rf = 0.33 (petroleum ether/EtOAc 2:1); [α]25D = +10.15 (c 1.245, MeOH); 1H NMR (400 MHz, chloroform-d) δ 4.07 (dd, J = 7.4, 5.6 Hz, 1H), 4.03–3.92 (m, 2H), 3.88 (ddd, J = 9.2, 8.2, 4.0 Hz, 1H), 3.67 (s, 3H), 3.63 (t, J = 8.4 Hz, 1H), 3.18 (br s, 1H), 2.78 (dd, J = 15.6, 4.2 Hz, 1H), 2.54 (dd, J = 15.6, 8.2 Hz, 1H), 1.47 (s, 3H), 1.32 (s, 3H), 1.27 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 172.0, 109.6, 78.4, 78.0, 72.5, 69.8, 69.1, 51.9, 37.4, 27.9, 25.8, 18.3; HRMS (ESI): calcd for C12H20O6Na+ [M + Na]+, 283.1153; found 283.1153.
For compound 17, Rf = 0.65 (petroleum ether/EtOAc 2:1); [α]25D = −0.06 (c 1.125, MeOH); 1H NMR (400 MHz, chloroform-d) δ 4.58 (dd, J = 2.8, 1.2 Hz, 1H), 4.49 (d, J = 5.2 Hz, 1H), 4.33 (dd, J = 5.0, 2.8 Hz, 1H), 3.77 (dd, J = 9.6, 5.2 Hz, 1H), 3.25 (dq, J = 9.6, 6.2 Hz, 1H), 2.73 (dd, J = 17.6, 4.8 Hz, 1H), 2.60 (d, J = 17.6 Hz, 1H), 1.44 (s, 3H), 1.36 (s, 3H), 1.22 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 174.3, 108.9, 78.1, 75.0, 72.1, 71.93, 71.90, 37.0, 28.1, 26.0, 17.9; HRMS (ESI): calcd for C11H16O6Na+ [M + Na]+, 251.0890; found 251.0891.
:1). [α]25D = +31.27 (c 0.1, MeOH); 1H NMR (400 MHz, methanol-d4) δ 4.52 (dt, J = 4.6, 2.4 Hz, 1H), 4.19 (dd, J = 4.6, 1.8 Hz, 1H), 4.04 (dq, J = 5.4, 2.0 Hz, 1H), 3.70 (dd, J = 9.8, 2.6 Hz, 1H), 3.64–3.53 (m, 1H), 3.01 (dd, J = 19.4, 5.2 Hz, 1H), 2.83 (dd, J = 19.4, 1.6 Hz, 1H), 1.30 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 171.3, 79.8, 70.8, 70.1, 68.3, 66.7, 36.3, 18.3; HRMS (ESI): calcd for C8H12O5Na+ [M + Na]+, 211.0577; found 211.0577.
:1) to afford pure compound 20 (105 mg, 92%) as an amorphous solid. Rf = 0.45 (petroleum ether/EtOAc 4:1); [α]25D = +1.843 (c 0.12, MeOH); 1H NMR (400 MHz, chloroform-d) δ 7.37–7.26 (m, 5H), 4.89 (d, J = 11.6 Hz, 1H), 4.60 (d, J = 11.6 Hz, 1H), 4.28 (dd, J = 7.0, 5.6 Hz, 1H), 4.07–3.89 (m, 3H), 3.64 (s, 3H), 3.49 (dd, J = 9.2, 7.0 Hz, 1H), 2.77 (dd, J = 15.4, 4.0 Hz, 1H), 2.47 (dd, J = 15.4, 8.4 Hz, 1H), 1.52 (s, 3H), 1.37 (s, 3H), 1.29 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 171.8, 138.2, 128.5 (d), 128.3 (d), 127.9, 109.4, 78.8, 78.4, 78.1, 72.9, 69.1, 68.5, 51.8, 37.7, 28.1, 26.0, 18.1; HRMS (ESI): calcd for C19H26O6Na+ [M + Na]+, 373.1622; found 373.1623.
:1) to obtain the physical data. For compound 21: colorless oil, Rf = 0.35 (petroleum ether/EtOAc 1:1); [α]25D = −13.27 (c 0.265, MeOH); 1H NMR (400 MHz, chloroform-d) δ 7.43–7.29 (m, 5H), 4.72 (d, J = 12.0 Hz, 1H), 4.69–4.66 (m, 1H), 4.61 (d, J = 12.1 Hz, 1H), 4.27–4.24 (m, 1H), 3.90 (dd, J = 4.5, 1.8 Hz, 1H), 3.75 (dd, J = 9.7, 2.7 Hz, 1H), 3.57 (dq, J = 9.6, 5.9 Hz, 1H), 2.95 (dd, J = 19.3, 1.7 Hz, 1H), 2.79 (dd, J = 19.3, 5.1 Hz, 1H), 2.23 (br s, 1H), 1.39 (d, J = 5.9 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 168.5, 137.0, 128.9 (d), 128.5, 128.0 (d), 76.3, 71.8, 71.4, 70.0, 67.8, 66.7, 35.7, 18.1; HRMS (ESI): calcd for C15H18O5Na+ [M + Na]+, 301.1047; found 301.1047.
To a solution of the crude compound 21 in pyridine (4 mL) were added Ac2O (53 μL, 0.56 mmol) and DMAP (2 mg). The mixture was stirred at room temperature for 2 h and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/EtOAc 1.5:1) to give compound 22 (51 mg, 87% for 2 steps) as a crystal. Rf = 0.45 (petroleum ether/EtOAc 1.5:1); [α]25D = +53.90 (c 0.12, MeOH); 1H NMR (400 MHz, chloroform-d) δ 7.42–7.29 (m, 5H), 5.03 (dd, J = 10.0, 2.6 Hz, 1H), 4.86 (dt, J = 4.6, 2.4 Hz, 1H), 4.70 (s, 2H), 4.26 (dq, J = 5.6, 1.8 Hz, 1H), 3.89–3.78 (m, 2H), 2.96 (dd, J = 19.2, 1.8 Hz, 1H), 2.80 (dd, J = 19.2, 5.2 Hz, 1H), 2.12 (s, 3H), 1.28 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 170.3, 168.1, 136.8, 128.9 (d), 128.5, 128.1 (d), 73.1, 71.5, 71.34, 71.26, 67.4, 64.6, 35.6, 21.1, 18.0; HRMS (ESI): calcd for C17H20O6Na+ [M + Na]+, 343.1153; found 343.1153.
:1 to 2:3) to afford pure (+)-protulactone B (2′) (22 mg, 31%) as a colorless oil and compounds 18 (28 mg, 39%) and 19 (7 mg, 8%) as colorless oils.
Procedure B (from compound 22): A solution of compound 22 (18 mg, 0.06 mmol) in EtOAc (1 mL) and MeOH (3 mL) was hydrogenated over palladium-loaded activated carbon (Pd/C, 20%). The mixture was stirred under a H2 atmosphere for 2 h and filtered. The filtrate was concentrated and purified on a silica gel column (petroleum ether/EtOAc 1:1.5) to afford (+)-protulactone B (2′) (12 mg, 96%) as a colorless oil. Rf = 0.35 (petroleum ether/EtOAc 1:1.5). [α]25D = +48.30 (c 0.16, MeOH); {Lit.2 [α]25D = −82 (c 0.57, MeOH)}; 1H NMR (400 MHz, chloroform-d) δ 4.96 (dd, J = 10.0, 2.6 Hz, 1H), 4.73 (ddd, J = 4.8, 2.4, 2.4 Hz, 1H), 4.14 (m, 2H), 3.79 (dq, J = 10.0, 6.0 Hz, 1H), 2.96 (dd, J = 19.4, 1.6 Hz, 1H), 2.86 (dd, J = 19.4, 5.0 Hz, 1H), 2.07 (s, 3H), 1.68 (br s, 1H), 1.22 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 170.4, 167.8, 74.8, 70.9, 69.9, 66.1, 65.0, 35.3, 21.1, 18.1; HRMS (ESI): calcd for C10H14O6Na+ [M + Na]+, 253.0683; found 253.0683.
1H NMR (400 MHz, chloroform-d with drops of methanol-d4) δ 4.96 (dd, J = 10.0, 2.6 Hz, 1H), 4.71 (ddd, J = 4.6, 2.2, 2.2 Hz, 1H), 4.17–4.05 (m, 2H), 3.78 (dq, J = 10.0, 5.8 Hz, 1H), 2.93 (dd, J = 19.2 Hz, 0.8 Hz, 1H), 2.85 (dd, J = 19.2, 4.6 Hz, 1H), 2.07 (s, 3H), 1.22 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, chloroform-d with drops of methanol-d4) δ 170.6, 168.5, 75.0, 71.1, 69.6, 65.5, 64.6, 35.3, 21.0, 17.8.
For compound 19, Rf = 0.35 (petroleum ether/EtOAc 2:1); [α]25D = +58.30 (c 0.14, MeOH); 1H NMR (400 MHz, chloroform-d) δ 5.16 (dd, J = 4.4, 1.6 Hz, 1H), 4.91 (dd, J = 10.0, 2.4 Hz, 1H), 4.79 (ddd, J = 4.8, 2.4, 2.4 Hz, 1H), 4.30 (dq, J = 5.0, 2.3 Hz, 1H), 3.86 (dq, J = 1.0, 6.0 Hz, 1H), 3.02–2.96 (m, 2H), 2.20 (s, 3H), 2.13 (s, 3H), 1.28 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 170.2, 169.9, 167.3, 72.6, 70.9, 67.4, 67.2, 64.6, 35.5(d), 21.1, 18.0; HRMS (ESI): calcd for C12H16O7Na+ [M + Na]+, 295.0789; found 295.0788. Data are consistent with the reported literature.2
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a grant from the Binzhou Institute of Technology (GYY-GDHX-2023-WT-001) and the STS Project of CAS (KFJ-STS-QYZD-201-5-1). We thank Dr Yong Li and Shuang Peng for HRMS measurements.References
- X. Bai, Y. Sheng, Z. Tang, J. Pan, S. Wang, B. Tang, T. Zhou, L. Shi and H. Zhang, Polyketides as Secondary Metabolites from the Genus Aspergillus, J. Fungi, 2023, 9, 261 CrossRef CAS
.
- J. H. Sohn and H. Oh, Protulactones A and B: Two New Polyketides from the Marine-derived Fungus Aspergillus sp. SF-5044, Bull. Korean Chem. Soc., 2010, 31, 1695–1698 CrossRef CAS
- X. P. Fang, J. E. Anderson, C. J. Chang, P. E. Fanwick and J. L. McLaughlin, Novel Bioactive Styryl-lactones: Goniofufurone, Goniopypyrone, and 8-Acetylgoniotriol from Goniofha/arnus giganteus (Annonaceae). X-Ray Molecular Structure of Goniofufurone and of Goniopypyrone, J. Chem. Soc., Perkin Trans. 1, 1990, 1655–1661 RSC
- M. Markovič, P. Kooš, T. Čarny, S. Sokoliova, N. Bohačikova, J. Moncol and T. Gracza, Total Synthesis, Configuration Assignment, and, Cytotoxic Activity Evaluation of Protulactone A, J. Nat. Prod., 2017, 80, 1631–1638 CrossRef
- Q. Lv, C. Chang, Y. Li, Y. Du and J. Liu, Stereoselective synthesis of (−)-protulactone A, Tetrahedron, 2020, 76, 131290 CrossRef CAS
- S. Djokić, J. Francuz, M. Popsavin, M. V. Rodić, V. Kojić, M. Stevanović and V. Popsavin, Natural product protulactone A: Total synthesis from D-galactose, X-ray analysis and biological evaluation, Bioorg. Chem., 2022, 127, 105980 CrossRef
- J. Francuz, S. Djokić, M. Popsavin, M. V. Rodić, V. Kojić, B. Krüger and V. Popsavin, New synthetic approach to protulactone a and structural analogues, Synlett, 2023, 34, 1699–1703 CrossRef CAS
- M. T. Ngo, M. V. Nguyen, J. W. Han, B. Kim, Y. K. Kim, M. S. Park, H. Kim and G. J. Choi, Biocontrol Potential of Aspergillus Species Producing Antimicrobial Metabolites, Front. Microbiol., 2021, 12, 804333 CrossRef PubMed
-
(a) Y. X. Liu, J. Liu, C. F. Zhao and Y. G. Du, Stereoselective Total Synthesis of Siladenoserinols A and D, Org. Lett., 2021, 23, 3264–3268 CrossRef CAS
- H. Kumamoto, K. Deguchi, T. Wagata, Y. Furuya, Y. Odanaka, Y. Kitade and H. Tanaka, Radical-mediated stannylation of vinyl sulfones: access to novel 4′-modified neplanocin A analogues, Tetrahedron, 2009, 65, 8007–8013 CrossRef CAS
-
(a) A. Bercier, R. Plantier-Royon and C. Portella, Domino reactions of 5-deoxy-5-iodo-d-xylo-and-l-arabinofuranose derivatives with organometallic reagents. A way towards polyfunctionalized building-blocks, Tetrahedron, 2010, 66, 4109–4114 CrossRef CAS
- T. L. Choi, D. P. Sanders and R. H. Grubbs, A general model for selectivity in olefin cross metathesis, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef PubMed
- M. Kanematsu, M. Yoshida and K. Shishido, Total Synthesis of Aspergillide A and B Based on the Transannular Oxy–Michael Reaction, Angew. Chem., Int. Ed., 2011, 50, 2618–2620 CrossRef CAS
- CCDC 2258703† contains the supplementary crystallographic data for this paper.
- See the ESI† for detailed comparisons of the 1H and 13C NMR spectra of natural and synthetic ent-protulactone A (1′) and ent-protulactone B (2′).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2258703. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01708g |
| This journal is © The Royal Society of Chemistry 2024 |