
Two-step, high-yielding total synthesis of antibiotic pyrones†
Akram
Hussain‡
a,
Revoju
Sravanthi‡
ab,
Sunitha
Katta
b and
Dhevalapally B.
Ramachary
 *a
*a
aCatalysis Laboratory, School of Chemistry, University of Hyderabad, Hyderabad-500 046, India. E-mail: ramsc@uohyd.ac.in; ramchary.db@gmail.com
bPharmacognosy and Phytochemistry Division, Gitam Institute of Pharmacy, Gitam Deemed to be University, Visakhapatnam, 530 045, Andhra Pradesh, India
First published on 14th December 2023
Abstract
A simple two-step dialkylation protocol was developed to synthesize biologically active antibiotics photopyrones, pseudopyronines, and violapyrones from bio-renewable triacetate lactone in excellent yields. These pyrones are functionally modified into another set of pyrone natural products by a single O-methylation reaction. The high-yielding gram scale synthesis of four natural products [pseudopyronine A, photopyrone A, pseudopyronine B and photopyrone C] demonstrated the viability for industrial applications.
Introduction
When we delve into nature's chemistry, we see several exciting chemical scaffolds, designed by a sequence of reactions catalysed by enzymes. For instance, polyketides, active metabolites synthesized by fungi, bacteria, plants and animals through various metabolic pathways,1 display a wide range of biological activities such as antiviral, antibiotic, antibacterial, antifungal, and antineoplastic properties. A plethora of interesting polyketides engineered by polyketide synthases, typically by sequential decarboxylative Claisen condensation reactions, produce α-pyrones, especially 3,6-dialkyl-2H-pyran-2-ones, which are inclusive structures of different classes of natural products.2 Hence, a myriad of attempts are being made to synthesize them either by chemical methods or by generating bio-catalytic pathways owing to their exciting activities (Fig. 1).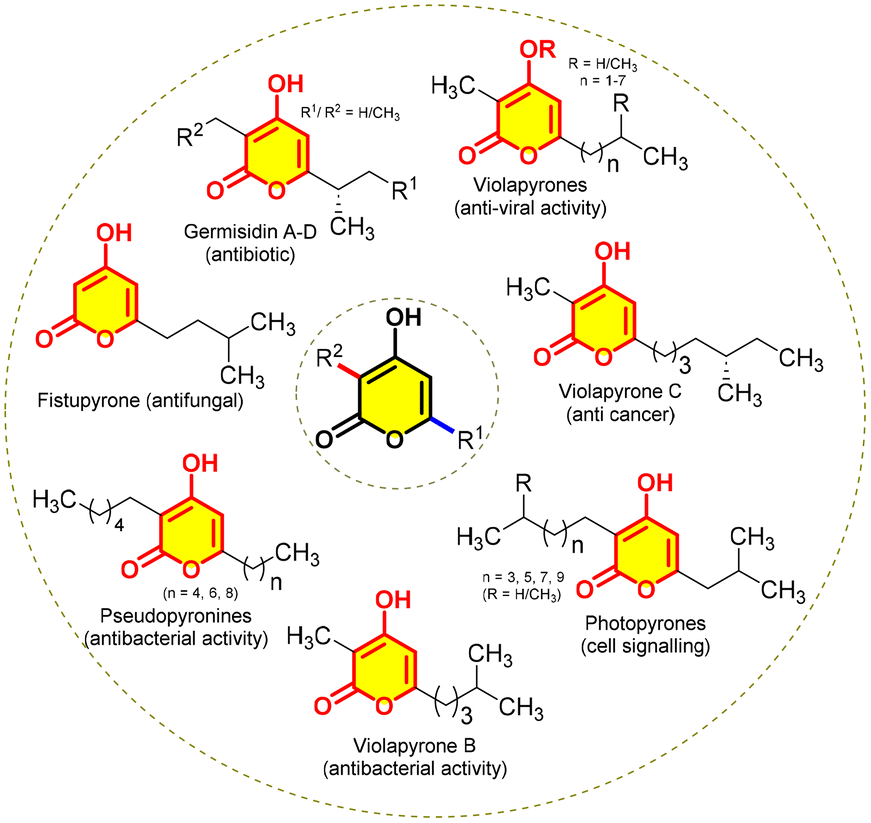 | ||
| Fig. 1 4-Hydroxy-2H-pyran-2-one-containing natural products. | ||
Among them, photopyrones have a unique ability to work as cell signalling molecules in bacteria enabling communication among them and thereby mediating a group-coordinated behaviour. This phenomenon, known as quorum sensing,3 enables them to interact with other species and regulate phenotypic behaviour such as virulence factor secretion, competence, bioluminescence, swarming motility, sporulation, and biofilm formation. Moreover, α-pyrones show an extensive range of biological activities, e.g. violapyrones4 present anti-H1N1, anti-H3N2, anticancer, antiviral, and antibacterial activities; pseudopyronines5 show antibiotic, algicidal, anti-atherosclerosis, and anti-tubercular activities; fistupyrone6 has antifungal activity; germicidin7 shows antibiotic activity and so on (Fig. 1).
Although the metabolic pathway enables the synthesis of a diverse range of 3,6-dialkyl-2H-pyran-2-ones, their laboratory synthesis is highly challenging owing to the lack of an operationally simple route to access them. Therefore, a simple one-pot method that generates a vast library of different α-pyrones smoothly is highly desired. Herein, we want to develop a common method to synthesize 3,6-dialkyl-2H-pyran-2-ones, as the known target-oriented methods suffer from drawbacks such as a large number of steps, low overall yields, extreme conditions, use of sensitive reagents, non-availability of starting materials and absence of cost-effectiveness (Scheme 1A).8–14
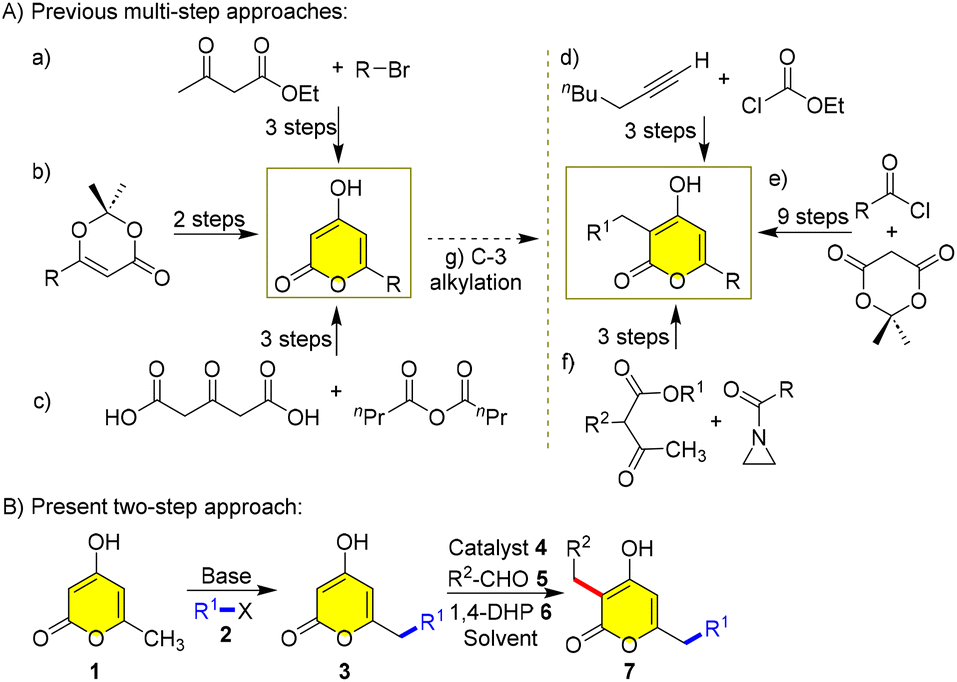 | ||
| Scheme 1 Previous and present strategies for the synthesis of 3,6-dialkyl-2H-pyrones. | ||
Herein, we envision to synthesize 3,6-dialkyl-2H-α-pyrones by a protection-free two-step dialkylation, starting from the triacetate lactone 1 (Scheme 1B). In the first step, an electrophilic substitution at the C-7 position of 1 with alkyl halide 2 through base-mediated dienolate occurs to generate 3. And then, Ramachary reductive C-alkylation of 3 with aldehyde 5, 1,4-DHP 6 and amine 4 catalyst affords C-3 alkylated products 7. Although this strategy was used by Kraus et al. previously for synthesizing photopyrone A, they have not elaborately screened the conditions, as indicated by their low overall yield14 and incomplete understanding of substrate scope, and the synthesis of only one derivative has made it worthwhile for us to revisit the screening of both C-7 and C-3 alkylations to exemplify the importance of this protocol and synthesize for the first time various natural pyrones and analogues 7 (Scheme 1B).
Results and discussion
Hence, our investigation of the three-component Ramachary reductive C-alkylation commenced,15 after synthesizing a library of C-7 alkylated products 3 from the commercially available triacetate lactone 1 with various aliphatic alkyl iodides or bromides 2 as starting materials using modified Hsung's protocol in very good to excellent yields (Scheme 2).16 First, C-7 alkylation was performed by addition of nBuLi at −78 °C to 1 in THF and HMPA (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) followed by addition of ethyl iodide 2a, and stirring at 25 °C for 15 h gave the desired selective C-7 alkylated pyrone 3a in a very good yield of 85% (Scheme 2). Further we tested the feasibility of this protection-free selective alkylation protocol by choosing different aliphatic alkyl halides 2b to (−)-2l having linear and branched chains to generate a library of pyrones 3b to (−)-3l in very good yields owing to the exciting finding of these structures in bioactive natural product synthesis (Scheme 2). Astonishingly, fistupyrone 3d, isolated from Streptomyces sp. TP-A0569,6 was synthesized from 1 and isobutyl iodide 2d in a single step. It is an antifungal agent that inhibits spore germination in the infection of Chinese cabbage by Alternaria brassicicola, the cause of leaf spot disease in cabbage,6 highlighting the importance of this regioselective C-7 alkylation protocol.
:1) followed by addition of ethyl iodide 2a, and stirring at 25 °C for 15 h gave the desired selective C-7 alkylated pyrone 3a in a very good yield of 85% (Scheme 2). Further we tested the feasibility of this protection-free selective alkylation protocol by choosing different aliphatic alkyl halides 2b to (−)-2l having linear and branched chains to generate a library of pyrones 3b to (−)-3l in very good yields owing to the exciting finding of these structures in bioactive natural product synthesis (Scheme 2). Astonishingly, fistupyrone 3d, isolated from Streptomyces sp. TP-A0569,6 was synthesized from 1 and isobutyl iodide 2d in a single step. It is an antifungal agent that inhibits spore germination in the infection of Chinese cabbage by Alternaria brassicicola, the cause of leaf spot disease in cabbage,6 highlighting the importance of this regioselective C-7 alkylation protocol.
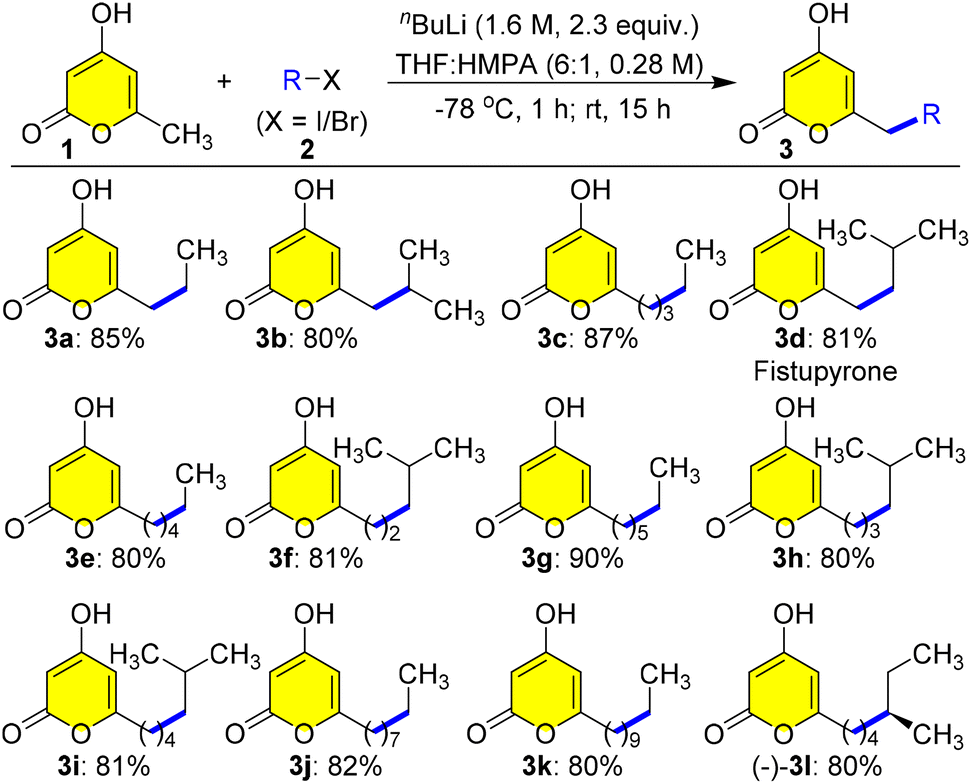 | ||
| Scheme 2 Synthesis of 7-alkyl-4-hydroxy-2H-pyran-2-ones 3 from triacetate lactone 1. Reaction conditions: the reaction mixture of 1 (5.0 mmol) in THF/HMPA (6:1, 17.5 mL) with nBuLi (1.6 M, 2.3 equiv.) at −78 °C was stirred for 1 h before addition of alkyl halide 2a–l (2.0 equiv.) followed by stirring at rt for 15 h. Yields refer to the column purified products. | ||
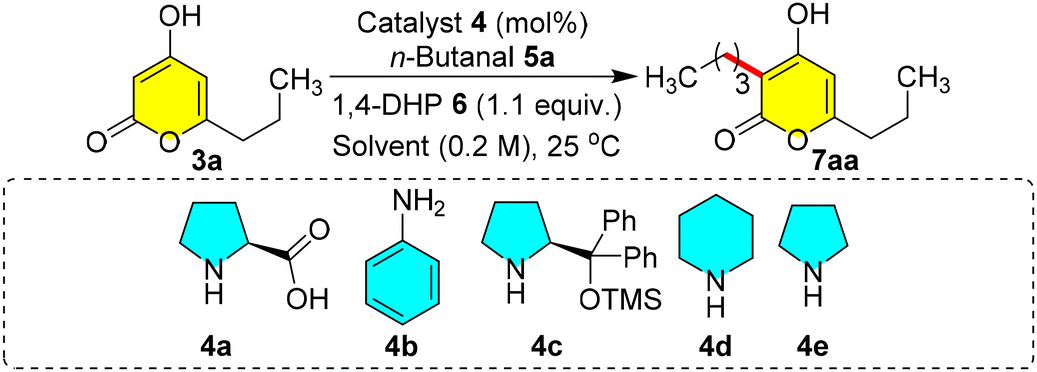
We commenced the optimization of C-3 alkylation through reductive coupling using pyrone 3a and n-butanal 5a as coupling partners. When 3a (0.2 mmol) was reacted with 5a and diethyl 1,4-dihydropyridine dicarboxylate (1,4-DHP) 6 in the presence of proline 4a (10 mol%) in EtOH (0.2 M) at 25 °C, an enthralling yield of 86% of 7aa was observed in 12 h (entry 1, Table 1). Furthermore, when we carried out the same reaction in a polar aprotic solvent such as DMSO (0.2 M), the reaction did not endure well, even after stirring for 24 h, producing only 46% yield of 7aa (entry 2, Table 1), but with chloroform and acetonitrile, there is a sharp increase in the yield to 93% and 95%, respectively, within 1.0 h (entries 3 and 4, Table 1). The reaction in THF also produced a high yield of 91% in 2.0 h (entry 5, Table 1). Similarly, when DCE and DCM were tried, 86% and 97% yields of 7aa were obtained, respectively, within 1.0 h (entries 6 and 7, Table 1). Based on these reactions, we concluded that polar aprotic solvents such as DCM/acetonitrile/chloroform are the most suitable solvents for 7aa without any side reactions. Changing the equivalence of 5a from 1.2 to 1.3 improved the yield slightly to 98% (entry 8, Table 1). And then a decrease in the equivalence of 5a from 1.2 to 1.0 showed a decrease in the yield from 97% to 84% (entry 9), and similarly decreasing the catalyst loading from 10 mol% to 5 mol% also showed a slight reduction in the yield to 93% (entry 10, Table 1). We further screened various catalysts such as primary and secondary amines. In the case of aniline 4b, the reaction was well tolerated with 87% yield, and with DPPOTMS 4c, we could notice a sharp plummet in the yield to 60% in 6 h (entries 11 and 12, Table 1). The use of piperidine 4d and pyrrolidine 4e along with co-catalyst acetic acid could not give any betterment compared to 4a (entries 13 and 14, Table 1). Therefore, 10 mol% of proline 4a was the most suitable catalyst for the promotion of olefination compared to other amines 4b–4e whose reactivity was high, resulting in self-aldol reactions of aldehydes rather than the olefination. With the suitable catalytic conditions for selective C-3 reductive alkylation, we endeavoured to address the scope of this one-pot method for various other derivatives.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 3a (equiv.) | 5a (equiv.) | 4 (10 mol%) | Solvent (0.2 M) | t (h) | 7aa yield (%) |
| a Reactions were carried out in solvent (1.0 mL, 0.2 M) with 1.2 equiv. of 5a (0.24 mmol) and 1.1 equiv. of 6 (0.22 mmol) relative to 3a (0.2 mmol) in the presence of 10 mol% of 4, and yields refer to the column-purified products. b 5 mol% of catalyst 4a was used. | ||||||
| 1 | 1 | 1.2 | 4a | EtOH | 12 | 86 |
| 2 | 1 | 1.2 | 4a | DMSO | 24 | 46 |
| 3 | 1 | 1.2 | 4a | CHCl3 | 1 | 93 |
| 4 | 1 | 1.2 | 4a | CH3CN | 1 | 95 |
| 5 | 1 | 1.2 | 4a | THF | 2 | 91 |
| 6 | 1 | 1.2 | 4a | DCE | 1 | 86 |
| 7 | 1 | 1.2 | 4a | DCM | 1 | 97 |
| 8 | 1 | 1.3 | 4a | DCM | 1 | 98 |
| 9 | 1 | 1.0 | 4a | DCM | 1 | 84 |
| 10b | 1 | 1.2 | 4a | DCM | 1 | 93 |
| 11 | 1 | 1.2 | 4b | DCM | 1 | 87 |
| 12 | 1 | 1.2 | 4c | DCM | 6 | 60 |
| 13 | 1 | 1.2 |
4d/AcOH (1:1) |
DCM | 1 | 88 |
| 14 | 1 | 1.2 |
4e/AcOH (1:1) |
DCM | 1 | 48 |
As a model study, first we investigated the role of alkyl chain lengths at the C-6 position of 3 in the reductive coupling using n-butanal 5a as a coupling partner in C-3 alkylation. Initially, a 4a-catalysed reaction of 3b with 5a was performed using 6 as a reducing source at 25 °C in DCM (0.2 M), which give 7ba in excellent yield (96%) within 1.0 h (Scheme 3). When we tried the catalytic 5a reaction with other pyrones containing various C-6 alkyl groups such as 3c, 3f, 3g, 3j and 3k, the reaction gave extremely good yields (90–97%) of natural pyrone analogues 7ca, 7fa, 7ga, 7ja and 7ka at 25 °C within 1.0 h without any steric factors of C-6 alkyl groups (Scheme 3).
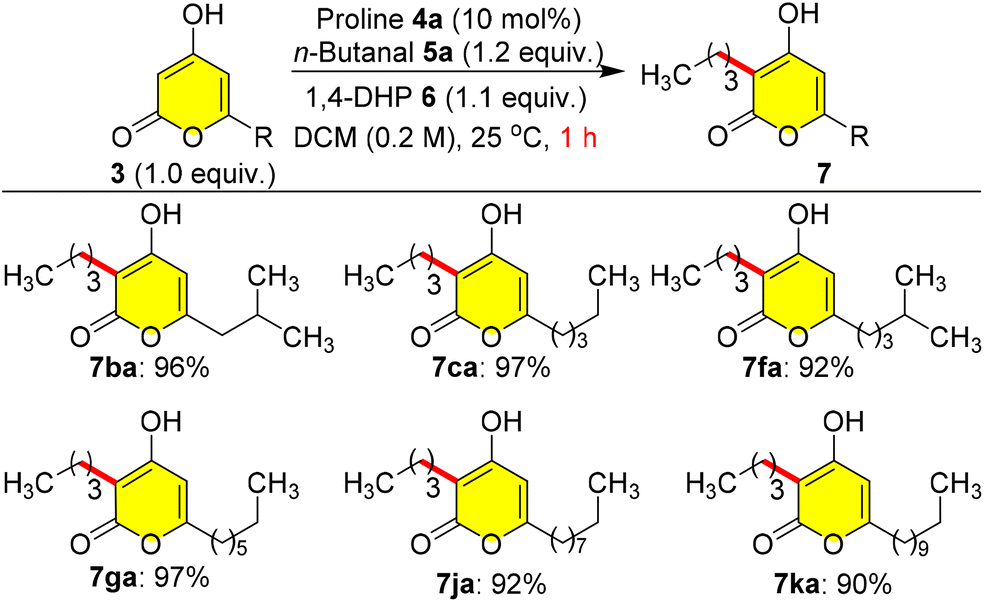 | ||
| Scheme 3 Synthesis of photopyrone analogues. | ||
Furthermore, the sensitivity of the reaction was tested using volatile acetaldehyde 5b as a coupling partner with various C-6 alkylated pyrones 3a–k; here we used 5.0 equiv. of 5b due to the volatile nature, and the reaction endured well with yields of 7ab–7kb ranging from 89 to 96% within 1.0 h at 25 °C (Scheme 4). Inadvertently, we ended up synthesizing isogermicidin A (7bb) and isogermicidin B (7ab), two natural products isolated from the fermentation broth of marine derived Streptomyces sp. MDW-06,7 as a first total synthesis highlighting the efficiency of this coupling protocol.
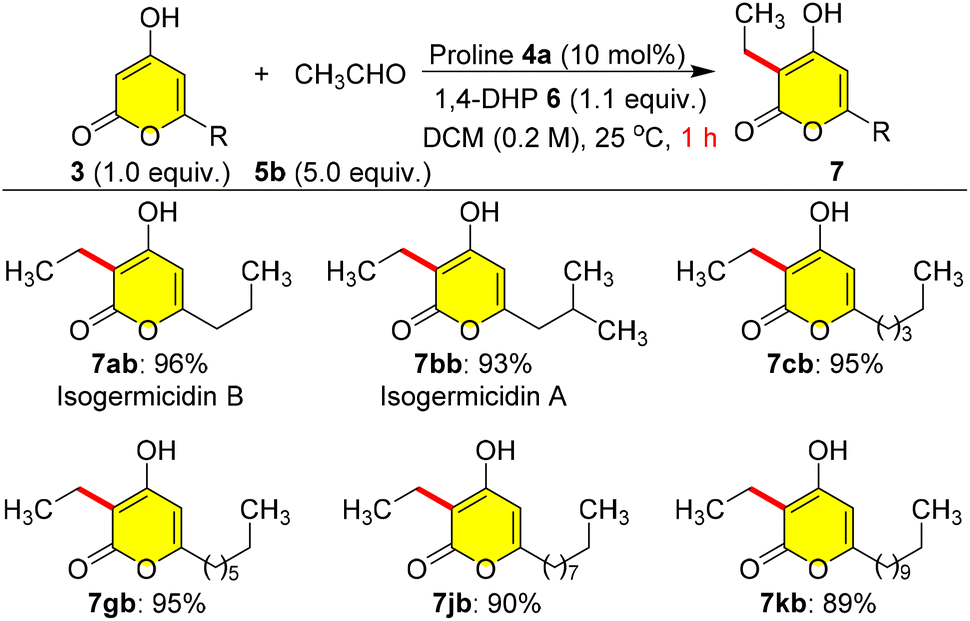 | ||
| Scheme 4 Synthesis of isogermicidin A, isogermicidin B and analogues. | ||
After these studies, we attempted to use n-hexanal 5c as an appropriate coupling partner with 3 for the synthesis of pseudopyronine and photopyrone natural products (Scheme 5). Therefore, we carried out a coupling reaction between 3a and 5c under the optimized conditions to synthesize the photopyrone A analogue 7ac in 91% yield within 1.0 h. Interestingly, when 3b was reductively coupled with 5c, we could obtain photopyrone A 7bc in 96% yield at 25 °C in 1.0 h. In a similar manner, we synthesized pseudopyronine A 7cc by coupling 3c with 5c in 94% yield, pseudopyronine B 7gc using 3g and 5c in 96% yield, pseudopyronine C 7jc by coupling 3j and 5c in 92% yield, and the pseudopyronine C analogue 7kc by coupling 3k and 5c in 90% yield (Scheme 5). Photopyrone A 7bc is a cell signalling molecule isolated from Photorhabdus luminescens, and pseudopyronines A–C were first isolated from Pseudomonas fluorescens and they showed interesting biological activities.3,5
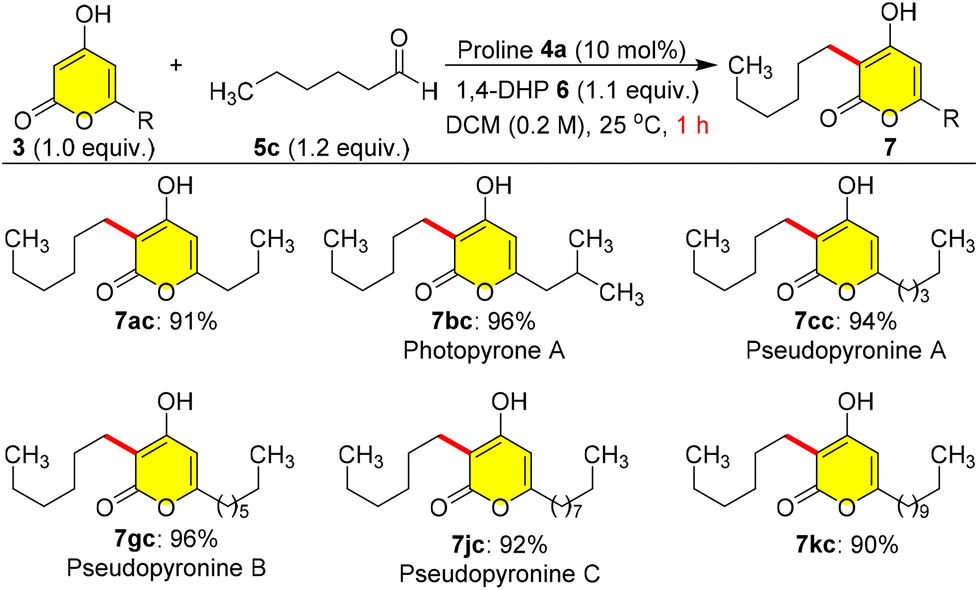 | ||
| Scheme 5 Total synthesis of pseudopyronines and photopyrones. | ||
As there were no earlier reports on methods for the total synthesis of photopyrones, herein we endeavoured to synthesize cell signalling natural products in high yields (Scheme 6). Hence, we employed pyrone 3b as a coupling partner, and treated it with different branched and long chain aliphatic aldehydes such as 5d, 5e, 5f, and 5g which furnished the corresponding natural products photopyrone B 7bd, photopyrone C 7be, photopyrone E 7bf, and photopyrone G 7bg in excellent yields ranging from 86% to 92% within 1.0 h at 25 °C (Scheme 6).
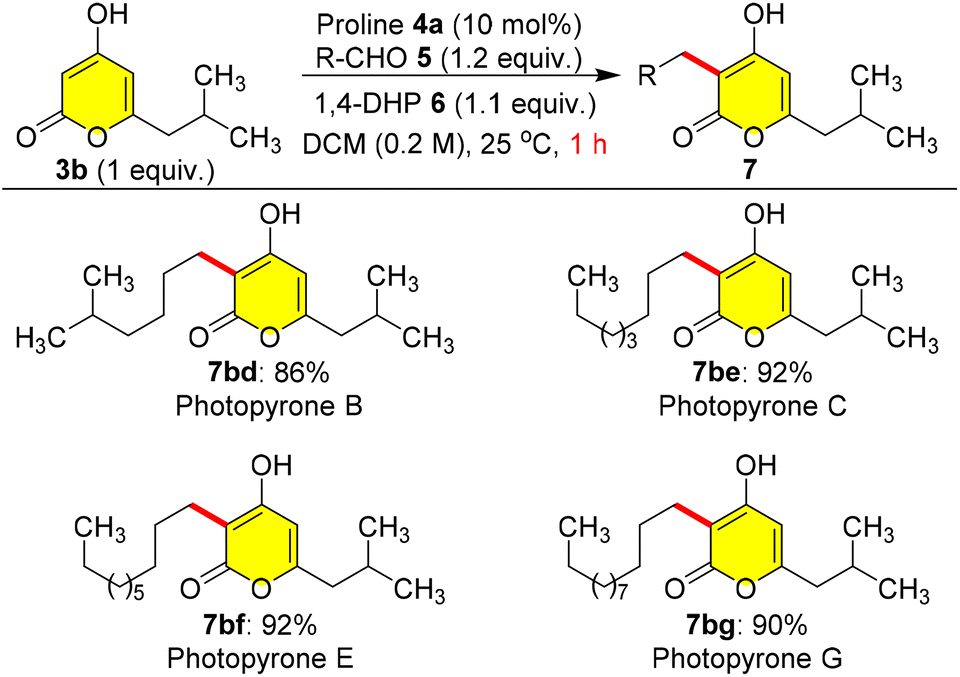 | ||
| Scheme 6 Total synthesis of photopyrones B-G. | ||
After the successful synthesis of photopyrones and pseudopyronines, we planned to synthesize violapyrones, a similar class of natural products having exciting pharmacological activities. Violapyrones are 4-hydroxy-2H-α-pyrones having a methyl group at C-3 and alkyl chains at C-6. At this juncture, we encountered an obstacle in our design, as our direct 4a-catalysed reductive C-3 alkylation of the pyrones 3 failed when employing formaldehyde 5h as a coupling partner and 1,4-DHP 6 as a hydrogen source, due to many side reactions. In order to address this drawback, we employed another three-component organocatalytic reaction,17 where thiophenol 8 was used to mask the in situ formed olefin from the reaction of pyrone 3 with paraformaldehyde 5h under piperidine/acetic acid-catalysis in ethanol at 55 °C for 6 h, which gave phenyl sulfide masked violapyrones 9 in very good yields (Scheme 7). Interestingly, the three-component reaction of pyrone 3b with paraformaldehyde 5h and thiophenol 8 under the catalysis of proline 4a (10 mol%) or piperidine/acetic acid in DCM (0.25 M) at 25 °C for 6 h furnished the expected phenyl sulfide masked germicidin-I 9bh in only <7% yield; but the same piperidine/acetic acid-catalysed reaction at 55 °C in a sealed tube for 6 h furnished 9bh in moderate (31%) yield (results not shown in Scheme 7). In a further development, the three-component organocatalytic reaction of pyrone 3b with paraformaldehyde 5h and thiophenol 8 under the catalysis of piperidine/acetic acid in ethanol at 55 °C for 6 h furnished the phenyl sulfide masked germicidin-I 9bh in 76% yield (Scheme 7). A protic polar (EtOH) solvent at 55 °C induced the three-component reaction rate in a better manner than an aprotic polar (DCM) solvent at 25 °C. In a similar manner, a series of different C-6 alkylated pyrones 3 having varied linear and branched alkyl chains were employed in the three-component organocatalytic reaction to synthesize the phenyl sulfide masked violapyrones 9ch to 9jh in very good yields within 6 h as shown in Scheme 7.
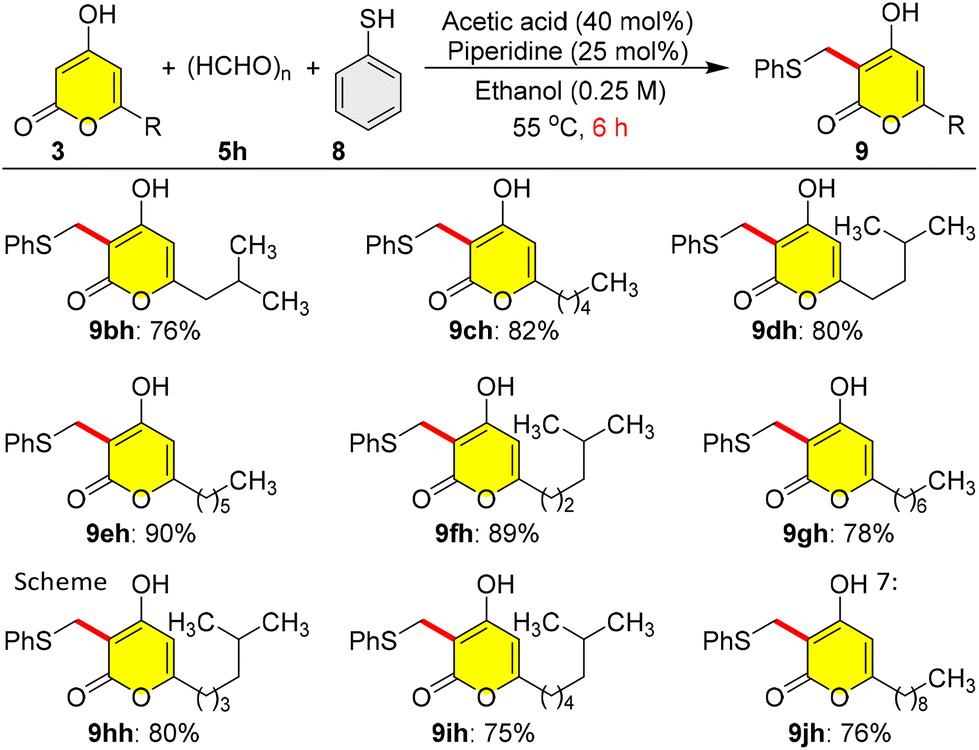 | ||
| Scheme 7 Synthesis of methyl-masked violapyrones 9via three-component C-3 alkylation of pyrones 3 with paraformaldehyde 5h and thiophenol 8. Reaction conditions: reactions were carried out by dissolving 3 (0.5 mmol) in ethanol (1.0 mL) and adding this solution to a mixture of 1.0 equiv. of 5h (0.5 mmol), 3.0 equiv. of 8 (1.5 mmol), 40 mol% of acetic acid, and 25 mol% of piperidine in ethanol (1.0 mL) at 55 °C and stirring for 6 h. Yields refer to the column-purified products. | ||
Masked violapyrones 9bh–9jh were converted into natural violapyrones 7bh–7jh by single transformation as first total synthesis. When we treated phenyl sulfide masked 9bh (0.2 mmol) with freshly prepared RANEY®-nickel (200 mg) in ethanol at 25 °C for 1.0 h, the germicidin I 7bh was furnished in 82% yield (Scheme 8).7 In a similar manner, treatment of other masked violapyrones 9ch–9jh with RANEY®-nickel at 25 °C for 1.0 h furnished the following medicinally important violapyrones (VLPs): VLP-A 7fh in 78%, VLP-B 7hh in 80%, VLP-I 7gh in 80%, VLP-J 7eh in 79%, VLP-J1 7dh in 81%, VLP-H 7ih in 79%, and VLP-L 7ch in 80% yields (Scheme 8).4 With this impetus, we further showed interest to synthesize chiral ent-(−)-violapyrone C and its analogue. Natural (+)-violapyrone C was isolated from the mass culture of an actinomycete, Streptomyces sp. 112CH148,18 and is known to possess cytotoxicity against a panel of six human tumor cell lines and has an inhibitory effect on the HIF (hypoxia-inducible factor) pathway, which is related to tumor progression, invasion, and metastasis.19 Therefore, we ventured on a route to synthesize ent-(−)-violapyrone C starting from (S)-(−)-citronellal, a commercially available starting material, which was transformed into the chiral pyrone (−)-3l in 8 steps with 25% overall yield (Scheme S1, see the ESI†). Chiral pyrone (−)-3l was then employed in a catalytic reductive C-3 alkylation with n-hexanal 5c and 1,4-DHP 6 to synthesize the violapyrone derivative (−)-7lc in 87% yield (Scheme 9). Similarly, sulphur masked three-component methylation of (−)-3l was performed to obtain (−)-9lh, followed by its desulfurization with freshly prepared RANEY®-nickel, which furnished the opposite enantiomer (−)-violapyrone C (−)-7lh in 80% yield (Scheme 9).
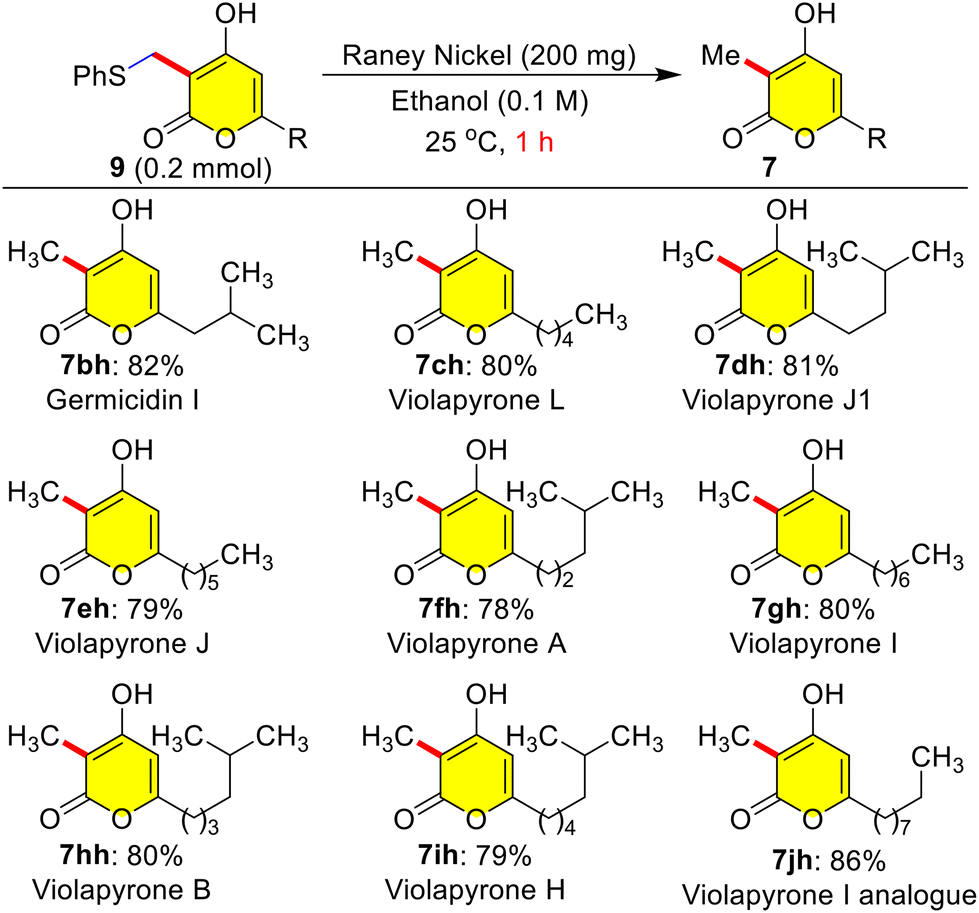 | ||
| Scheme 8 Total synthesis of violapyrones A–L. | ||
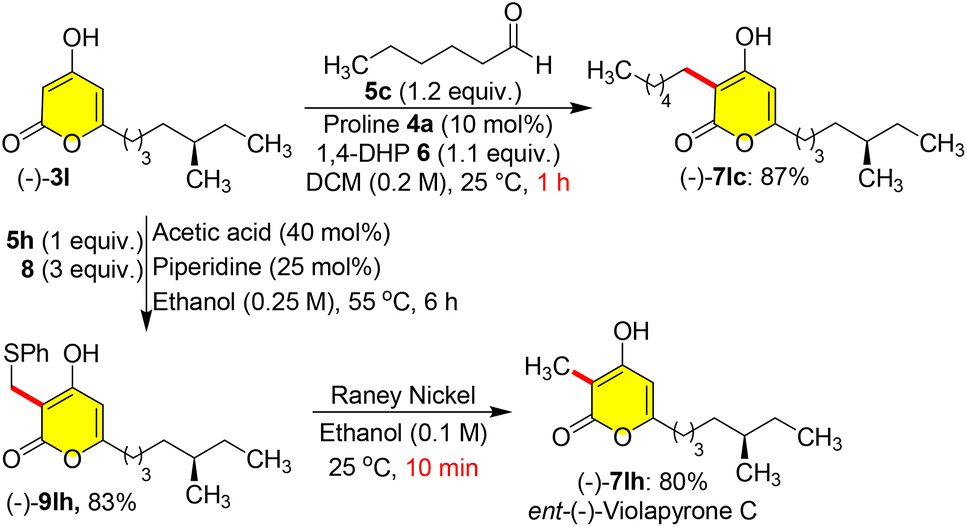 | ||
| Scheme 9 Total synthesis of (−)-violapyrone C and its analogue. | ||
With further applications in mind, we transformed the natural products 7 into another set of natural products 10–12 by simple acetylation/methylation (Scheme 10). The reaction of photopyrone A 7bc with Ac2O in DCM at 25 °C for 0.5 h gave the protected photopyrone A 10bc in 93% yield. Interestingly, the reaction of violapyrone L 7ch with ethereal diazomethane at 0 °C for 30 min gave the α-pyrone childinin G2011ch in 63% yield as a major product and γ-pyrone 12ch in 34% yield as a minor product. Childinins are natural products isolated from the fruiting bodies of Daldinia childiae. Similarly, treatment of violapyrones VLP-A 7fh, VLP-I 7gh and VLP-B 7hh with ethereal diazomethane at 0 °C for 30 min gave the α-pyrones VLP-Q 11fh in 56%, VLP-S 11gh in 50%, and VLP-R 11hh in 50% yields as major products and γ-pyrones 12fh in 31%, 12gh in 31%, and 12hh in 30% yields as minor products, respectively (Scheme 10). These α- and γ-pyrones 11/12 are well separated and differences in their carbonyl peaks in 13C NMR clearly evidence the structural differences between them (see the ESI† for more information). A simple O-methylation of 7 unveiled the synthesis of two distinct classes of natural products α- and γ-pyrones 11/12 highlighting the potential of the present protocol (Scheme 10).
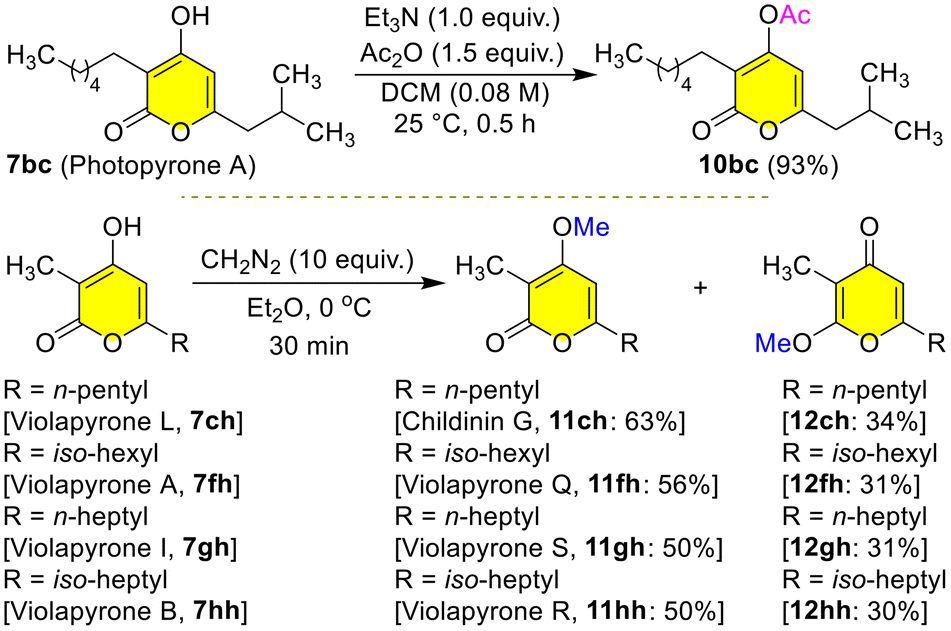 | ||
| Scheme 10 Total synthesis of childinin G and violapyrones Q–S. | ||
Furthermore, to demonstrate the robustness of our protection-free total synthesis,21 we performed the gram scale synthesis of four natural products pseudopyronine A 7cc, photopyrone A 7bc, pseudopyronine B 7gc and photopyrone C 7be with very good overall yields from the corresponding starting materials pyrones 3b, 3c or 3g and n-hexanal 5c or n-octanal 5e (Scheme 11). In these organocatalytic reductive C-3 alkylation steps, it was feasible to obtain the final natural products 7cc, 7bc, 7gc and 7be as fine off-white solids by simple filtration after washing the reaction mixture with hexanes to get rid of the side products without column purification (Fig. S2, see the ESI†).
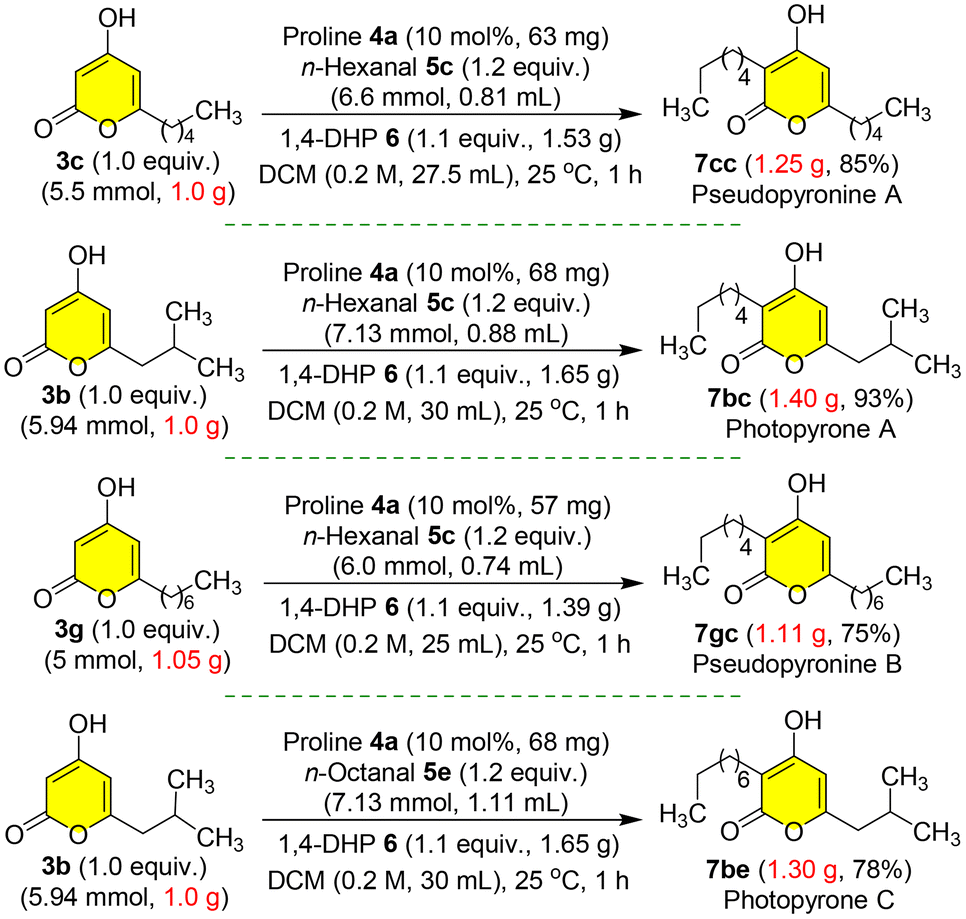 | ||
| Scheme 11 Gram-scale synthesis of pyrones. | ||
Conclusions
In summary, a two-step, C-dialkylation strategy was developed for the construction of a library of natural products of pseudopyronines, photopyrones, violapyrones and their analogues in a highly selective manner with excellent yields. A single O-methylation reaction was revealed for the synthesis of 4-methoxy-2H-α-pyrones and 2-methoxy-4H-γ-pyrones covering a colossal niche of natural products, showing extraordinary biological properties. The gram scale synthesis of four natural products demonstrated the method's viability for industrial applications. A study of systematic screening of structure activity relationships (SAR) of the synthesized molecules to reveal their biological properties is under progress.Data availability
General information, experimental procedures, and characterization data of all new compounds, correlation data of synthetic compounds with natural products and NMR spectra are provided in the ESI.† Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Center (CCDC) under the deposition numbers CCDC 2292620 (7bb), CCDC 2292621 (7bc), and CCDC 2292622 (7jb).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was made possible by a grant from the Department of Science and Technology (DST), SERB, New Delhi [Grant No.: CRG/2018/000775], and a UoH-IoE grant [Grant No.: UoH/IoE/RC1/RC1-20-002]. A. H. thanks the University Grants Commission (UGC), New Delhi, for a Maulana Azad National Fellowship. R. S. thanks the UoH-IoE for her research fellowship. We thank Dr P. Raghavaiah, Central University of Karnataka, Gulbarga, for assistance with X-ray structural analysis.References
-
(a) Y. Huang, S. Hoefgen, F. Gherlone and V. Valiante, Angew. Chem., Int. Ed., 2022, 61, e202206851 CrossRef CAS PubMed
and references cited therein; (b) D. Kresovic, F. Schempp, Z. Cheikh-Ali and H. B. Bode, Beilstein J. Org. Chem., 2015, 11, 1412 CrossRef CAS PubMed
- A. Goel and V. J. Ram, Tetrahedron, 2009, 65, 7865 CrossRef CAS
- A. O. Brachmann, S. Brameyer, D. Kresovic, I. Hitkova, Y. Kopp, C. Manske, K. Schubert, H. B. Bode and R. Heermann, Nat. Chem. Biol., 2013, 9, 573 CrossRef CAS PubMed
-
(a) L. Hou, H. Huang, H. Li, S. Wang, J. Ju and W. Li, Microb. Cell Fact., 2018, 17, 61 CrossRef PubMed
-
(a) A. C. Giddens, L. Nielsen, H. I. Boshoff, D. Tasdemir, R. Perozzo, M. Kaiser, F. Wang, J. C. Sacchettini and B. R. Copp, Tetrahedron, 2008, 64, 1242 CrossRef CAS
- Y. Igarashi, M. Ogawa, Y. Sato, N. Saito, R. Yoshida, H. Kunoh, H. Onaka and T. Furumai, J. Antibiot., 2000, 53, 1117 CrossRef CAS PubMed
- X.-M. Zhang, A.-H. Peng, W.-D. Xie, M. Wang, D. Zheng and M.-K. Feng, Chem. Biodivers., 2020, 17, e2000140 CrossRef CAS PubMed
- J. S. Yadav, B. Ganganna, P. Dutta and K. K. Singarapu, J. Org. Chem., 2014, 79, 10762 CrossRef CAS PubMed
- Y. Liu, Q. Zhang, L.-H. Chen, H. Yang, W. Lu, X. Xie and F.-J. Nan, ACS Med. Chem. Lett., 2016, 7, 579 CrossRef CAS PubMed
- C. A. Brandenburg, C. A. Castro and A. A. Blacutt,
et al., J. Nat. Prod., 2020, 83, 1810 CrossRef CAS PubMed
- Gayyur, S. Choudhary, A. Saxena and N. Ghosh, Org. Biomol. Chem., 2020, 18, 8716 RSC
- I. P. Lokot, F. S. Pashkovsky and F. A. Lakhvich, Tetrahedron, 1999, 55, 4783 CrossRef CAS
- D. Schmidt, J. Conrad, I. Klaiber and U. Beifuss, Chem. Commun., 2006, 45, 4732 RSC
- G. A. Kraus, K. Basemann and T. Guney, Tetrahedron Lett., 2015, 56, 3494 CrossRef CAS
- For the three-component reductive C-alkylation reaction, see:
(a) D. B. Ramachary and M. Kishor, J. Org. Chem., 2007, 72, 5056–5068 CrossRef CAS PubMed
- X. Zhang, M. McLaughlin, R. L. P. Munoz, R. P. Hsung, J. Wang and J. Swidorski, Synthesis, 2007, 749 Search PubMed
- P. de March, M. Moreno-Manas, R. Pi and A. Trius, J. Heterocycl. Chem., 1982, 19, 335 CrossRef CAS
- H. J. Shin, H.-S. Lee, J. S. Lee, J. Shin, M. A. Lee, H.-S. Lee, Y.-J. Lee, J. Yun and J. S. Kang, Mar. Drugs, 2014, 12, 3283 CrossRef CAS PubMed
- J. S. Lee, J. Shin, H. J. Shin, H.-S. Lee, Y.-J. Lee, H.-S. Lee and H. Won, Eur. J. Org. Chem., 2014, 4472 CrossRef CAS
- Z.-Z. Zhao, H.-P. Chen, Y. Huang, S.-B. Zhang, Z.-H. Li, T. Feng and J.-K. Liu, Phytochemistry, 2017, 142, 68 CrossRef CAS PubMed
- For the total synthesis of natural products and drugs from organocatalytic domino reactions, see:
(a) D. B. Ramachary, M. Kishor and G. B. Reddy, Org. Biomol. Chem., 2006, 4, 1641–1646 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2292620–2292622 for 7bb, 7bc and 7jb. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01923c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |