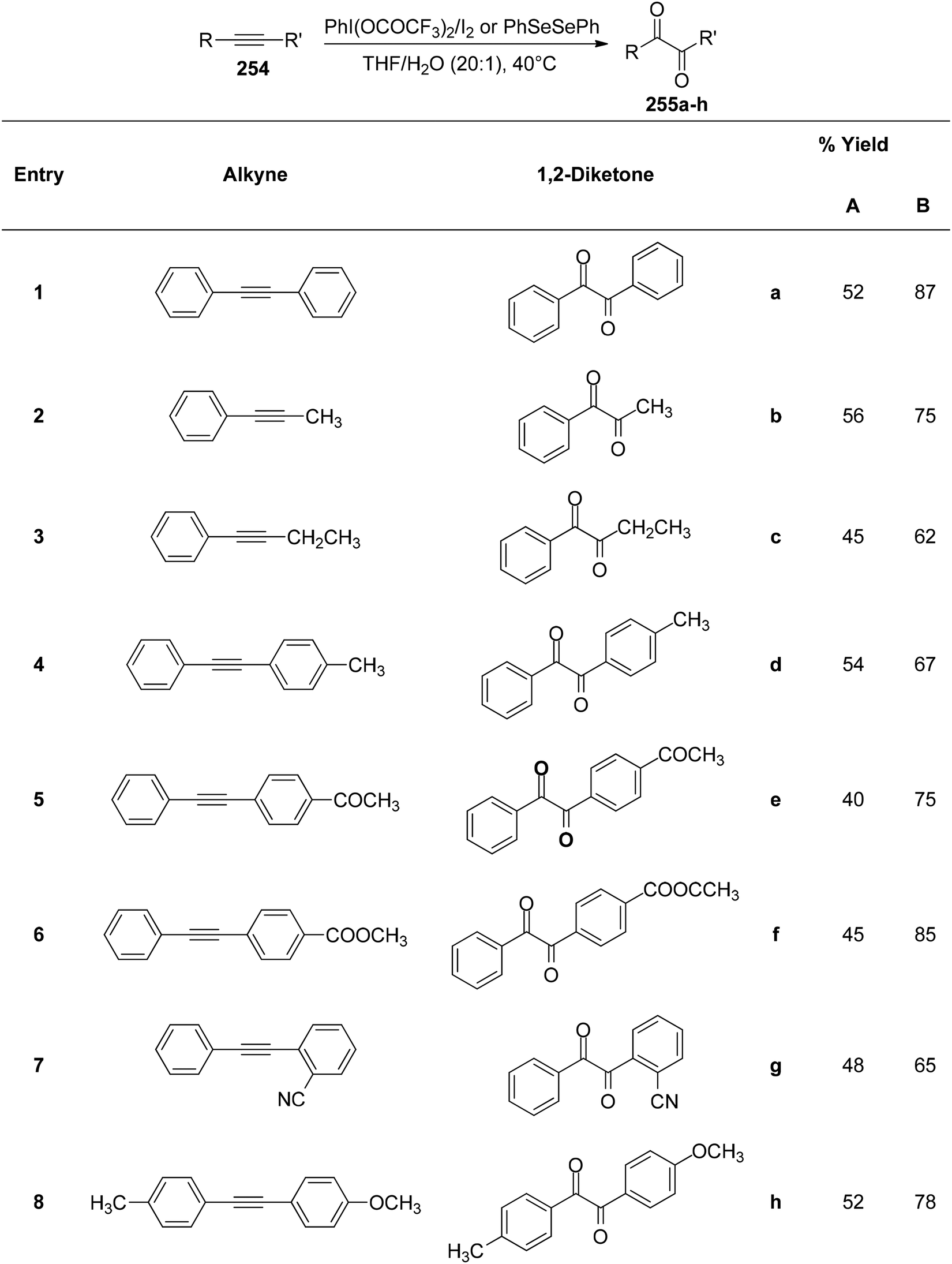
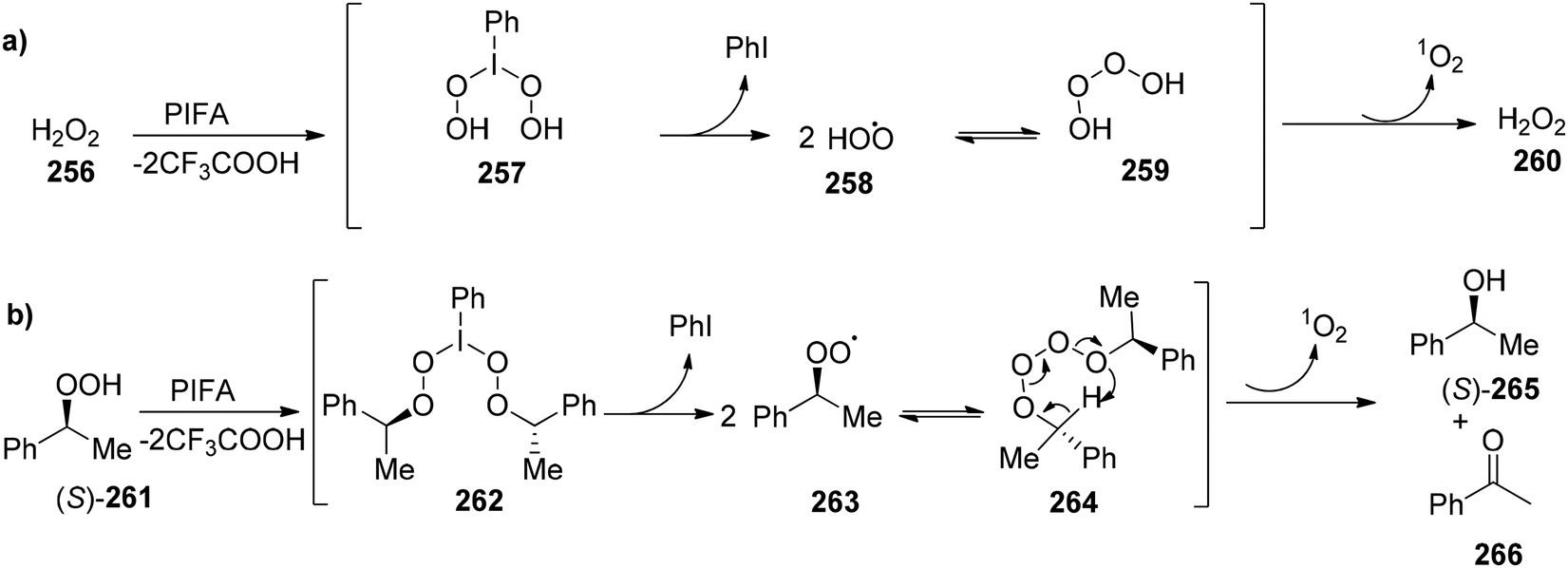
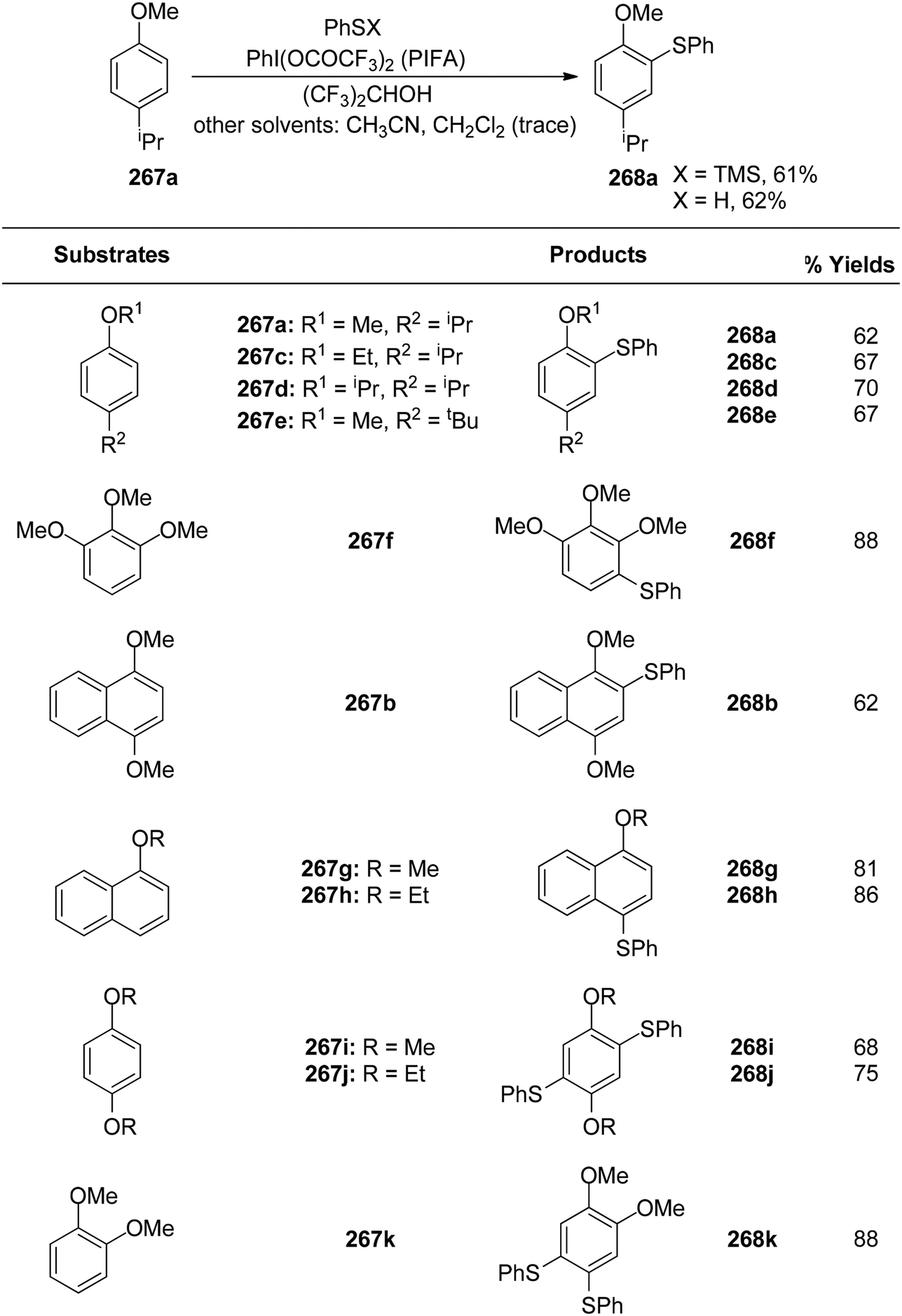
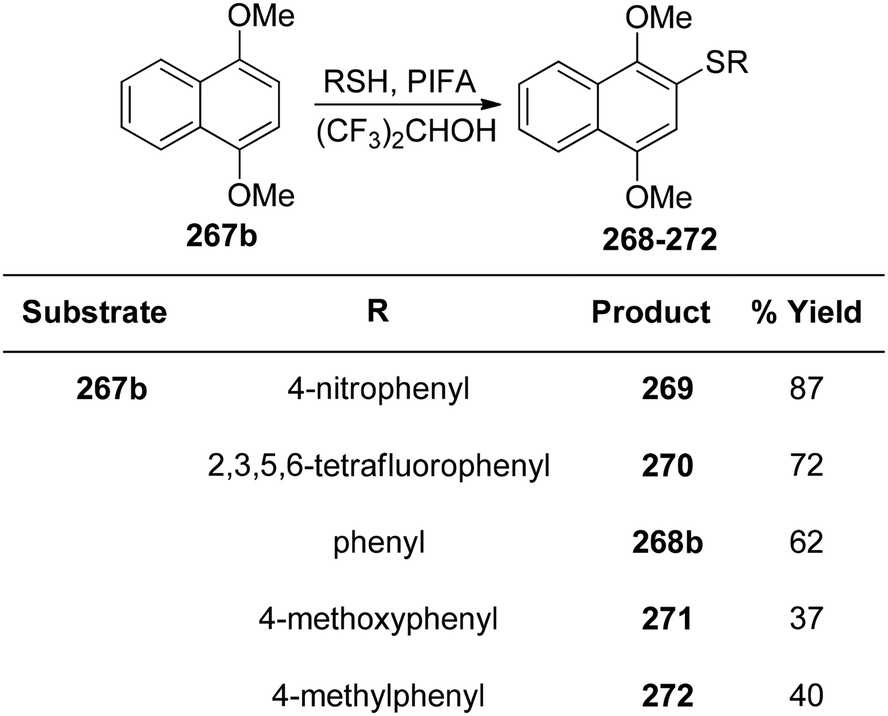
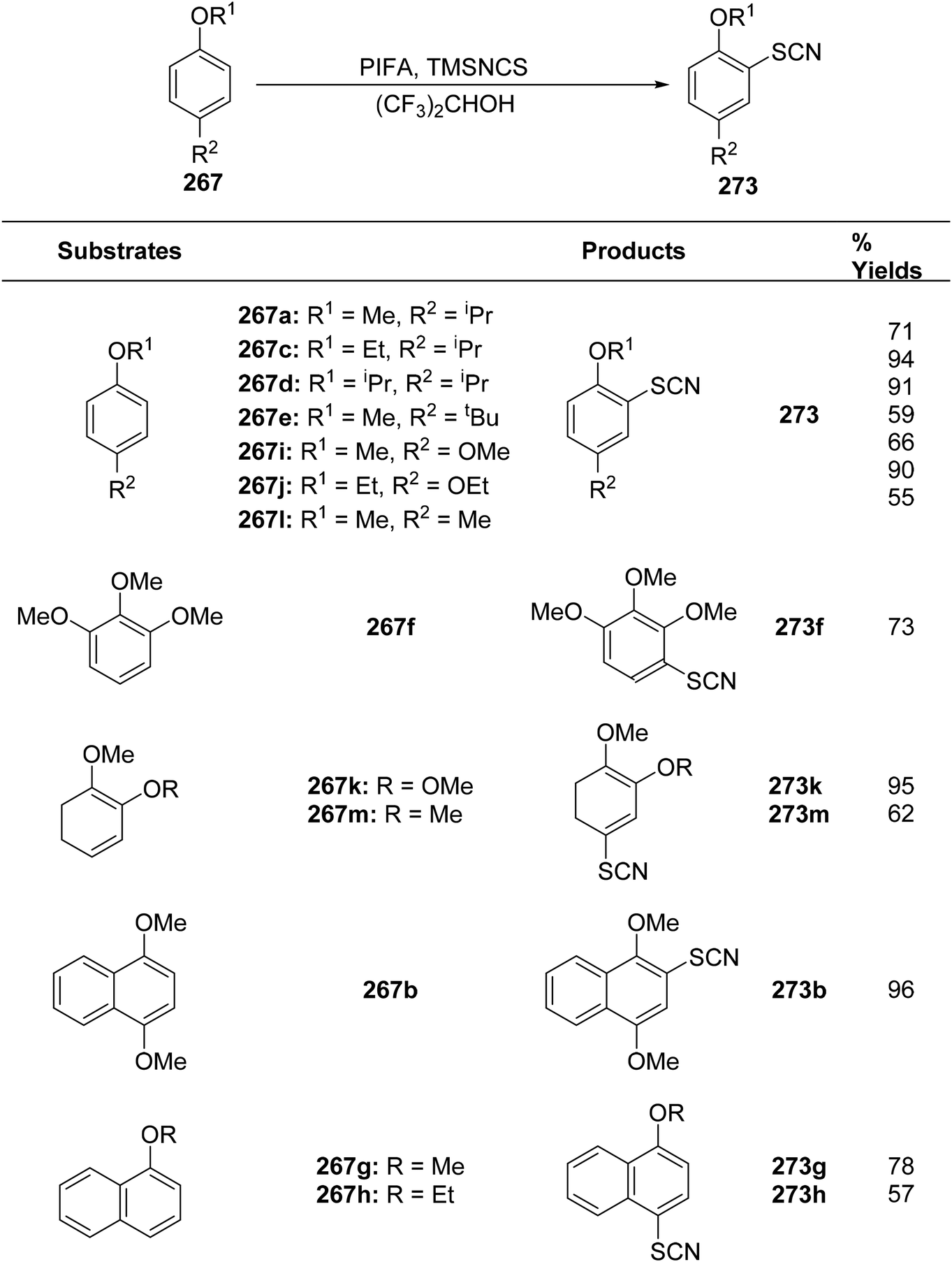
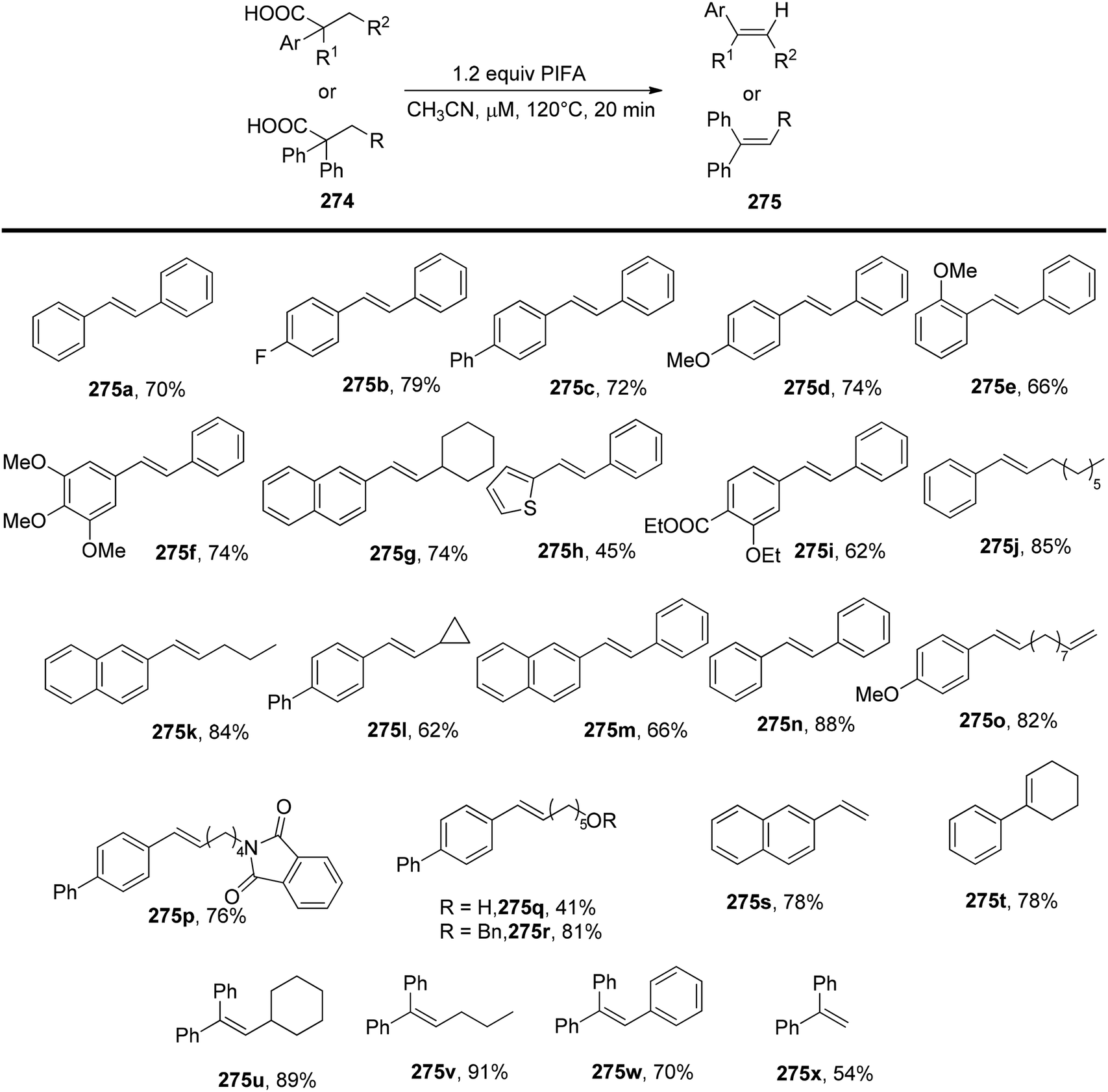
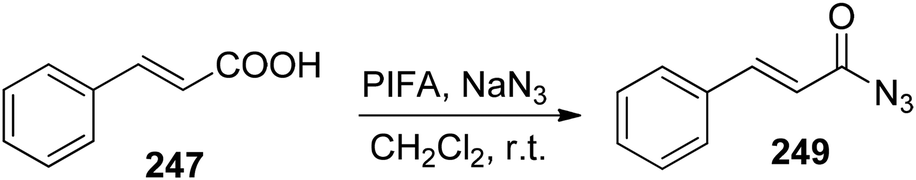DOI:
10.1039/D3OB01964K
(Review Article)
Org. Biomol. Chem., 2024,
22, 3109-3185
Phenyliodine bis(trifluoroacetate) as a sustainable reagent: exploring its significance in organic synthesis
Received
3rd December 2023
, Accepted 15th March 2024
First published on 15th March 2024
Abstract
Iodine-containing molecules, especially hypervalent iodine compounds, have gained significant attention in organic synthesis. They are valuable and sustainable reagents, leading to a remarkable surge in their use for chemical transformations. One such hypervalent iodine compound, phenyliodine bis(trifluoroacetate)/bis(trifluoroacetoxy)iodobenzene, commonly referred to as PIFA, has emerged as a prominent candidate due to its attributes of facile manipulation, moderate reactivity, low toxicity, and ready availability. PIFA presents an auspicious prospect as a substitute for costly organometallic catalysts and environmentally hazardous oxidants containing heavy metals. PIFA exhibits remarkable catalytic activity, facilitating an array of consequential organic reactions, including sulfenylation, alkylarylation, oxidative coupling, cascade reactions, amination, amidation, ring-rearrangement, carboxylation, and numerous others. Over the past decade, the application of PIFA in synthetic chemistry has witnessed substantial growth, necessitating an updated exploration of this field. In this discourse, we present a concise overview of PIFA's applications as a ‘green’ reagent in the domain of synthetic organic chemistry. A primary objective of this article is to bring to the forefront the scientific community's awareness of the merits associated with adopting PIFA as an environmentally conscientious alternative to heavy metals.
 Sumit Kumar | Sumit Kumar obtained his Master's Degree in Organic Chemistry from the Department of Chemistry, University of Delhi, in 2020 and Honours Degree from Kirori mal College, University of Delhi, in 2018. Currently, he is pursuing a Ph.D. from the Department of Chemistry, University of Delhi, under the supervision of Prof. Brajendra K. Singh. His research interests include design and synthesis of sugar-modified heterocyclic motifs of therapeutic importance. He visited Medgar Evers College, City University of New York (CUNY) in the Chemistry and Environmental Science Department as a Research Scholar. He has published several research articles in various reputable journals such as European Journal of Polymer Chemistry, New Journal of Chemistry, Beilstein Journal of Organic Chemistry, Synthesis, Organic and Biomolecular Chemistry and others. |
 Aditi Arora | Aditi Arora obtained her Honours Degree from Miranda House, University of Delhi, in 2018. She completed her Master's in Organic Chemistry from the Department of Chemistry, University of Delhi, in 2020. She was a Gold Medalist during her Master's. At present, she is pursuing a Ph.D. from the Department of Chemistry, University of Delhi. Her research interests include design and synthesis of carbohydrate and coumarin-modified molecules. She has published several research articles in various reputable journals. |
 Sunil K. Singh | Dr Sunil K Singh is an Associate Professor at the Department of Chemistry, Kirori Mal College, University of Delhi. He received his Ph.D. from the Department of Chemistry, University of Delhi, India, in the area of synthesis of modified nucleosides and biocatalytic organic transformations. He has published 32 articles in the journals of international repute. He has also published three book chapters. |
 Rajesh Kumar | Dr Rajesh Kumar received his Master of Science degree in Chemistry from the University of Delhi in 2010. He joined the same Department for a Ph.D. and completed his Ph.D. in 2017, and during his Ph.D. Dr Kumar visited the University of Southern Denmark as a Research Assistant from December 2014 to August 2015. After completion of his Ph.D., he joined as Assistant Professor in Chemistry at B.R.A. Bihar University, India. He has published more than 30 research papers in reputable national and international journals such as Journal of Organic Chemistry, Theranostics, European Polymer Journal, etc. His research interest lies in Metal Complexes, Biotransformations, Catalysis, Green Chemistry, Nucleoside and Heterocyclic Chemistry. |
 Bhawni Shankar | Dr Bhawani Shankar is an Associate Professor at the Department of Chemistry, Deshbandhu College, University of Delhi. He obtained his Ph.D. in Chemistry from the Department of Chemistry, University of Delhi, in 2022 under the supervision of the late Professor Ashok K. Prasad. His area of research includes synthesis of modified sugars, nucleosides, heterocyclic compounds and hydroxamic acids for different applications. |
 Brajendra K. Singh | Professor Brajendra K. Singh currently holds the position of Professor in the Department of Chemistry, University of Delhi, Delhi, India. He visited the Department of Chemistry, University of Leuven, Leuven, Belgium, as International Scholar for the period November 2005–April 2007 and then as post-doctoral fellow until November 2009. His key interest areas are the study and investigation of bio-catalysis and metal-catalysis, C–H activation, specifically in the design, synthesis and evaluation of bioactive compounds and LED-mediated synthesis. He has published more than 85 research articles in internationally reputable journals. He has delivered around 30 Invited Lectures in various National and International Conferences at different Universities and Institutes. He has served and is serving in different capacities in administration at Delhi University. |
1. Introduction
Iodine, the heaviest element in the Periodic Table, is typically classified as a non-metal and is one of the most massive elements found on Earth. Due to its substantial size, the bonding behavior of iodine-containing compounds differs significantly from its lighter counterparts in the same group. The term ‘hypervalent iodine compounds’ refers to iodine compounds in higher oxidation states. Iodine, being the most polarizable and least electronegative halogen, exhibits unique reactivity. Various oxidation states of iodine, including I(III), I(V), and I(VII), can form hyper-coordinate compounds. In hyper-coordinate iodine(III) compounds, a linear 3-center-4-electron (3c–4ē) bonding arrangement results, with electronegative ligands occupying the axial positions and giving these compounds a T-shaped geometry.1
Hypervalent iodine compounds exhibit structural and reactivity characteristics akin to transition metal elements. Notably, their reaction mechanisms often align with those attributed to transition metals, encompassing phenomena like ligand coupling, oxidative addition, ligand exchange, and reductive elimination. Iodine, as an element, offers economic feasibility, with bulk prices ranging from $20 to $100 per kilogram over the past decade, and it boasts environmental safety credentials, unlike heavy metals. Consequently, the employment of hypervalent iodine compounds for effecting synthetic organic transformations has experienced rapid proliferation.2–4 Over the past two decades, the functionalization of a wide array of organic molecules, within the broader context of organic synthesis, has witnessed significant utilization of organic hypervalent iodine compounds as environmentally friendly and potential oxidizing agents.5–12 These hypervalent compounds serve as environmentally conscious alternatives to heavy metal oxidants like Pb(IV), Hg(II), and Tl(III). Their attributes, including ready availability, mild reactivity, enhanced efficiency, moisture stability, minimal toxicity, and ease of handling, contribute to their green chemistry appeal.
(Bis(trifluoroacetoxy)iodo)benzene/phenyliodine bis(trifluoroacetate), commonly referred to as PIFA, stands out as a prominent example of a hypervalent iodine compound extensively employed in synthetic chemistry. PIFA's versatility extends to its successful application as a viable replacement for hazardous organometallic catalysts and heavy metal oxidants, among other roles.13 Commercially, PIFA is acquired as a light-sensitive, colorless solid that maintains stability at room temperature.
PIFA catalyzes a wide range of organic transformations, including ring-rearrangements, amidations, sulfenylations, carboxylations, and various others.14,15 These reactions result in the synthesis of N-arylated and N-alkylated heterocyclic compounds,16 oxidative deprotection reactions,17 and the formation of pyrrolidinone and lactone frameworks,18 alongside various other transformations.19,20 Additionally, under acidic conditions, PIFA can facilitate the Hofmann rearrangement.21 Notably, in 1990, Kikugawa et al.22 elucidated a synthetic pathway for N-acylated nitrenium ions through PIFA-mediated amide oxidation. Substituted nitrenium ions, as consequential intermediates, serve as versatile precursors for constructing N–N, N–C, and N–S bond connections.23,24
PIFA demonstrates versatility in catalyzing various coupling reactions, leveraging its highly electrophilic iodine core.25,26 Among these reactions, intramolecular phenolic coupling processes mediated by PIFA serve as effective biomimetic methodologies for the synthesis of fused and spirocyclic molecules.27,28 PIFA also participates in ligand-exchange reactions with diverse nucleophiles, followed by reductive elimination.29,30 Furthermore, PIFA has been established as a catalyst for the cyclo-isomerization of propargylamides,31 oxidative amidation reactions of alkynes,32 and carboxylation processes.33 In these reactions, PIFA activates the triple bond, enabling cyclization.
Owing to the potent electron-withdrawing attributes of the fluorine atom, PIFA exhibits higher reactivity in comparison with its counterpart, phenyliodine diacetate (PIDA). In the presence of basic olefin substrates, PIFA-mediated alkene functionalization facilitates the swift introduction of diverse functional groups into organic molecules, thereby enabling efficient and expedited molecular complexity enhancement.34,35 PIFA frequently acts as an electrophilic species in metal-free reactions, generating an intermediate iodonium ion. This intermediate subsequently undergoes transformation into a leaving group, facilitating nucleophilic attack.36 Recently, the field of alkene functionalization reactions has experienced renewed vigor through the synergistic combination of transition metal catalysts with iodine-based reagents.37
An update in this field is warranted, considering PIFA's significant diversification in organic chemistry in the past decade. This report offers a concise overview of PIFA's role as a green reagent in synthetic organic chemistry, with the aim of highlighting its potential as an eco-friendly substitute for heavy metals to the scientific community.
2. Synthesis
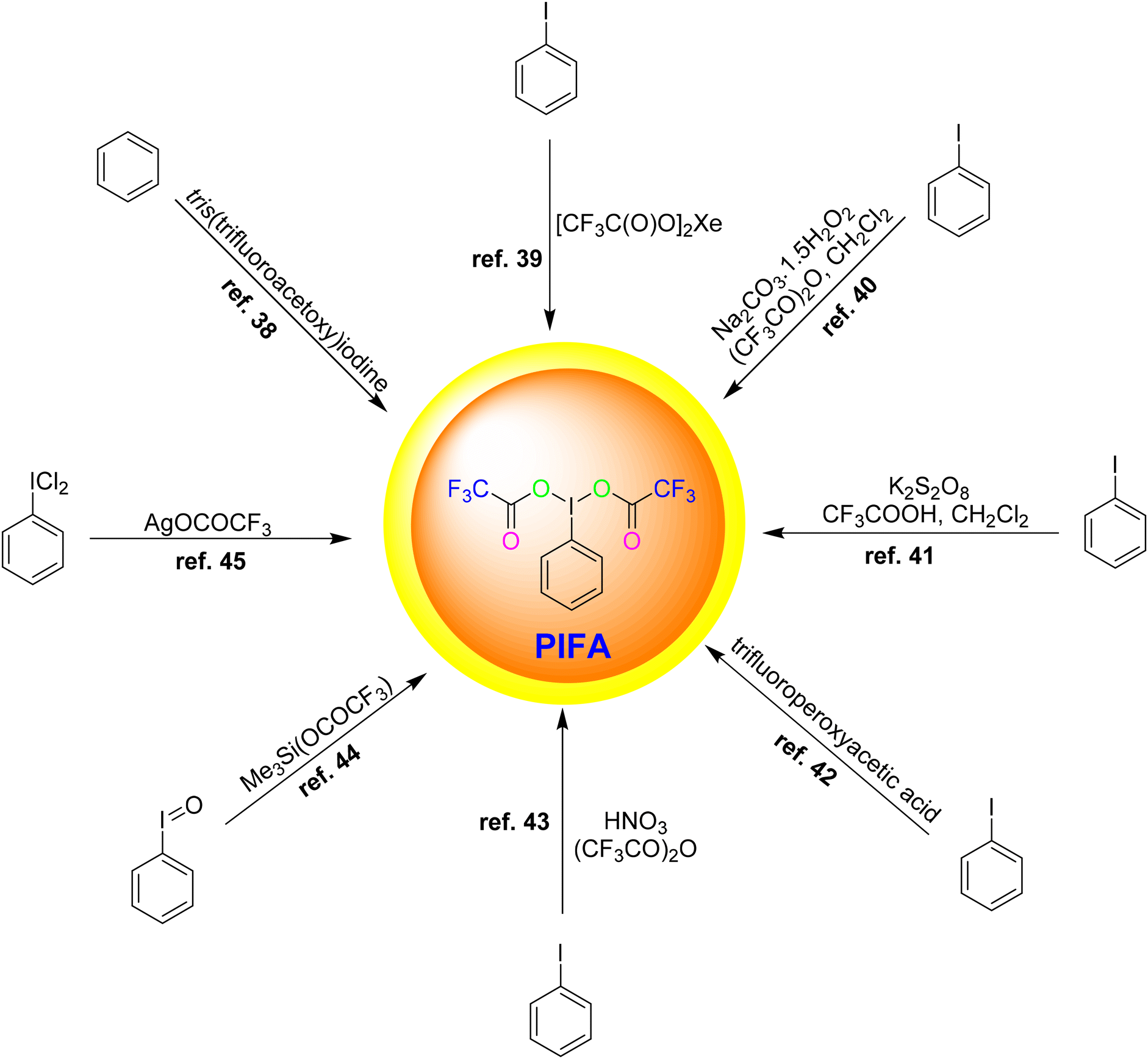
Various methodologies exist for the synthesis of PIFA. Herein, we outline some commonly employed procedures as shown in Fig. 1:
• Reaction of benzene with tris(trifluoroacetoxy)iodine38
• Oxidation of iodobenzene with xenon bis(trifluoroacetate)39
• Oxidation of iodobenzene with sodium percarbonate/(CF3CO)2O/CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40
40
• Oxidation of iodobenzene with using a K2S2O8/CF3COOH/CH2Cl2 system41
• Oxidation of iodobenzene with trifluoroperoxyacetic acid42
• Oxidation of iodobenzene with HNO3/(CF3CO)2O43
• Reaction of iodosylbenzene with Me3Si(OCOCF3)44
• Reaction of (dichloroiodo)benzene with AgOCOCF345
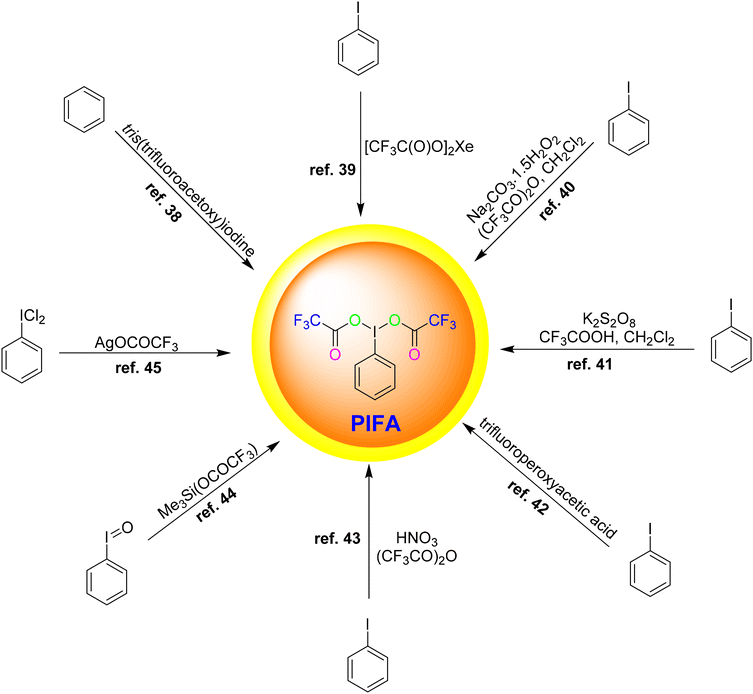 |
| | Fig. 1 Synthesis of PIFA. | |
3. PIFA in organic synthesis
3.1. Amidation reaction
The amide functionality constitutes a substantial structural element in a majority of natural products and pharmaceutical compounds, playing a pivotal role in biologically and pharmaceutically relevant molecules.46 Notably, approximately 25% of medicinal compounds feature amide groups.47 Beyond their pharmaceutical significance, amides find diverse applications in organic chemistry as dyes, polymers, and agrochemicals. Amides also serve as crucial functionalities in peptides and proteins, with the N-formyl group being readily removable without affecting the peptide linkage.48 Moreover, fatty acid amides hold promise for their potential anti-inflammatory, anti-bacterial, anti-tubercular, and antiproliferative properties.49 Fundamentally, amides serve as a central scaffold for synthesizing a vast spectrum of organic compounds. Due to the paramount importance of the amide functionality, numerous synthetic strategies have evolved over time.
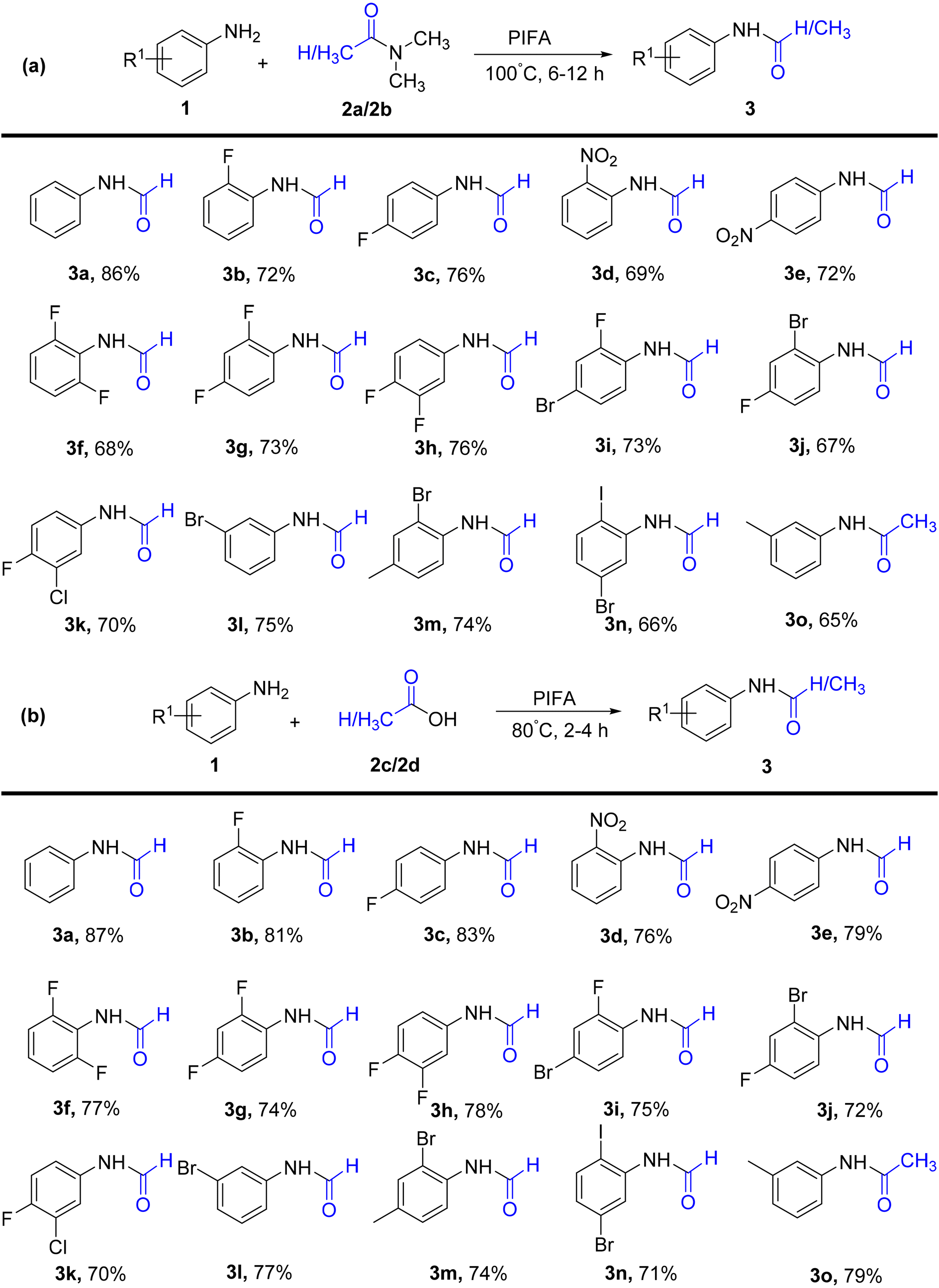
Kamble et al.50 presented an effective, mild, metal-free procedure for catalyzing the transamidation reaction using PIFA (Scheme 1). High yields of amide analogs 3 were achieved by employing various aromatic amines 1, predominantly those featuring fluoro substituents. In this synthetic protocol, DMF/DMAc 2 underwent transamidation with amines. PIFA was involved in the reaction via an ionic pathway, actively participating in the process through its contribution to the ionic mechanism.
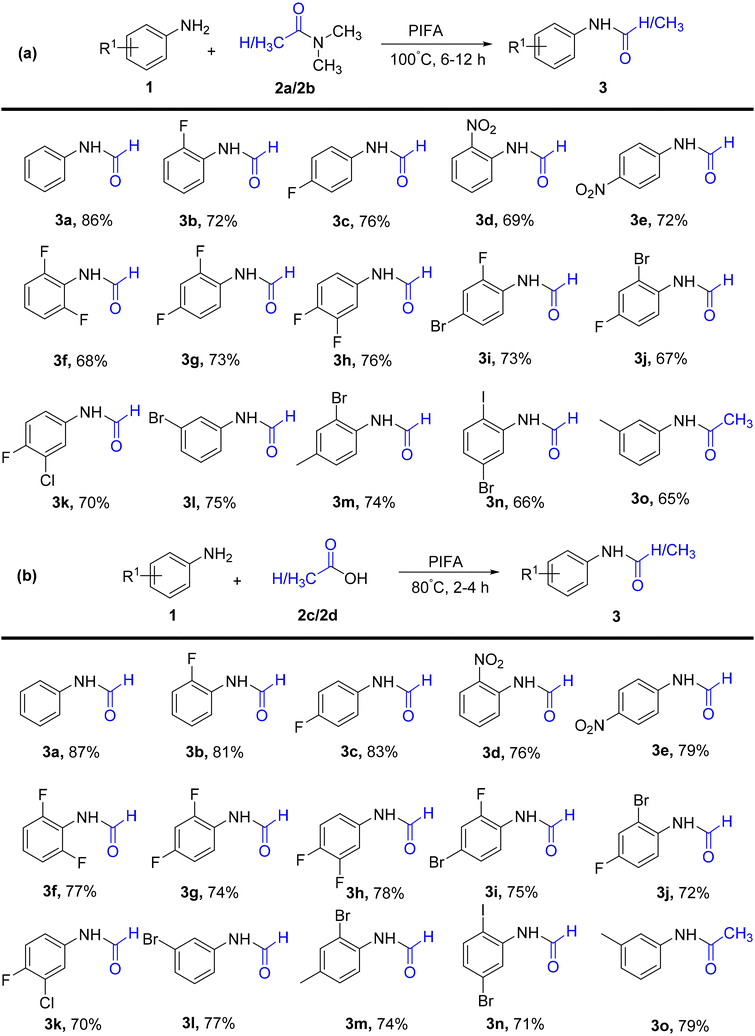 |
| | Scheme 1 a) Synthetic scheme for amides using DMF and DMAc. (b) Synthesis of amides using carboxylic acids. | |
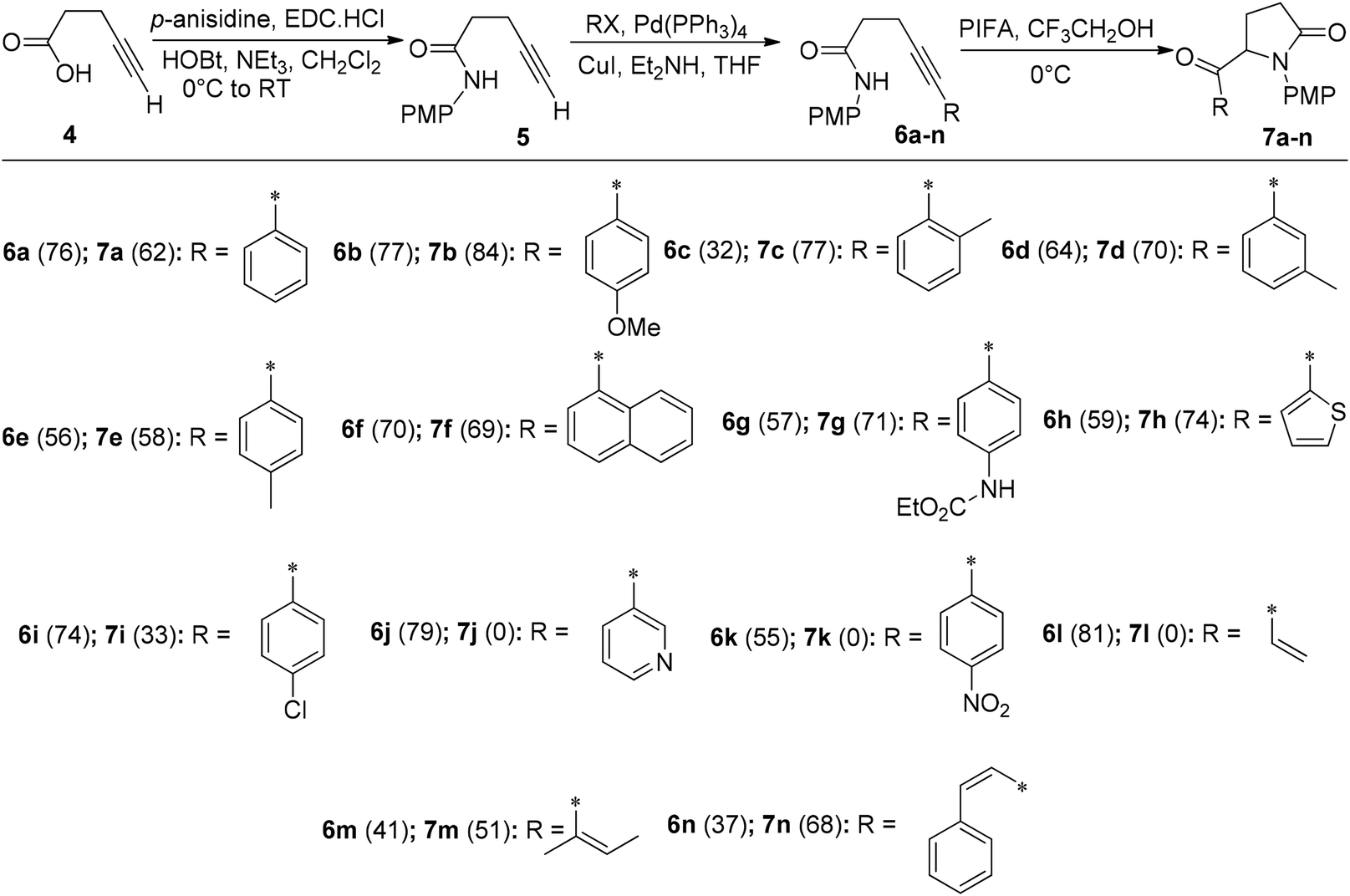
The intramolecular insertion of a nitrogen atom represents an efficacious approach for the synthesis of nitrogen-containing heterocyclic compounds. Alkyne hydroamination stands as an attractive synthetic route for accessing both linear and cyclic amines/imine functionalities through intra/intermolecular reactions.51 Serna et al.52 reported a simple intramolecular amidation reaction for the synthesis of 5-substituted pyrrolidinone analogues 7a–n catalyzed by PIFA with overall yields of 58–84% (Scheme 2). Firstly, the β-alkynylamide precursors 6a–n were obtained by following a two-step procedure as described in Scheme 2. Treatment of p-anisidine with 4-pentynoic acid 4 in the presence of EDC·HCl/HOBt53 resulted in the formation of amide 5. Finally, a Sonogashira cross-coupling reaction54 was employed to substitute the terminal alkyne functionality55 of amide 5 in 32–81% yields. The desired compounds 7a–n were further obtained by treatment of precursors 6a–n with PIFA in acidic media at 0 °C. PIFA engaged in the reaction by following the ionic pathway.
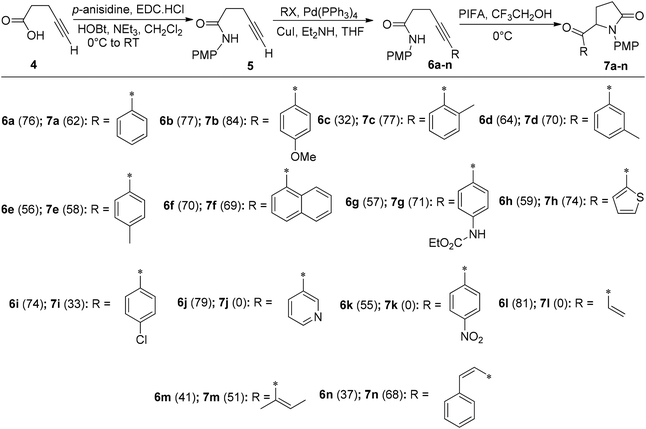 |
| | Scheme 2 Aminohydroxylation of alkyne catalyzed by PIFA. | |
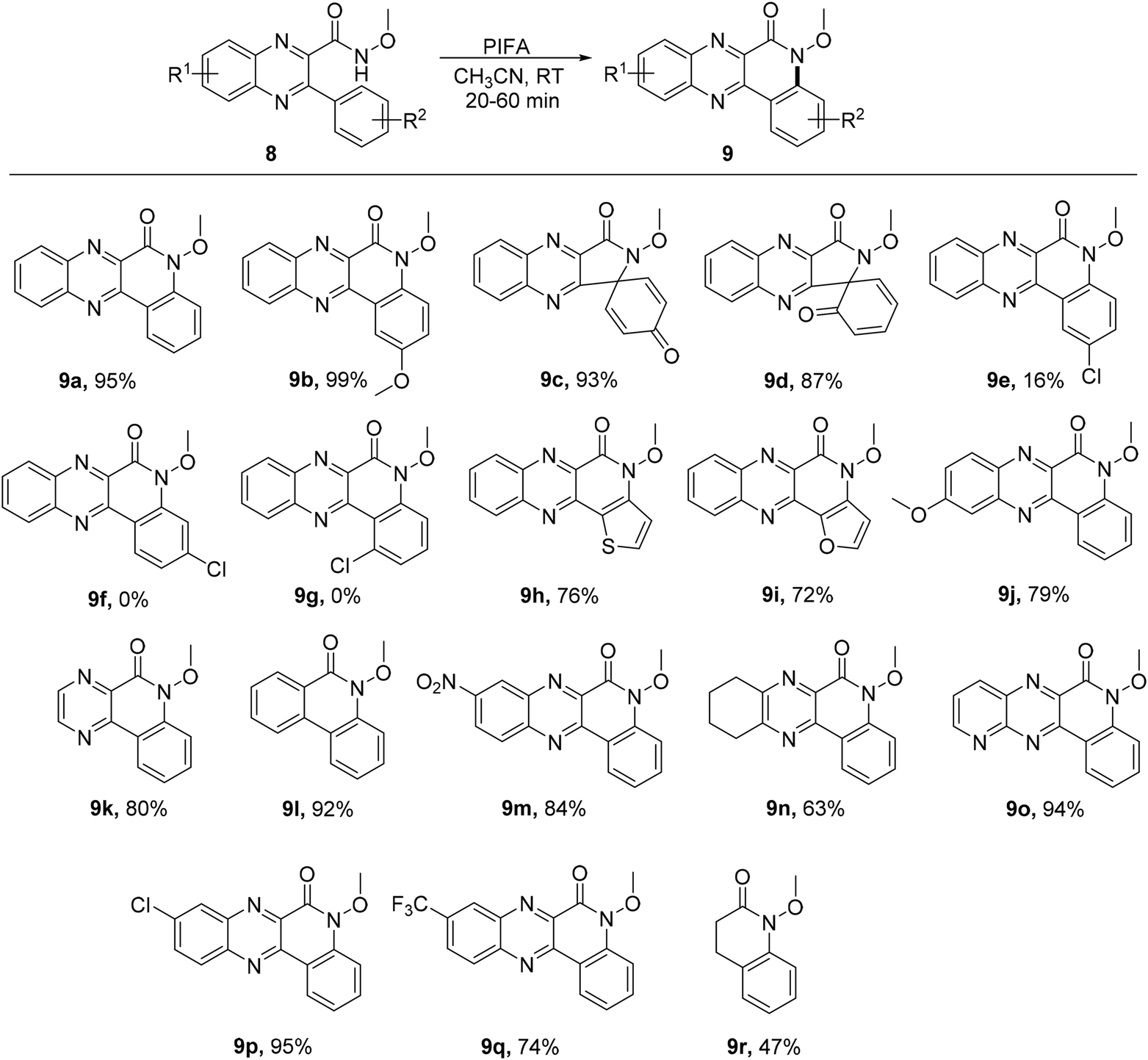
Quinolino and pyrazino analogs of quinoxalin-6(5H)-ones represent intriguing heterocyclic structures with notable applications.56 A swift, metal-free, and effective intramolecular reaction was described by Hu et al.57 for the synthesis of quinoline-quinoxalinones 9 in satisfactory yields using readily accessible starting materials 8. The ring-closure C–H amidation was conducted at ambient temperature with PIFA serving as the catalyst (Scheme 3). PIFA engaged in the reaction by following the ionic pathway, actively participating in the process through its involvement in ionic mechanisms.
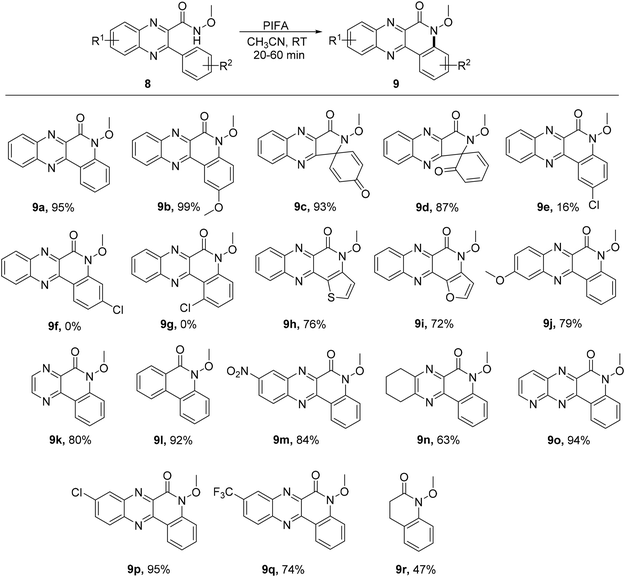 |
| | Scheme 3 Synthesis of quinolino[3,4-b]quinoxalin-6(5H)-ones 9a–r. | |
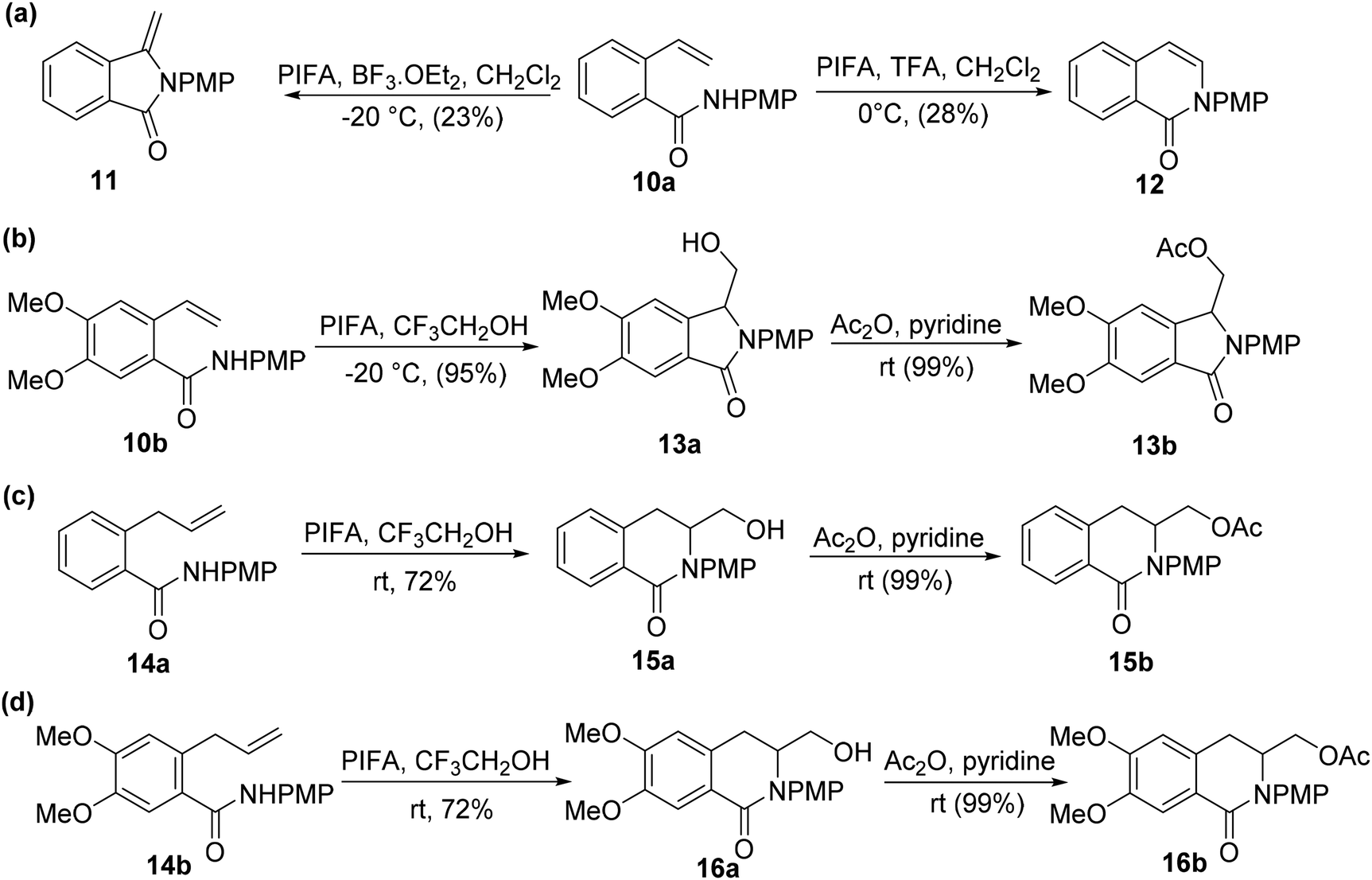
Serna et al.58 reported PIFA-mediated reaction on aromatic substrates to yield isoindolinones as well as isoquinolin-2-ones. Isoindolinone 11 was obtained in 23% yield from amide 10a on treatment with PIFA/BF3·OEt2 in DCM at −20 °C (Scheme 4a). In contrast, isoquinolinone 12 was produced in 28% yield by employing TFA rather than BF3·OEt2 through a less preferred 6-endo-trigcyclization mechanism. They hypothesized that the enhanced nucleophilicity of the styrene fragment would enable nitrogen atom to readily undergo cyclization, leading to improved results. To achieve this, a low-temperature condition was maintained for the treatment of amide 10b with PIFA/CF3CH2OH in the absence of an activating agent (Scheme 4b). As expected, the 5-exo-trig cyclization went without a hitch, resulting in a 95% yield of the isoindolinone 13a. For a complete structural identification, this molecule was later acetylated to produce derivative 13b. The behavior of amides 14a–b was then examined in order to increase the viability of the intended synthesis (Scheme 4c and d). A 6-exo-trig cyclization occurred on treating CF3CH2OH solution of amide 14a with PIFA, producing isoquinolinone 15a in 72% yield. Similarly, amide 14b resulted in the formation of isoquinolinone 16a at room temperature in excellent yield. Furthermore, these motifs were acetylated to yield compound 15b and 16b, respectively, in order to obtain a complete structural identification. PIFA was involved in the reaction via an ionic pathway. Future research in the area of heterocyclic chemistry will benefit from the introduction of this subsidiary hydroxyl functionality which will enable the synthesis of advanced heterocycles.
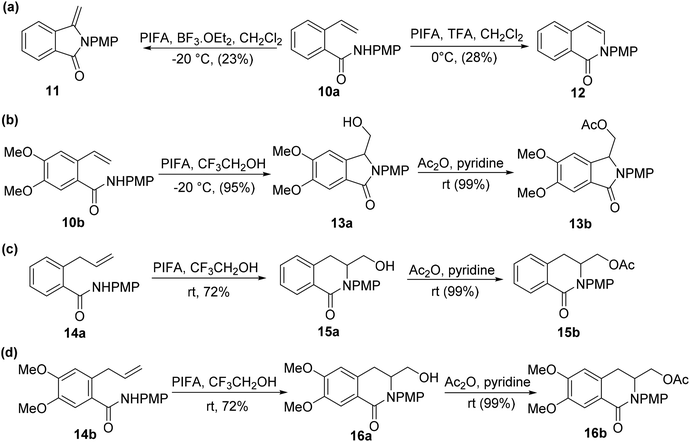 |
| | Scheme 4 Synthesis of isoindolinones as well as isoquinolin-2-ones from suitably modified aromatic substrates. | |
In addition to serving as a pivotal functionality in tryptophan-containing compounds, indole and indoline scaffolds constitute integral components of biologically significant natural products.59 Due to their compelling therapeutic applications, substantial research efforts have been dedicated to the development of innovative and efficacious synthetic methodologies for these heterocyclic structures.
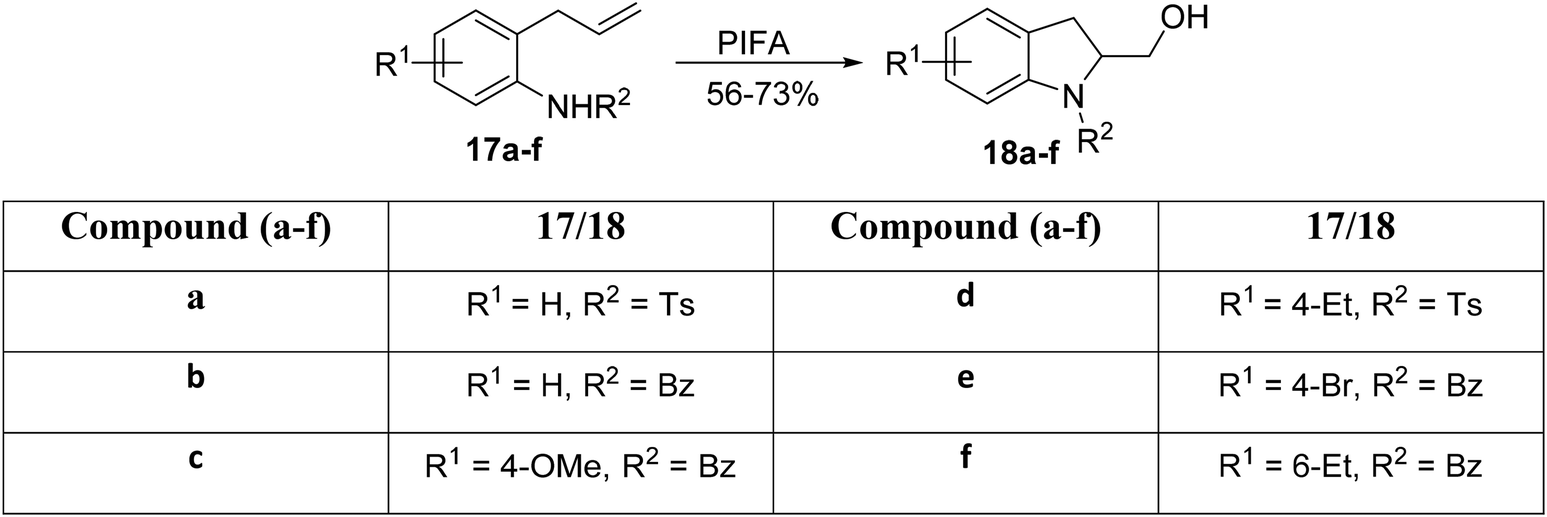
Correa et al.60 introduced an innovative synthetic strategy for the synthesis of indoline derivatives. Their work demonstrates that the PIFA-mediated amidohydroxylation reaction represents a rapid and efficient route to access indoline derivatives. Notably, compared with other metal-based approaches, this method allows for the dual functionalization of the alkene moiety, leading to the incorporation of both a hydroxyl group and a C–N linkage in the final product. This capability enables the synthesis of complex heterocycles through subsequent transformations. The critical cyclization step involves the PIFA-catalyzed generation of N-acylated nitrenium ions, followed by their intramolecular trapping with olefins. PIFA contributed to the reaction by engaging in the ionic pathway, playing a role in the process through its involvement. Optimization of reaction conditions revealed that treating substrate 17 with PIFA in trifluoroethanol for 3 hours at room temperature yields superior results in the amidation process. The broad applicability of this reaction was demonstrated by preparing a diverse set of indoline derivatives 18a–f (Scheme 5).
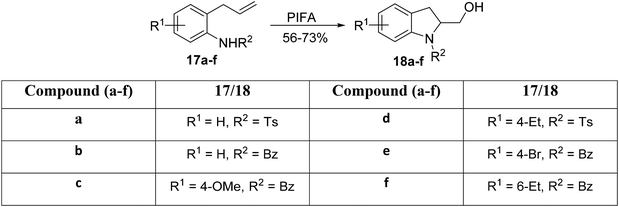 |
| | Scheme 5 Synthesis of indoline derivatives 18a–f. | |
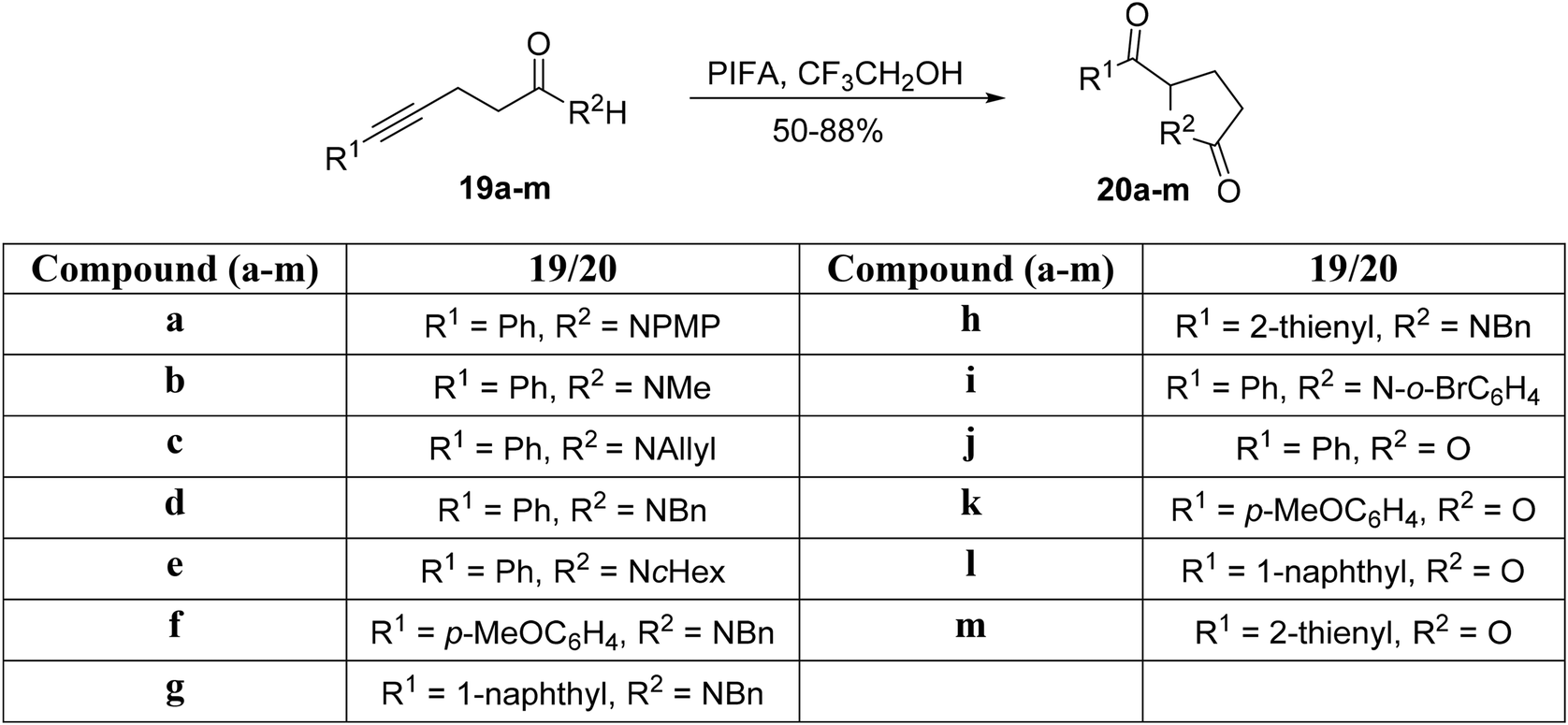
Tellitu et al.61 conducted an intramolecular electrophilic cyclization reaction catalyzed by PIFA using readily available alkynyl-amides 19a–i and alkynyl carboxylic acids 19j–m as substrates. This process led to the formation of pyrrolidinones 20a–i and lactone derivatives 20j–m, respectively. Specifically, amides 19a–i, when dissolved in CF3CH2OH and subjected to the reaction at 0 °C, yielded the corresponding pyrrolidinones 20a–i in a brief reaction time (Scheme 6). Similarly, the reaction of carboxylic acids 19j–m with PIFA in the presence of CF3CH2OH at 0 °C resulted in the formation of the predicted furanones 20j–m in excellent yields (Scheme 6). PIFA was involved in the reaction via an ionic pathway.
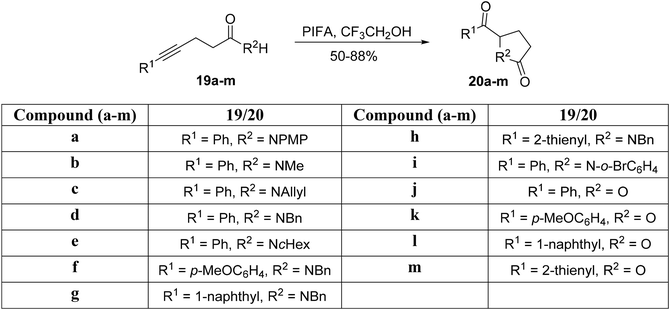 |
| | Scheme 6 Synthetic scheme for pyrrolidinone as well as lactone derivatives. | |
3.2. Amination reaction
Azides serve as efficient amino sources for catalyzing amination reactions on aromatic C–H bonds with transition metals. Their effectiveness is attributed to their ready availability, diverse structural versatility, and the environmentally friendly production of N2 gas as a byproduct of the reaction.62 The selective functionalization of double bonds has become increasingly important as a versatile method for synthesizing complex molecules using a minimal number of steps.63–65
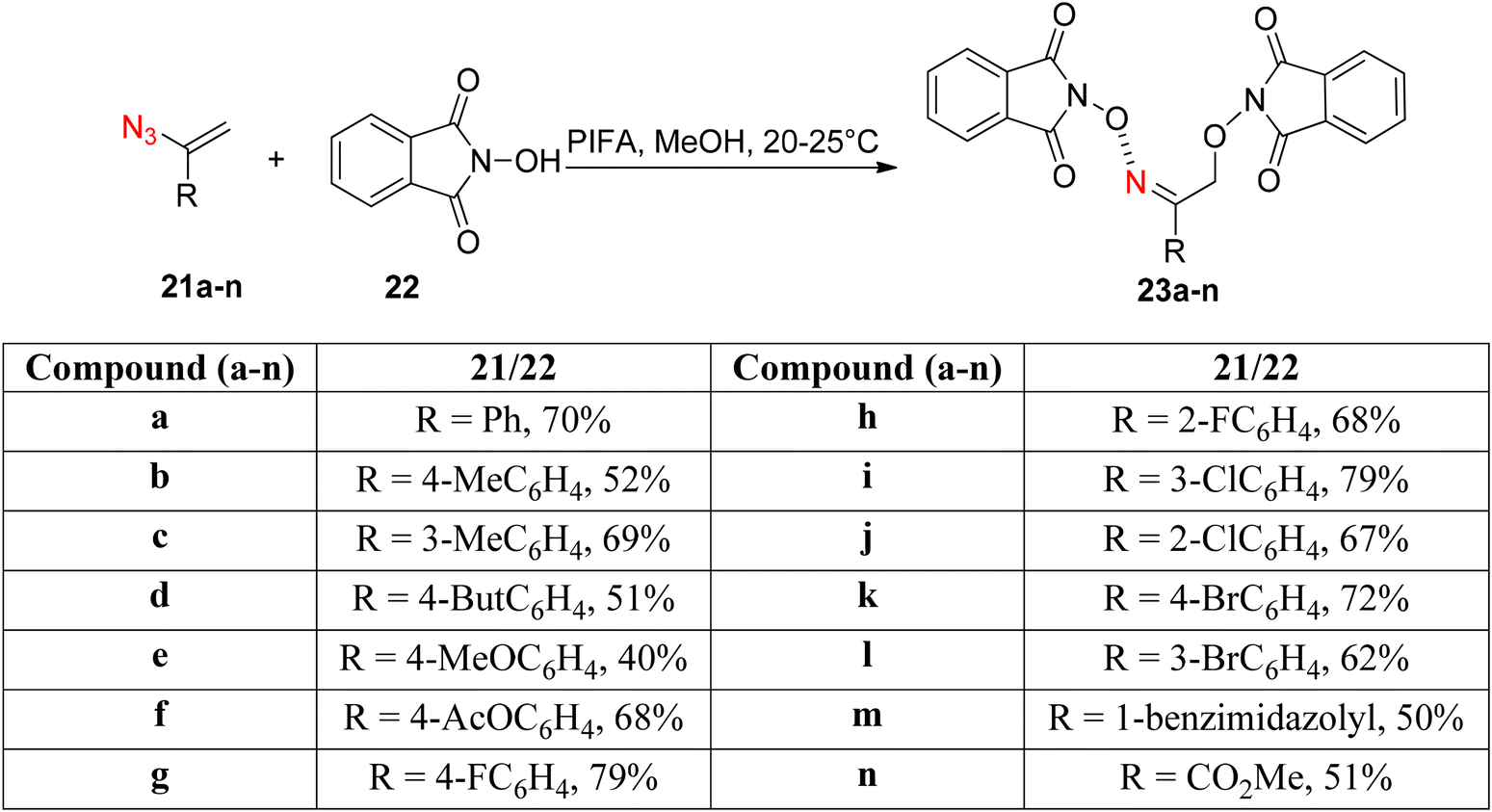
Paveliev et al.66 synthesized analogs of 2-(hydroxyamino)ethanols 23a–nvia a PIFA-catalyzed reaction between azides 21a–n and NHPI 22 (Scheme 7). PIFA actively engaged in the reaction by following a free radical pathway, specifically through hydrogen abstraction. The mild reaction conditions offer a broad substrate scope for vinyl azides. The reaction mechanism initiates with the formation of an N-oxyl radical through an oxidative pathway. Subsequently, it reacts with the vinyl azide. Finally, another N-oxyl radical molecule captures the resulting iminyl radical.
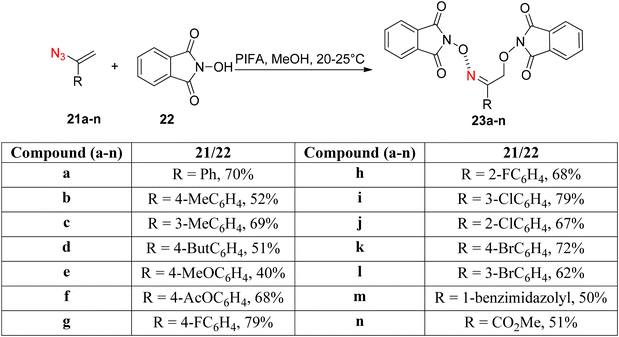 |
| | Scheme 7 Synthesis of analogues of 2-(hydroxyimino)ethanols. | |
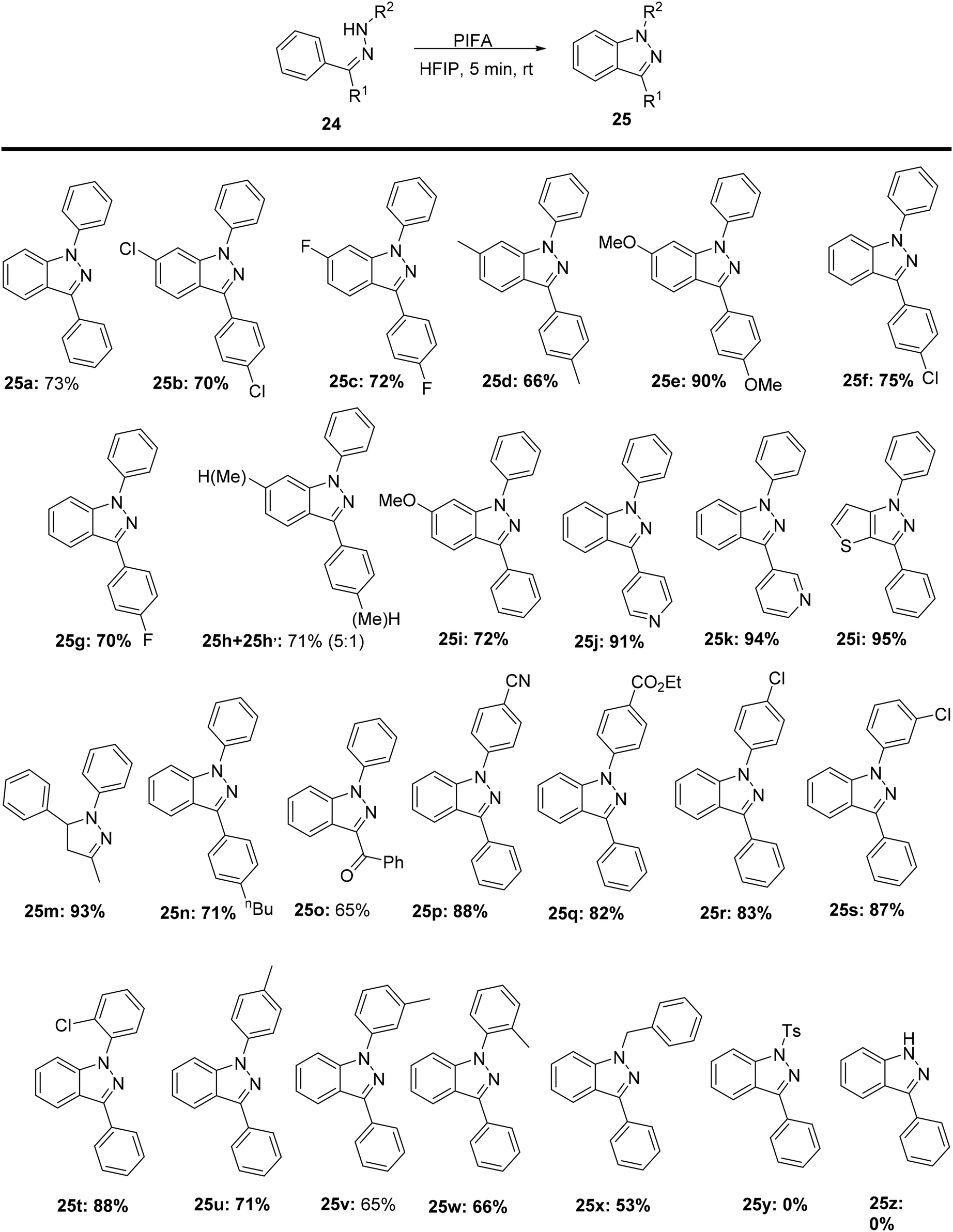
1H-Indazole scaffolds hold great importance as key components in biologically active compounds, synthetic medicines, and potential drug candidates.67,68 Zhang et al.69 reported a method for synthesizing substituted heterocycles 25 through a PIFA-mediated C–N coupling reaction, using readily available substrates 24 without the need for any metal catalyst (Scheme 8). PIFA was involved in the reaction via an ionic pathway. In addition, the cyclization of starting materials 24y bearing a tosyl group at the nitrogen atom and 24z did not react under the current optimal conditions. This reaction demonstrates excellent tolerance for various functional groups and substrates.
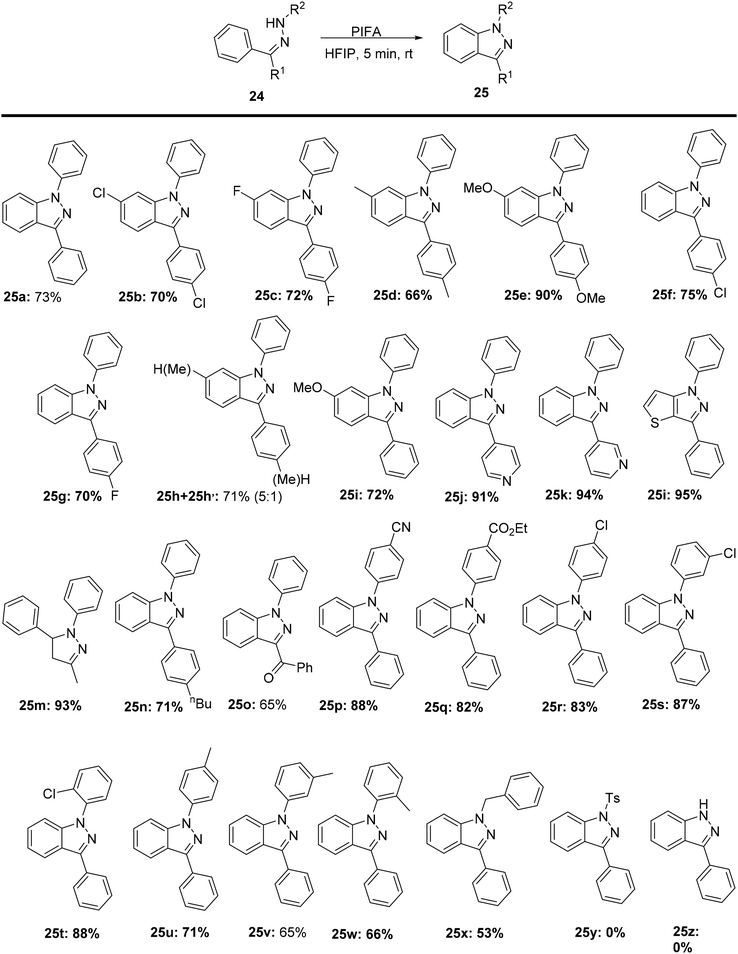 |
| | Scheme 8 Synthetic pathway for construction of 1H-indazole analogues. | |
3.3. C–H functionalization
In modern synthetic organic chemistry, the functionalization of sp3 C-atoms through C–H bonds is a significant methodology for accessing C–C, C–O, and C–N linkages.70
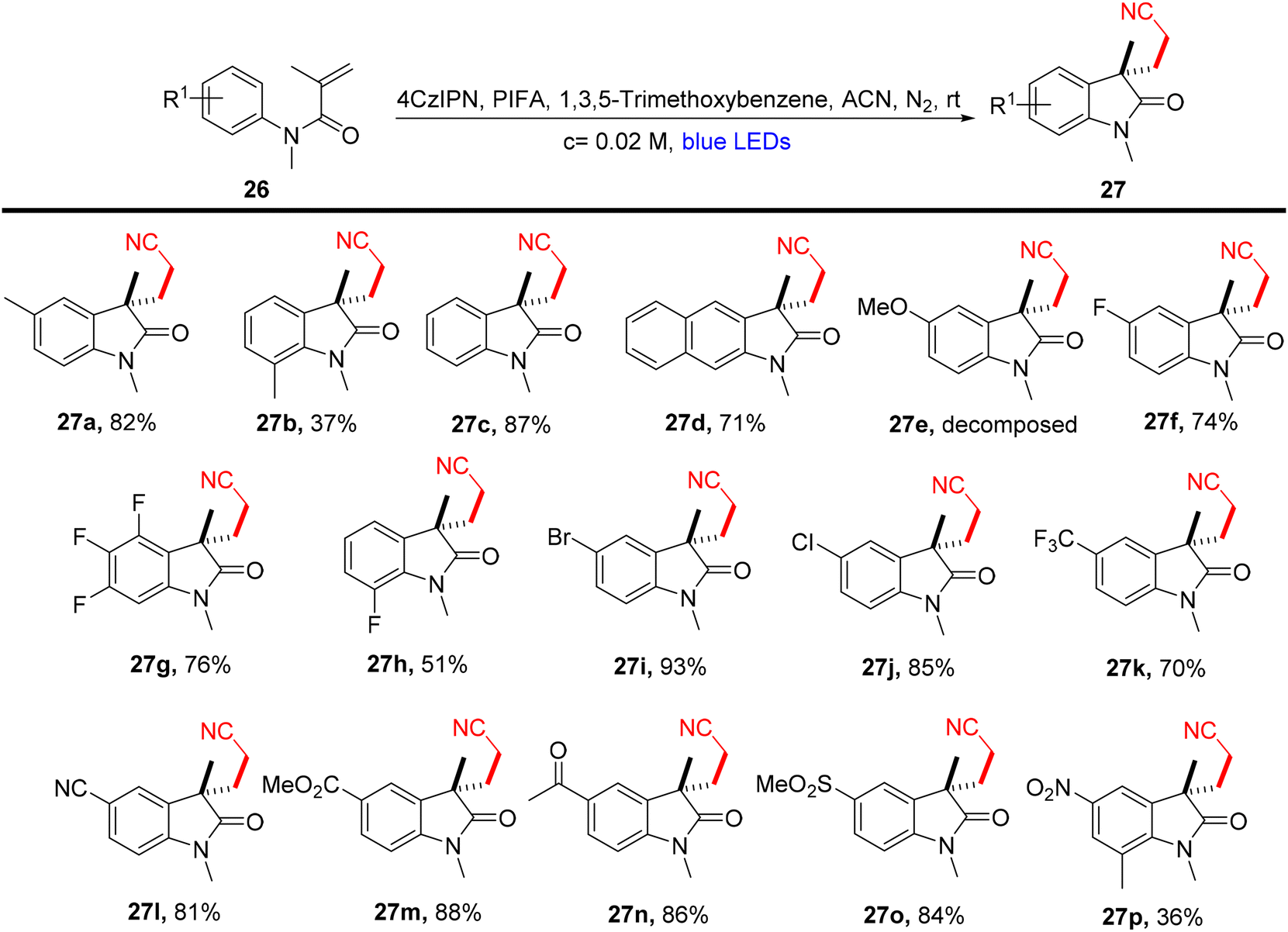
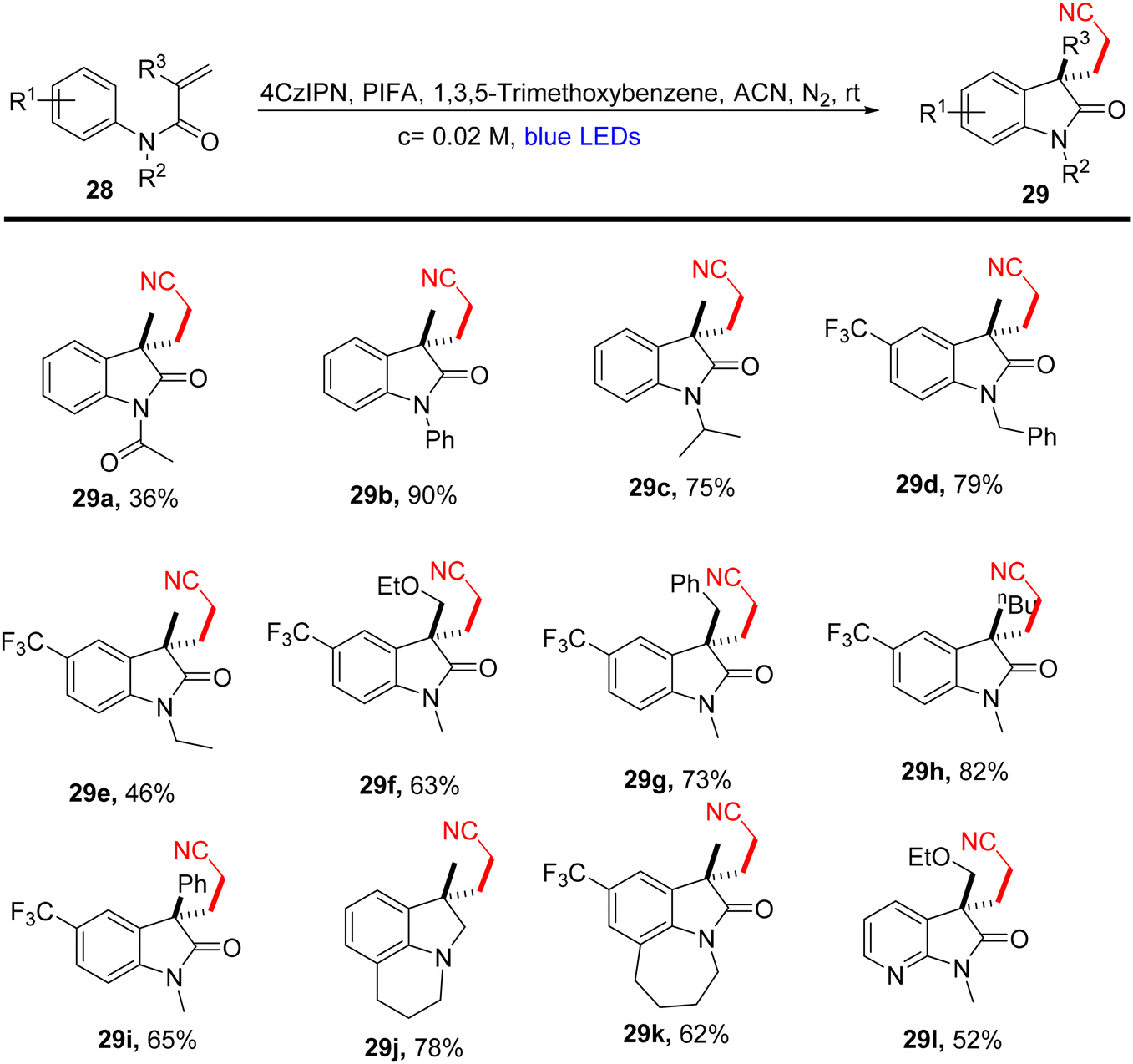
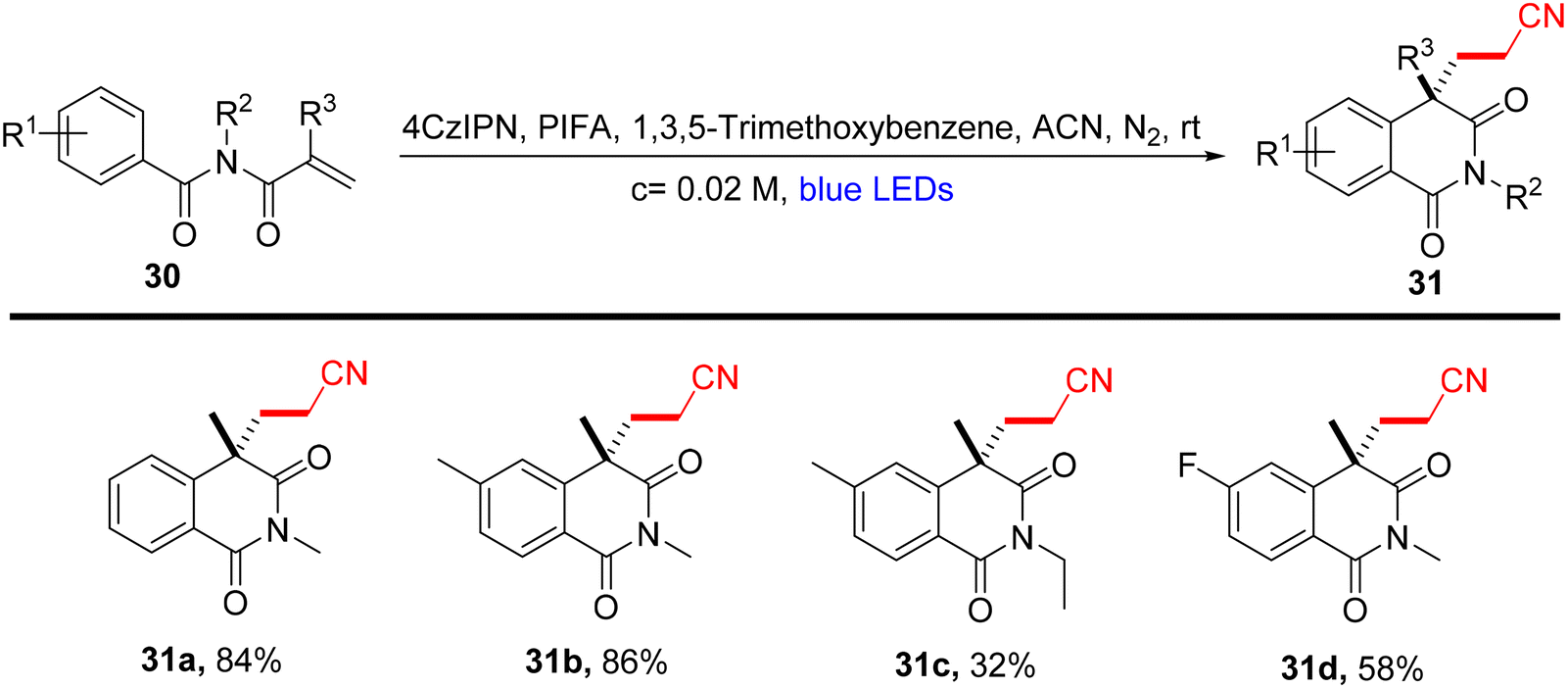
Lu et al.71 employed visible light-enabled photocatalysis in the presence of 4CzIPN and PIFA to perform a radical cascade oxidation of N-aryl/benzoyl acrylamides 26/28/30, yielding both oxindole 27/29 and isoquinolinedione 31 analogs in good yields. This reaction exhibited several noteworthy features, including a broad substrate scope, mild reaction conditions, and excellent tolerance for diverse functional groups (Scheme 9). PIFA actively engaged in the reaction by following a free radical pathway, specifically through single-electron oxidation. To expand the applicability of this photocatalytic pathway, the researchers investigated various substrates by modifying substituents at the R2/R3 positions, resulting in the facile synthesis of desired products 29a–l with high to excellent yields (Scheme 10). In addition, they delved into the synthesis of cyano-functionalized isoquinolinedione 31 (Scheme 11), a pivotal structural motif commonly encountered in pharmaceuticals and agrochemicals.72
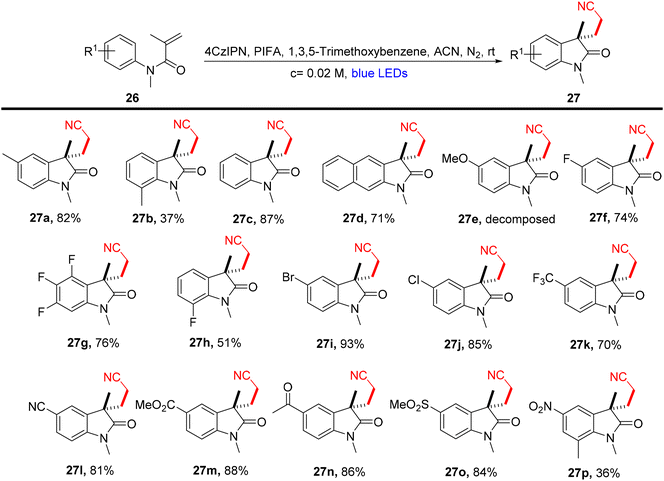 |
| | Scheme 9 Synthesis of functionalized oxindoles 27a–p. | |
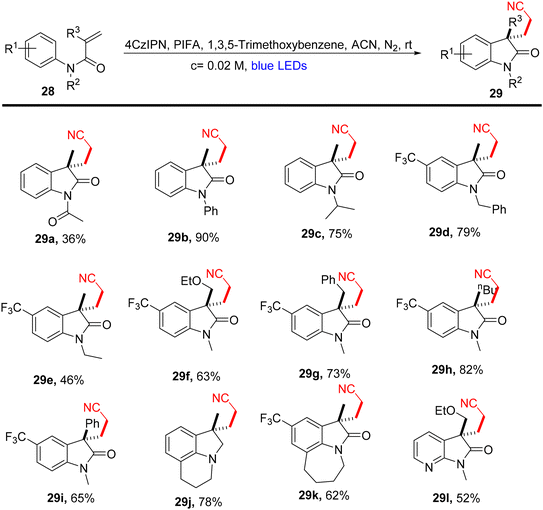 |
| | Scheme 10 Synthesis of functionalized oxindoles 29a–l. | |
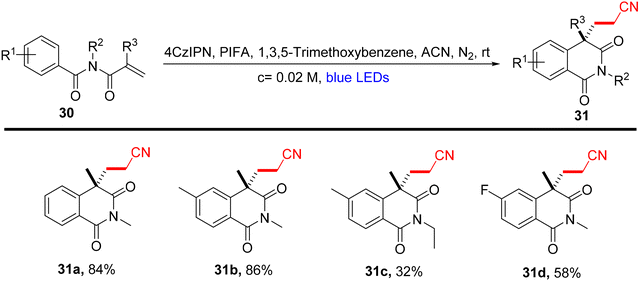 |
| | Scheme 11 Synthesis of isoquinolinediones 31a–d. | |
Organochalcogenides have consistently demonstrated substantial potential as prospective candidates for therapeutic applications, attributable to their inherent versatility as molecular scaffolds capable of accommodating an extensive array of functional groups.73 Furthermore, they find broader utility across diverse domains, including agrochemistry, catalysis, materials science, and organic synthesis.74
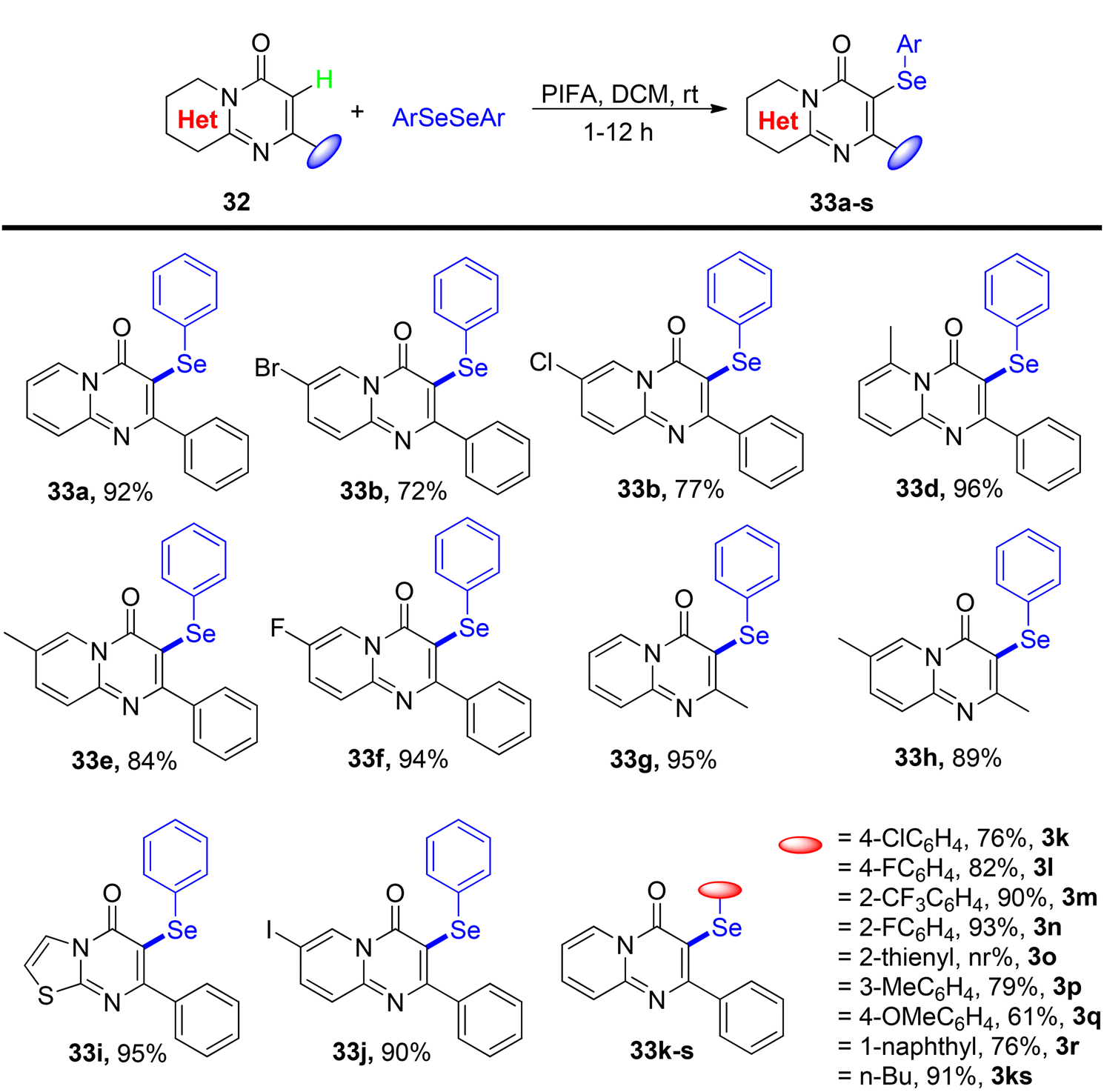
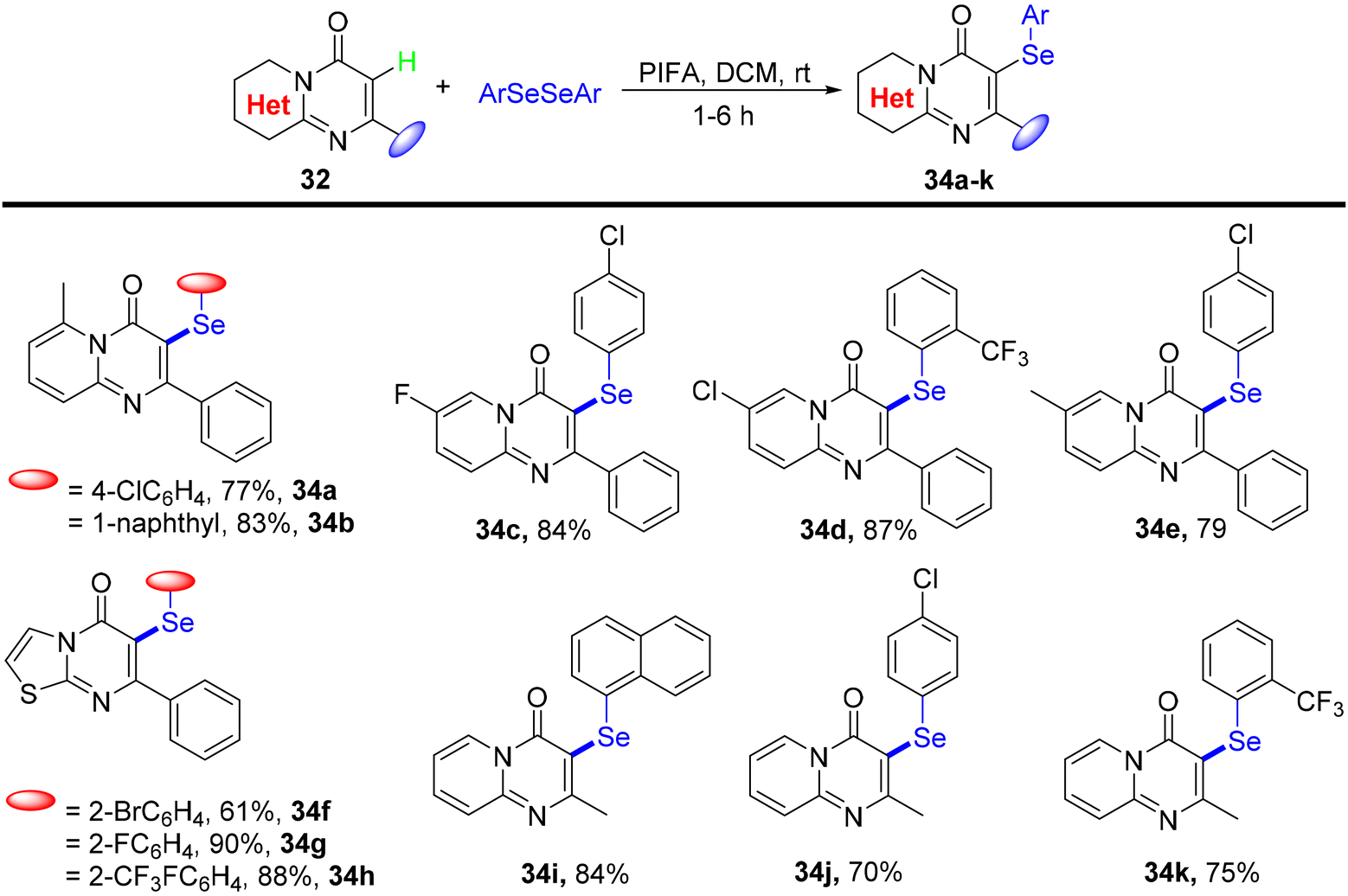
Ghosh et al.75 presented a more environmentally friendly approach employing PIFA (PhI(OCOCF3)2) for the C–H functionalization of pyrimidine-4-one 32, enabling the direct selenylation process to afford the desired products 33a–s in high yields, as depicted in Scheme 12. PIFA actively engaged in the reaction by following a free radical pathway, specifically through hydrogen abstraction, contributing to the process through this mechanism. Notably, this reaction exhibited remarkable tolerance towards a wide array of functional groups and structural variations, consistently delivering excellent yields of the target compounds. Furthermore, the applicability of the reaction scheme was extended to encompass various diaryl diselenides, as illustrated in Scheme 13.
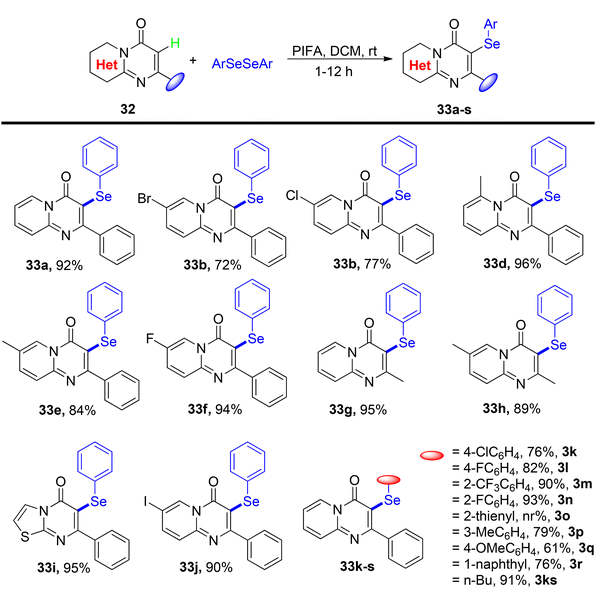 |
| | Scheme 12 Synthesis of selenylated-pyrimidin-4-ones 33a–s. | |
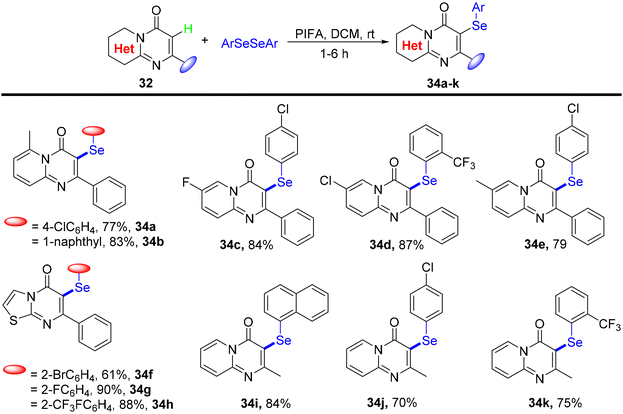 |
| | Scheme 13 Synthetic of selenylated-pyrimidin-4-ones 34a–k. | |
The structural components of benzothiazoles serve as essential constituents of various naturally occurring molecules, agricultural chemicals, synthetic compounds as well as pharmaceuticals.76C-2-Arylacylated benzothiazole derivatives are such compounds that have drawn considerable attention because of their therapeutic importance.
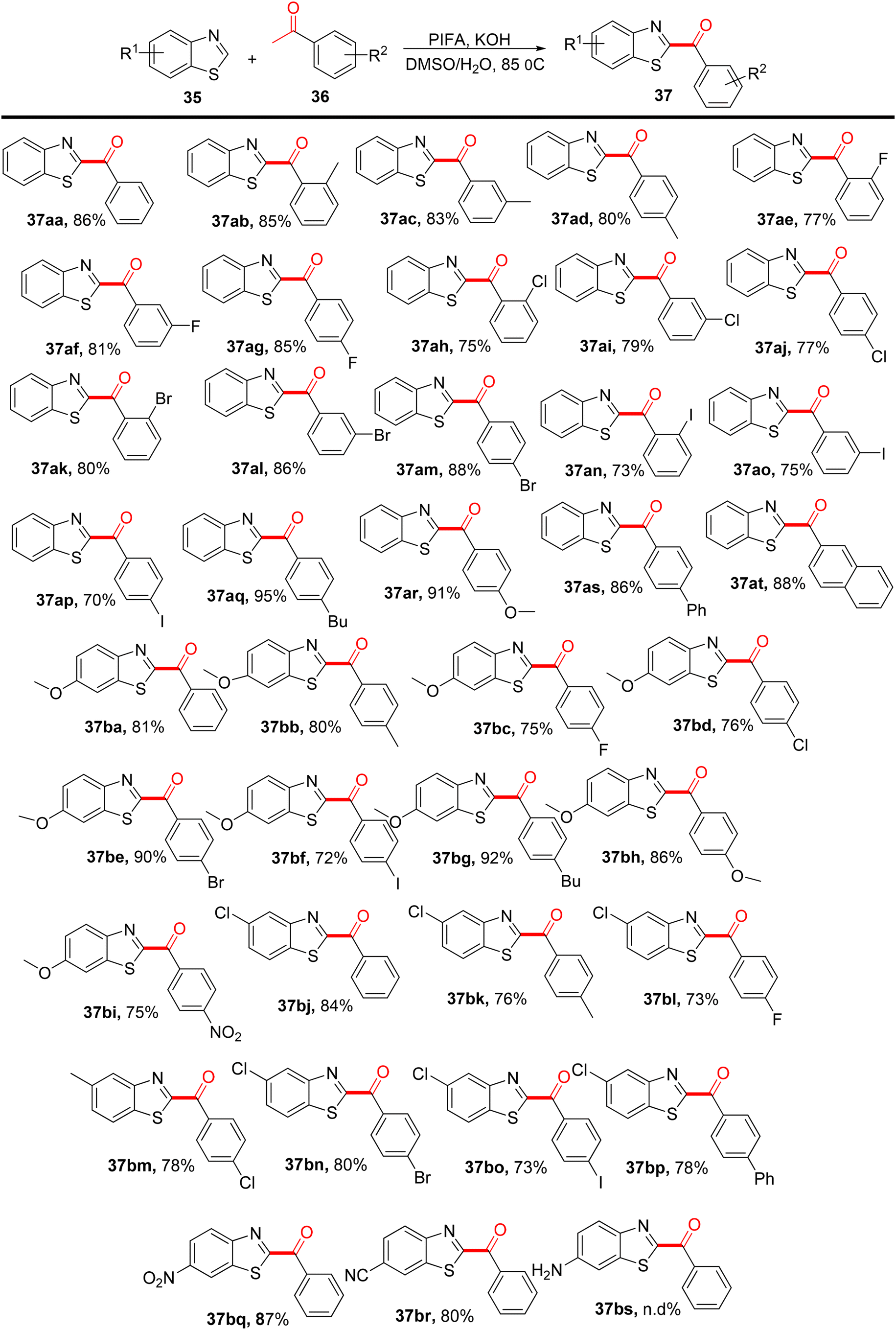
Sun et al.77 developed an efficient method using PIFA as a mediator to synthesize distinct C-2-arylacylated benzothiazoles 37 with yields ranging from 70% to 95%. These compounds show promise as potential therapeutic scaffolds and are obtained through a ring-opening reaction of 2H-benzothiazoles 35 (Scheme 14). PIFA actively engaged in the reaction through a free radical pathway, involving hydrogen abstraction as a mechanism for its participation in the process. This protocol is versatile, accommodating various substrates under favorable conditions, offering a faster and simpler route compared with prior methods for producing C-2-arylacylated benzothiazoles 37.
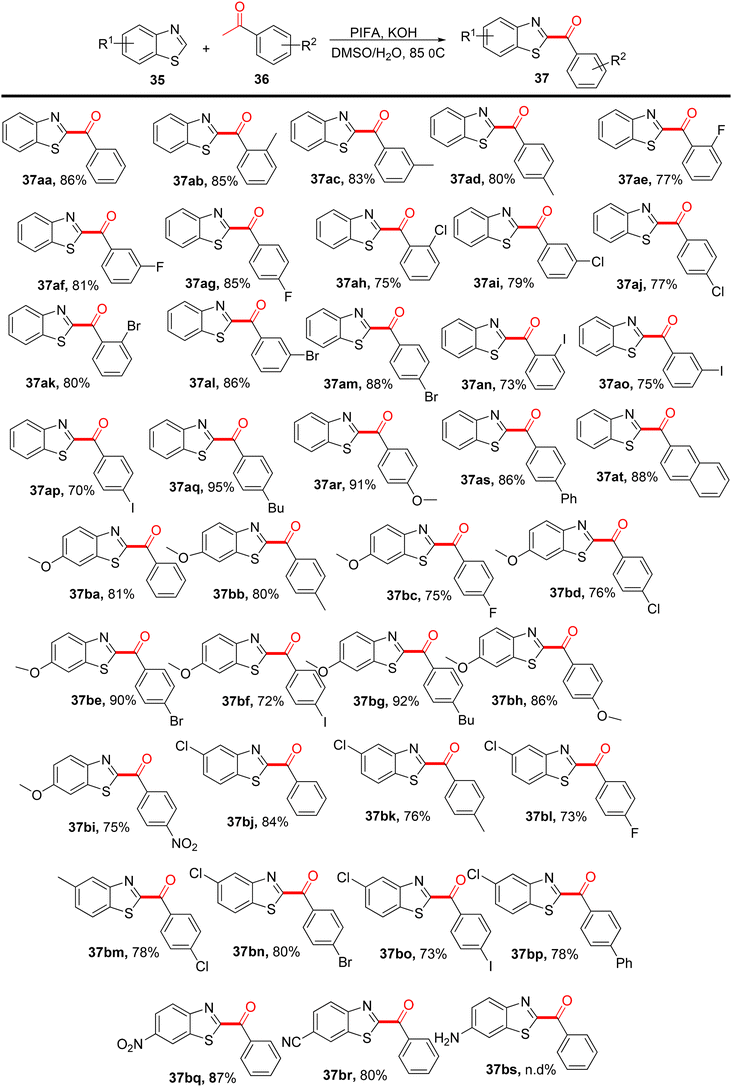 |
| | Scheme 14 Synthesis of C-2-arylacylated benzothiazoles 37. | |
Numerous methodologies have been documented for converting alkylbenzenes into their respective benzyl esters. However, a significant portion of the recently reported esterification processes involving benzylic C–H bonds necessitate the use of excessive acid or elevated temperatures (>60 °C).78
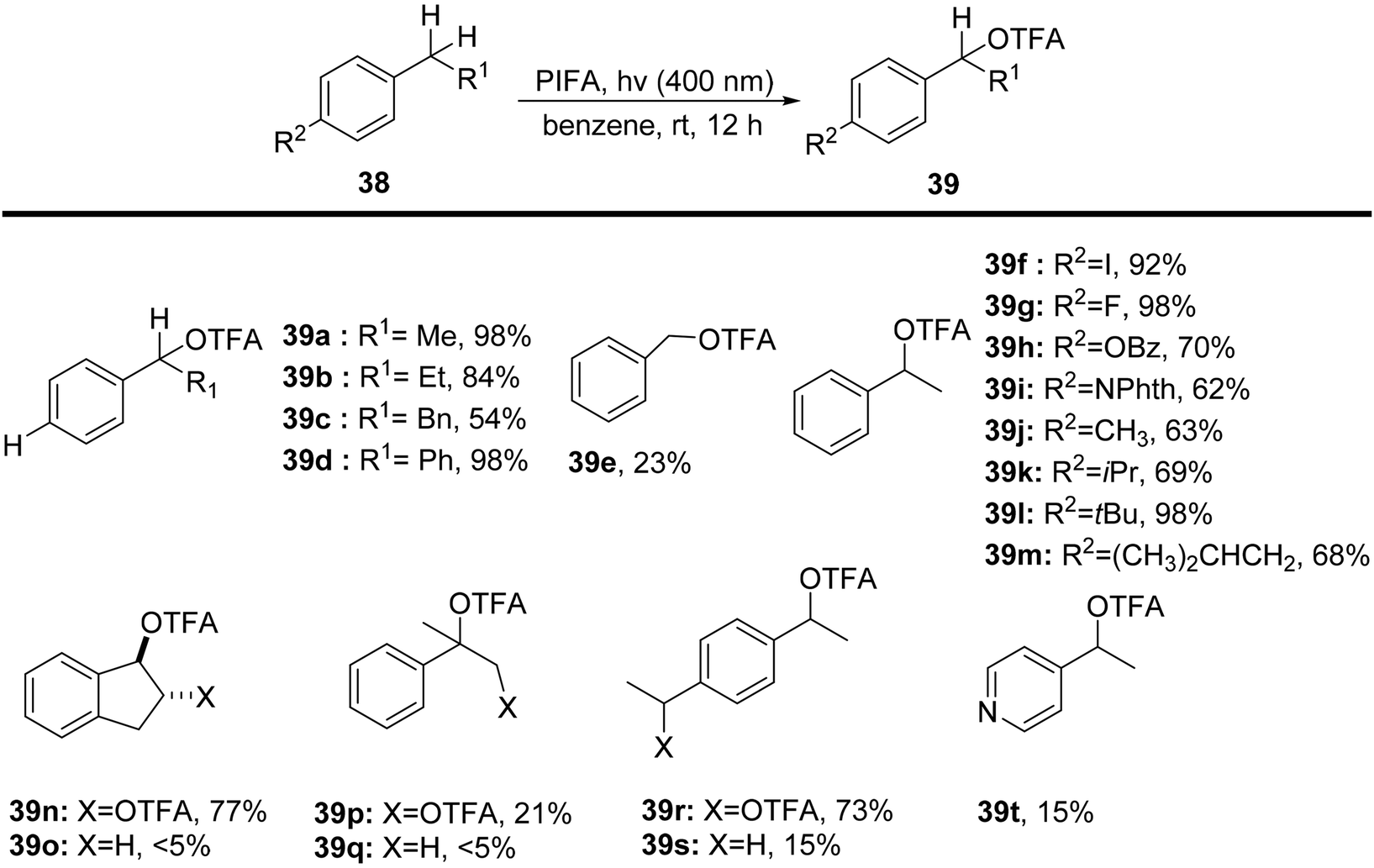
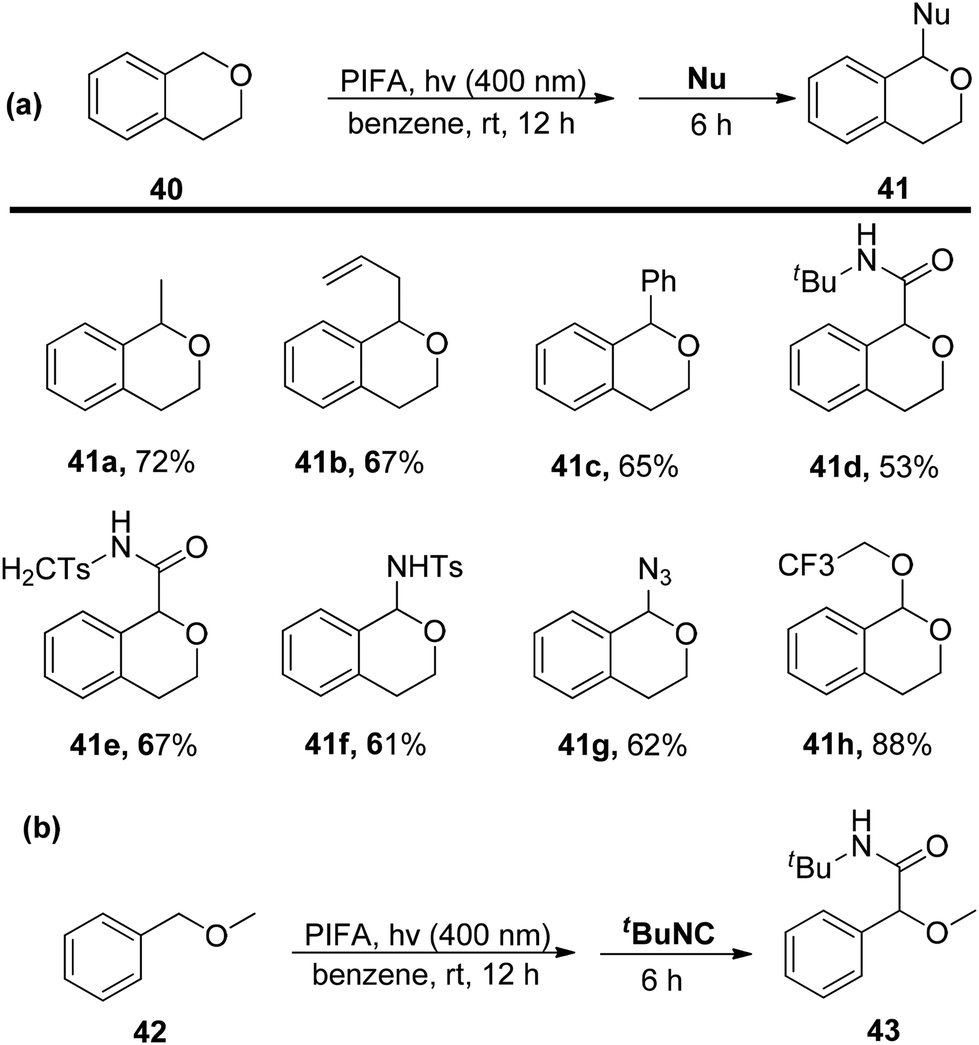
Sakamoto et al.79 accomplished the esterification of alkylbenzenes through a combination of PIFA catalysis and visible light irradiation. This methodology yielded favorable results, providing high yields of desired benzyl ester derivatives 39 from various alkylbenzenes 38 (Scheme 15). PIFA actively engaged in the reaction through a free radical pathway, involving hydrogen abstraction as a mechanism for its participation in the process. The versatility of this approach was demonstrated through an exploration of its substrate scope, as illustrated in Schemes 15 and 16a. Furthermore, the acyclic analogue 42 underwent a straightforward reaction, resulting in the production of α-hydroxy carbonyl compound 43 with a yield of 54% (Scheme 16b).
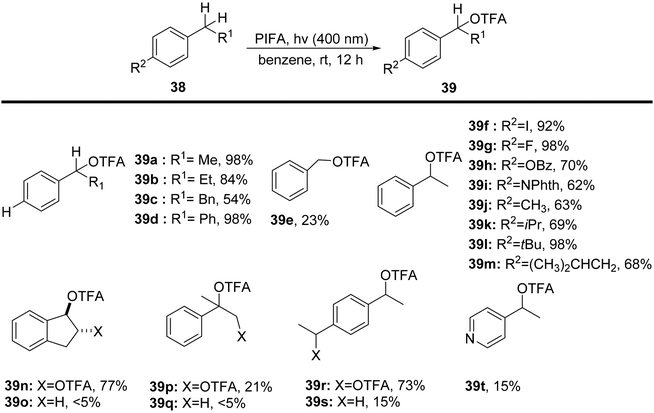 |
| | Scheme 15 C–H functionalization of alkylbenzenes in mild conditions using PIFA. | |
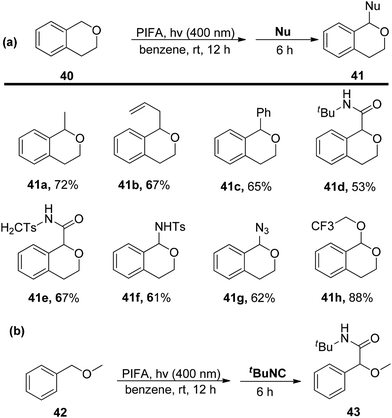 |
| | Scheme 16 C–H functionalization of alkylbenzenes in mild conditions using PIFA. | |
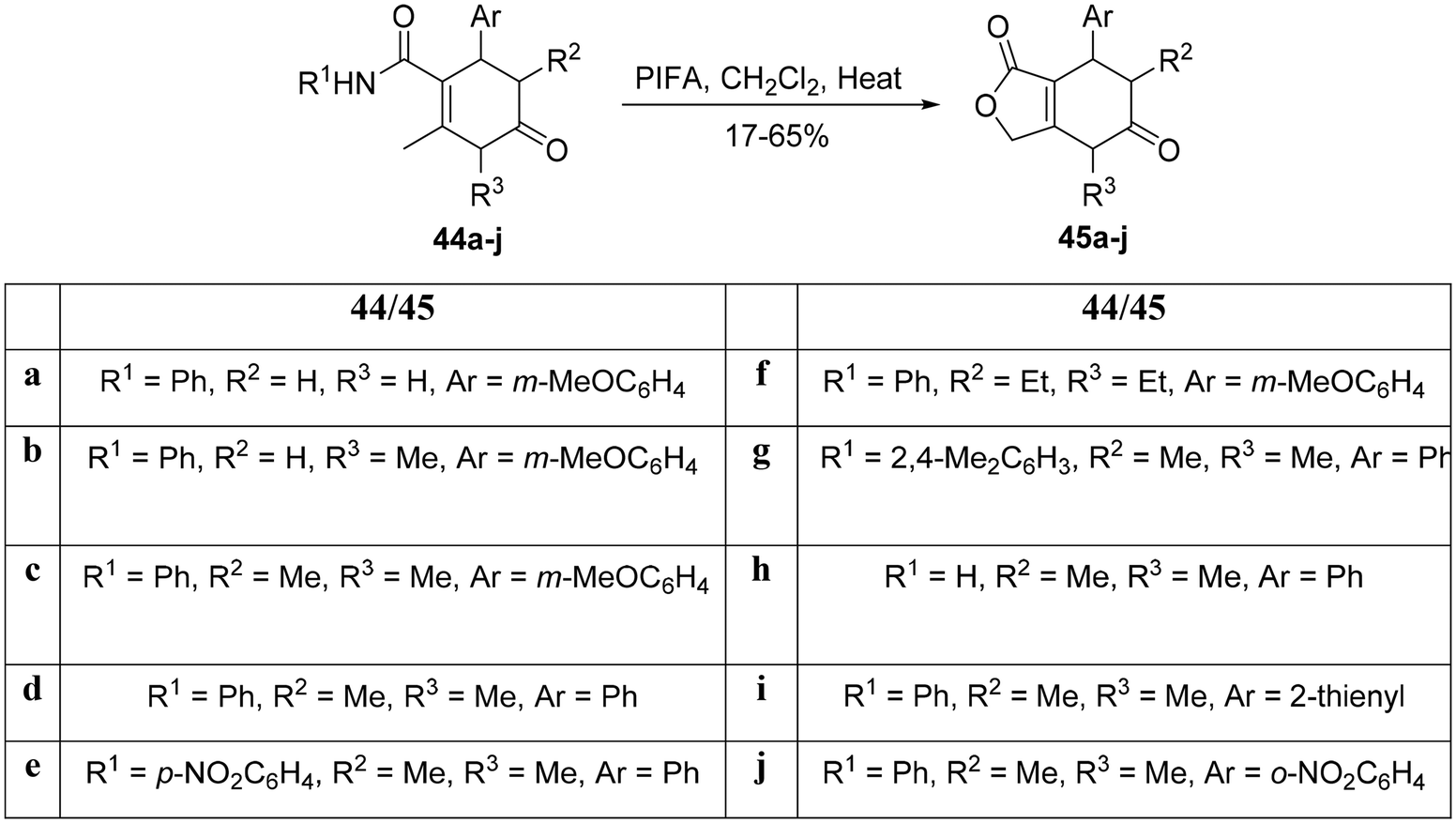
As reported by Couto et al.,80 the fused lactone was synthesized via a metal-free C–H activation reaction. Subsequently, the adducts 44a–j underwent a transformation into the corresponding pyrimidinones 45a–j through an intramolecular reaction mediated by PIFA (Scheme 17). PIFA was involved in the reaction via an ionic pathway, actively participating in the process through its contribution to the ionic mechanism.
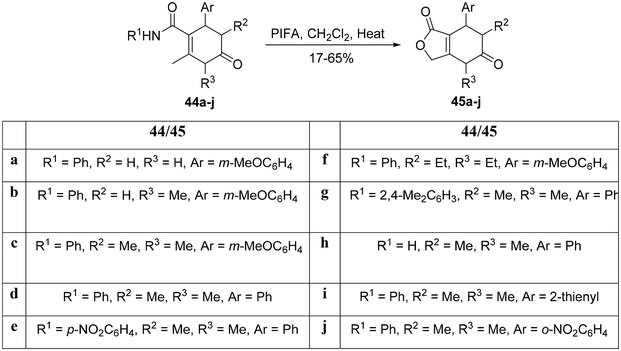 |
| | Scheme 17 Synthetic scheme for pyrimidine-2,5-dione analogues 45a–j. | |
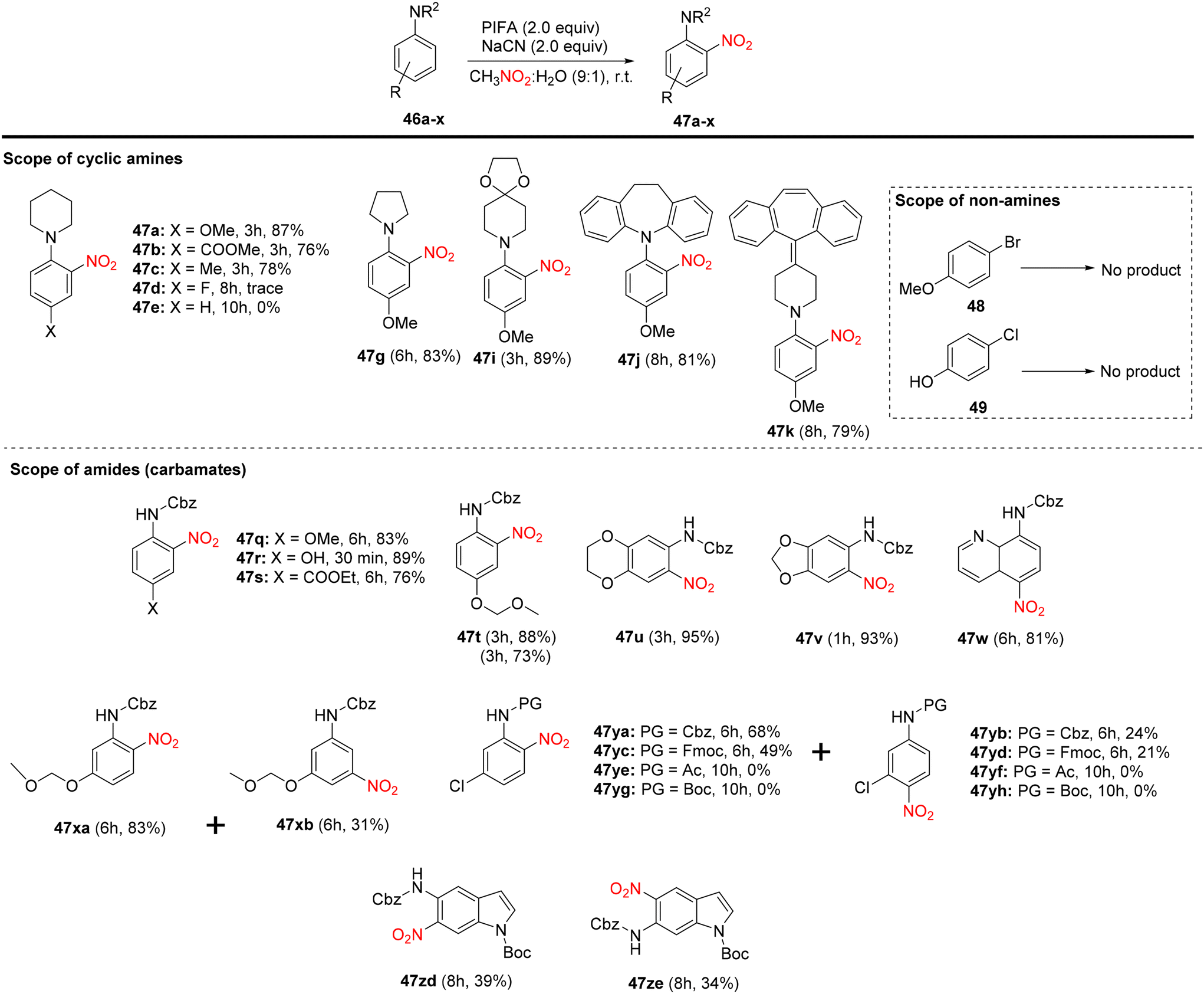
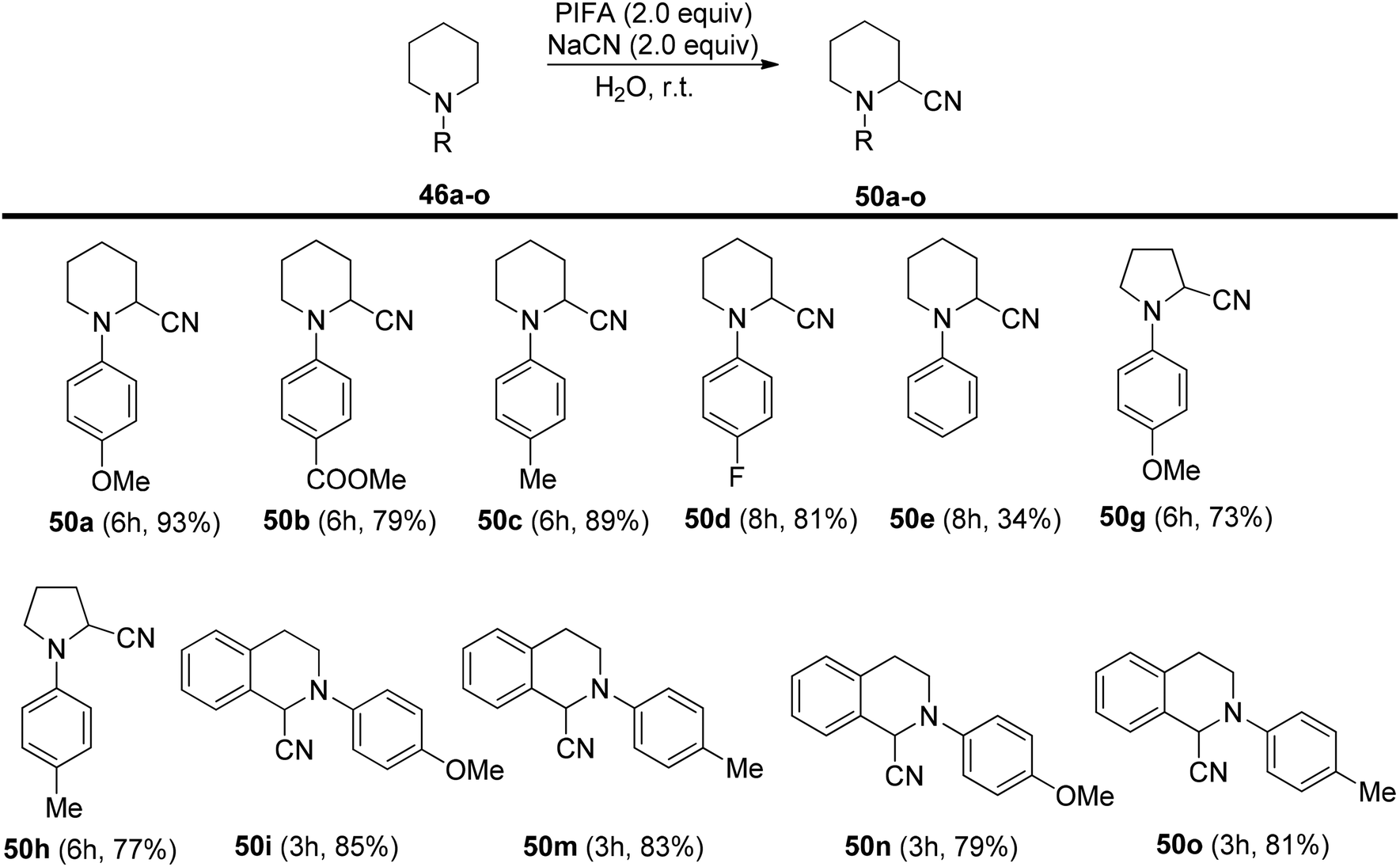
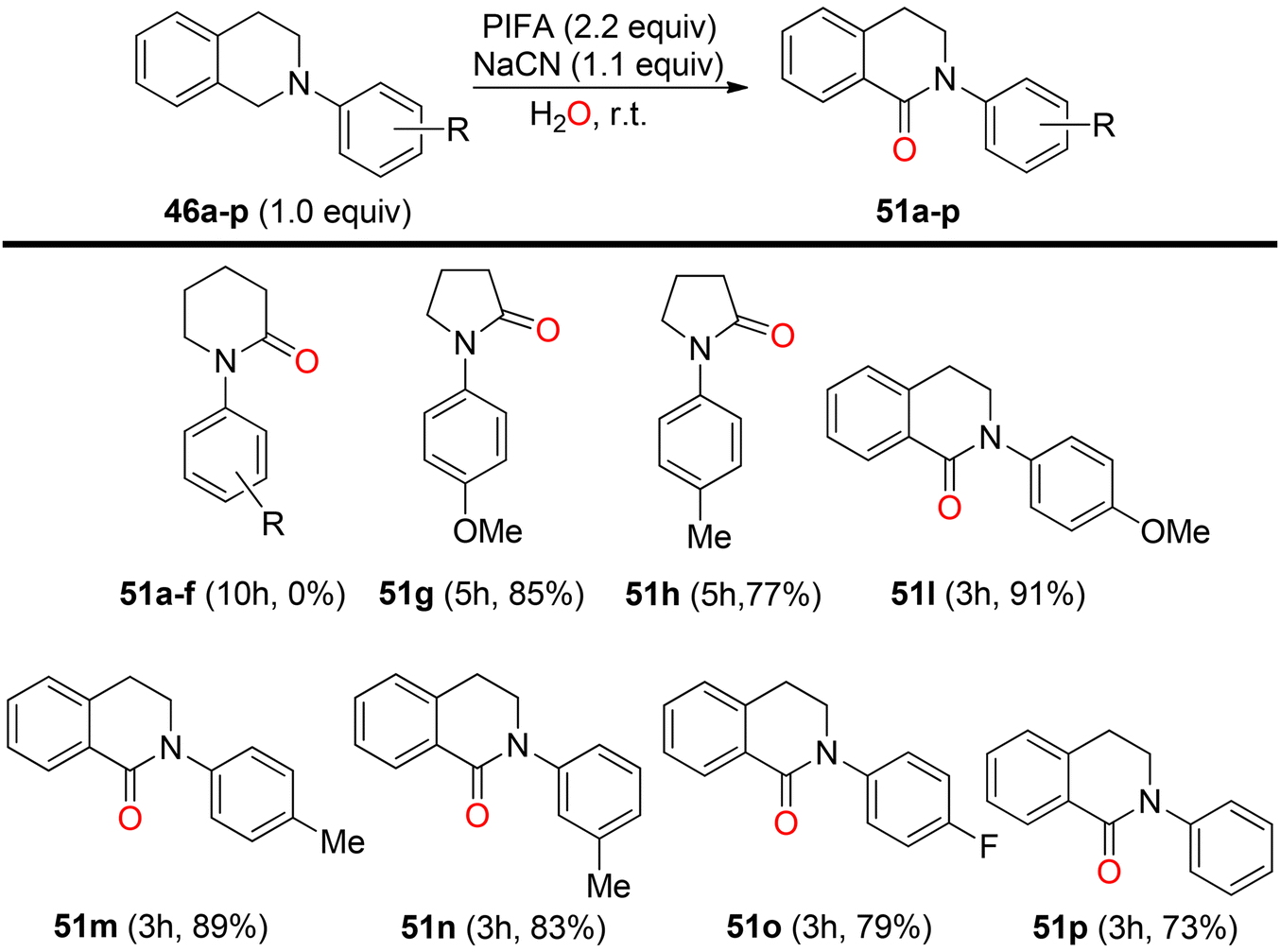
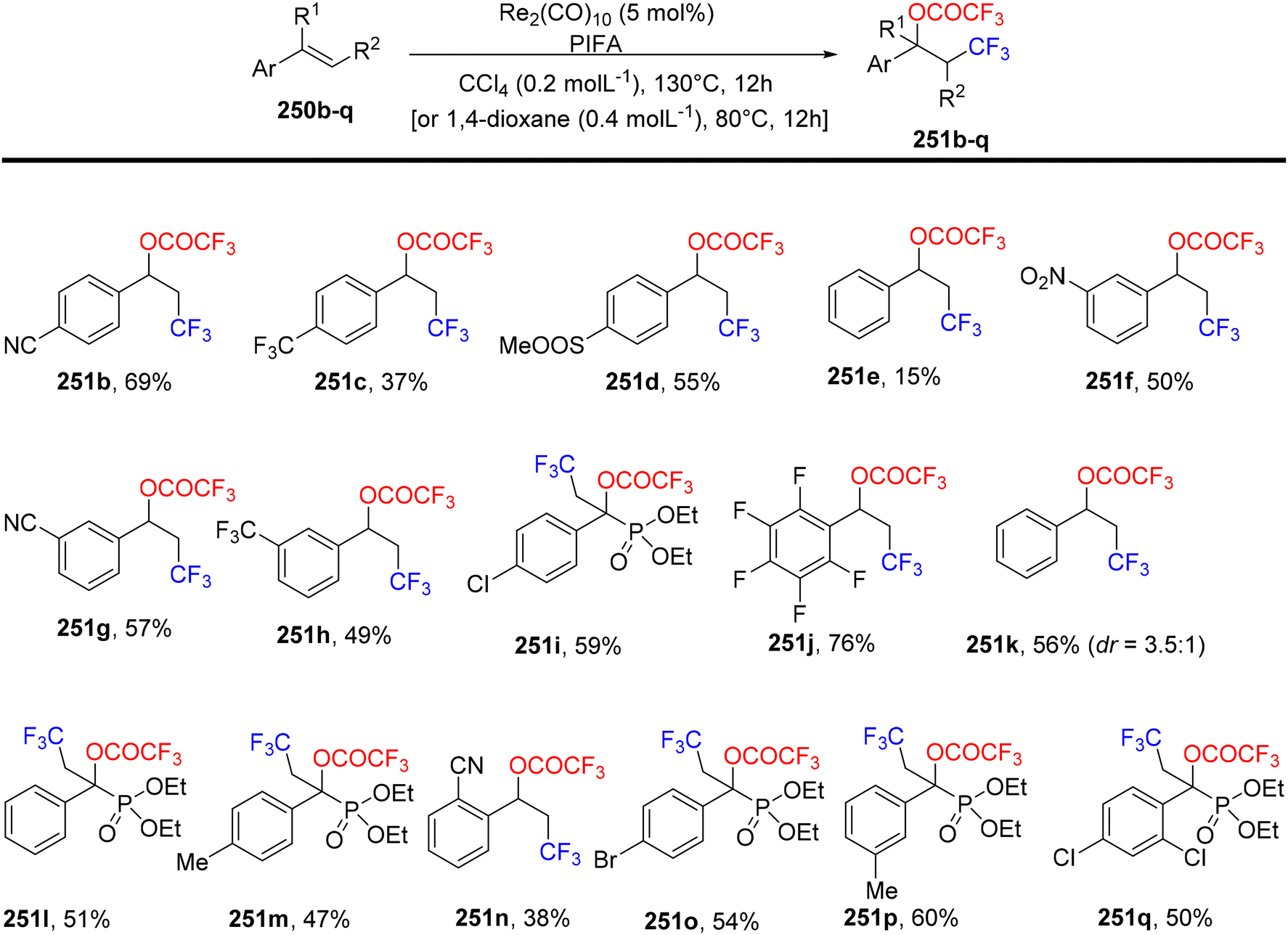
Mudithanapelli et al.81 introduced a PIFA-catalyzed, metal-free/NaCN reaction capable of effecting oxygenation, cyanation, and nitration through sp3 and sp2 C–H activation processes, respectively. This versatile methodology was successfully applied to a range of amine derivatives 46, as outlined in Scheme 18. Under the influence of CH3NO2/H2O (9:1), all conversions were efficiently catalyzed by PIFA/NaCN (Scheme 18). Notably, in cyanation complete conversion of substrates 46a–o to the desired products 50a–o was achieved (Scheme 19). The oxidized compounds 51a–p were readily obtained from cyclic amines 46a–p in yields ranging from 73% to 91% (Scheme 20). However, it is worth noting that none of the oxygenation products 51a–f were obtained when 6-membered amines were employed as substrates. This methodology stands out due to the cost-effectiveness and compatibility of PIFA as an oxidant. Remarkably, a single catalytic system, namely PIFA/NaCN, proved highly effective in facilitating three distinct types of functionalizations. PIFA was involved in the reaction via an ionic pathway.
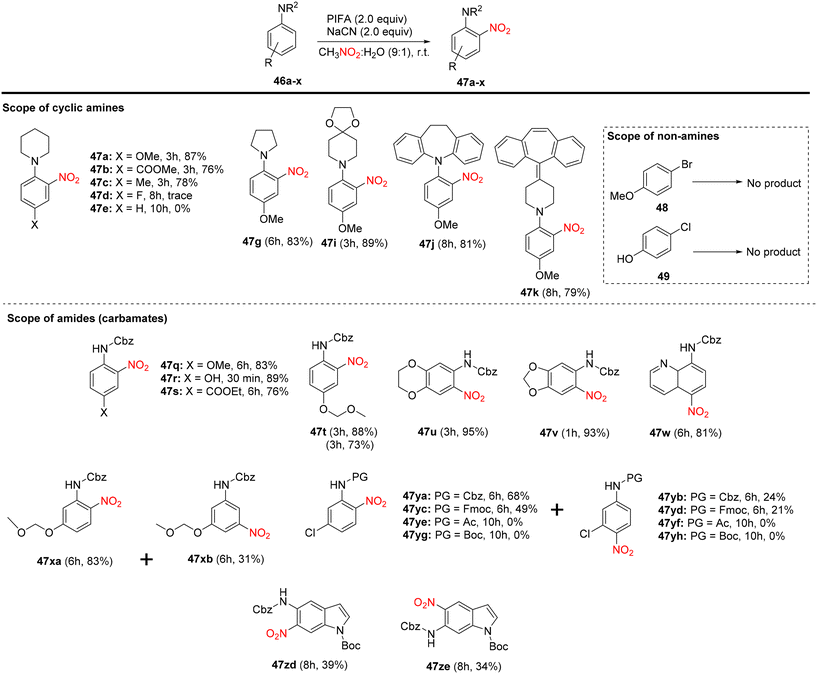 |
| | Scheme 18 Variation of substrates in nitration reaction. | |
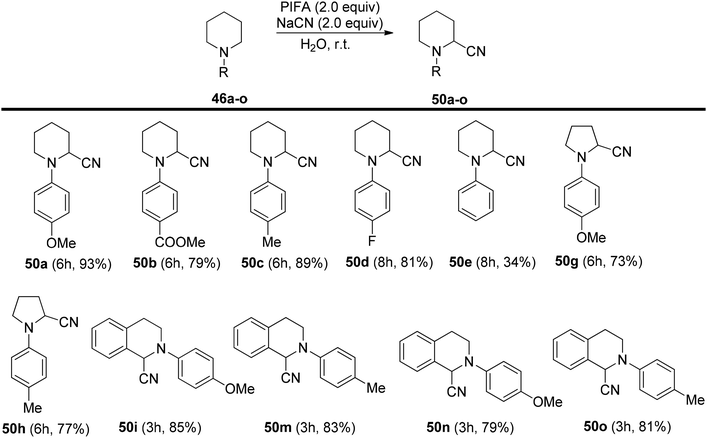 |
| | Scheme 19 Scope of cyanation reaction. | |
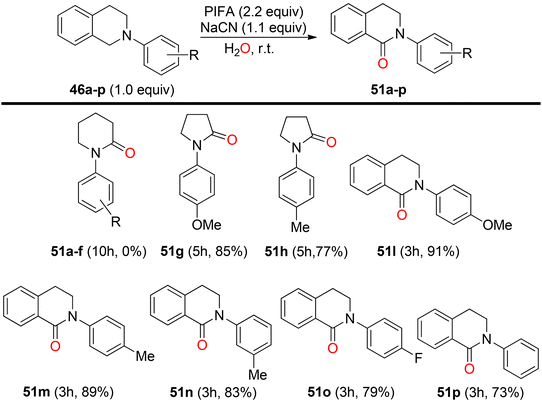 |
| | Scheme 20 Oxygenation reaction with varied substrates. | |
3.4. Coupling reaction
Owing to their myriad applications in sensors, photosynthetic systems, and nonlinear optical (NLO) devices, porphyrin and chlorin derivatives have garnered significant scientific attention.82
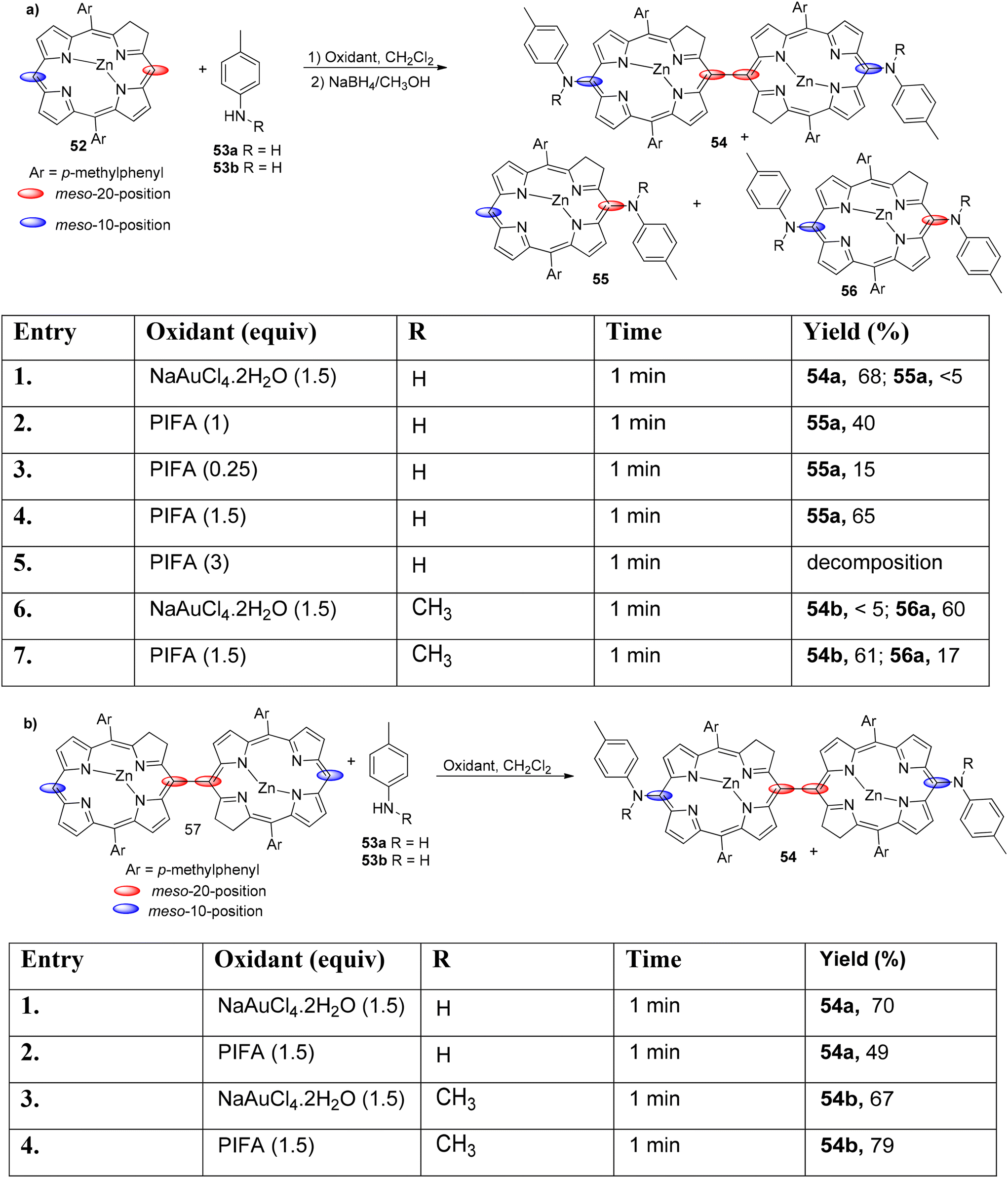
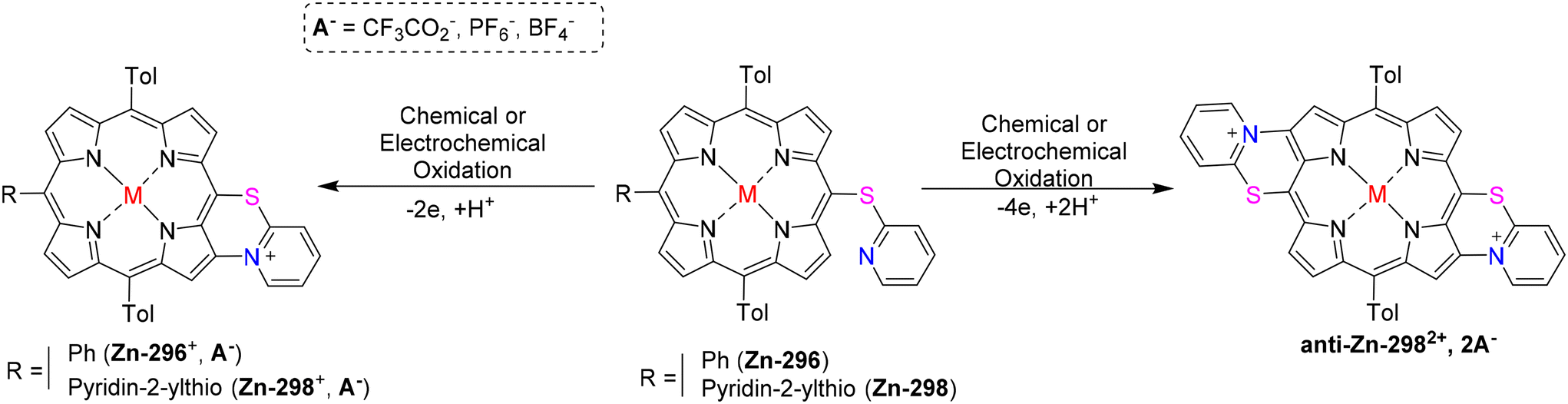
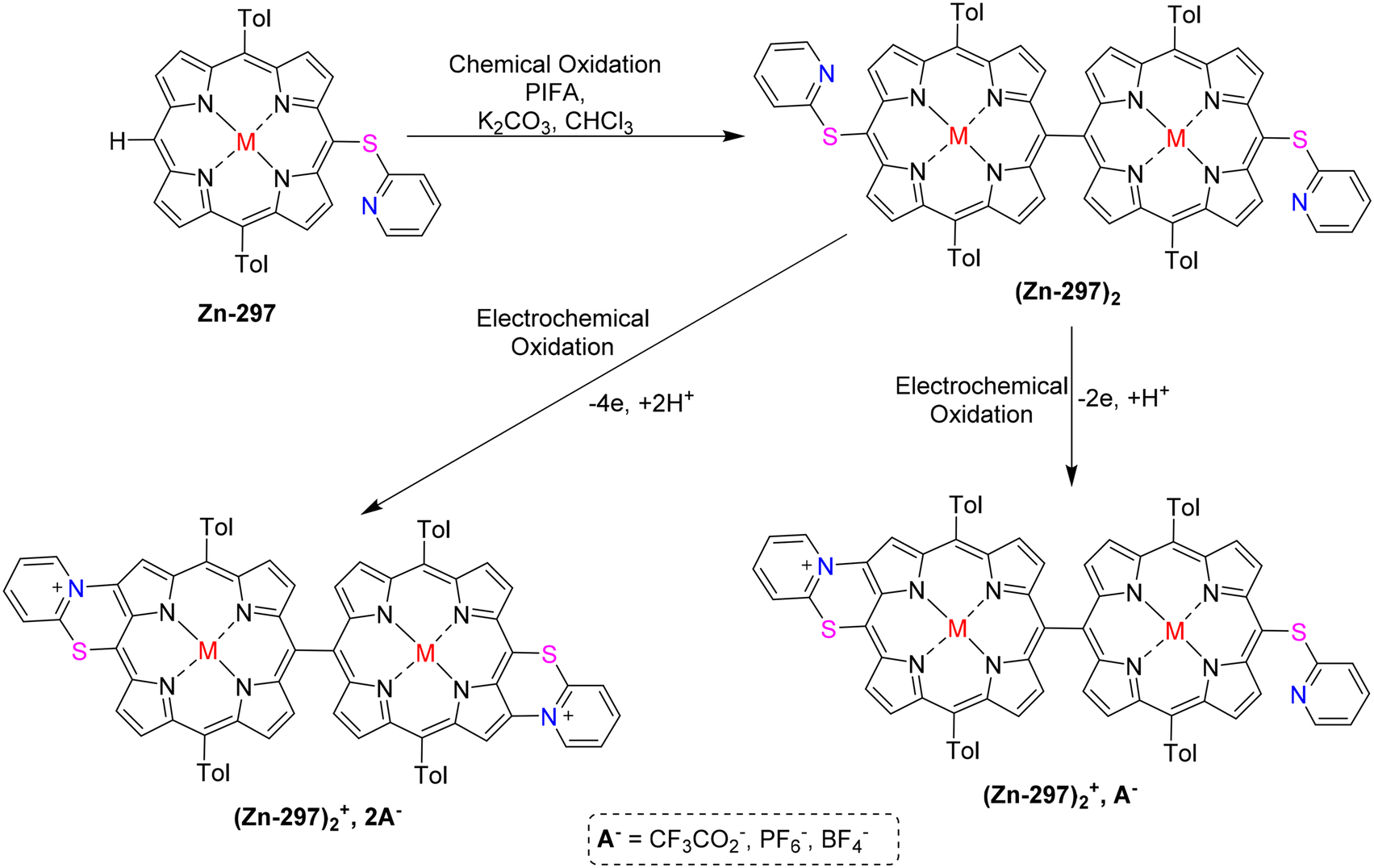
Cheng et al.83 used different oxidants to study the interactions between anilines 53 and chlorins 52. Aminated compounds were obtained by performing the oxidation reaction of anilines as well as chlorins in presence of PIFA. NaAuCl4·2H2O was used to successfully carry out the amination reaction (Scheme 21). It is interesting to note that when monomeric Zn(II) chlorin was treated with p-toluidine in the presence of NaAuCl4·2H2O and PIFA as the catalyst, diaminated chlorin dimers 54 were obtained (Scheme 21). Moreover, the inactive 10th position also undergoes amination reaction. Further, these chlorin analogues can be developed to function as photosensitizers.
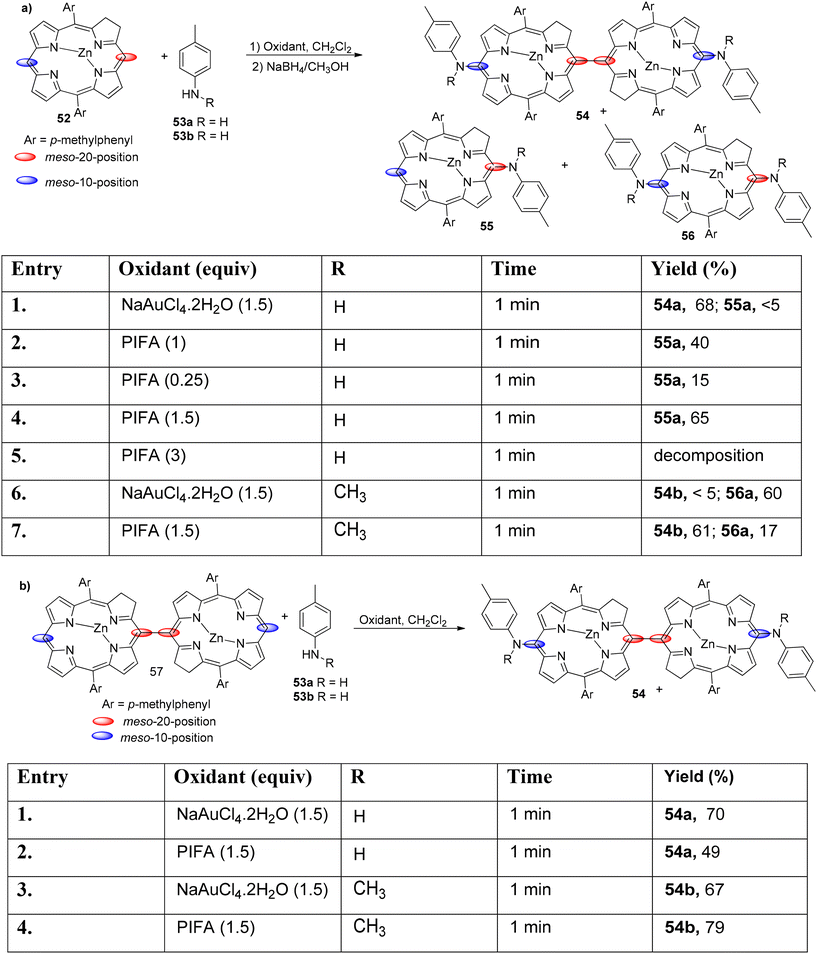 |
| | Scheme 21 Amination reaction of chlorins as well as N-unsubstituted anilines. | |
Nitriles represent a notable class of functional groups in organic synthesis, typically serving as precursors for the conversion into various other functional groups, including amines, amides, carboxylic analogs, heterocyclic compounds, and aldehydic moieties.84
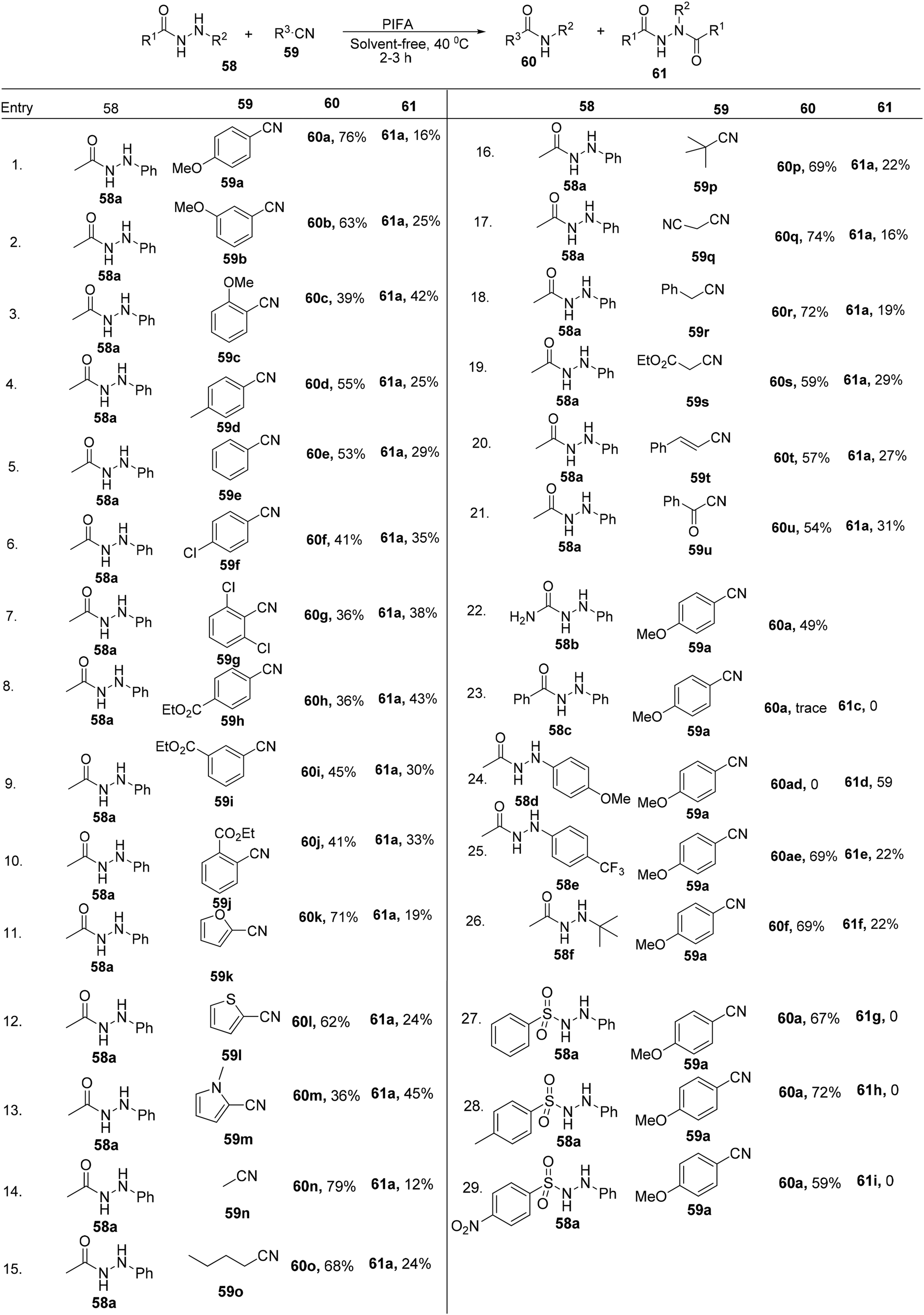
Yan et al.85 effectively converted nitriles 59 into their respective N-phenyl amides 60 through a PIFA-catalyzed intermolecular cross-coupling mechanism conducted in the absence of any solvent. PIFA actively engaged in the reaction by following a free radical pathway, specifically through single-electron oxidation, contributing to the process through this mechanism. This method represents a mild oxidative reaction scheme for achieving high yields of N-phenyl amides 60. It encompasses the cleavage of sp2 C–N linkages concomitant with the formation of new C–N bonds, as illustrated in Scheme 22.
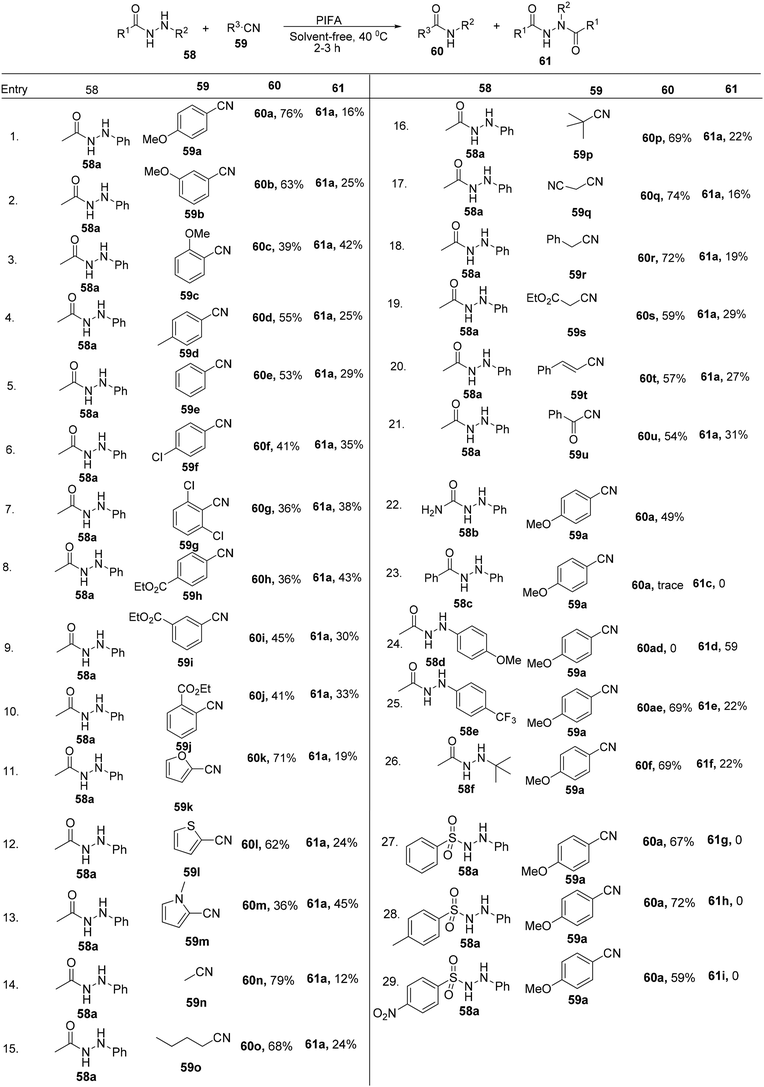 |
| | Scheme 22 Synthesis of N-phenyl amides. | |
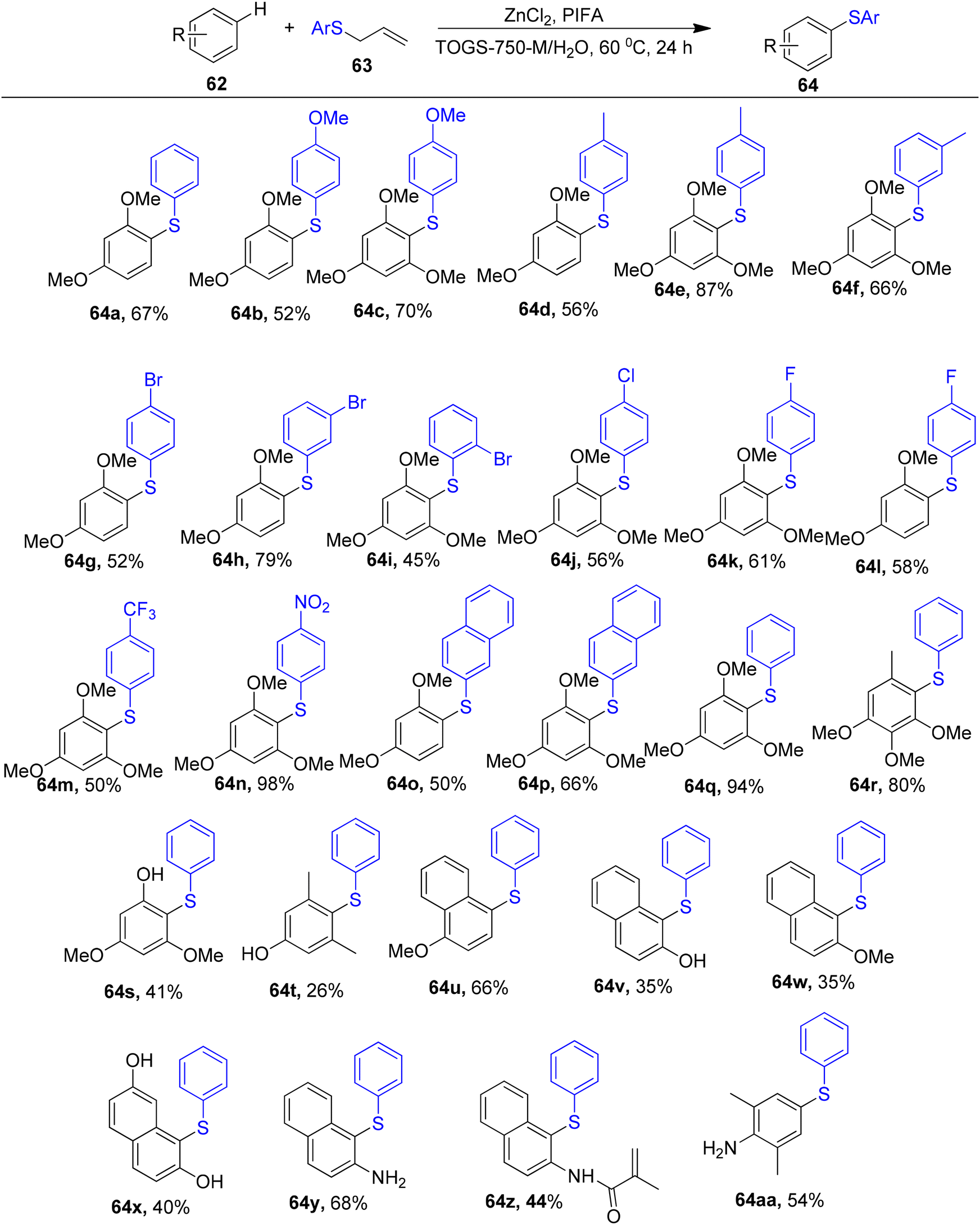
By C–H functionalization of electron-rich aromatic hydrocarbons in water and further coupling with an equal stoichiometry of allyl sulphides, Feng et al.86 described a methodology to obtain aryl sulphides 64. The Lipshutz surfactant, TPGS-750-M, which utilizes water as the recyclable reaction media, facilitates this arylthiolation. Varied allyl aryl sulphides were useful cross-coupling partners for m-dimethoxybenzene or 1,3,5-trimethoxybenzene 62 in the case of allyl sulphides (Scheme 23). Allyl phenyl sulphide 63 was successfully coupled with diverse electron-rich arenes 62. The fact that this reaction took place in an aqueous medium rather than an organic one and allowed for the recycling of water, surfactant and catalyst is perhaps its most notable feature. PIFA actively engaged in the reaction by following a radical pathway, specifically through single-electron oxidation.
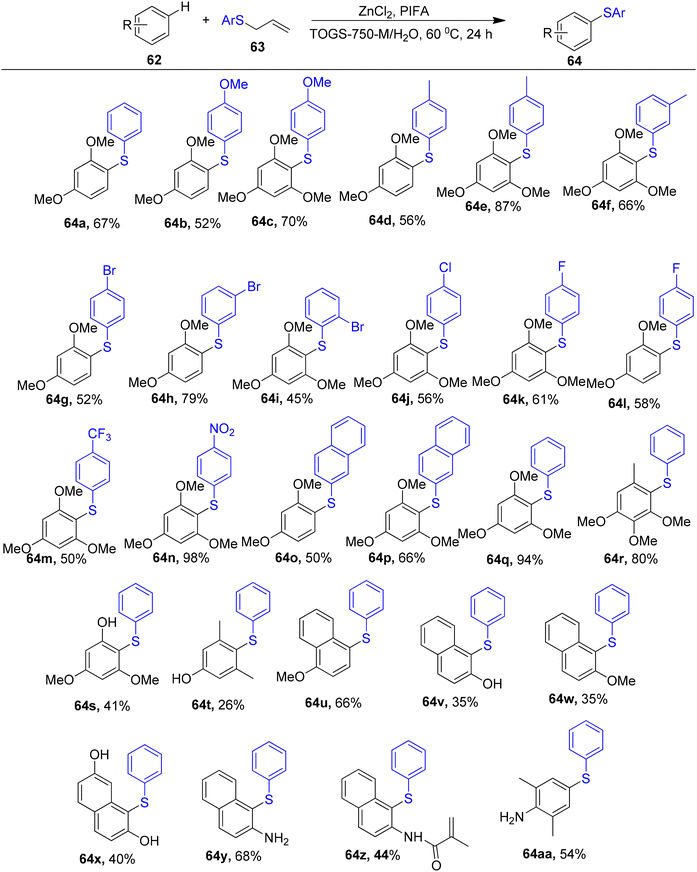 |
| | Scheme 23 Synthesis of aryl sulfides. | |
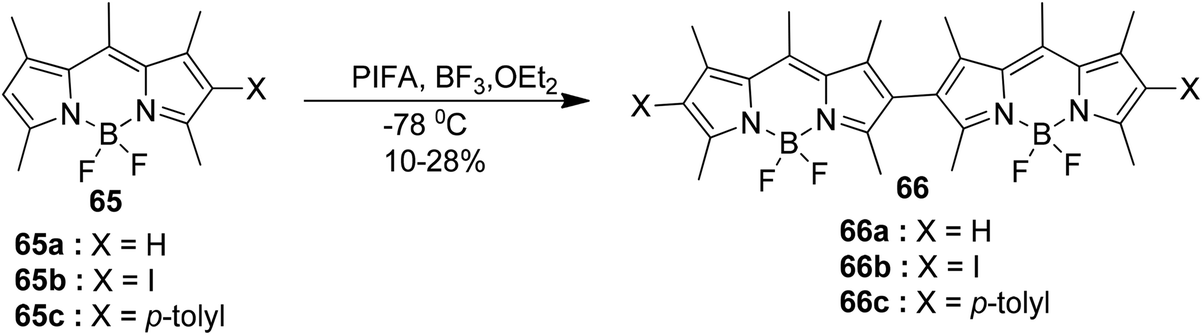
Bodipy-based dyes hold significant importance due to their exceptional optical properties, enhanced solubility, outstanding photochemical stability, high absorption coefficients, and ease of functionalization.87 While Bodipy dimers possess intriguing characteristics related to charge delocalization, their potential utility as fluorescent or redox probes has not been a primary area of research focus.
Rihn et al.88 reported the coupling reaction of monomeric Bodipy 65, mediated by both a Lewis acid and PIFA, leads to the formation of a mixture of various oligomers. The formation of dimers is favored when one of the positions is occupied by either an iodine(I) or phenyl (Ph) substituent, as shown in Scheme 24. PIFA actively participated in the reaction by employing a free radical pathway, particularly through single-electron oxidation. These dimers exhibit two consecutive oxidative and reductive waves, with an approximate separation of 260 mV and 130 mV, respectively. Interestingly, the three dimers, namely 66a, 66b, and 66c, were found to display near identical stability and reactivity compared with Bodipy monomer units.
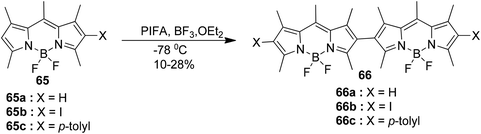 |
| | Scheme 24 Synthesis of bodipy dimers. | |
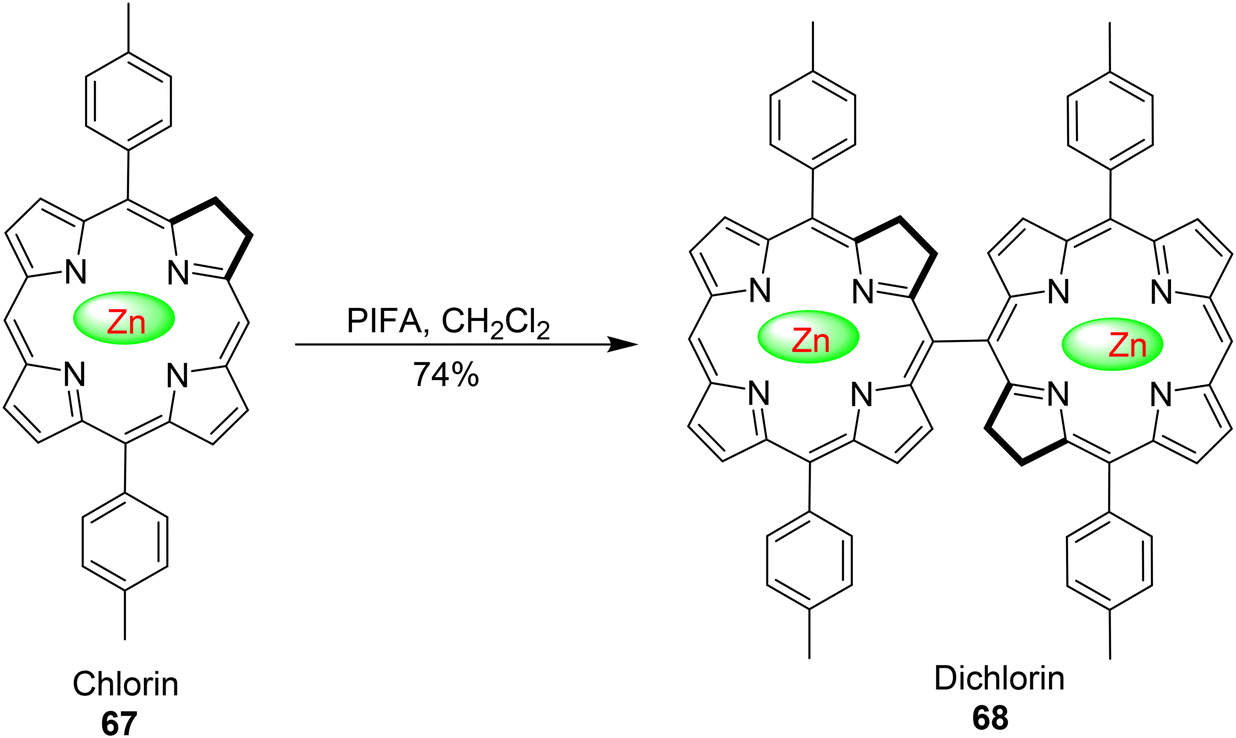
Porphyrin and chlorin dimers hold considerable significance due to their applications in nonlinear optical (NLO) devices, sensors, and photosynthetic systems.89 Ouyang et al.90 provided the inaugural account of synthesizing dimeric zinc chlorin by efficiently oxidizing zinc chlorin monomers 67 using PIFA, as outlined in Scheme 25. This process yielded a 74% yield, demonstrating a selective formation of dichlorin 68. Importantly, the reaction exhibited regioselectivity at the 20th position of the pyrrole ring. PIFA actively participated in the reaction by employing a free radical pathway, particularly through single-electron oxidation.
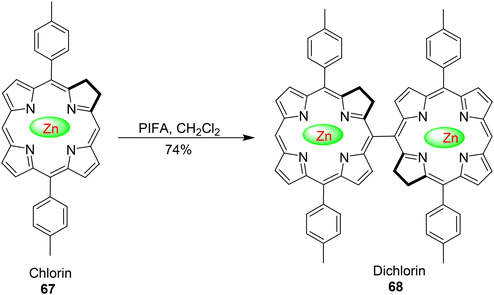 |
| | Scheme 25 Synthesis of zinc chlorin dimer. | |
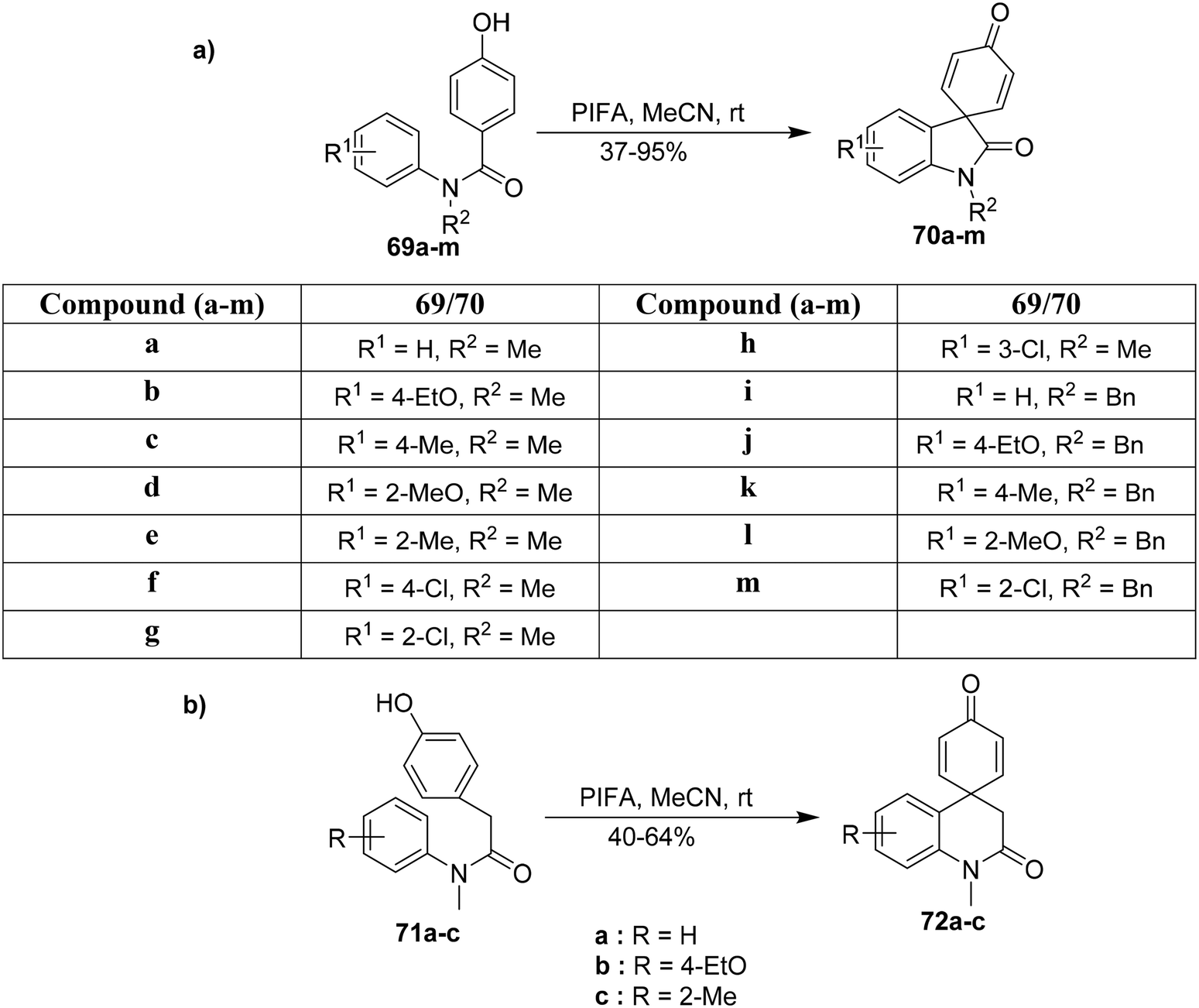
Spirooxindoles serve as essential intermediates for the synthesis of biologically significant molecules.91 PIFA functions as an effective catalyst for the synthesis of diversely functionalized N-phenylbenzamides by oxidative coupling. Yu et al.92 described an effective methodology for synthesizing such molecules 70a–m in moderate yields from 69a–m as demonstrated in Scheme 26a. This approach can further be utilized to obtain functionalized spirooxindole analogues. Also, the spirooxindoles 72a–c were synthesized from the N-acetamides 71a–c (Scheme 26b). PIFA was involved in the reaction via an ionic pathway.
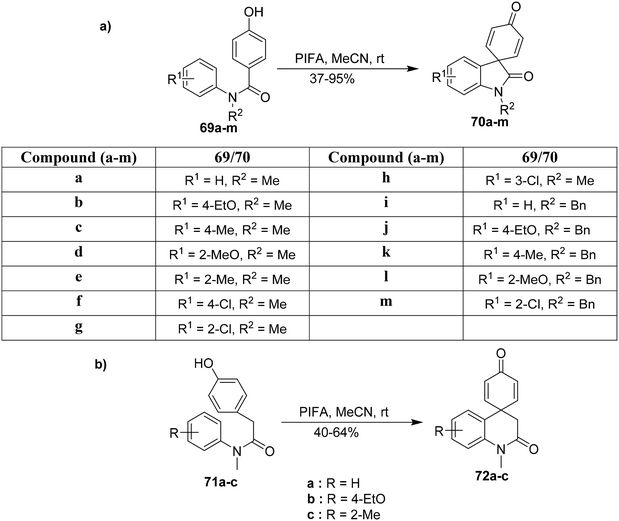 |
| | Scheme 26 Synthesis of zinc chlorin dimer. | |
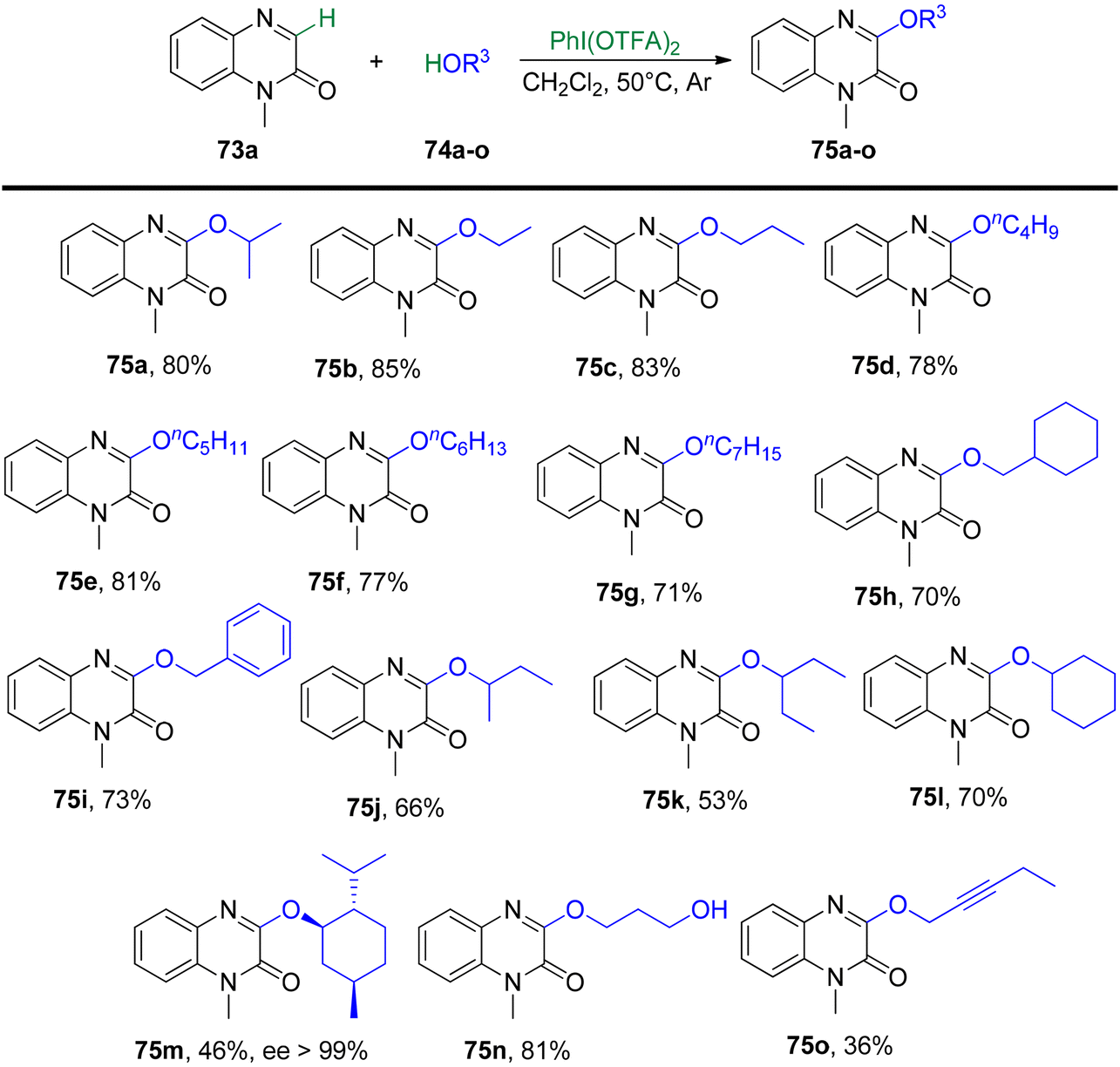
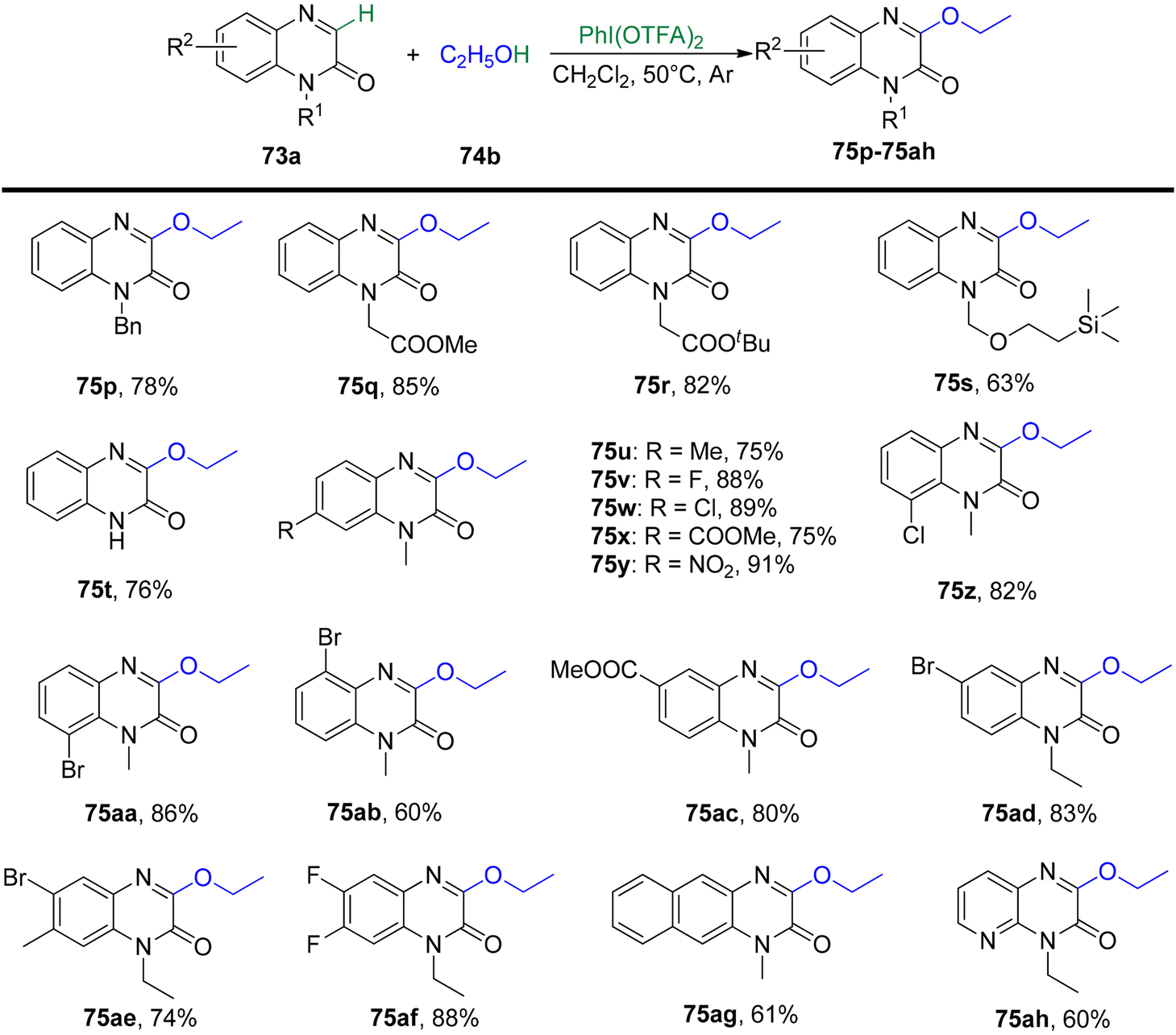
Yang et al.93 have unveiled a straightforward and effective alkoxylation method for quinoxalin-2(1H)-ones 73 using primary or secondary alcohols 74 (Schemes 27 and 28). This process operates under catalyst-free conditions and involves cross-dehydrogenative coupling. By employing PIFA as an oxidant, this technique offers a convenient means to access 3-alkoxylquinoxalin-2(1H)-ones 75 in good to excellent yields. PIFA actively engaged in the reaction by following a free radical pathway, specifically through hydrogen abstraction. The method is characterized by its user-friendliness, simplicity of the reaction system, and a broad tolerance for various functional groups. Consequently, it provides a pathway to effortlessly produce a range of potential drug molecules containing 3-alkoxylquinoxalinone frameworks in excellent yields.
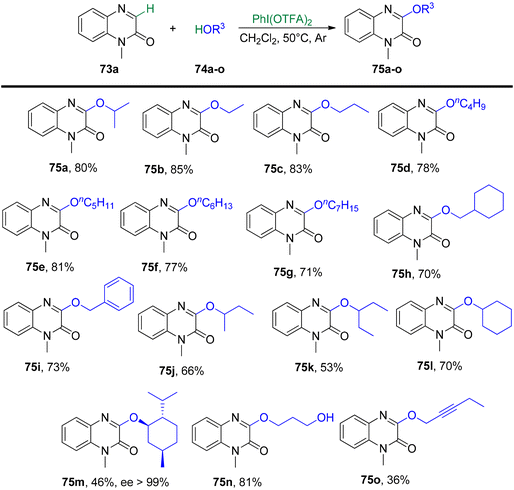 |
| | Scheme 27 Synthesis of 3-alkoxylquinoxalin-2(1H)-ones. | |
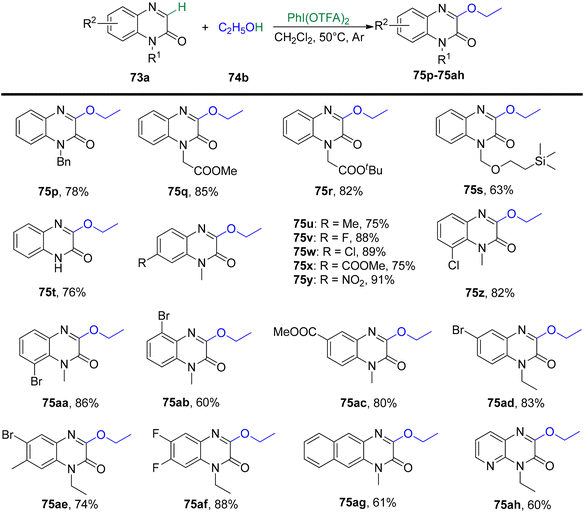 |
| | Scheme 28 Synthesis of 3-alkoxylquinoxalin-2(1H)-ones. | |
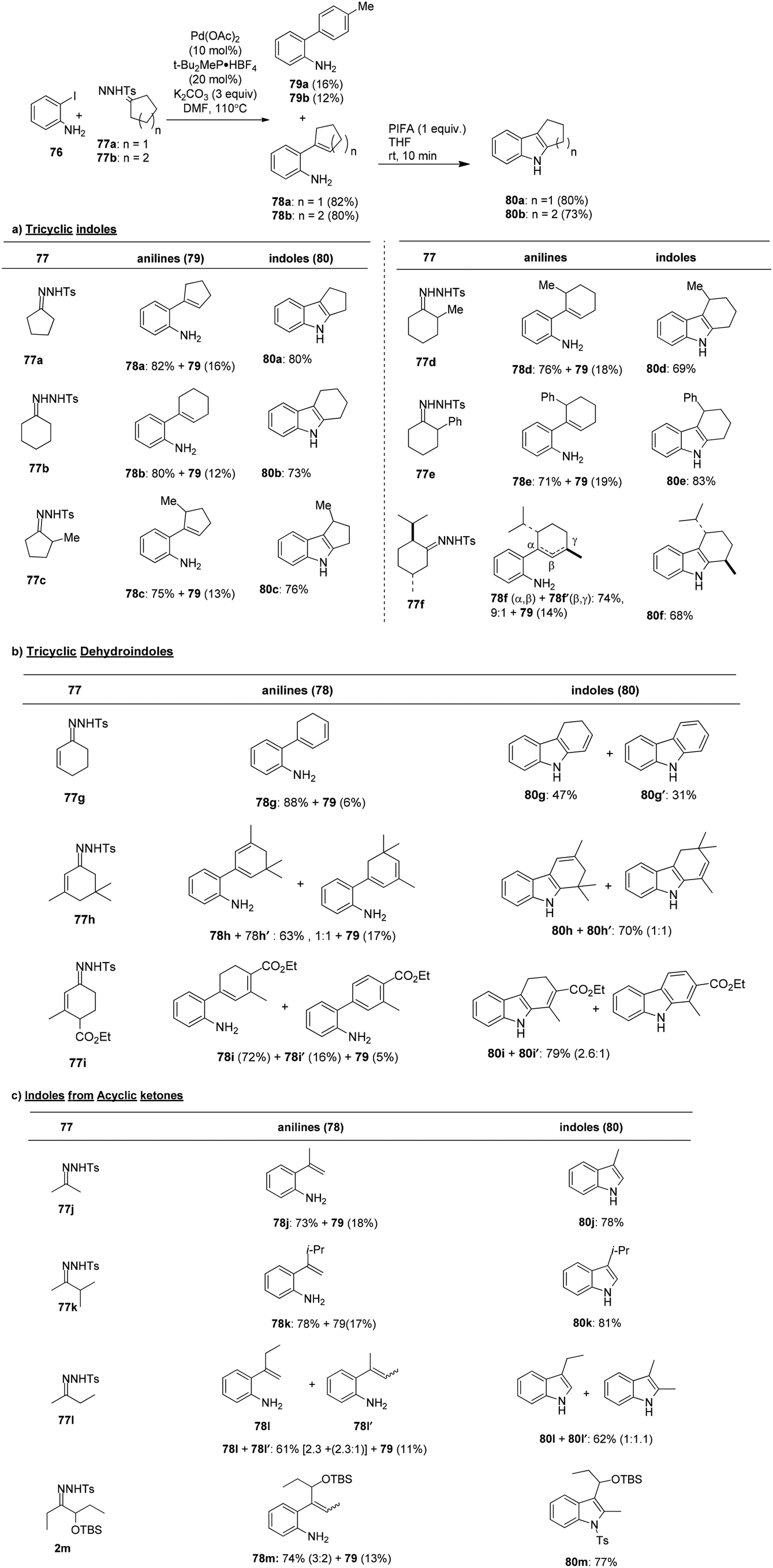
Chebieb et al.94 have introduced an efficient method for synthesizing indoles 80 by orchestrating a two-step process that involves the cross-coupling reaction of o-haloanilines 76 and the PIFA oxidation of the resulting 2-alkenylanilines 78. A notable feature of this sequential approach is its modular strategy, which is applicable to both acyclic and cyclic starting materials. The regiochemistry of this method complements the Fischer indole synthesis and related approaches. Another advantage of this method is that it enables the direct preparation of N–H indoles without the need for N-protecting groups. The scope of this indole synthesis was further explored using tosylhydrazones derived from cyclopentanone and cyclohexanone, with a specific focus on achieving the regioselective formation of indole products 80a–f (Scheme 29). The formation of dehydroindoles (dihydrocarbazoles) was also investigated, starting with cyclohexenone-derived tosylhydrazones and proceeding to the oxidative cyclization of 1,3-dienyanilines 78g–i (Scheme 29). Finally, the versatility of the method was demonstrated by synthesizing indoles 80j–m from acyclic substrates, showcasing a broad substrate scope (Scheme 29).
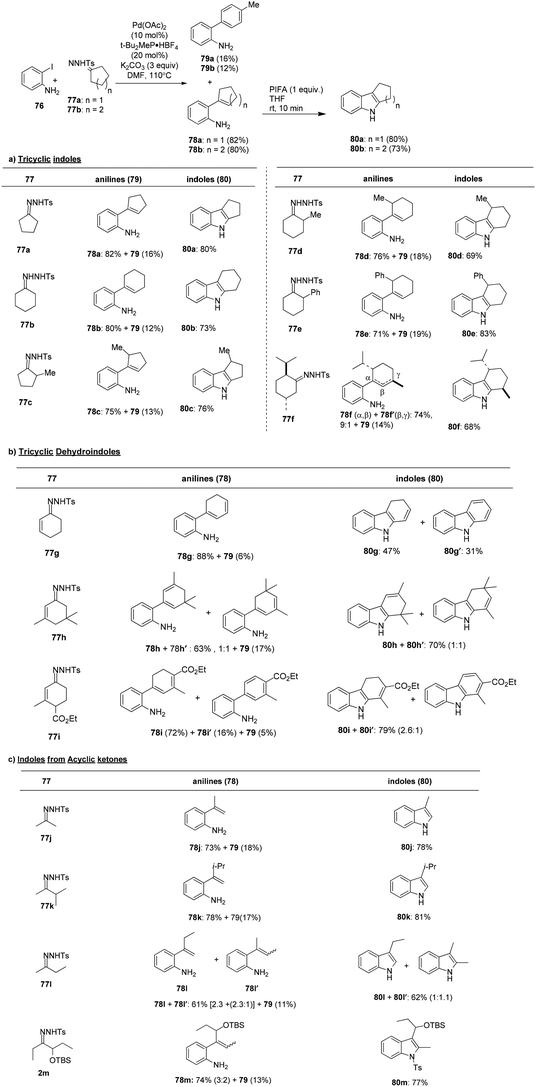 |
| | Scheme 29 An efficient method for synthesizing indoles 80. | |
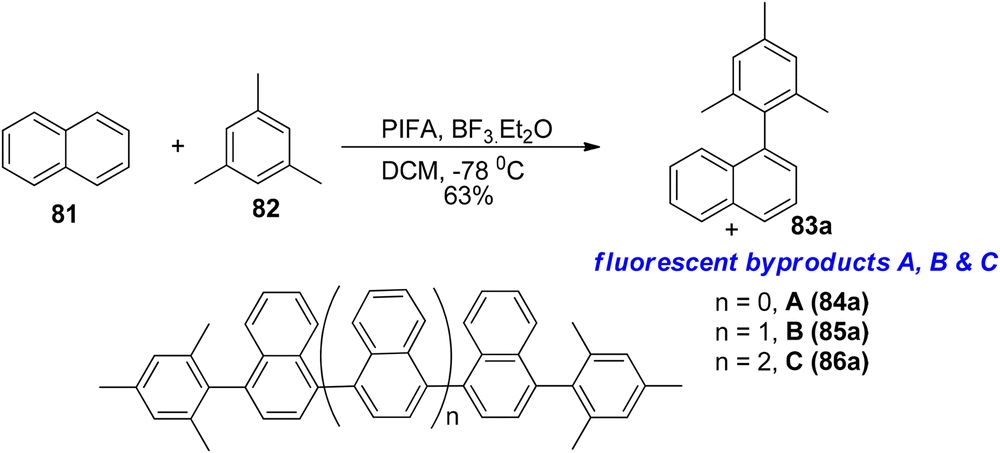
A direct oxidative Kita-type coupling reaction was observed between naphthalene 81 and substituted benzenes reported by Faggi et al.,95 resulting in the formation of a linear tetraarene with a binaphthalene core, 83a, in a 63% yield after 3 hours at −78 °C when using PIFA/BF3·Et2O as a catalyst (Scheme 30). The ratio of 83a to potential overoxidation products like 84a, 85a, and 86a seemed to be influenced by the quantity of PIFA oxidant utilized.
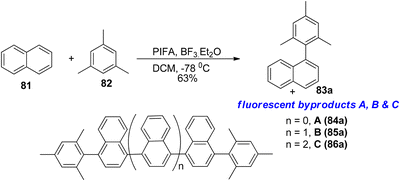 |
| | Scheme 30 Kita-type coupling reaction between naphthalene 81 and substituted benzenes. | |
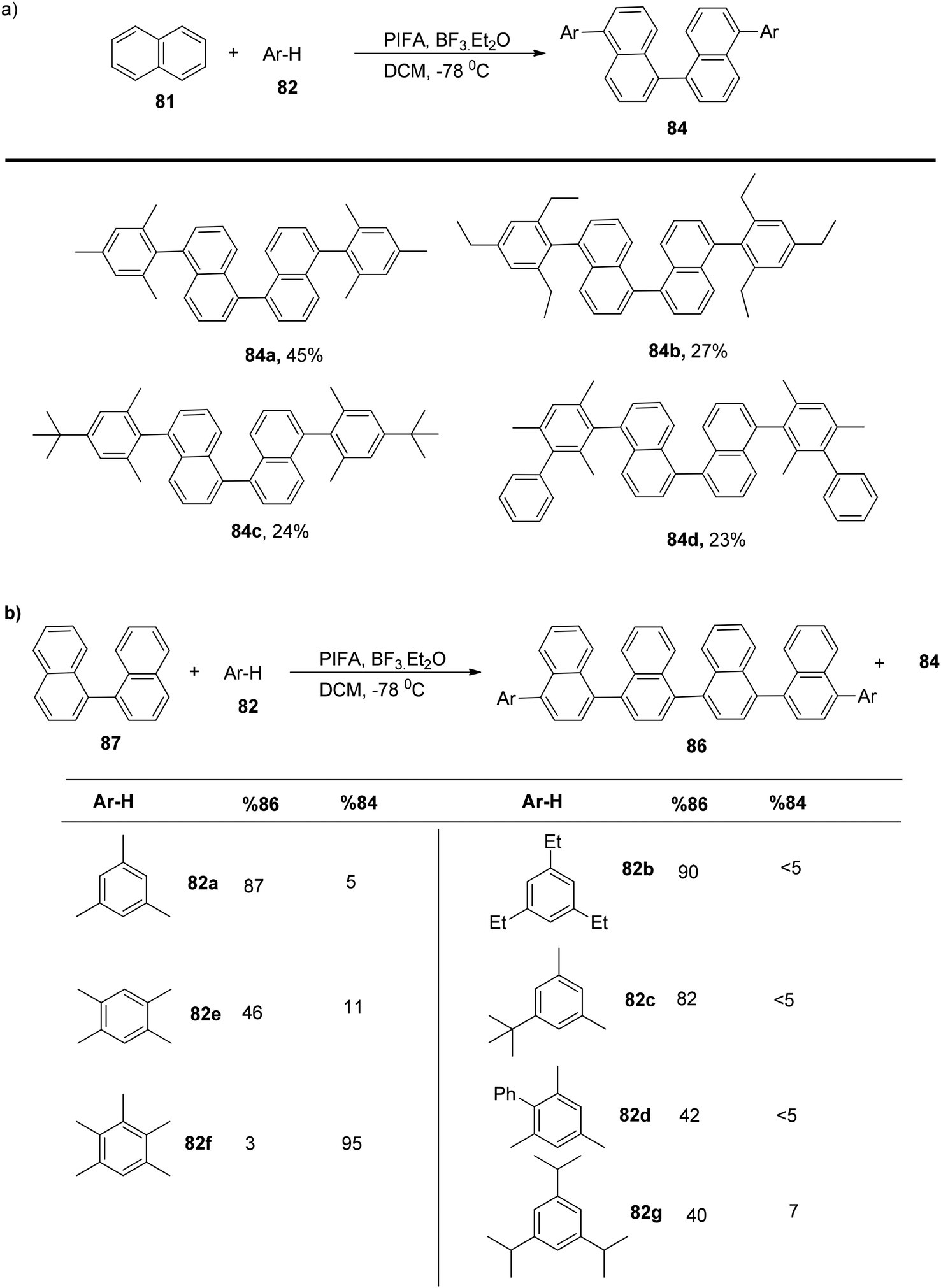
To explore this, an excess of PIFA was employed, allowing the extension of the method to various electron-rich arenes, yielding tetraaryl products 84a–d in the range of 45–23% yields (Scheme 31a). This methodology could also be applied to the coupling of unfunctionalized 1,1′-binaphthalene 87 with mesitylene 82, resulting in the selective formation of a linear hexaarene product with an impressive 87% yield (Scheme 31b).
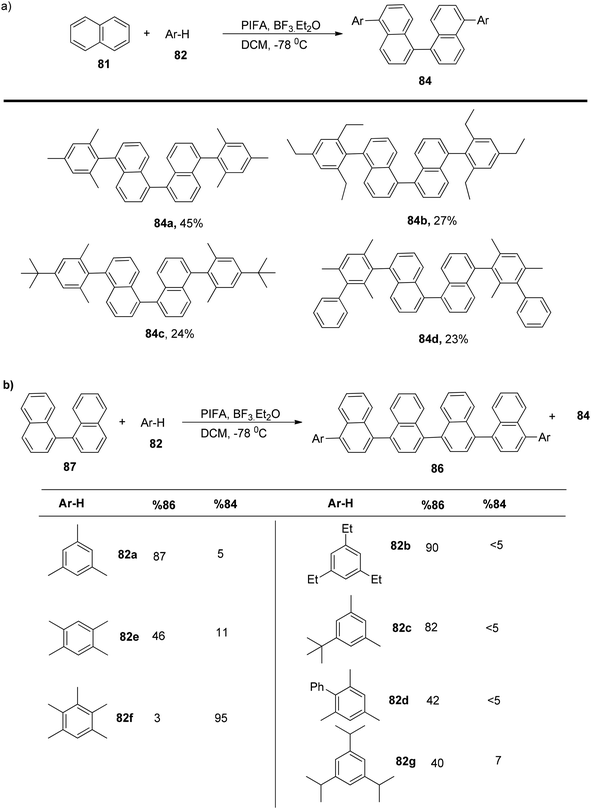 |
| | Scheme 31 Oxidative four-component Kita-type coupling reaction. | |
3.5. Substituted pyrroles
Owing to their paramount importance as the principal structural constituents within various alkaloids and pharmacologically active compounds, highly substituted pyrrole derivatives have been the focal point of considerable scientific interest in recent years.96 Oxidative dimerization emerges as a pivotal synthetic route for the acquisition of symmetric compounds.
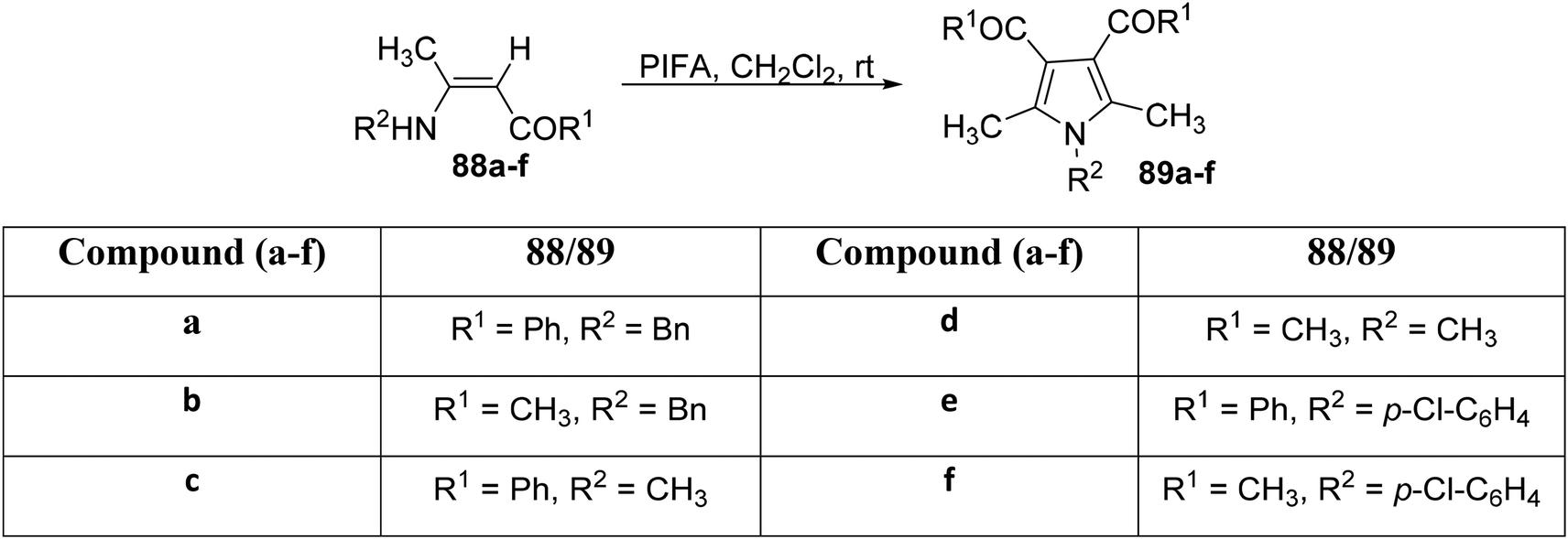
Zhang et al.97 elucidated a tandem dimerization–cyclocondensation strategy for the synthesis of highly substituted pyrrole analogues utilizing enaminones in the presence of PIFA. This method demonstrates efficiency and offers multiple advantages, including readily available starting materials, non-toxic reagents, and mild reaction conditions. PIFA was involved in the reaction via an ionic pathway, actively participating in the process through its contribution to the ionic mechanism. To assess the scope of this reaction, several enaminones 88a–f were subjected to experimentation, as outlined in Scheme 32. Product characterization involved a comprehensive array of analytical techniques, including melting point analysis, infrared spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and mass spectrometry (MS), leading to the characterization of products 89a–f.
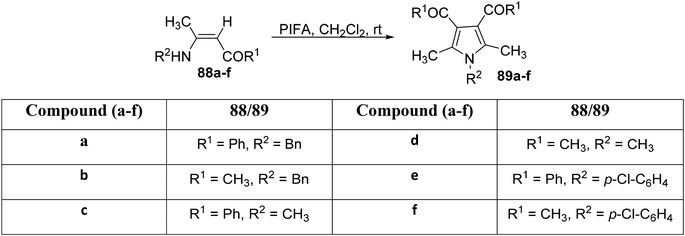 |
| | Scheme 32 Synthesis of substituted pyrroles 89a–f. | |
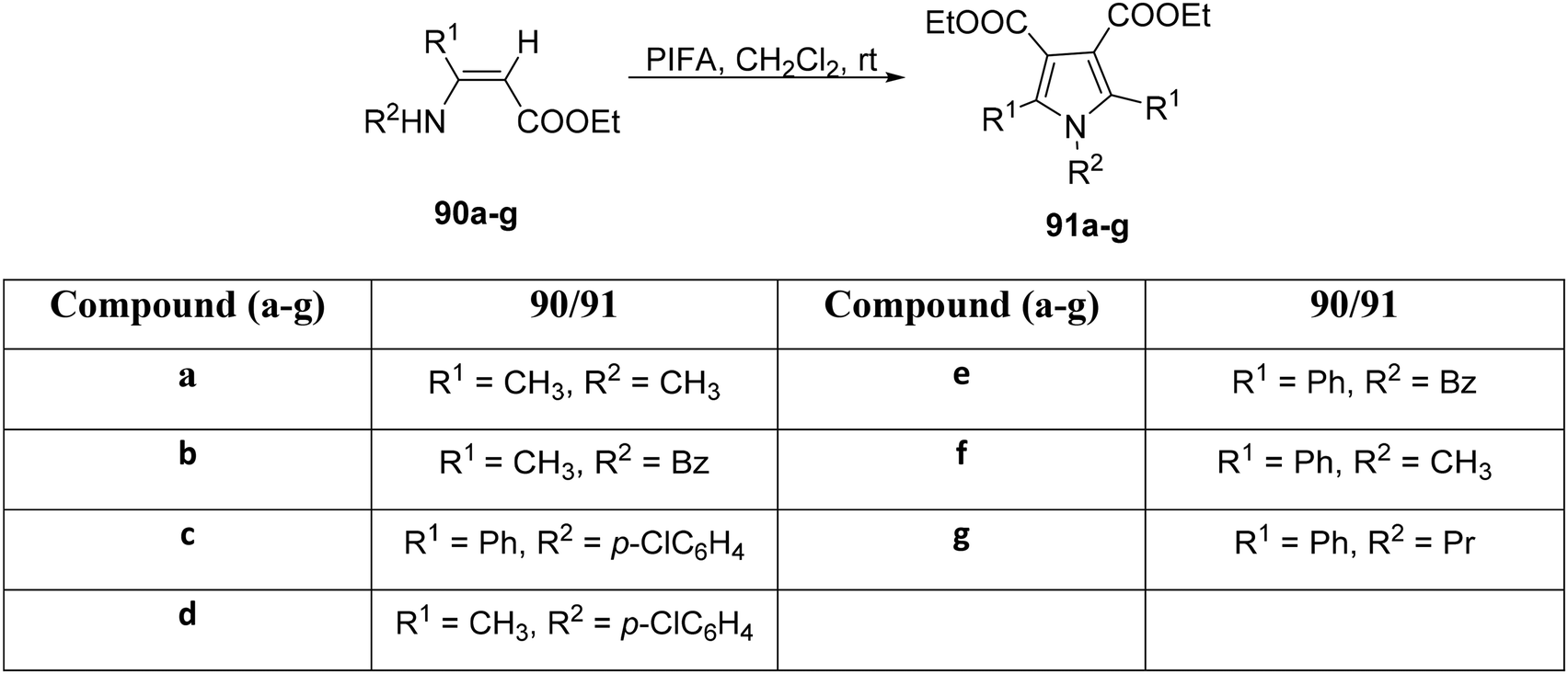
Furthermore, within the same year, Zhang et al.98 explored the applicability of this reaction to various enamine-esters (Scheme 33). Utilizing PIFA as a catalyst, the transformation of enamine-esters 90a–g into pyrrole derivatives 91a–g was achieved with ease. PIFA was involved in the reaction via an ionic pathway. Subsequently, the compounds underwent comprehensive structural characterization, encompassing analyses such as melting point determination, MS, infrared spectroscopy, as well as NMR spectroscopic investigations.
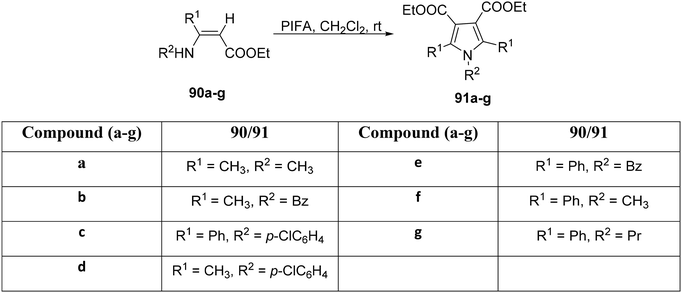 |
| | Scheme 33 Synthesis of substituted pyrroles 91a–f. | |
3.6. Cyclization
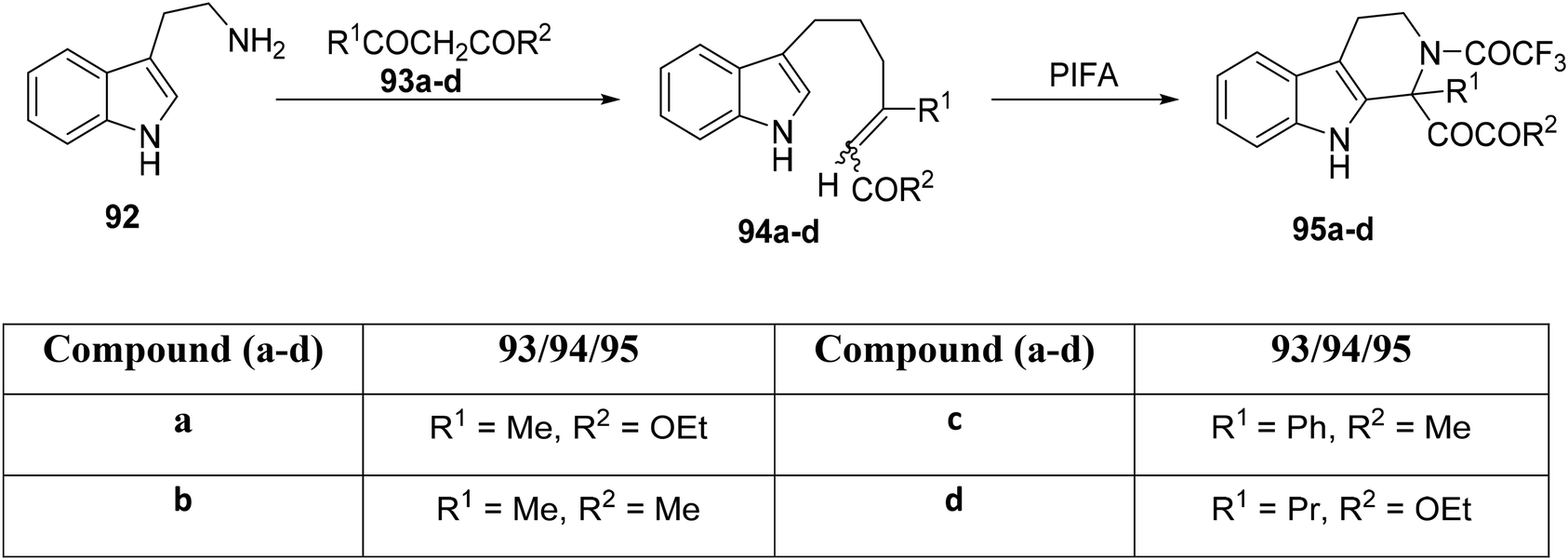
The Pictet–Spengler methodology is a recognized synthetic approach for the construction of both indole and isoquinoline alkaloids. Papadopoulou et al.99 demonstrated its application in the derivatization of tryptophan and tryptamine derivatives to yield 1,2,3,4-tetrahydro-β-carbolines 95a–d (Scheme 34). The straightforward combination of tryptamine 92 with β-dicarbonyl compounds 93a–d leads to the formation of carbonyl compounds of the type 94a–d.100 Under controlled reaction conditions ranging from 0 °C to room temperature, the synthesis of carboline analogues 95a–d was achieved in the presence of PIFA, serving as the cyclization agent.
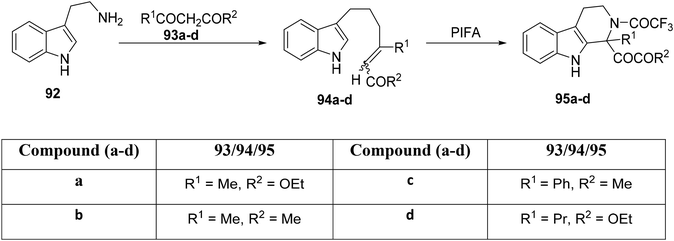 |
| | Scheme 34 Synthetic methodology for carboline analogues 95a–d. | |
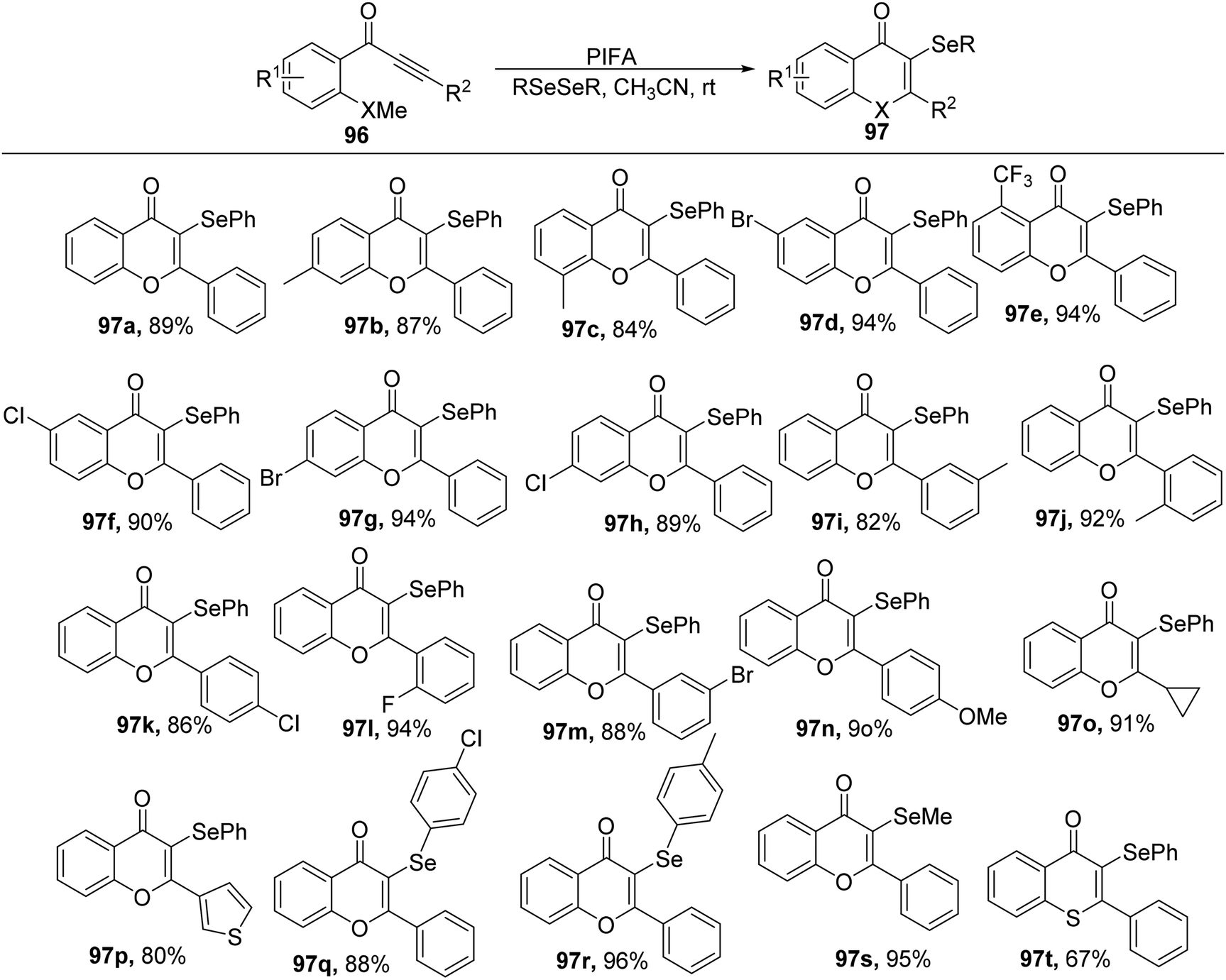
Chromones, characterized by their unique heterocyclic framework, are commonly encountered in both pharmaceuticals and natural compounds.101 The introduction of sulfur or selenium functionalities into the chromone structure imparts intriguing biological properties to the resultant molecules, thereby attracting significant scientific interest over recent decades.102
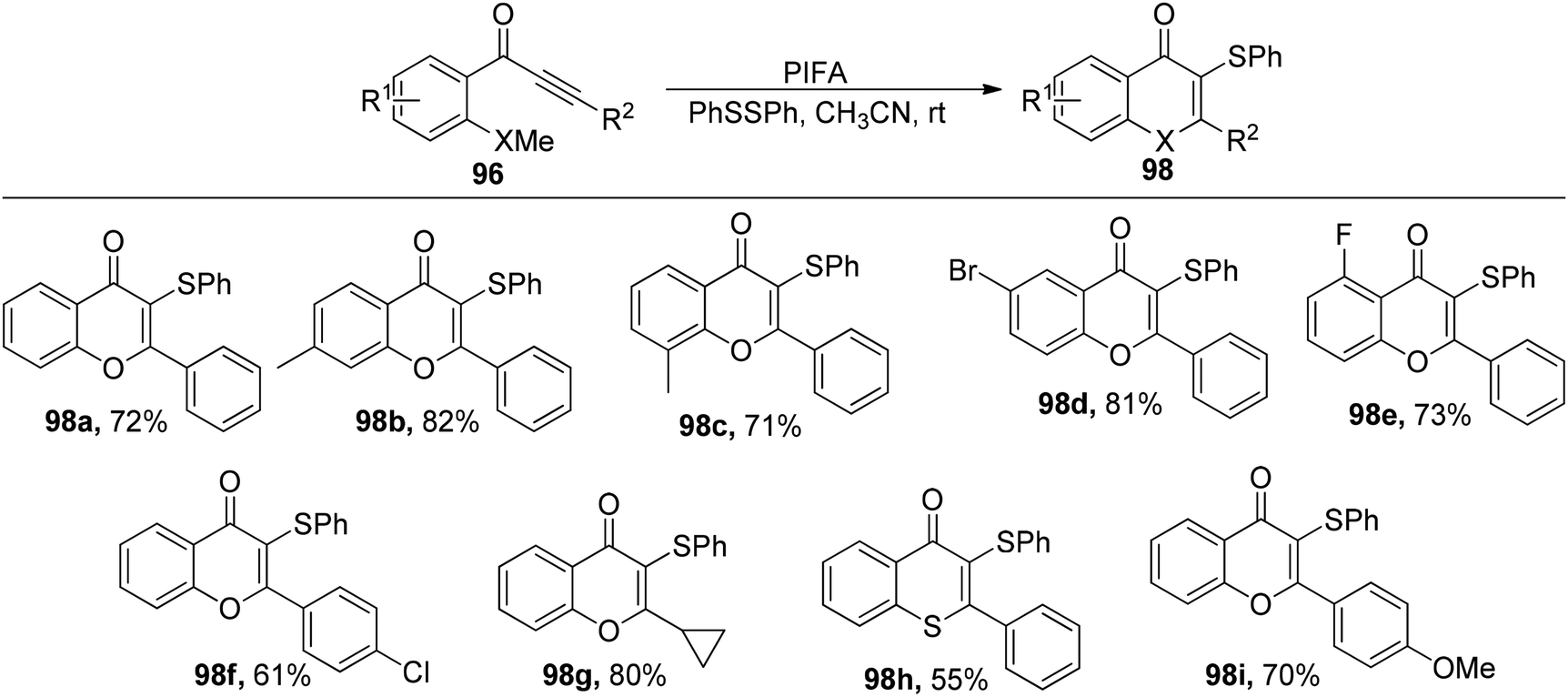
Ai et al.103 accomplished the synthesis of 3-selenyl/sulfenyl chromones/thiochromones 97/98 through the reaction of alkynyl aryl ketonic substrates 96 with diselenides/disulfides, employing PIFA as the catalytic agent (Scheme 35). Furthermore, the synthesis of sulfenylated chromone derivatives was explored using PIFA/PhSSPh (Scheme 36). PIFA was involved in the reaction via an ionic pathway. Notably, this methodology is distinguished by its metal-free nature, wide substrate applicability, mild reaction conditions, and the production of high-yielding products.
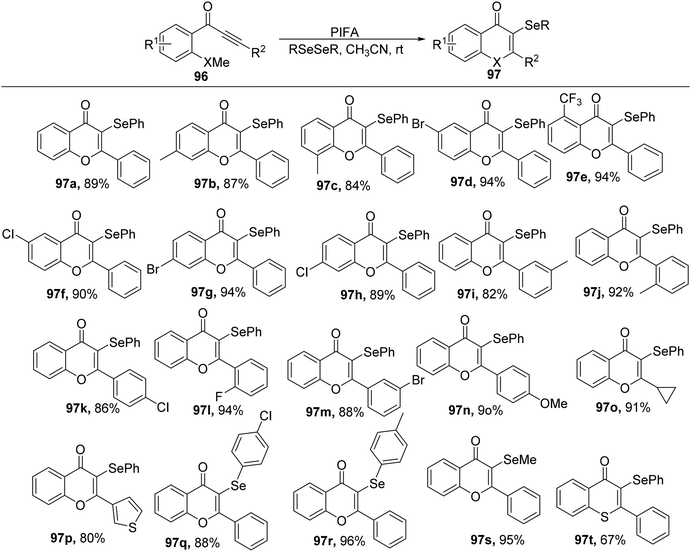 |
| | Scheme 35 Reaction scheme for synthesizing 3-Se chromones/thiochromones. | |
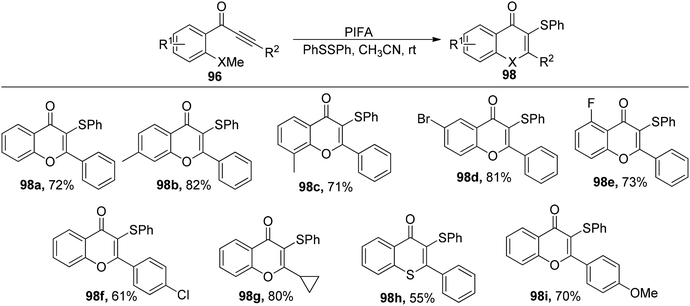 |
| | Scheme 36 Reaction scheme for synthesizing 3-S chromones/thiochromones. | |
Enaminones, arising from the union of nucleophilic enamines and electrophilic enones, represent pivotal synthetic intermediates due to their auspicious structural attributes.104 Pyrrolin-4-one analogues, nitrogen-containing heterocycles, find extensive utility as fundamental building blocks in the design and synthesis of anti-cancer, anti-thrombotic, and antibacterial pharmaceutical agents.105
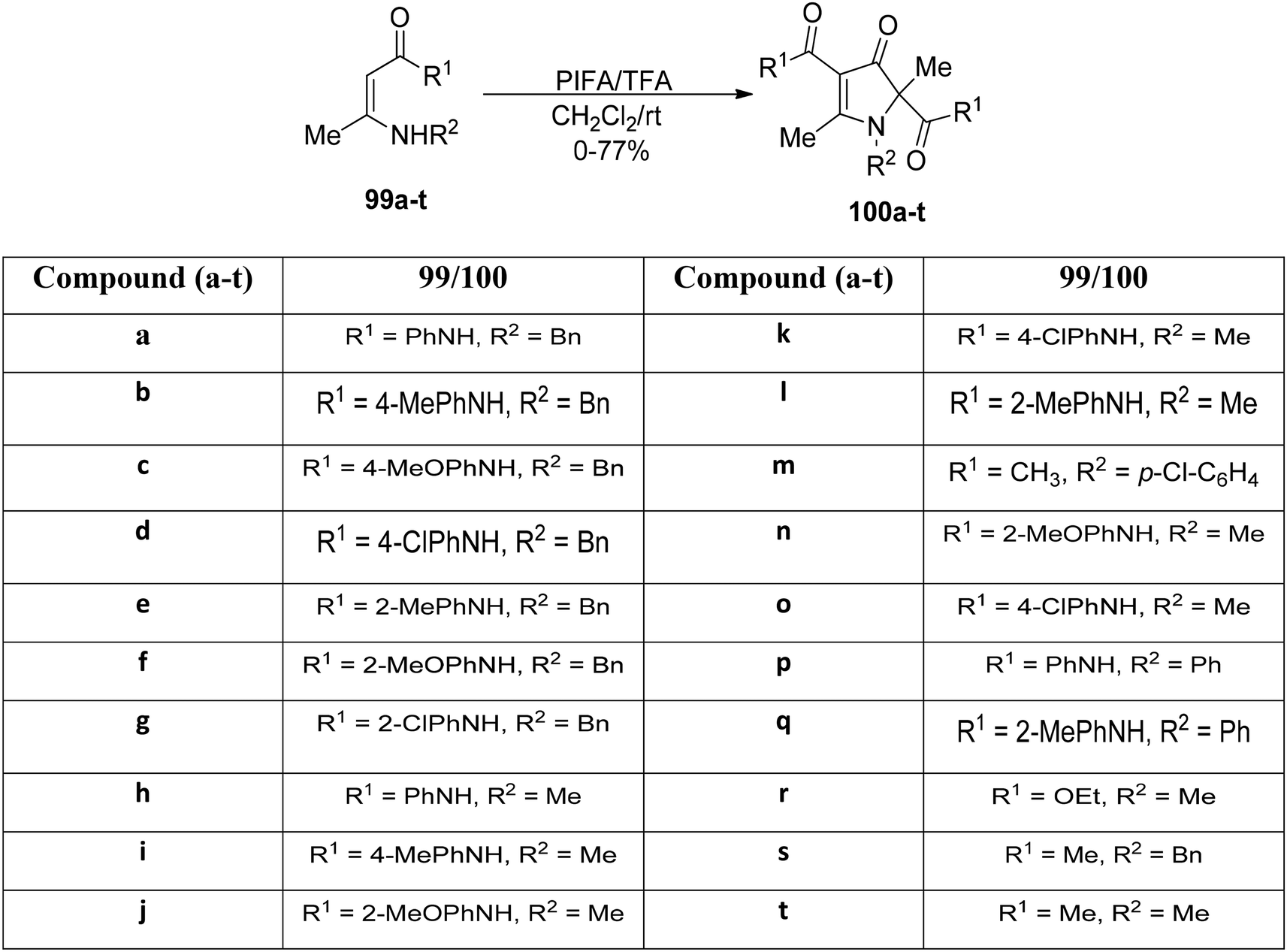
The readily accessible enaminones 99a–t underwent PIFA-mediated cyclization reactions to yield highly substituted pyrrolin-4-ones 100a–t, as reported by Huang et al.106 This rearrangement reaction entails concurrent cleavage and formation of C–N and C–C bonds (Scheme 37). PIFA engaged in reactions via the ionic pathway. Noteworthy advantages of this process include its mild reaction conditions, wide substrate versatility, accessibility of starting materials, and the attainment of excellent product yields.
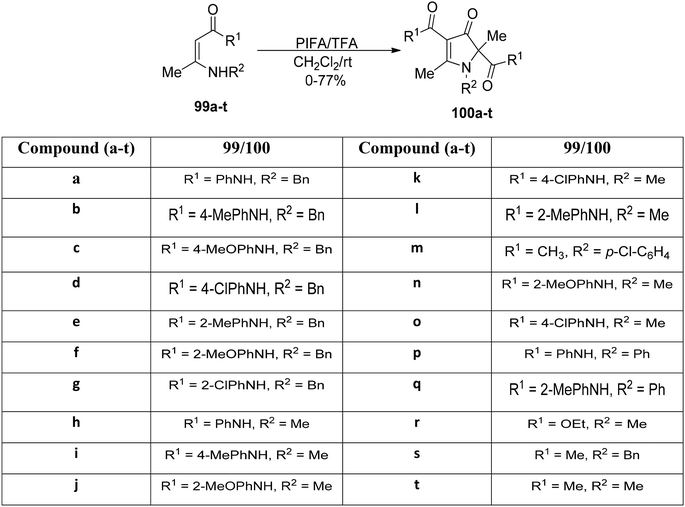 |
| | Scheme 37 Synthesis of pyrrolin-4-one derivatives 85a–t. | |
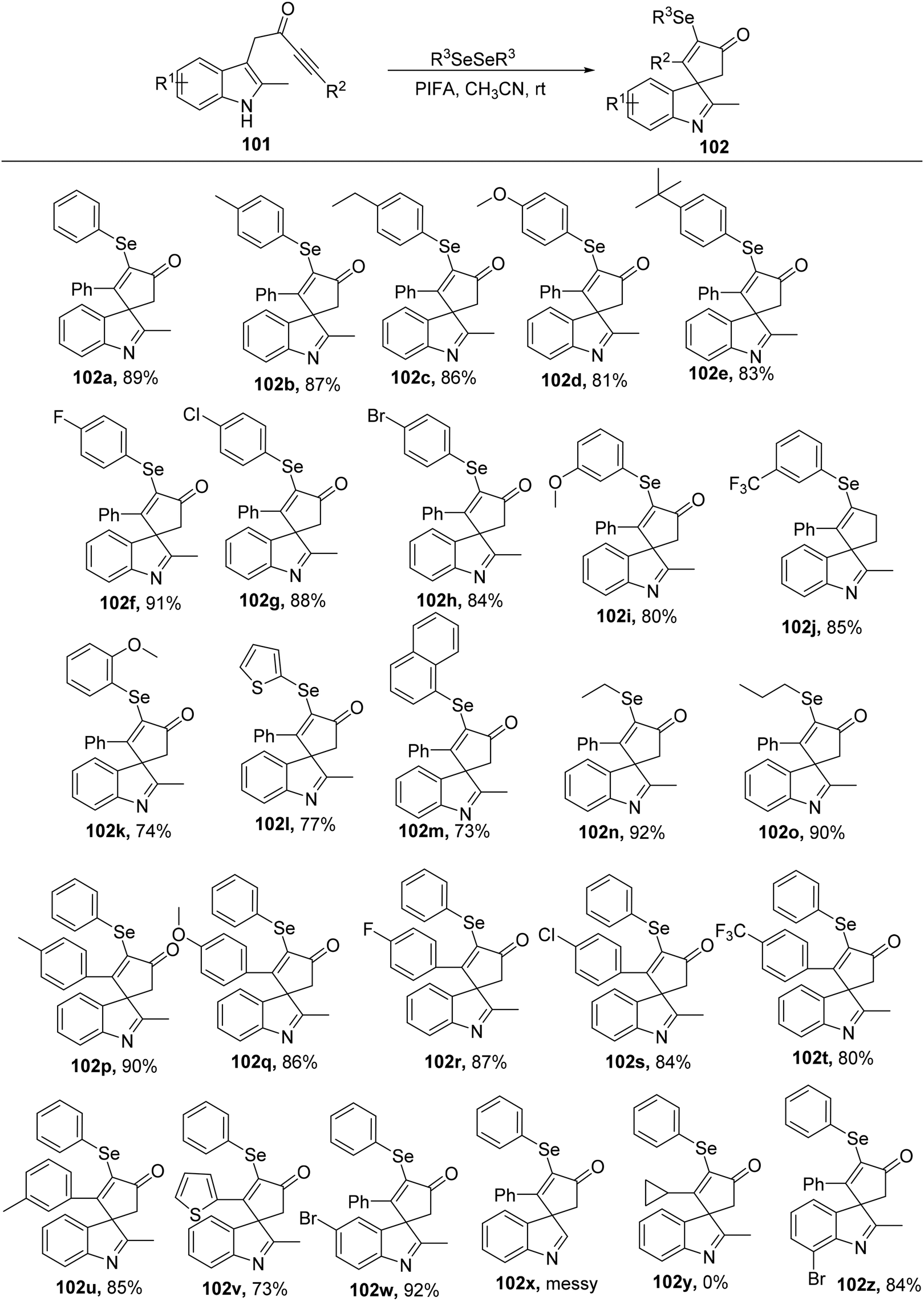
Spirocycles are integral components of pharmaceuticals, natural products, and valuable materials.107 Among these spirocycles, spiroindolenines have garnered significant attention among organic chemists due to their diverse array of biological properties.108 The key class of bioactive chemicals known as organoselenides, on the other hand, are widely present in physiologically active substances and medications.109 Organo-selenide synthesis has been the subject of extensive research up to this point.110 Chen et al.111 described a rapid, mild selenylative spirocyclization of indolyl ynones 101via the use of PIFA that involved an ionic route and an electrophilic cyclization to obtain selenated spiroindolenines 102a–z. Indolyl ynones undergo an electrophilic dearomative cascade cyclization catalyzed by PIFA leading to the in situ generation of RSeOCOCF3 from diselenides. PIFA engaged in reactions via the ionic pathway. This approach offers a rapid and effective pathway to develop selenated spiro[cyclopentenone-1,3′-indoles] with diverse functional group tolerance (Scheme 38).
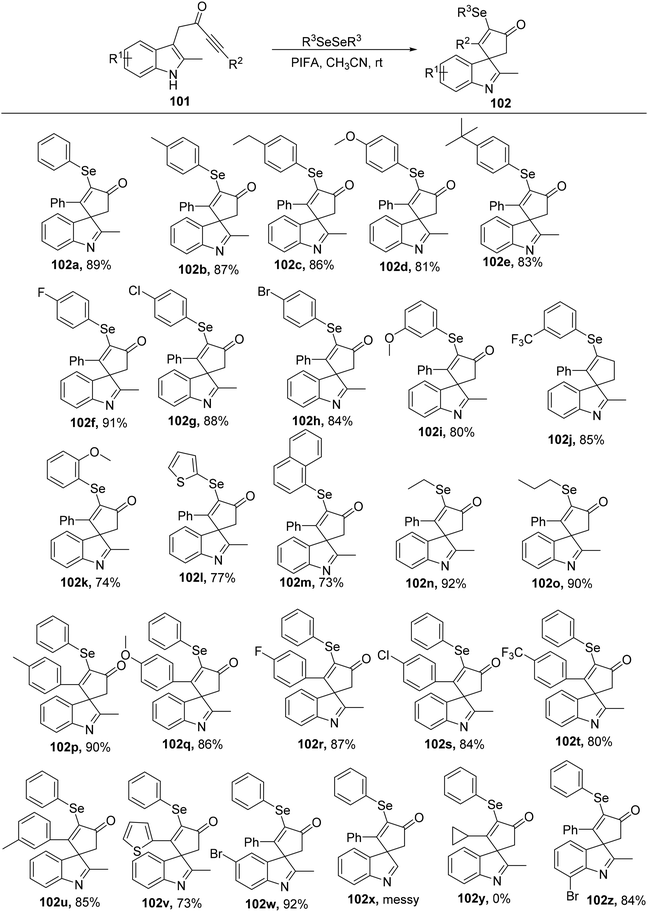 |
| | Scheme 38 Synthesis of selenated spiro[cyclopentenone-1,3′-indoles]. | |
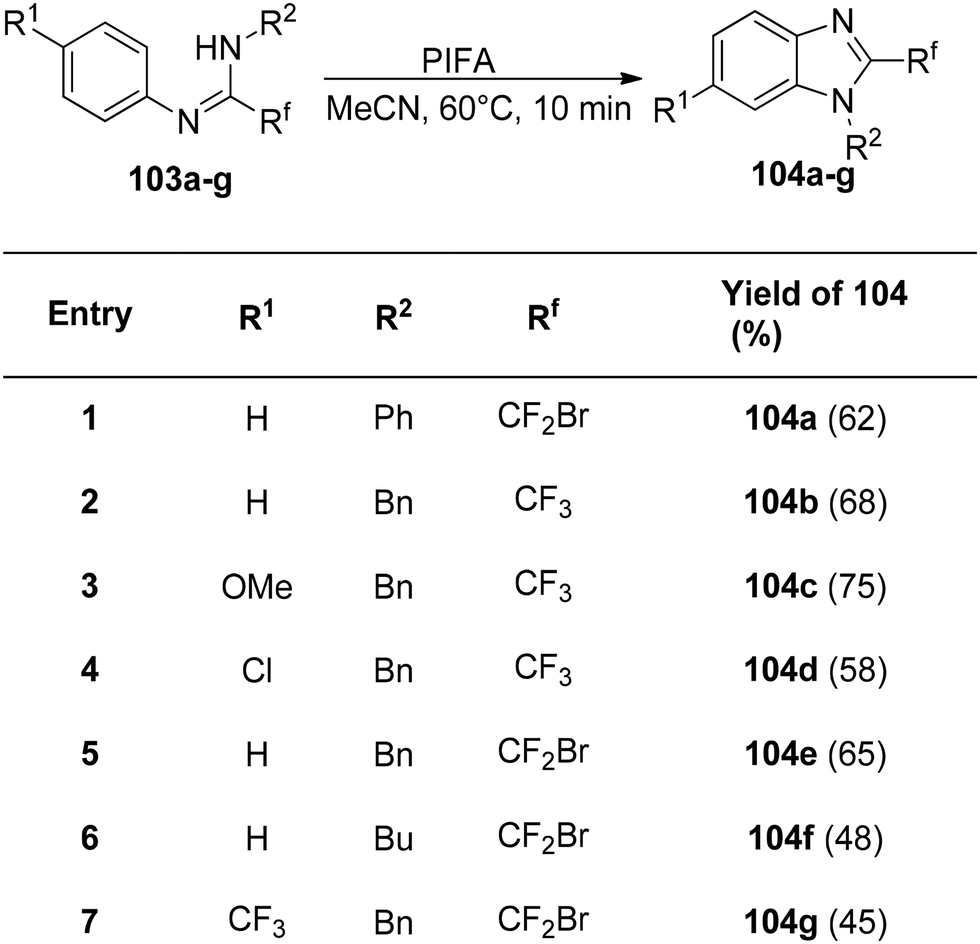
Fluorine exhibits distinct properties, and consequently, organofluorine compounds, particularly those containing fluorine heterocyclic motifs, hold significant importance in the fields of medicine, agriculture, and materials chemistry.112 Notably, 2-fluoroalkylbenzimidazoles are recognized for their potency as insecticides and herbicides.113
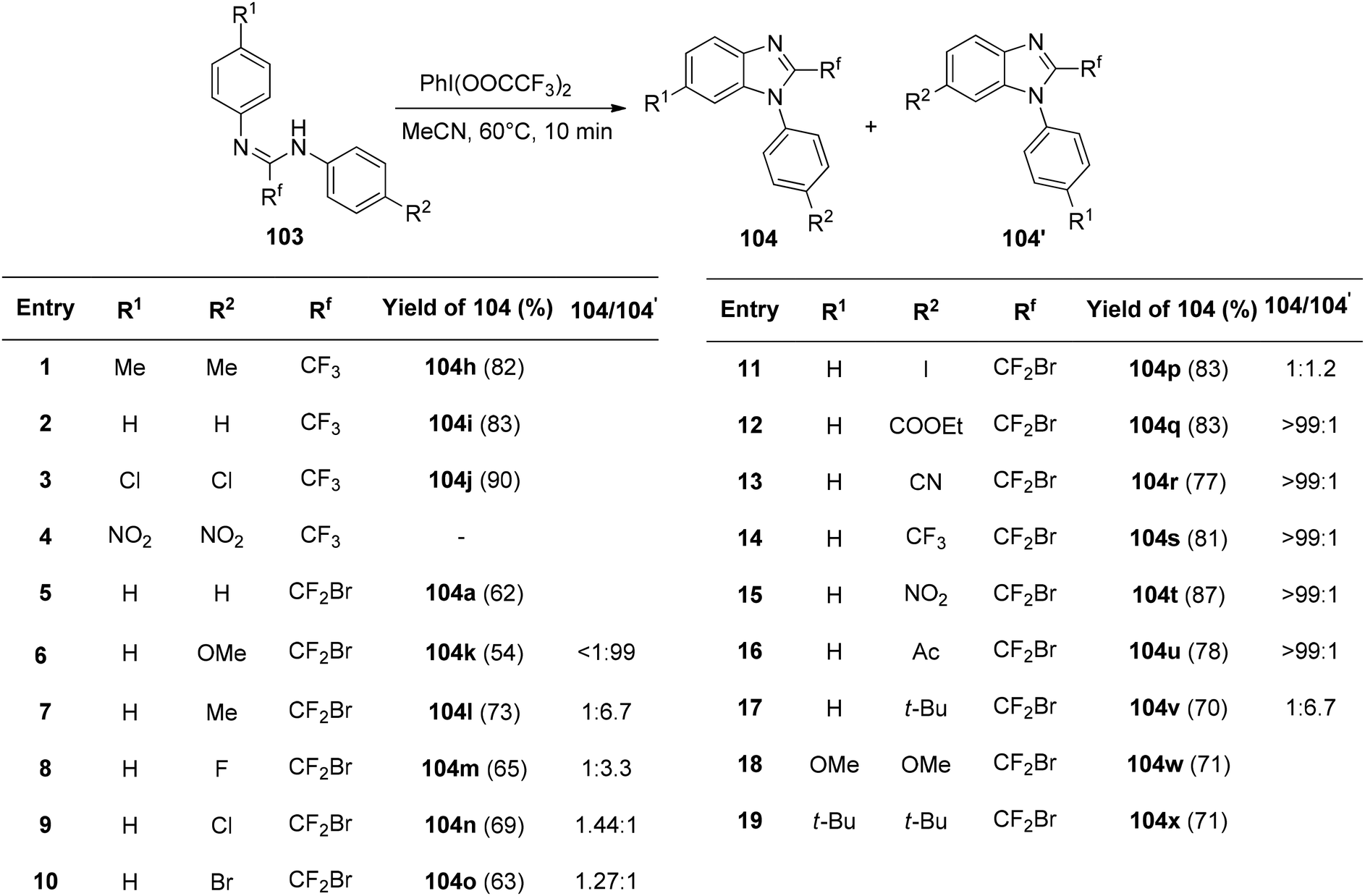
The interaction of PIFA with N,N-disubstituted fluoroethanimidamides 103 to produce N-substituted 2-fluoromethylbenzimidazoles was disclosed by Zhu et al.114 as a novel methodology for the synthesis of these compounds. The group investigated the range and limitations of this transformation after determining the ideal conditions. As a result of the initial testing of a number of N-aryl-N′-alkylfluoroethanimidamides 103, fluoromethylbenzimidazoles 104b–g were obtained in moderate yields (Scheme 39). Furthermore, N,N′-diarylfluoroethanimidamides were utilized, either symmetrically or asymmetrically, and the outcomes are compiled in Scheme 40. PIFA engaged in reactions via the ionic pathway.
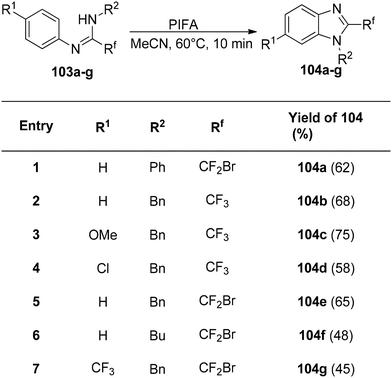 |
| | Scheme 39 Synthetic scheme for development of N-alkyl-2-fluoromethylbenzimidazoles. | |
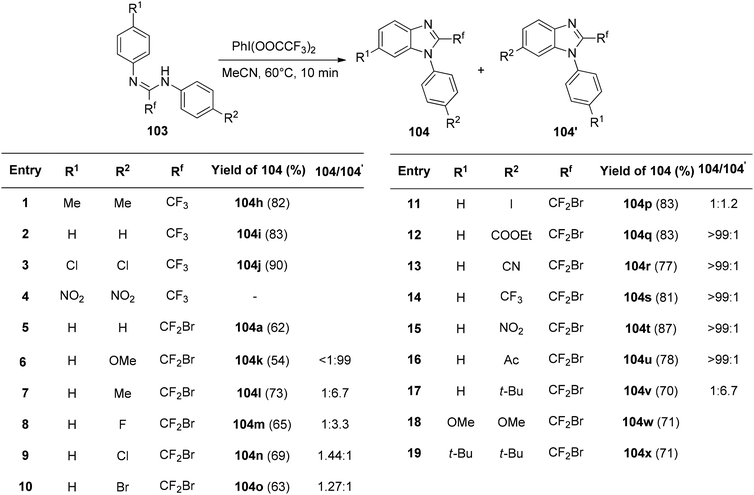 |
| | Scheme 40 Synthesis of N-aryl-2-fluorobenzimidazoles. | |
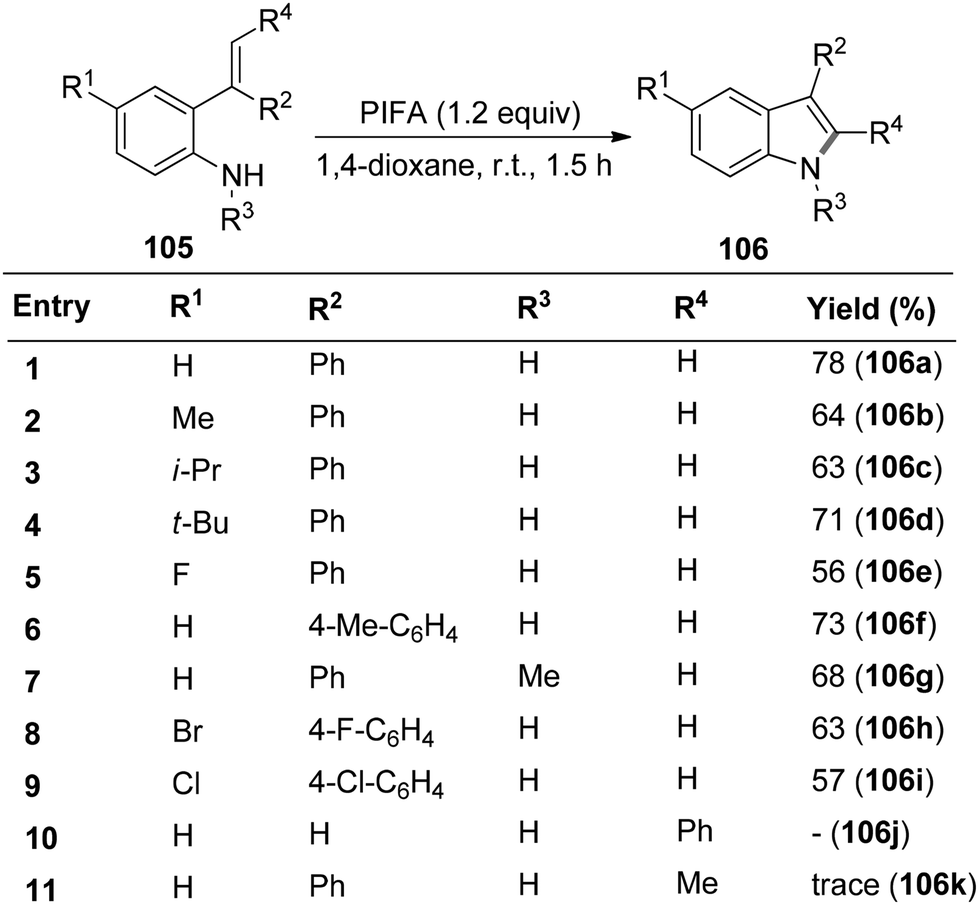
Wu et al.115 introduced a method for synthesizing different indoles 106via cyclization reactions involving 2-vinylanilines 105 in the presence of PIFA, all conducted under mild reaction conditions. This strategy exhibits notable versatility and works well with a diverse array of substituents, both electron-withdrawing and electron-donating groups on the aromatic ring and the nitrogen atom of 2-vinylanilines 105, resulting in the desired products with moderate to good yields (Scheme 41). PIFA engaged in reactions via the ionic pathway.
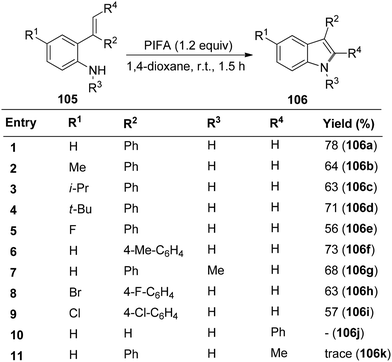 |
| | Scheme 41 Synthesis of indole derivatives 106 with 2-vinylanilines 105 and PIFA. | |
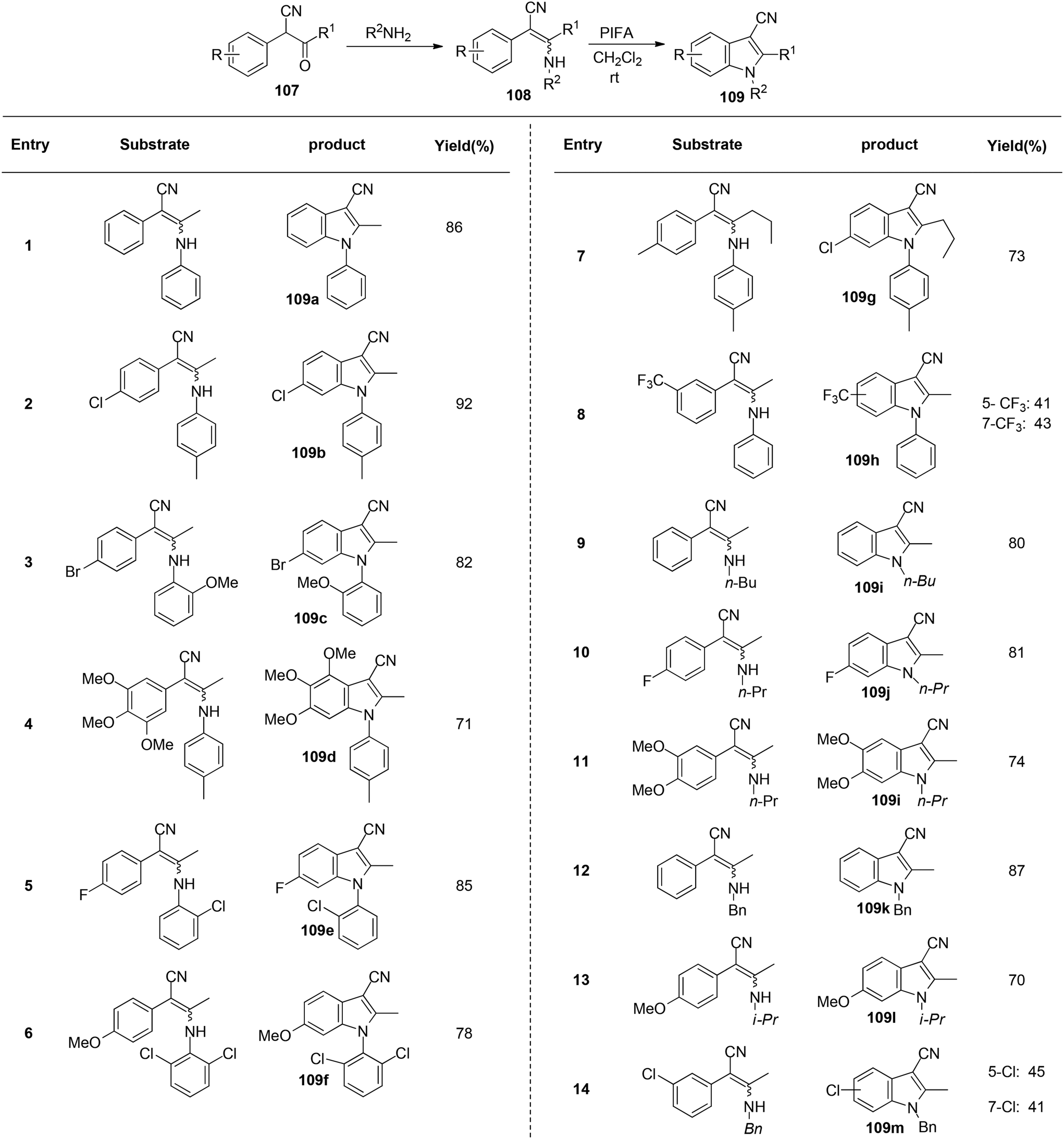
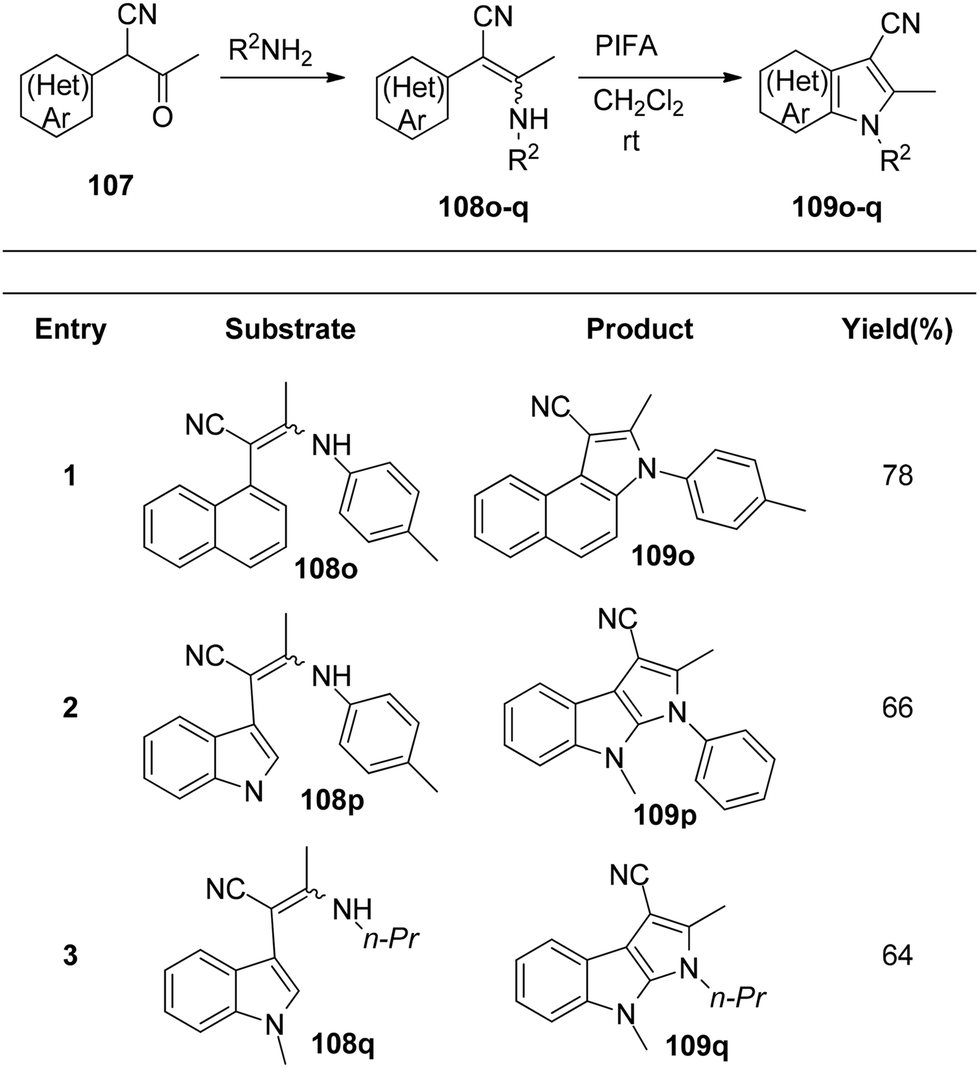
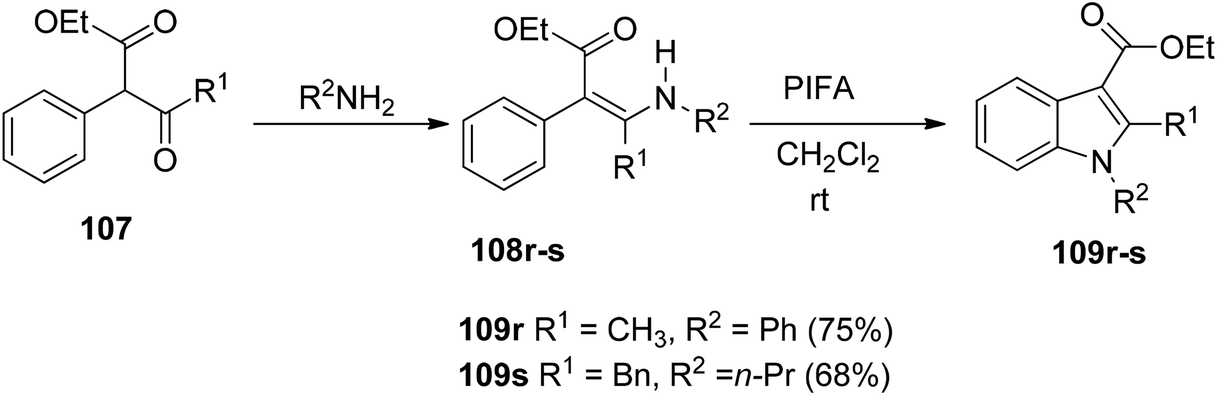
Du et al.116 have developed a method for the synthesis of a range of N-arylated and N-alkylated indole derivatives 109 through a PIFA-mediated intramolecular cyclization (Scheme 42). This innovative approach enables the formation of an indole framework by linking the N-atom in the side chain to the benzene ring 108 in the final step of synthesis. Furthermore, this method can also be employed to produce novel pyrrole-fused aromatic compounds 109o–q (Scheme 43) and N-substituted indole-3-carboxylic acid ester 109r–s (Scheme 44). PIFA actively engaged in the reaction by following a free radical pathway, specifically through single-electron oxidation. The creation of substituted indole frameworks has garnered significant interest over the years, as the indole unit has found increasingly diverse applications in both natural products and designed medicinal agents.117 This method stands out for its accessible starting materials, mild reaction conditions, and straightforward workup process.
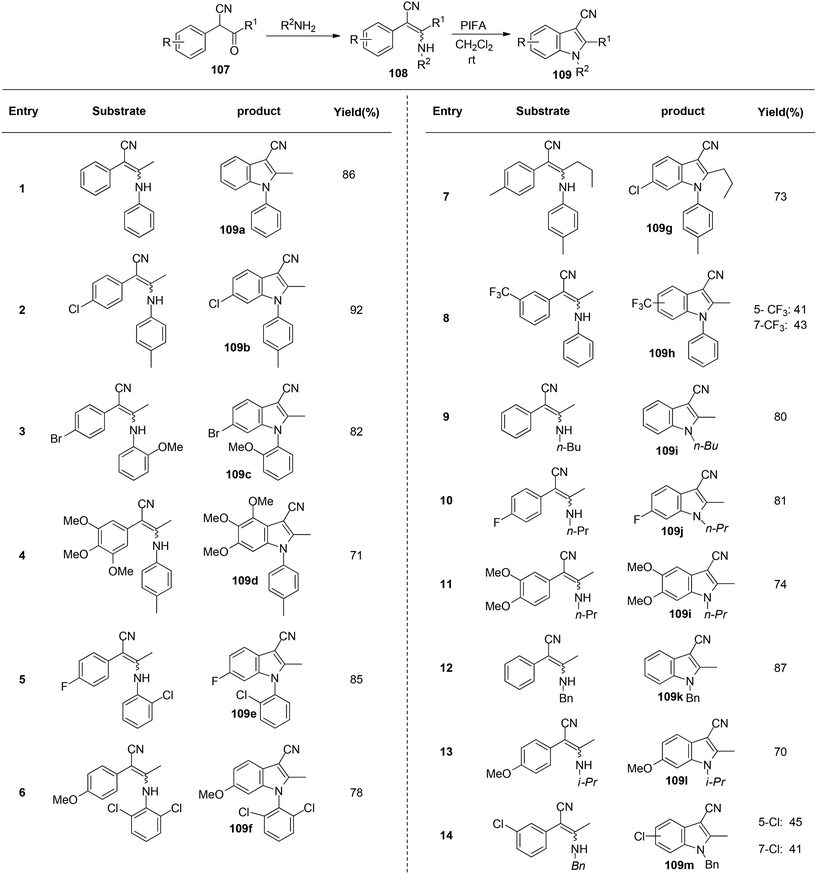 |
| | Scheme 42 Synthesis of a range of N-arylated and N-alkylated indole derivatives 109. | |
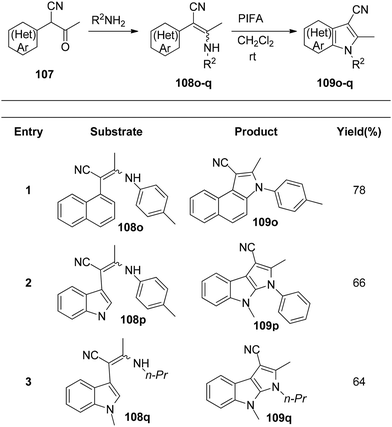 |
| | Scheme 43 Synthesis of novel pyrrole-fused aromatic compounds 109o–q. | |
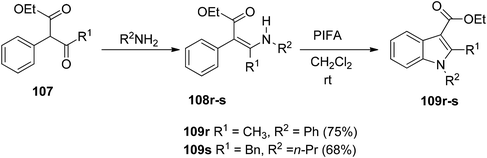 |
| | Scheme 44 Synthesis of N-arylated and N-alkylated indole derivatives 109r–s. | |
3.7. Oxidative cyclization
Some compounds, both naturally occurring and synthetic, feature isoxazolones and their heterocyclic counterparts as central structural motifs. These compounds also encompass a diverse array of bio-, physio-, and pharmacological properties.118,119 Analogues of isoxazol-3(2H)-ones are commonly utilized in agrochemistry, materials chemistry, and organic chemistry.120
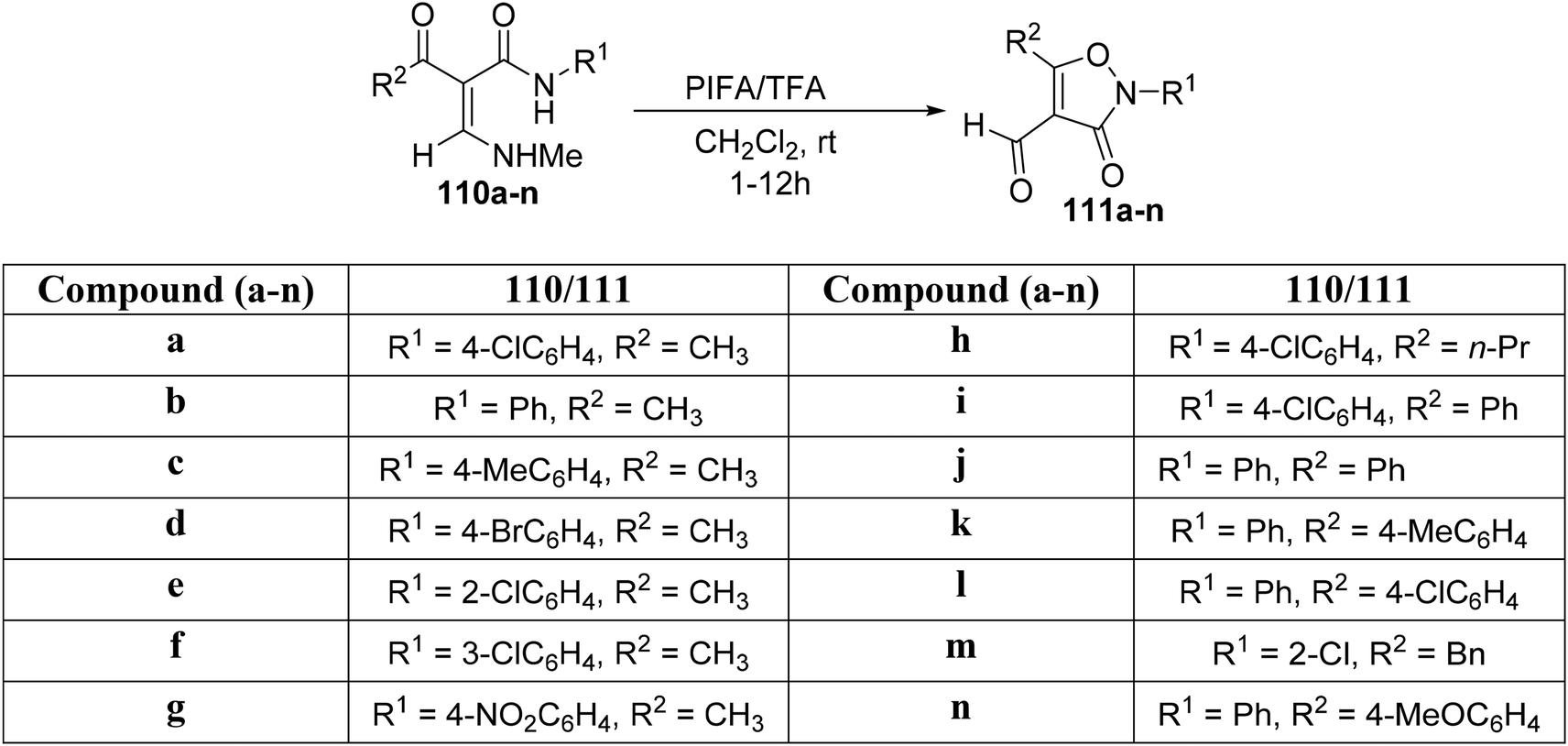
Using PIFA/TFA as the catalytic system, Yuan et al.121 observed that α-acyl acrylamides 110a–n underwent intramolecular cyclization at room temperature (Scheme 45). This chemoselective and mild oxidative transformation, free of metal catalysts, provides an alternative methodology to create O–N linkages, ultimately resulting in the formation of isoxazolone analogues 111a–n. PIFA engaged in the reaction by following the ionic pathway.
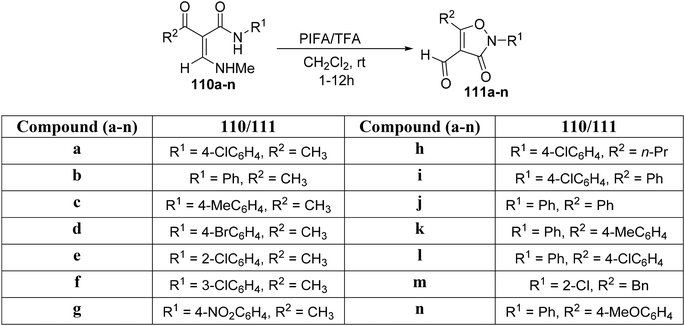 |
| | Scheme 45 Reaction scheme for isoxazol-3(2H)-ones 111a–n synthesis. | |
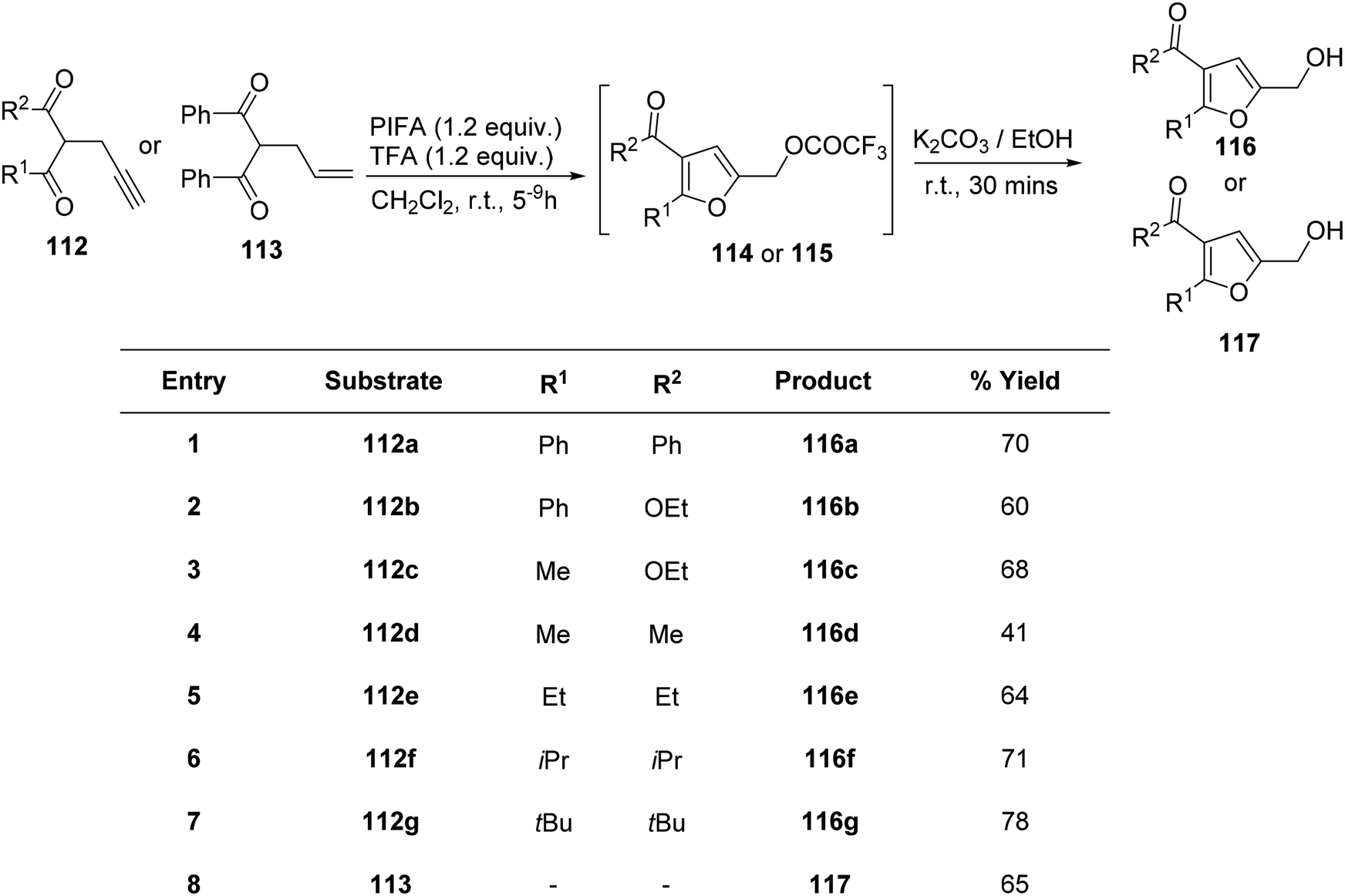
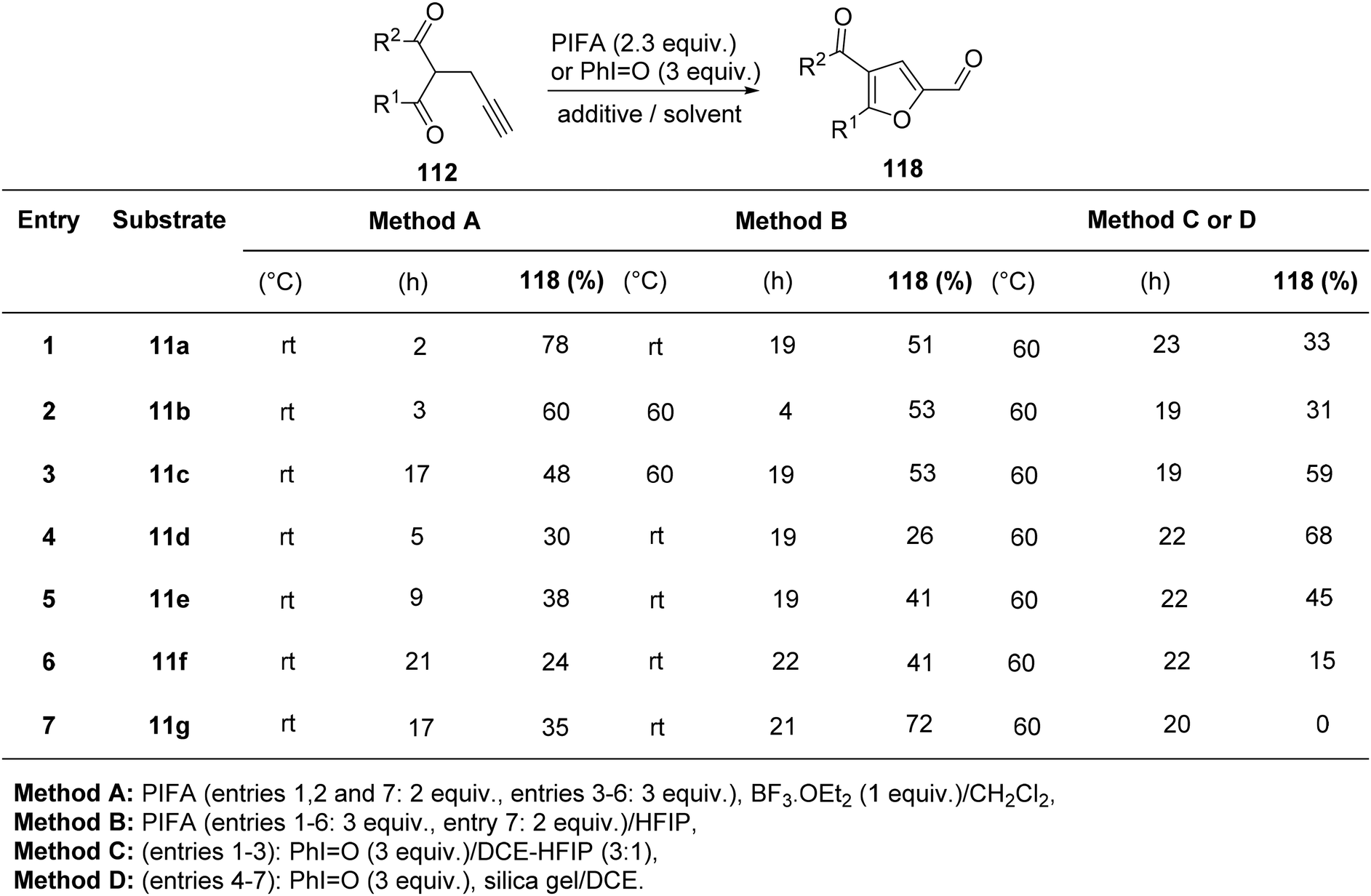
Saito et al.122 employed PIFA in the oxidative cycloisomerization of propargylated/allylated β-keto esters 112/113, leading to the formation of furfuryl alcohol analogues 116/117 (Scheme 46) as well as furfurals 118 (Scheme 47). The direct synthesis of furfurals 118 from 2-propargyl 1,3-dicarbonyl compounds was accomplished by PIFA in hexafluoroisopropanol (HFIP) or PIFA–BF3–OEt2 in CH2Cl2.
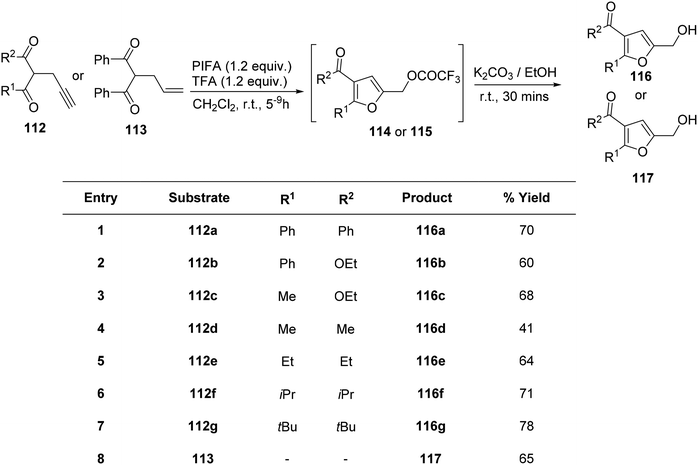 |
| | Scheme 46 The oxidative cyclo-isomerization of propargylated/allylated-1,3-diketones 112/113 to obtain analogues of furfuryl alcohols 116/117. | |
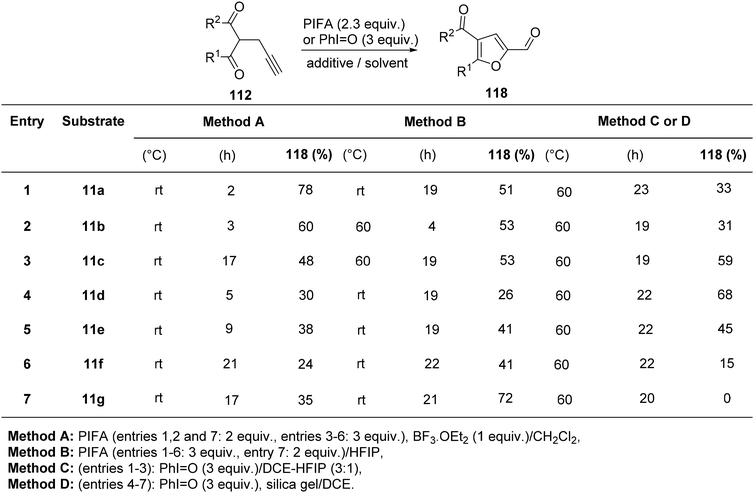 |
| | Scheme 47 The oxidative cyclo-isomerization of different propargylated-1,3-diketones 112 to obtain analogues of furfurals 118. | |
To cultivate novel motifs with enhanced potential for generating potent lead compounds featuring advantageous physicochemical attributes, structural modifications of naturally occurring compounds represent a fundamental design approach in contemporary drug discovery.123 This approach facilitates the synthesis of analogues based on natural compounds, a critical endeavor for synthetic chemists.
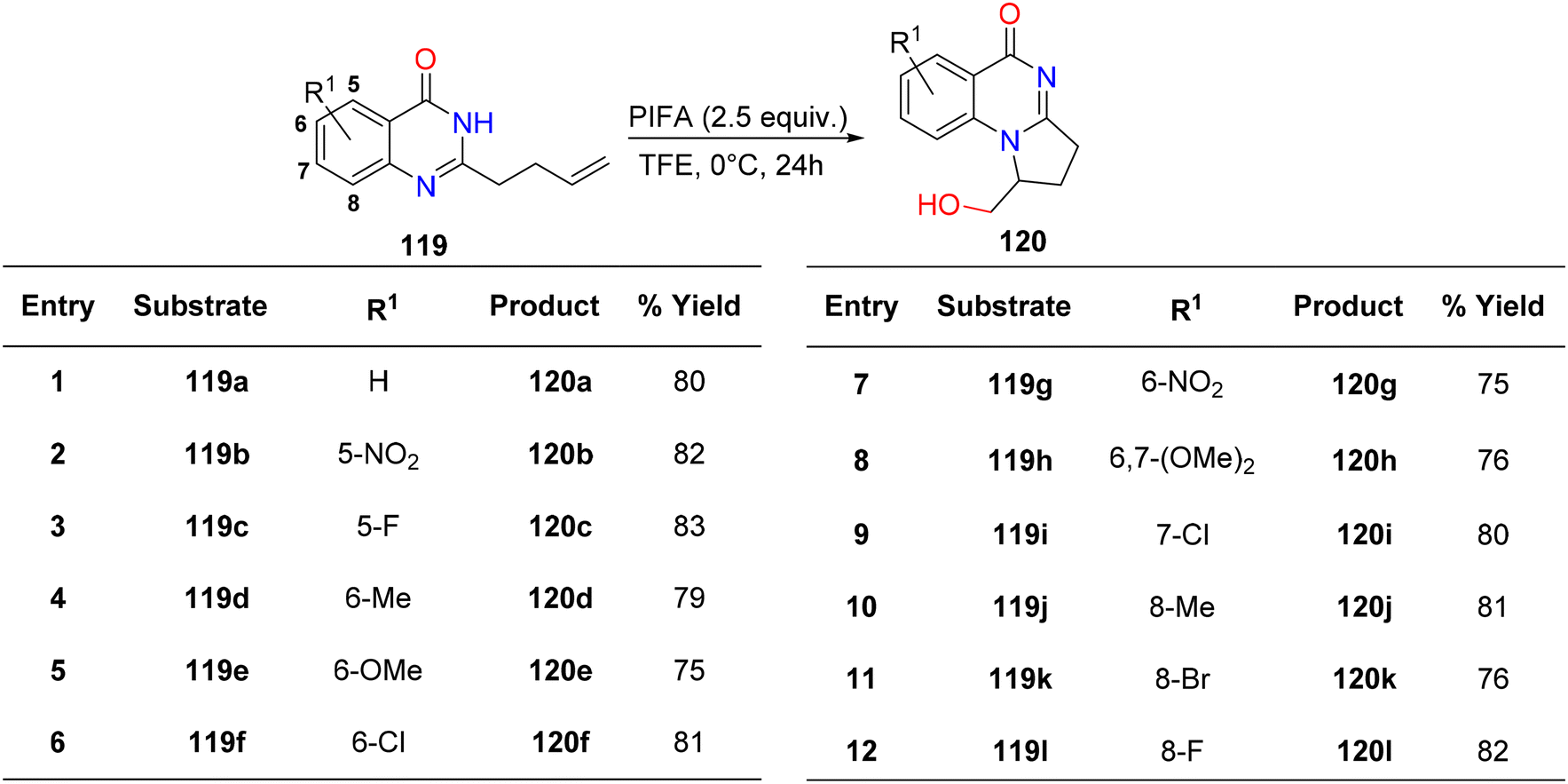
Vaskevych et al.124 detailed an oxidative and regioselective PIFA-mediated synthetic methodology for the synthesis of dihydropyrroloquinazolinones 120a–l, structural analogues of vasicinone alkaloids, derived from quinazolinones 119a–l. All quinazolones 119a–l underwent 5-exo-trig cyclization, resulting in the desired products 120a–l obtained with yields ranging from 75–83% (Scheme 48). PIFA engaged in the reaction by following the ionic pathway.
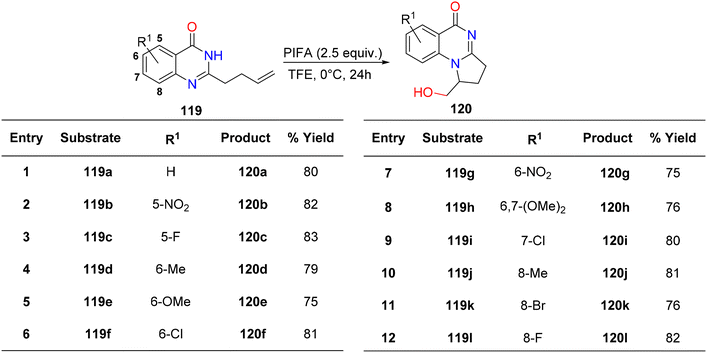 |
| | Scheme 48 Synthetic scheme for analogues of dihydropyrroloquinazolinones 120a–l. | |
Various domains within the field of chemistry have significantly contributed to the advancement of organic synthesis over the years, with a major emphasis on identifying efficient methodologies to facilitate complex transformations.125
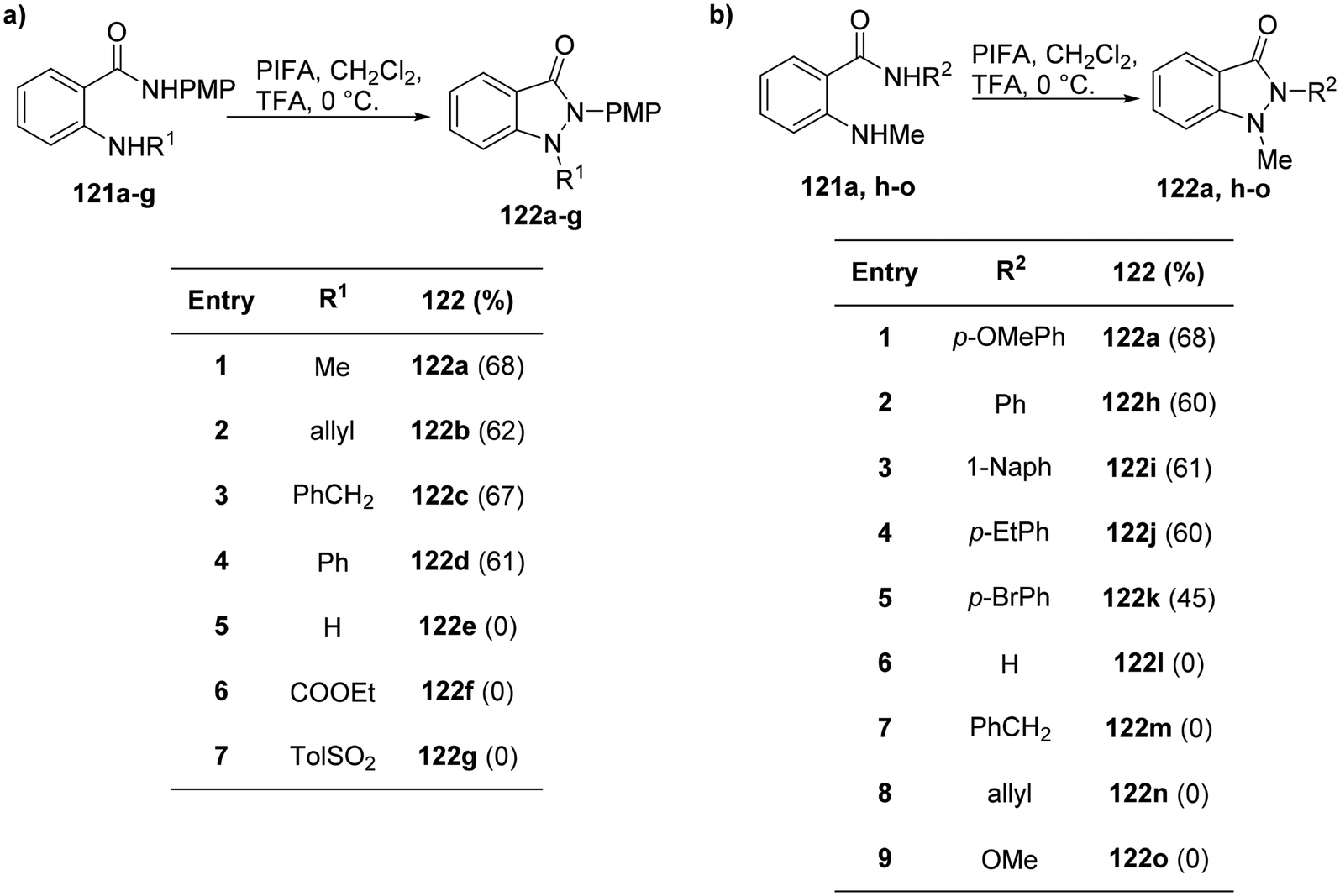
Correa et al.126 effectively harnessed the potential of cost-effective, easy-to-handle PIFA to synthesize functionalized indazolones 122 (Scheme 49). The use of PIFA on amide derivatives 121 led to the generation of N-acylated nitrenium ions, representing the pioneering instance of successful intramolecular trapping of these ions by the amine group. This breakthrough allowed for the synthesis of N-heterocycles through N–N bond formation.
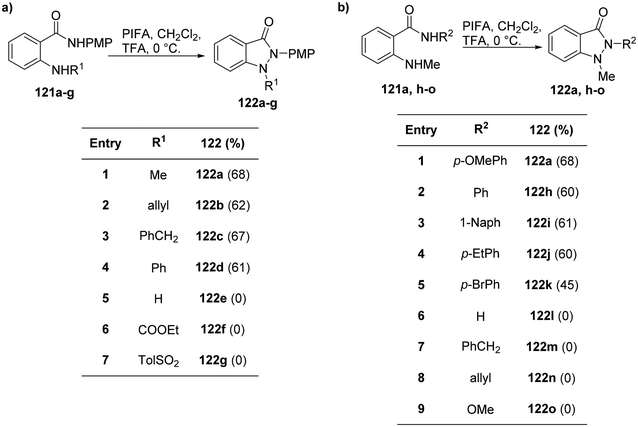 |
| | Scheme 49 a) Synthesis of indazolones 122a–g. (b) Synthesis of indazolones 122a,h–o. | |
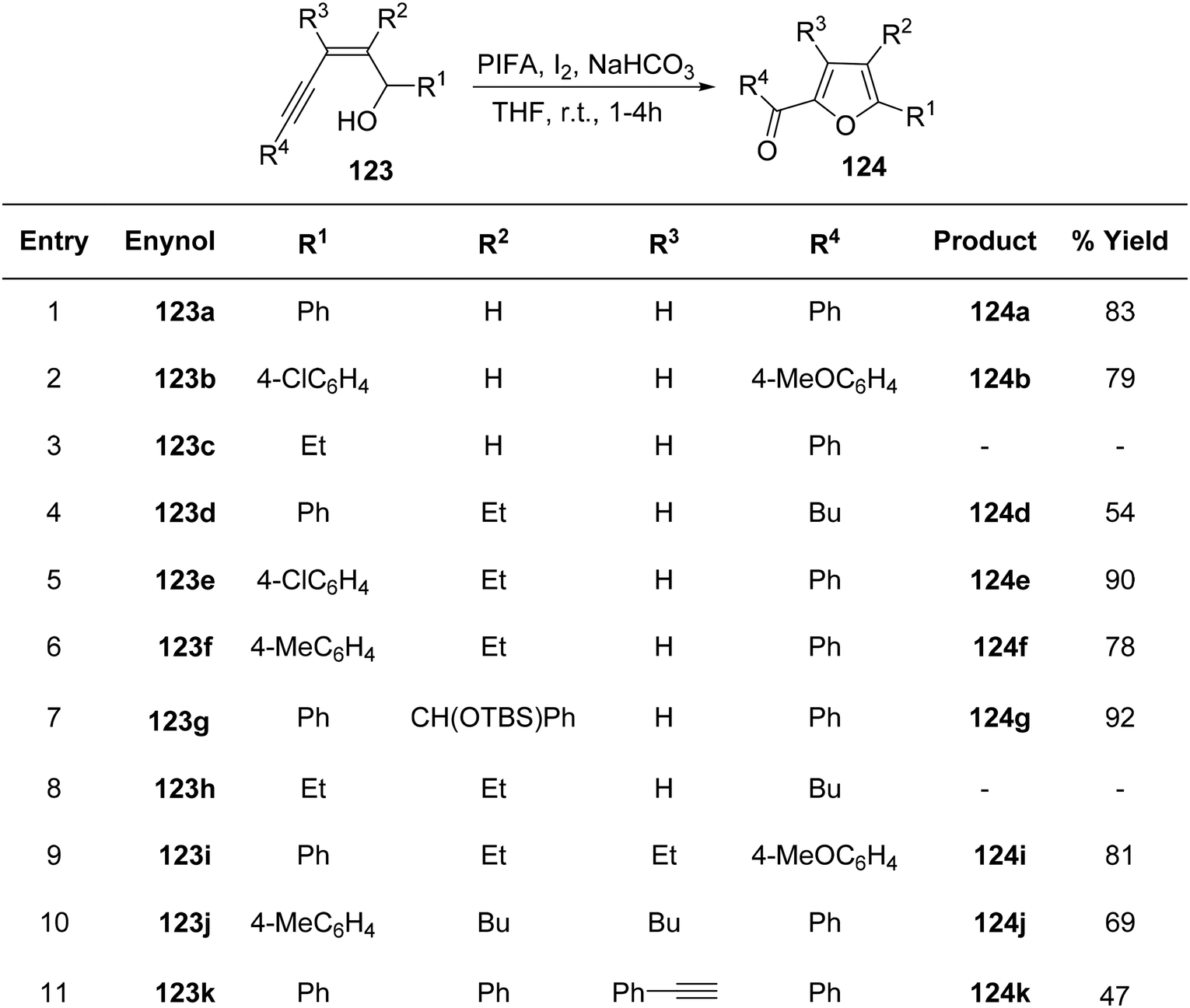
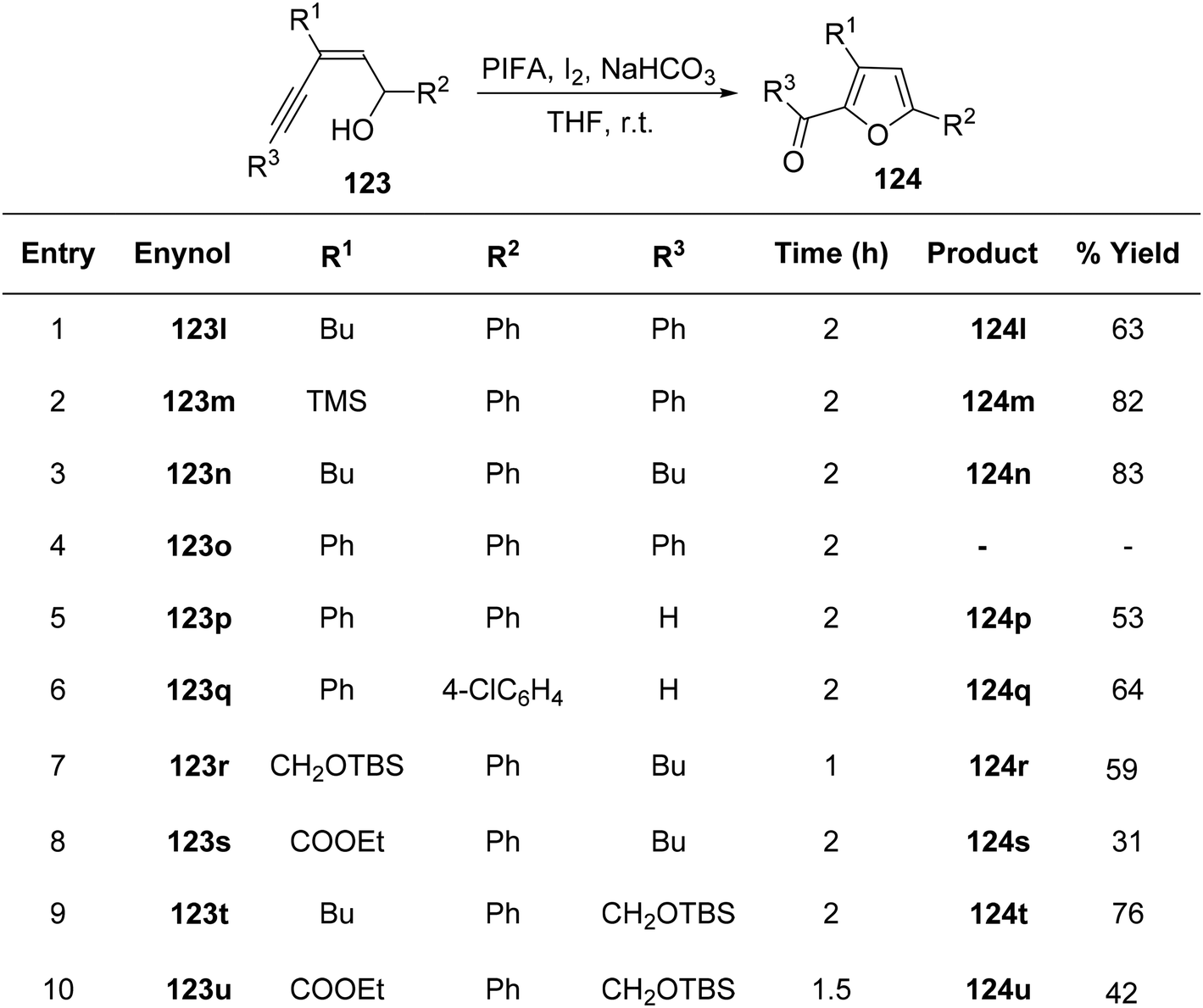
To achieve the synthesis of 2-acyl furans at room temperature, Du et al.127 employed an oxidative cycloisomerization approach using cis-enynols in the presence of PIFA/molecular iodine. These reactions demonstrate efficacy with a diverse range of substrates, making them valuable for the synthesis of furan analogues. The proposition is that trifluoroacetylhypoiodite, generated in situ, serves as the activator for alkyne 123 in these reactions. Employing this methodology, C-1/C-5 functionalized cis-enynols have been processed, yielding 2-acyl furan analogues 124 in notably high yields (Schemes 50 and 51). PIFA engaged in the reaction by following the ionic pathway.
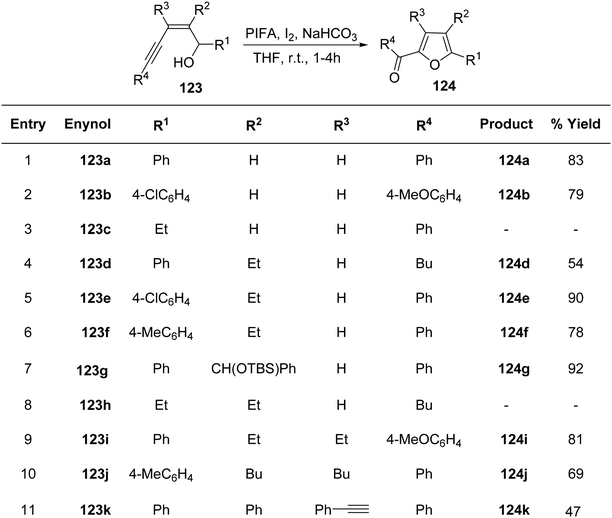 |
| | Scheme 50 Synthetic pathway for 2-acyl furan analogues 124a–k in presence of PIFA/I2. | |
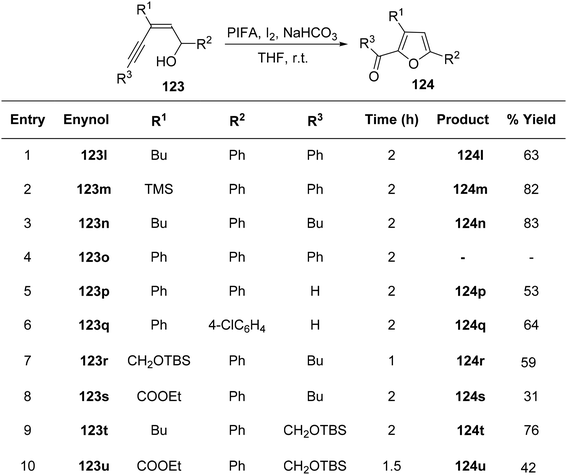 |
| | Scheme 51 Synthetic methodology for 2-acyl furans 124l–u catalyzed by PIFA/I2. | |
This method surpasses the IBX synthetic pathway, showcasing superior attributes such as mild reaction conditions and shortened reaction durations.
1,3,4-Oxadiazole analogues constitute prominent heterocyclic motifs with diverse therapeutic properties,128,129 serving as crucial intermediates in the synthesis of organic compounds.130
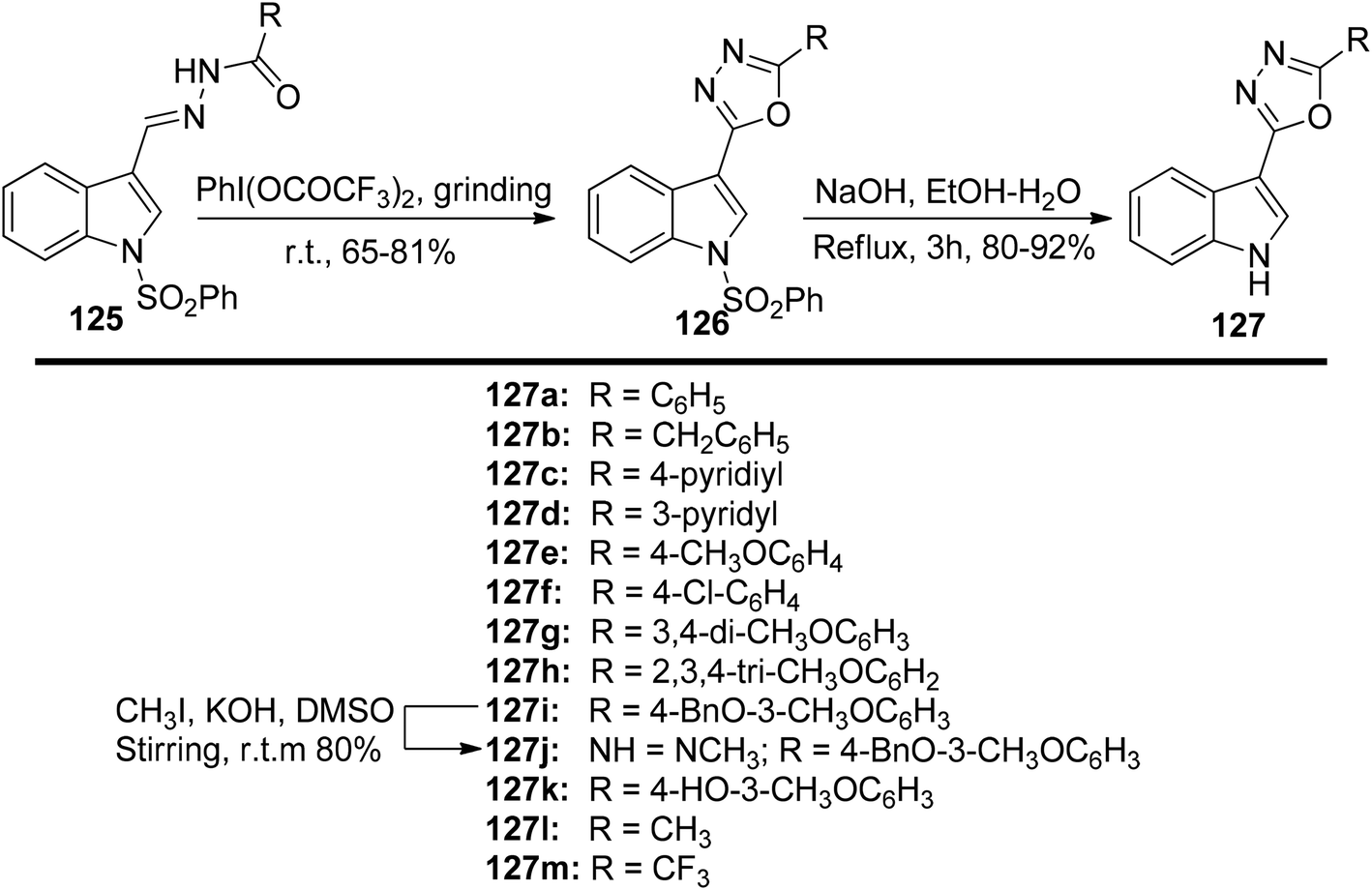
In a solvent-free environment, Kumar et al.131 presented a PIFA-mediated synthetic methodology for obtaining analogues of 1,3,4-oxadiazoles 127 (Scheme 52). The key step in this approach involved PIFA-mediated solvent-free cyclization of N-acylated hydrazones 125. The in vitro anti-cancer activity of the synthesized 1,3,4-oxadiazoles 127a–m was assessed for their potential against various cancer cell lines (PC3, LnCaP, DU145, PaCa2, MD-MDA231, and MCF7).
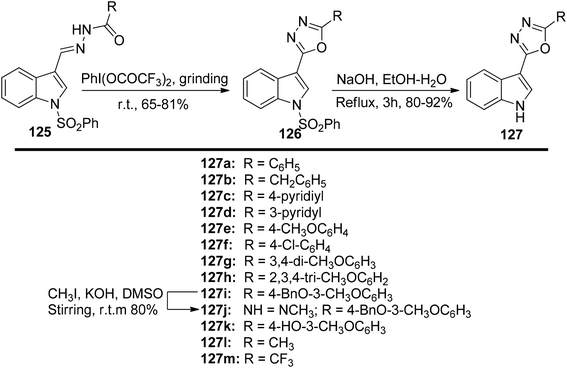 |
| | Scheme 52 Synthetic scheme for indolyl-1,3,4-oxadiazoles 126 as well as 5-(3′-indolyl)-1,3,4-oxadiazole analogues 127a–m. | |
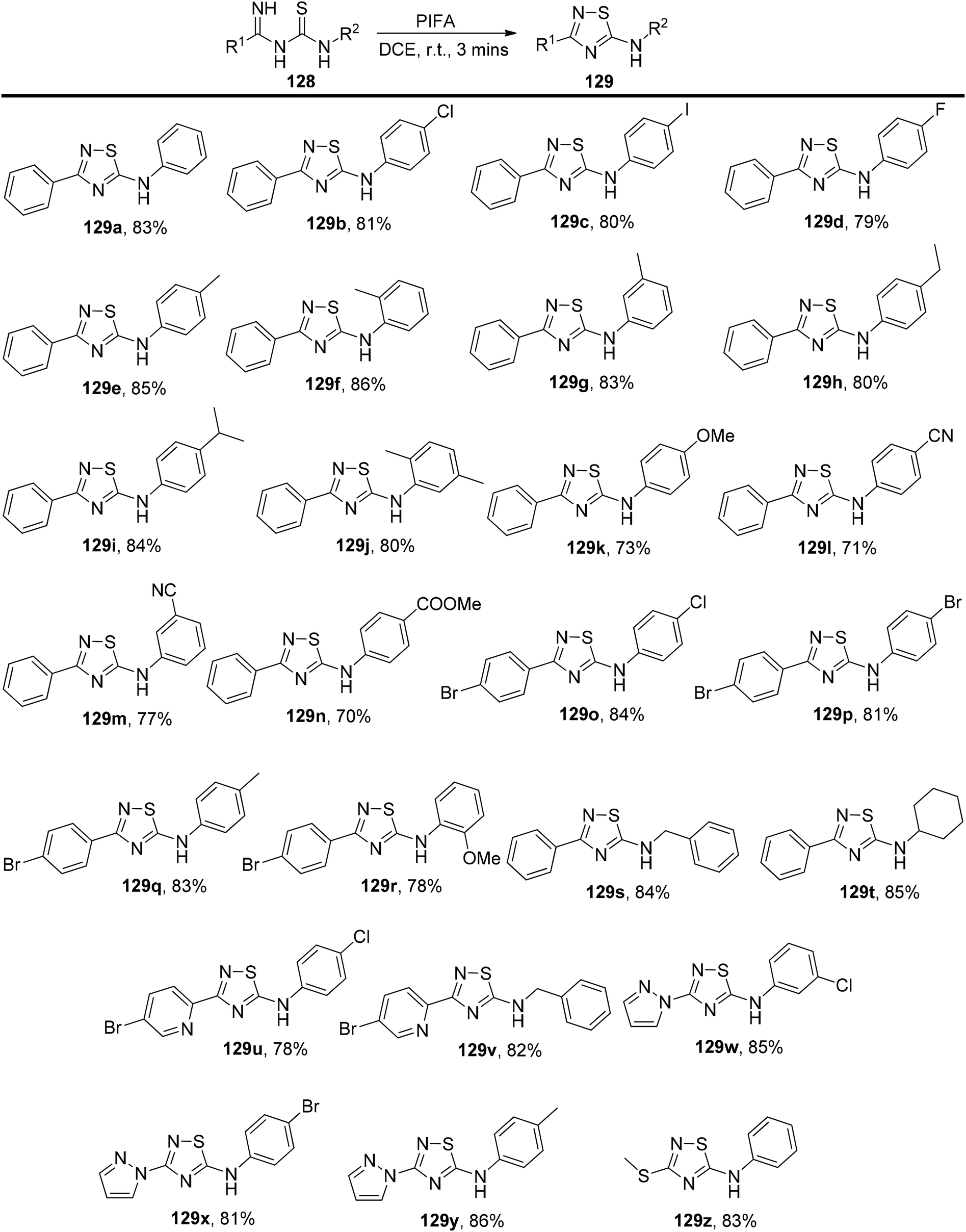
Mariappan et al.132 have introduced an exceptionally efficient method for producing 3-substituted-5-arylamino-1,2,4-thiadiazoles 129. This innovative process involves the intramolecular oxidative formation of S–N bonds within imidoyl thioureas 128, with PIFA acting as the catalyst. PIFA engaged in the reaction by following the ionic pathway. This method stands out due to its metal-free character, broad applicability across different substrates, rapid reaction kinetics, and its capacity to deliver good to excellent yields, all while using readily available starting materials. It represents a convenient and gentle approach for synthesizing 3-substituted 5-arylamino-1,2,4-thiadiazoles 129 in a remarkably short timeframe at room temperature, as showcased in Scheme 53.
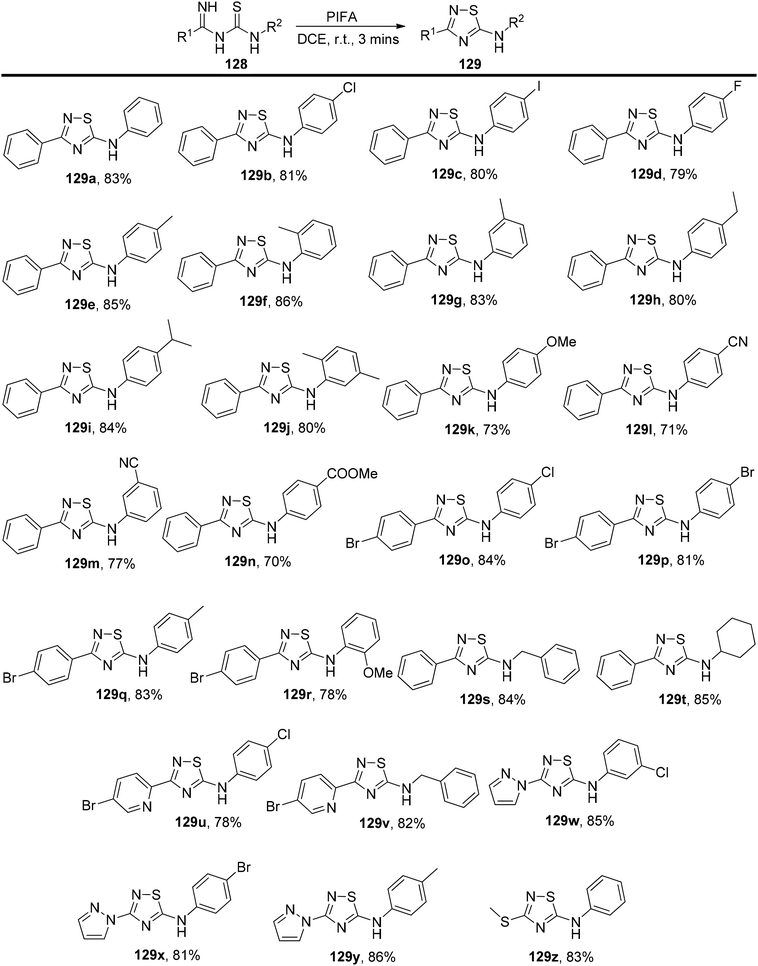 |
| | Scheme 53 Synthesis of compounds 129a–z. | |
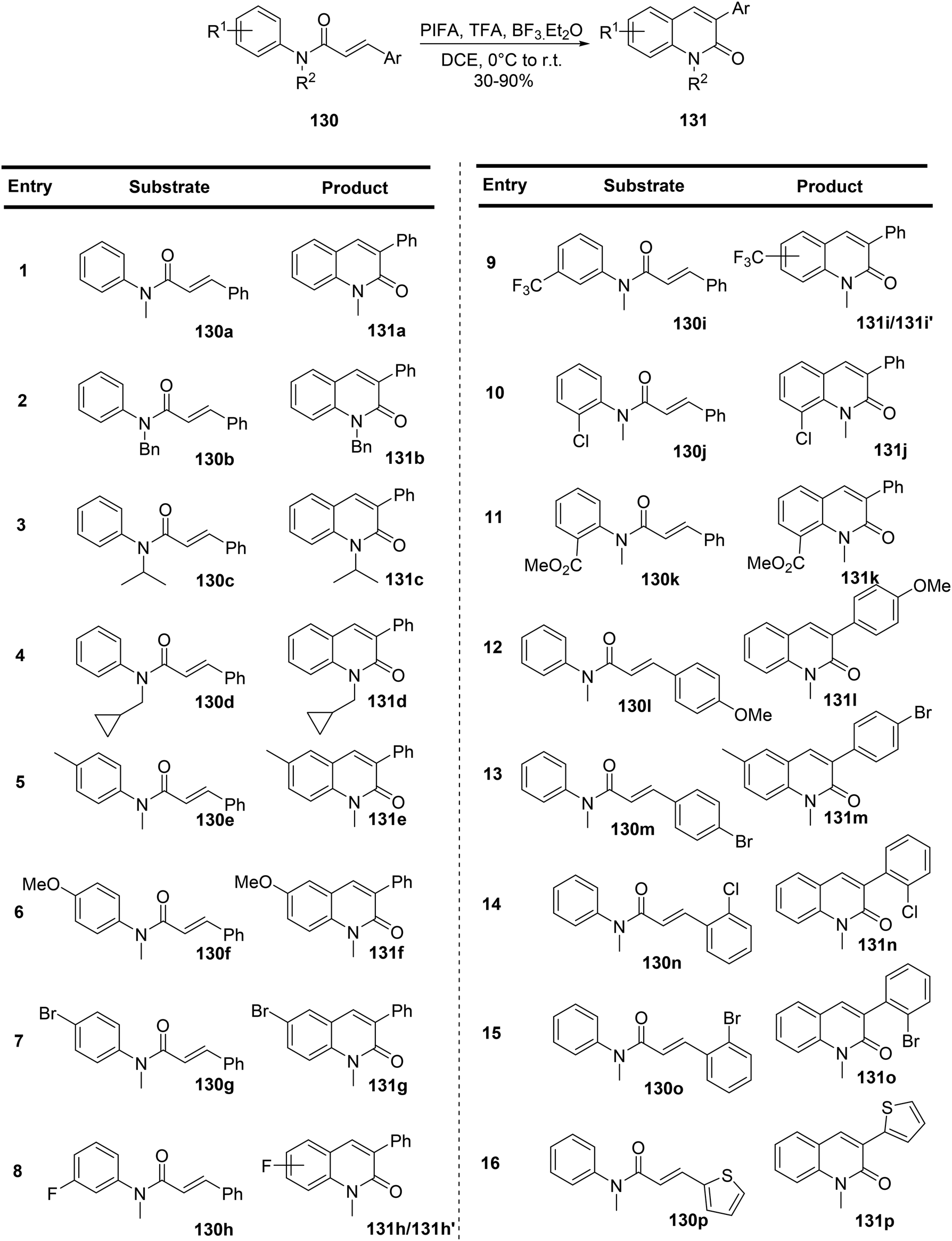
Liu et al.133 have introduced a novel and versatile method for synthesizing a range of 3-arylquinolin-2-one compounds 131. This method involves the reaction of readily available N-methyl-N-phenylcinnamamides 130 with PIFA in the presence of Lewis acids (Scheme 54). PIFA engaged in the reaction by following the ionic pathway. What sets this approach apart is its ability to facilitate metal-free oxidative C(sp2)–C(sp2) bond formation and a distinctive 1,2-aryl migration.
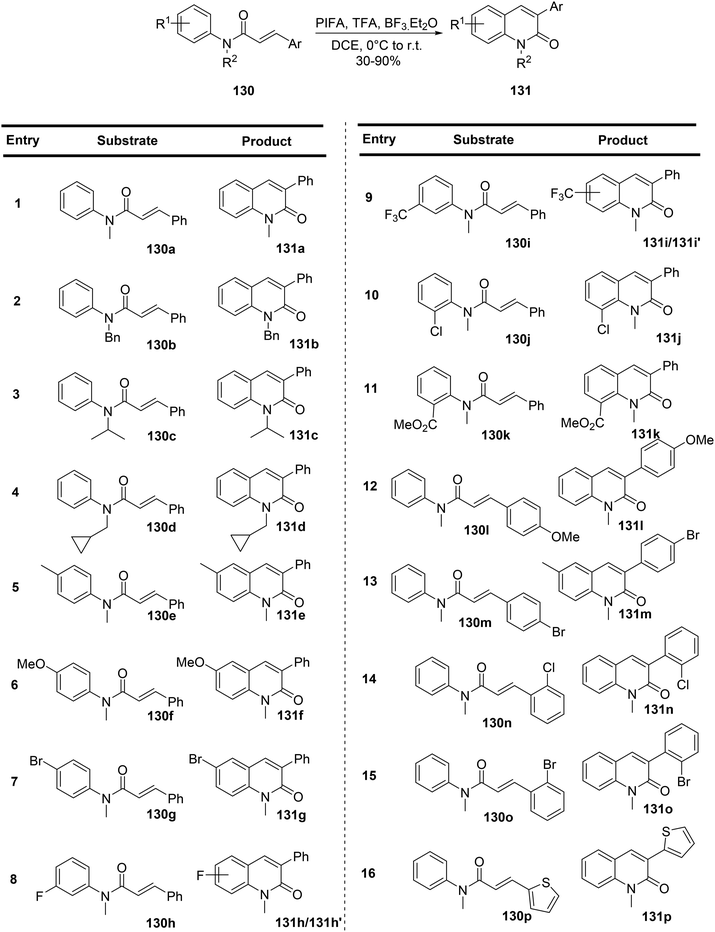 |
| | Scheme 54 Synthesis of 4-unsubstituted 3-arylquinol-2-one 131via PIFA-mediated C–C bond formation and 1,2-aryl shift. | |
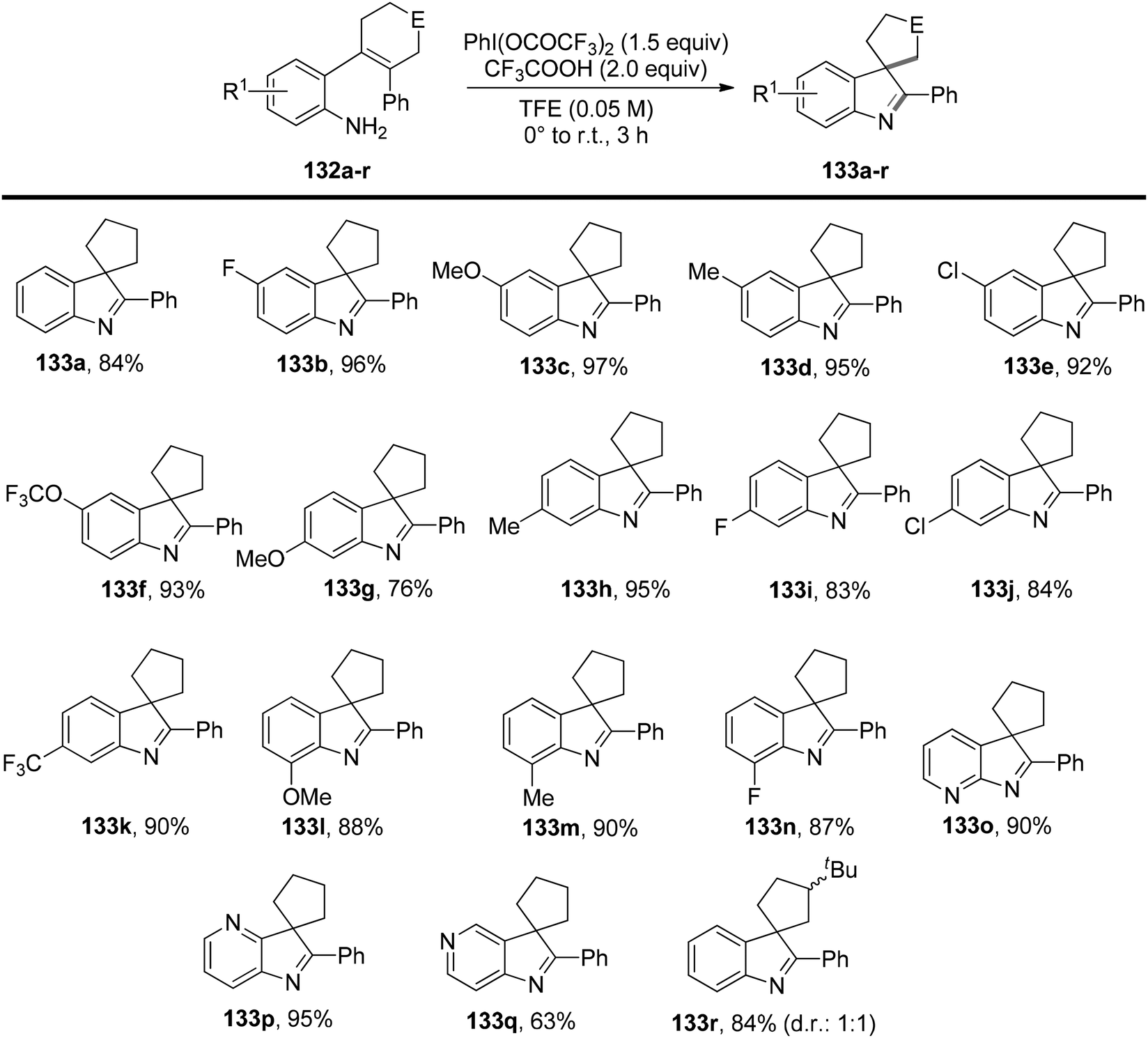
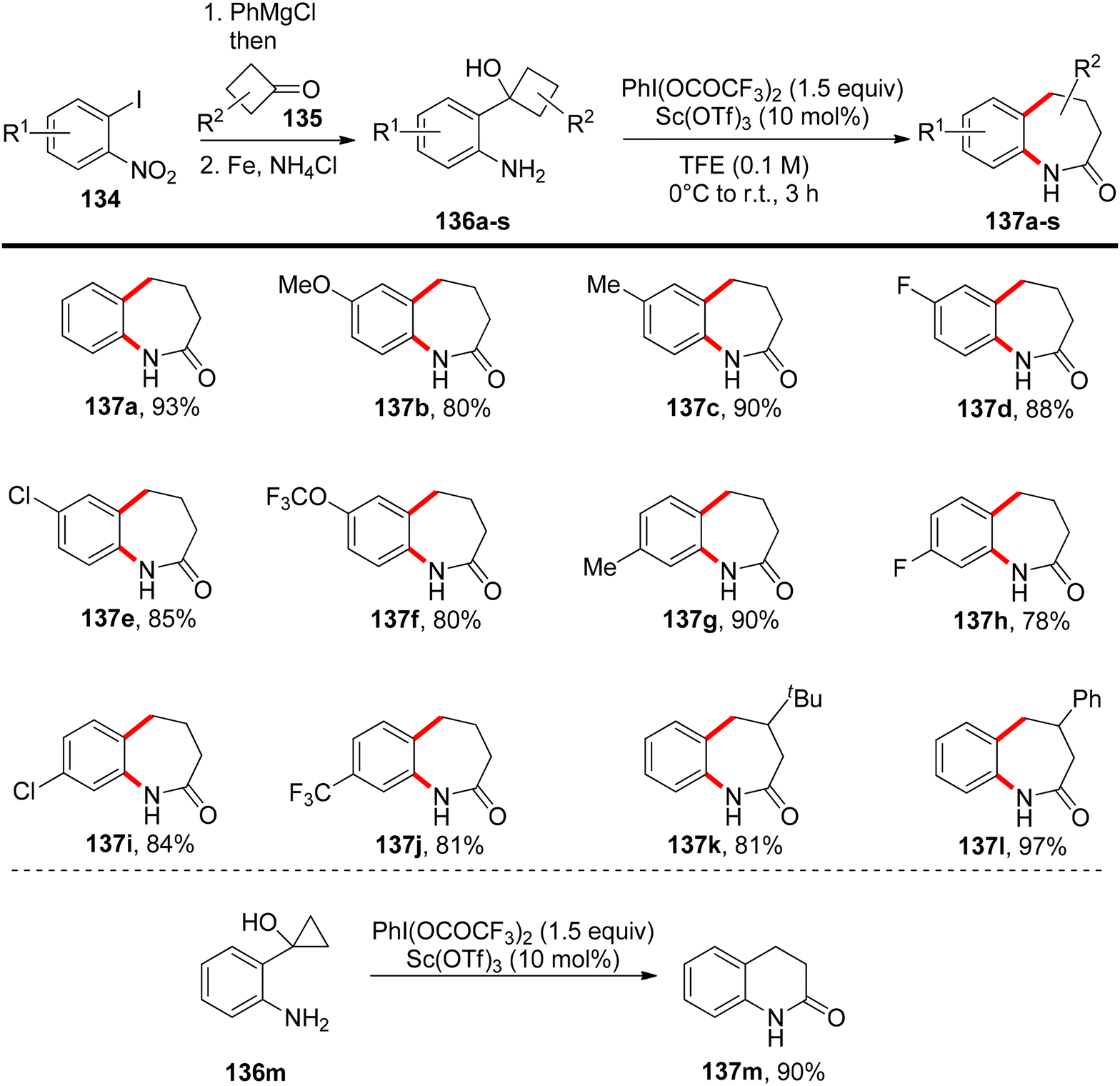
Deng et al.134 have introduced a method for oxidizing 2-substituted anilines 132 at low temperatures without the need for protecting groups. This process generates an electrophilic N-aryl nitrenoid intermediate capable of participating in the formation of C–NAr bonds, enabling the construction of functionalized N-heterocycles 133 (Scheme 55). When 2-substituted anilines are exposed to PIFA and trifluoroacetic acid, or 10 mol% Sc(OTf)3, it triggers the formation of the nitrenoid intermediate. Subsequently, this leads to the selective and efficient formation of C–NAr and C–C bonds, resulting in the creation of spirocyclic or bicyclic 3H-indoles or benzazepinones 133 or 137. PIFA engaged in the reaction by following the ionic pathway. The generation of 3H-indoles occurs as the N-aryl nitrogen reactive intermediate is captured by the ortho-alkenyl substituent, triggering a 1,2 shift. The direction of this shift is controlled by the stabilization of the partial positive charge and the reduction of interactions between the aryl C–H and hydrogen atoms on the contracting ring. By altering the identity of the ortho substituent on a cyclobutanol 136, access to benzazepinones 137 is achieved through a selective and stereospecific ring-expansion reaction (Scheme 56).
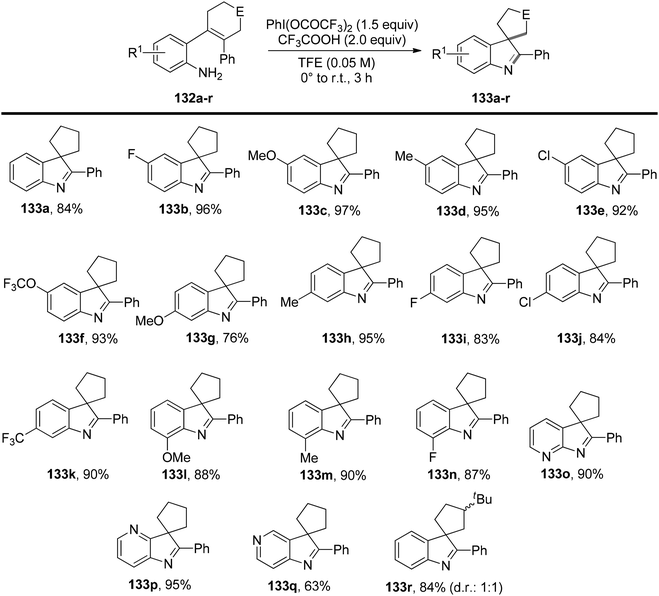 |
| | Scheme 55 Investigation of the scope and limitations of the oxidative generation of N-aryl nitrenes. | |
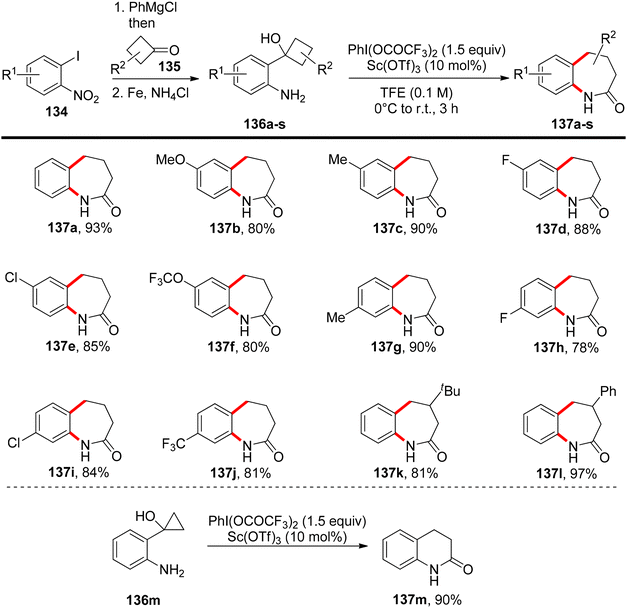 |
| | Scheme 56 Construction of benzazepinones 137 from 2-cyclobutanol-substituted anilines 136. | |
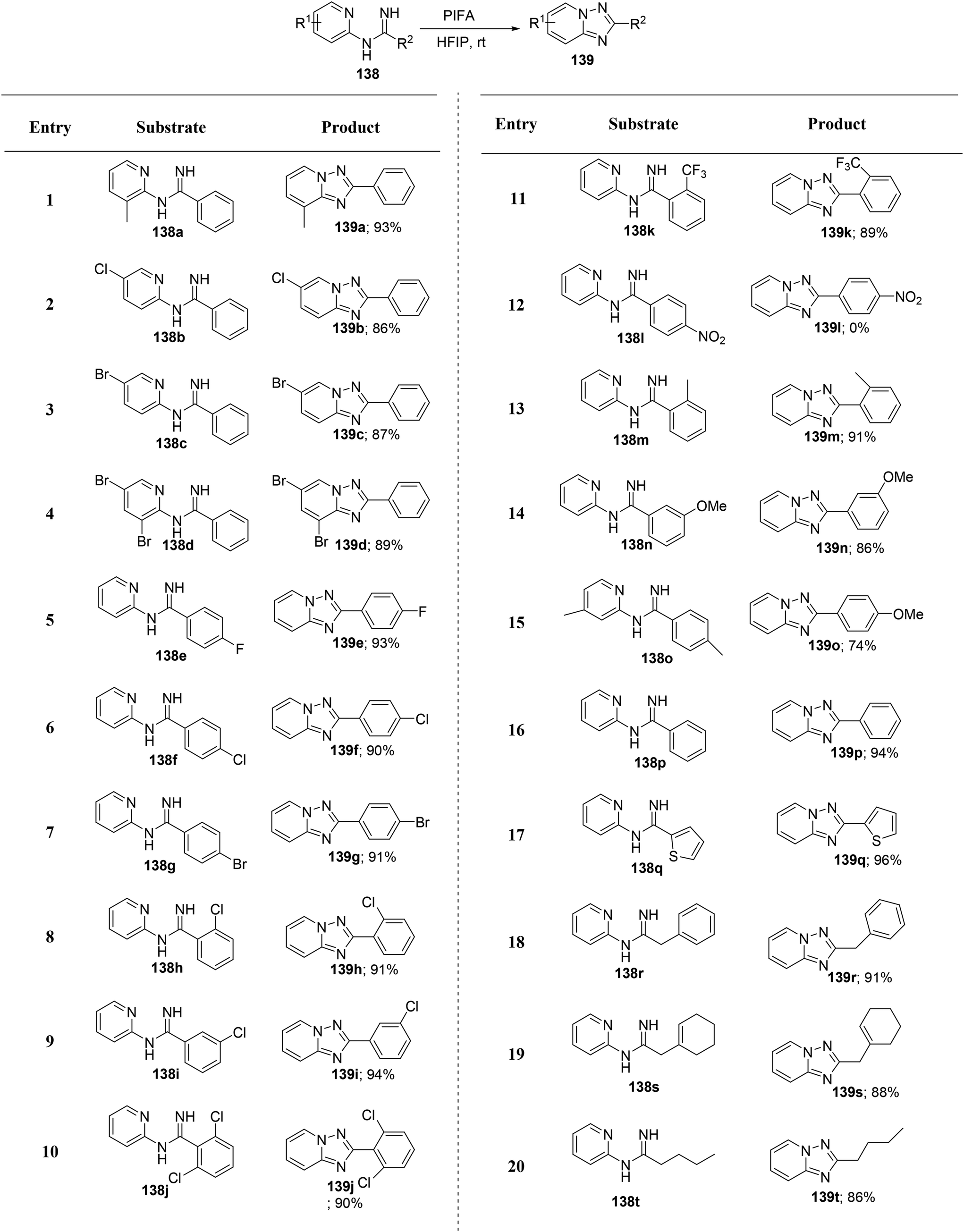
Zheng et al.135 have introduced an efficient method for synthesizing biologically significant 1,2,4-triazolo[1,5-a]pyridines 139 from N-(pyridin-2-yl)benzimidamides 138, utilizing PIFA-mediated intramolecular annulation (Scheme 57). This innovative approach enables the convenient construction of the 1,2,4-triazolo[1,5-a]pyridine framework through direct metal-free oxidative N–N bond formation. The method stands out for its brief reaction time and high yields, representing a novel pattern of N–N bond formation mediated by PIFA. PIFA engaged in reactions via the ionic pathway, contributing to the process through its involvement in the ionic mechanisms. Remarkably, this method accommodates a broad range of substrates, offers mild reaction conditions, and delivers high yields, rendering it exceptionally efficient for click chemistry applications.
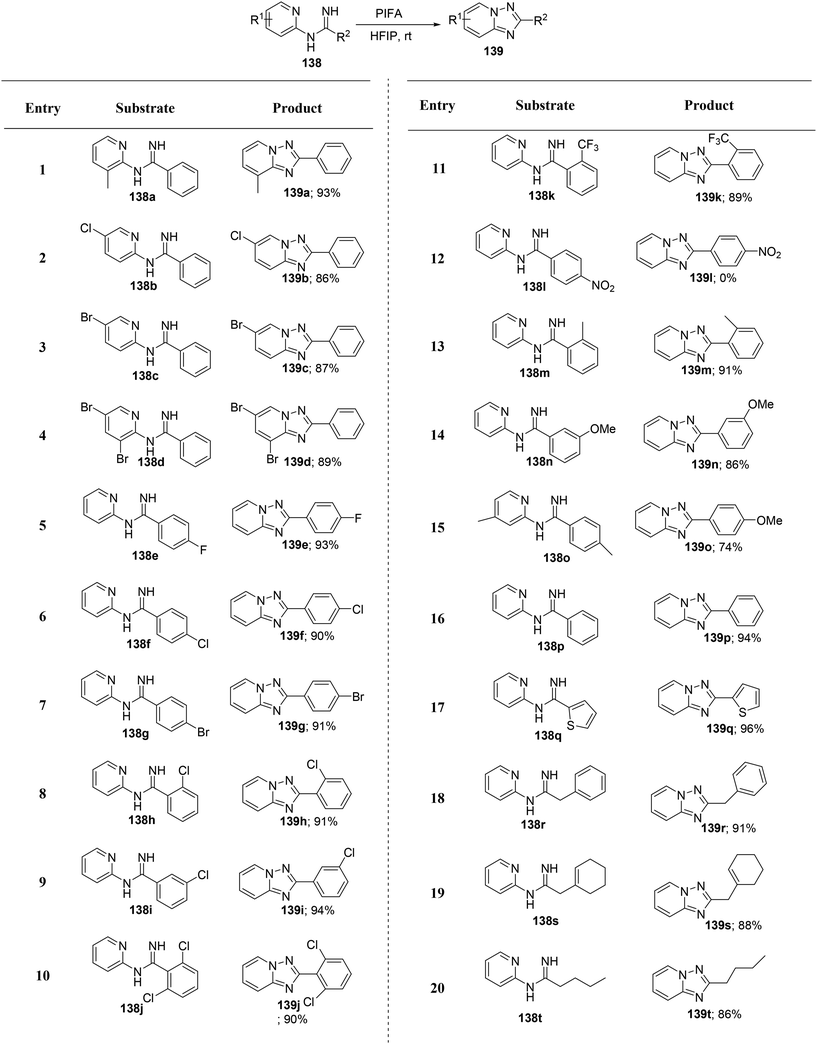 |
| | Scheme 57 Synthesis of 1,2,4-triazolo[1,5-a]pyridines 139. | |
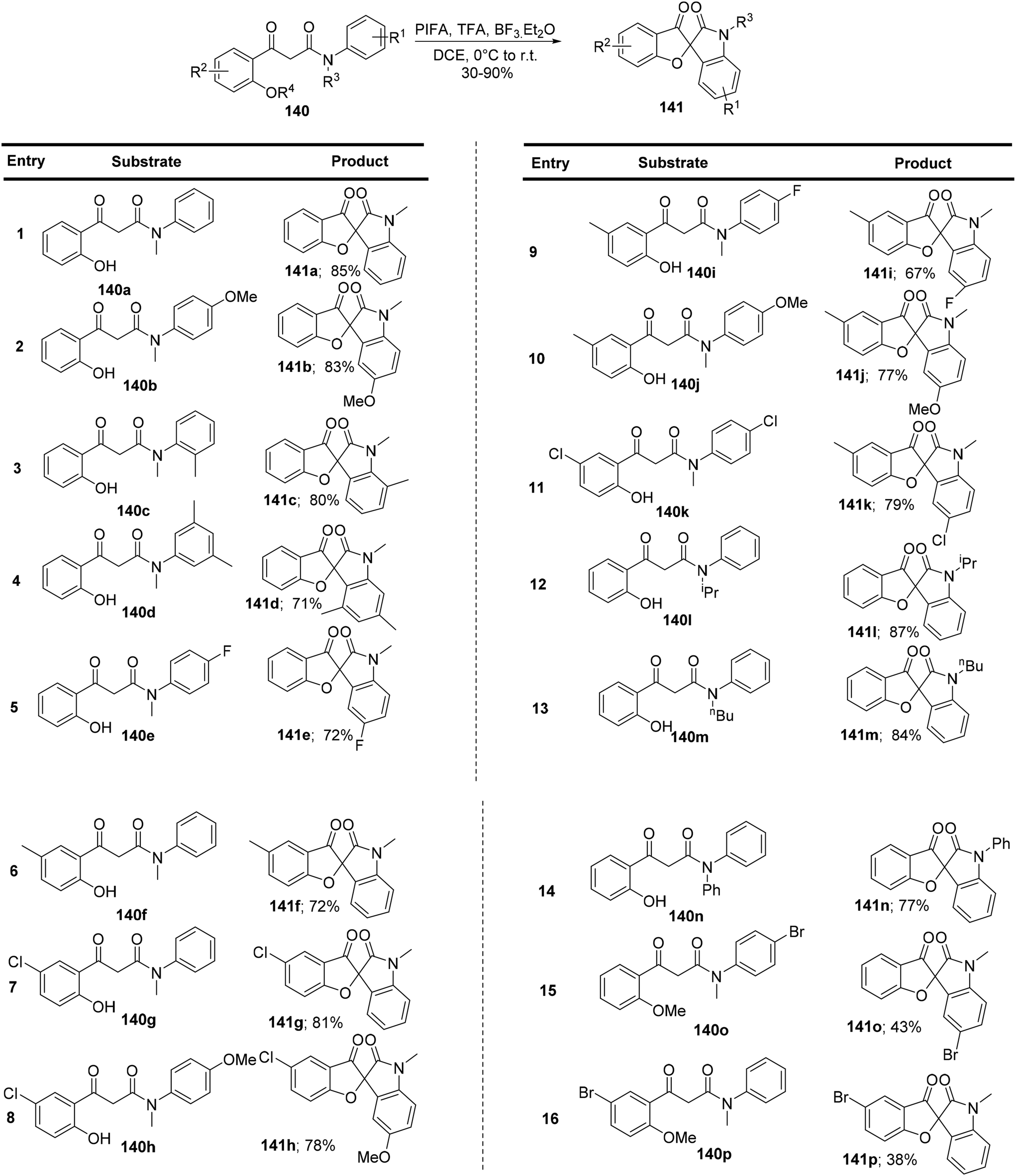
Sun et al.136 have successfully developed an innovative method for synthesizing a novel class of spirofurooxindole molecules 141. This process, mediated by hypervalent iodine, involves a cascade of C–O and C–C bond formations under metal-free and mild reaction conditions. They achieved this by conveniently converting 3-(2-hydroxyphenyl)-3-oxo-N-phenyl propanamides 140 and their derivatives into previously undocumented spirofurooxindoles 141 (Scheme 58). PIFA engaged in reactions via the ionic pathway. Control experiments substantiated that this spirocyclization process unfolds as a sequence of oxidative reactions, with the formation of a C–O bond preceding the formation of a C–C bond.
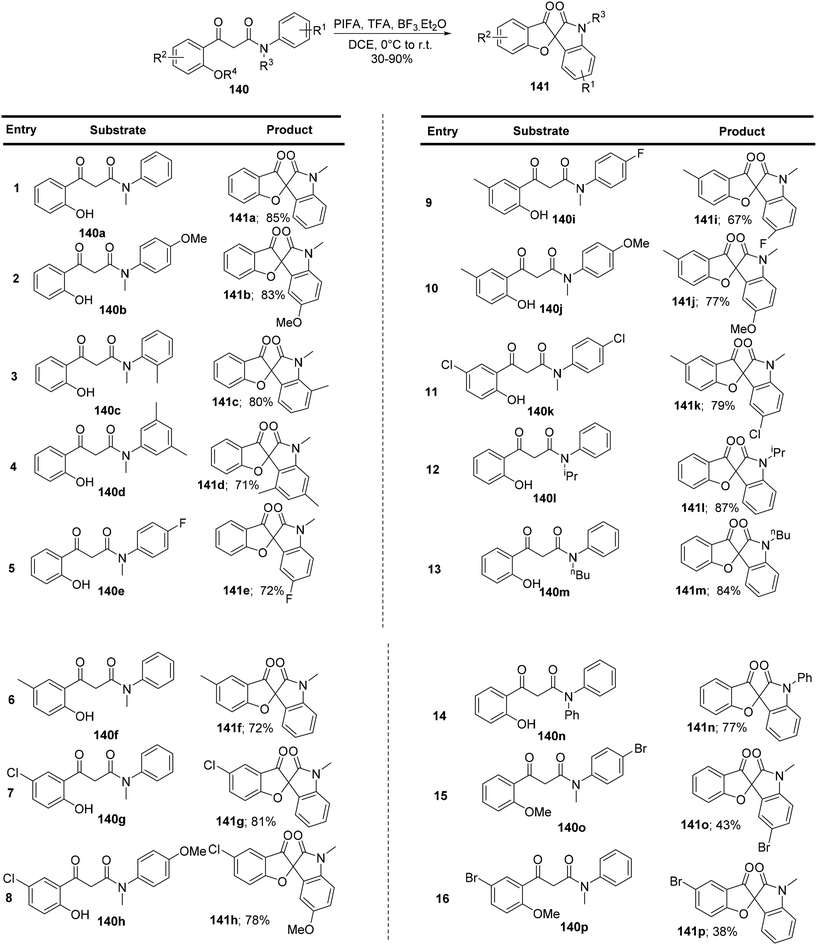 |
| | Scheme 58 Synthesis of spirofurooxindole molecules 141. | |
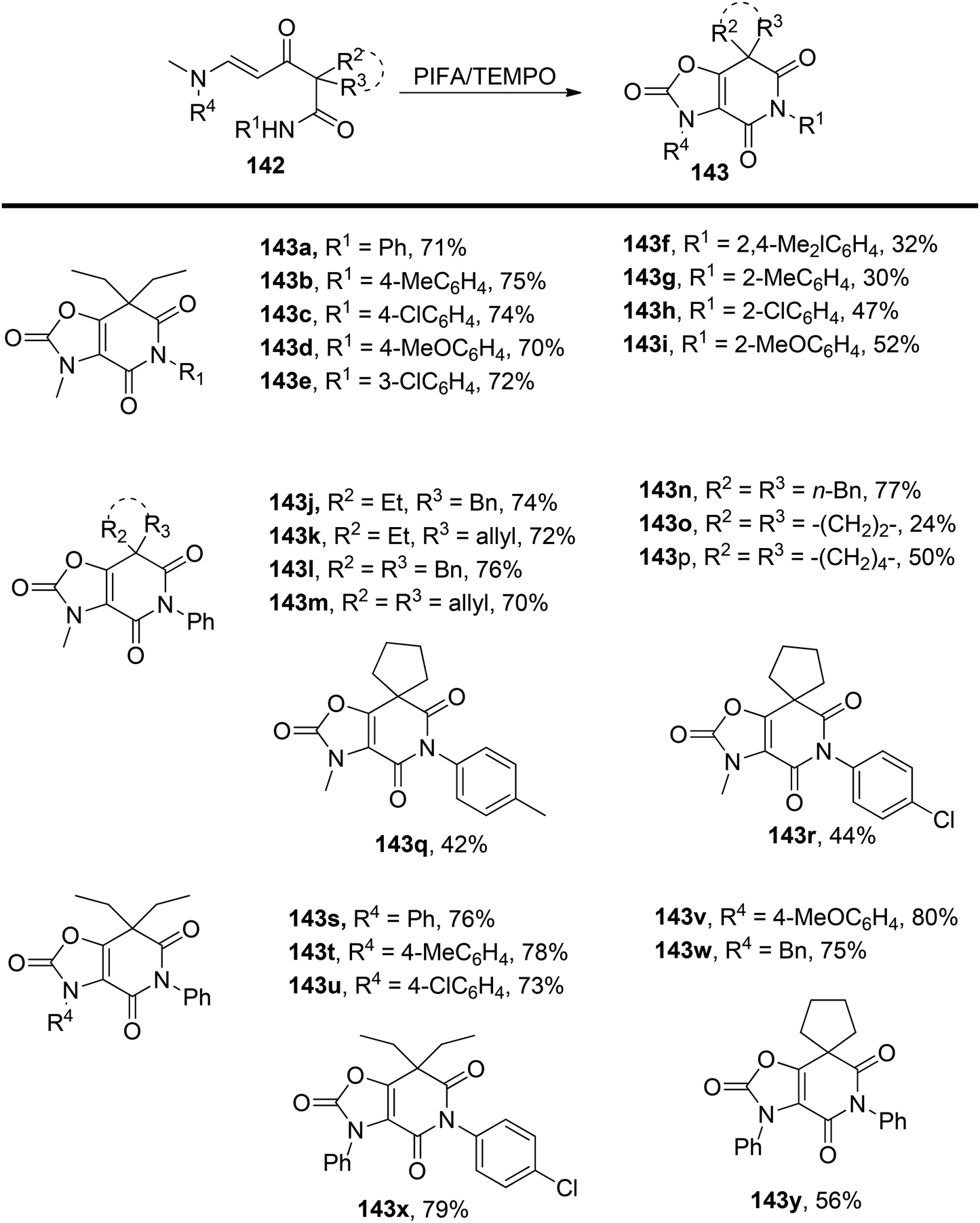
Yuan et al.137 have introduced an innovative oxidative cascade cyclization process involving α-[(β-dimethylamino)propenoyl]-alkyl amides 142, under the mediation of PIFA and TEMPO. This transformation comprises a radical-induced aziridination, ring-opening reaction, and intramolecular O-alkylation sequence (Scheme 59). PIFA actively engaged in the reaction by following a free radical pathway, specifically through hydrogen abstraction. The noteworthy features of this reaction are its mild reaction conditions, straightforward execution, and its capacity to yield good results while maintaining high levels of chemoselectivity and regioselectivity. As a result, it offers an efficient and convenient approach for synthesizing polysubstituted 3,7-dihydro-oxazolo[4,5-c]pyridine-2,4,6-(5H)-triones 143.
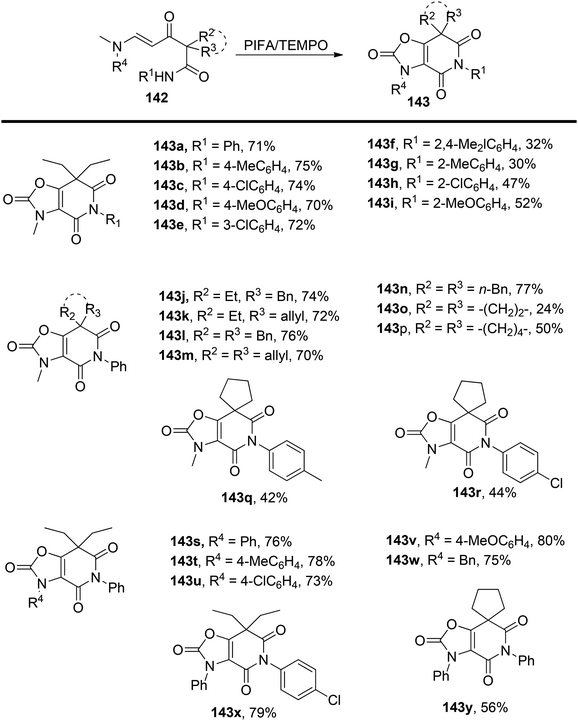 |
| | Scheme 59 Synthesis of polysubstituted 3,7-dihydro-oxazolo[4,5-c]pyridine-2,4,6-(5H)-triones 143. | |
3.8. Oxidative reaction
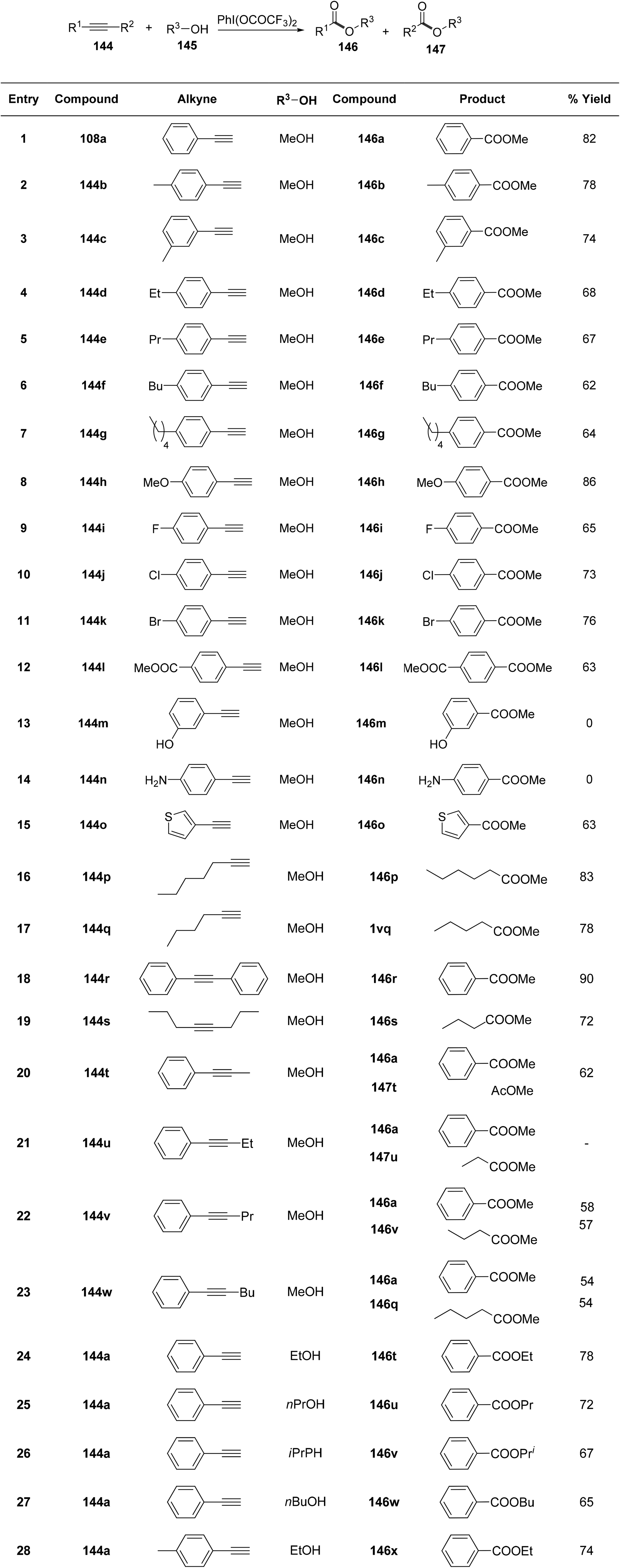
Jiang et al.138 elucidated a mild, PIFA-mediated practical synthetic pathway for the conversion of alkyne 144 or alcohol 145 into carboxylic ester 146 or 147. The reaction exhibited tolerance to a wide array of substrates with diverse functionalities, resulting in the synthesis of carboxylic esters in high yields (Scheme 60). PIFA actively engaged in the reaction by following an ionic pathway. Mechanistic inquiries disclosed that the transformation proceeds through intermediates such as hydroxyethanones and ethanediones.
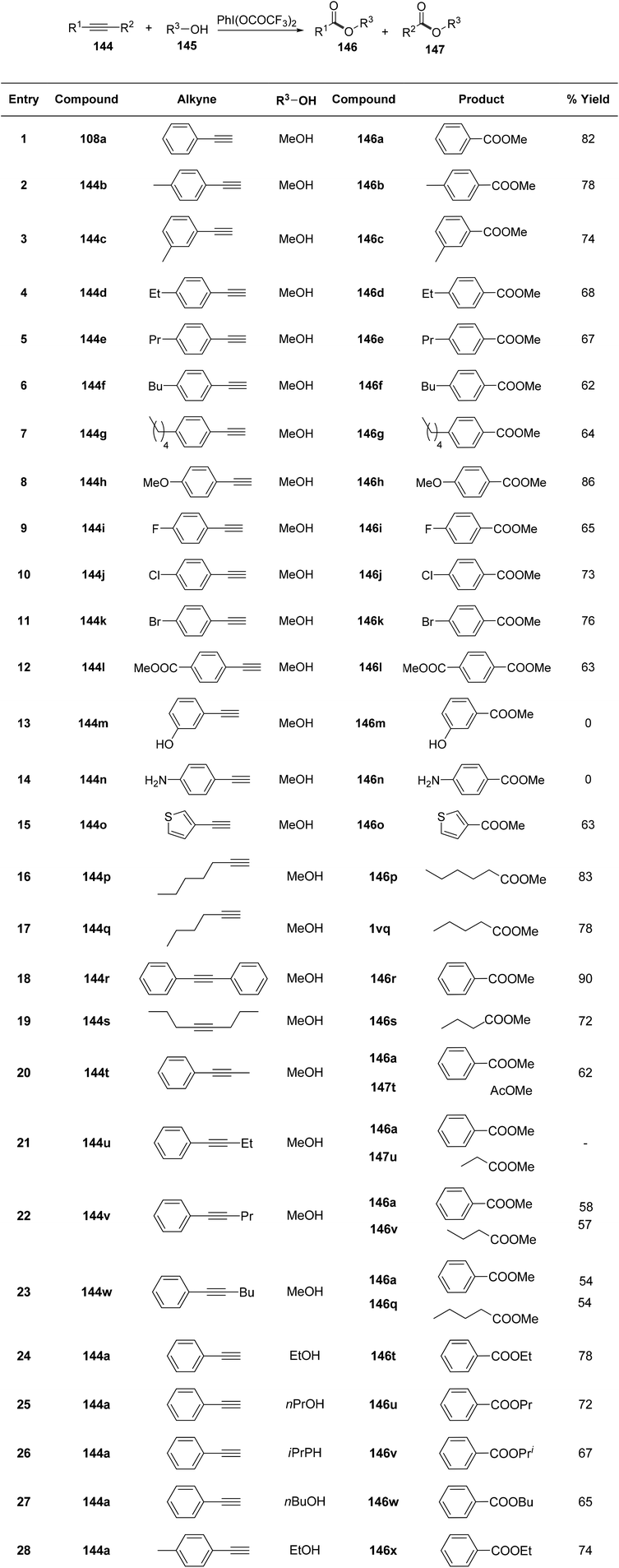 |
| | Scheme 60 Synthetic pathway for oxidative esterification with varied substrates. | |
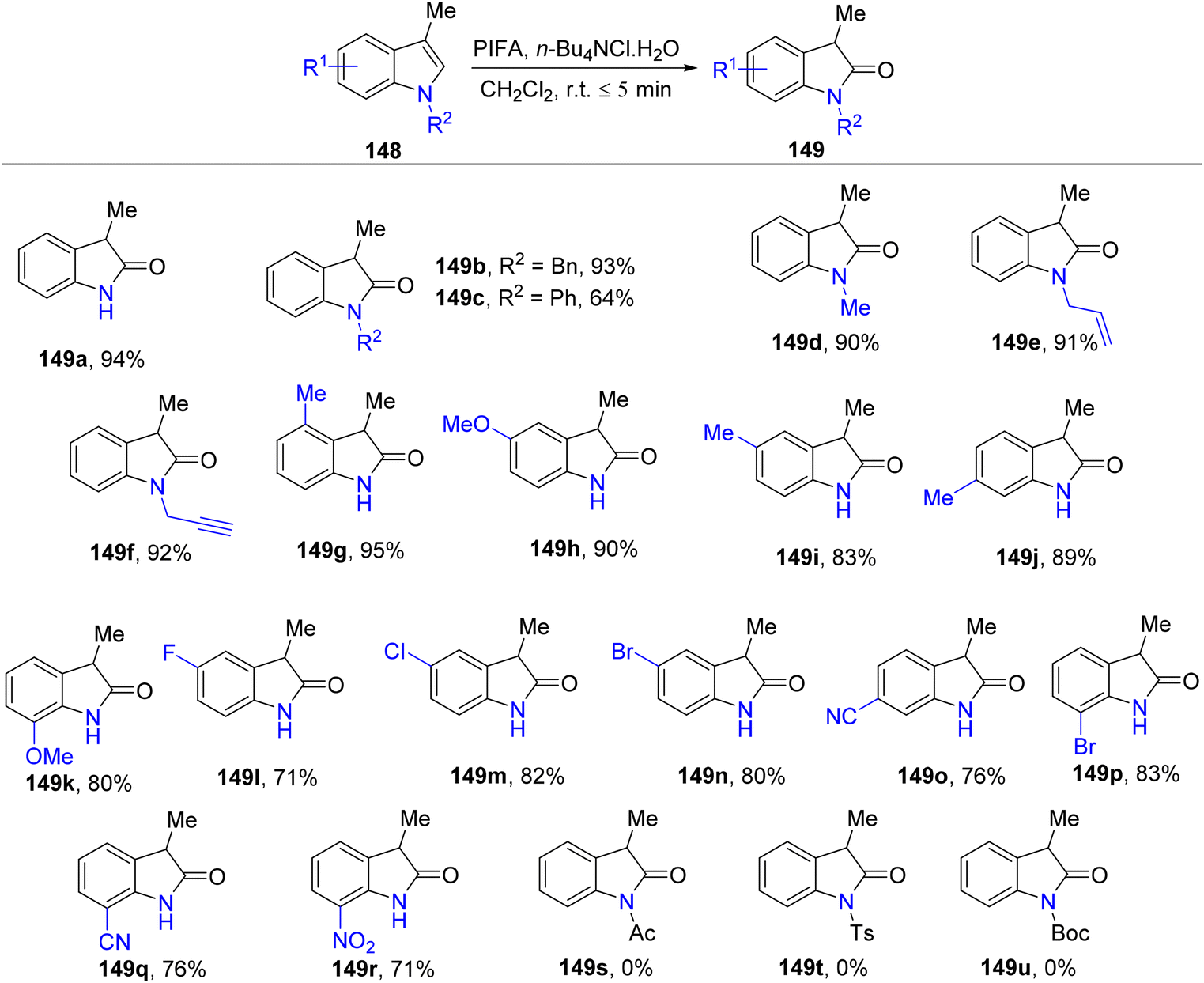
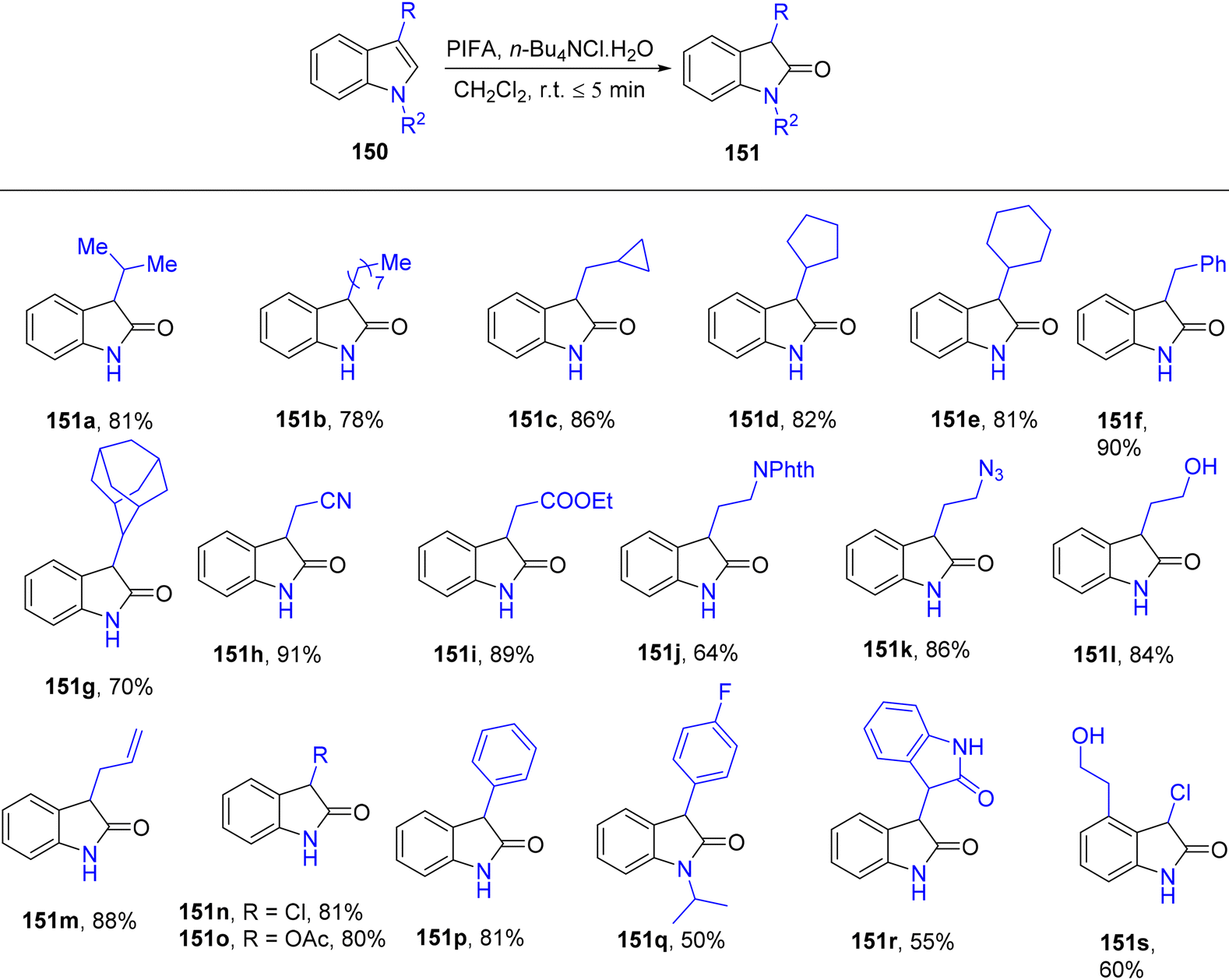
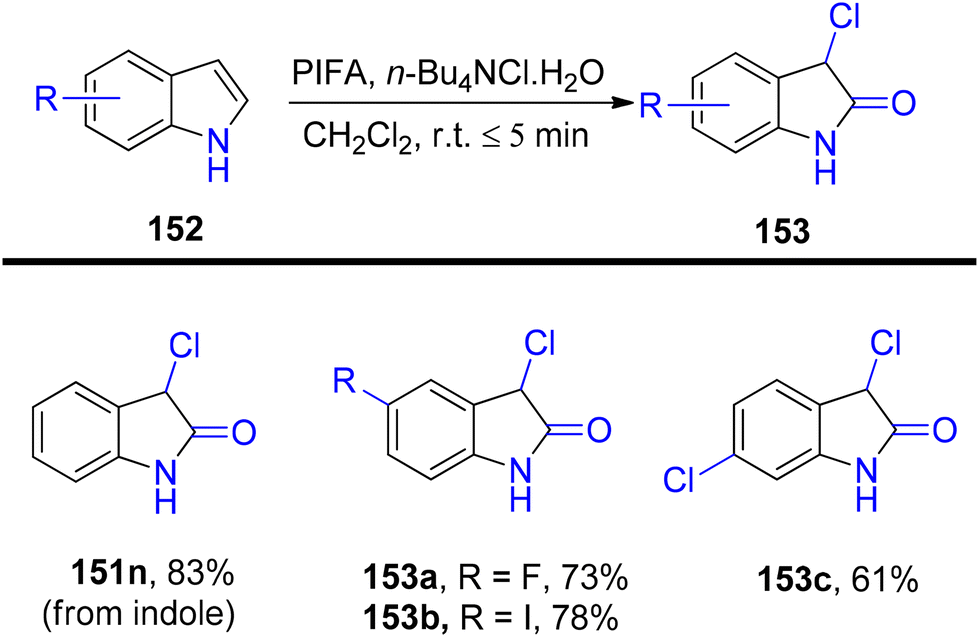
3-Substituted 2-oxindole moiety is a versatile structure employed in the synthesis of biologically relevant indole compounds.139 Liang et al.140 established an efficient methodology catalyzed by PIFA/n-Bu4NCl·H2O for the rapid in situ transformation of indoles into their respective 2-oxindoles. The reaction exhibited broad functional group tolerance and could furnish 2-oxindoles 149/150 in yields of up to 95% within just 5 minutes. Notably, this methodology was applied successfully in the synthesis of (−)-folicanthine and demonstrated gram-scale preparation capability. The findings of their investigation involving substituted indole derivatives 148 are summarized in Scheme 61. To extend the applicability of these transformations, reports on 3-substituted indoles 150 were also presented (Scheme 62). Moreover, the synthetic methodology was utilized to convert indole derivatives 152 into oxindole analogues 153 with moderate yields (Scheme 63). PIFA actively engaged in the reaction by following an ionic pathway. This reaction sequence boasts advantages such as minimal reaction times (<5 minutes), high functional group tolerance, and excellent yields.
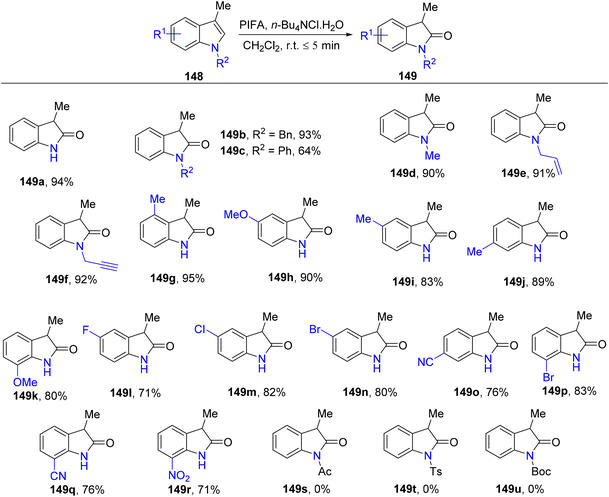 |
| | Scheme 61 Synthetic scheme with substituted indole derivatives 148. | |
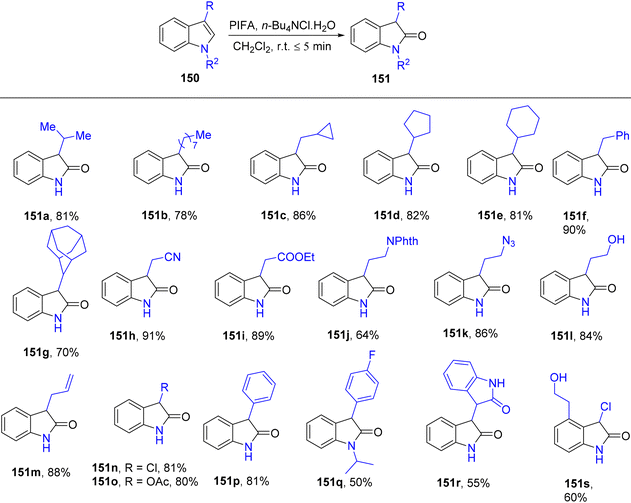 |
| | Scheme 62 Synthesis from 3-substituted indole derivatives 150. | |
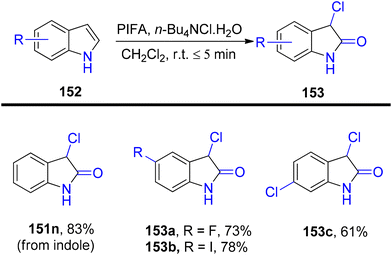 |
| | Scheme 63 Transformation of indole derivatives 152 into oxindole analogues 153. | |
Given the extensive range of applications in which nitroarenes have been employed over the past century, including insecticides, dyes, agrochemicals, and medications, they are recognized as significant structural motifs.141
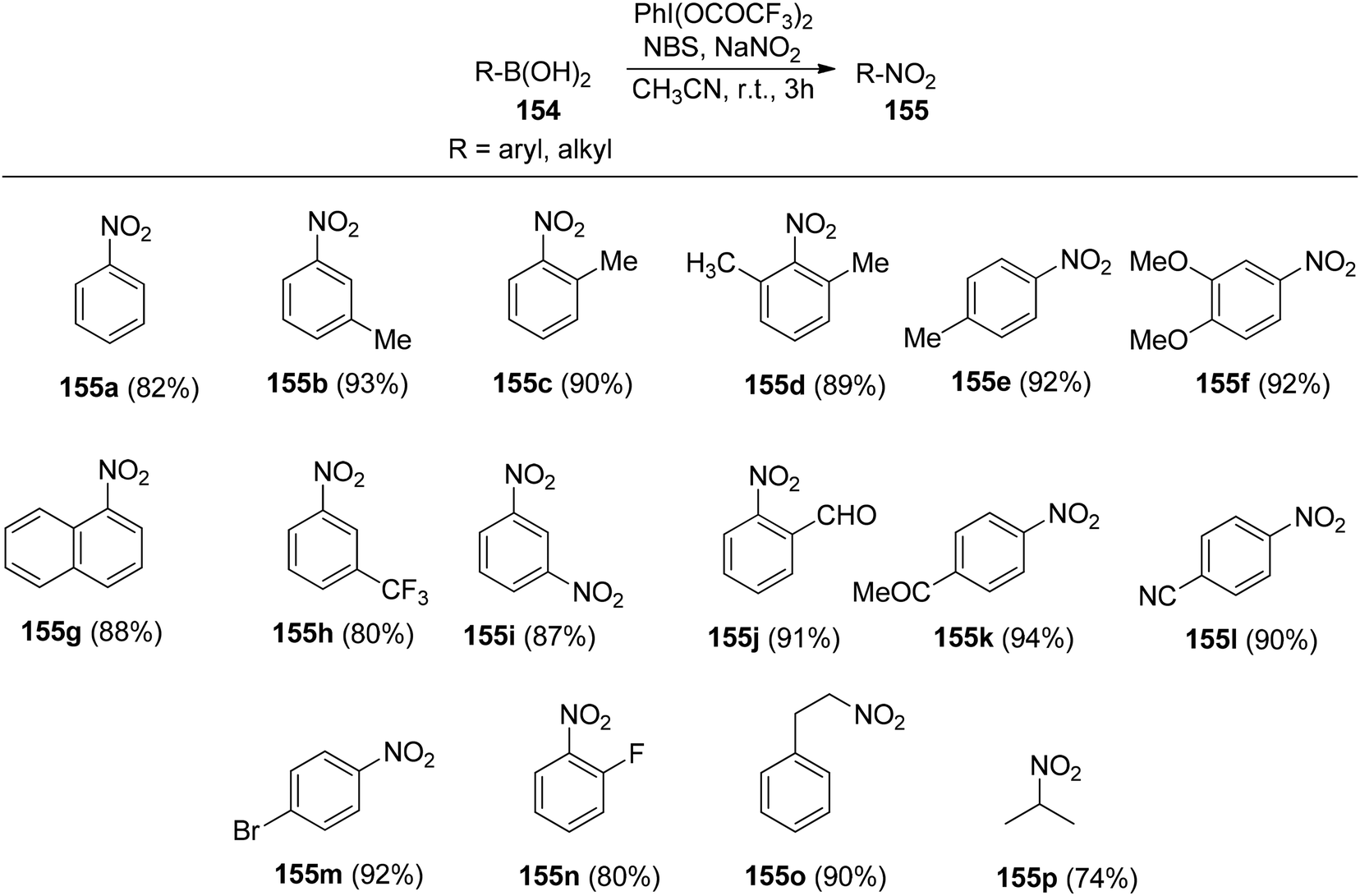
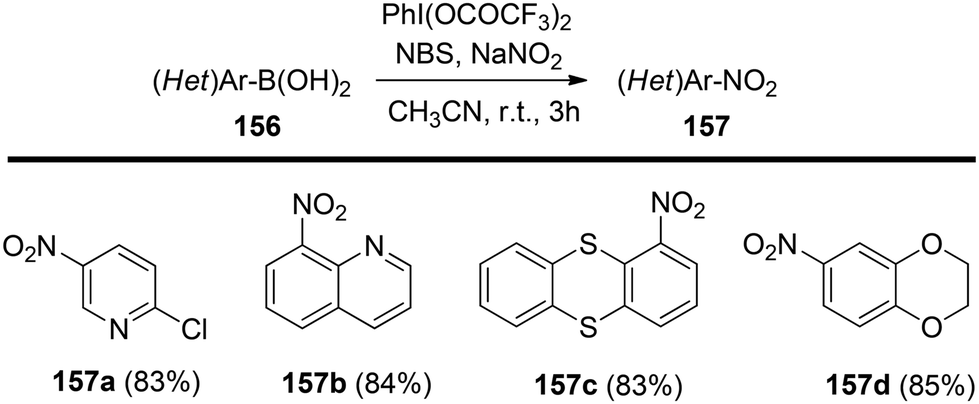
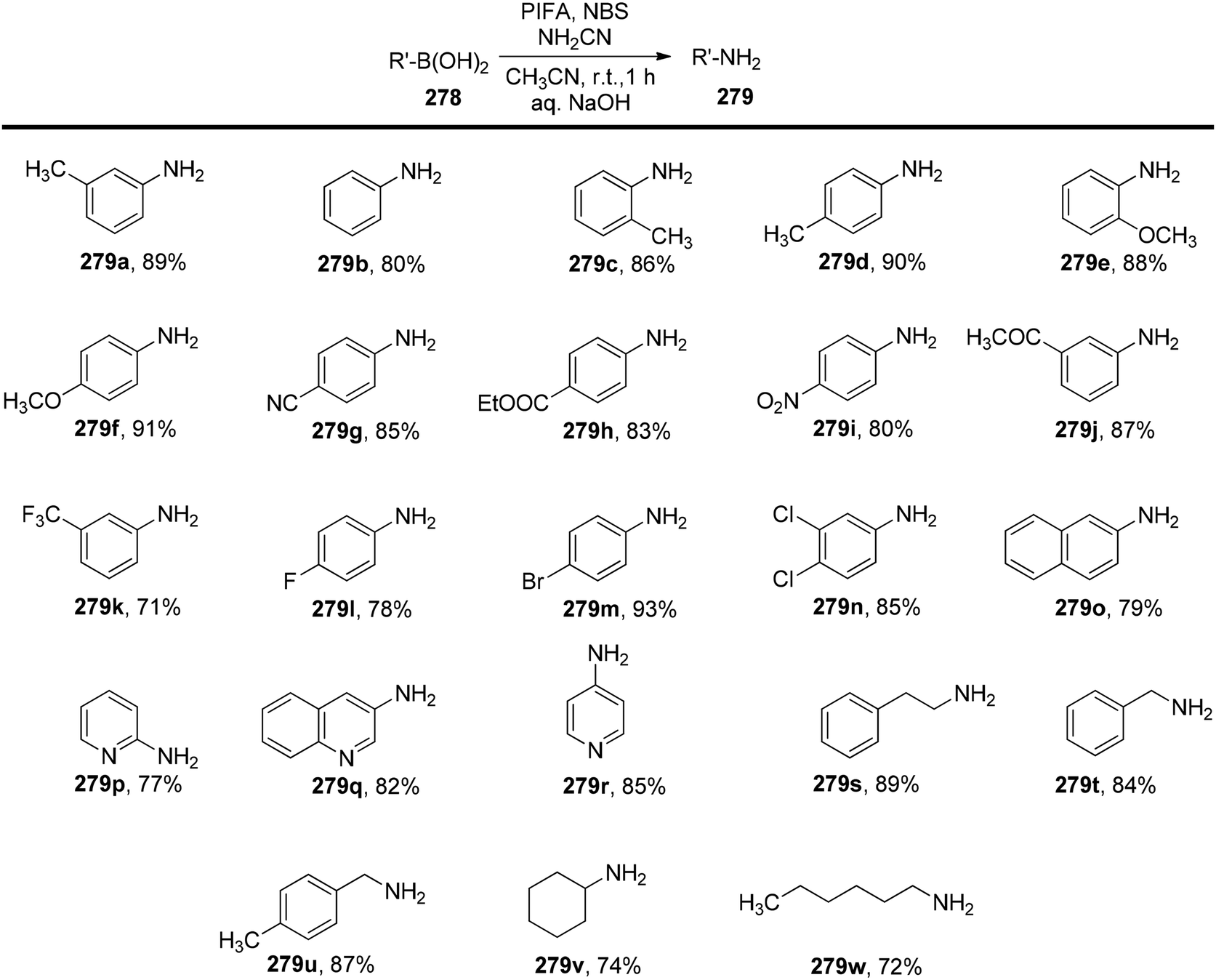
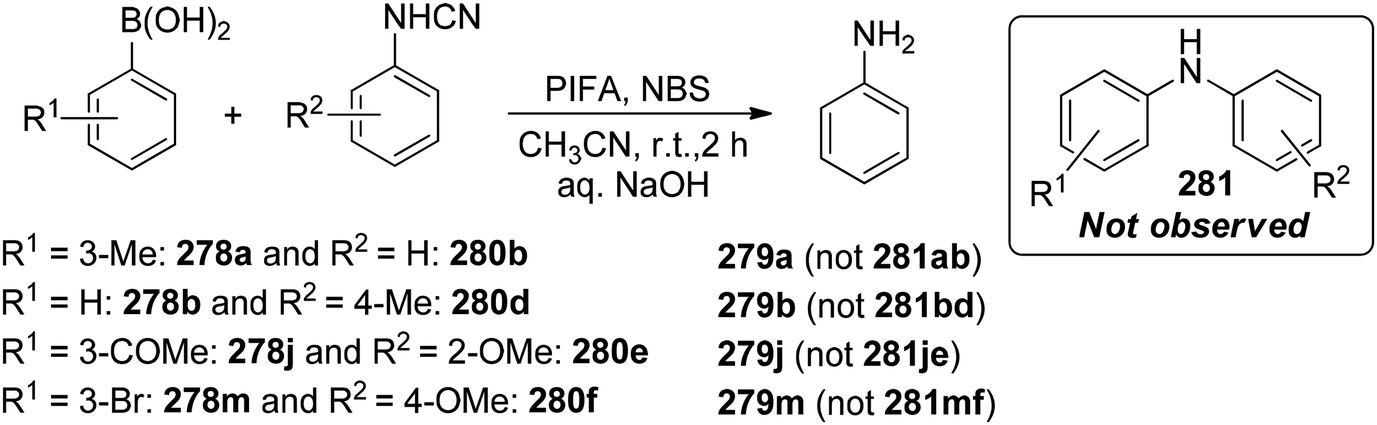
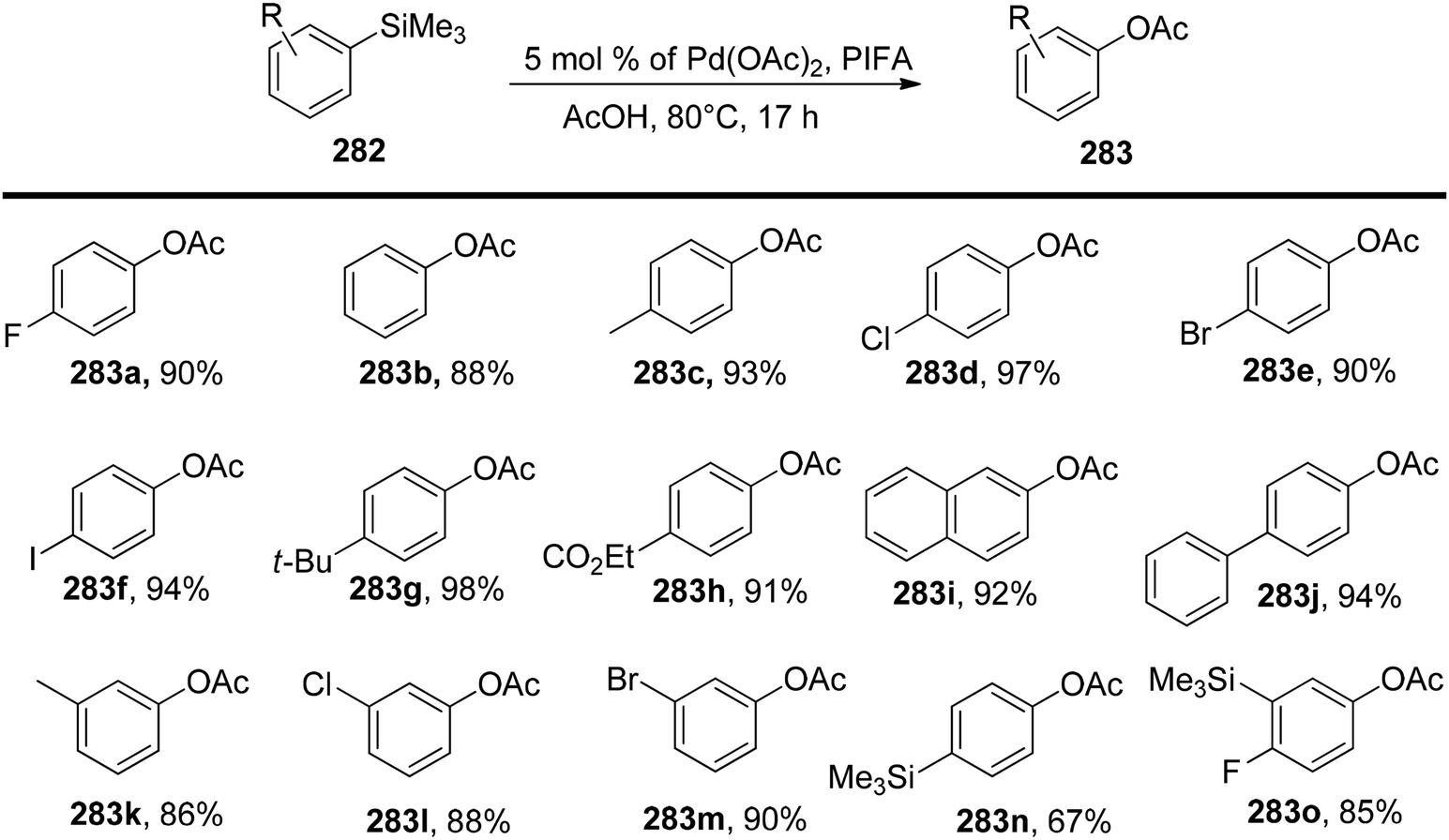
Chatterjee et al.142 introduced a metal-free synthetic methodology based on PIFA/NBS to functionalize organoboronic acids 154 with a –NO2 group 155 at room temperature. The envisioned reaction involved the generation of NO2 as well as organoboronic acid radicals, which subsequently combine to establish the O–N linkage. Following this, the –NO2 group experiences a 1,3-aryl migration, ultimately becoming incorporated into the aryl moiety. This approach is distinguished by its simplicity, utilization of readily accessible substrates, and exceptional efficiency. Nitration was successfully demonstrated with a diverse array of starting materials (Scheme 64). PIFA actively engaged in the reaction by following a free radical pathway. Additionally, the typically challenging task of nitration for hetero-arylboronic acids 156 was achieved conveniently through this methodology, yielding highly favorable results (Scheme 65).
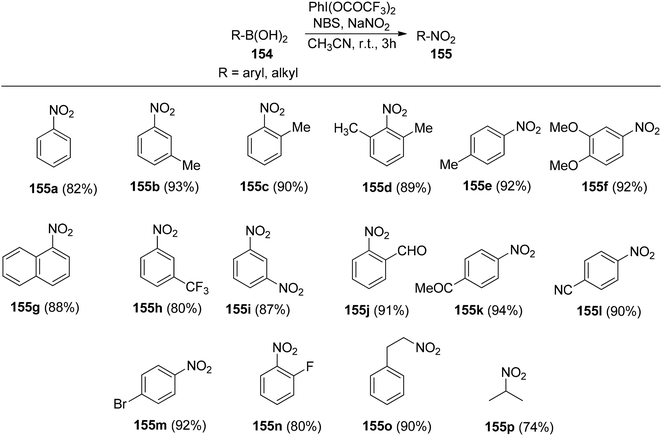 |
| | Scheme 64 PIFA-catalyzed nitration reaction of aryl- and alkylboronic acids. | |
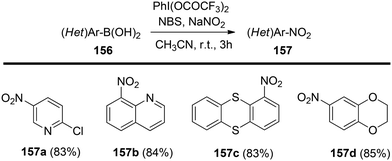 |
| | Scheme 65 PIFA-mediated ipso nitration of heteroarylboronic acids. | |
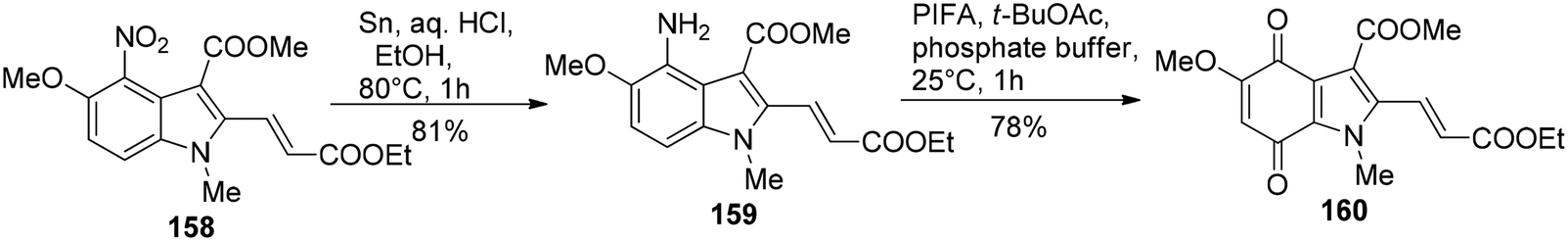
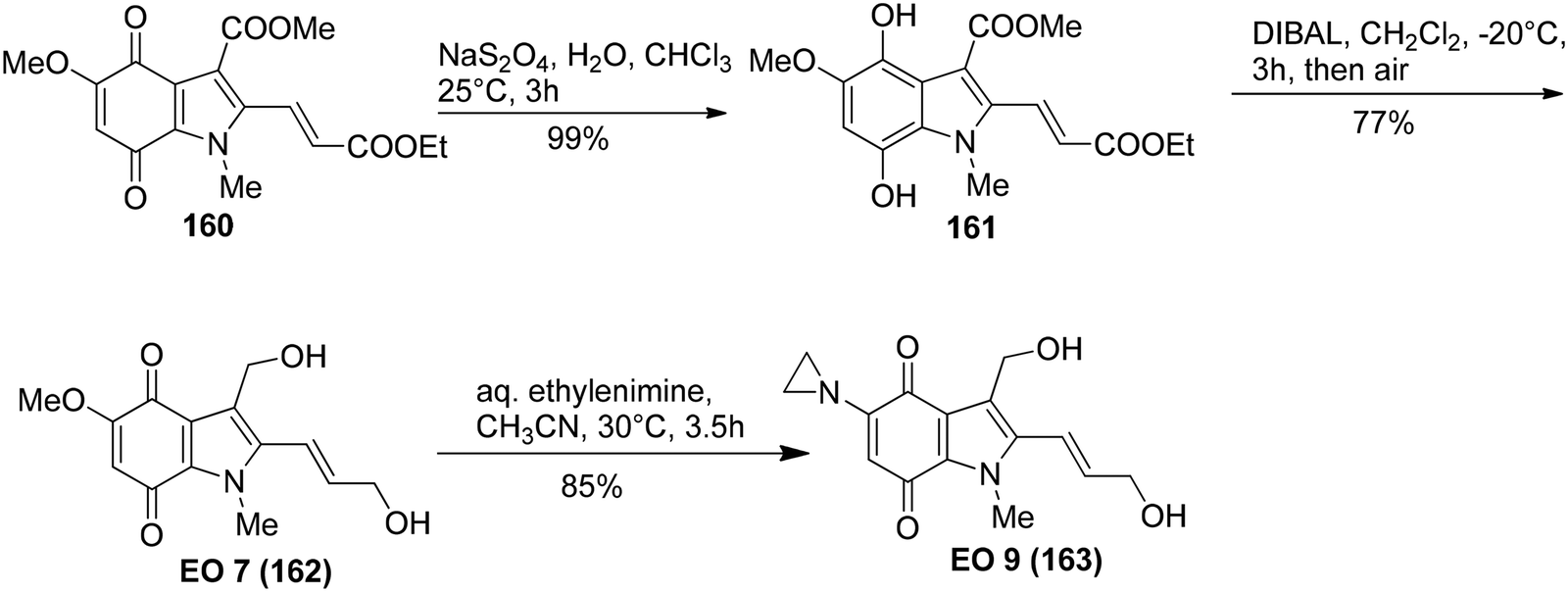
Kinugawa et al.143 reported the convenient synthesis of the indoloquinone anti-tumor agent EO 9 (163)144 on a large scale. The aminoindole 159 was obtained from 158 upon treatment with tin and HCl in 81% yield. 4-Aminoindole 159 then underwent oxidation in the presence of PIFA to result in the formation of the corresponding indoloquinone 160 (Scheme 66). EO 7 (162) was derived from 160 by reduction, followed by diisobutylaluminium hydride reduction and then finally air oxidation (Scheme 67). In the final step, substitution of methoxy group with ethylenimine results in the incorporation of aziridinyl group to give indoloquinone 163 (Scheme 67).
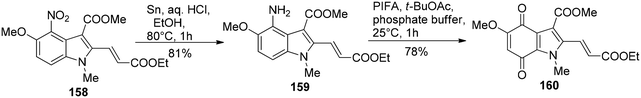 |
| | Scheme 66 Synthesis of indoloquinone 160. | |
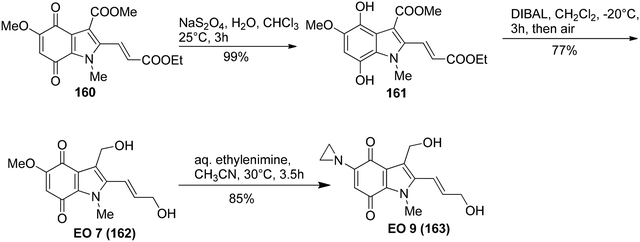 |
| | Scheme 67 Synthetic scheme for hydroquinone 161, indoloquinone EO 7 (162) and indoloquinone antitumor agent EO 9 (163). | |
Covalently linked multiporphyrin arrays represent important functionalities due to their wide range of applications, including nonlinear optical (NLO) devices, molecular wires, photosynthetic systems, and sensors.145
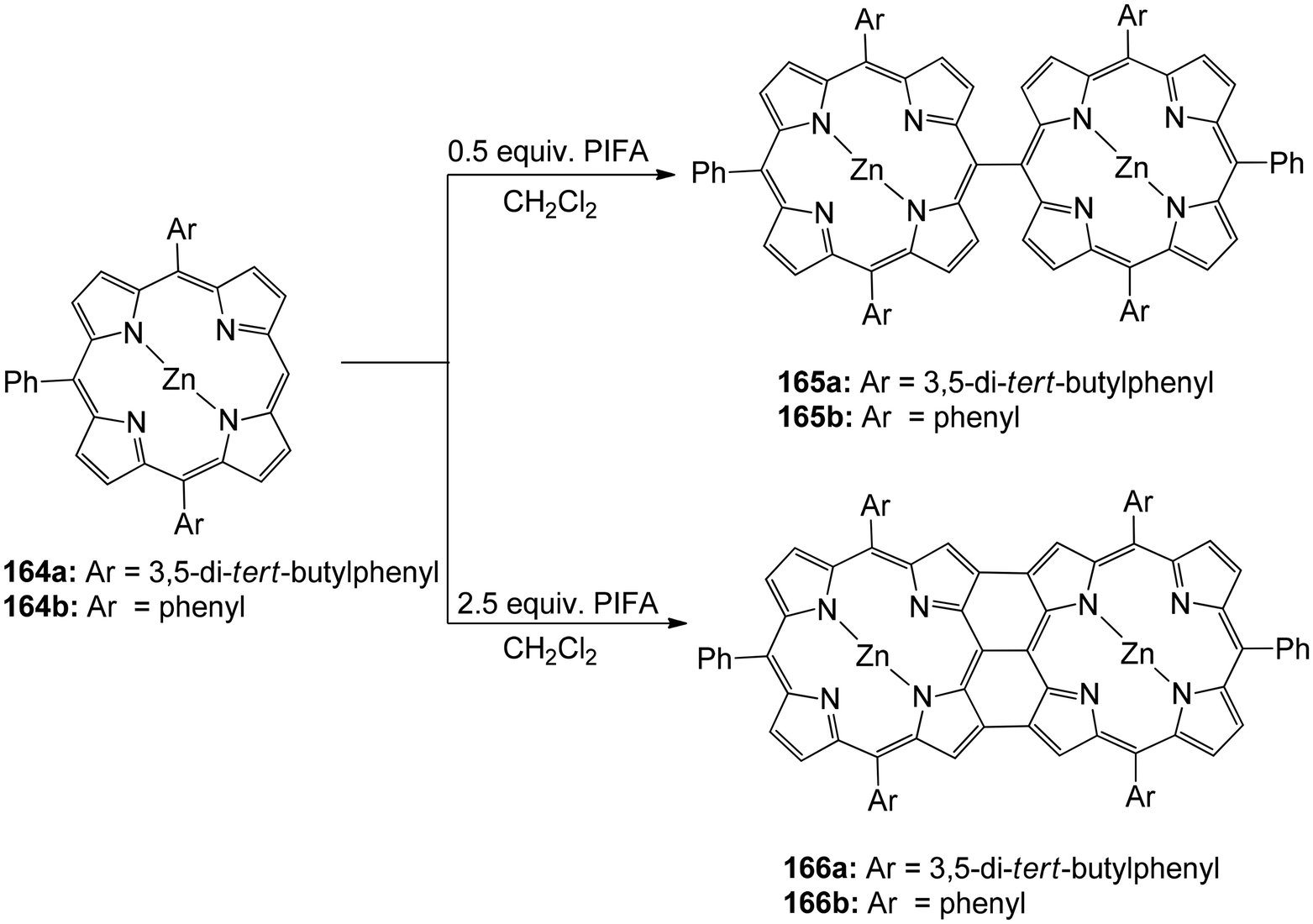
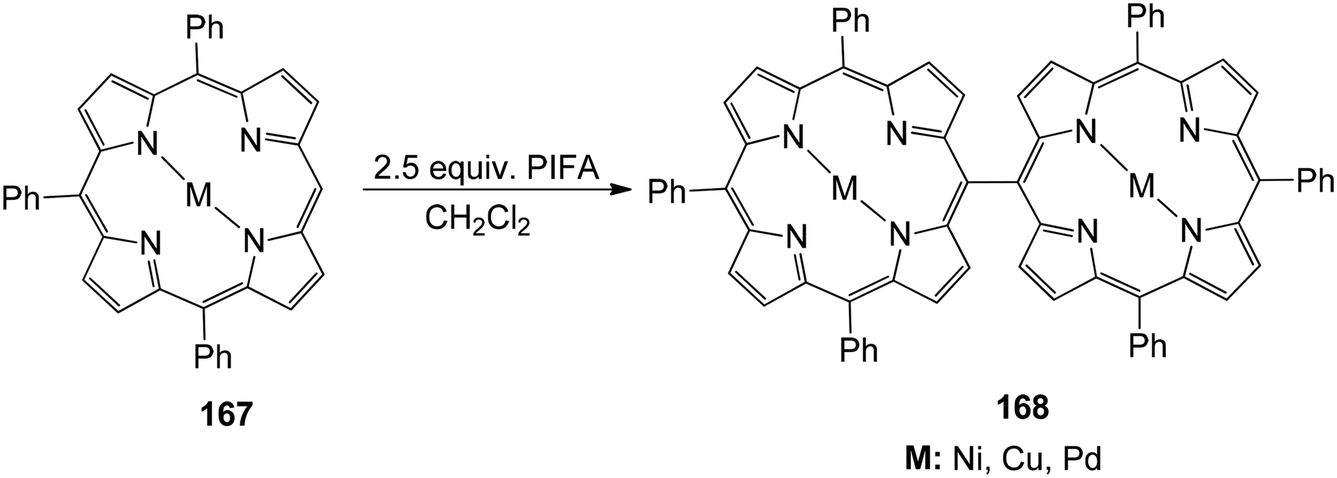
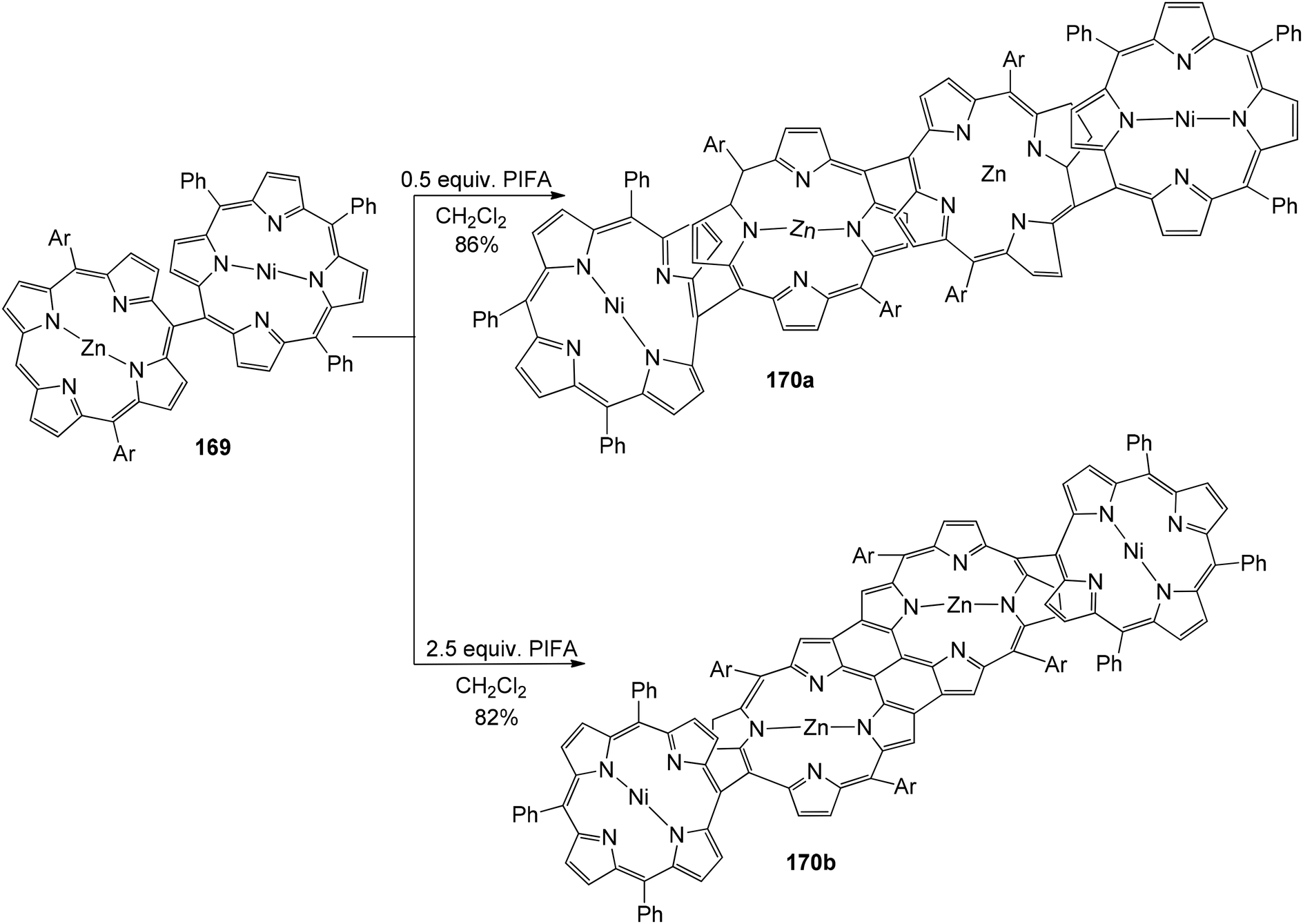
Ouyang et al.146 described an effective methodology utilizing PIFA to obtain diporphyrins 165, 166 and 168 in the absence of a metal. The porphyrin array exhibited strong selectivity for Zn(II) porphyrins 164 (Scheme 68). The reaction was also performed with Cu(II), Pd(II) as well as Ni(II) porphyrins 167 (Scheme 69). The diporphyrin precursor 169 was chosen because of its straightforward synthetic methodology with a 91% yield.147,148 Singly linked tetraporphyrin 170a was obtained with an 86% yield by the addition of 0.5 equiv. of PIFA. On increasing the equivalents of PIFA to 2.5, the porphyrin array 170b was produced in 82% yield. It consisted of one triple linkage and two single linkages (Scheme 70).
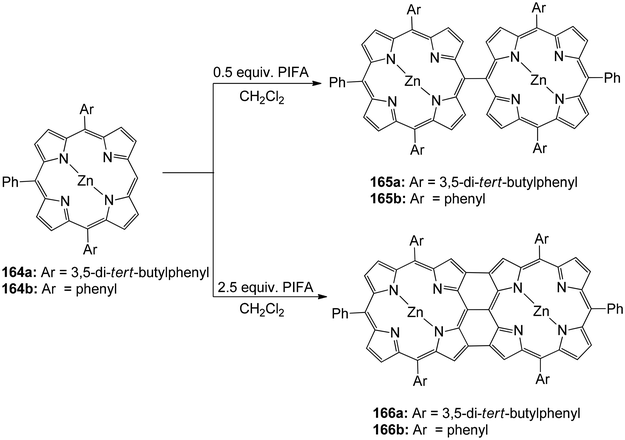 |
| | Scheme 68 Synthesis of diporphyrins 165 and 166. | |
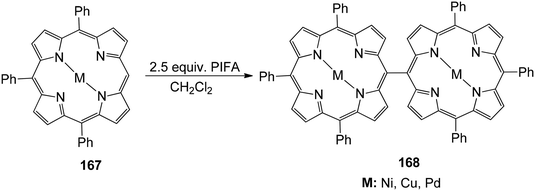 |
| | Scheme 69 Reaction pathway for Cu(II), Ni(II) and Pd(II) porphyrin coupling. | |
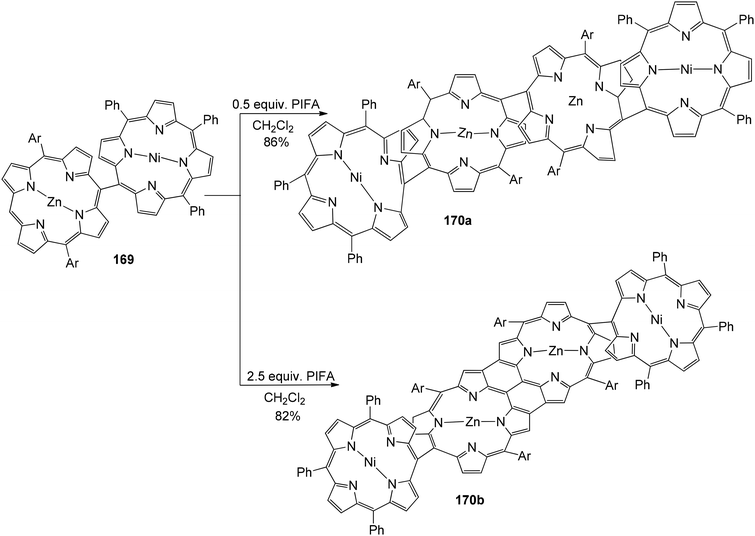 |
| | Scheme 70 PIFA-mediated coupling of hybdrid diphorphyrin. | |
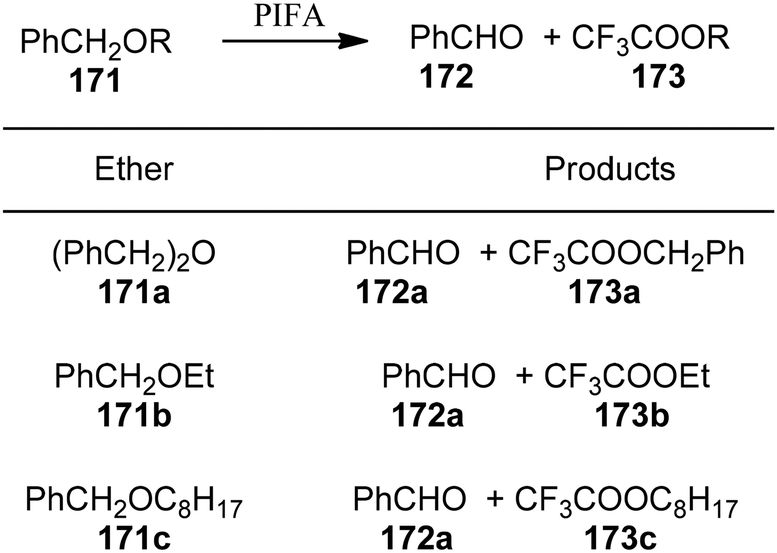
Spyroudis et al.149 reported the oxidation of benzyl ethers 171 to their respective carbonyls 172 and benzyl trifluoroacetates 173 using PIFA (Scheme 71). PIFA actively engaged in the reaction by following an ionic pathway.
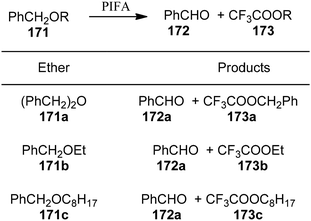 |
| | Scheme 71 Oxidation of benzyl ethers 171. | |
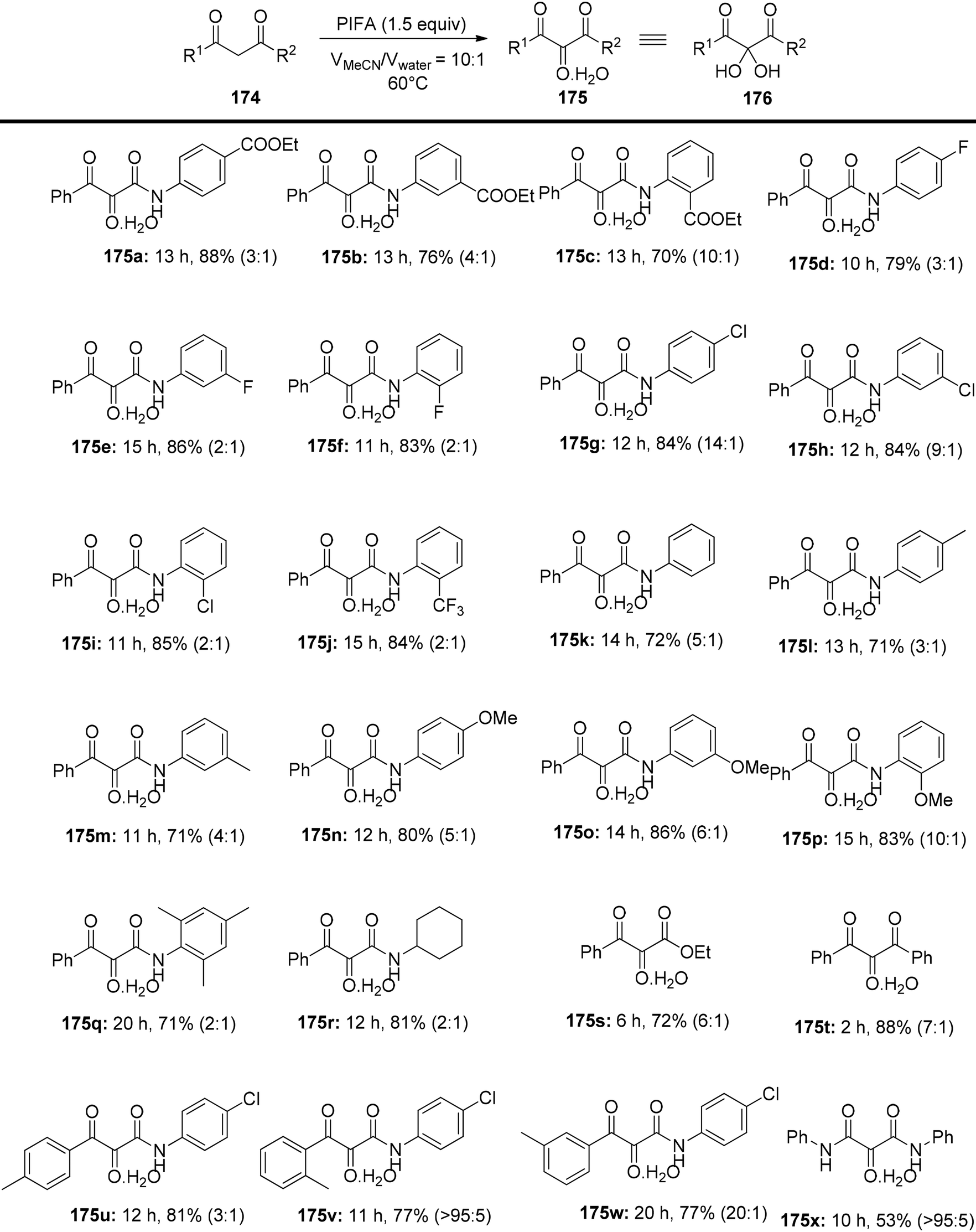
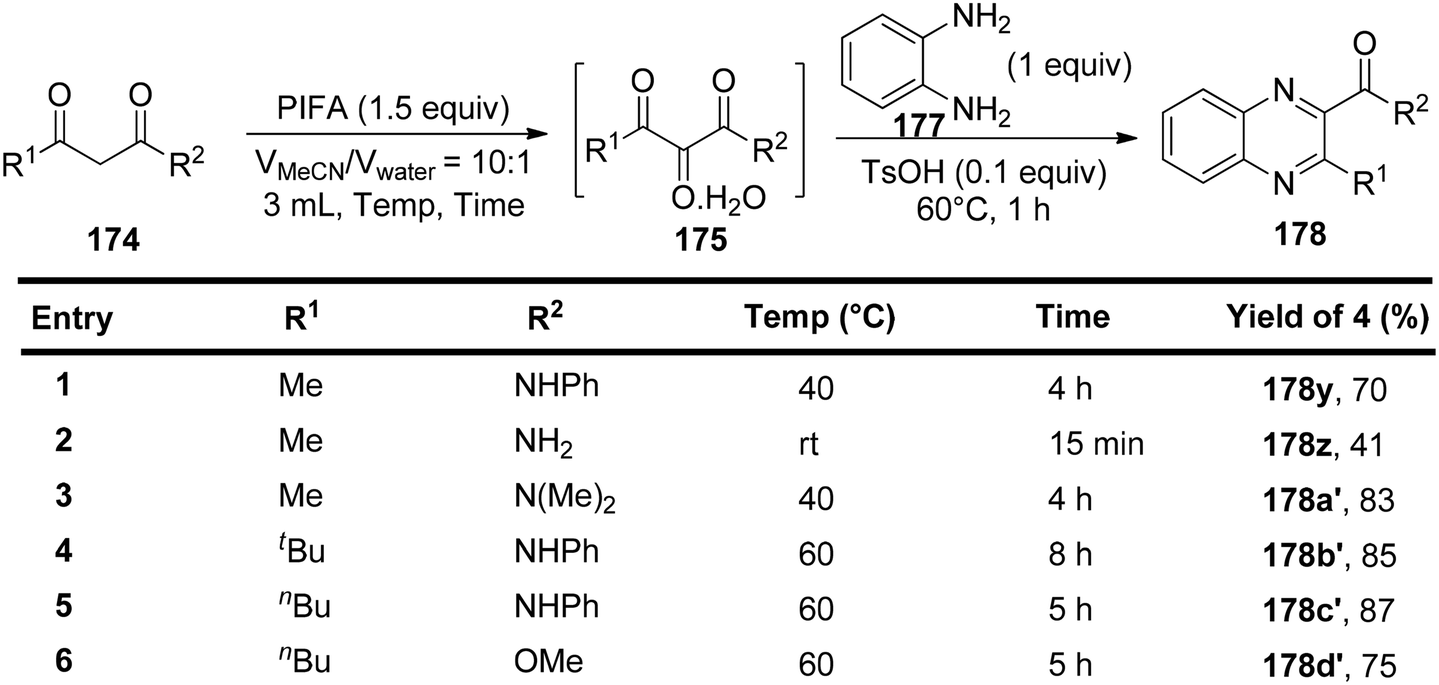
Liu et al.150 have introduced a simple and direct oxidative method for producing vicinal tricarbonyl amides 175, yielding from moderate to excellent results, in the range of 53% to 88%. This process commences with the use of readily accessible β-ketoamides 174 in the presence of PIFA, as outlined in Scheme 72. The advantages of this approach encompass the accessibility of starting materials, the attainment of high yields, the flexibility to introduce various substitutions, and the substantial synthetic potential of the resultant products. To further expand the versatility of this method, subsequent modifications were executed using benzene-1,2-diamine 177 in conjunction with 4-methylbenzenesulfonic acid at 60 °C in a one-pot reaction. Encouragingly, this approach proved effective for a range of aliphatic β-ketoamides, spanning from 174z to 174c′, and even extended to aliphatic β-keto ester 174d′, yielding their respective derivatives 178z to 178d′ with cumulative yields spanning from 41% to 87% (Scheme 73).
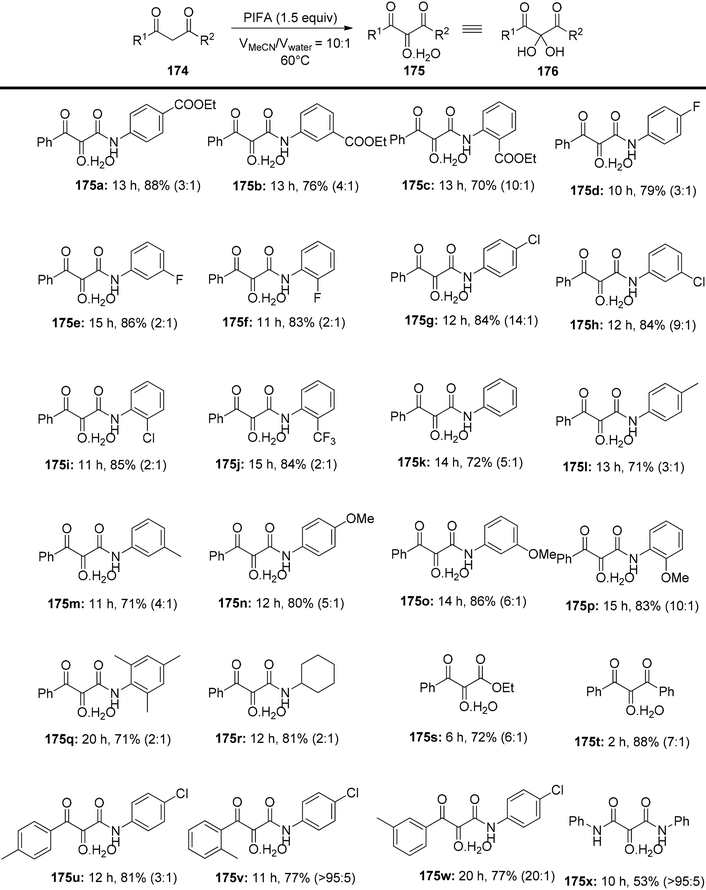 |
| | Scheme 72 Synthesis of vicinal tricarbonyl amides 175. | |
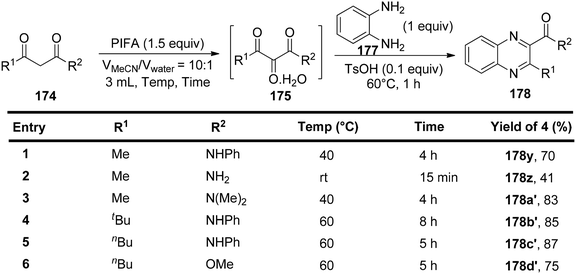 |
| | Scheme 73 One-pot synthesis of quinoxaline derivative 178. | |
1,2-Diols play a crucial role in the synthesis of pharmaceuticals and natural compounds. The direct addition of oxygen atoms to adjacent positions (vicinal dioxygenation) of olefins is a simple and widely employed approach for the preparation of 1,2-diols. Numerous effective methods have been developed and are extensively utilized in contemporary chemistry.151
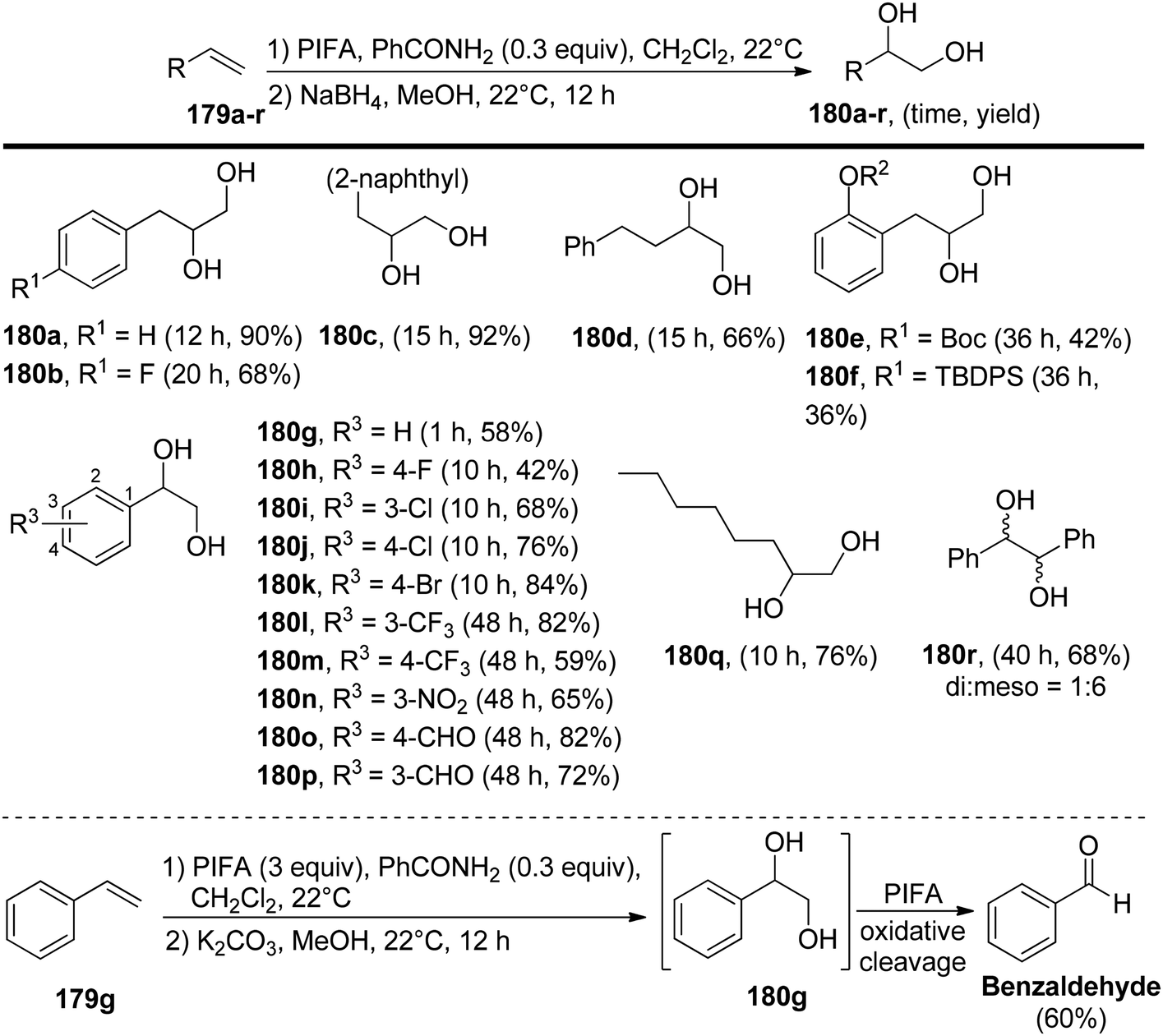
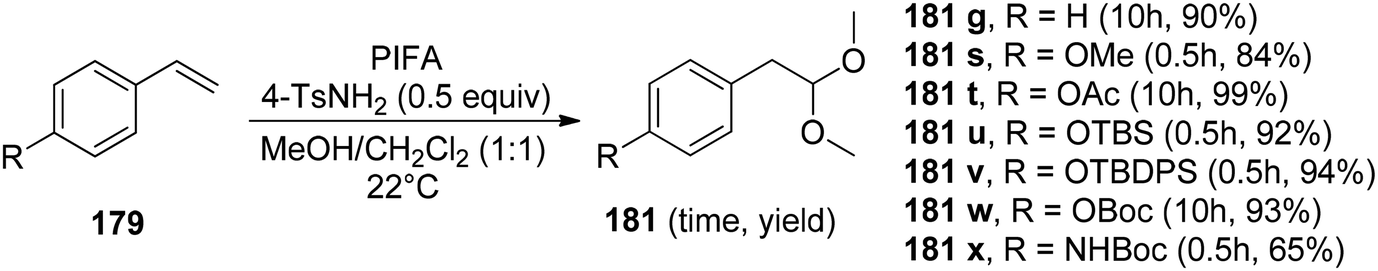
Pan et al.152 have introduced a catalytic method for accomplishing the vicinal dioxygenation of olefins 179 (Scheme 74). They employed hypervalent iodine reagents with Lewis bases as catalysts. These reaction conditions are gentle and versatile, making them suitable for a wide array of functional groups. After fine-tuning the reaction conditions, they investigated the practicality of this dioxygenation process in a one-step approach, leading to the synthesis of the corresponding diols 180. Significantly, the authors were pleased to discover that styrenes 179 carrying diverse substituents, such as ethers, esters, silyl ethers, OBoc, and NHBoc, were well tolerated, yielding the desired products 181 with robust yields and exceptional selectivity, as illustrated in Scheme 75. PIFA actively engaged in the reaction by following an ionic pathway. Crucially, this method is compatible with compounds that are sensitive to both acids and oxidants.
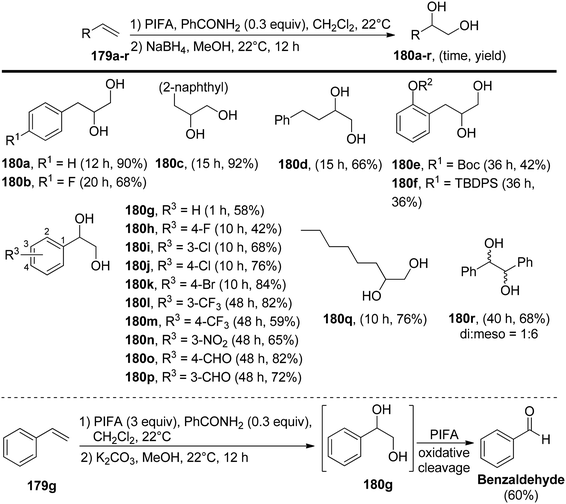 |
| | Scheme 74 One-pot synthesis of vicinal diols 180a–r. | |
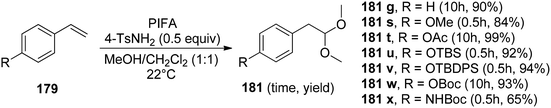 |
| | Scheme 75 Synthesis of anti-Markovnikov methyl acetals from styrene derivatives using Lewis base-activated HIR. | |
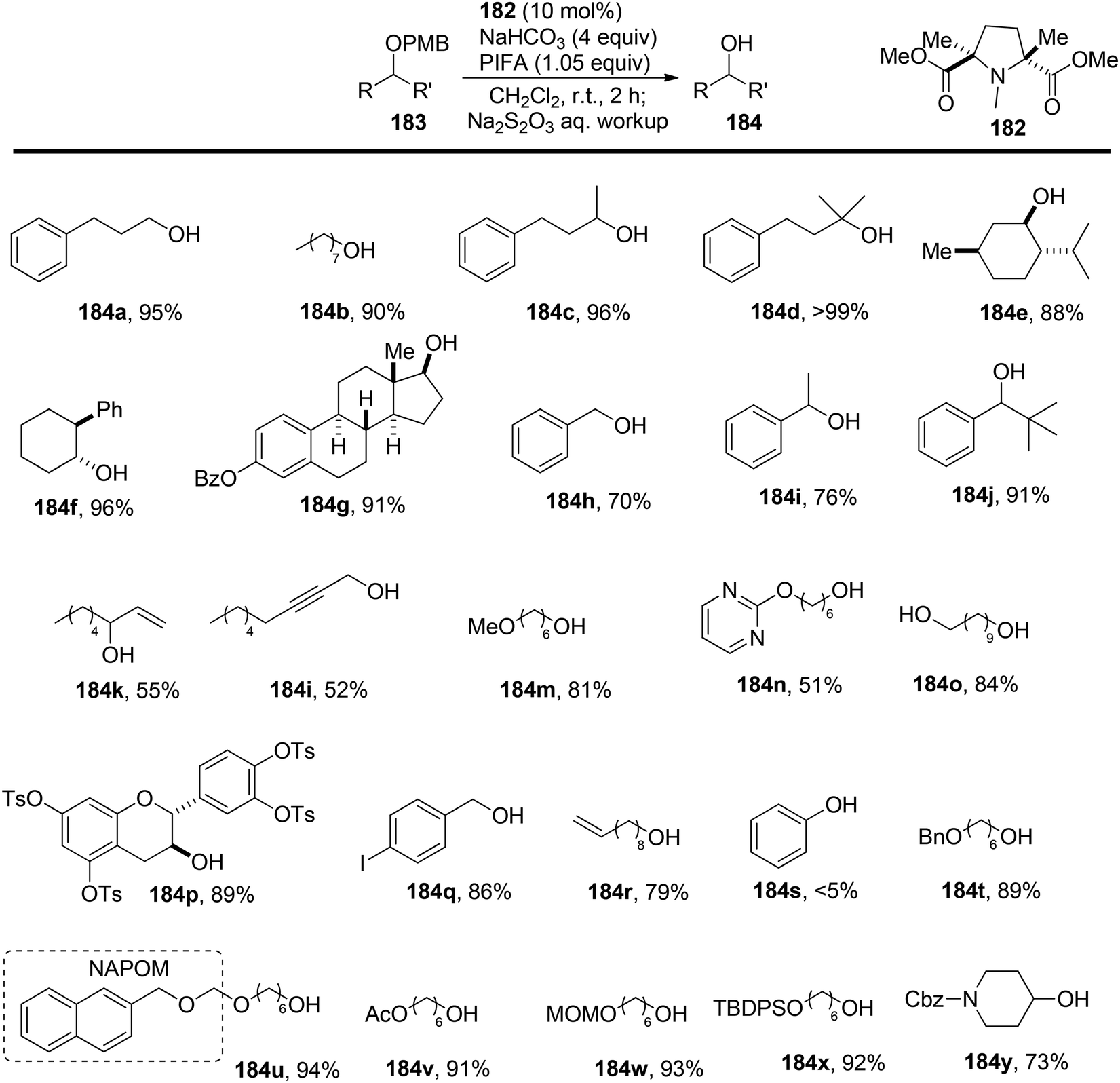
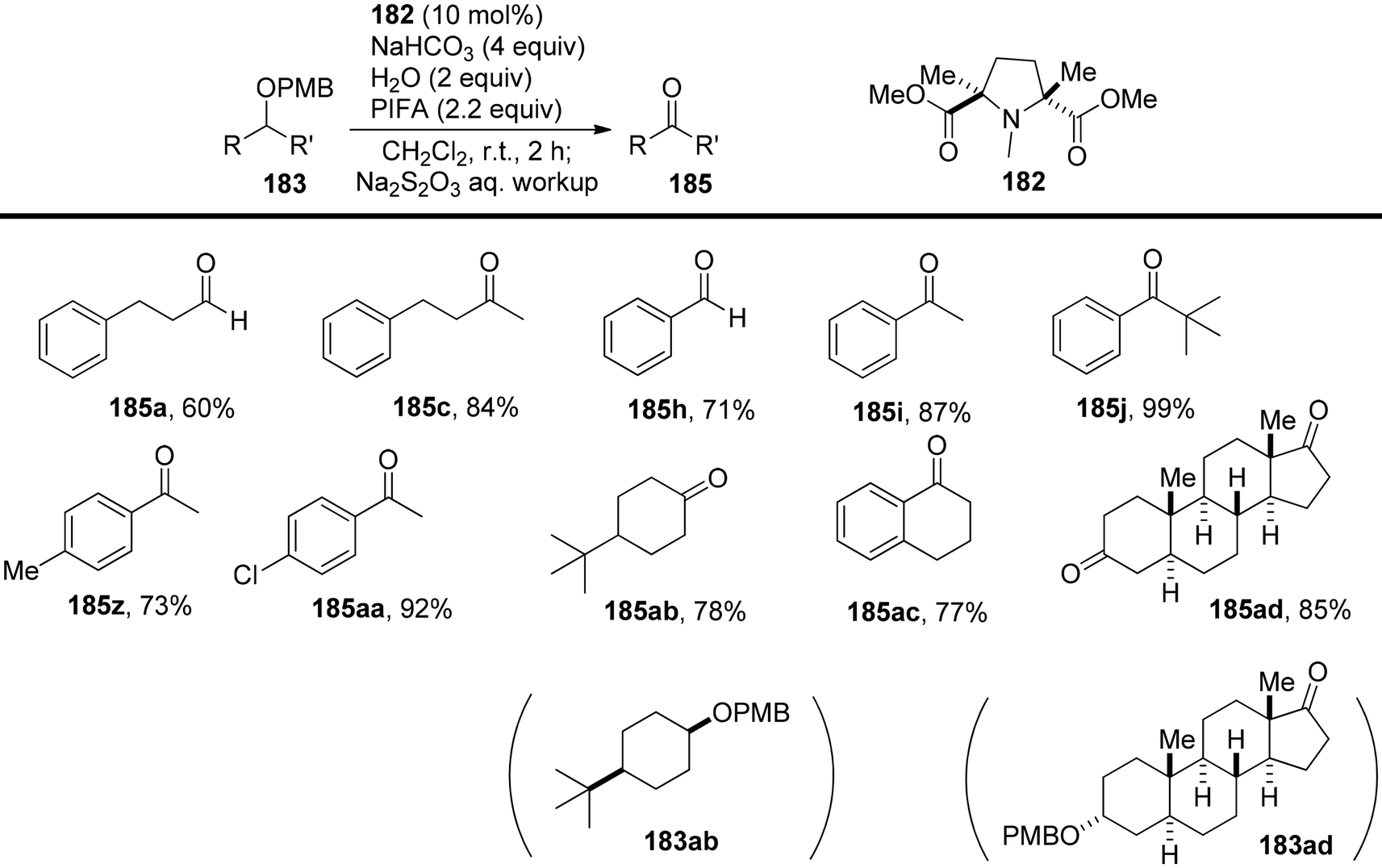
Hamada et al.153 achieved the oxidation of p-methoxy benzyl (PMB) ethers 183 using a nitroxyl radical catalyst labeled as catalyst 182. This catalyst features electron-withdrawing ester groups positioned in close proximity to the nitroxyl group. The removal of PMB protective groups from hydroxyl compounds occurred through oxidative deprotection when catalyst 182 was treated with an equivalent of the co-oxidant PIFA (Scheme 76). This system displayed remarkable selectivity in deprotecting PMB ethers, even in the presence of a diverse range of functional groups, some of which are susceptible to oxidation. The desired carbonyl compounds 185 were obtained by reacting the PMB-protected alcohols 183 with catalyst 182 and an excess of PIFA, as illustrated in Scheme 77. PIFA actively engaged in the reaction by following a free radical pathway, specifically through single-electron oxidation.
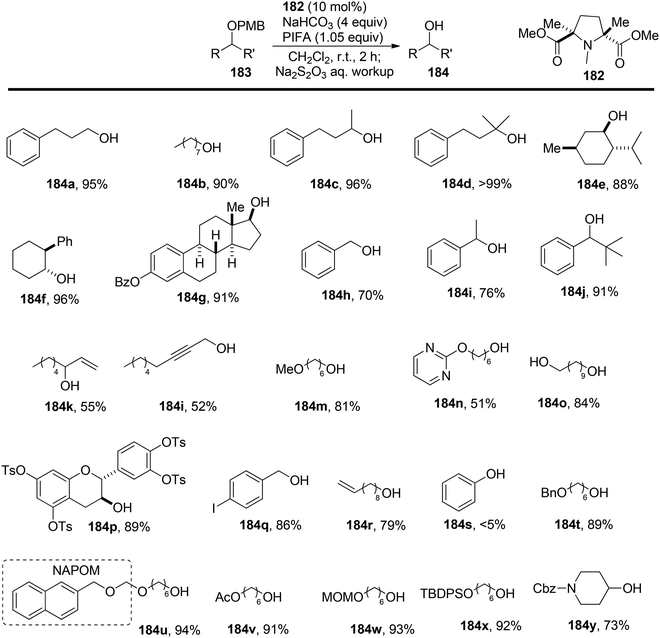 |
| | Scheme 76 Oxidative deprotection of PMB groups by I/PIFA. | |
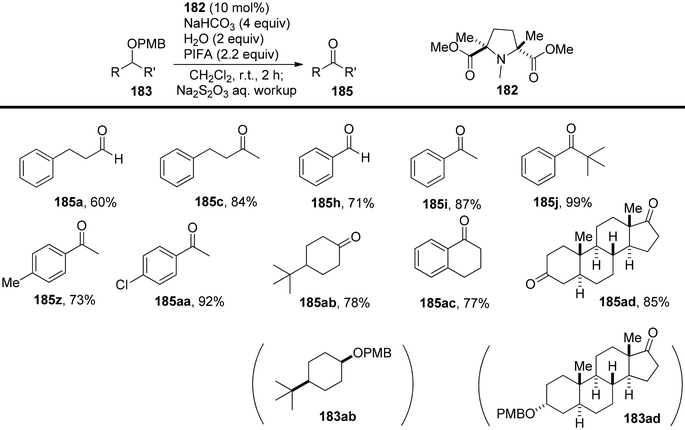 |
| | Scheme 77 Direct synthesis of ketones and aldehydes from PMB-protected alcohols by I/PIFA. | |
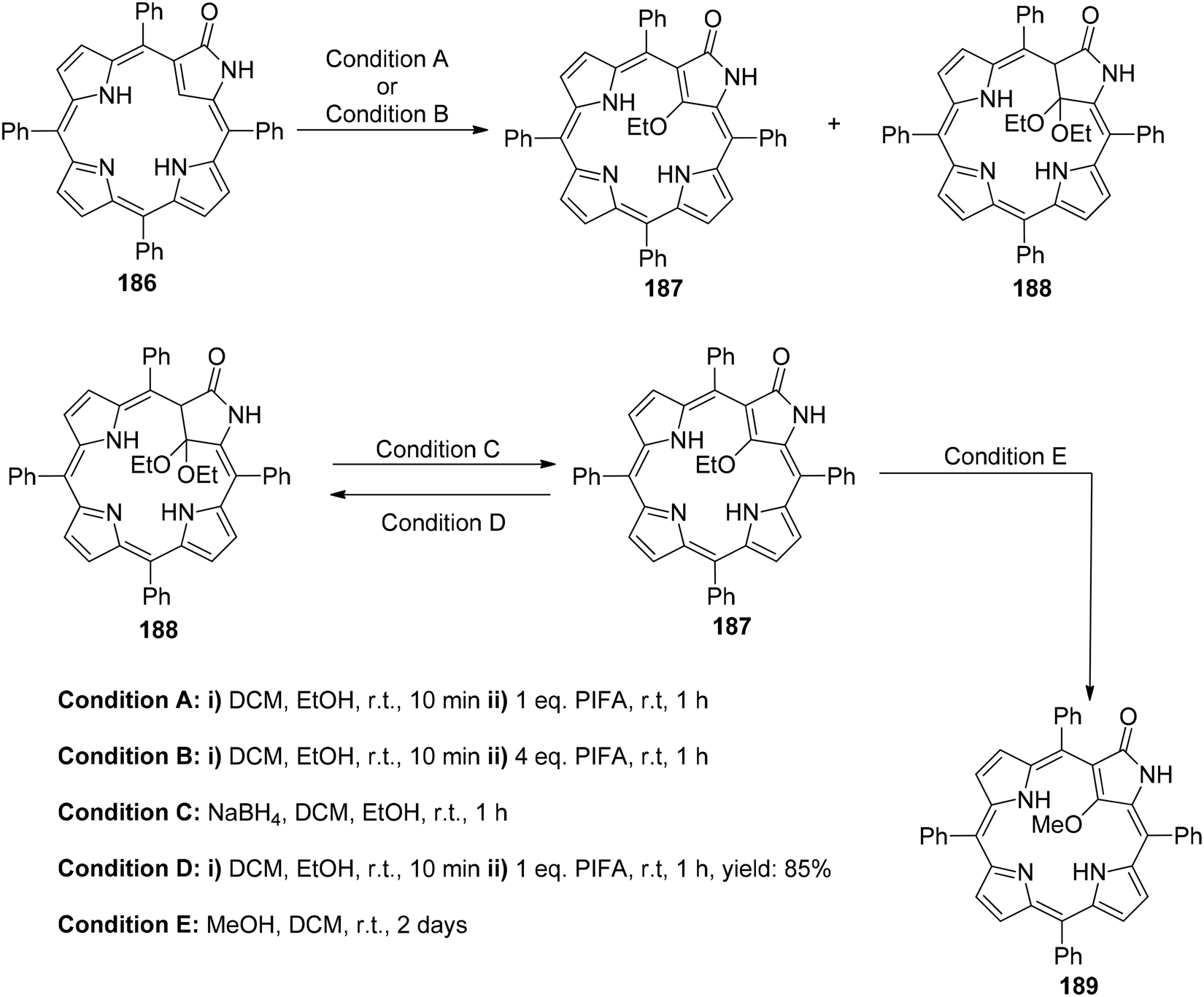
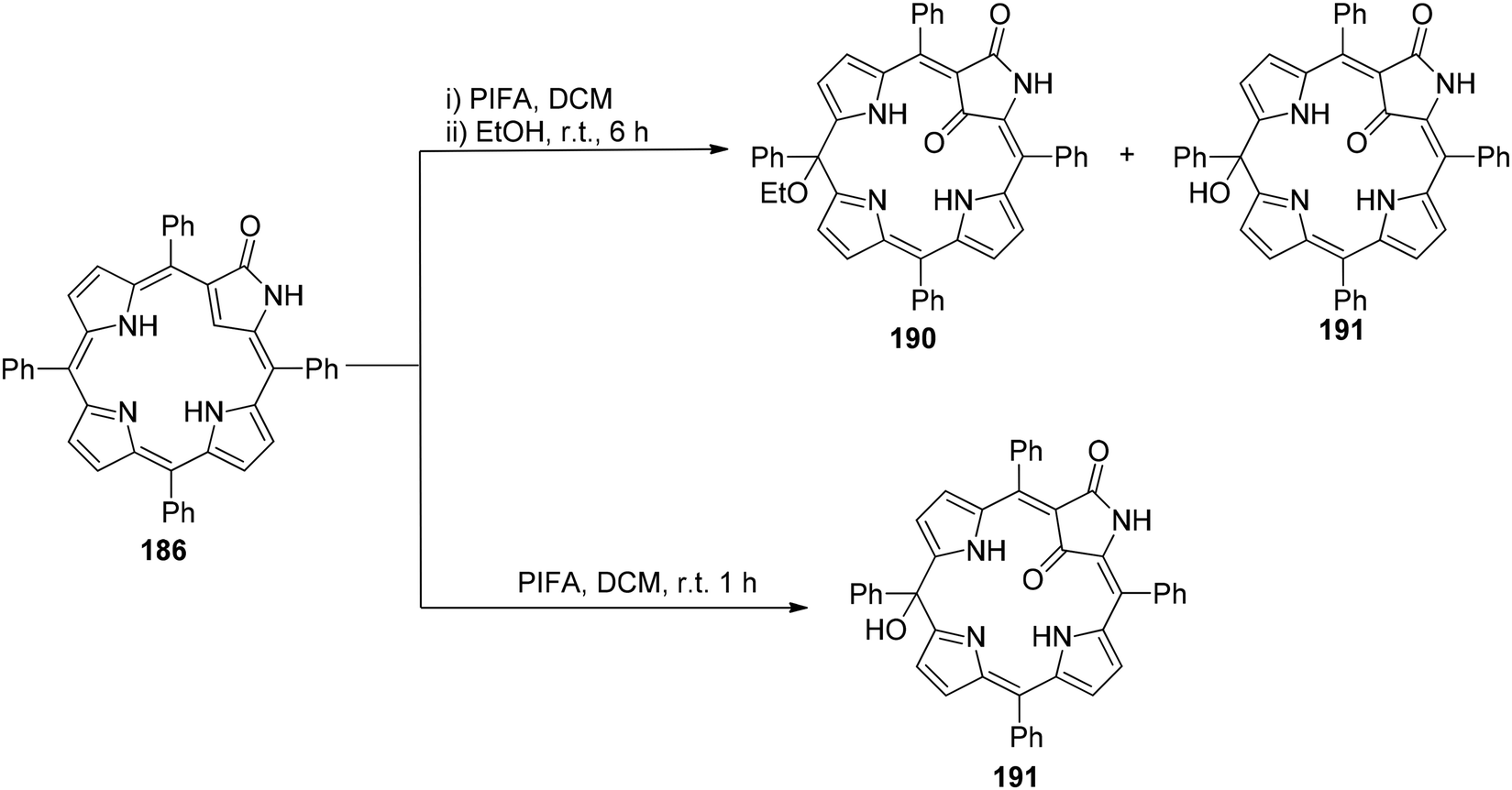
Zhang et al.154 have reported the synthesis of 21-ethoxy- and 21,21-diethoxy-ONCPs 187 and 188 through a sequence involving the conjugate addition of ethanol followed by oxidation with PIFA. This process enabled the inner ethoxylation of 3-oxo-N-confused tetraphenylporphyrin 186 (ONCP) (Scheme 78). The initial step involved the oxidation of ONCP with PIFA, followed by treatment with ethanol, resulting in the generation of two additional N-confused phlorin derivatives, denoted as 190 and 191 (Scheme 79). The transformation from 186 to 187, 188, 189, 190 and 191 has been well established through the use of PIFA and ethanol. This signifies the versatility of the conjugated lactam structural feature for making modifications at both the C21 (inner core) and the C10 (outer meso-position) of these molecules. The formation of phlorins 190 and 191 highlights that ONCPs serve as an intriguing platform for meso modifications. N-Confused porphyrins (NCPs), introduced by the Furuta and Latos-Grażyński groups,155,156 exhibit fascinating properties distinct from regular porphyrins due to the presence of a unique pyrrole arrangement.
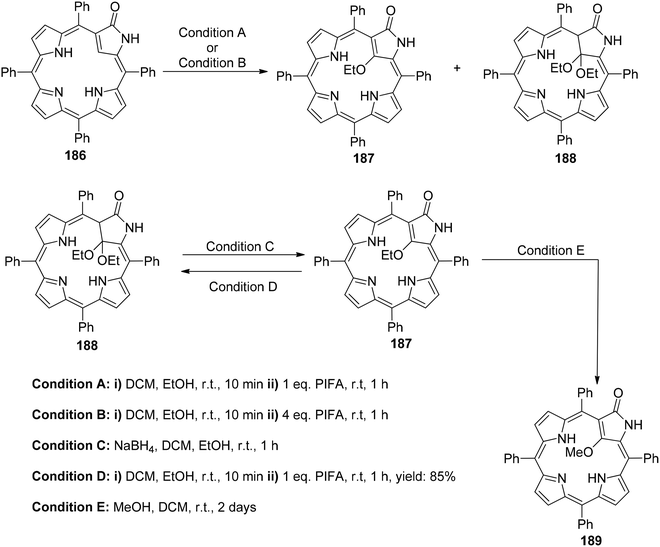 |
| | Scheme 78 Synthesis of 187, 188 & 189. | |
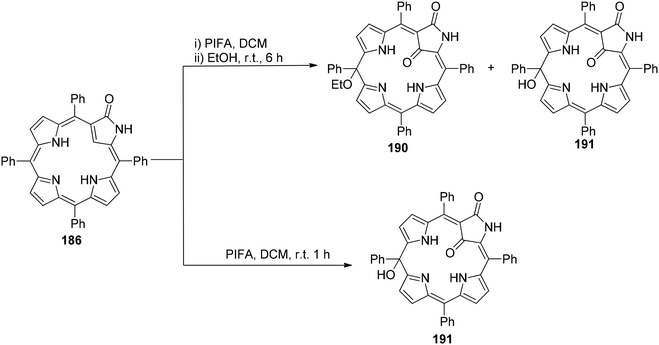 |
| | Scheme 79 Synthesis of N-confused phlorins derivatives, denoted as 190 and 191. | |
3.9. Hoffmann reaction
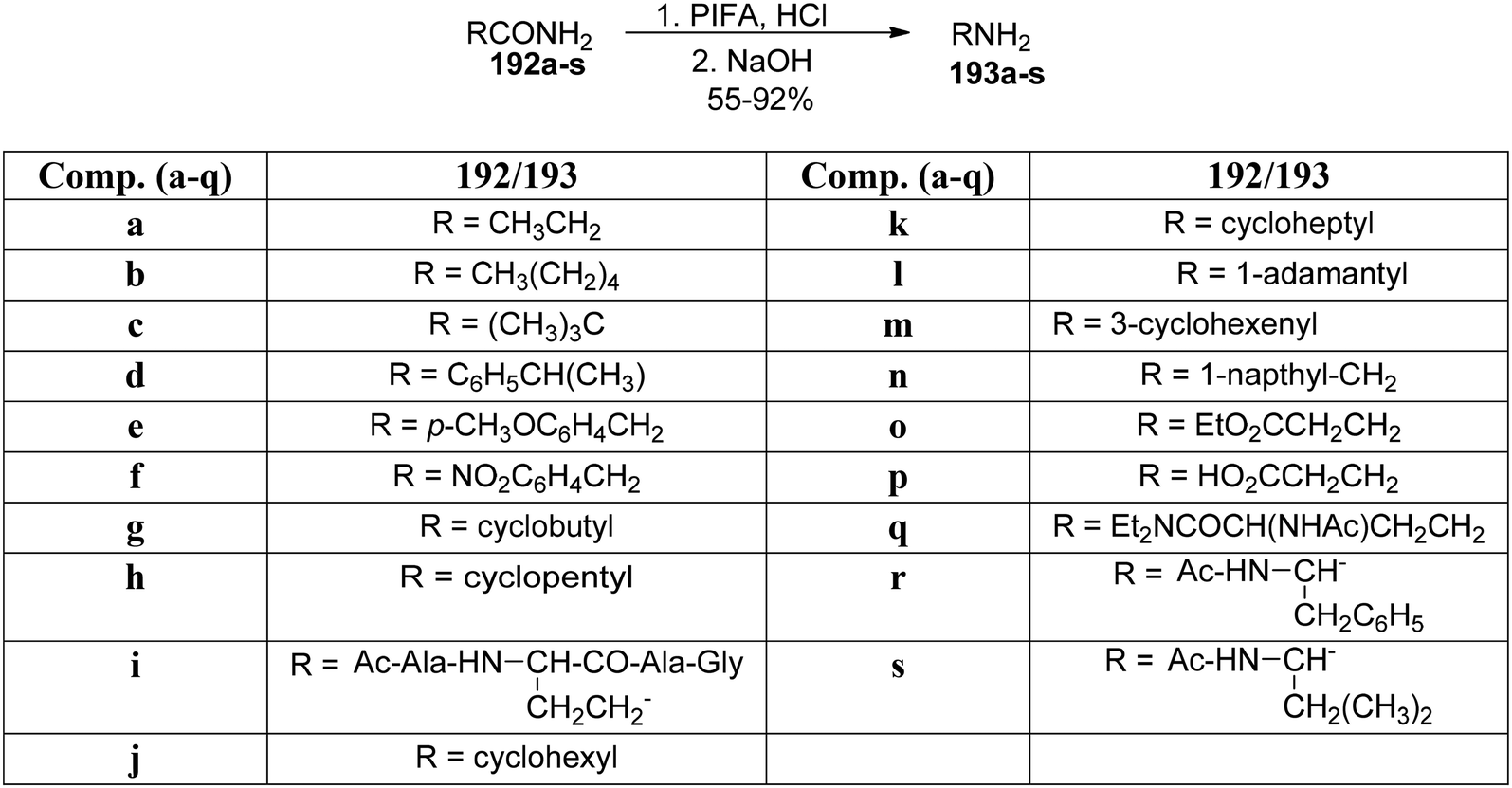
Organic fluorescent molecules are significant molecules in varied scientific domains, including chemical biology, photocatalytic chemistry and materials research owing to their specificity, easy accessibility, and stability as well as sensitivity.157 Gao et al.158 reported an effective and simple methodology to construct pyrido-quinazolin-6-ones 193 by subjecting readily accessible carboxamide derivatives 192 to Hofmann rearrangement. The PIFA-catalyzed reaction was performed at room temperature for 1 hour to obtain the pyridoquinazzolin-6-ones 193a–s in 55–92% yields (Scheme 80). PIFA engaged in reactions via the ionic pathway. This straightforward methodology allows the development of excellent yields of fluorophores chemoselectively under mild conditions. The reaction also tolerates a range of functional groups.
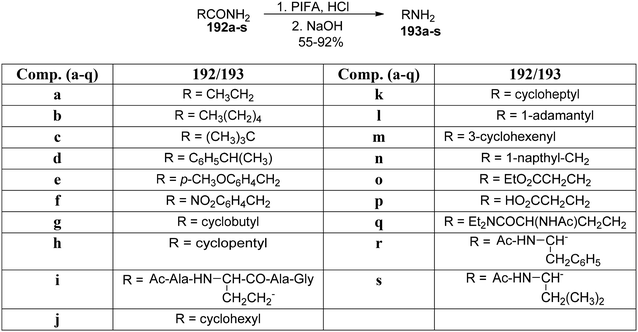 |
| | Scheme 80 Synthesis of pyridoquinazolin-6-ones 193a–s. | |
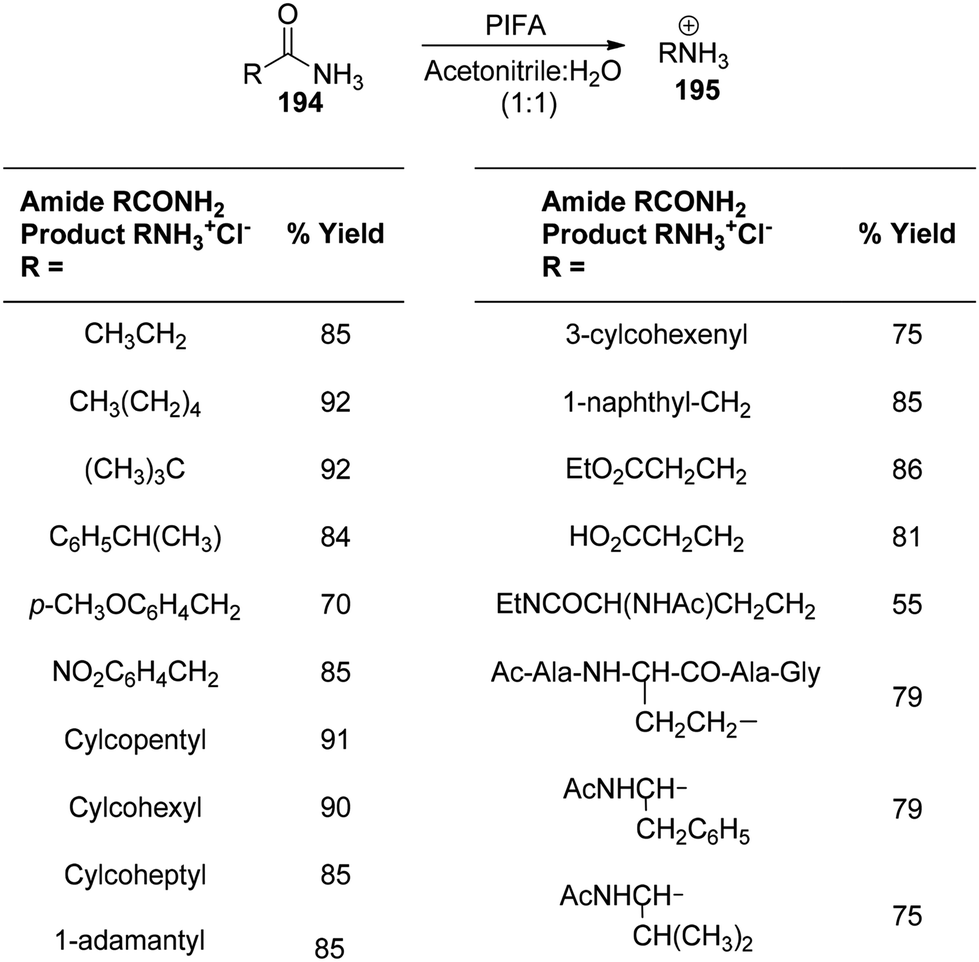
Loudon et al.159 employed PIFA under mild acidic conditions to catalyze the rearrangement of aliphatic amides 194, yielding amines 195. The reaction, known as the “acidic Hofmann rearrangement”, proceeds with complete retention of configuration for the migrating group. Although the reaction involves isocyanates as intermediate species, under mildly acidic conditions, they rapidly degrade to yield amines. The resultant amines 195 are protected from urea formation due to the degradation of isocyanate intermediates in an acidic environment. The addition of pyridine to maintain a pH of approximately 3 expedites the reaction. Scheme 81 provides additional visual representations of the reaction. PIFA engaged in reactions via the ionic pathway.
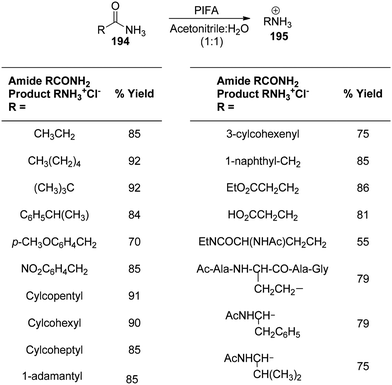 |
| | Scheme 81 Transformation of 1° amides to amines by PIFA-mediated rearrangement reaction. | |
3.10. PIFA in carbohydrate chemistry
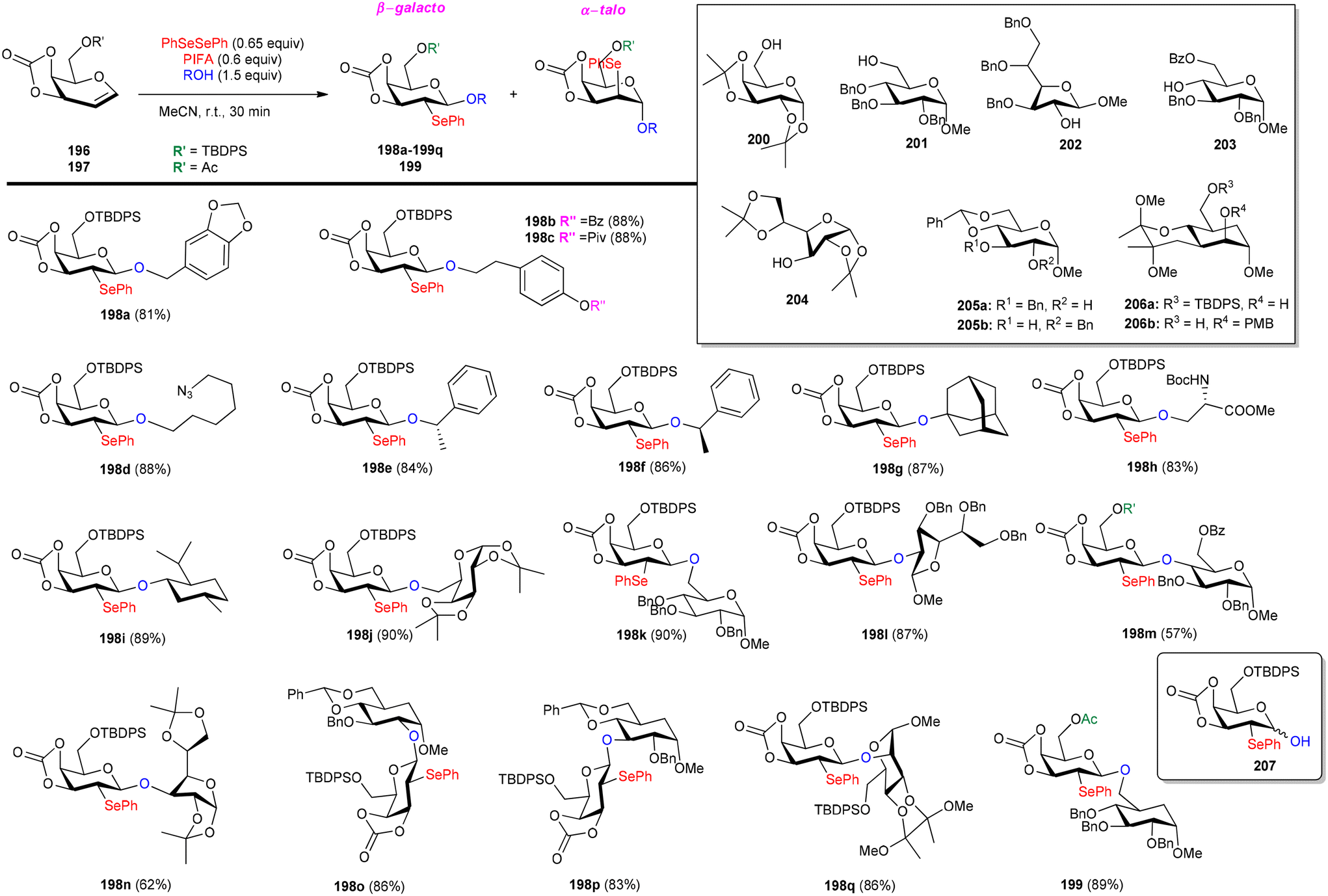
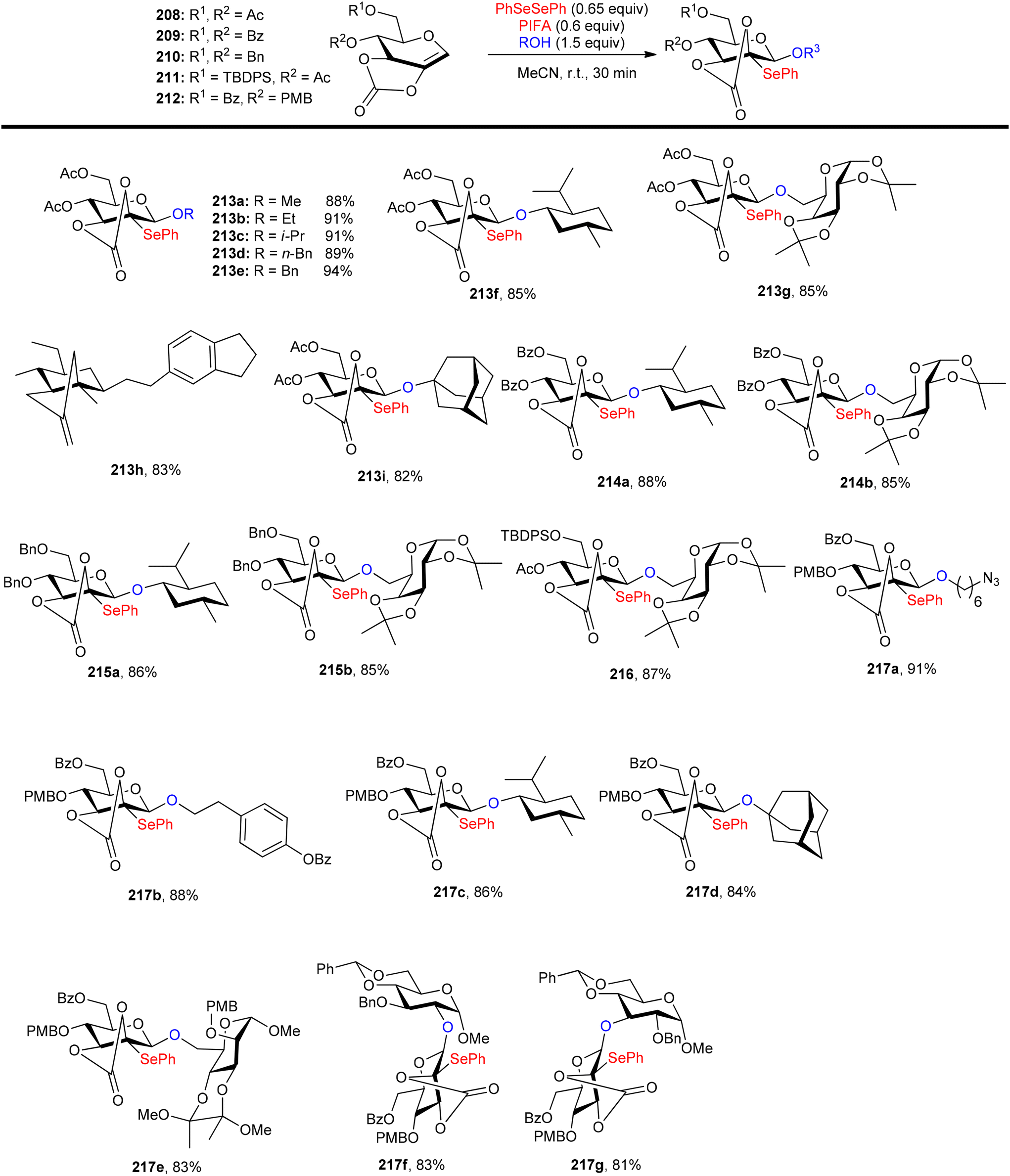
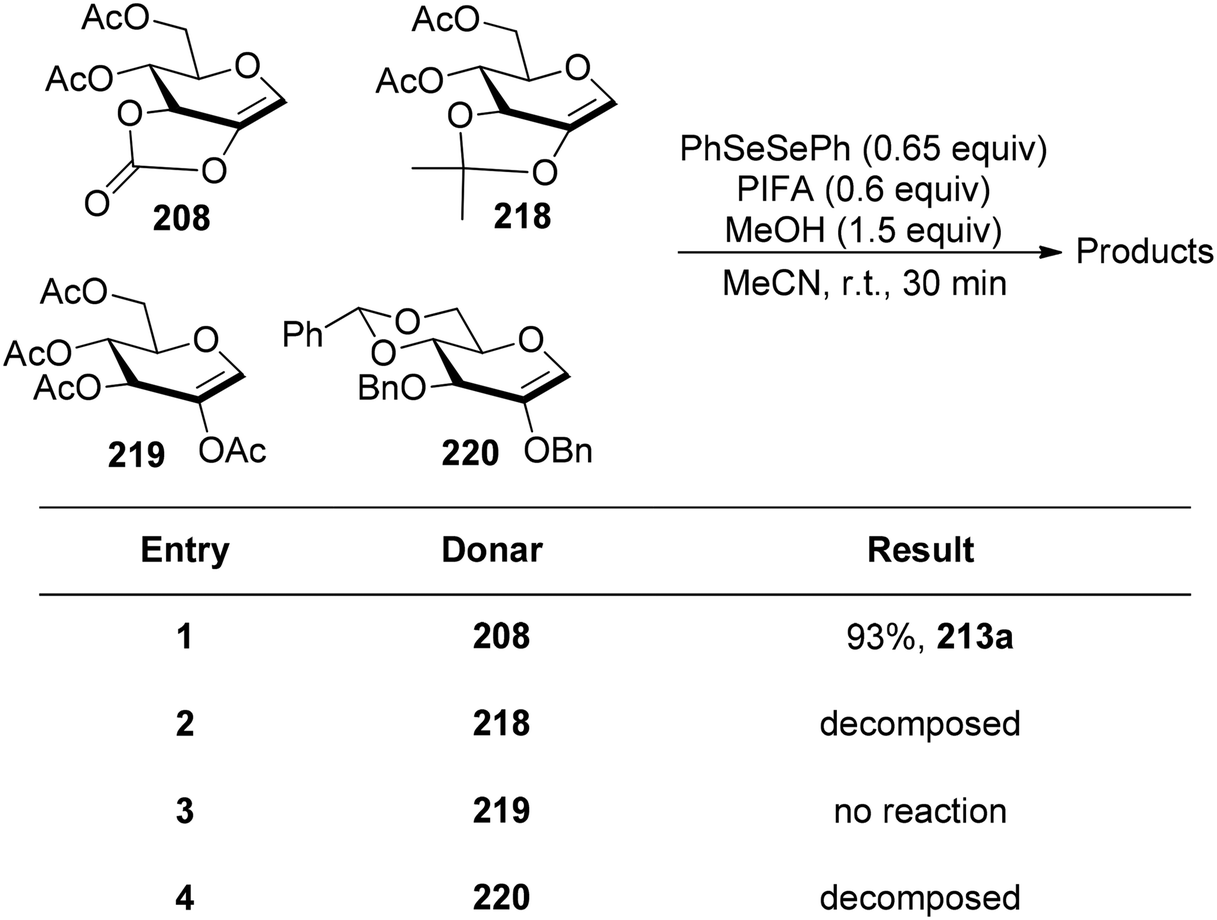
Glycoconjugates and oligosaccharides are crucial components for numerous biological processes.160,161 Meng et al.162 described a PIFA-mediated phenylselenoglycosylation of glycol analogues under benign conditions. By attachment of fused carbonate to the glycols, stereo-selective glycosylation was accomplished. Galactal 196 resulted in the formation of 2-phenylseleno-2-deoxy-β-galactosides in all of these noncarbohydrate acceptor processes in excellent yields (Scheme 82). Additionally, they reported the glycosylation of compound 196 with various acceptors derived from sugars 200–206. The majority of primary alcohols and several secondary alcohols exhibited efficient reactivity with 196, yielding the corresponding β-linked disaccharides in yields ranging from 83% to 90%. When coupling with sugars 203 and 204, disaccharides 198m and 198n were obtained with yields of 57% and 62%, respectively, and lactol 207 was isolated in yields of 36% and 30%, respectively (Scheme 82). The glycosylation of 197 with acceptor 201 under the optimal condition provided β-linked disaccharide 200 in 89% yield. The phenylselenoglycosylation process was applied to a variety of 2,3-O-carbonate-2-hydroxyglucal 208–212 donors to produce the 2-phenylseleno-2,3-O-carbonate-mannosides 213–217 in yields of 81–94% (Scheme 83). For comparison, donors protected by 2,3-O-carbonate 208, 4,6-O-benzylidene 220, 2,3-O-isopropylidene 218 and peracetylation 219 with structural flexibility were also synthesised. The best conditions were utilized to glycosylate these four donors in methanolic media (Scheme 84). The majority of donor 219 was retrieved after 24 hours, likely due to its limited reactivity attributed to the electron-withdrawing impact of 2-O-Ac. Conversely, the electron-rich olefins in 218 and 220 could be overly responsive to electrophilic species, possibly causing them to react directly with PIFA, leading to the formation of intricate byproducts. Intriguingly, only the reaction involving donor 208 yielded promising results, with the identification of product 213a as a β-glycoside. PIFA engaged in reactions via the ionic pathway.
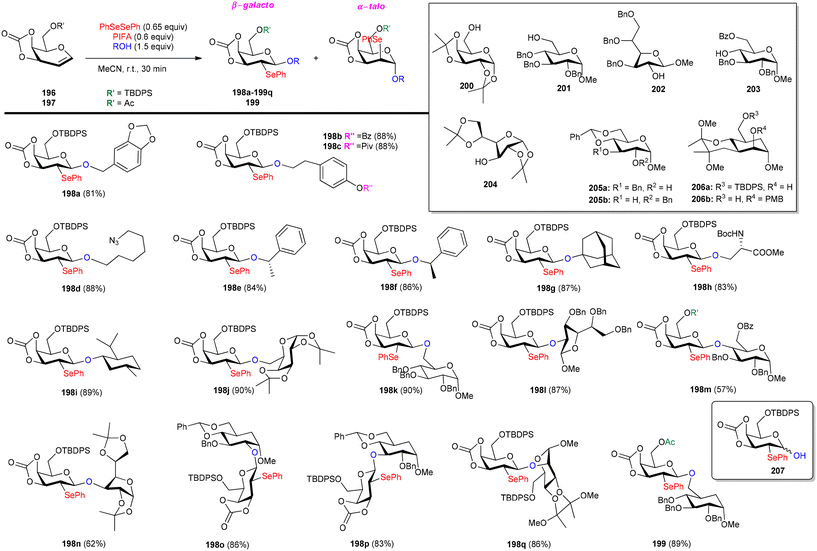 |
| | Scheme 82 Synthetic scheme for stereoselective formation of 2-phenylseleno-2-deoxy-β-galactosides. | |
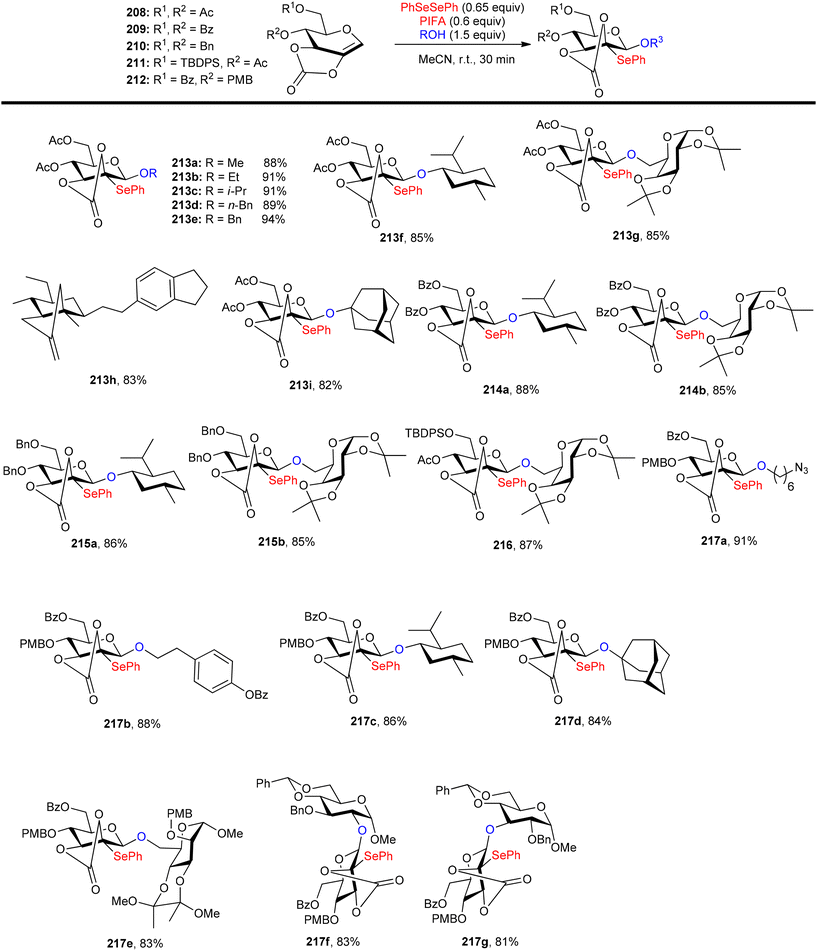 |
| | Scheme 83 Phenylselenoglycosylation reaction of 2,3-O-carbonate-2-hydroxyglucals. | |
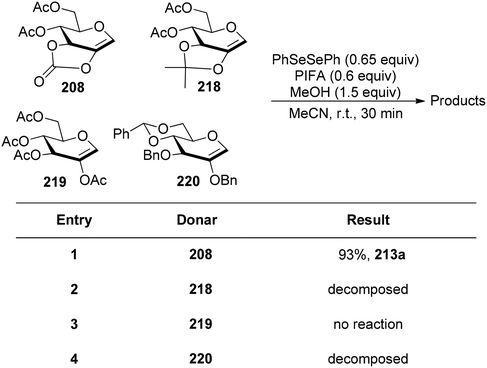 |
| | Scheme 84 Glycosylation of 2-hydroxyglucals 208, 218–220. | |
The complex oligosaccharides located on the cell surface play crucial roles in various biological processes, particularly in cell-to-cell adhesions, as evidenced by advancements in the field of glycosciences. Drugs containing glycoside linkages are now commonly employed in the treatment of diabetes, influenza virus, and cardiovascular ailments.163
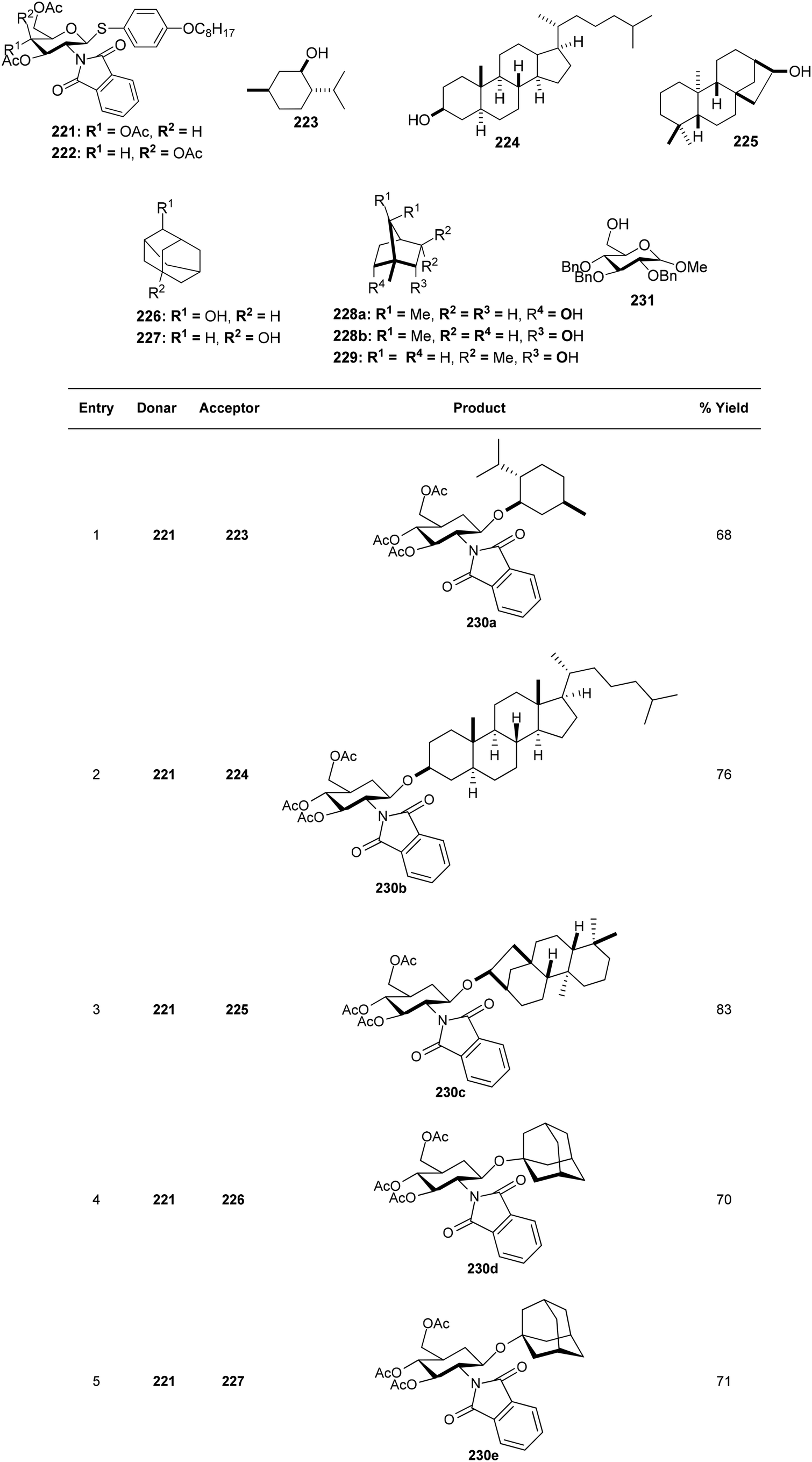
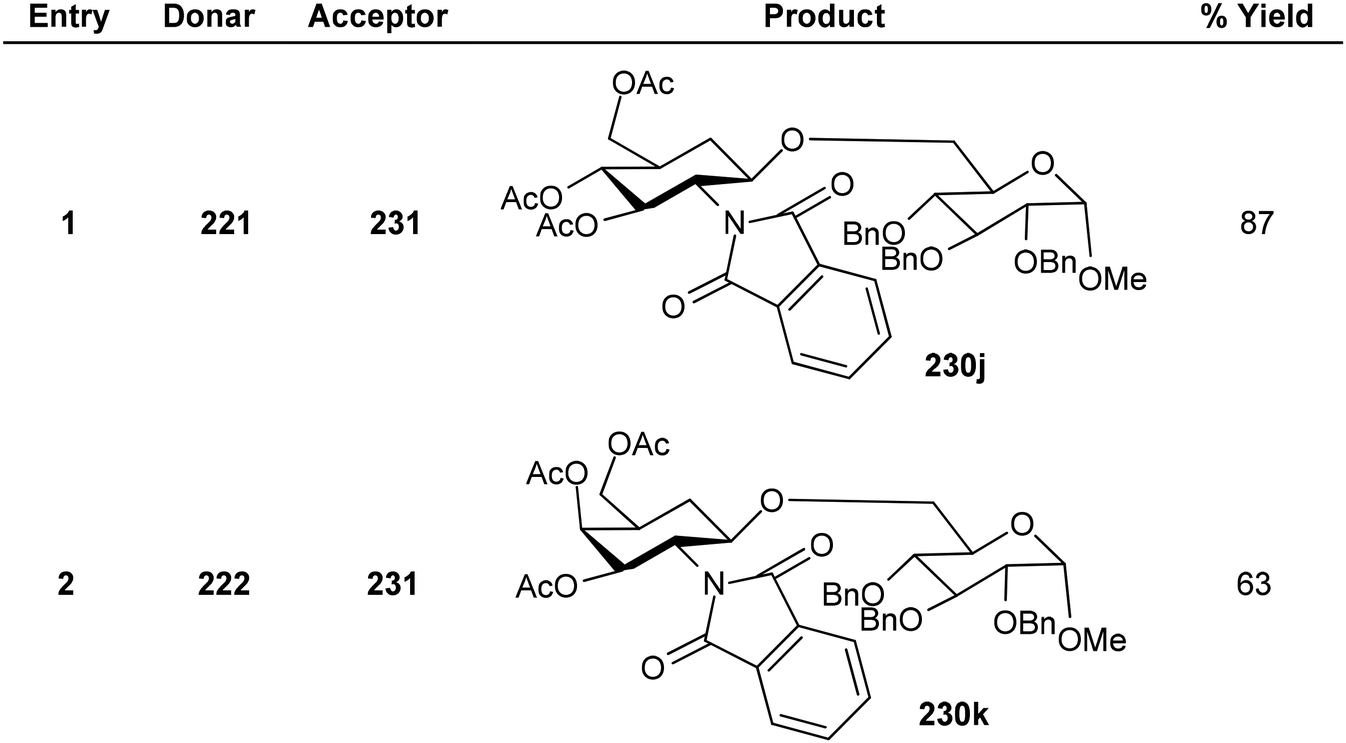
Substituted analogues of D-glucopyranoside as well as D-galactopyranoside were utilized as the glycosyl donors in the study by Kajimoto et al.164 The reaction gave good yields with cholestanol, 1-,2-adamantanols as well as (−)-menthol acceptor molecules. Thioglycosides 221, and 222 and a range of alkanols 223–230 were glycosylated by activating PIFA with TfOH (Scheme 85). Additionally, utilizing this reaction scheme, disaccharides with a D-glucosamine/galactosamine 231j–k functionality at the non-reducing end can also be synthesized (Scheme 86).
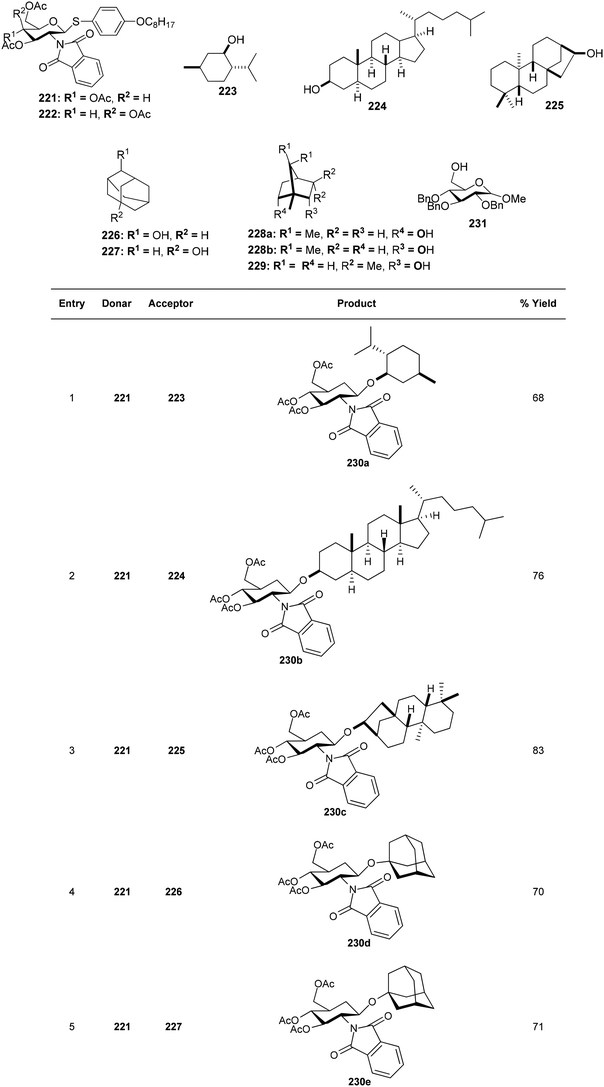 |
| | Scheme 85 PIFA/TfOH catalyzed glycosylation reaction of thioglycosides as well as various alkanols. | |
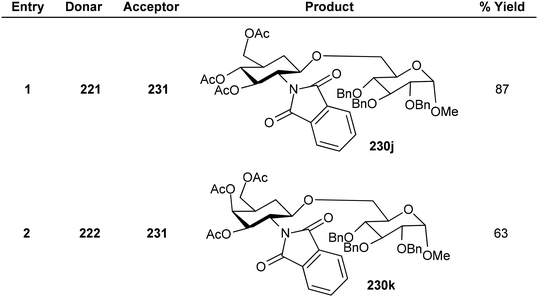 |
| | Scheme 86 Disaccharide synthetic methodology via thioglycosylation in presence of PIFA/TfOH. | |
3.11. One-pot reaction
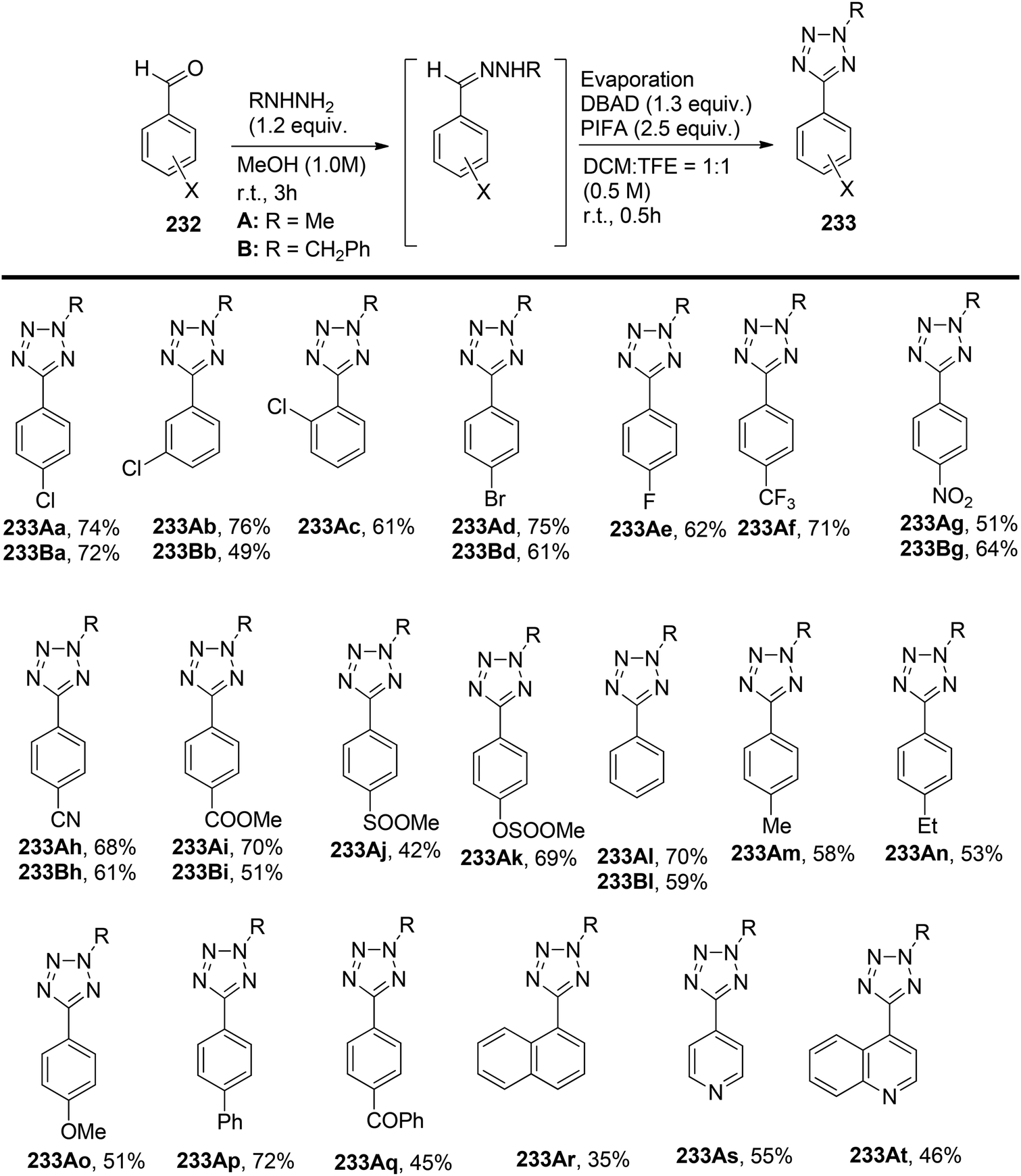
Tetrazoles, known for their high lipophilicity, serve as important heterocyclic motifs and bioisosteres of carboxylic acids and amides.165 Imai et al.166 achieved the direct synthesis of various 5-aryl-2-methyltetrazoles and 5-aryl-2-benzyltetrazoles. The researchers prepared a library of arylated tetrazoles through a two-step process starting from substituted aromatic aldehydes 232. In the initial step, the aldehydic substrates were reacted with hydrazones A and B. This was followed by a reaction with DBAD/PIFA, resulting in the desired tetrazoles 233 (Scheme 87).
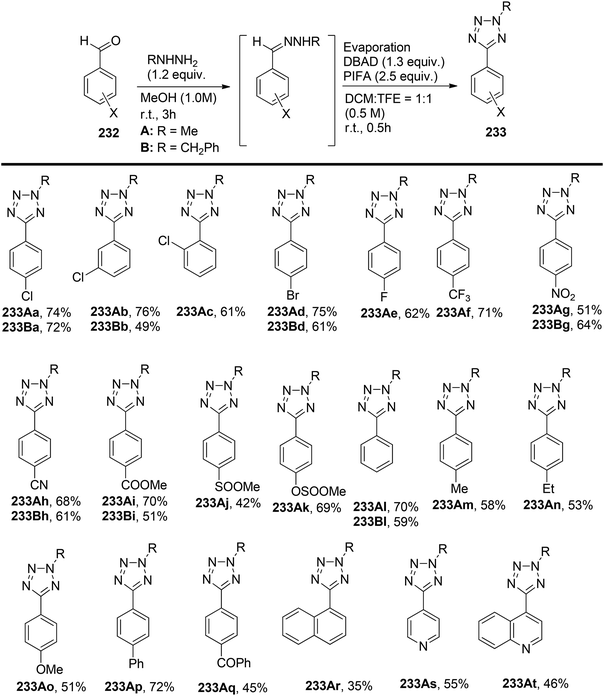 |
| | Scheme 87 Synthetic scheme for transforming aromatic aldehydes to 2-alkyl-5-aryltetrazoles. | |
Recently, selenosulfonates have garnered significant attention in chemical synthesis. These compounds undergo selenosulfonations with diazomethane, olefins, acetylenes, and allenes through electrophilic and free radical reactions. Chen et al.167 introduced an innovative one-pot methodology for synthesizing selenosulfonates 236 using diaryl diselenides 235, sodium sulfinates 234, and PIFA (Scheme 88).
 |
| | Scheme 88 Preparation of selenosulfonates 236. | |
3.12. Miscellaneous
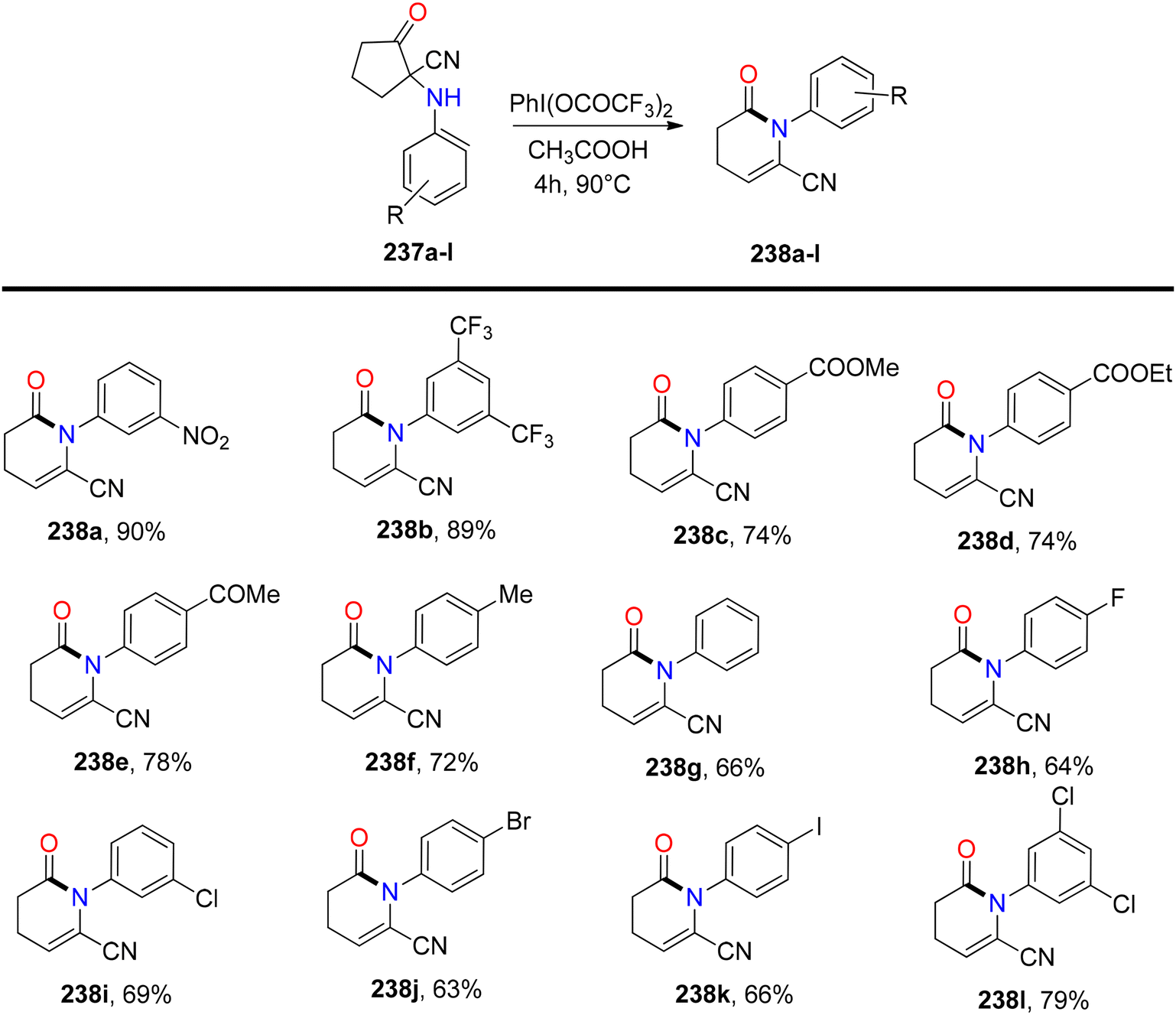
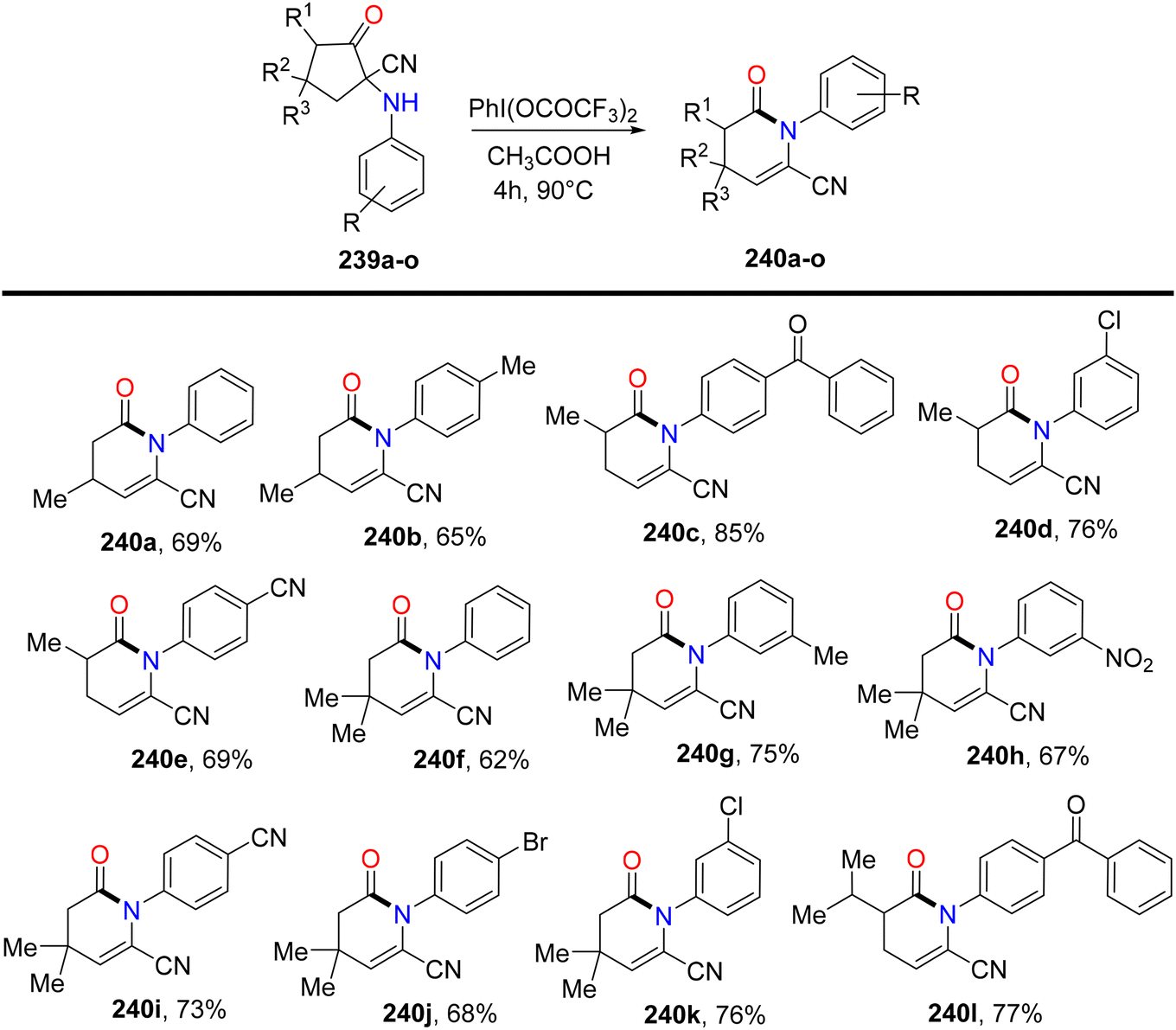
Bhattacherjee et al.168 reported the first synthetic methodology for regioselective formation of N-aryl-valerolactams 238a–lvia PIFA-catalyzed rearrangement of carbonitriles 237a–l. The reaction involves the rearrangement of the C–N bond, elimination of the C–5H atom, and opening of the C1–C2 linkage. Structural integrity of the lactam was further confirmed through single-crystal XRD measurements. PIFA actively participated in the reaction by employing an ionic pathway. The reaction conditions exhibited tolerance to various functionalities, yielding lactams in good to exceptional yields. Remarkably, electron-deficient cyclopentanones resulted in outstanding product yields due to stabilized positive charge (Scheme 89). Subsequently, the reaction was explored with variably substituted cyclopentanone derivatives 239 (Scheme 90). This reaction scheme also provides a novel pathway for obtaining biologically significant nitrile-functionalized lactams.
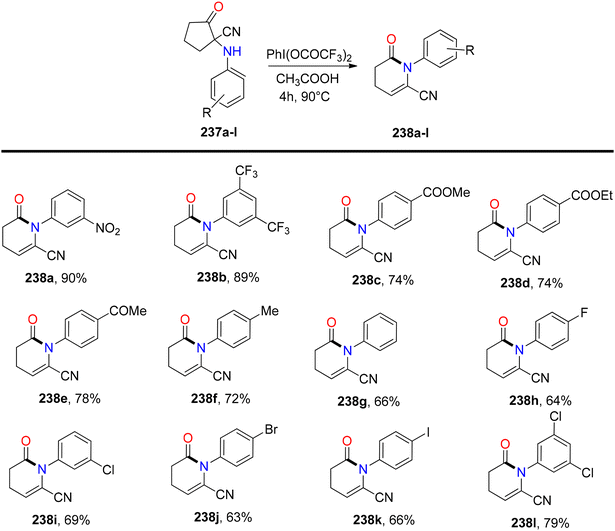 |
| | Scheme 89 Synthesis of N-aryl-valerolactams 238a–lvia PIFA-catalyzed rearrangement of carbonitriles 237a–l. | |
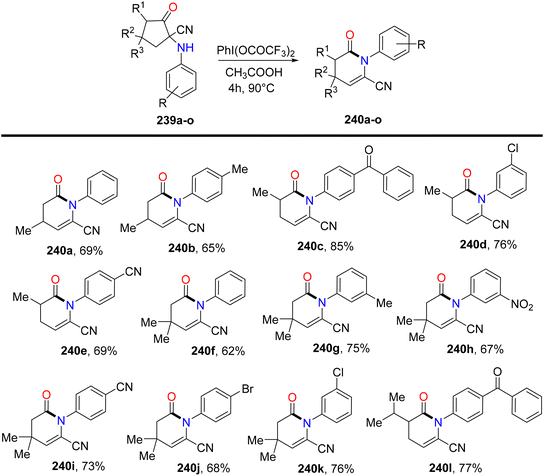 |
| | Scheme 90 Examination of the lactamization reaction with variably substituted substrates. | |
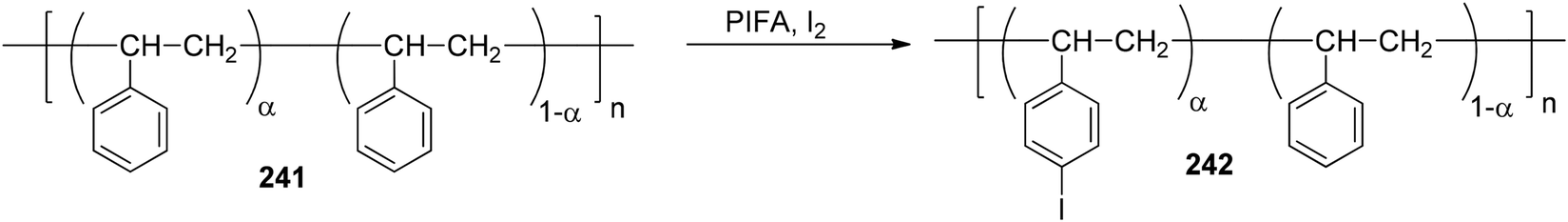
Yudina et al.169 investigated the room temperature PIFA-mediated direct iodination of polystyrene 241 (PS) (Scheme 91). The resulting poly(p-iodostyrene) 242 exhibited a molecular weight distribution (MWD) quite similar to that of the original PS. Varying the substrate to reagent ratio resulted in the production of iodinated PS samples with a degree of substitution ranging from 0.5 to 1.
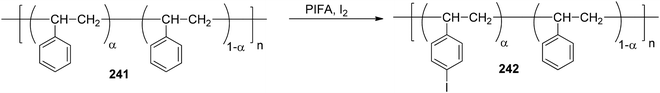 |
| | Scheme 91 Iodination of polystyrene (PS) 241. | |
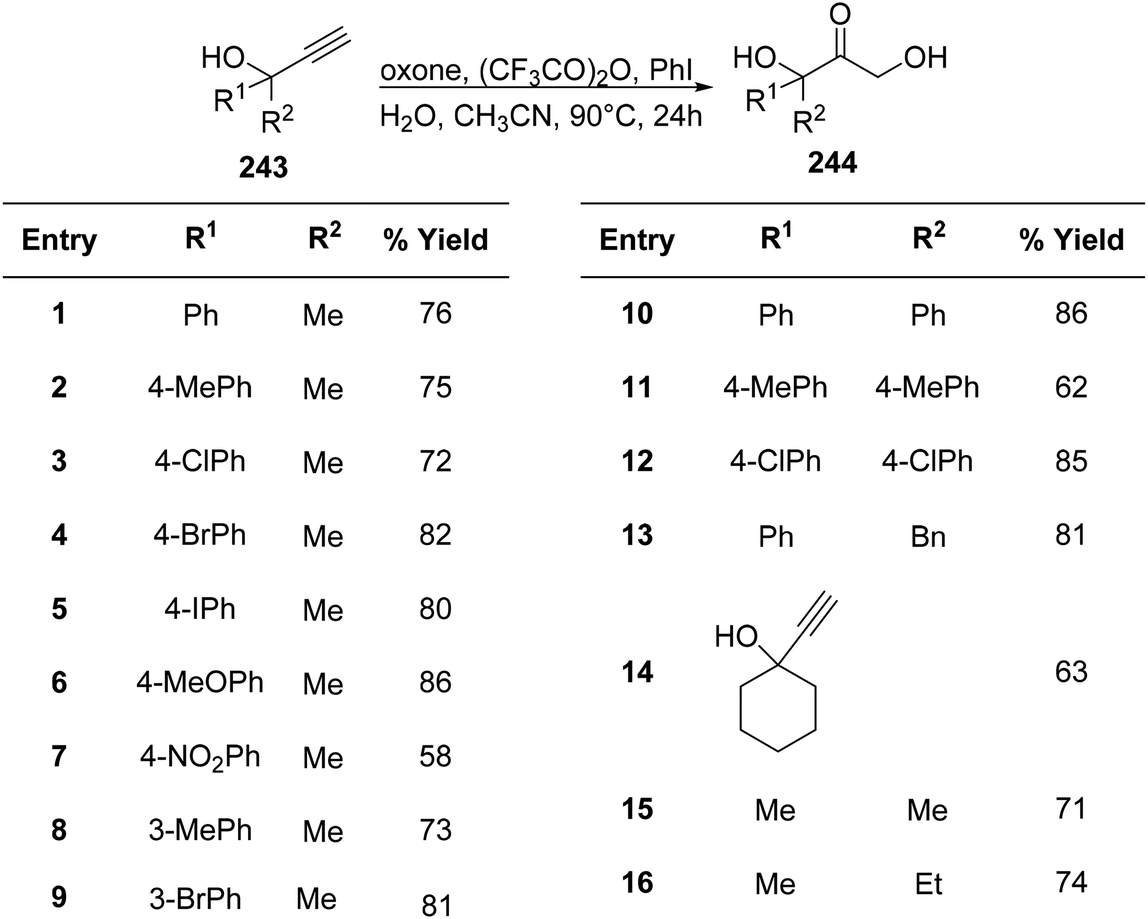
The α,α′-dihydroxy ketone 243 moiety is a significant component of many naturally occurring molecules such as betamethasone and pirarubicin. Thus, its synthetic protocol is of great interest.170 Chen et al.171 utilized a PIFA/oxone/(CF3CO)2O system to obtain a series of α,α′-dihydroxy ketones 244 (Scheme 92). PIFA actively participated in the reaction by employing an ionic pathway. This methodology is easy to handle and does not utilize stoichiometric ratios of PIFA.
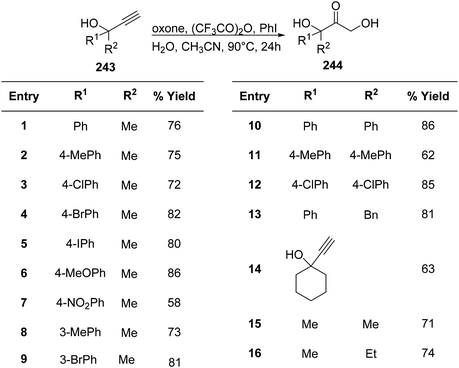 |
| | Scheme 92 Reaction scheme for converting ethynyl carbinols to α,α′-dihydroxy ketones in the presence of oxone/(CF3CO)2O system. | |
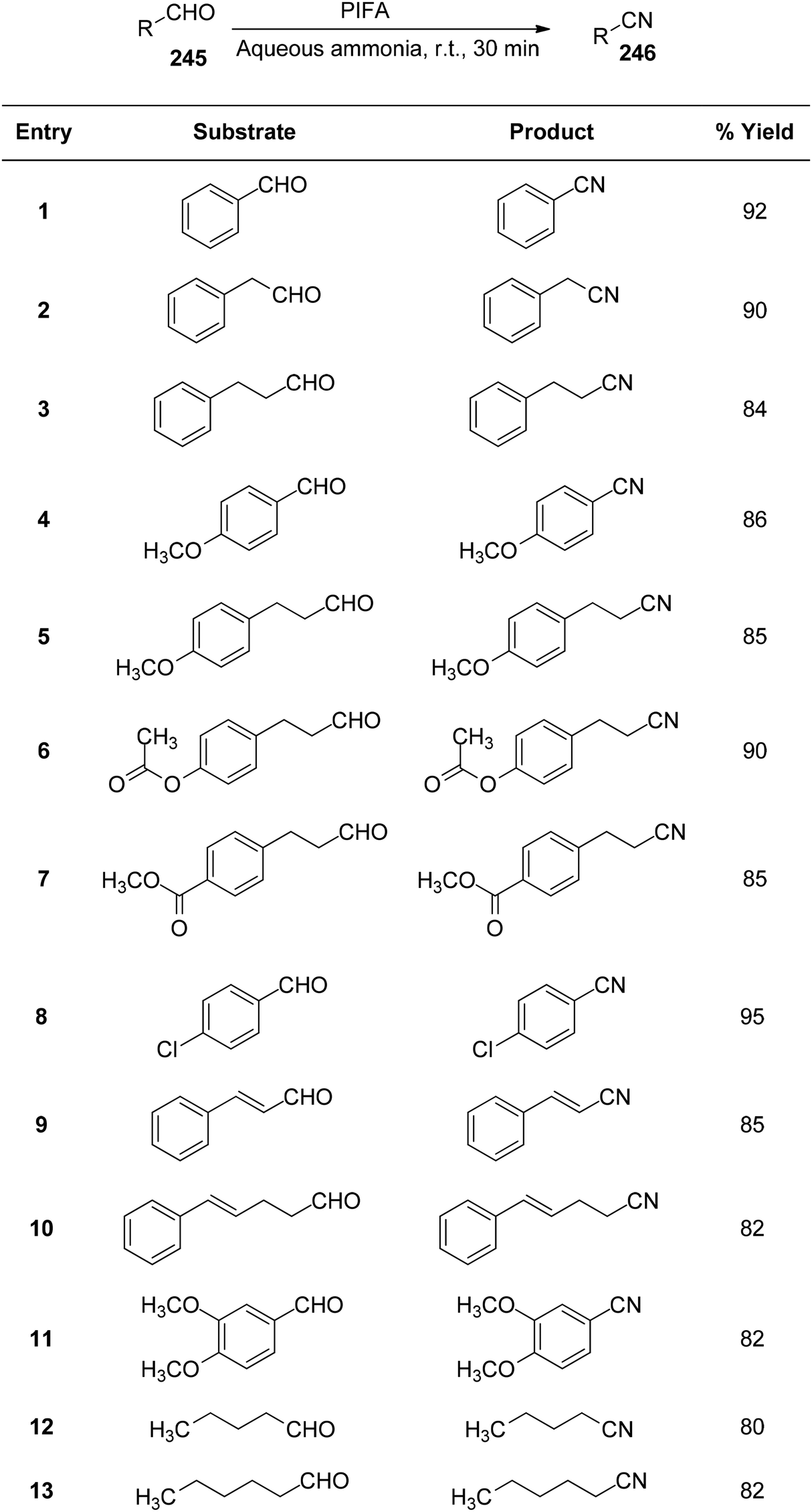
Telvekar et al.172 presented a straightforward and mild process to convert aldehydes 245 with various substitutions into nitriles 246 at room temperature, utilizing aqueous ammonia and PIFA. This system demonstrated rapid reaction times, a simple setup, and yielded nitriles in average to good yields. Both aliphatic and aromatic substrates showed successful formation of nitriles using this methodology, with yields ranging from good to outstanding (Scheme 93).
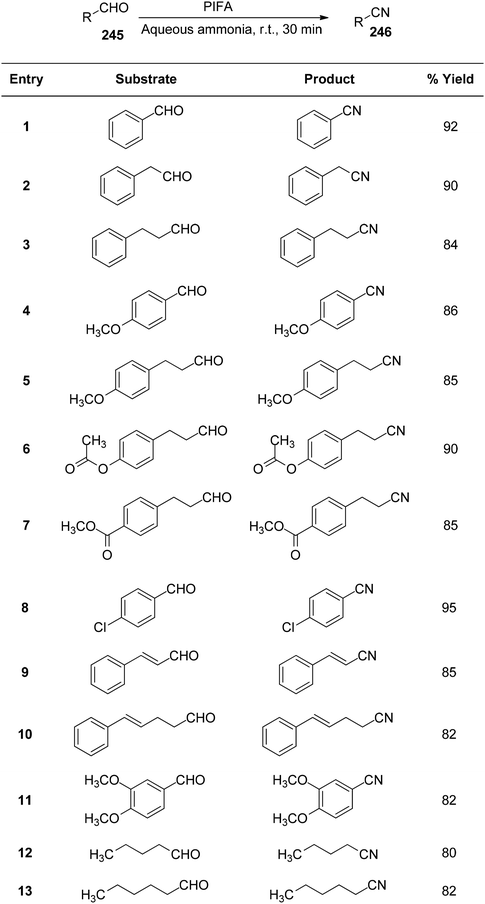 |
| | Scheme 93 Preparation of nitriles 246 from aldehydes 245. | |
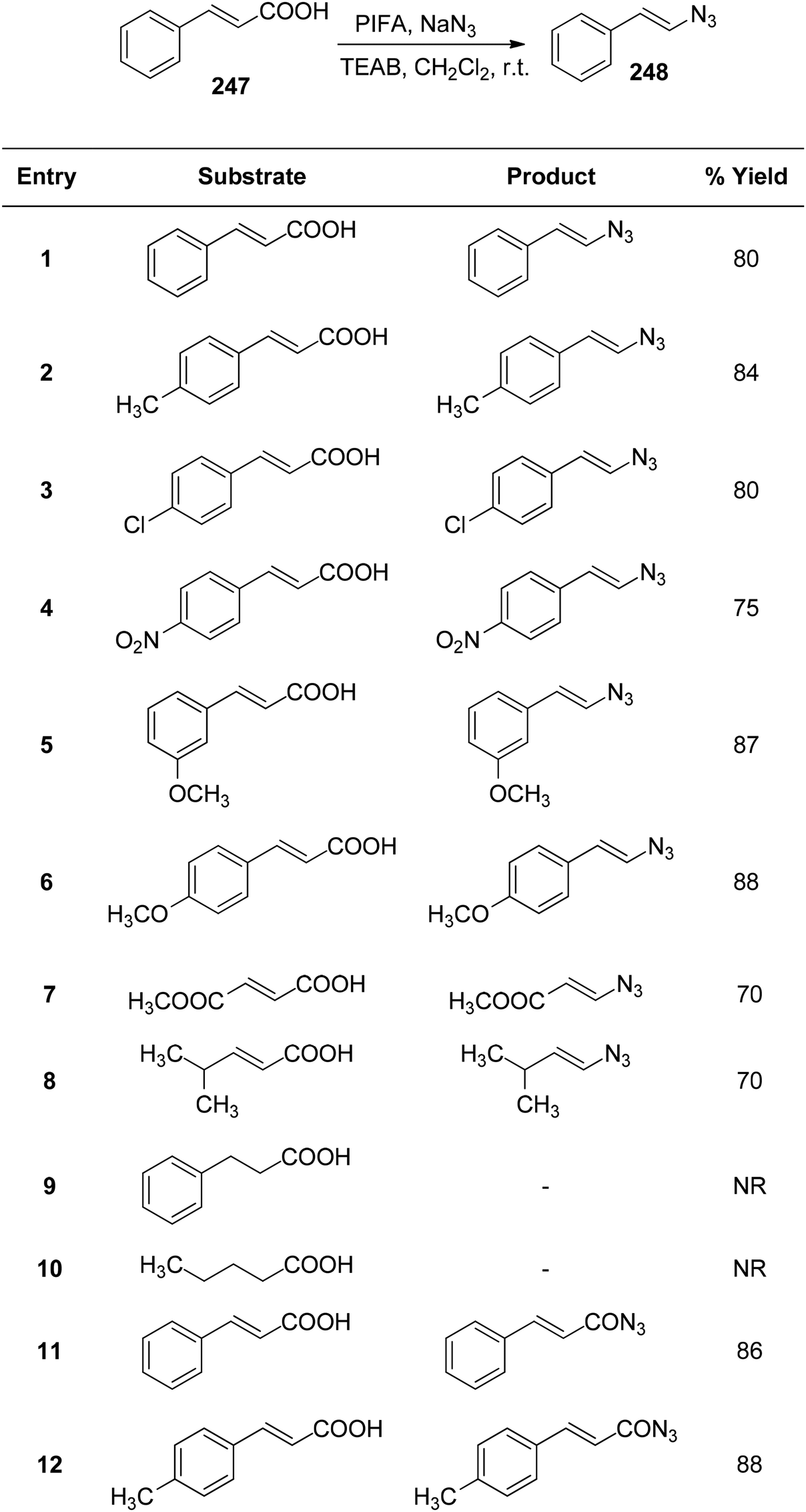
Telvekar et al.173 employed PIFA/TEAB in the presence of sodium azide to synthesize vinyl azides 248 from α,β-unsaturated carboxylic acids 247 with various substitutions. This mild reaction exhibited broad substrate tolerance, consistently yielding excellent yields of vinyl azides. Diverse α,β-unsaturated carboxylic acids 247 were successfully transformed into vinyl azides 248, obtaining yields ranging from 70–88% (Scheme 94).
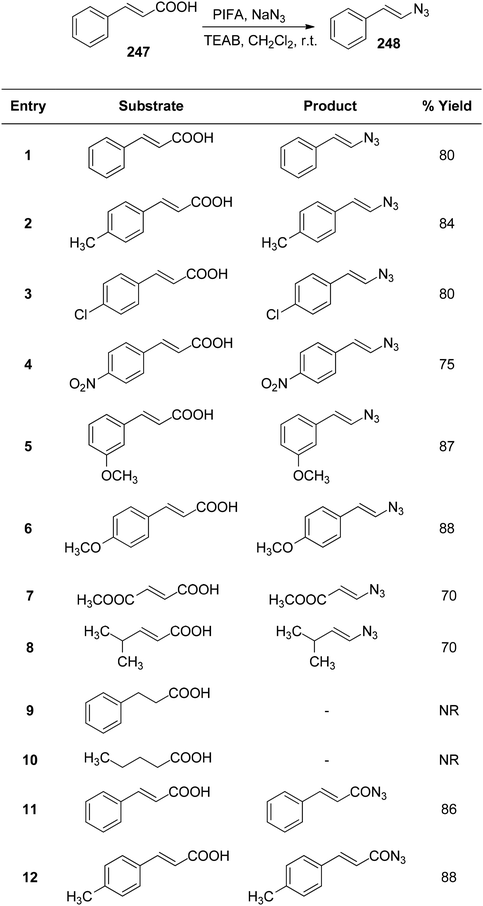 |
| | Scheme 94 Carboxylic acid reaction with PIFA and NaN3. | |
Shorter reaction times as well as the use of non-toxic substrate are the associated advantages for this synthetic scheme. Furthermore, it can also be used to obtain the acyl azides 249 (Scheme 95).
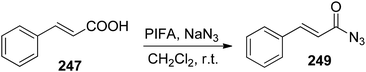 |
| | Scheme 95 Cinnamic acid converted into acyl azide using PIFA and sodium azide. | |
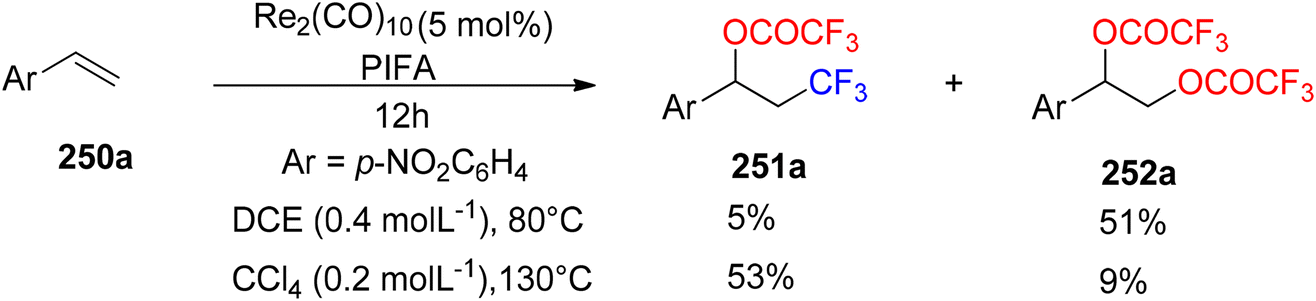
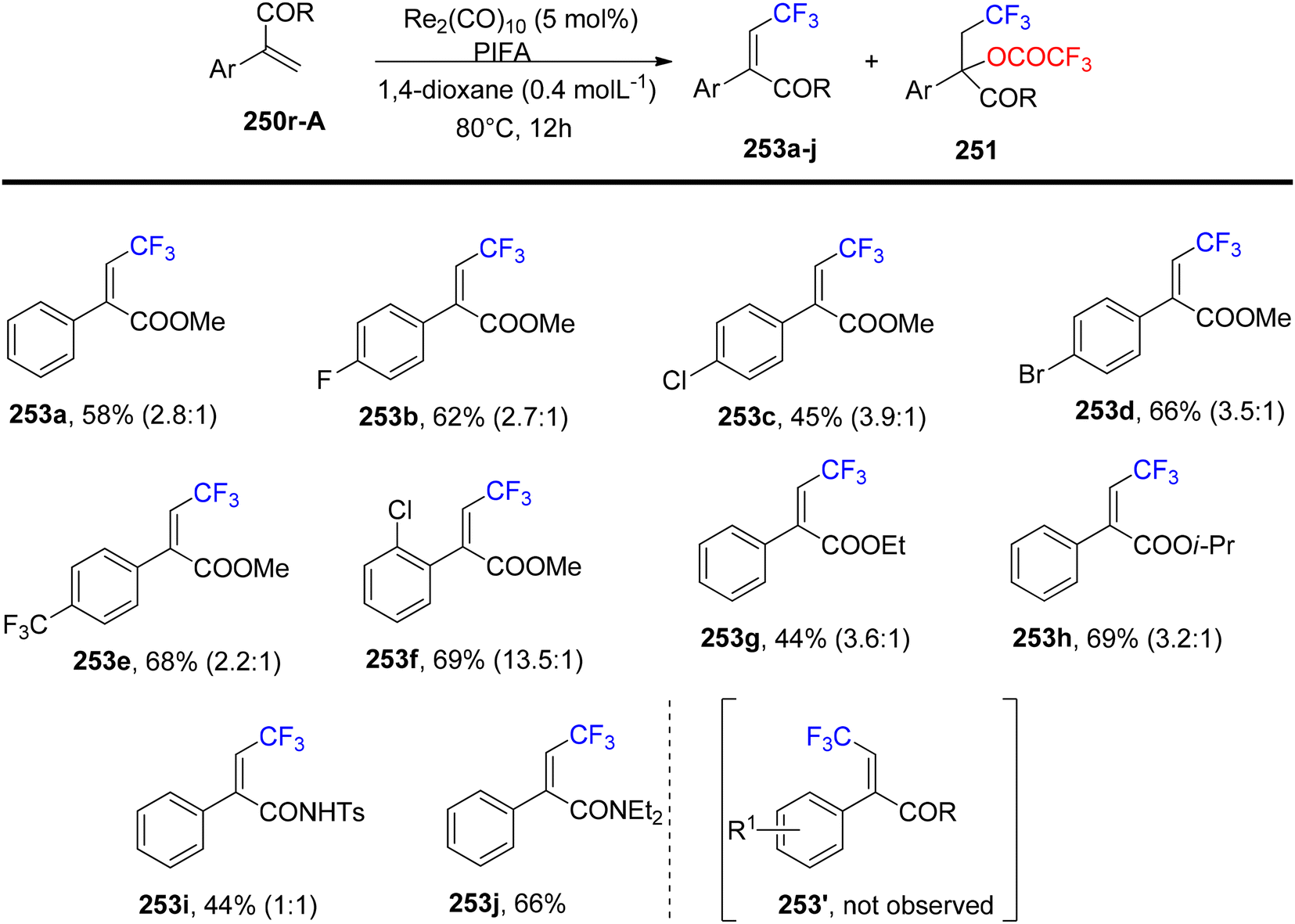
Wang et al.174 employed PIFA to achieve the inaugural oxytrifluoromethylation of styrenes 250a utilizing rhenium as the catalyst (Scheme 96). Following optimization, they investigated the reactivity of different alkenes 250b–q with PIFA as their reaction counterpart (Scheme 97). Notably, when they subjected α,β-unsaturated esters and amides 250r-A to the reaction conditions, the primary product formed was the Heck-type trifluoromethylation product 253a–j, while the concurrent generation of the oxytrifluoromethylation product 251 occurred but in a relatively minor quantity (Scheme 98). PIFA actively participated in the reaction by employing a free radical pathway, particularly through hydrogen abstraction.
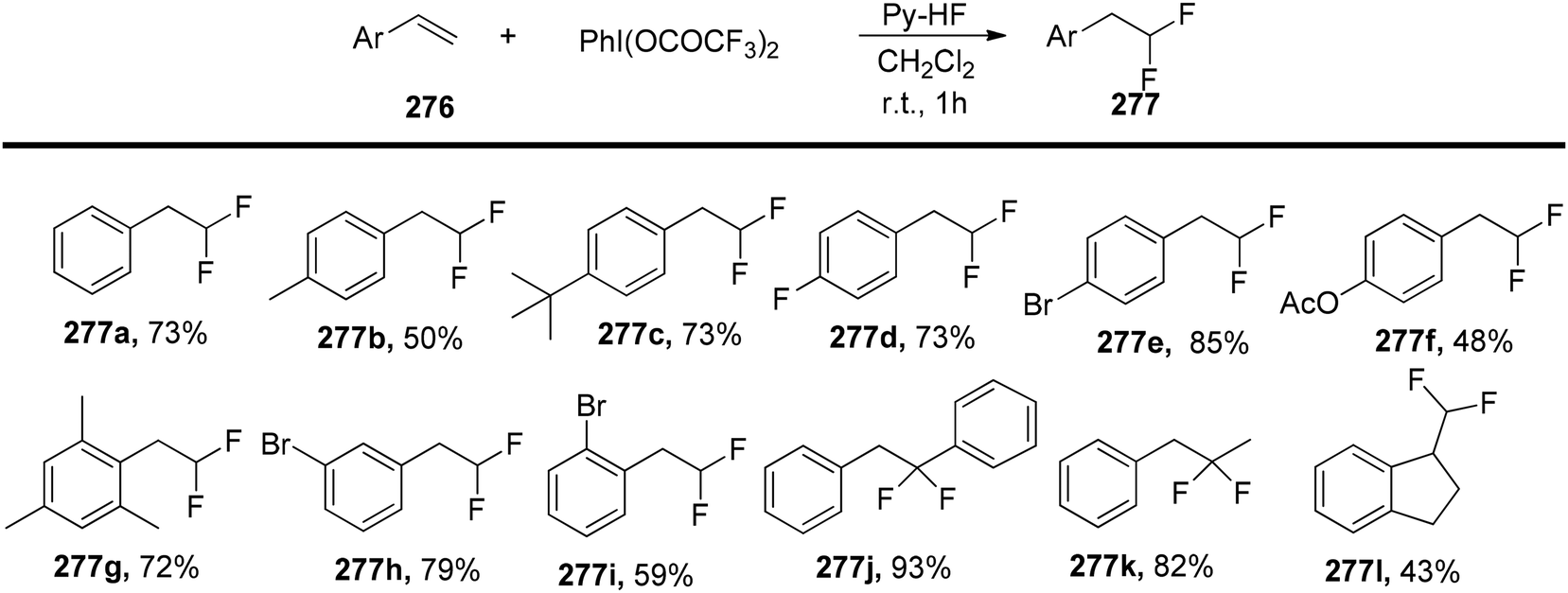
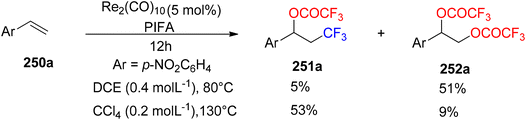 |
| | Scheme 96 Re-catalyzed decarboxylative oxytrifluoromethylation of styrene using PIFA. | |
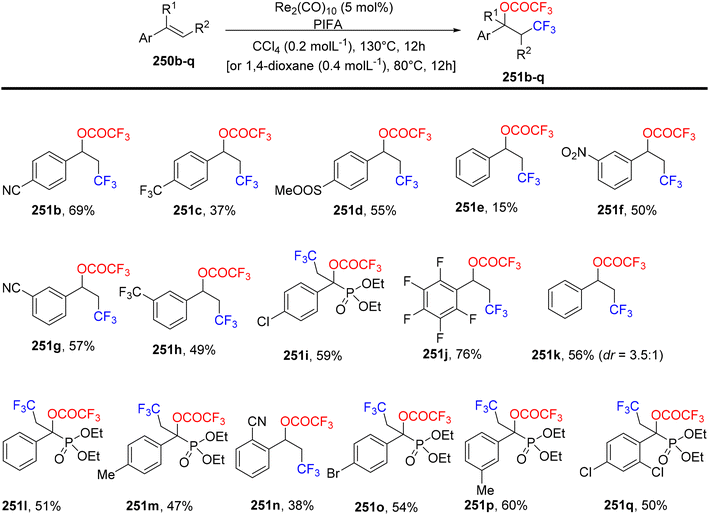 |
| | Scheme 97 Oxytrifluoromethylation of styrenes. | |
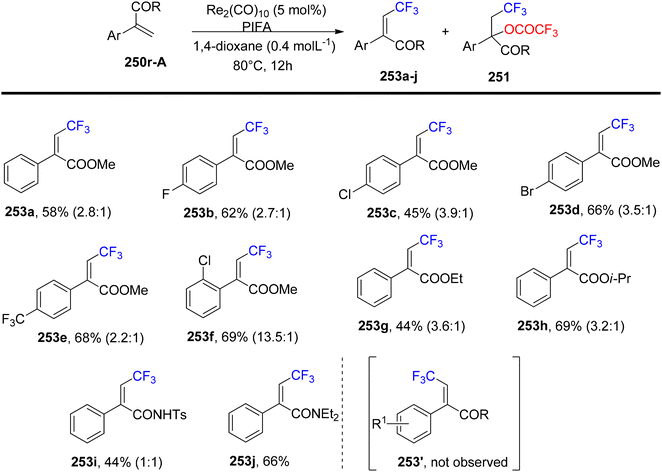 |
| | Scheme 98 Trifluoromethylation reaction of α,β-unsaturated esters as well as amides stereoselectively. | |
In order to synthesize 1,2-dicarbonyls 255 from variably substituted alkynes 254, Tingoli et al.175 investigated the usage of in situ-produced iodonium and selenium species (Scheme 99). PIFA enables the synthesis of 1,2-diketones 255, utilizing diphenyl diselenides as well as elemental iodine, from internal alkynes 254. Unfortunately, initial reactions with iodine resulted in incomplete conversion. Diphenyl diselenide, on the other hand, was more effective and resulted in the formation of the desired product in excellent yields. Furthermore, in order to synthesize quinoxaline derivatives, a second condensation procedure was utilized, thus validating the dicarbonyls as vicinal.
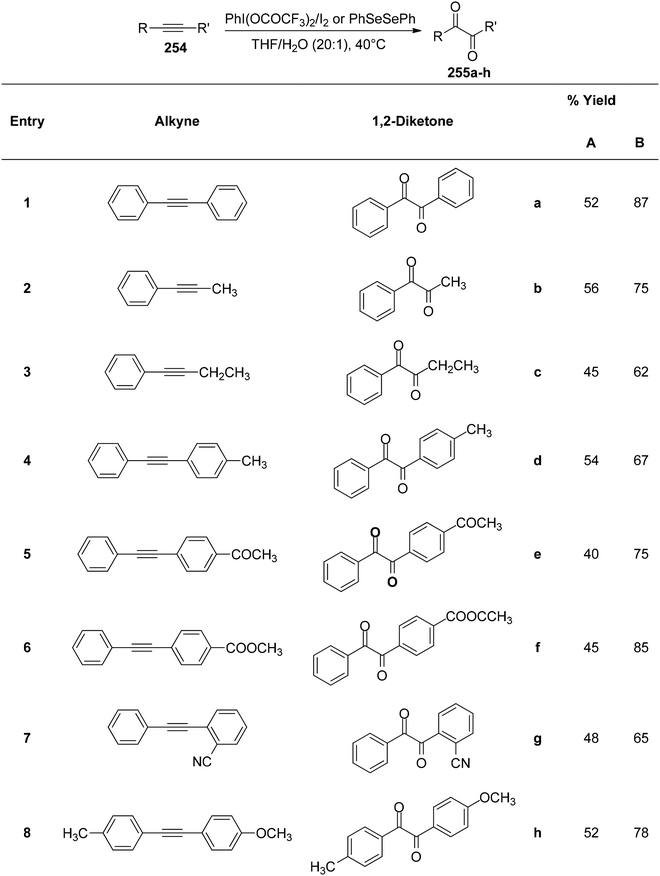 |
| | Scheme 99 Synthetic scheme for 1,2-diketones from alkynes in presence of (A) PhI(OCOCF3)2 or (B) PhSeSePh. | |
Catir et al.176 utilized a PIFA/H2O2 system and singlet oxygen acceptor molecules to generate singlet oxygen products.177 PIFA was used to study the breakdown of hydrogen peroxide 256. PIFA actively participated in the reaction by employing a free radical pathway. The outcomes of the study exhibited that hydrogen peroxide treatment on a PIFA molecule results in generation of 1O2 (Scheme 100a). Furthermore, the production of 1O2 was confirmed by chemiluminescence detection at 1270 nm as well as trapping experiments. There is strong evidence to support the idea that the hydroperoxyl radical, formed when hydrogen peroxide and PIFA combine, decomposes into singlet molecular oxygen through a tetraoxidane intermediate. The decomposition of the experiment has been also reported with optically pure (1S)-1-phenylethyl hydroperoxide 261 instead of hydrogen peroxide 256 (Scheme 100b).
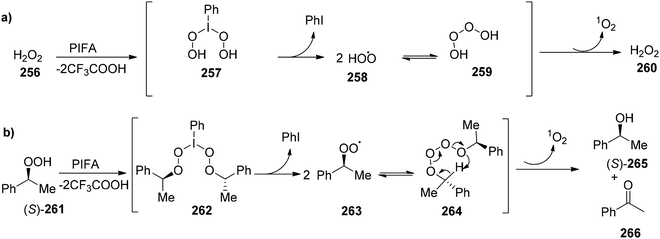 |
| | Scheme 100 Decomposition of 256 and (S)-261 with PIFA. | |
Kita et al.178 conducted innovative and direct nucleophilic reactions using PIFA for sulfenylation (Schemes 101 and 102) and thiocyanation (Scheme 103) of phenol ethers and related compounds 267. This study successfully synthesized numerous unsymmetrical diaryl sulfides 268 and aryl thiocyanates 273 in high yields, which can serve as synthons for sulfur aromatics. Unlike other solvents such as CH3CN and CH2Cl2, which produced only minimal sulfenylation products, (CF3)2–CHOH, a polar, protic, and weakly nucleophilic solvent, facilitated the reaction effectively. Additionally, variably substituted thiophenols were employed as sulfur nucleophiles. Under similar conditions, arylthiol molecules 269 and 270 were obtained by treating 267b with thiophenols, achieving excellent yields. However, substrates with electron-releasing substituents like OMe/Me resulted in reduced yields of the sulfenylated products 271 and 272. PIFA actively participated in the reaction by employing a free radical pathway, particularly through single-electron oxidation, contributing to the process through this mechanism. Moreover, this methodology proved useful for the synthesis of diverse sulfur-containing aromatic molecules.
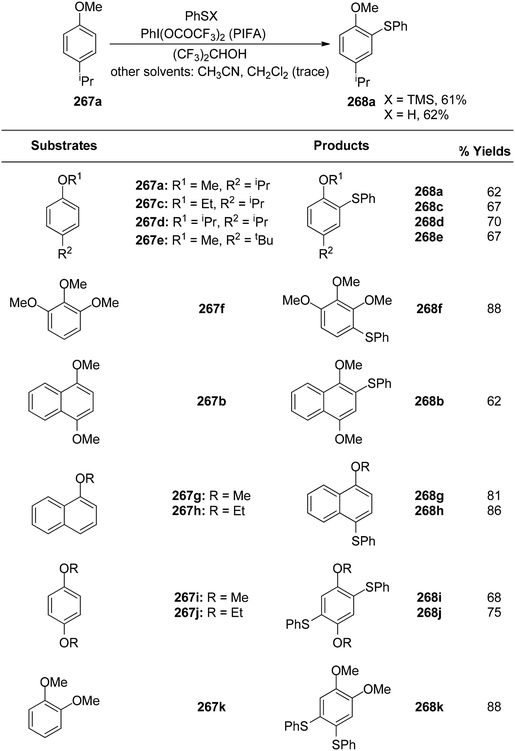 |
| | Scheme 101 Synthesis of direct sulfenylation compound 267. | |
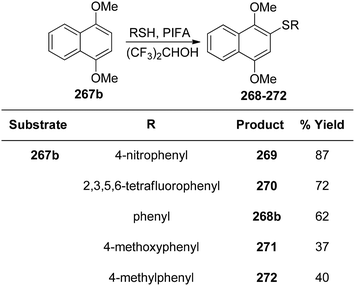 |
| | Scheme 102 Sulfenylation of 267b by various thiophenols. | |
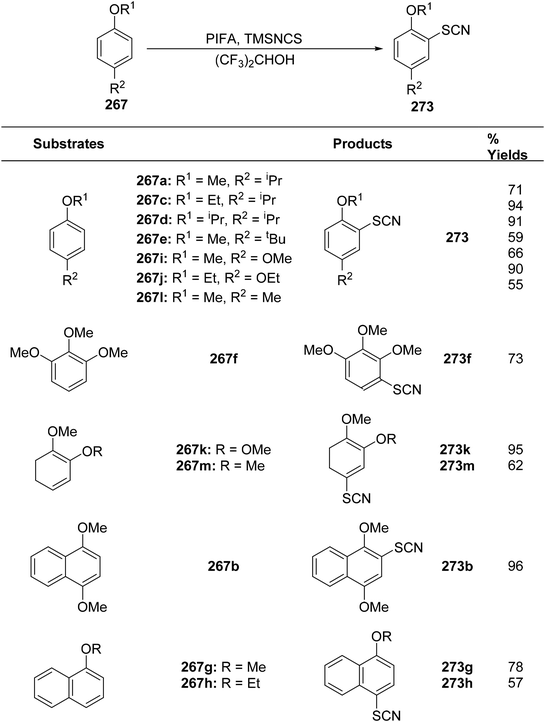 |
| | Scheme 103 Thiocyanation of phenol ethers and alkoxynaphthalenes. | |
Wu et al.179 have introduced a novel metal-free approach for the synthesis of olefins 275 through microwave-assisted direct decarboxylative elimination of arylacetic acids 274, as illustrated in Scheme 104. This method, employing commercially available PIFA, demonstrates a high level of efficiency in generating a variety of aryl/diphenylvinyl compounds 275 with yields ranging from moderate to good. PIFA actively engaged in the reaction by following a radical pathway, specifically through hydrogen abstraction. It is noteworthy that this decarboxylation process exclusively produces E-olefins, which could potentially be the thermodynamically favored products. Preliminary mechanistic inquiries suggest the involvement of a radical process in the decarboxylative elimination reaction.
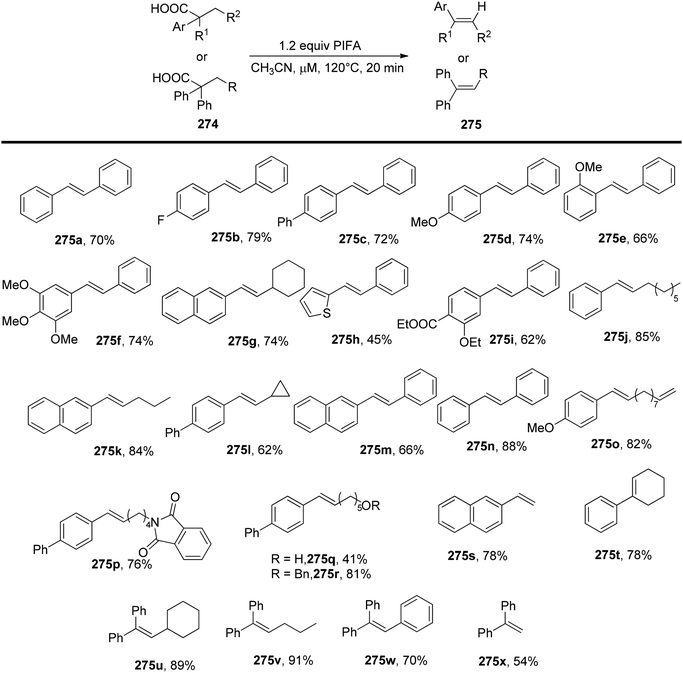 |
| | Scheme 104 Substrate scope of aryl/diphenylacetic acids 274. | |
Kitamura et al.180 have introduced an efficient and practical method for synthesizing 2,2-difluoroethylarenes 277 through the use of a hypervalent iodine-mediated fluorination reaction involving various styrene derivatives 276. They conducted a systematic exploration of different styrene derivatives 276 to investigate the scope of substrates, as outlined in Scheme 105.
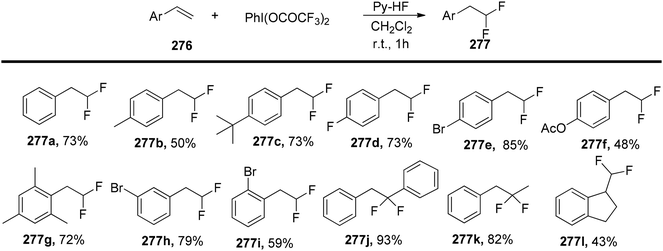 |
| | Scheme 105 Synthesis of 2,2-difluoroethylarenes 277. | |
Chatterjee et al.181 have introduced an effective, metal-free, and base-free approach for the precise synthesis of primary amines 279 derived from boronic acids 278, encompassing various types such as aryl, heteroaryl, and alkyl. This reaction takes place at room temperature and relies on the combined action of PIFA and NBS, employing cyanamidyl/arylcyanamidyl radicals as pivotal intermediates for the amination process. PIFA actively engaged in the reaction by following a radical pathway. It is noteworthy that these reactions are remarkably fast, typically completing within just one hour. Furthermore, to gain a deeper insight into the underlying reaction mechanism, the researchers conducted computational studies using DFT. These quantum chemical calculations, combined with experimental observations, validate that the ipso amination of substituted boronic acids 278 involves the generation of cyanamidyl/arylcyanamidyl radicals, followed by their regiospecific interaction with the boron atom of the boronic acids. This specific interaction leads to the selective formation of primary amines 279, as exemplified in Schemes 106 and 107.
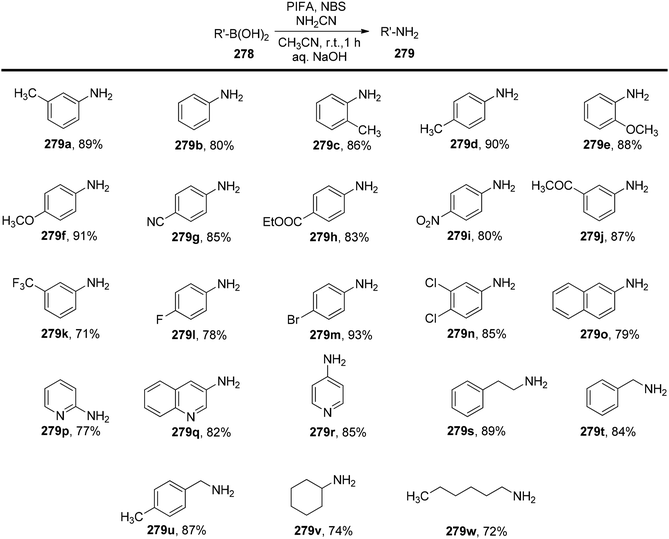 |
| | Scheme 106 PIFA–NBS–NH2CN mediated ipso-amination of different boronic acids 278. | |
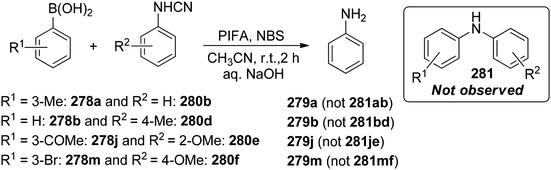 |
| | Scheme 107 Reaction with arylcyanamides 280 and arylboronic acids 278 mediated by PIFA–NBS. | |
Gondo et al.182 have introduced a method for desilylative acetoxylation of (trimethylsilyl)-arenes 282, effectively converting them into acetoxyarenes 283 (Scheme 108). The direct acetoxylation is achieved by employing 5 mol% of Pd(OAc)2 and 1.5 equiv. of PIFA in AcOH at 80 °C for 17 hours, resulting in the formation of acetoxyarenes 283 in good to high yields (ranging from 67% to 98%). The practicality of this approach is demonstrated through a one-pot transformation of (trimethylsilyl)arenes 282 into phenols 284 by carrying out successive acetoxylation and hydrolysis (Scheme 109a). Furthermore, the desilylative acyloxylation of 2-(trimethylsilyl)-naphthalene 282 is achieved using various carboxylic acids (Scheme 109b). The utilization of “non-activated” (trimethylsilyl)arenes for acetoxylation or acyloxylation offers an efficient and practical means of synthesizing phenol derivatives.
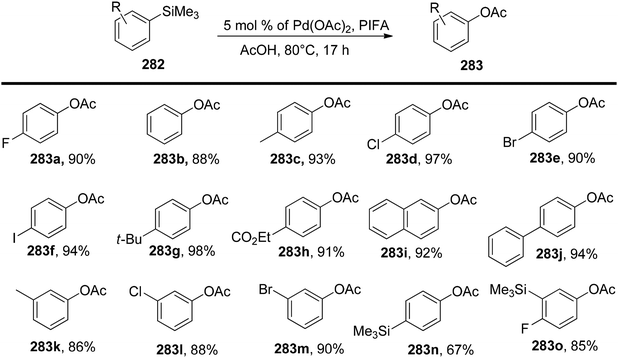 |
| | Scheme 108 Desilylative acetoxylation of (trimethylsilyl)-arenes 282. | |
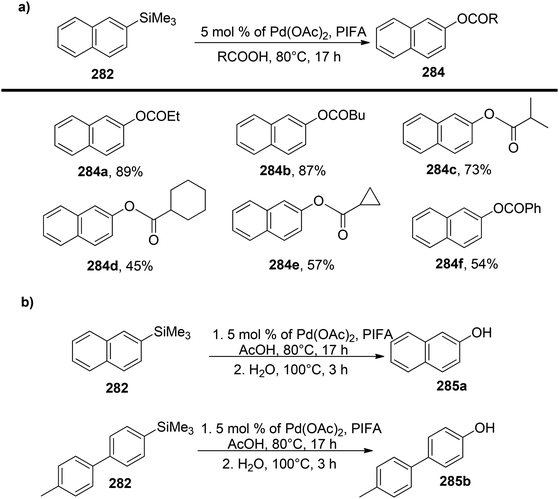 |
| | Scheme 109 a) One-pot transformation of (trimethylsilyl)arenes 282 into phenols 284. (b) Desilylative acyloxylation of 2-(trimethylsilyl)-naphthalene 282 using various carboxylic acids. | |
Pan et al.183 have developed a mechanochemical and solvent/catalyst-free method for functionalizing olefins 286 using PIFA reagents, facilitating the synthesis of 1,3-dioxygenated compounds 287 (Scheme 110) and 1,4-iodoalcohols 289 (Scheme 111). The process, challenging with conventional methods, involves an intermolecular cascade triggered by the active monomeric form of PIFA through an ionic pathway, as revealed by mechanistic studies. The synthetic efficiency of the described method is exemplified by the facile two-step synthesis of the natural product (±)-calyxolane A (290a)184,185 from styrene (286a). Additionally, the calyxolane A analogue 290b (85%) was obtained from 3-nitrostyrene (286b), showcasing the advantages of the ball-milling protocol over traditional multi-step syntheses,186 yielding 290a in just two steps with a 30% overall yield.
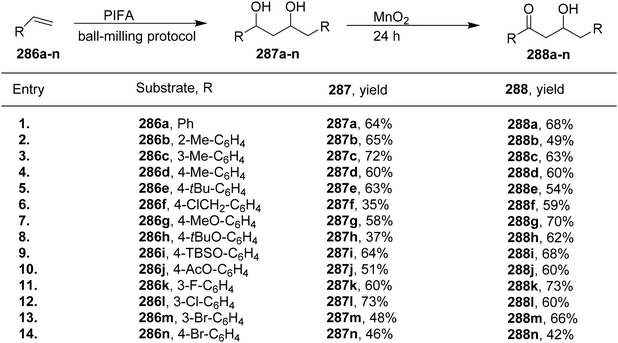 |
| | Scheme 110 Synthesis of 1,3-dioxygenated compounds. | |
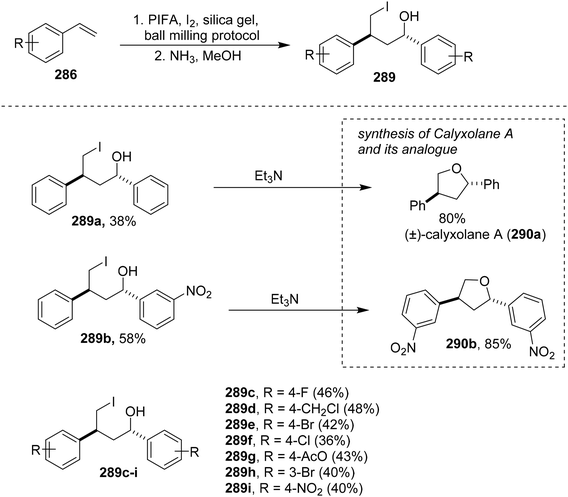 |
| | Scheme 111 Synthesis of 1,4-iodoalcohols 289 and (±)-calyxolane A. | |
Berthelot et al.187 have detailed the synthesis and characterization of zinc(II) meso-pyridin-2-ylthio-porphyrins 296–298 (Scheme 112). The electrochemical oxidation of zinc(II) porphyrins, specifically [5-(pyridin-2-ylthio)-10,20-bis(p-tolyl)-15-phenylporphyrinato] zinc(II) Zn-296 or [5,15-bis(pyridin-2-ylthio)-10,20 bis(p-tolyl)porphyrinato] zinc(II) Zn-298, results in the intriguing formation of one or two C–N bonds (Scheme 113). This transformation occurs through an intramolecular nucleophilic attack of the peripheral thiopyridinyl fragments on the adjacent β-pyrrolic positions, leading to a C–N fusion reaction. Moreover, the oxidation of [5-(pyridin-2-ylthio)-10,20-bis(p-tolyl)-porphyrinato] zinc(II) Zn-297, which possesses a vacant meso position, predominantly yields the meso,meso-dimer (Zn-297)2. Sequential electrochemical oxidation steps then result in the selective formation of both mono and bis C–N fused meso,meso-dimer products (Scheme 114). The planarization of the molecules and the generation of positive charges during the C–N fusion reaction lead to significant electronic alterations. These changes manifest in observable shifts in the UV–vis absorption spectra, including a bathochromic shift of the Soret and Q bands, as well as the splitting of the Soret bands. Additionally, the cyclic voltammograms demonstrate modifications such as the reduction of the pyridinium moiety-(ies) occurring between −0.5 and −1.0 V.
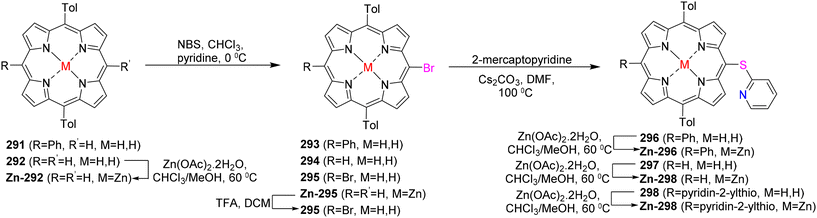 |
| | Scheme 112 Synthesis of porphyrins Zn-296, Zn-297, and Zn-298. | |
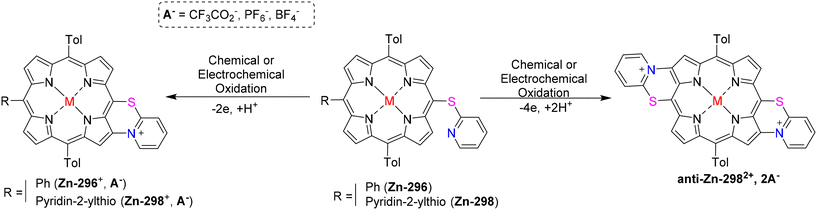 |
| | Scheme 113 Intramolecular C–N oxidative coupling. | |
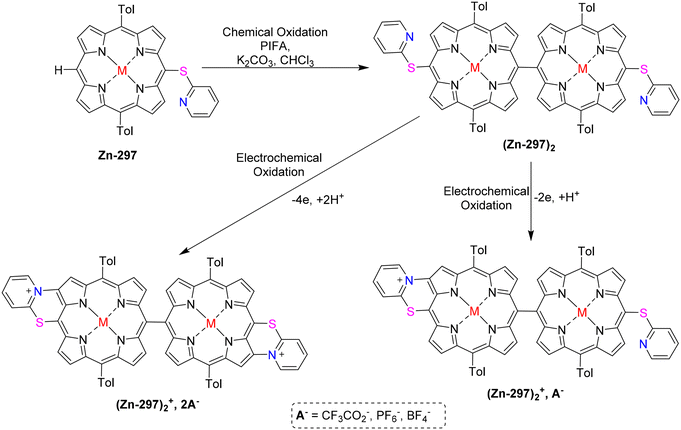 |
| | Scheme 114 Oxidative behaviour of Zn-297. | |
4. Conclusion
Iodine, characterized as the most polarizable, least electronegative, and largest halogen, exhibits unique reactivity. Iodine manifests itself in various oxidation states, forming hypercoordinate compounds such as I(III), I(V), and I(VII). Over time, hypercoordinate iodine compounds have gained significance as reagents in synthesis due to their low toxicity, cost-effectiveness, environmental friendliness, and transition metal-like reactivity under mild conditions. Hypervalent iodine reagents, recognized for their low toxicity, accessibility, ease of handling, efficiency, stability to air and moisture, and safe substitution for toxic heavy metal reagents like lead(IV), thallium(III), and mercury(II), have been widely embraced as green oxidants in organic synthesis. Among them, [bis(trifluoroacetoxy)iodo]arenes (PIFA) stand as pivotal compounds extensively employed as chemoselective oxidants in contemporary organic synthesis. The pronounced electron-withdrawing nature of the fluorine atom renders PIFA more reactive than its counterpart, phenyliodine diacetate (PIDA). PIFA is known to facilitate reactions that may not proceed efficiently with PIDA. Moreover, PIFA is recognized as a potent and attractive catalyst for numerous crucial chemical processes, encompassing amination, sulfenylation, oxidative coupling, amidation, carboxylation, ring-rearrangement, cascade reactions, alkylarylation, and more. It also finds application in simple oxidation of nitrogen compounds, phenolic oxidation, oxidation of alkynes, ketones, sulfur compounds, and oxidative biaryl coupling, among other processes.
The utilization of PIFA reagents in chemical synthesis is continuously evolving. This review comprehensively summarizes synthetic methodologies and various PIFA-catalyzed oxidative reactions for the production and functionalization of heterocyclic compounds, providing illustrative examples. The diverse applications of PIFA in organic synthesis, including amidation, amination, C–H functionalization, coupling, cyclization, oxidative and Hoffmann reactions, as well as in carbohydrate chemistry, one-pot reactions, and more, are systematically presented. We anticipate that researchers in synthetic organic chemistry and related fields will find this compilation to be a valuable addition.
Abbreviations
| PIFA | (Bis(trifluoroacetoxy)iodo)benzene |
| PIDA | (Diacetoxyiodo)benzene |
| Kg | Kilogram |
| DMF | Dimethylformamide |
| DMAc | Dimethylacetamide |
| HOBt | Hydroxybenzotriazole |
| EDC·HCl | (3-Dimethylamino-propyl)-ethyl-carbodiimide·hydrochloride |
| TFA | 2,2,2-Trifluoroacetic acid |
| NHPI |
N-Hydroxyphthalimide |
| 4CzIPN | 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene |
| KOH | Potassium hydroxide |
| NLO | Non-linear optical |
| mV | Millivolt |
| IR | Infra-red |
| NMR | Nuclear magnetic resonance |
| MS | Mass spectrum |
| m.p. | Melting point |
| HFIP | Hexafluoroisopropanol |
| DCE | Dichloroethane |
| TFE | Tetrafluoroethylene |
| IBX | 2-Iodoxybenzoic acid |
| THF | Tetrahydrofuran |
| SAR | Structure–activity relationship |
| IC50 | Half maximal inhibitory concentration |
| μM | Micromolar |
| EtOH | Ethanol |
| HOCl | Hypochlorous acid |
| CH2Cl2 | Dichloromethane |
| NBS |
n-Bromo succinimide |
| PS | Polystyrene |
| DIBAL | Diisobutylaluminium hydride |
| TfOH | Triflic acid |
| NaOH | Sodium hydroxide |
| DBAD | Di-tert-butyl azodicarboxylate |
| TFE | Tetrafluoroethylene |
| XRD | X-ray diffraction |
| MWD | Molecular weight distributions |
| TEAB | Tetraethylammonium bromide |
Author contributions
SK, AA, and SKS conceived and gathered the data. The initial manuscript was composed by SK, AA, and SKS, and later revised by RK and BS. SKS and BKS provided critical reviews of the manuscript and played a pivotal role in its finalization. The final version of the manuscript received approval from all authors.
Conflicts of interest
Authors declare no potential conflict of interest.
Acknowledgements
We thank the Institute of Eminence at the University of Delhi for contributing funds to promote research and development. A Senior Research Fellowship (SRF) was awarded to Sumit Kumar (File No.: 09/045(1798)/2020-EMR-I) and Aditi Arora (File No.: 09/0045(11270)/2021-EMR-I) by the Council of Scientific and Industrial Research (CSIR), New Delhi, India.
References
- T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656–3665 CrossRef CAS PubMed.
- T. Dohi and Y. Kita, Chem. Commun., 2009, 16, 2073–2085 RSC.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- L. Long, J. Wang, L. Gu, S. Yang, L. Qiao, G. Luo and Z. Chen, J. Org. Chem., 2021, 86, 12084–12092 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- M. Arisawa, S. Utsumi, M. Nakajima, N. G. Ramesh, H. Tohma and Y. Kita, Chem. Commun., 1999, 5, 469–471 RSC.
- M. T. Alam, S. Maiti and P. Mal, Beilstein J. Org. Chem., 2018, 14, 2396–2403 CrossRef CAS PubMed.
- A. Bal, S. Maiti and P. Mal, Chem. – Asian J., 2020, 15, 624–635 CrossRef CAS PubMed.
- K. Choudhuri, S. Maiti and P. Mal, Adv. Synth. Catal., 2019, 361, 1092–1101 CrossRef CAS.
- T. Kumar Achar and P. Mal, Adv. Synth. Catal., 2015, 357, 3977–3985 CrossRef CAS.
- S. Maiti and P. Mal, Adv. Synth. Catal., 2015, 357, 1416–1424 CrossRef CAS.
- R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402–4404 CrossRef CAS PubMed.
- V. V. Zhdankin, ARKIVOC, 2009, 1, 1–62 Search PubMed.
- T. Wirth, M. Ochiai, V. V. Zhdankin, G. F. Koser, H. Tohma and Y. Kita, Top. Curr. Chem., 2003, 224, 99–136 CrossRef.
- Y. Du, R. Liu, G. Linn and K. Zhao, Org. Lett., 2006, 8, 5919 CrossRef CAS PubMed.
- F. Fleming, L. Funk, R. Altundas and Y. Tu, J. Org. Chem., 2001, 66, 6502 CrossRef CAS PubMed.
- I. Tellitu, S. Serna, M. T. Herrero, I. Moreno, E. Domínguez and R. SanMartin, J. Org. Chem., 2007, 72, 1526 CrossRef CAS PubMed.
- R. M. Moriarty, B. A. Berylund and R. Penmasta, Tetrahedron Lett., 1992, 33, 6065 CrossRef CAS.
- O. Karam, J. C. Jacquesy and M. P. Jouannetand, Tetrahedron Lett., 1994, 35, 2541 CrossRef CAS.
-
J. Aubé, C. Fehl, R. Liu, M. C. McLeod and H. F. Motiwala, Heteroatom Manipulations. Comprehensive Organic Synthesis II, 1993, vol. 6, pp. 598–635 Search PubMed.
- Y. Kikugawa and M. Kawase, Chem. Lett., 1990, 19, 581–582 CrossRef.
- D. J. Wardrop and W. Zhang, Org. Lett., 2001, 3, 2353 CrossRef CAS PubMed.
- N. Itoh, T. Sakamoto, E. Miyazawa and Y. Kikugawa, J. Org. Chem., 2002, 67, 7424 CrossRef PubMed.
- T. Dohi, M. Ito, M. Iwata and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 1301 CrossRef CAS PubMed.
- A. Kar, N. Mangu, H. M. Kaiser, M. Beller and M. K. Tse, Chem. Commun., 2008, 3, 386 RSC.
- Y. Kita, T. Tanaka, M. Gyoten, H. Tohma, M. H. Zenk and J. Eichhorn, J. Org. Chem., 1996, 61, 5857 CrossRef CAS.
- T. Honda and H. Shigehisa, Org. Lett., 2006, 8, 657 CrossRef CAS PubMed.
- P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS PubMed.
- A. Saito, A. Matsumoto and Y. Hanzawa, Tetrahedron Lett., 2010, 51, 2247 CrossRef CAS.
- S. Serna, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartin, Org. Lett., 2005, 7, 3073 CrossRef CAS PubMed.
- I. Tellitu, S. Serna, M. T. Herrero, I. Moreno, E. Domínguez and R. SanMartin, J. Org. Chem., 2007, 72, 1526 CrossRef CAS PubMed.
- A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142–1152 RSC.
- S. R. Chemler and P. H. Fuller, Chem. Soc. Rev., 2007, 36, 1153–1160 RSC.
- H. M. Lovick and F. E. Michael, J. Am. Chem. Soc., 2010, 132, 1249–1251 CrossRef CAS PubMed.
- J. Streuff, C. H. Hövelmann, M. Nieger and K. Muñiz, J. Am. Chem. Soc., 2005, 127, 14586–14587 CrossRef CAS PubMed.
- I. I. Maletina, V. V. Orda and L. M. Yagupolskii, J. Org. Chem. USSR, 1974, 10, 294 CAS.
- T. M. Kasumov, V. K. Brel, Y. K. Grishin, N. S. Zefirov and P. J. Stang, Tetrahedron, 1997, 53, 1145 CrossRef CAS.
- P. Kazmierczak and L. Skulski, Molecules, 2002, 7, 810 CrossRef CAS.
- M. D. Hossain,
et al.
, Bull. Chem. Soc. Jpn., 2006, 79, 1 CrossRef.
- V. V. Zhdankin, M. C. Scheuller and P. J. Stang, Tetrahedron Lett., 1993, 34, 6853 CrossRef CAS.
- L. M. Yagupolskii, I. I. Maletina, N. V. Kondratenko and V. V. Orda, Synthesis, 1977, 574 CrossRef CAS.
- N. S. Zefirov, S. O. Safronov, A. A. Kaznacheev and V. V. Zhdankin, J. Org. Chem. USSR, 1989, 25, 1807 CAS.
- N. W. Alcock and T. C. Waddington, J. Chem. Soc., 1963, 4103 RSC.
- B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437–4492 CrossRef CAS PubMed.
- J. Carey, D. Laffan, C. Thomson and T. Mike, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- T. Cupido, J. Tulla-Puche, J. Spengler and A. Fernando, Curr. Opin. Drug Discovery Dev., 2007, 10, 768–783 CAS.
- J. Kothandapani, A. Ganesan and S. S. Ganesan, Synthesis, 2017, 685–692 CAS.
- O. S. Kamble, R. Chatterjee and R. Dandela, ARKIVOC, 2022, 5, 270–281 Search PubMed.
- F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104 RSC.
- S. Serna, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartín, Org. Lett., 2005, 7, 3073–3076 CrossRef CAS PubMed.
- N. Sukekatsu, Chem. Lett., 1997, 26, 1 CrossRef.
- K. J. Sonogashira, Organomet. Chem., 2002, 653, 46 CrossRef CAS.
- Y. Tamura, T. Yakura, J.-I. Haruta and Y. Kita, Tetrahedron Lett., 1985, 26, 3837 CrossRef CAS.
- F. Palluotto, A. Sosic and O. Pinato,
et al.
, Eur. J. Med. Chem., 2016, 123, 704–717 CrossRef CAS PubMed.
- C. Hu, Z. Zhang, W. Gao, G. Zhang, T. Liu and Q. Liu, Tetrahedron, 2018, 74, 665–671 CrossRef CAS.
- S. Serna, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartín, Tetrahedron Lett., 2003, 44, 3483–3486 CrossRef CAS.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- A. Correa, I. Tellitu, E. Domínguez and R. SanMartin, J. Org. Chem., 2006, 71, 8316–8319 CrossRef CAS PubMed.
- I. Tellitu, S. Serna, M. T. Herrero, I. Moreno, E. Domínguez and R. SanMartin, J. Org. Chem., 2007, 72, 1526–1529 CrossRef CAS PubMed.
- D.-G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802 CrossRef CAS PubMed.
- T. Koike and M. Akita, Org. Chem. Front., 2016, 3, 1345 RSC.
- G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed.
- L. Lin, Q. Liang, X. Kong, Q. Chen and B. Xu, J. Org. Chem., 2020, 85, 15708 CrossRef CAS PubMed.
- S. A. Paveliev, O. O. Segida, U. V. Fedorova, O. M. Mulina and A. O. Terent'ev, Mendeleev Commun., 2022, 32, 167–169 CrossRef CAS.
- M. J. Haddadin, W. E. Conrad and M. J. Kurth, Mini-Rev. Med. Chem., 2012, 12, 1293 CrossRef CAS PubMed.
- N. A. S. Ali, B. A. Dar, V. Pradhan and M. Farooqui, Mini-Rev. Med. Chem., 2013, 13, 1792 CrossRef PubMed.
- Z. Zhang, Y. Huang, G. Huang, G. Zhang and Q. Liu, J. Heterocycl. Chem., 2017, 54, 2426–2433 CrossRef CAS.
- R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed.
- S. Cai, M. Lu, T. Zhang, D. Tan, C. Chen, Y. Zhang and M. Huang, Adv. Synth. Catal., 2019, 361, 4237–4242 CrossRef.
- S. Tang, Y.-L. Deng, J. Li, W. Wang, Y. Wang, Z.-Z. Li, L. Yuan, S.-L. Chen and R.-L. Sheng, Chem. Commun., 2016, 52, 4470 RSC.
- I. L. Martins, C. Charneira, V. J. L. Ferreira da Silva, G. C. Justino, J. P. Telo, A. J. S. C. Vieira, C. Marzano and A. M. M. Antunes, J. Med. Chem., 2015, 58, 4250–4265 CrossRef CAS PubMed.
- F. Frankel, M. Priven, E. Richard, C. Schweinshault, O. Tongo, A. Webster, E. Barth, K. Slejzer and S. Edelstein, Int. J. Food Prop., 2016, 19, 537–548 CrossRef CAS.
- P. Ghosh, G. Chhetri, E. Perl and S. Das, Adv. Synth. Catal., 2021, 363, 2148–2156 CrossRef CAS.
- Sumit, A. Kumar and A. K. Mishra, Mini-Rev. Med. Chem., 2021, 21, 314–335 CrossRef CAS PubMed.
- X.-T. Sun, Z.-G. Hu, Z. Huang, L.-L. Zhou and J.-Q. Weng, Molecules, 2022, 27, 726 CrossRef CAS PubMed.
- G. Majji, S. Guin, A. Gogoi, S. K. Rout and B. K. Patel, Chem. Commun., 2013, 49, 3031 RSC.
- R. Sakamoto, T. Inada, S. Selvakumar, S. A. Moteki and K. Maruoka, Chem. Commun., 2016, 52, 3758–3761 RSC.
- I. Couto, I. Tellitu and E. Domínguez, J. Org. Chem., 2010, 75, 7954–7957 CrossRef CAS PubMed.
- C. Mudithanapelli, L. P. Dhorma and M. Kim, Org. Lett., 2019, 21, 3098–3102 CrossRef CAS PubMed.
- Y. Ding, W.-H. Zhu and Y. Xie, Chem. Rev., 2017, 117, 2203 CrossRef CAS PubMed.
- Q. Cheng, Y.-H. Qiu, S.-L. Luo, L. Shuai, Y. Yuan, Y.-C. Chen and Q. Ouyang, Org. Lett., 2017, 19, 3871–3874 CrossRef CAS PubMed.
- S. F. Rach and F. E. Kühn, Chem. Rev., 2009, 109, 2061 CrossRef CAS PubMed.
- Y. Yan, Z. Zhang, Y. Wan, G. Zhang, N. Ma and Q. Liu, J. Org. Chem., 2017, 82, 7957–7963 CrossRef CAS PubMed.
- Q. Feng, D. Chen, M. Hong, F. Wang and S. Huang, J. Org. Chem., 2018, 83, 7553–7558 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 31, 496 Search PubMed.
- S. Rihn, M. Erdem, A. De Nicola, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 1916–1919 CrossRef CAS PubMed.
- P. F. H. Schwab, M. D. Levin and J. Michl, Chem. Rev., 1999, 99, 1863 CrossRef CAS PubMed.
- Q. Ouyang, K.-Q. Yan, Y.-Z. Zhu, C.-H. Zhang, J.-Z. Liu, C. Chen and J.-Y. Zheng, Org. Lett., 2012, 14, 2746–2749 CrossRef CAS PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed.
- W. Yu, Z. Yu, X. Ju and J. Wang, Synthesis, 2011, 860–866 CrossRef.
- Q. Yang, X. Han, J. Zhao, H. Y. Zhang and Y. Zhang, J. Org. Chem., 2019, 84, 11417–11424 CrossRef CAS PubMed.
- A. Chebieb, Y. G. Kim and J. K. Cha, J. Org. Chem., 2023, 88, 10164–10170 CrossRef CAS PubMed.
- E. Faggi, R. M. Sebastián, R. Pleixats, A. Vallribera, A. Shafir, A. Rodríguez-Gimeno and C. Ramirez de Arellano, J. Am. Chem. Soc., 2010, 132, 17980–17982 CrossRef CAS PubMed.
-
R. A. Jones and G. P. Bean, The Chemistry of Pyrroles, Academic, London, 1977, vol. 9, pp. 525 Search PubMed.
- P. F. Zhang and Z. C. Chen, J. Chem. Res., 2001, 2001, 150–152 CrossRef.
- P.-F. Zhang and Z.-C. Chen, Synth. Commun., 2001, 31, 1619–1624 CrossRef CAS.
- D. Papadopoulou, I. Papoutsis, S. Spyroudis and A. Varvoglis, Tetrahedron Lett., 1998, 39, 2865–2866 CrossRef CAS.
- M. A. Sucari and J. M. Vernon, Tetrahedron, 1983, 39, 793 CrossRef.
- K. Zhou, F. Zhao, Z. Liu, Y. Zhuang, L. Chen and F. Qiu, J. Nat. Prod., 2009, 72, 1563 CrossRef CAS PubMed.
- C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed.
- Z. Ai, J. Xiao, Y. Li, B. Guo, Y. Du and K. Zhao, Org. Chem. Front., 2020, 7, 3935–3940 RSC.
- B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433–2480 CrossRef CAS PubMed.
- M. N. S. Saudi, M. M. A. El Semary and G. El Sawaf, Pharmazie, 2002, 57, 519–522 CAS.
- J. Huang, Y. Liang, W. Pan, Yang and D. Dong, Org. Lett., 2007, 9, 5345–5348 CrossRef CAS PubMed.
- W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC.
- J. Bariwal, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3831 RSC.
- B. Banerjee and M. Koketsu, Coord. Chem. Rev., 2017, 339, 104 CrossRef CAS.
- S. Wu, J. Shi and C.-P. Zhang, Org. Biomol. Chem., 2019, 17, 7468 RSC.
- Z. Chen, J. Li, W. Weng, X. Xie and J. Lei, RSC Adv., 2022, 12, 28800–28803 RSC.
- K. Uneyama, H. Amii, T. Katagirl, T. Kobayashi and T. Hosokawa, J. Fluorine Chem., 2005, 126, 165 CrossRef CAS.
- K. C. Joshi, R. Jain, A. Dandia and K. Sharma, J. Fluorine Chem., 1992, 56, 1 CrossRef CAS.
- Y. Wu, J. Zhu, H. Xie, Z. Chen and S. Li, Synlett, 2009, 2009, 3299–3302 CrossRef.
- M. Wu and R. Yan, Synlett, 2017, 28, 729–733 CAS.
- Y. Du, R. Liu, G. Linn and K. Zhao, Org. Lett., 2006, 8, 5919–5922 CrossRef CAS PubMed.
- B. P. Smart, R. C. Oslund, L. A. Walsh and M. H. Gelb, J. Med. Chem., 2006, 49, 2858–2860 CrossRef CAS PubMed.
- A. A. Jensen, N. Plath, M. H. F. Pedersen, V. Isberg, J. Krall, P. Wellendorph, T. B. Stensbol, D. E. Gloriam, P. Krogsgaard-Larsen and B. Frolund, J. Med. Chem., 2013, 56, 1211 CrossRef CAS PubMed.
- L. Cheng, D. Pettersen, B. Ohlsson, P. Schell, M. Karle, E. Evertsson, S. Pahlén, M. Jonforsen, A. T. Plowright, J. Boström, T. Fex, A. Thelin, C. Hilgendorf, Y. Xue, G. Wahlund, W. Lindberg, L.-O. Larsson and D. Gustafsson, ACS Med. Chem. Lett., 2014, 5, 538 CrossRef CAS PubMed.
- M. D. Cullen, T. Sarkar, E. Hamel, T. L. Hartman, K. M. Watson, R. W. Buckheit Jr., C. Pannecouque, E. De Clercqd and M. Cushman, Bioorg. Med. Chem. Lett., 2008, 18, 469 CrossRef CAS PubMed.
- R. Zhang, D. Dong, J. Yuan, C. Rao, Q. Zhang, Y. Liang and N. Zhang, Synthesis, 2018, 1875–1882 CrossRef.
- A. Saito, T. Anzai, A. Matsumoto and Y. Hanzawa, Tetrahedron Lett., 2011, 52, 4658–4661 CrossRef CAS.
- O. O. Grygorenko, D. M. Volochnyuk, S. V. Ryabukhin and D. B. Judd, Chem. – Eur. J., 2020, 26, 1196–1237 CrossRef CAS PubMed.
- A. I. Vaskevych, N. O. Savinchuk, R. I. Vaskevych, E. B. Rusanov and O. O. Grygorenko, Beilstein J. Org. Chem., 2021, 17, 2787–2794 CrossRef CAS PubMed.
-
Classics in Total Synthesis, ed. K. C. Nicolaou and E. J. Sorensen, VCH Publishers, New York, 1995 Search PubMed.
- A. Correa, I. Tellitu, E. Domínguez and R. SanMartin, J. Org. Chem., 2006, 71, 3501–3505 CrossRef CAS PubMed.
- Y. Liu, X. Du, H. Chen, Y. Chen and J. Chen, Synlett, 2011, 1010–1101 CrossRef CAS.
- M. T. Khan, M. I. Choudhary, K. M. Khan, M. Rani and A. U. Rahman, Bioorg. Med. Chem., 2005, 13, 3385 CrossRef CAS PubMed.
- A. S. Aboraia, H. M. Abdel-Rahman, N. M. Mahfouz and M. A. Gendy, Bioorg. Med. Chem. Lett., 2006, 14, 1236 CrossRef CAS PubMed.
- R. N. Warrener, Eur. J. Org. Chem., 2000, 3363 CrossRef CAS.
- D. Kumar, S. Sundaree, E. O. Johnson and K. Shah, Bioorg. Med. Chem. Lett., 2009, 19, 4492–4494 CrossRef CAS PubMed.
- A. Mariappan, K. Rajaguru, N. Merukan Chola, S. Muthusubramanian and N. Bhuvanesh, J. Org. Chem., 2016, 81, 6573–6579 CrossRef CAS PubMed.
- L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 2906–2909 CrossRef CAS PubMed.
- T. Deng, W. Mazumdar, R. L. Ford, N. Jana, R. Izar, D. J. Wink and T. G. Driver, J. Am. Chem. Soc., 2020, 142, 4456–4463 CrossRef CAS PubMed.
- Z. Zheng, S. Ma, L. Tang, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2014, 79, 4687–4693 CrossRef CAS PubMed.
- D. Sun, X. Zhao, B. Zhang, Y. Cong, X. Wan, M. Bao, X. Zhao, B. Li, D. Zhang-Negrerie and Y. Du, Adv. Synth. Catal., 2018, 360, 1634–1638 CrossRef CAS.
- J. Yuan, B. Deng, Y. Liang, C. B. Rao, R. Zhang, Y. Zhao and D. Dong, Adv. Synth. Catal., 2019, 361, 4324–4333 CrossRef CAS.
- Q. Jiang, A. Zhao, B. Xu, J. Jia, X. Liu and C. Guo, J. Org. Chem., 2014, 79, 2709–2715 CrossRef CAS PubMed.
- A. P. Sakla, P. Kansal and N. Shankaraiah, Eur. J. Org. Chem., 2021, 757–772 CrossRef CAS.
- P. Liang, H. Zhao, T. Zhou, K. Zeng, W. Jiao, Y. Pan and H. Shao, Adv. Synth. Catal., 2021, 363, 3532–3538 CrossRef CAS.
- K. S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed.
- N. Chatterjee, D. Bhatt and A. Goswami, Org. Biomol. Chem., 2015, 13, 4828–4832 RSC.
- M. Kinugawa, Y. Masuda, H. Arai, H. Nishikawa, T. Ogasa, S. Tomioka and M. Kasai, Synthesis, 1996, 633–636 CrossRef CAS.
- E. A. Oostveen and W. N. Speckamp, Tetrahedron, 1987, 43, 255 CrossRef CAS.
-
J.-H. Chou, M. E. Kosal, H. S. Nalwa, N. A. Rakow and K. S. Suslick, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 6, pp. 43–131 Search PubMed.
- Q. Ouyang, Y.-Z. Zhu, C.-H. Zhang, K.-Q. Yan, Y.-C. Li and J.-Y. Zheng, Org. Lett., 2009, 11, 5266–5269 CrossRef CAS PubMed.
- M. A. Miller, R. K. Lammi, S. Prathapan, D. Holten and J. S. Lindsey, J. Org. Chem., 2000, 65, 6634 CrossRef CAS PubMed.
- N. Aratani and A. Osuka, Org. Lett., 2001, 3, 4213 CrossRef CAS PubMed.
- S. Spyroudis and A. Varvoglis, J. Chem. Soc., Chem. Commun., 1979, 14, 615–616 RSC.
- Y. Liu, Z. Zhang, Y. Wan, G. Zhang, Z. Li, J. Bi, N. Ma, T. Liu and Q. Liu, J. Org. Chem., 2017, 82, 3901–3907 CrossRef CAS PubMed.
- C. J. R. Bataille and T. J. Donohoe, Chem. Soc. Rev., 2011, 40, 114–128 RSC.
- L. Pan, Z. Ke and Y. Y. Yeung, Org. Lett., 2021, 23, 8174–8178 CrossRef CAS PubMed.
- S. Hamada, K. Sugimoto, E. E. Elboray, T. Kawabata and T. Furuta, Org. Lett., 2020, 22, 5486–5490 CrossRef CAS PubMed.
- Y. Zhang, H. Liu, H. He, B. Zou, Z. Zhai, T. Yu, A. Osuka and H. W. Jiang, Eur. J. Org. Chem., 2023, e202300666 CrossRef CAS.
- H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS.
- P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Glowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779–781 CrossRef.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- W. Gao, Y. Wan, Z. Zhang, H. Wu, T. Liu and G. Zhang, Green Chem., 2020, 22, 7955–7961 RSC.
- G. M. Loudon, A. S. Radhakrishna, M. R. Almond, J. K. Blodgett and R. H. Boutin, J. Org. Chem., 1984, 49, 4272–4276 CrossRef CAS.
- A. Varki, Glycobiology, 2017, 27, 3 CrossRef CAS PubMed.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS PubMed.
- S. Meng, W. Zhong, W. Yao and Z. Li, Org. Lett., 2020, 22, 2981–2986 CrossRef CAS PubMed.
- T. Kajimoto and M. Node, Curr. Top. Med. Chem., 2009, 9, 13–33 CrossRef CAS PubMed.
- T. Kajimoto, K. Morimoto, R. Ogawa, T. Dohi and Y. Kita, Eur. J. Org. Chem., 2015, 2138–2142 CrossRef CAS.
- J. Roh, K. Vavrova and A. Hrabalek, Eur.
J. Org. Chem., 2012, 6101 CrossRef CAS.
- T. Imai, R. Harigae, K. Moriyama and H. Togo, J. Org. Chem., 2016, 81, 3975–3980 CrossRef CAS PubMed.
- D. W. Chen and Z. C. Chen, Tetrahedron Lett., 1994, 35, 7637–7638 CrossRef CAS.
- D. Bhattacherjee, S. Kang, A. Kumar, A. Sharma, R. Purohit and P. Das, Org. Biomol. Chem., 2020, 18, 745 RSC.
- N. D. Yudina, V. S. Raida, O. L. Vasil'eva, V. V. Deniskin, M. P. Stepanets and A. S. Sitnikov, Polym. Sci. USSR, 1989, 31, 1318–1323 CrossRef.
- M. E. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99 CrossRef CAS.
- C. Chen, M. You and H. Chen, Synth. Commun., 2015, 46, 73–78 CrossRef.
- V. N. Telvekar, R. A. Rane and T. V. Namjoshi, Synth. Commun., 2010, 40, 494–497 CrossRef CAS.
- V. N. Telvekar, B. S. Takale and H. M. Bachhav, Tetrahedron Lett., 2009, 50, 5056–5058 CrossRef CAS.
- Y. Wang, Y. Yang and C. Wang, Chin. J. Chem., 2019, 37, 1229–1233 CrossRef CAS.
- M. Tingoli, M. Mazzella, B. Panunzi and A. Tuzi, Eur. J. Org. Chem., 2011, 399–404 CrossRef CAS.
- M. Catir, H. Kilic, V. Nardello-Rataj, J.-M. Aubry and C. Kazaz, J. Org. Chem., 2009, 74, 4560–4564 CrossRef CAS PubMed.
- M. Catir and H. Kilic, Synlett, 2004, 12, 2151–2154 Search PubMed.
- Y. Kita, T. Takada, S. Mihara, B. A. Whelan and H. Tohma, J. Org. Chem., 1995, 60, 7144–7148 CrossRef CAS.
- S. W. Wu, J. L. Liu and F. Liu, Org. Lett., 2016, 18, 1–3 CrossRef CAS PubMed.
- T. Kitamura, K. Muta and J. Oyamada, J. Org. Chem., 2015, 80, 10431–10436 CrossRef CAS PubMed.
- N. Chatterjee, M. Arfeen, P. V. Bharatam and A. Goswami, J. Org. Chem., 2016, 81, 5120–5127 CrossRef CAS PubMed.
- K. Gondo, J. Oyamada and T. Kitamura, Org. Lett., 2015, 17, 4778–4781 CrossRef CAS PubMed.
- L. Pan, L. Zheng, Y. Chen, Z. Ke and Y. Y. Yeung, Angew. Chem., Int. Ed., 2022, 61, e202207926 CrossRef CAS PubMed.
- A. D. Rodríguez, O. M. Cóbar and O. L. Padilla, J. Nat. Prod., 1997, 60, 915–919 CrossRef PubMed.
- A. M. Bernard, A. Frongia, P. P. Piras, F. Secci and M. Spiga, Org. Lett., 2005, 7, 4565–4568 CrossRef CAS PubMed.
- S. J. Gharpure, D. S. Vishwakarma and S. K. Nanda, Org. Lett., 2017, 19, 6534–6653 CrossRef CAS PubMed.
- M. Berthelot, F. Akhssas, A. K. Dimé, A. Bousfiha, J. Echaubard, G. Souissi, H. Cattey, D. Lucas, P. Fleurat-Lessard and C. H. Devillers, Inorg. Chem., 2022, 61, 7387–7405 CrossRef CAS PubMed.
Footnote |
| † Authors have equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Rajesh
Kumar
c,
Bhawani
Shankar
*b,
Rajesh
Kumar
c,
Bhawani
Shankar