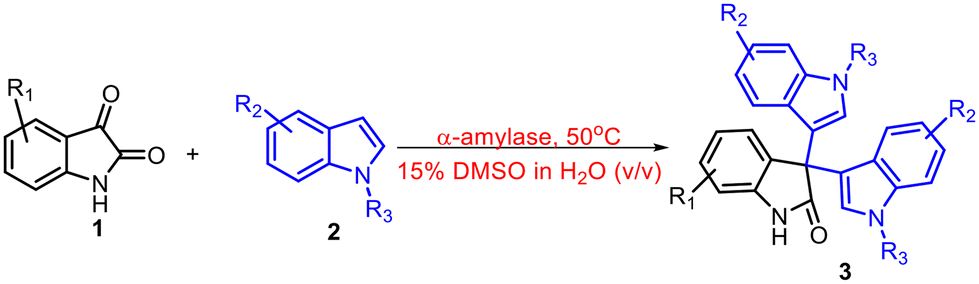
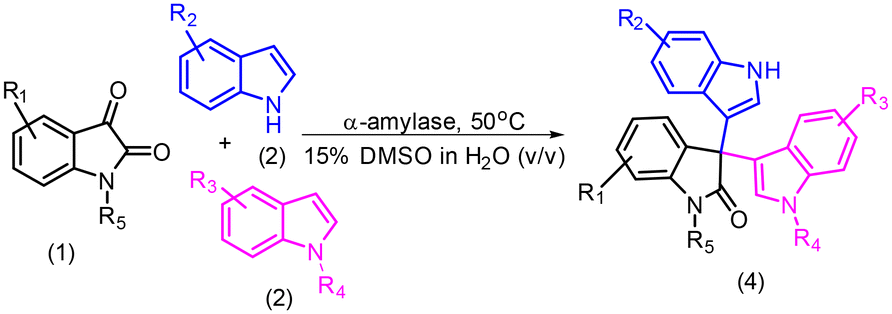
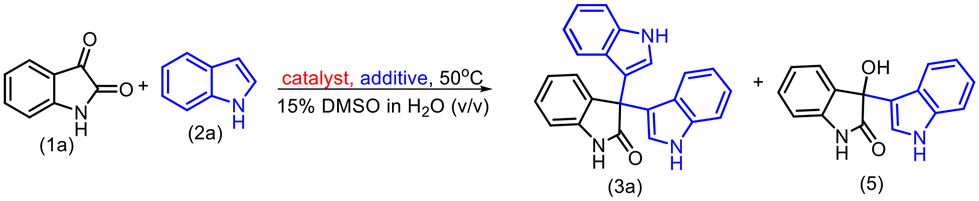
Experimental and computational investigation of the α-amylase catalyzed Friedel–Crafts reaction of isatin to access symmetrical and unsymmetrical 3,3′,3′′-trisindoles†
Received
27th November 2023
, Accepted 2nd February 2024
First published on 2nd February 2024
Abstract
Trisindoles are of tremendous interest due to their wide range of biological activities. In this context, a number of methods have been reported in the past to synthesize 3,3′,3′′-trisindoles. However, most of the methods are only able to produce symmetrical 3,3′,3′′-trisindoles. Herein, we develop a sustainable and efficient approach to synthesize symmetrical as well as unsymmetrical 3,3′,3′′-trisindoles in a very selective manner using the α-amylase enzyme as a catalyst. Furthermore, various differently substituted isatin and indoles were used to prove the generality of the protocol and symmetrical or unsymmetrical 3,3′,3′′-trisindoles were obtained in 43–97% isolated yields. Next, a probable mechanism is proposed and investigated using molecular dynamics (MD) investigation to gain more insight into the role of residues available in the active site of the α-amylase enzyme. These studies revealed that Glu230, Lys209, and Asp206 in the active site of α-amylase play an important role in this catalysis. Moreover, the DFT studies suggested the formation of bisindole and alkylideneindolenine intermediates during the transformation. We synthesized four different biologically important 3,3′,3′′-trisindoles on a gram scale, which proved the robustness and scalability of this protocol.
Introduction
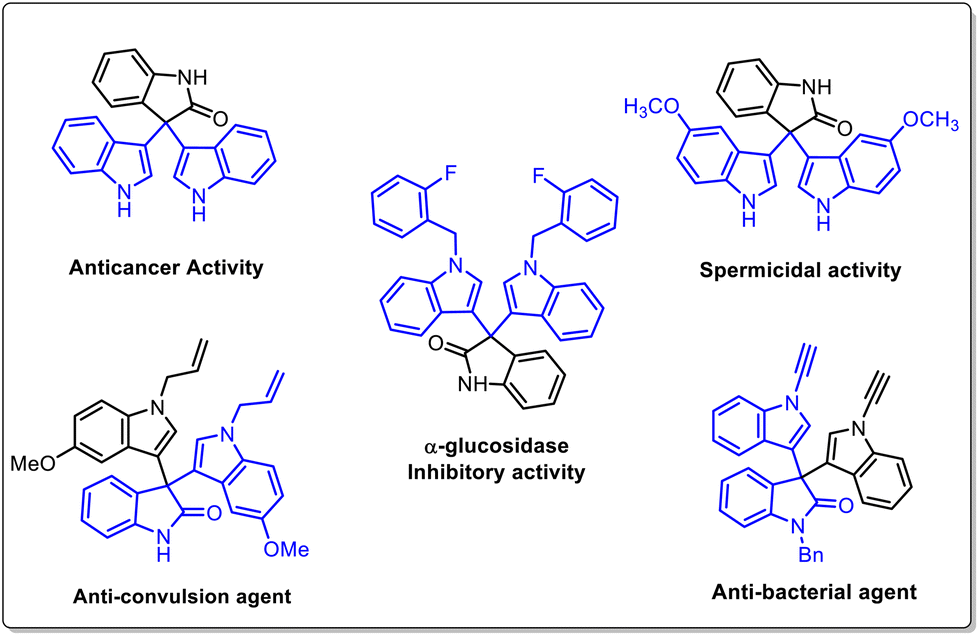
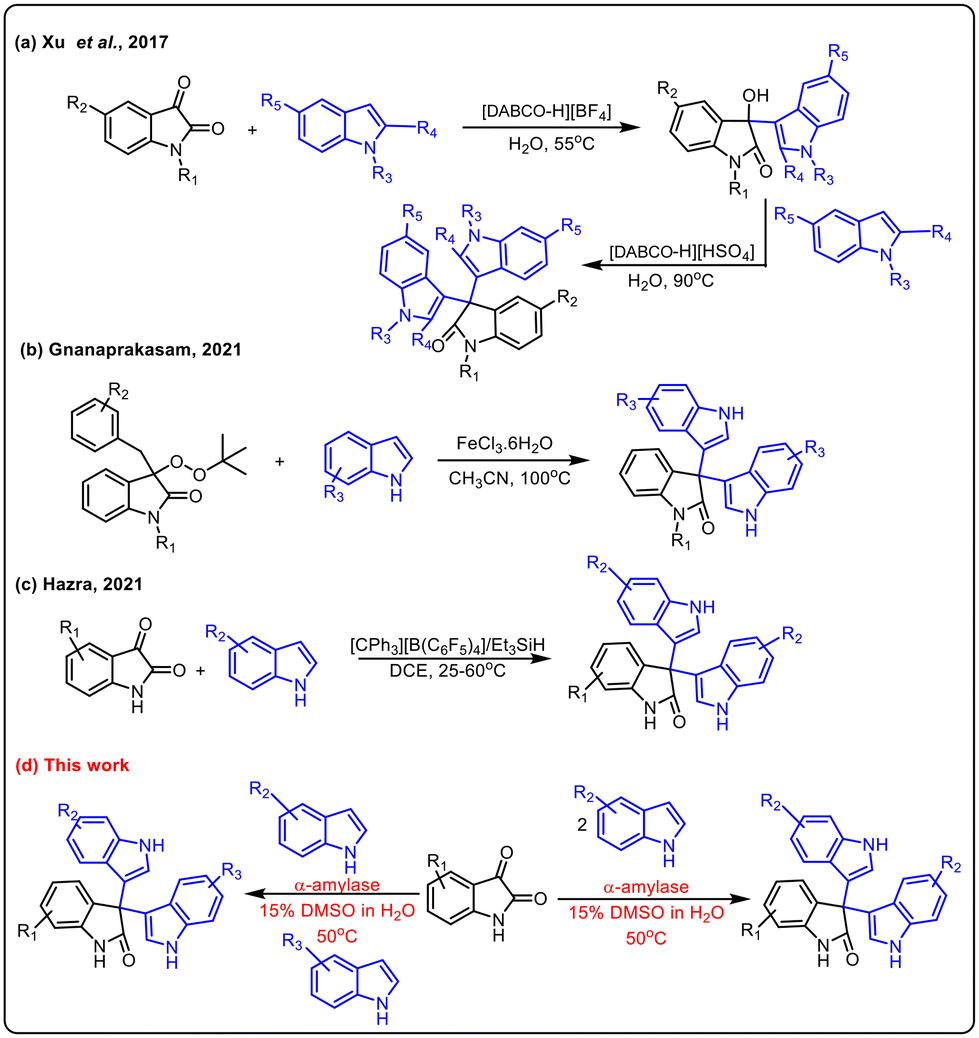
Indole derivatives have been identified as an important class of compounds due to their wide range of pharmaceutical applications and they can be used as versatile building blocks for constructing indole-containing natural products.1 In particular, 3,3′,3′′-trisindoles are of great interest and display a broad spectrum of biological activities such as anti-bacterial, anti-cancer, anti-inflammatory, α-glucosidase inhibitory, spermicidal, and anti-convulsant activities (Fig. 1).2 Also, these compounds have some other industrial applications like use as a hydride acceptor and dyes for physicochemical studies. Notwithstanding a variety of pharmaceutical and industrial applications, smaller numbers of methods are available to synthesize this class of compounds. The Friedel–Crafts type electrophilic substitution reaction of isatin and indole catalysed by Lewis or protic acids is one of the traditional methods to synthesize 3,3′,3′′-trisindole derivatives.3 In 2017, Xu and co-workers used DABCO-based ionic liquids to catalyze Friedel–Crafts alkylation for synthesizing 3,3-diindolyloxindoles (Scheme 1a).4 Furthermore, Gnanaprakasam developed FeCl3·6H2O mediated oxidative cleavage of peroxyoxindole and further reaction with indole to afford trisindoline derivatives (Scheme 1b).5 Recently, Lambert's salt-initiated synthesis of symmetrical/unsymmetrical 3,3′,3′′-trisindole has been developed by Hazra and group members (Scheme 1c).6
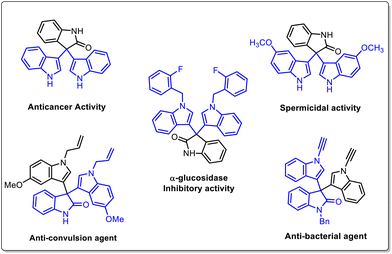 |
| | Fig. 1 Examples of biologically important 3,3′,3′′-trisindoles. | |
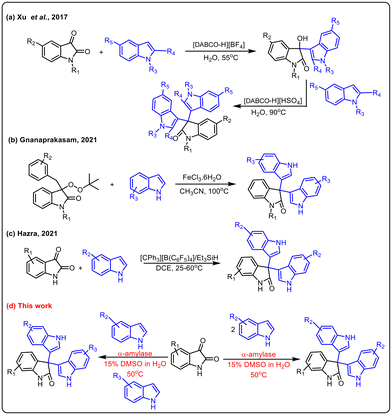 |
| | Scheme 1 Different methodologies for the synthesis of 3,3′,3′′-trisindoles. | |
Despite this progress, most of the methods suffer from several disadvantages such as the use of harsh reaction conditions, longer reaction times, laborious work-up procedures, and the use of expensive and/or toxic catalysts and solvents. Also, methods for the selective synthesis of unsymmetrical 3,3′,3′′-trisindoles are rare. Therefore, the development of an operationally simple and environment-friendly method that can overcome the disadvantages of previously reported protocols, and be able to selectively produce symmetrical as well as unsymmetrical 3,3′,3′′-trisindoles, is highly required.
Nowadays, biocatalysis which mainly uses enzymes as a catalyst in chemical transformations has evolved into a widely accepted technology in the area of chemical and pharmaceutical synthesis.7 Furthermore, the use of enzymes in chemical catalysis offers a number of advantages, e.g. can work under mild reaction conditions, easy reaction workup procedure, higher product selectivity, less or no toxic waste generation, etc.8 A number of new-to-nature chemical transformations using enzymes as a catalyst have been reported in the last few years.9 In this context, Arnold's group has reported the application of engineered heme-proteins in numerous abiotic chemical transformations.10 Besides, some other research groups are also exploring the applications of different enzymes as a catalyst in abiotic organic transformations.11 Hydrolase enzymes are still being widely used in the search for new-to-nature chemical transformations due to their various advantages like higher stability under reaction conditions, easy accessibility, and longer shelf life.12 Previously, we have also reported a number of novel enzymatic transformations using lipase and α-amylase as a catalyst.13,14 Recently, we reported the application of the α-amylase enzyme in the gram-scale synthesis of bis-indole containing bioactive molecules.15 Keeping in view the diverse applications of 3,3′,3′′-trisindoles and limitations of the previously reported methods, herein, we report the highly selective and efficient synthesis of symmetrical and unsymmetrical 3,3′,3′′-trisindoles using the α-amylase enzyme catalysed Friedel–Crafts type reaction of isatin and indoles.
Results and discussion
Enzyme screening for suitable α-amylase
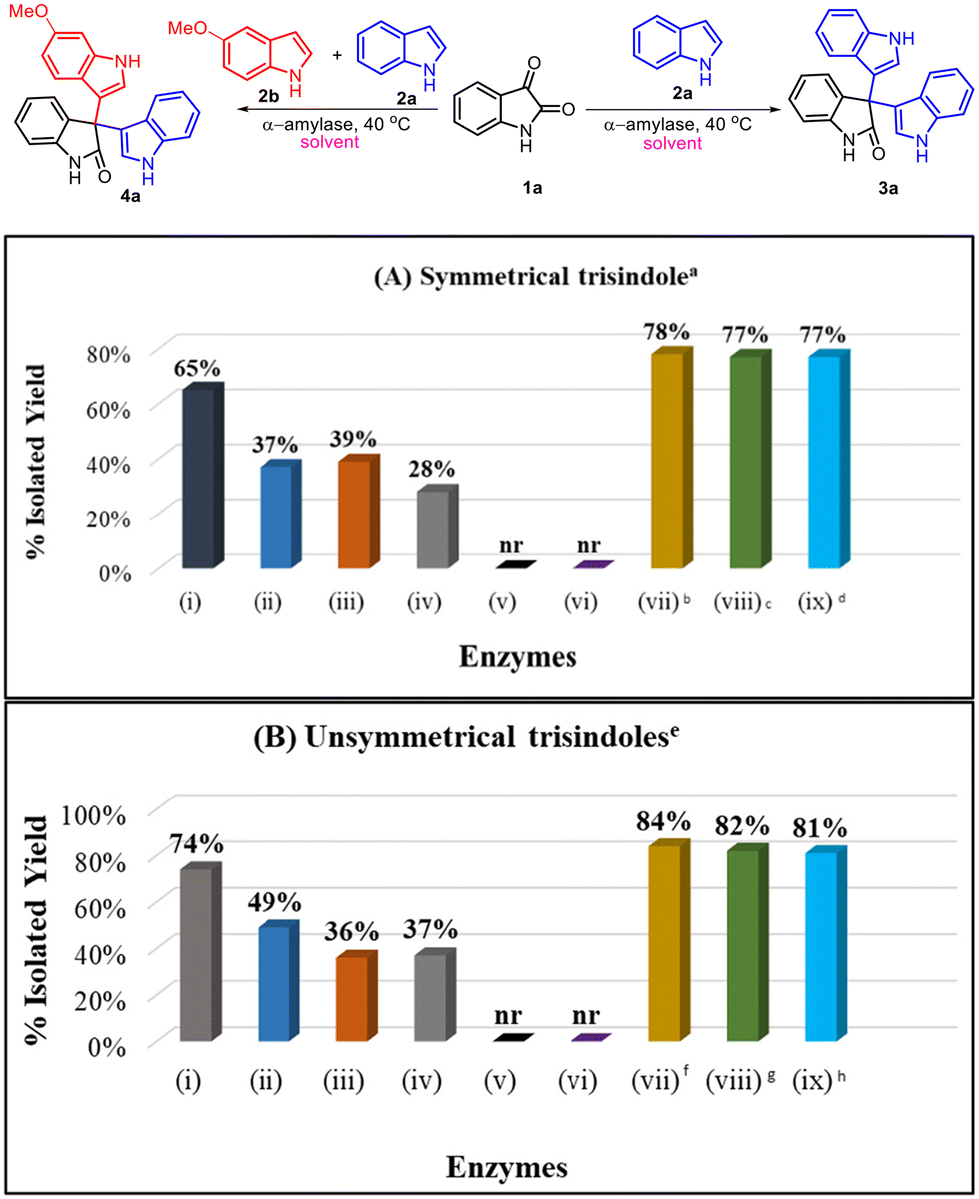
We began our investigation by selecting isatin (1a) and indole (2a) as model substrates and screened a library of amylase enzymes to synthesize symmetrical 3,3′,3′′-trisindole (3a). In this context, α-amylase from Aspergillus oryzae furnished a good yield i.e. 65% for the model reaction (entry i, Scheme 2A). Interestingly, α-amylase obtained from other sources provided moderate conversion in the model reaction (entries ii–iv, Scheme 2A). However, there was no reaction observed either in the presence of the β-amylase enzyme or in the absence of the enzyme catalyst (entries v and vi, Scheme 2A). Furthermore, we optimized the loading of the catalyst and first increased the concentration of α-amylase from 2 mg ml−1 to 3 mg ml−1 and then to 5 mg ml−1, and the product (3a) was obtained in a slightly improved yield i.e. 78% while using 3 mg ml−1 of the enzyme (entries vii and viii, Scheme 2A). Also, we observed that the change in the molar ratio of isatin and indole from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 1:3 did not lead to any improvement in the outcome of the model reaction (entry ix, Scheme 2A). Next, we screened the same library of amylase enzymes for the synthesis of unsymmetrical 3,3′,3′′-trisindole by using isatin (1a), indole (2a) and 5-methoxyindole (2b) as the model substrates. α-Amylase from Aspergillus oryzae was still the best enzyme and provided the product (4a) in 74% isolated yield (entry i, Scheme 2B). Interestingly, we obtained an improved yield i.e. 84% for unsymmetrical 3,3′,3′′-trisindole (4a) when 3 mg ml−1 of α-amylase from Aspergillus oryzae was employed (entry vii, Scheme 2B).
:2 to 1:3 did not lead to any improvement in the outcome of the model reaction (entry ix, Scheme 2A). Next, we screened the same library of amylase enzymes for the synthesis of unsymmetrical 3,3′,3′′-trisindole by using isatin (1a), indole (2a) and 5-methoxyindole (2b) as the model substrates. α-Amylase from Aspergillus oryzae was still the best enzyme and provided the product (4a) in 74% isolated yield (entry i, Scheme 2B). Interestingly, we obtained an improved yield i.e. 84% for unsymmetrical 3,3′,3′′-trisindole (4a) when 3 mg ml−1 of α-amylase from Aspergillus oryzae was employed (entry vii, Scheme 2B).
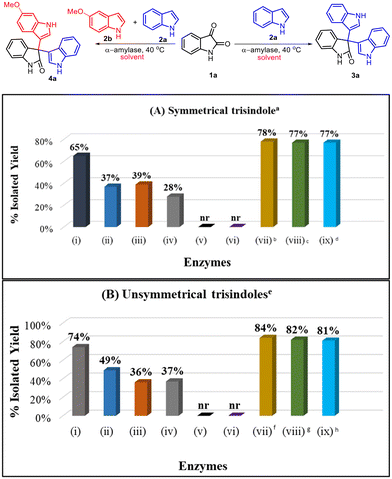 |
| | Scheme 2 Screening of different α-amylase enzymes for the model reactions. Enzymes: (i) α-amylase from Aspergillus oryzae, (ii) α-amylase from Bacillus sp., (iii) α-amylase from Bacillus amyloliquefaciens, (iv) α-amylase from hog pancreas, (v) β-amylase, (vi) no enzyme, and (vii–ix) α-amylase from Aspergillus oryzae. aReaction conditions for 3a: indole (2 equiv.), isatin (1 equiv.), catalyst (2 mg ml−1), 2.5 ml of 10% DMSO in water (v/v) as a solvent at 40 °C for 12 hours, benzyme = 3 mg ml−1, cenzyme = 5 mg ml−1, dindole = 3 equiv. eReaction conditions for 4a: indole (1 equiv., 0.67 mmol), 5-methoxyindole (1 equiv., 0.68 mmol), isatin (1 equiv., 0.68 mmol), catalyst (2 mg ml−1), 2.5 ml of 10% DMSO in water (v/v) as a solvent at 40 °C for 12 hours, fenzyme = 3 mg ml−1, genzyme = 5 mg ml−1, hindole and 5-methoxyindole = 1.5 equiv. each. | |
Solvent screening to improve the conversion
After selecting the best amylase enzyme, we screened different solvents for the formation of symmetrical as well as unsymmetrical 3,3′,3′′-trisindoles (Scheme 3). Unpredictably, there was no product formation when DCM, toluene, hexane, 1,4-dioxane, and H2O were employed as solvents in the model reactions (Scheme 3A and B). Also, the conversion was found to be lower in the case of EtOH, MeOH, ACN, and DMSO (Scheme 3A and B). It has been observed previously that water is the best solvent for enzymatic reactions; however, the solubility of reactants in water is an issue. To overcome this issue, we tried different combinations of organic solvents along with water (v/v) like EtOH/water, MeOH/water, ACN/water and DMSO/water and observed that in the presence of DMSO in water the reactants were fully soluble and the conversion increased significantly. In this context, the use of 5 or 10% DMSO in water did not lead to increased yields significantly due to the limited solubility of the reactants. On the other hand, the utilization of 15% DMSO in water resulted in the total solubility of the reactants. However, further increasing the amount of DMSO did not have any additional impact on the yield of the reaction. Therefore, the best conversion was obtained in the case of 15% DMSO in H2O (v/v) (Scheme 3).
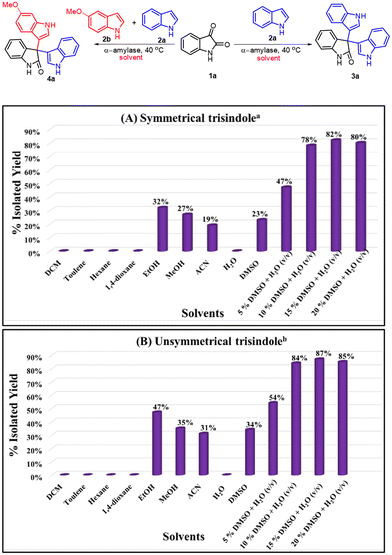 |
| | Scheme 3 Screening of different solvents for the synthesis of trisindoles. aReaction conditions for 3a: indole (2 equiv., 0.68 mmol), isatin (1 equiv., 0.34 mmol), α-amylase from Aspergillus oryzae (3 mg ml−1), 2.5 ml of solvent at 40 °C for 12 hours. bReaction conditions for 4a: indole (1.0 equiv.), 5-methoxyindole (1.0 equiv.), isatin (1.0 equiv.), α-amylase from Aspergillus oryzae (3 mg ml−1), 2.5 ml of solvent at 40 °C for 12 hours. | |
The effect of reaction temperatures on the conversion
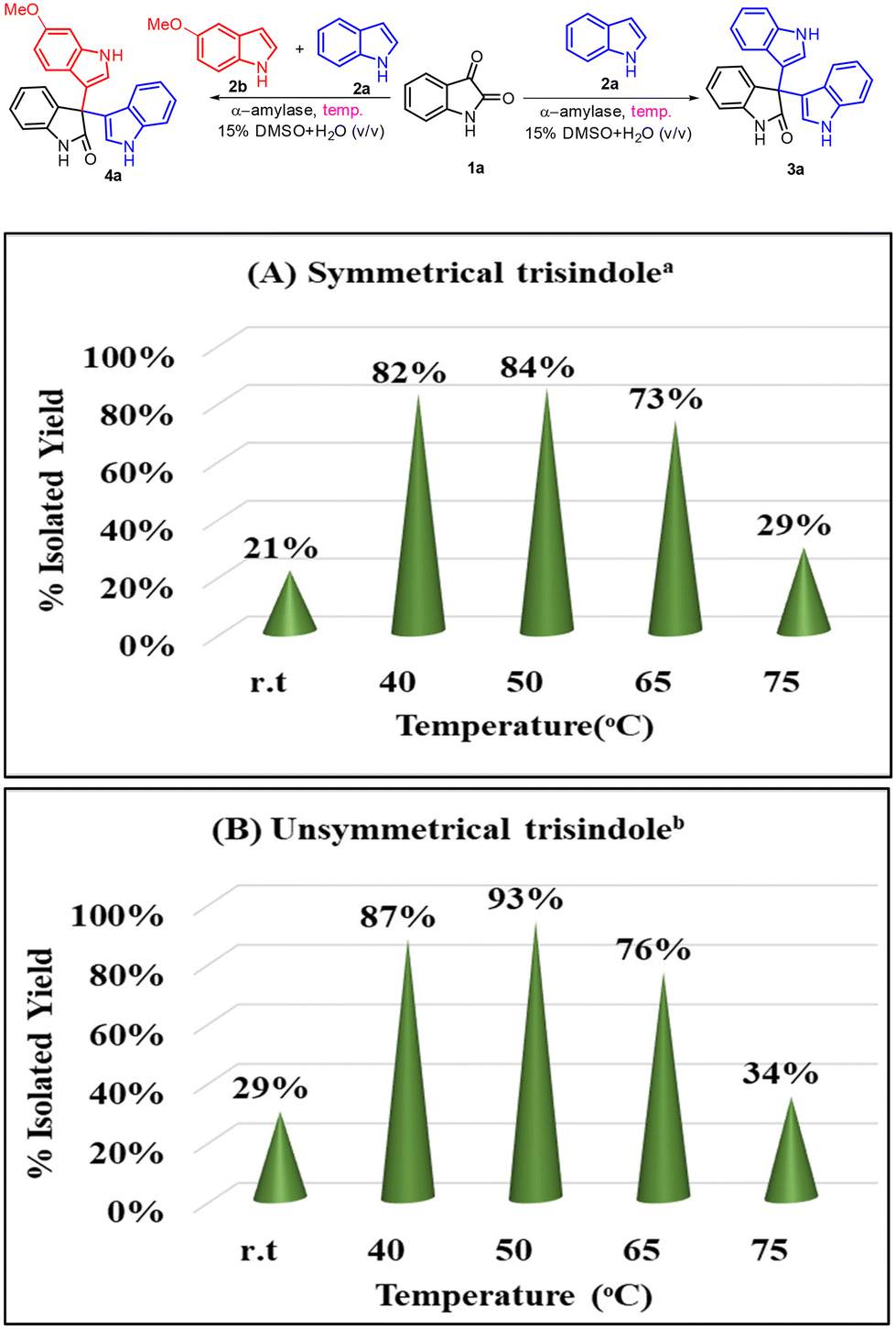
Afterwards, we examined the effect of temperature on the outcome of this transformation (Scheme 4A and B). In this context, we observed that the reaction works better at elevated temperatures (40–50 °C); however, when we increased the temperature higher than 65 °C, the yield of the reaction decreased significantly and this might be due to the denaturation of the enzyme at this temperature. Also, we checked the frequency of this transformation and found that the reaction was getting completed in 10–12 h.
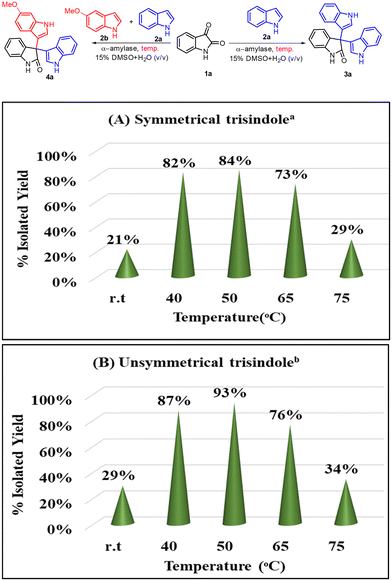 |
| | Scheme 4 Screening of the reaction temperature for the α-amylase catalyzed reaction. aReaction conditions for 3a: indole (2 equiv., 0.68 mmol), isatin (1 equiv., 0.34 mmol), α-amylase from Aspergillus oryzae (3 mg ml−1), 2.5 ml of 15% DMSO in water (v/v) as a solvent, reaction time = 12 h. bReaction conditions for 4a: indole (1.0 equiv.), 5-methoxyindole (1.0 equiv.), isatin (1.0 equiv.), α-amylase from Aspergillus oryzae (3 mg ml−1), 2.5 ml of 15% DMSO in water (v/v) as a solvent, reaction time = 12 h. | |
Substrate scope
With the optimized reaction conditions in hand, we examined the substrate scope to prove the robustness and generality of the enzymatic protocol (Tables 1 and 2). First, we screened the effect of different substitutions on isatin and found that there was no effect of electron-donating groups like 5-OMe and 5-CH3 on the outcome of the reaction and the products 3b and 3c were obtained in 82% and 87% yields respectively which were close to the yield of product 3a (entries 1–3, Table 1). Next, we screened the effect of halides such as 5-Cl and 5-Br on isatin and observed a slightly inferior yield; however, in the presence of an electron-withdrawing group i.e., 5-NO2, the isolated yield dropped significantly (entries 4–6, Table 1). Also, we installed –OCH3, –CH3, and –NO2 groups at the C-5 position of indole and obtained the desired products 3g and 3h in good yields; however, there was a decrement in the yield when NO2 substitution was employed (entries 7–9, Table 1). Moreover, different substitutions like –OMe, –Me, and –Br at the C-5 position of indoles were tested along with substituted isatin and interestingly the corresponding products were obtained in 83–86% isolated yields (entries 10–12, Table 1). There was no reaction in the case of nitro substitution at the C-5 positions of both isatin and indole (entry 13, Table 1). Finally, N1-substituted indoles were employed in the reaction and the symmetrical 3,3′,3′′-trisindole products 3n and 3o were obtained in 86% and 77% isolated yields respectively (entries 14 and 15, Table 1). These results demonstrated the robustness of the protocol since no significant effect of the substitutions either on isatin or indole in the synthesis of symmetrical 3,3′,3′′-trisindole was observed.
Table 1 Substrate scope for the enzymatic synthesis of symmetrical trisindolesa
|
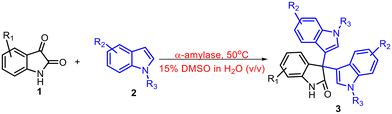
|
| Entry |
R1 |
R2 |
R3 |
Product, yieldb |
|
Reaction conditions: indole (2.0 equiv.), isatin (1.0 equiv.), α-amylase from Aspergillus oryzae (3 mg ml−1), 5 ml of 15% DMSO in water (v/v) as a solvent at 50 °C, reaction time = 12 h.
Isolated yields.
nr = no reaction.
|
| 1 |
H |
H |
H |
3a, 87% |
| 2 |
5-OMe |
H |
H |
3b, 82% |
| 3 |
5-Me |
H |
H |
3c, 87% |
| 4 |
5-Br |
H |
H |
3d, 79% |
| 5 |
5-Cl |
H |
H |
3e, 81% |
| 6 |
5-NO2 |
H |
H |
3f, 63% |
| 7 |
H |
5-OMe |
H |
3g, 79% |
| 8 |
H |
5-Me |
H |
3h, 74% |
| 9 |
H |
5-NO2 |
H |
3i, 43% |
| 10 |
5-OMe |
5-OMe |
H |
3j, 86% |
| 11 |
5-Me |
5-OMe |
H |
3k, 84% |
| 12 |
5-Br |
5-Br |
H |
3l, 83% |
| 13 |
5-NO2 |
5-NO2 |
H |
3m,c nr |
| 14 |
H |
H |
CH3 |
3n, 86% |
| 15 |
H |
5-OMe |
CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
3o, 77% |
Table 2 Substrate scope for the enzymatic synthesis of unsymmetrical trisindolesa
|
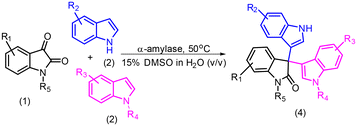
|
| Entry |
R1 |
R2 |
R3 |
R4 |
R5 |
Product, yieldb |
|
Reaction conditions: indole (1.0 equiv.), isatin (1.0 equiv., 0.68 mmol), α-amylase from Aspergillus oryzae (3 mg ml−1), 5 ml of 15% DMSO in water (v/v) as a solvent at 50 °C, reaction time = 12 h.
Isolated yields.
nr = no reaction.
|
| 1 |
H |
H |
5-OMe |
H |
H |
4a, 96% |
| 2 |
H |
H |
5-CH3 |
H |
H |
4b, 81% |
| 3 |
H |
H |
5-Br |
H |
H |
4c, 75% |
| 4 |
H |
H |
5-Cl |
H |
H |
4d, 79% |
| 5 |
H |
H |
5-CN |
H |
H |
4e, 89% |
| 6 |
H |
H |
5-NO2 |
H |
H |
4f, 79% |
| 7 |
H |
5-NO2 |
5-OMe |
H |
H |
4j, 73% |
| 8 |
H |
5-NC |
5-OMe |
H |
H |
4h, 79% |
| 9 |
5-OMe |
H |
5-OMe |
H |
H |
4i, 97% |
| 10 |
5-CH3 |
H |
5-OMe |
H |
H |
4j, 93% |
| 11 |
5-Cl |
H |
5-OMe |
H |
H |
4k, 84% |
| 12 |
5-NO2 |
H |
5-OMe |
H |
H |
4l, 82% |
| 13 |
5-Br |
H |
5-OMe |
H |
H |
4m, 92% |
| 14 |
5-CH3 |
5-Cl |
5-Br |
H |
H |
4n, 83% |
| 15 |
5-OMe |
5-NC |
5-Cl |
H |
H |
4o, 71% |
| 16 |
H |
4-Br |
5-Cl |
H |
H |
4p, 73% |
| 17 |
7-CF3 |
5-OMe |
H |
H |
H |
4q, 83% |
| 18 |
6-OMe |
5-OMe |
H |
H |
H |
4r, 79% |
| 19 |
H |
H |
H |
CH3 |
CH3 |
4s, 82% |
In the next phase of our endeavour, we explored the scope of substrates to synthesize unsymmetrical 3,3′,3′′-trisindoles (4a–s). As depicted in Table 2, there was no significant effect of substitution either on isatin or indole and the reaction provided the corresponding products in very good yields i.e., 71–97%. However, the yield decreased slightly in the case of electron-withdrawing substitutions on indole (entries 7, 8 and 15, Table 2). Also, the substitutions at different positions of isatin or indole like C-4, C-5, C-6, or C-7 were tolerated well (entries 16–18, Table 2). Gratifyingly, we found that the reaction was very selective towards the synthesis of unsymmetrical 3,3′,3′′-trisindoles, since no formation of symmetrical 3,3′,3′′-trisindoles was observed.
Control experiments
Furthermore, we set up a number of control experiments to get more insight into the role of the active site of enzymes in this transformation (Table 3). First, starch was introduced in equimolar quantities with isatin in the reaction, as the hydrolysis of starch is a known enzymatic reaction facilitated by α-amylase. Consequently, α-amylase catalyses the hydrolysis of starch in a competitive manner, leading to a higher frequency of the natural starch reaction compared to non-natural reactions. As a result, the desired trisindole product (3a) was obtained with an isolated yield of only 20% (entry 2, Table 3). Subsequently, we set up the reaction in the presence of urea which is an α-amylase inhibitor; as a result, the trisindole product (3a) did not form (entry 3, Table 3). In addition, we used BSA or the denatured α-amylase enzyme as a catalyst and in both cases, only the bisindole product was obtained. All these control experiments suggested that the residues present in the active site of amylase play an important role in catalyzing trisindole synthesis. To get more insight into the residues of active sites, we further conducted computational studies to confirm the role played by the active sites of amylase.
Table 3 Control experiments to confirm the role of the α-amylase enzymea
|
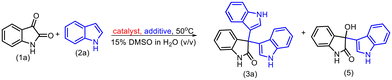
|
| Entry |
Catalyst |
Additive |
% yield of trisindoles (3a) |
% yield of bisindoles (5) |
|
Reaction conditions: indole (2.0 equiv., 1.35 mmol), isatin (1 equiv., 0.68 mmol), catalyst (3 mg ml−1), 5 ml of 15% DMSO in water (v/v) as a solvent at 50 °C, reaction time = 12 h.
Starch (1.0 equiv., 0.001 mmol).
Urea (1.0 equiv., 0.75 mmol).
|
| 1 |
α-Amylase from Aspergillus oryzae |
— |
96% |
— |
| 2 |
α-Amylase from Aspergillus oryzae |
Starchb |
20% |
— |
| 3 |
α-Amylase from Aspergillus oryzae |
Ureac |
— |
23% |
| 4 |
Denatured α-amylase |
— |
— |
19% |
| 5 |
BSA |
— |
— |
27% |
Mechanistic investigation
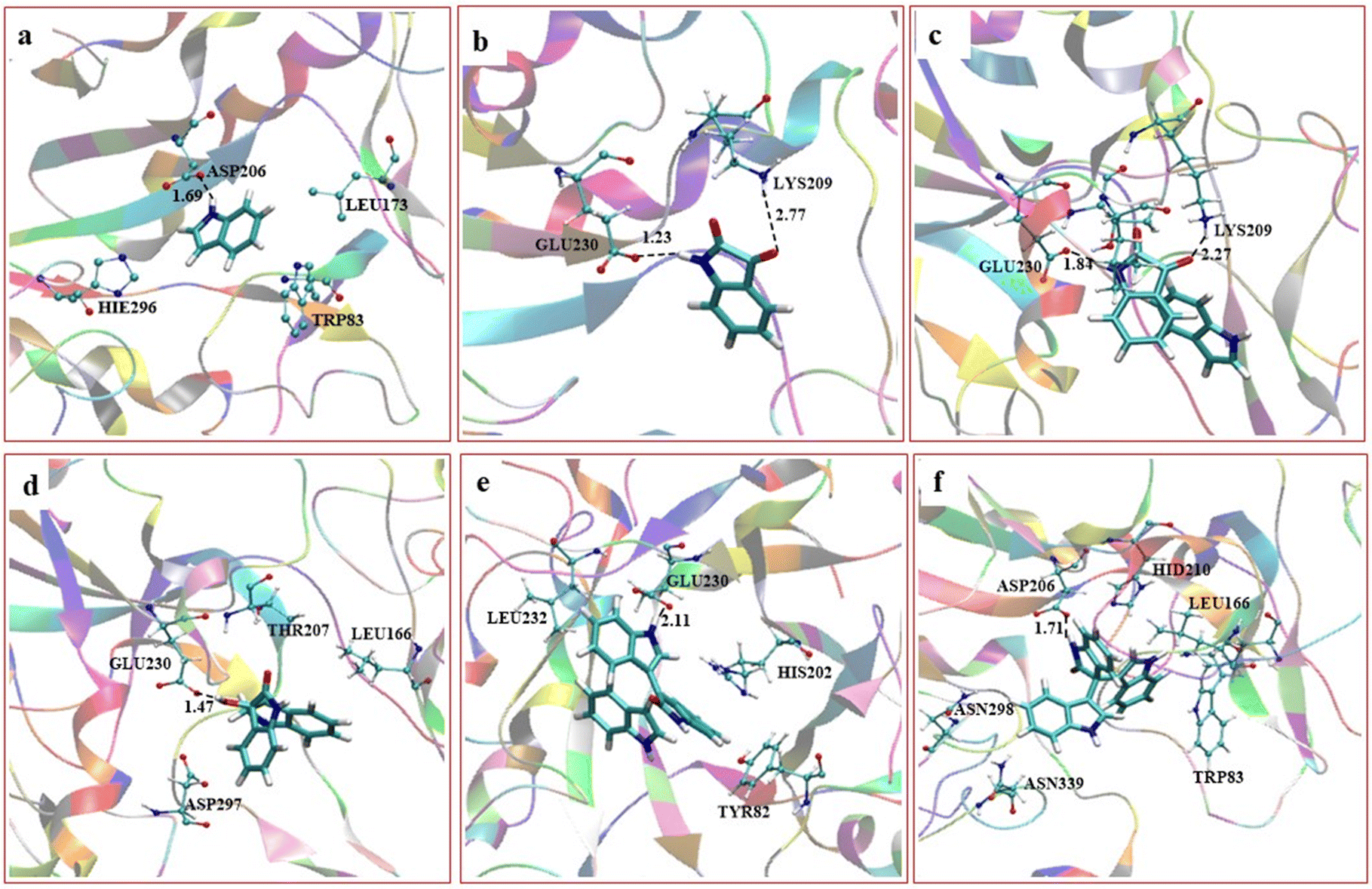
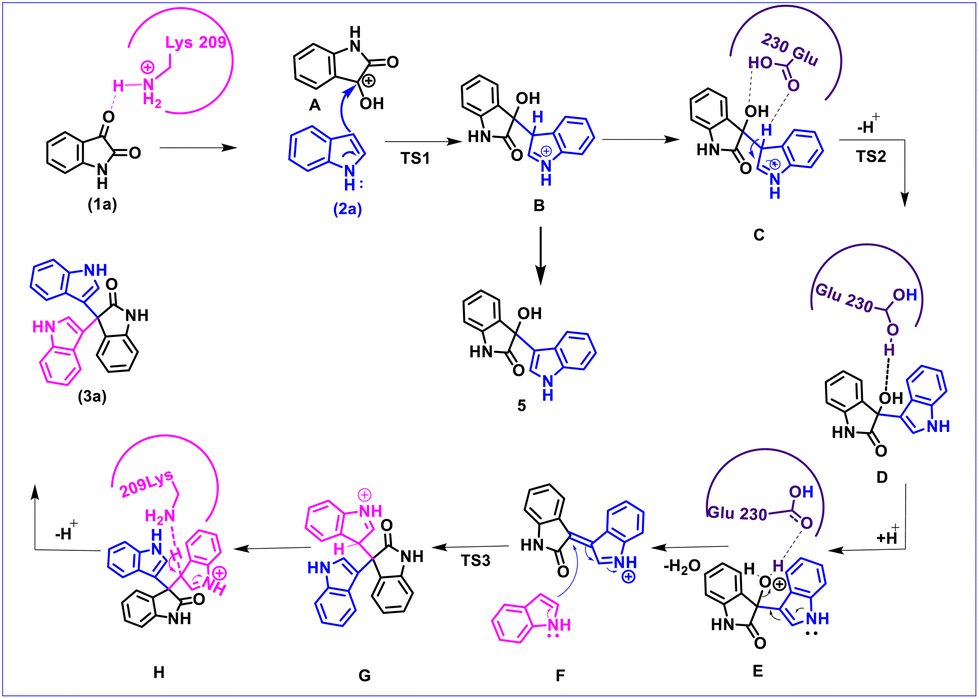
We further performed a careful molecular dynamics (MD) simulation and consequent density functional theory (DFT) investigation to establish the mechanism of trisindole formation by the α-amylase enzyme-catalyzed reaction of isatin and indole (the details of computational protocols have been presented in the ESI†). In brief, we predicted the active site, docked the substrates there, and then conducted extensive MD simulation. This study was extended beyond traditional MD simulations to find out the stability and probable interactions of the substrates as well as the intermediates and products with the amino acid residues available in the active site of the enzyme during the progress of the reaction. The most feasible MD snapshots consisting of orientation and interactions of substrates, intermediates, and products in the active site of the α-amylase enzyme have been presented in Fig. 2.
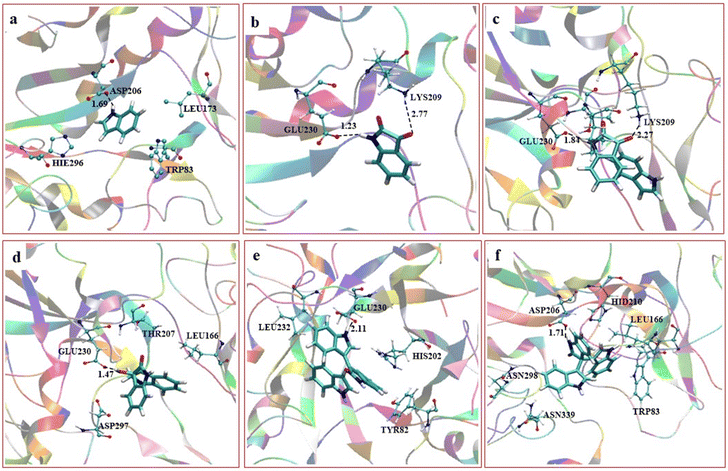 |
| | Fig. 2 Orientation and interactions of substrates, intermediates and products in the active site of the α-amylase enzyme: (a) indole interacting with Asp206, (b) isatin interacting with Lys209 and Glu230, (c) both isatin and indole interacting with Lys209, (d) bisindole intermediate B interacting with Glu230, (e) dehydrated bisindole and the second indole moiety and (f) the final trisindole product. | |
Based on the MD results, we observed that the carbonyl group of isatin forms a strong hydrogen bond with Lys209 (CO⋯H3N distance 2.77 Å) as shown in Fig. 2b, which makes the C-3 carbon of isatin more electrophilic. On the other hand, Fig. 2a shows that stabilization by a strong hydrogen bond with Asp206 (NH⋯OC distance 1.69 Å) makes indole a stronger nucleophile. It is also obvious that Lys209 and Asp206 play significant roles in activating the substrates which lead to the formation of the bisindole intermediate B (Scheme 5). The interaction pattern of the bisindole intermediate (B) with the active site of α-amylase is shown in Fig. 2d. In continuation, the interactions of intermediate F and the final trisindole product with the active sites are presented in Fig. 2e and f, respectively. From these insightful interaction patterns, we proposed the most plausible mechanism for this transformation (Scheme 5) and further verified the mechanism using a standard density functional theory computation and the corresponding potential energy surface has been presented in Fig. 3.
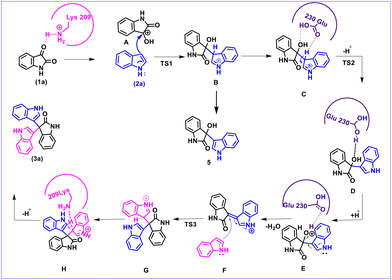 |
| | Scheme 5 Proposed mechanism of α-amylase catalyzed trisindole synthesis. | |
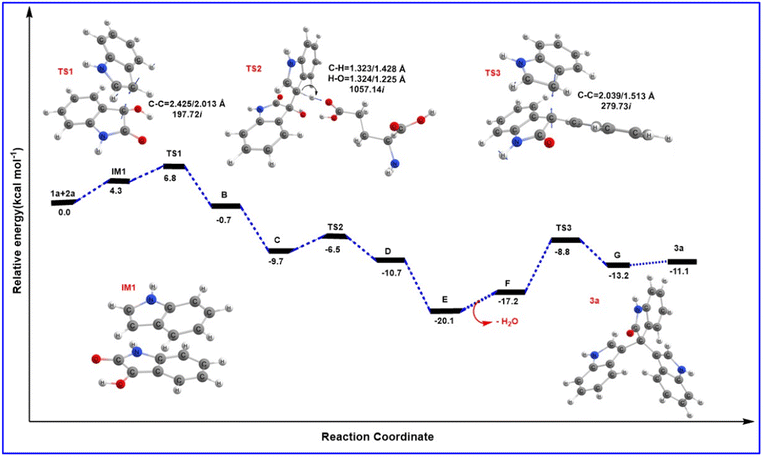 |
| | Fig. 3 The potential energy path represents ΔE + ZPE for the proposed trisindole formation calculated with the B3LYP/6-31+G** level of theory with respect to the reactant in kcal mol−1. | |
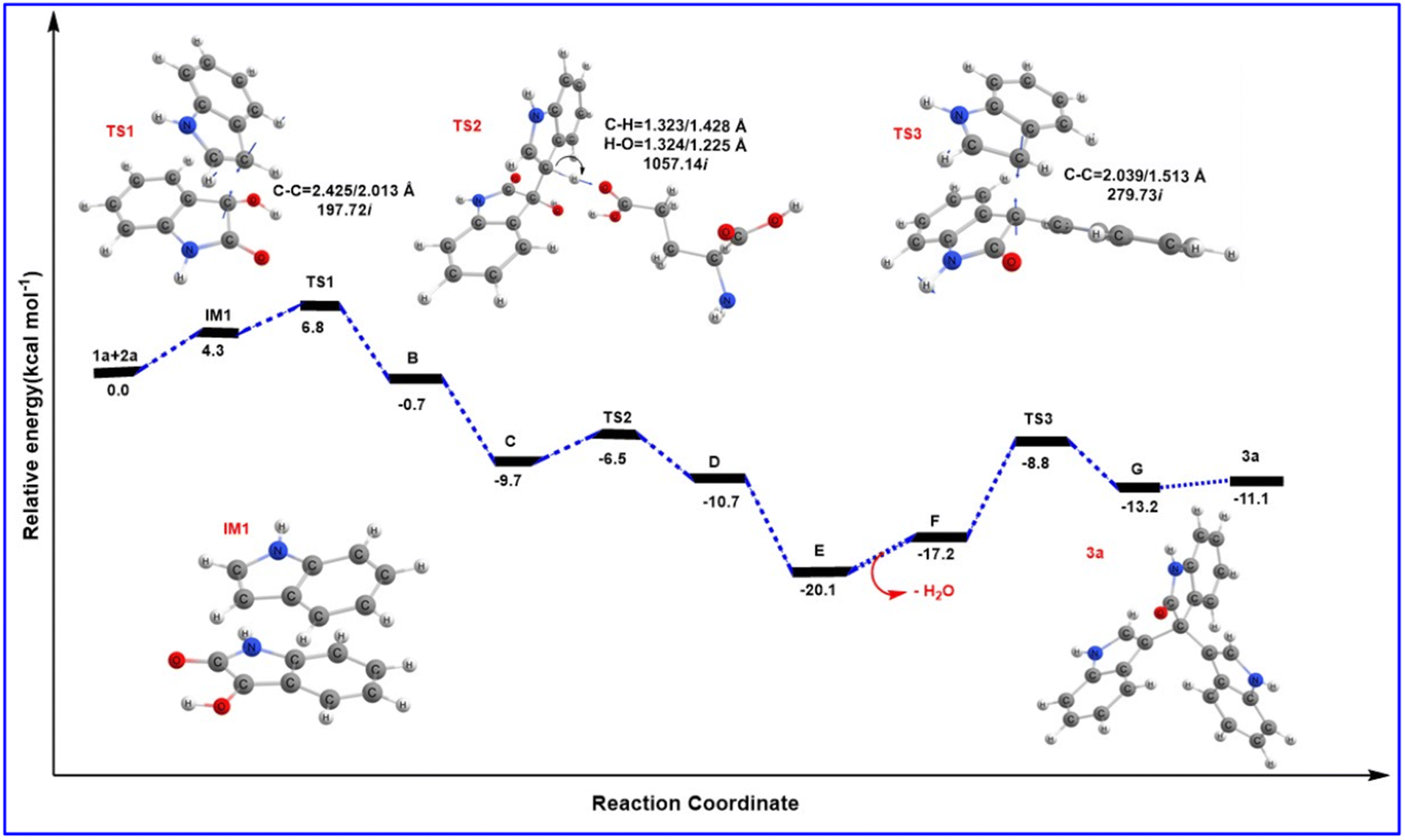
From the MD results, we started the initial mechanism where a proton from Lys transfers to isatin to form compound A. This transformation takes place without any transition state via a barrierless path which can also be correlated by the relaxed PES graph of O–H bond formation presented in Fig. S3† available in the ESI.† Next, compound A interacts with indole (2a) and forms complex IM1. IM1 further transforms into intermediate B through C–C bond formation via the transition state TS1, with a barrier of 6.8 kcal mol−1 with respect to the initial reactant. Furthermore, intermediate B transforms into intermediate C upon complexation with Glu230 which promotes proton transfer and the removal of a water molecule. C then leads to D through H-transfer from the C3 position of indole to Glu230 which then forms intermediate D. This pathway involves a transition state TS2, favoured by only 3.1 kcal mol−1 energy of activation with respect to C. The bisindole intermediate (D) then remains as a complex with Glu230 along with another indole (2a) molecule represented as intermediate E. In continuation, the release of H2O from E leads to the formation of intermediate F. However, we couldn't obtain a transition state for the O–H proton transfer by Glu230 during the formation of intermediate F which can also be correlated by a relaxed PES scan in favour of proton transfer from Glu230 presented in Fig. S4 available in the ESI.† Intermediates E and F are found to be quite stable (20.1 and 17.2 kcal mol−1, respectively) compared to the other species in the PES. Apparently, this looks like a sink and the reaction will not proceed further. But the unusual stability of both E and F may be due to the incapability of gas phase optimisation that ignores the protein environment and the appropriate solvent network. Intermediate F is then reoriented and forms a hydrogen bond with Glu230 (NH⋯OC distance 2.11 Å) (shown in Fig. 2e) to further increase the electrophilicity of the indole moiety, which allows the addition of a second indole molecule to produce the 2′-oxo-[3,3′:3′,3′′-terindolin]-1′′-ium intermediate mentioned as G in the PES. The formation of intermediate G proceeds through the transition state TS3, which possesses 8.4 kcal mol−1 higher energy than intermediate F. Finally, re-aromatization via proton abstraction from the C3 position of intermediate G by Lys-H(deprotonated)209 provides the final product trisindole (3a). It is worth noting that the Lys209 involved in the first step of the mechanism abstracts the proton from intermediate G and continues the natural enzymatic cycle.
Hammett study
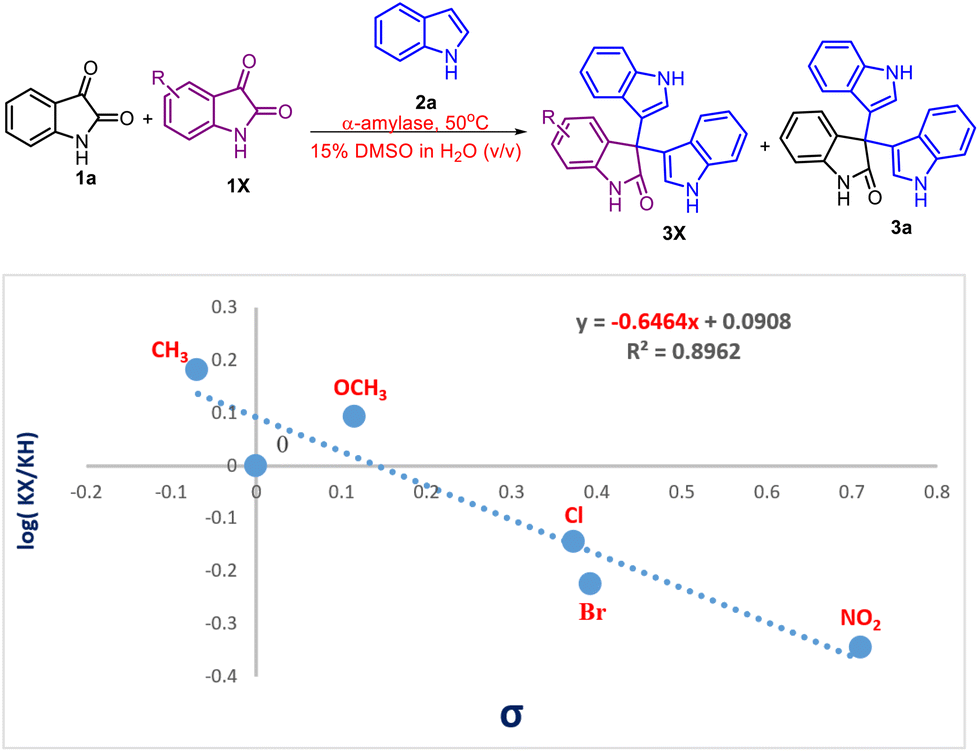
Furthermore, to demonstrate the electronic effect of the substituents on the rate of the reaction, a Hammett study was performed (Scheme 6). A good correlation (R2 = 0.89) between the log[kX/kH] and σp values of the respective substituents was obtained. The resulting negative ρ value of −0.64 indicates that there is a buildup of positive charge during the rate-determining step.
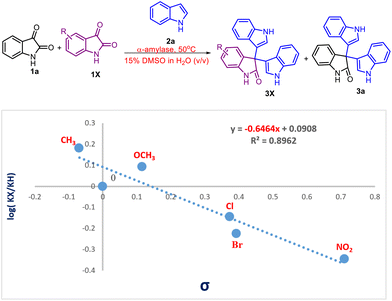 |
| | Scheme 6 Hammett plot for the reaction of indole with electronically varied isatin (X = Br, Cl, OMe, and NO2 groups). | |
Application in the gram scale synthesis of bioactive compounds
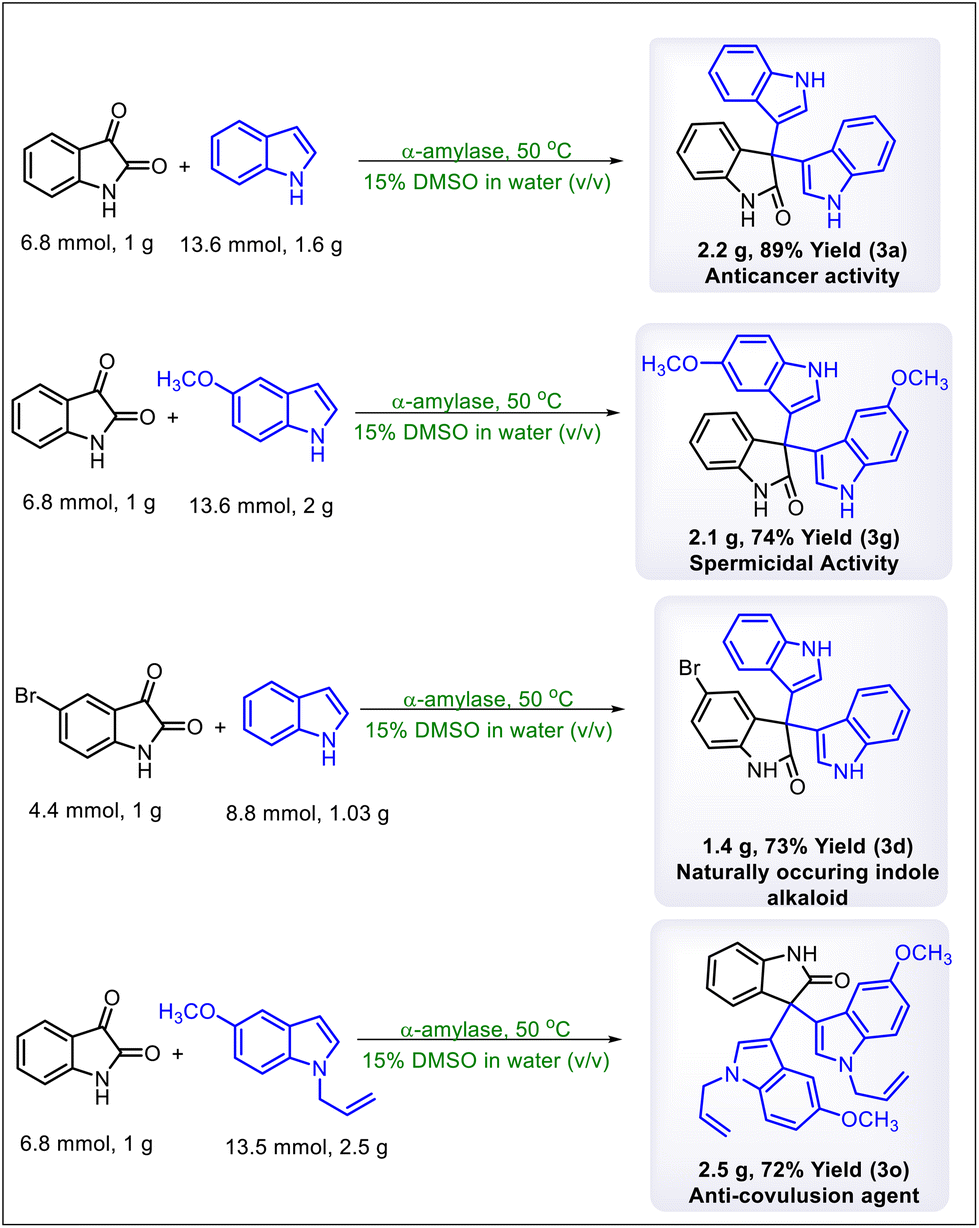
In the last phase of our study, we synthesized four biologically important 3,3′,3′′-trisindoles on a gram scale to prove the robustness and scalability of this transformation. First, we synthesized product 3a on a 2.2 g scale which has been reported previously as an anticancer agent and synthesized by using various chemical catalysts.16 Furthermore, the anti-spermicidal 3,3′,3′′-trisindole (3 g) was synthesized on a 2.1 g scale by using isatin (1 g) and 5-methoxyindole (2 g).17 A naturally occurring indole alkaloid (3d) was synthesized on a 1.4 g scale using isatin and 5-bromo indole.18 Finally, the anti-convulsion agent 3o was produced on a 2.5 g scale by using isatin and N-substituted indole (Scheme 7).
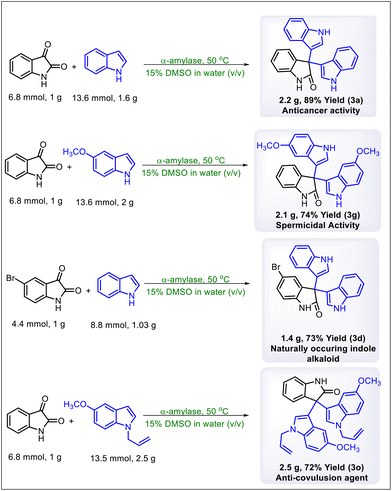 |
| | Scheme 7 Gram scale synthesis of biologically active 3,3′,3′′-trisindoles. | |
Conclusion
In summary, we have developed a highly sustainable and efficient approach to synthesize symmetrical as well as unsymmetrical 3,3′,3′′-trisindoles in a selective manner by using the α-amylase enzyme as a catalyst in water. To check the generality and sturdiness of this transformation, differently substituted isatin and indoles were screened and as a result the corresponding products were obtained in good to excellent yields. Also, a number of control experiments were performed, and they confirmed the usefulness of the α-amylase enzyme in this transformation. Furthermore, the synthetic utility was revealed by synthesizing four different biologically important trisindole derivatives on a gram scale. Finally, a probable mechanism for the enzymatic reaction was proposed and investigated using molecular dynamics and density functional theory investigations, which suggested that Glu230, Lys209, and Asp206 in the active site of α-amylase play an important role in this catalysis.
Experimental
General information
All solvents, chemicals and enzymes were purchased from commercial suppliers and used without any extra purification. All the reactions were conducted using oven-dried glassware with magnetic stirring. The silica gel chromatography purification procedures employed silica gel 60–120 meshes. The 1H and 13C NMR spectra were collected on a JEOL or Bruker NMR (400 MHz) using DMSO-d6 as a solvent with TMS as an internal reference. The coupling constant (J) is presented in hertz (Hz), and the chemical shift (δ) is reported in parts per million (ppm). Multiplicities are abbreviated as s: singlet, d: doublet, dd: doublet of doublet, t: triplet, br s: broad singlet, and m: multiplet.
General procedure for the preparation of 3,3′,3′′-trisindoles 3a–o and 4a–s
In a Teflon tube having a stirrer bar were added 4.25 ml of deionized water and 3 mg ml−1 of the enzyme (α-amylase from Aspergillus oryzae). Furthermore, the reactants i.e., isatin (0.68 mmol, 1.0 equiv.) and indole (1.35 mmol, 2.0 equiv.) in the case of symmetrical 3,3′,3′′-trisindoles or indole (0.67 mmol, 1.0 equiv.) and substituted indole (0.68 mmol, 1.0 equiv.) in the case of unsymmetrical 3,3′,3′′-trisindoles were dissolved in 0.75 ml of DMSO and added to the reaction tube. The resulting reaction mixture was slowly stirred at 50 °C for 12 hours. After completion of the reaction, the mixture was extracted with ethyl acetate (3 × 10 ml). Next, volatiles were evaporated using high vacuum and the resulting crude mixture was purified through column chromatography by using silica (mesh size 60–120) as a stationary phase and the mixture of ethyl acetate and hexane as a mobile phase to obtain 3,3′,3′′-trisindoles (3a–o) in 43–87% isolated yields.
NMR characterization data
[3,3′:3′,3′′-Terindolin]-2′-one (3a)19.
1H NMR (400 MHz, DMSO): δ 10.94 (s, 2H), 10.59 (s, 1H), 7.33 (d, J = 8 Hz, 2H), 7.20 (d, J = 4 Hz, 4H), 7.00–6.94 (m, 3H), 6.90–6.87 (m, 1H), 6.82 (d, J = 2.4 Hz, 2H), 6.78–6.74 (m, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 53.09, 110.10, 112.13, 114.81, 118.75, 121.30, 121.48, 121.99, 124.81, 125.43, 126.22, 128.37, 135.12, 137.44, 141.84, 179.29 ppm.
5′-Methoxy-[3,3′:3′,3′′-terindolin]-2′-one (3b)19.
1H NMR (400 MHz, DMSO): δ10.94 (s, 2H), 10.43 (s, 1H), 7.33 (d, J = 8 Hz, 2H), 7.22 (d, J = 8 Hz, 2H), 7.00–6.96 (m, 2H), 6.87–6.85 (m, 2H), 6.79–6.75 (m, 5H), 3.57 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 53.55, 55.81, 110.39, 112.14, 112.61, 114.76, 118.76, 121.30, 121.47, 124.88, 126.20, 135.20, 136.48, 137.44, 155.14, 179.15 ppm.
5′-Methyl-[3,3′:3′,3′′-terindolin]-2′-one (3c)20.
1
H NMR (400 MHz, DMSO): δ 10.93 (s, 2H), 10.49 (s, 1H), 7.34 (d, J = 8 Hz, 2H), 7.24 (d, J = 8 Hz, 2H), 7.01–6.99 (m, 2H), 6.97 (s, 1H), 6.87 (m, 1H), 6.83 (d, J = 2.4 Hz, 2H), 6.80–6.76 (m, 3H), 2.96 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 21.35, 53.16, 109.85, 112.13, 114.93, 118.75, 121.40, 121.46, 124.90, 126.00, 126.25, 128.66, 130.72, 135.17, 137.46, 139.42, 179.34 ppm.
5′-Bromo-[3,3′:3′,3′′-terindolin]-2′-one (3d)19.
1H NMR (400 MHz, DMSO): δ 11.02 (s, 2H), 10.77 (s, 1H), 7.40 (d, J = 8 Hz, 1H), 7.35 (d, J = 8 Hz, 2H), 7.27 (s, 1H), 7.18 (d, J = 8.4 Hz, 2H), 7.02–6.98 (m, 2H), 6.94 (d, J = 8 Hz, 1H), 6.86 (d, J = 4 Hz, 2H), 6.81–6.77 (m, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 75.44, 112.16, 112.29, 113.87, 115.15, 119.19, 120.52, 121.72, 124.11, 125.17, 127.85, 132.27, 136.37, 137.30, 141.44, 178.44 ppm.
5′-Chloro-[3,3′:3′,3′′-terindolin]-2′-one (3e)19.
1H NMR (400 MHz, DMSO): δ 10.99 (s, 2H), 10.73 (s, 1H), 7.34 (d, J = 8 Hz, 2H), 7.26 (d, J = 4 Hz, 1H), 7.16 (s, 1H), 7.14–7.13 (m, 2H), 7.01–6.96 (m, 3H), 6.84 (d, J = 2.8 Hz, 2H), 6.80–6.76 (m, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 53.30, 112.21, 112.28, 113.67, 118.95, 121.02, 121.60, 124.95, 125.99, 127.87, 131.23, 137.45, 141.19, 178.78 ppm.
5′-Nitro-[3,3′:3′,3′′-terindolin]-2′-one (3f)19.
1H NMR (400 MHz, DMSO): δ 11.33 (s, 1H), 11.08 (s, 2H), 8.20 (d, J = 4 Hz, 1H), 7.95 (s, 1H), 7.36 (d, J = 8 Hz, 2H), 7.20 (s, 1H), 7.18 (t, J = 8 Hz, 2H), 7.01 (t, J = 4 Hz, 2H), 6.94 (d, J = 4 Hz, 2H), 6.80 (t, J = 8 Hz, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 53.05, 110.47, 112.40, 113.31, 119.13, 120.61, 120.85, 121.76, 125.14, 125.89, 126.02, 135.78, 137.54, 142.73, 148.31, 179.46 ppm.
5,5′′-Dimethoxy-[3,3′:3′,3′′-terindolin]-2′-one (3g)20.
1H NMR (400 MHz, DMSO): δ 10.78 (s, 2H), 10.58 (s, 1H), 7.23–7.20 (m, 3H), 7.18 (d, J = 8 Hz, 1H), 6.95 (dd, J = 12.8 Hz, 2H), 6.82 (d, J = 4 Hz, 2H), 6.67–6.64 (m, 4H), 3.48 (s, 6H) ppm. 13C NMR (100 MHz, DMSO): δ 53.02, 55.59, 103.78, 109.96, 110.92, 112.60, 114.10, 122.03, 125.49, 125.68, 126.61, 128.37, 132.68, 135.05, 141.94, 152.91, 179.29 ppm.
5,5′′-Dimethyl-[3,3′:3′,3′′-terindolin]-2′-one (3h)20.
1H NMR (400 MHz, DMSO): δ 10.20 (s, 2H), 10.17 (s, 1H), 7.70 (s, 1H), 7.24 (s, 1H), 7.20 (s, 2H), 7.13 (d, J = 8 Hz, 2H), 7.01 (d, J = 8 Hz, 1H), 6.91 (t, J = 8 Hz, 1H), 6.88–6.84 (m, 4H), 2.27 (s, 6H) ppm. 13C NMR (100 MHz, DMSO): δ 21.44, 52.77, 109.47, 110.98, 113.83, 120.46, 121.27, 122.55, 124.60, 125.88, 126.77, 127.36, 134.65, 135.36, 141.44, 179.45 ppm.
5,5′′-Dinitro-[3,3′:3′,3′′-terindolin]-2′-one (3i)20.
1H NMR (400 MHz, DMSO): δ 11.71 (s, 2H), 10.92 (s, 1H), 8.19 (s, 2H), 7.93 (d, J = 8 Hz, 2H), 7.54 (s, J = 8 Hz, 2H), 7.27 (t, J = 8 Hz, 2H), 7.18 (t, J = 8 Hz, 2H), 7.05 (d, J = 8 Hz, 1H), 6.98 (t, J = 8 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO): δ 52.60, 110.71, 113.03, 117.23, 118.02, 122.71, 125.12, 125.47, 128.84, 129.21, 133.45, 140.76, 141.72, 178.55 ppm.
5,5′,5′′-Trimethoxy-[3,3′:3′,3′′-terindolin]-2′-one (3j)21.
1H NMR (400 MHz, DMSO): δ 10.77 (s, 2H), 10.41 (s, 1H), 7.23 (d, J = 4 Hz, 1H), 7.21 (s, 1H), 6.89 (d, J = 8 Hz, 1H), 6.83 (d, J = 2.8 Hz, 2H), 6.79 (dd, J = 4 Hz, 1H), 6.73 (d, J = 4 Hz, 1H), 6.68 (d, J = 4 Hz, 1H), 6.66 (d, J = 4 Hz, 3H), 3.58 (s, 3H), 3.49 (s, 6H) ppm. 13C NMR (100 MHz, DMSO): δ 53.48, 55.64, 55.84, 103.81, 110.27, 110.93, 112.52, 112.61, 112.71, 114.07, 125.75, 126.59, 132.69, 135.34, 136.35, 152.92, 155.16, 179.14 ppm.
5,5′′-Dimethoxy-5′-methyl-[3,3′:3′,3′′-terindolin]-2′-one (3k)21.
1H NMR (400 MHz, DMSO): δ 10.78 (s, 2H), 10.50 (s, 1H), 7.25 (d, J = 8 Hz, 2H), 7.00 (t, J = 8 Hz, 2H), 6.88 (d, J = 8 Hz, 1H), 6.84 (s, 2H), 6.69 (s, 3H), 6.67 (s, 1H), 3.50 (s, 6H), 2.16(s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 21.35, 53.11, 55.64, 103.89, 109.74, 110.95, 112.60, 114.24, 125.81, 126.08, 126.65, 128.66, 130.74, 132.71, 135.06, 139.56, 152.94, 179.37 ppm.
5,5′,5′′-Tribromo-[3,3′:3′,3′′-terindolin]-2′-one (3l)21.
1H NMR (400 MHz, DMSO): δ 11.26 (s, 2H), 10.88 (s, 1H), 7.43 (dd, J = 4 Hz, 1H), 7.35 (s, 1H), 7.33 (s, 1H), 7.30 (s, 2H), 7.21 (s, 1H), 7.15 (s, 1H), 7.12 (s, 1H), 6.97 (d, J = 8 Hz, 1H), 6.91 (d, J = 4 Hz, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 52.85, 111.71, 112.48, 113.50, 114.02, 114.52, 122.94, 124.30, 126.73, 127.654, 127.98, 131.69, 136.22, 136.40, 141.08, 178.53 ppm.
1,1′′-Dimethyl-[3,3′:3′,3′′-terindolin]-2′-one (3n)22.
1H NMR (400 MHz, DMSO): δ 10.58 (s, 1H), 7.34 (d, J = 8 Hz, 2H), 7.21–7.17 (m, 4H), 7.04 (t, J = 8 Hz, 2H), 6.93 (d, J = 8 Hz, 1H), 6.87 (t, J = 4 Hz, 1H), 6.84 (s, 2H), 6.79 (t, J = 8 Hz, 2H), 3.66 (s, 6H) ppm. 13C NMR (100 MHz, DMSO): δ 32.86, 52.90, 110.37, 113.94, 118.92, 121.45, 122.09, 126.53, 128.42, 128.97, 135.01, 137.83, 141.76, 179.07 ppm.
1,1′′-Diallyl-5,5′′-dimethoxy-[3,3′:3′,3′′-terindolin]-2′-one (3o)23.
1H NMR (400 MHz, DMSO): δ 10.63 (s, 1H), 7.22 (q, J = 8 Hz, 8 Hz, 4H), 6.97–6.90 (m, 2H), 6.85 (s, 2H), 6.70 (s, 1H), 6.67 (s, 3H), 5.93–5.84 (m, 2H), 5.06 (d, J = 8 Hz, 2H), 4.94 (d, J = 16 Hz, 2H), 4.69 (s, 4H), 3.47 (s, 6H) ppm. 13C NMR (100 MHz, DMSO): δ 48.49, 52.86, 55.56, 103.87, 110.08, 111.31, 111.36, 113.58, 116.88, 122.22, 125.40, 127.11, 128.48, 128.66, 132.58, 134.83, 135.15, 141.81, 153.20, 178.97 ppm.
5-Methoxy-[3,3′:3′,3′′-terindolin]-2′-one (4a)24.
1H NMR (400 MHz, DMSO): δ 10.93 (s, 1H), 10.77 (s, 1H), 10.57 (s, 1H), 7.33 (d, J = 8 Hz, 1H), 7.23 (s, 2H), 7.21–7.18 (m, 2H), 6.98–6.95 (m, 2H), 6.90–6.86 (m, 1H), 6.82 (s, 1H), 6.79–6.74 (m, 2H), 6.68–6.65 (m, 2H), 3.48 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 53.07, 55.61, 103.82, 110.03, 110.94, 112.12, 112.60, 114.13, 114.29, 114.66, 114.82, 118.75, 121.45, 121.99, 124.81, 125.47, 126.23, 126.63, 128.36, 132.68, 135.13, 137.45, 141.91, 152.92, 179.29 ppm.
5-Methyl-[3,3′:3′,3′′-terindolin]-2′-one (4b)27.
1H NMR (400 MHz, DMSO): δ 10.94 (d, J = 8 Hz, 1H), 10.81 (d, J = 8 Hz, 1H), 10.58 (t, J = 4 Hz, 1H), 7.33 (d, J = 8 Hz, 1H), 7.22–7.16 (m, 4H), 7.05–7.01 (m, 1H), 6.96 (t, J = 8 Hz, 2H), 6.89 (t, J = 8 Hz, 1H), 6.83–6.81 (m, 2H), 6.78–6.74 (m, 2H), 2.17 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 22.00, 53.10, 110.04, 111.87, 112.13, 114.23, 114.84, 118.75, 120.90, 121.38, 121.46, 121.95, 123.10, 124.81, 125.05, 125.46, 126.22, 126.41, 126.86, 128.34, 135.14, 135.87, 137.44, 141.85, 179.34 ppm.
5-Bromo-[3,3′:3′,3′′-terindolin]-2′-one (4c)25.
1H NMR (400 MHz, DMSO): δ 11.21 (d, J = 8 Hz, 1H), 10.96 (d, J = 8 Hz, 1H), 10.66 (t, J = 28 Hz, 1H), 7.42–7.30 (m, 3H), 7.25–7.17 (m, 2H), 7.15–7.09 (m, 2H), 7.00–6.93 (m, 3H), 6.92–6.87 (m, 1H), 6.86–6.81 (m, 1H), 6.78–6.74 (q, J = 8 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO): δ 52.88, 110.24, 111.59, 112.13, 114.31, 114.81, 118.74, 121.30, 122.16, 123.18, 124.17, 124.81, 125.42, 126.22, 126.53, 127.78, 128.36, 134.11, 134.59, 135.13, 136.21, 137.41, 141.73, 178.98 ppm.
5-Chloro-[3,3′:3′,3′′-terindolin]-2′-one (4d)26.
1H NMR (400 MHz, DMSO): δ 10.20 (d, J = 8 Hz, 1H), 10.96 (d, J = 8 Hz, 1H), 10.65 (t, J = 28 Hz, 1H), 7.38 (d, J = 8 Hz, 1H), 7.33 (t, J = 8 Hz, 1H), 7.25–7.23 (m, 2H), 7.21–7.15 (m, 2H), 7.01–6.98 (m, 2H), 6.96–6.94 (m, 1H), 6.92–6.87 (m, 2H), 6.84 (d, J = 8 Hz, 1H), 6.76 (t, J = 8 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO): δ 53.08, 110.09, 112.12, 113.88, 114.63, 114.81, 118.74, 120.15, 120.14, 121.31, 121.44, 123.49, 124.80, 125.42, 126.22, 126.65, 127.09, 128.34, 134.62, 135.13, 135.99, 137.44, 141.93, 179.29 ppm.
2′-Oxo-[3,3′:3′,3′′-terindoline]-5-carbonitrile (4e)28.
1H NMR (400 MHz, DMSO): δ 10.95 (s, 2H), 10.59 (s, 1H), 7.33 (d, J = 8 Hz, 2H), 7.21 (s, 1H), 7.19 (s, 2H), 7.00–6.95 (q, J = 8 Hz, 3H), 6.91 (t, J = 8 Hz, 1H), 6.83 (d, J = 8 Hz, 2H), 6.76 (t, J = 4 Hz, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 53.07, 110.07, 112.11, 114.81, 118.72, 121.29, 121.43, 121.96, 124.79, 125.42, 126.21, 128.34, 135.12, 137.43, 141.84, 179.30 ppm.
5-Nitro-[3,3′:3′,3′′-terindolin]-2′-one (4f)24.
1H NMR (400 MHz, DMSO): δ 10.92 (s, 1H), 10.91 (s, 1H), 10.56 (s, 1H), 7.32 (d, J = 8 Hz, 2H), 7.19 (s, 1H), 7.17 (s, 1H), 6.99–6.93 (q, J = 8 Hz, 8 Hz, 4H), 6.90–6.87 (t, J = 8 Hz, 1H), 6.80 (d, J = 4 Hz, 2H), 6.77–6.73 (t, J = 8 Hz, 2H) ppm. 13C NMR (100 MHz, DMSO): δ 53.01, 110.08, 112.12, 114.81, 118.73, 121.29, 121.44, 121.97, 124.80, 125.43, 126.22, 128.35, 135.12, 137.44, 141.84, 179.27 ppm.
5-Methoxy-5′′-nitro-[3,3′:3′,3′′-terindolin]-2′-one (4g).
1H NMR (400 MHz, DMSO): δ 10.84 (s, 1H), 10.77 (s, 1H), 10.58 (s, 1H), 8.34 (s, 1H), 7.92 (d, J = 4 Hz, 1H), 7.54 (d, J = 8 Hz, 1H), 7.25–7.19 (m, 3H), 7.12 (s, 1H), 6.99–6.92 (m, 1H), 6.85 (t, J = 8 Hz, 1H), 6.68–6.66 (m, 2H), 6.56 (s, 1H), 3.49 (s, 3 H) ppm. 13C NMR (100 MHz, DMSO): δ 53.04, 55.62, 103.01, 103.84, 109.96, 110.94, 112.60, 114.14, 117.07, 117.52, 119.04, 122.02, 122.37, 125.70, 126.64, 128.36, 128.78, 132.71, 134.22, 135.07, 140.60, 141.96, 152.94, 179.31 ppm.
5′′-Methoxy-2′-oxo-[3,3′:3′,3′′-terindoline]-5-carbonitrile (4h).
1H NMR (400 MHz, DMSO): δ 10.77 (s, 2H), 10.58 (s, 1H), 7.70–7.52 (m, 1H), 7.37 (t, J = 8 Hz, 1H), 7.24–7.22 (m, 2H), 7.21–7.19 (m, 1H), 7.07 (d, J = 12 Hz, 1H), 7.01–6.89 (m, 2H), 6.84 (d, J = 4 Hz, 1H), 6.70–6.67 (m, 1H), 6.66–6.55 (m, 2H), 3.49 (s, 3 H) ppm. 13C NMR (100 MHz, DMSO): δ 53.05, 55.82, 100.85, 103.84, 109.97, 110.95, 112.60, 114.15, 115.84, 121.33, 122.03, 124.18, 125.51, 125.70, 126.36, 126.65, 127.24, 128.37, 132.71, 134.33, 135.08, 139.35, 141.97, 152.95, 179.32 ppm.
5,5′-Dimethoxy-[3,3′:3′,3′′-terindolin]-2′-one (4i).
1H NMR (400 MHz, DMSO): δ 10.96 (s, 1H), 10.81 (s, 1H), 10.45 (s, 1H), 7.37 (d, J = 8 Hz, 2H), 7.25 (s, 1H), 7.23 (s, 1H), 7.04 (t, J = 8 Hz, 2H), 6.92 (d, J = 4 Hz, 1H), 6.88 (s, 1H), 6.83 (d, J = 4 Hz, 1H), 6.80 (s, 2H), 6.71–6.69 (m, 1H), 3.61 (s, 3H), 3.52 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 53.53, 55.83, 103.86, 110.37, 110.94, 112.13, 112.57, 114.24, 114.60, 114.77, 118.75, 121.45, 124.88, 125.67, 126.20, 126.59, 132.68, 135.29, 135.28, 136.42, 136.47, 137.45, 152.92, 155.14, 179.14 ppm.
5-Methoxy-5′-methyl-[3,3′:3′,3′′-terindolin]-2′-one (4j).
1H NMR (400 MHz, DMSO): δ 10.91 (s, 1H), 10.76 (s, 1H), 10.46 (s, 1H), 7.33 (d, J = 8 Hz, 2H), 7.22 (d, J = 8 Hz, 2H), 6.98 (t, J = 8 Hz, 4H), 6.86–6.83 (m, 1H), 6.81 (s, 1H), 6.78–6.75 (m, 1H), 6.67 (s, 1H), 3.49 (s, 3H), 2.15 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 21.34, 53.13, 55.65, 103.89, 109.82, 110.95, 112.11, 112.58, 114.39, 114.77, 114.94, 118.72, 121.43, 124.88, 125.69, 126.03, 126.24, 126.62, 128.64, 130.69, 132.68, 135.18, 137.45, 139.42, 152.91, 179.31 ppm.
5′-Chloro-5-methoxy-[3,3′:3′,3′′-terindolin]-2′-one (4k).
1H NMR (400 MHz, DMSO): δ 11.02 (s, 1H), 10.88 (s, 1H), 10.76 (s, 1H), 7.38 (d, J = 8 Hz, 1H), 7.30–7.26 (m, 2H), 7.21 (d, J = 8 Hz, 1H), 7.18–7.16 (m, 1H), 7.03–7.02 (m, 1H), 7.01–6.99 (m, 1H), 6.93–6.88 (dd, J = 4, 8 Hz, 1H), 6.85 (t, J = 4 Hz, 1H), 6.83–6.80 (m, 1H), 6.73 (d, J = 8 Hz, 1H), 6.65 (s, 1H), 3.36 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 53.36, 55.65, 103.59, 111.07, 111.60, 112.27, 112.78, 113.46, 113.86, 114.01, 118.94, 121.03, 121.59, 124.95, 125.20, 125.96, 126.02, 126.40, 128.37, 132.69, 137.13, 137.46, 140.86, 153.04, 178.91 ppm.
5-Methoxy-5′-nitro-[3,3′:3′,3′′-terindolin]-2′-one (4l).
1H NMR (400 MHz, DMSO): δ 11.32 (s, 1H), 11.06 (s, 1H), 10.92 (s, 1H), 8.22 (d, J = 12 Hz, 1H), 7.95 (s, 1H), 7.36 (d, J = 8 Hz, 1H), 7.26 (d, J = 8 Hz, 1H), 7.18 (t, J = 8 Hz, 2H), 7.00 (t, J = 8 Hz, 1H), 6.93 (t, J = 8 Hz, 2H), 6.80 (d, J = 8 Hz, 1H), 6.71–6.64 (q, J = 8 Hz, 2H), 3.49 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 53.02, 55.66, 103.43, 110.39, 112.24, 112.39, 112.79, 112.93, 113.11, 113.32, 119.11, 120.62, 120.85, 121.74, 125.14, 125.29, 125.90, 126.30, 132.85, 135.78, 137.55, 142.73, 148.35, 153.15, 179.44 ppm.
5′-Bromo-5-methoxy-[3,3′:3′,3′′-terindolin]-2′-one (4m).
1H NMR (400 MHz, DMSO): δ 10.98 (s, 1H), 10.84 (s, 1H), 10.74 (s, 1H), 7.40–7.38 (d, J = 8 Hz, 1H), 7.35–7.32 (d, J = 8 Hz, 1H), 7.26 (s, 1H), 7.17–7.15 (d, J = 8 Hz, 1H), 7.01–6.98 (t, J = 8 Hz, 1H), 6.94–6.92 (d, J = 8 Hz, 1H), 6.89 (s, 1H), 6.81 (s, 1H), 6.79–6.76 (m, 2H), 6.69–6.66 (d, J = 8 Hz, 1H), 6.61 (s, 1H), 3.49 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 53.29, 55.85, 103.57, 111.09, 112.15, 112.28, 112.79, 113.45, 113.66, 113.86, 114.01, 118.95, 120.92, 121.60, 125.07, 125.73, 126.01, 126.38, 127.92, 131.22, 132.66, 137.45, 141.26, 153.04, 178.79 ppm.
5-Bromo-5′′-chloro-5′-methyl-[3,3′:3′,3′′-terindolin]-2′-one (4n).
1H NMR (400 MHz, DMSO): δ 11.17 (s, 2H), 10.60 (s, 1H), 7.37 (d, J = 12 Hz, 2H), 7.34 (d, J = 12 Hz, 1H), 7.19 (s, 1H), 7.13 (d, J = 8 Hz, 1H), 7.02 (t, J = 8 Hz, 2H), 6.92 (s, 1H), 6.88 (s, 2H), 6.86 (s, 1H), 2.17 (s, 3 H) ppm. 13C NMR (100 MHz, DMSO): δ 21.32, 52.73, 110.10, 111.56, 113.87, 114.35, 114.43, 114.53, 120.23, 121.62, 123.29, 123.46, 124.13, 125.97, 126.63, 127.08, 127.78, 129.67, 131.14, 134.13, 135.99, 136.22, 139.30, 179.03 ppm.
5′′-Chloro-5′-methoxy-2′-oxo-[3,3′:3′,3′′-terindoline]-5-carbonitrile (4o).
1H NMR (400 MHz, DMSO): δ 11.18 (s, 2H), 10.54 (s, 1H), 7.37 (d, J = 12 Hz, 2H), 7.19 (s, 2H), 7.02 (d, J = 8 Hz, 2H), 6.92 (t, J = 8 Hz, 3H), 6.82 (d, J = 4 Hz, 1H), 6.70 (s, 1H), 3.60 (s, 3 H) ppm. 13C NMR (100 MHz, DMSO): δ 53.12, 55.86, 110.74, 112.48, 113.12, 113.89, 114.36, 120.17, 121.63, 123.47, 126.77, 127.06, 135.05, 135.44, 135.99, 155.34, 178.86 ppm.
4-Bromo-5′′-chloro-[3,3′:3′,3′′-terindolin]-2′-one (4p).
1H NMR (400 MHz, DMSO): δ 11.18 (s, 2H), 10.70 (s, 1H), 7.39 (s, 1H), 7.37 (s, 1H), 7.35 (s, 1H), 7.25 (t, J = 8 Hz, 1H), 7.16 (s, 1H), 7.07 (d, J = 8 Hz, 1H), 7.01 (d, J = 8 Hz, 2H), 6.97 (d, J = 8 Hz, 1H), 6.94–6.90 (m, 1H), 6.90 (s, 1H), 6.85 (d, J = 8 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO): δ 52.87, 110.38, 112.01, 113.89, 114.42, 120.14, 121.65, 122.35, 122.72, 123.35, 123.49, 125.39, 126.64, 127.10, 128.78, 134.16, 135.98, 139.43, 141.72, 178.99 ppm.
5-Methoxy-7′-(trifluoromethyl)-[3,3′:3′,3′′-terindolin]-2′-one (4q).
1H NMR (400 MHz, DMSO): δ 11.12 (s, 1H), 11.02 (s, 1H), 10.87 (s, 1H), 7.52 (d, J = 8 Hz, 1H), 7.43 (t, J = 8 Hz, 1H), 7.37 (d, J = 8 Hz, 1H), 7.26 (d, J = 8 Hz, 2H), 7.09 (t, J = 4 Hz, 1H), 7.01 (t, J = 8 Hz, 1H), 6.88 (d, J = 12 Hz, 1H), 6.81 (s, 1H), 6.69 (d, J = 4 Hz, 1H), 6.63–6.58 (m, 2H), 3.34 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 52.81, 55.47, 102.98, 103.15, 111.35, 112.30, 112.86, 113.14, 113.43, 113.60, 118.99, 121.20, 121.67, 122.37, 124.85, 125.13, 125.92, 126.31, 129.49, 132.69, 136.75, 137.56, 139.22, 153.12, 170.61, 179.64 ppm.
5,6′-Dimethoxy-[3,3′:3′,3′′-terindolin]-2′-one (4r).
1H NMR (400 MHz, DMSO): δ 10.89 (s, 1H), 10.74 (s, 1H), 10.51 (s, 1H), 7.32 (d, J = 8 Hz, 1H), 7.21 (d, J = 8 Hz, 2H), 7.07 (d, J = 4 Hz, 1H), 6.97 (t, J = 8 Hz, 2H), 6.83 (d, J = 8 Hz, 2H), 6.78–6.74 (m, 1H), 6.67–6.64 (m, 1H), 6.51 (s, 1H), 6.47–6.43 (m, 1H), 3.71 (s, 3H), 3.33 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 52.58, 55.67, 96.80, 104.01, 106.86, 110.87, 112.08, 112.54, 115.26, 118.70, 121.32, 121.41, 124.73, 126.06, 126.24, 126.64, 127.17, 132.71, 137.46, 142.96, 152.89, 159.74, 179.77 ppm.
1,1′-Dimethyl-[3,3′:3′,3′′-terindolin]-2′-one (4s)29.
1H NMR (400 MHz, DMSO): δ 10.94 (s, 1H), 7.31 (q, J = 4 Hz, 4H), 7.25 (d, J = 8 Hz, 1H), 7.16 (s, 1H), 7.14 (d, J = 4 Hz, 1H), 7.11 (d, J = 8 Hz, 1H), 7.04 (t, J = 8 Hz, 1H), 6.99–6.95 (m, 2H), 6.82 (s, 1H), 6.77 (q, J = 8 Hz, 2H), 3.66 (s, 3H), 3.22 (s, 3H) ppm. 13C NMR (100 MHz, DMSO): δ 26.79, 32.87, 52.56, 109.22, 110.31, 112.15, 113.76, 114.44, 118.89, 120.99, 121.51, 121.55, 121.61, 122.76, 124.89, 125.08, 126.06, 126.49, 128.55, 128.94, 134.13, 137.42, 137.84, 143.20, 177.31 ppm.
Conflicts of interest
There is no conflict of interest to report.
Acknowledgements
The financial support from SERB, India (CRG/2021/002077) is extremely acknowledged.
References
-
(a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed;
(b) Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed;
(c) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742 RSC;
(d) F.-T. Sheng, J.-Y. Wang, W. Tan, Y.-C. Zhang and F. Shi, Org. Chem. Front., 2020, 7, 3967 RSC.
-
(a) E. Mahmoud, A. M. Hayallah, S. Kovacic, D. Abdelhamid and M. Abdel-Aziz, Pharmacol. Rep., 2022, 74, 570–582 CrossRef CAS PubMed;
(b) P. Paira, A. Hazra, S. Kumar, R. Paira, K. B. Sahu, S. Naskar, S. Mondal, A. Maity, S. Banerjee, N. B. Mondal and P. Saha, Bioorg. Med. Chem. Lett., 2009, 19, 4786–4789 CrossRef CAS PubMed;
(c) A. Kamal, Y. Srikanth, M. N. A. Khan, T. B. Shaik and M. Ashraf, Bioorg. Med. Chem. Lett., 2010, 20, 5229–5231 CrossRef CAS PubMed;
(d) G. Wang, J. Wang, Z. Xie, M. Chen, L. Li, Y. Peng, S. Chen, W. Li and B. Deng, Bioorg. Chem., 2017, 72, 228–233 CrossRef CAS PubMed.
- A. Kumar, R. D. Shukla, D. Yadav and L. P. Gupta, RSC Adv., 2015, 5, 52062–52065 RSC.
- J. Tong, L. S. Huang and D. Z. Xu, New J. Chem., 2017, 41, 3966–3974 RSC.
- M. A. Shaikh, A. S. Ubale and B. Gnanaprakasam, Adv. Synth. Catal., 2021, 363, 4876–4882 CrossRef CAS.
- J. Khan, A. Tyagi, N. Yadav, R. Mahato and C. K. Hazra, J. Org. Chem., 2021, 86, 17833–17847 CrossRef CAS PubMed.
-
(a) S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed;
(b) G. W. Huisman and S. J. Collier, Curr. Opin. Chem. Biol., 2013, 17, 284–292 CrossRef CAS PubMed;
(c)
J. M. Blamey, F. Fischer, H. P. Meyer, F. Sarmiento and M. Zinn, Biotechnology of microbial enzymes, 2017, pp. 347–403 Search PubMed.
-
(a) M. S. Humble and P. Berglund, Eur. J. Org. Chem., 2011, 19, 3391–3401 CrossRef;
(b) M. López-Iglesias and V. Gotor-Fernández, Chem. Rec., 2015, 15, 743–759 CrossRef PubMed;
(c) M. Kapoor and M. N. Gupta, Process Biochem., 2012, 47, 555–569 CrossRef CAS;
(d) E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero and K. S. Ryan, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- I. Oroz-Guinea and E. García-Junceda, Curr. Opin. Chem. Biol., 2013, 6, 1039 CrossRef.
-
(a) A. M. Knight, S. J. Kan, R. D. Lewis, O. F. Brandenberg, K. Chen and F. H. Arnold, ACS Cent. Sci., 2018, 4, 372–377 CrossRef CAS PubMed;
(b) O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102–111 CrossRef CAS PubMed.
-
(a) M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS;
(b) R. Mu, Z. Wang, M. C. Wamsley, C. N. Duke, P. H. Lii, S. E. Epley, L. C. Todd and P. J. Roberts, Catalysts, 2020, 10, 832 CrossRef CAS;
(c) M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 CrossRef CAS.
-
(a) M. López-Iglesias and V. Gotor-Fernández, Chem. Rec., 2015, 15, 743–759 CrossRef PubMed;
(b) R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC;
(c) B. A. Sandoval and T. K. Hyster, Curr. Opin. Chem. Biol., 2020, 55, 45–51 CrossRef CAS PubMed.
-
(a) S. Dutt, V. Goel, N. Garg, D. Choudhury, D. Mallick and V. Tyagi, Adv. Synth. Catal., 2020, 362, 858–866 CrossRef CAS;
(b) S. Dutt and V. Tyagi, Tetrahedron Lett., 2021, 87, 153527 CrossRef CAS;
(c) S. Dutt and V. Tyagi, Biocatal. Biotransform., 2023, 1–11 CrossRef.
-
(a) M. Budhiraja, R. Kondabala, A. Ali and V. Tyagi, Tetrahedron, 2020, 76, 131643 CrossRef CAS;
(b) M. Budhiraja, A. Ali and V. Tyagi, New J. Chem., 2022, 46, 4837–4849 RSC;
(c) M. Budhiraja, A. Ali and V. Tyagi, Eur. J. Org. Chem., 2023, 26, 202201426 CrossRef;
(d) M. Budhiraja, B. Chudasama, A. Ali and V. Tyagi, RSC Adv., 2022, 12, 31734–31746 RSC.
- P. Kamboj and V. Tyagi, Asian J. Org. Chem., 2023, 12, 202200669 CrossRef.
- G. Wang, J. Wang, Z. Xie, M. Chen, L. Li, Y. Peng, S. Chen, W. Li and B. Deng, Bioorg. Chem., 2017, 72, 228–233 CrossRef CAS PubMed.
- A. Bahuguna, A. Kumar, S. Kumar, T. Chhabra and V. Krishnan, ChemCatChem, 2018, 10, 3121–3132 CrossRef CAS.
- G. Mohammadi Ziarani, R. Moradi, A. Badiei, N. Lashgari, B. Moradi and A. Abolhasani Soorki, J. Taibah Univ. Sci., 2015, 9, 555–563 CrossRef.
- R. Gupta, M. Yadav, R. Gaur, G. Arora, P. Rana, P. Yadav, A. Adholeya and R. K. Sharma, ACS Omega, 2019, 4, 21529–21539 CrossRef CAS PubMed.
- L. Xing, Y. Hui, J. Yang, X. Xing, Y. Hou, Y. Wu, K. Fan and W. Wang, J. Chem., 2018, 2018, 1–6 CrossRef.
- S. B. Gohain, M. Basumatary, P. K. Boruah, M. R. Das and A. J. Thakur, Green Chem., 2020, 22, 170–179 RSC.
- X. Yuan, S. Wang, J. Cheng, B. Yu and H. M. Liu, Chin. Chem. Lett., 2020, 31, 2465–2468 CrossRef CAS.
- M. A. Shaikh, A. S. Ubale and B. Gnanaprakasam, Adv. Synth. Catal., 2021, 363, 4876–4882 CrossRef CAS.
- P. K. Vuram, C. Kabilan and A. Chadha, Int. J. Org. Chem., 2015, 5, 108–118 CrossRef CAS.
- M. Nikpassand, M. Mamaghani, K. Tabatabaeian and H. A. Samimi, Synth. Commun., 2010, 40, 3552–3560 CrossRef CAS.
- Y. Li, D. Liang, X. Li, W. Huang, L. Yuan, B. Wang and P. Cheng, Heterocycl. Commun., 2017, 23, 29–34 CrossRef CAS.
- S. Y. Wang and S. J. Ji, Tetrahedron, 2006, 62, 1527–1535 CrossRef CAS.
- K. Rad-Moghadam, M. Sharifi-Kiasaraie and H. Taheri-Amlashi, Tetrahedron, 2010, 66, 2316–2321 CrossRef CAS PubMed.
- S. F. Hojati and A. S. Kaheh, Jordan J. Chem., 2019, 14, 17–27 CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Vikas
Tyagi
and
Vikas
Tyagi