
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation of N-trifluoromethylthio sulfoximines using NaOCl·5H2O†
Žan
Testen
 and
Marjan
Jereb
*
and
Marjan
Jereb
*
University of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia. E-mail: marjan.jereb@fkkt.uni-lj.si
First published on 12th January 2024
Abstract
N-Trifluoromethylthio sulfoximines are biologically interesting compounds, but their potential is still poorly understood. The oxidation of N-trifluoromethylthio sulfoximines led to their corresponding sulfoxide derivatives as a new class of compounds, when using sodium hypochlorite pentahydrate (NaOCl·5H2O) as a green and relatively unexplored reagent. The reactions took place with a small excess of oxidant under environmentally friendly conditions in EtOAc for 16 h at room temperature. Noteworthy distinctions of this transformation are the simplicity, high selectivity, energy and cost efficiency, minimal amounts of non-hazardous waste, isolation of most of the products without the additional need for chromatographic purification, and simple scalability to gram reactions without deterioration of the yield. The reaction exhibited excellent green chemistry metrics with high atom economy (82.0%), actual atom economy (79.5%), reaction mass efficiency (79.7%), E-factor (16.48) and a very high EcoScale score (84.5). Competitive experiments demonstrated that electron-rich substrates are more reactive than their electron-poor counterparts. Furthermore, the Suzuki–Miyaura functionalization of N-trifluoromethylsulfaneylidene sulfoximine could be achieved depending on the conditions, resulting in coupling products with or without an introduced sulfoxide moiety. Sonogashira coupling of N-trifluoromethylsulfaneylidene sulfoximine furnished the expected acetylene derivative in high yield, and the reaction conditions are compatible with the newly introduced sulfaneylidene functionality. Bromine and nickel catalysts were also shown to be deprotecting agents of the sulfoxide group. A selected N-trifluoromethylsulfaneylidene sulfoximine demonstrated its stability in water in the presence of air and in dilute hydrochloric acid, while it converted back to the parent sulfoximine under basic conditions.
Introduction
Sulfoximines and their derivatives have seen rapid development in recent years.1 This effort is in no small part due to the biological activities that these compounds exhibit.2 Some biologically active sulfoximines, trifluoromethylthio compounds, sulfoxides and trifluoromethyl sulfoxides (TFMSs) are presented in Fig. 1.3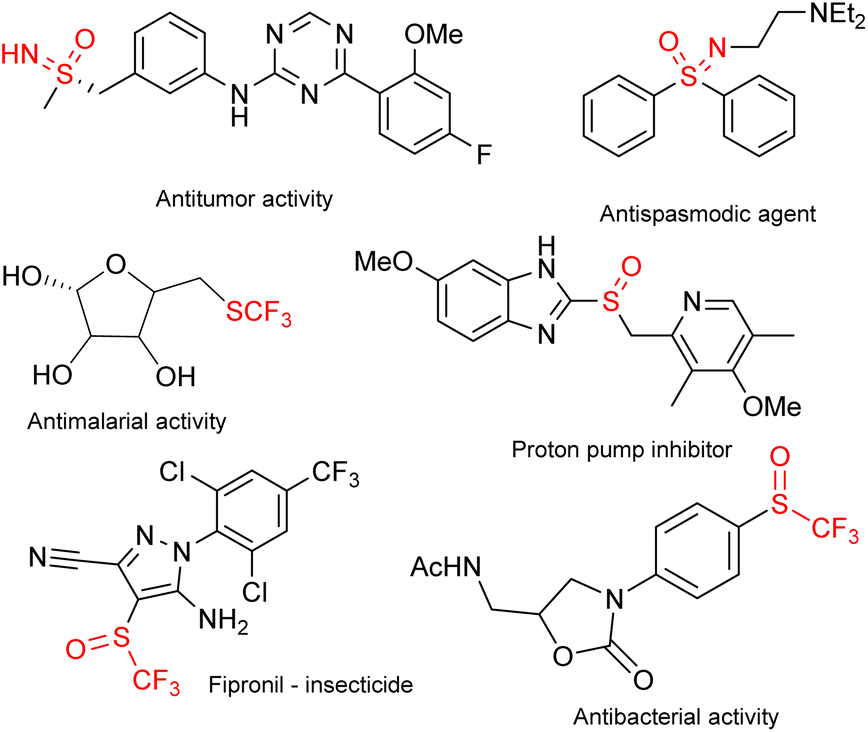 | ||
| Fig. 1 A selection of biologically active sulfides, sulfoximines and sulfoxides. | ||
Several pathways to synthesize sulfoximines have been reported. Some examples include N-alkylation of sulfides followed by oxidation,4 iridium-catalyzed regio- and enantioselective C–H borylation of N-silyl diaryl sulfoximines using a chiral bidentate boryl ligand with a bulky side arm,5 ruthenium-catalyzed synthesis of β-keto sulfoximines from N-tosyl-protected sulfoximidoyl chlorides and aryl alkynes under visible light,6 and enantioenriched preparation of sulfonimidoyl fluorides and their stereospecific reaction on sulfur with Grignard reagents.7 Various N-arylations have been developed, such as Ni/Ir photocatalyzed arylation of N-H sulfoximines with bromoarenes,8 Ni-catalyzed N-arylation of N-H sulfoximines via paired electrolysis,9 copper-catalyzed photoredox N-arylation of N-H sulfoximines with arylboronic acids,10 a stereospecific SNAr approach for the introduction of sulfonimidoyl functionalities into heterocyclic systems11 and Pd-catalyzed arylation of N-H sulfoximines with aryl bromides in micellar media.12 Pd-catalyzed introduction of lipophilic side chains into N-H sulfoximines using butadienes and alkyl bromides took place under blue light irradiation.13 Stereospecific S-alkylation of chiral sulfinamides with alkyl halides and NaOH in DME gave products with high optical purity.14 An Ir-catalyzed reaction of N-SCN with alkenes produced alkyl-substituted sulfoximines under photochemical conditions.15 Functionalization of N-H sulfoximines with gem-difluoroalkenes and NBS under blue LED conditions led to the corresponding α-ketoacyl-substituted derivatives.16N-acylation of N-H-sulfoximines with thioacids in the presence of a photoredox catalyst could be accomplished under blue LED irradiation.17 Similarly, the transformation of N-H sulfoximine with aryl aldehydes18 or ketones19 in the presence of NBS yielded the corresponding N-acyl derivatives. The introduction of a halogen atom20 or a halogenated moiety21 produced various interesting sulfoximines.
N-Trifluoromethylthio sulfoximines exhibit increased lipophilicity, which further increases their bioavailability.21b The addition of an oxygen atom to the trifluoromethylthio group would increase the polar character of the compound and potentially confer improved pharmacological properties to the products. The cLog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (calculated logP) decreased by about two points for most compounds, indicating that a higher amount of the compound would be found in the aqueous phase of an octanol/H2O mixture, which is more in line with Lipinski's rules. An example of these values is shown in Fig. 2 for sulfoximine 1, N-trifluoromethylthio sulfoximine 2 and N-trifluoromethylsulfaneylidene sulfoximine 3.
P (calculated logP) decreased by about two points for most compounds, indicating that a higher amount of the compound would be found in the aqueous phase of an octanol/H2O mixture, which is more in line with Lipinski's rules. An example of these values is shown in Fig. 2 for sulfoximine 1, N-trifluoromethylthio sulfoximine 2 and N-trifluoromethylsulfaneylidene sulfoximine 3.
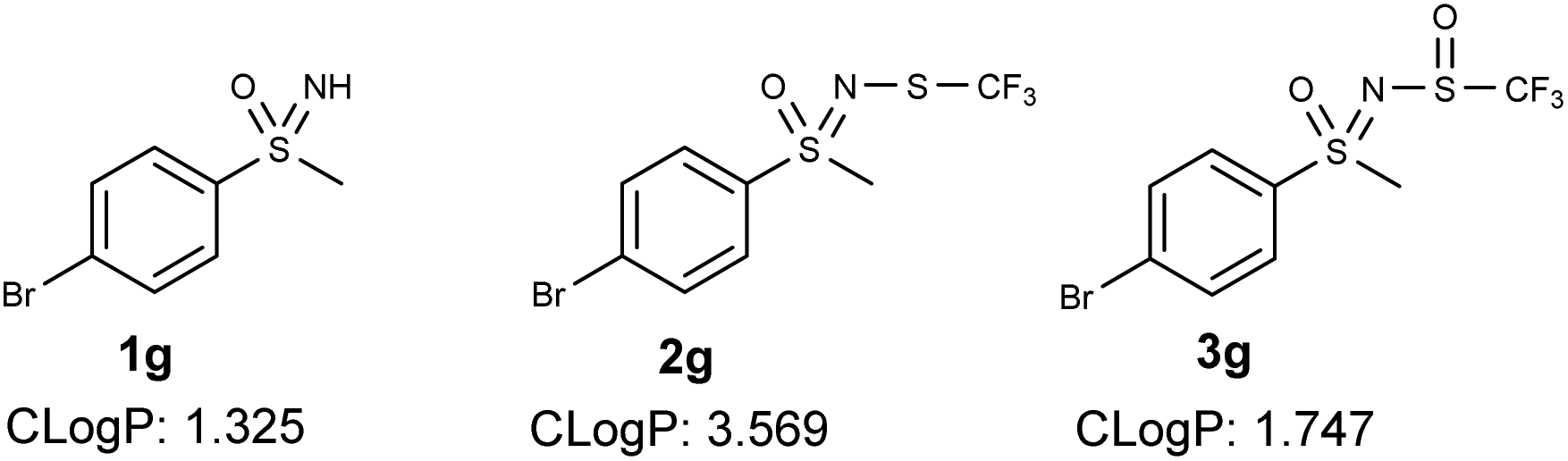 | ||
| Fig. 2 cLogP values for 1g, 2g and 3g obtained from ChemDraw. | ||
The trifluoromethylsulfinyl moiety22 (CF3SO) is a vital part of several important biologically active molecules, such as the insecticide fipronil (Fig. 1) and its derivatives, as well as other pharmacologically active compounds.23 TFMSs are reagents for metal-free C–H activation,24 as well as precursors of Yagupolskii and Umemoto-type reagents.23 Several synthetic approaches have been developed to introduce the trifluoromethylsulfinyl functionality. The functionalization of sulfite esters using TMSCF3/CsF at elevated temperature led to TFMS derivatives.25 Direct sulfinylation of common aromatic compounds by using triflinate salts in triflic acid led to TFMSs with preferential para-selectivity.26 Sodium triflinate and phosphoryl chloride (2/1) are capable of transferring CF3SO+ into organic molecules, yielding trifluoromethanesulfinates or trifluoromethanesulfinamides.27 Direct electrophilic trifluoromethylsulfinylation of activated indoles, pyrroles, anilines, and phenols took place with trifluoromethanesulfonyl chloride/tricyclohexyl phosphine.28 A combination of sodium triflinate and POCl3 afforded indole-derived TFMSs.29 Recently, N-trifluoromethylsulfinylphthalimide was introduced, which was able to transfer the CF3SO group into indoles, pyrroles and other electron-rich (hetero)aromatics. Alkyl, aromatic and heterocyclic amines were N-functionalized, while alcohols and phenols furnished the corresponding trifluoromethanesulfinate esters.30 The most common methodology for the synthesis of TFMSs is probably the oxidation of parent trifluoromethyl sulfides. The main challenge with oxidation to sulfoxides is the potential overoxidation to sulfones, which in some cases is as rapid as oxidation to sulfoxides. While the oxidation of non-halogenated sulfides is very well known and has through the years become increasingly green,31 the oxidation of the trifluoromethylthio functional group is mostly achieved using halogenated media32 or halogenated agents such as m-CPBA33 or TCCA,34 generating considerable amounts of harmful waste, or using metal catalysts.35 To this end, we have used solid sodium hypochlorite pentahydrate (NaOCl·5H2O) as a benign and selective oxidant.36 Its crystals are easy to use in stoichiometric quantities and eliminate the need for titration and other methods for determining the concentration of sodium hypochlorite in solution. It contains about 44 wt% NaOCl as an active oxidizing agent and only up to 0.08 wt% NaOH and up to 0.5 wt% NaCl. Furthermore, the enhanced purity of these crystals is paramount to the success of the reactions described herein, as they generate products that require little to no purification, whereas using a commercial aqueous solution of NaOCl produces a large amount of impurities and is less selective towards sulfoxides. Since its introduction in 2013, it has been used in several interesting oxidations.37
To the best of our knowledge, no oxidations of N-trifluoromethylthio sulfoximines have been reported, and while it is known that reactions of N-alkyl, N-aryl, and N-H sulfoximines with m-chloroperbenzoic acid cleanly give the corresponding sulfones in high yield,38 this was not the case for N-trifluoromethylthio sulfoximines. Herein we explore the optimization of the corresponding sulfoxide formation, the substrate scope, some mechanistic insights, some additional modifications and stability parameters for this novel functionalization of sulfoximines. The reaction proceeded at room temperature in ethyl acetate using a small excess of NaOCl·5H2O (0.1 equiv.) with excellent selectivity, yielding products in high yield and a small amount of non-hazardous waste (NaCl). A remarkable advantage of the present method is that column chromatography could be avoided to a great extent due to the NMR purity of the vast majority of the crude products.
Results and discussion
We tested various oxidants and solvents with the goal of making the process as green and sustainable as possible. The first success in obtaining an adequate amount of sulfoxide was with the use of hydrogen peroxide and tungstic acid in water, which unfortunately required a stoichiometric amount of tungstic acid (Table 1, entry 1). Reducing this amount at the expense of increasing the amount of H2O2 (Table 1, entry 2) proved beneficial, but the method was still prone to overoxidation to sulfone 4a. The reaction without tungstic acid (Table 1, entry 3) was more selective, but we observed a considerable amount of by-products that would require further purification. Substitution of tungsten with vanadium (Table 1, entry 4) resulted in a lower conversion, as 70% of the starting substrate 2a remained unreacted. The combination of H2O2 and acetic acid (Table 1, entries 5 and 6) eliminated the need for metal catalysts, but still produced the undesirable sulfone 4a in modest amounts. TBHP (Table 1, entry 7) did not yield any oxidized species in any of the tested solvents. Oxone (Table 1, entry 8) in water proved to be quite efficient, but there was always a non-negligible amount of 2a and 4a present, which would have required additional purification. Grinding neat 2a with oxone without solvent (Table 1, entry 9) proved unsuccessful. Oxidation with m-CPBA in DCM produced a relatively good result (Table 1, entry 10), but we ultimately did not consider it due to our green criteria.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Oxidant system | Temperature | Solvent | Relative ratiob (%) | ||
| 2a | 3a | 4a | ||||
| a Reaction conditions: a flask was charged with 2a (0.1 mmol), solvent and an oxidant and the mixture was stirred for 16 h. b Determined by 1H and 19F NMR. c Other side-products were observed. | ||||||
| 1 | H2O2 (1.2 equiv.)/H2WO4 (1 equiv.) | 60 °C | H2O | — | 79 | 21 |
| 2 | H2O2 (6 equiv.)/H2WO4 (10 mol%) | 60 °C | H2O | — | 75 | 25 |
| 3 | H2O2 (6 equiv.) | 60 °C | H2O | — | 91c | 9 |
| 4 | H2O2 (6 equiv.)/V2O5 (1 equiv.) | 60 °C | H2O | 70 | 30 | Trace |
| 5 | H2O2 (1.2 equiv.) | 60 °C | AcOH | — | 93 | 7 |
| 6 | H2O2 (1.2 equiv.) | r.t. | AcOH | 17 | 73 | 10 |
| 7 | TBHP | r.t. | H2O, MeOH, MeCN, EtOAc, AcOH, DMSO | 100 | — | — |
| 8 | Oxone (1.2 equiv.) | r.t. | H2O | 5 | 93 | 2 |
| 9 | Oxone (1.2 equiv.) | r.t. | — | 100 | — | — |
| 10 | m-CPBA (1.1 equiv.) | r.t. | DCM | 17 | 83 | — |
To our delight, NaOCl·5H2O exhibited a most selective reaction profile. The reaction in water with 1.1 equiv. of NaOCl·5H2O yielded the sulfoxide 3a, whereby the sulfone 4a was present only in trace amounts (Table 2, entry 1). Lowering the temperature by means of an ice bath did not change the result (Table 2, entry 2). As can be judged from 1H NMR spectra, about 3% of N-chloro sulfoximine was also present in the reaction; this by-product was only detected for 2a and for none of the other substrates. Reducing the amount of NaOCl·5H2O to 1.0 equiv. remedied this. The same reaction in DCM (Table 2, entry 3) afforded 3a in about 36% yield, with traces of 4a. In methanol, both oxidation products were found in traces, while no reaction was observed in ethanol (Table 2, entries 4 and 5). The same reaction in acetonitrile gave a mixture of sulfoxide 3a and sulfone 4a, with a portion of the starting material 2a still present (Table 2, entry 6). The oxidation in THF proceeded selectively to provide sulfoxide 3a, but there were many by-products that we assume were related to the oxidation/polymerization of THF itself (Table 2, entry 7). In hexane (Table 2, entry 8), two-thirds of 2a were converted to 3a, with traces of 4a. The reaction in EtOAc was comparable to that in water, with all the starting materials consumed and minimal 4a present, including traces of the corresponding N-chloro sulfoximine (Table 2, entry 9). Grinding NaOCl·5H2O and neat 2a without solvent (Table 2, entry 10) was found to be less effective and selective than using a solvent. Interestingly, the reaction in a commercial bleach solution was not selective and in addition produced unknown by-products (Table 2, entry 11). Although both water and EtOAc gave similar results, we ultimately decided in favour of EtOAc because it was significantly more selective towards sulfoxides for substituted phenyl substrates. This decision also allowed us to create a homogeneous reaction environment.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Temperature | Solvent | Relative ratiob (%) | ||
| 2a | 3a | 4a | |||
| a Reaction conditions: a flask was charged with 2a (0.1 mmol), solvent (0.3 mL) and an oxidant (1.1 equiv.) and the mixture was stirred for 16 h. b Determined by 1H and 19F NMR. c N-Chloro side-products were observed. d Commercial bleach solution (ca. 10%; 4 equiv.). | |||||
| 1 | r.t. | H2O | — | 96c | 1 |
| 2 | 0 °C | H2O | — | 96c | 1 |
| 3 | r.t. | DCM | 64 | 36 | Trace |
| 4 | r.t. | MeOH | 100 | Trace | Trace |
| 5 | r.t. | EtOH | 100 | — | — |
| 6 | r.t. | MeCN | 33 | 56 | 11 |
| 7 | r.t. | THF | Trace | 100c | Trace |
| 8 | r.t. | Hexane | 34 | 66 | Trace |
| 9 | r.t. | EtOAc | — | 99 | 1 |
| 10 | r.t. | — | 56 | 41 | 3 |
| 11 | r.t. | NaOCl (aq.)d | 34 | 48c | 18 |
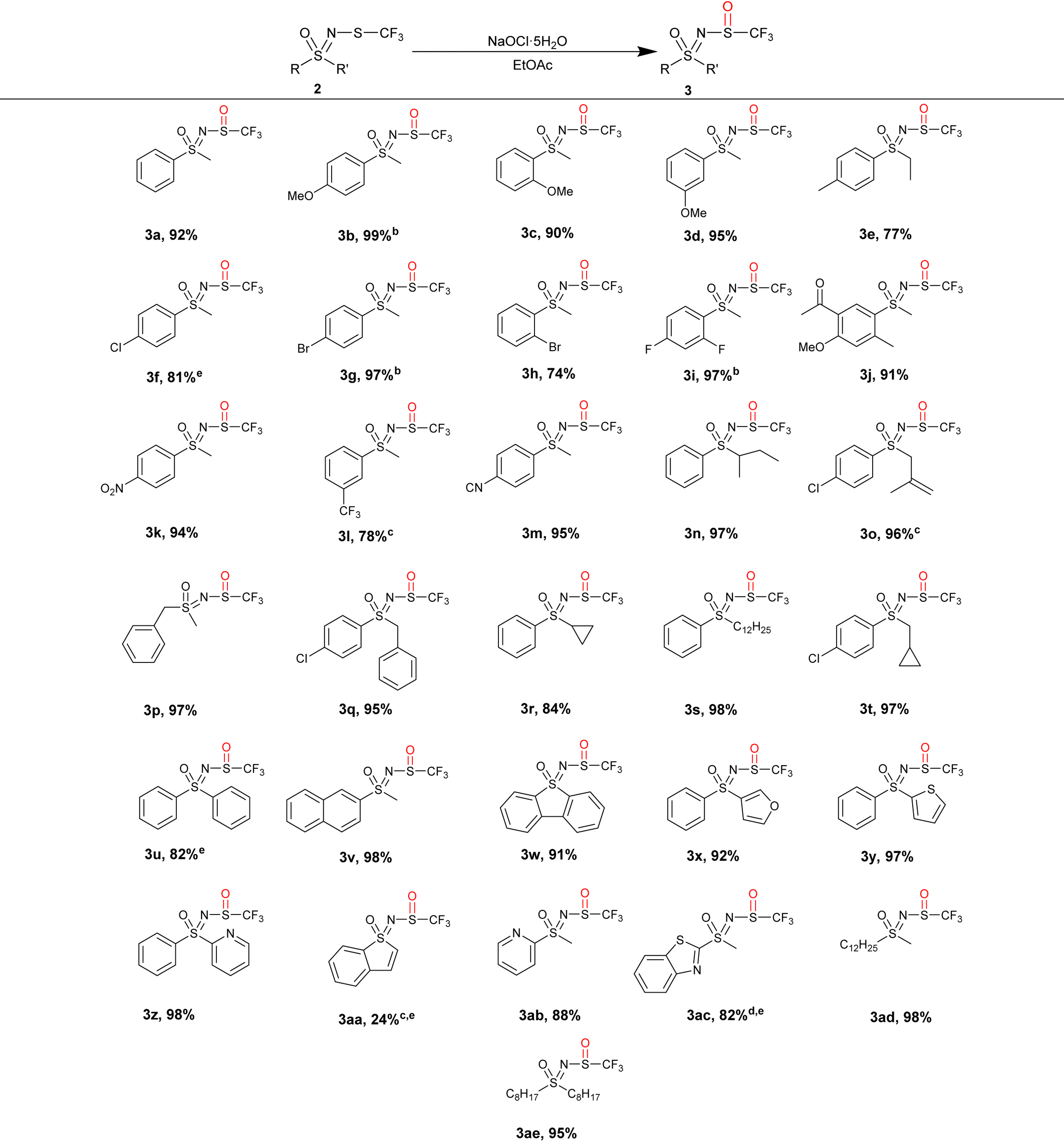
A number of N-trifluoromethylthio sulfoximines were prepared and oxidized in EtOAc with 1.1 equiv. of NaOCl·5H2O (Table 3). Due to the weak absorption of the sulfoxide moiety 3, TLC analysis was often inconclusive, so the reaction was stirred overnight (16 h) for convenience. Since only NaCl is formed during the reaction, the products could be purified by simple water/EtOAc extraction, which gave NMR-pure N-trifluoromethyl sulfoxides 3 in high yield without the need for additional purification.
| a Reaction conditions: a flask was charged with 2 (0.3 mmol) and EtOAc (0.3 mL) and lastly NaOCl·5H2O (1.1 equiv.) was added. The mixture was stirred for 16 h at r.t. After extraction from EtOAc/water, the organic phase was dried, and the solvent was removed. b 1.25 equiv. of NaOCl·5H2O were needed. c 1.5 equiv. of NaOCl·5H2O were needed. d 2 equiv. of NaOCl·5H2O were needed. e Column chromatography was needed for additional purification. |
|---|
|
The reaction worked well with aryl alkyl-substituted sulfoximines, regardless of whether they possessed an electron-rich or electron-poor aromatic ring, and gave the products 3a–3m in 74–99% yields. The starting substrate 2n furnished the expected 3n in 97% yield despite its branched alkyl side chain. We were pleased to find that the alkene moiety in 2o was compatible with the oxidizing reaction conditions, giving 3o in 96% yield. Benzyl-, cyclopropyl- and dodecyl-substituted substrates 2p–2t produced the corresponding sulfoxides 3p–3t in excellent yields. Diaryl, naphthyl, dibenzothienyl, furyl, thienyl, pyridyl, benzothienyl and benzothiazolyl trifluoromethylthio sulfoximines 2u–2ac produced the corresponding 3u–3ac sulfoxide derivatives in notable yields of up to 98%, except for 3aa. Substrates dodecyl methyl 2ad and dioctyl 2ae gave the corresponding sulfoxides 3ad and 3ae in 98% and 95% yields, respectively.
Most of the products were isolated in high yields. Products 3f, 3u, 3aa, and 3ac required additional purification via column chromatography to remove sulfones/starting materials or by-products, while in the case of 3a, a small amount of N-Cl derivative was observed. In further experiments, the N-Cl derivative was only detected when an excess of NaOCl·5H2O was added, suggesting that the sulfone 4a was somehow responsible for its formation. Excess NaOCl·5H2O should be avoided in the case of substrate 2a to minimize the quantity of the N-Cl derivative. Product 3o exhibited signs of decomposition since a fine white precipitate was found in the NMR sample tube. In fact, most of the product had already decomposed to many by-products at this point. This process appeared to be faster in deuterated chloroform, as undissolved 3o showed less decomposition after 2 months. Complete conversion to the products 3b, 3g and 3i was achieved with 1.25 equiv. of NaOCl·5H2O, 3l, 3o and 3aa with 1.5 equiv. of NaOCl·5H2O, and 3ac with 2 equiv. of NaOCl·5H2O. Overall, this method proved to be effective for (het)aryl-alkyl as well as (het)aryl-aryl and alkyl-alkyl substrates. Notably, several functional groups such as the keto- 3j, nitro- 3k, cyano- 3m, cyclopropyl- 3r and alkenyl moieties do not interfere with the oxidation, making this method suitable for late-stage functionalization.
Sulfoximines 1 were synthesised according to standard procedures39 and were racemic. Subsequent modification to N-trifluoromethylthio sulfoximines 2 and their oxidation to the corresponding sulfoxides 3 introduced an additional chiral center, leading to the formation of diastereoisomers, appearing as two sets of signals in the NMR spectra. For compound 3f, we succeeded in separating and characterizing the diastereoisomers, while the other products were characterized as mixtures of both diastereoisomers. While the diastereoisomers are mostly formed in a 1:1 ratio, the reactions of some substrates (most notably 3h) were more stereoselective, most likely due to their bulky substituents.
A gram-scale reaction experiment was also performed, in which aliquots were taken at 15 min intervals and analyzed by 1H and 19F NMR spectroscopy (Scheme 1). The reaction proceeded smoothly, with substrate 2g no longer observable after 1 h. No by-products or degradation of 3g were observed after a further 30 min. As with smaller scale reactions, ethyl acetate/water extraction was performed and the product 3g was isolated in a nearly stoichiometric amount (97%). Most of the EtOAc was recovered during the evaporation process.
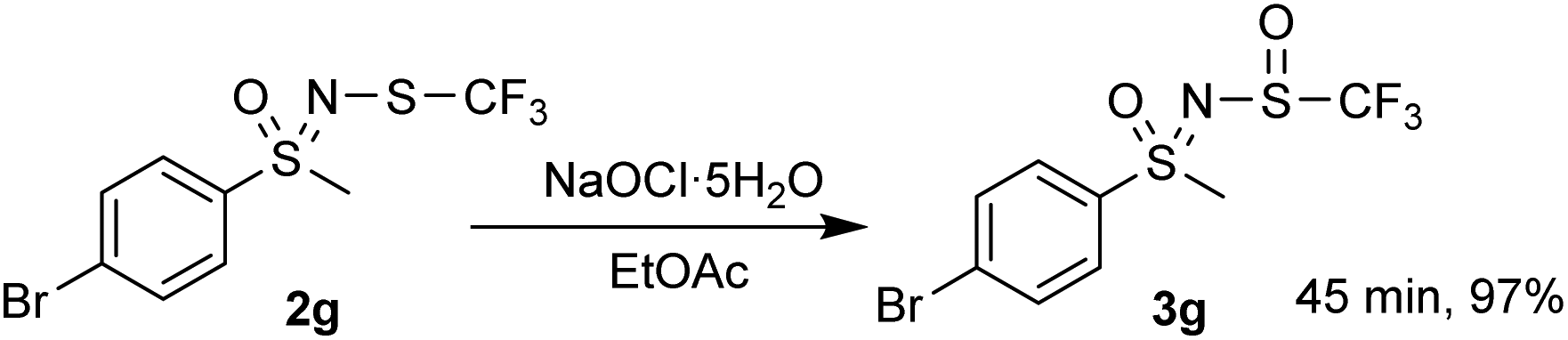 | ||
| Scheme 1 Gram-scale reaction. | ||
The gram-scale reaction was also used to determine the green chemistry metrics for this oxidation. The mass of the five moles of water in NaOCl·5H2O was not included in the calculations as it is not ecologically problematic. The atom economy was calculated to be 82%, while the actual atom economy and reaction mass efficiency were also high with 79.5% and 79.7%, respectively. The E-factor of 16.48 is mainly derived from water, EtOAc and salts (NaCl and Na2SO4), which are not particularly environmentally problematic. While the E-factor is still far from the ideal value of 0 or the acceptable value of 1–5, but at least the waste does not have to be specially disposed of. A result of 84.5 was achieved on the EcoScale,40 which corresponds to a “great synthesis” according to the criteria of the method (Table 4). The calculations can be found in the ESI.†
|
||
| Atom economy |
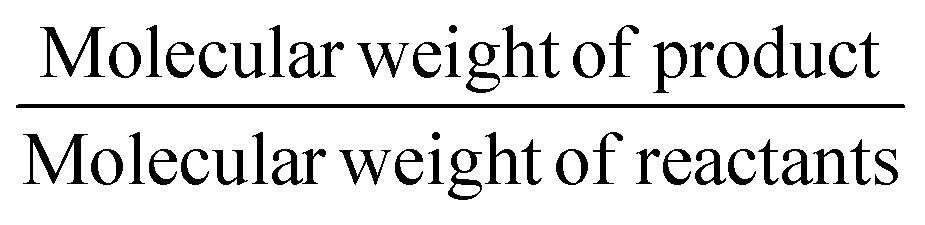
|
82% |
| Actual atom economy | Yield of product × atom economy | 79.5% |
| Reaction mass efficiency |
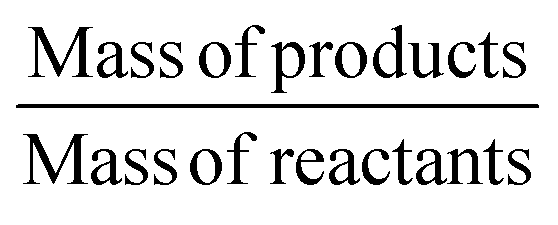
|
79.7% |
| E-Factor |
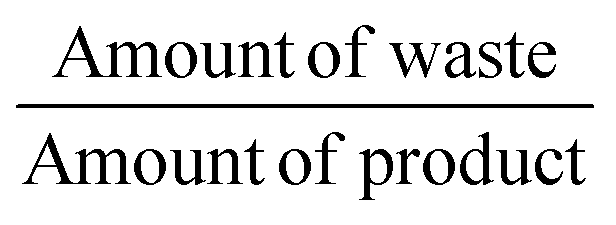
|
16.48 |
| EcoScale |
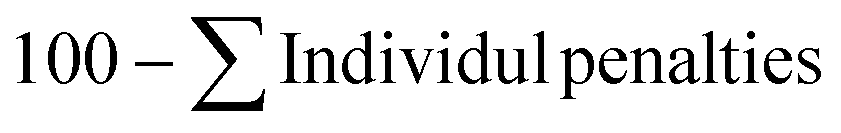
|
84.5 |
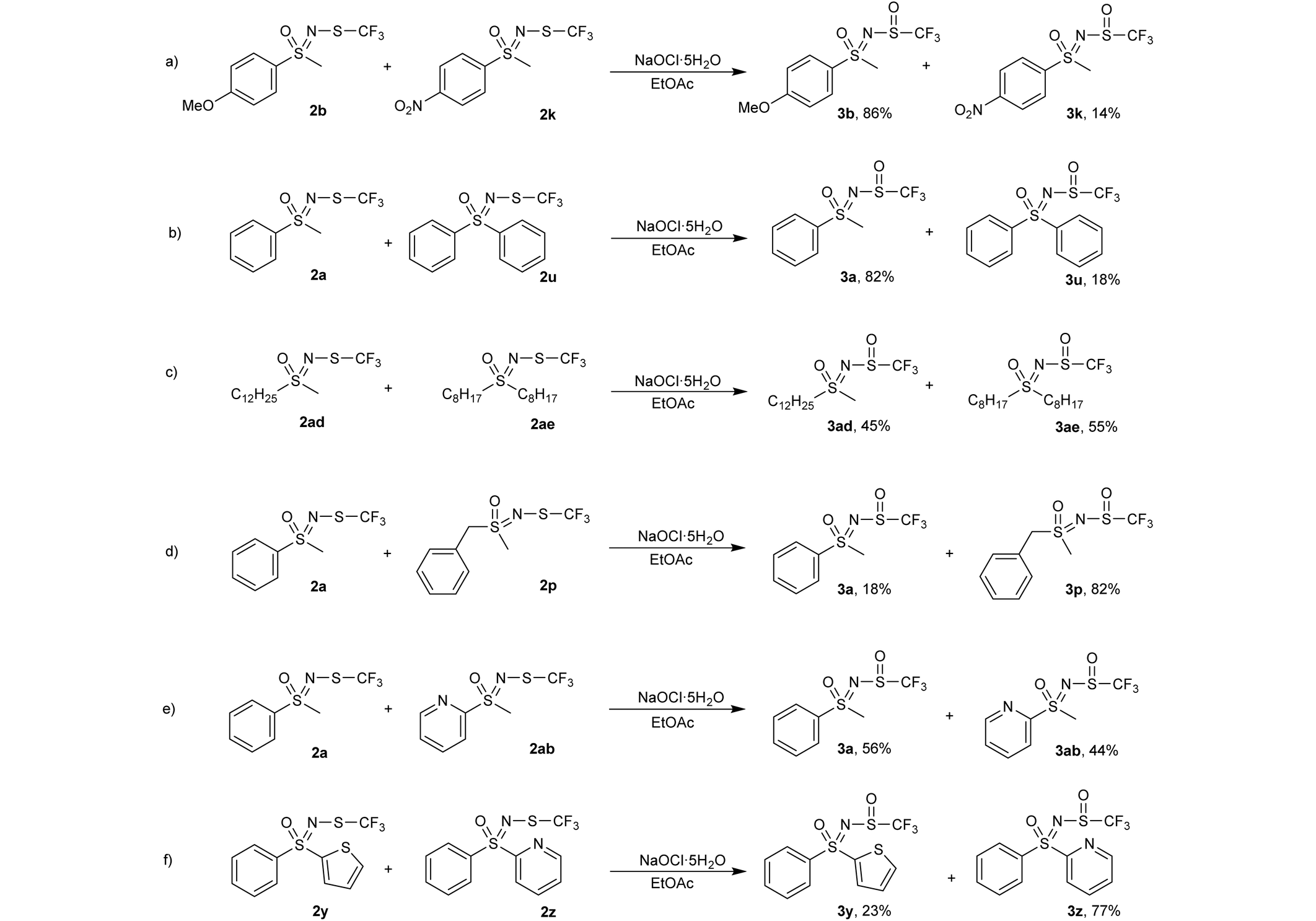
To gain a better understanding of the reactivity of our substrates, multiple competitive reactions were carried out in which two N-trifluoromethylthio sulfoximines 2 were mixed in a 1:1 ratio and 1 equiv. of oxidant was added. The reactions were stirred for 16 h, then an internal standard (1,3,5-trimethoxybenzene) was added and the mixture was analysed by 1H and 19F NMR spectroscopy. These reactions were repeated twice and only minimal deviations were observed.
In a competitive reaction between 4-methoxy-substituted 2b and 4-nitro-substituted 2k (Table 5, a), 86% of the oxidized species was attributed to the 4-methoxy product 3b, indicating that electron-donating substituents on the phenyl ring promote this type of oxidation. The reaction between the phenyl methyl substrate 2a and diphenyl 2u (Table 5, b) favored the formation of 3a, possibly due to steric hindrance or the electron-withdrawing effect of the second phenyl ring. Dodecyl methyl substrate 2ad and dioctyl substrate 2ae (Table 5, c) reacted similarly, both utilizing roughly half of the available oxidant, suggesting that the length of the alkyl chains is not an important factor in determining the reaction rate. Benzyl substrate 2p was more reactive than its phenyl 2a counterpart (Table 5, d), again indicating that the electron-withdrawing effect of the phenyl ring hinders oxidation. Phenyl-substituted 2a and 2-pyridyl-substituted 2ab (Table 5, entry e) produced similar amounts of oxidized products, although pyridine was thought to have a stronger electron-withdrawing effect. The substituted 2-pyridyl 3z product was substantially favoured over the 2-thienyl 3y product (Table 5, f), which again was somewhat surprising since the 2-thienyl moiety is more electron-donating and is less bulky than the 2-pyridyl group. The reaction mechanism for sulfide oxidations with NaOCl·5H2O has been proposed previously36 and involves the chlorination of the sulfide followed by nucleophilic attack by the hydroxide ion (Scheme 2). Our work supports this proposal as the electron-donating substituents were found to be more reactive and thus promote the nucleophilic attack of sulfur on the chloride atom.
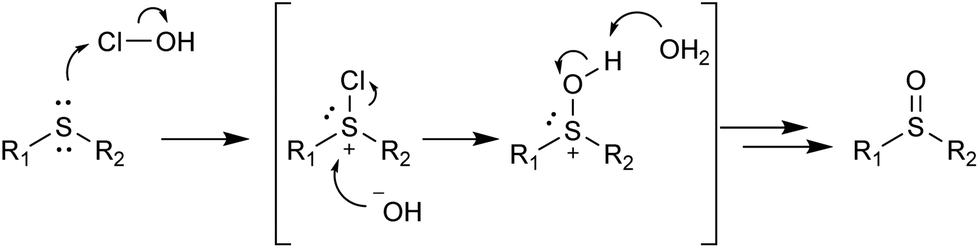 | ||
| Scheme 2 Proposed mechanism for the oxidation of sulfides with NaOCl·5H2O. | ||
| a Reaction conditions: a flask was charged with both substrates 2 (0.1 mmol each) and EtOAc (0.2 mL) and lastly NaOCl·5H2O (1 equiv.) was added. The mixture was stirred for 16 h at r.t. 1,3,5-Trimethoxy benzene was added and after extraction from EtOAc/water, the organic phase was dried and the solvent was removed under reduced pressure. |
|---|
|
The effect of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was also investigated. The reaction of 1 equiv. of TEMPO, 1 equiv. of 2a and 1.1 equiv. of NaOCl·5H2O gave product 3a with 58% conversion, leaving 42% of the starting material 2a unreacted. Upon reducing the amount of TEMPO to 0.2 equiv. the proportion of 3a decreased to 43%. From this it can be concluded that the reaction between NaOCl·5H2O and TEMPO takes place before the oxidation of the substrate and the oxidized species is then less reactive towards the substrate 2a.
The newly formed sulfinyl moiety was tested in some post-modification transformations. The reaction of 3g with 4-methylbenzeneboronic acid in boiling water in the presence of Pd/C led to Suzuki–Miyaura coupling with concomitant removal of the N-trifluoromethylsulfaneylidene moiety, yielding the sulfoximine 1ag (Scheme 3). A similar reaction in 1,4-dioxane under milder conditions, which had already been used for sulfoximines,41 furnished the coupling product 3af with an intact N-trifluoromethylsulfaneylidene group (Scheme 3).
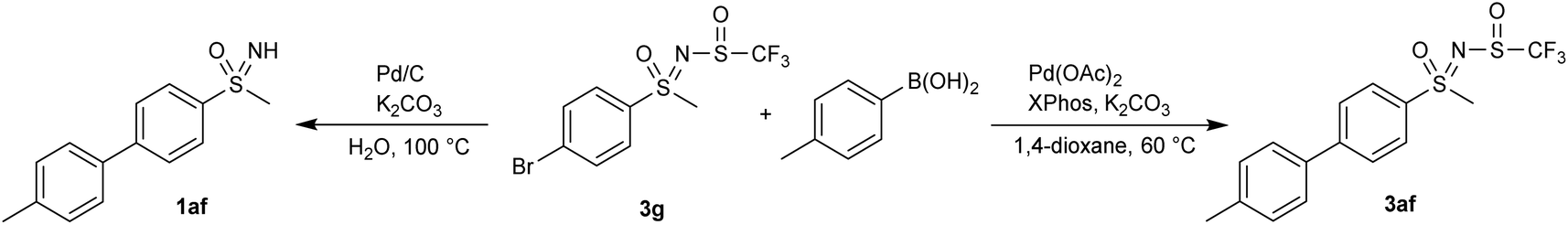 | ||
| Scheme 3 Condition-dependent Suzuki–Miyaura coupling reactions. | ||
Sonogashira coupling of 3g with phenylacetylene successfully furnished 3ag using catalytic amounts of PdCl2(PPh3)2 and CuI in Et3N (Scheme 4).
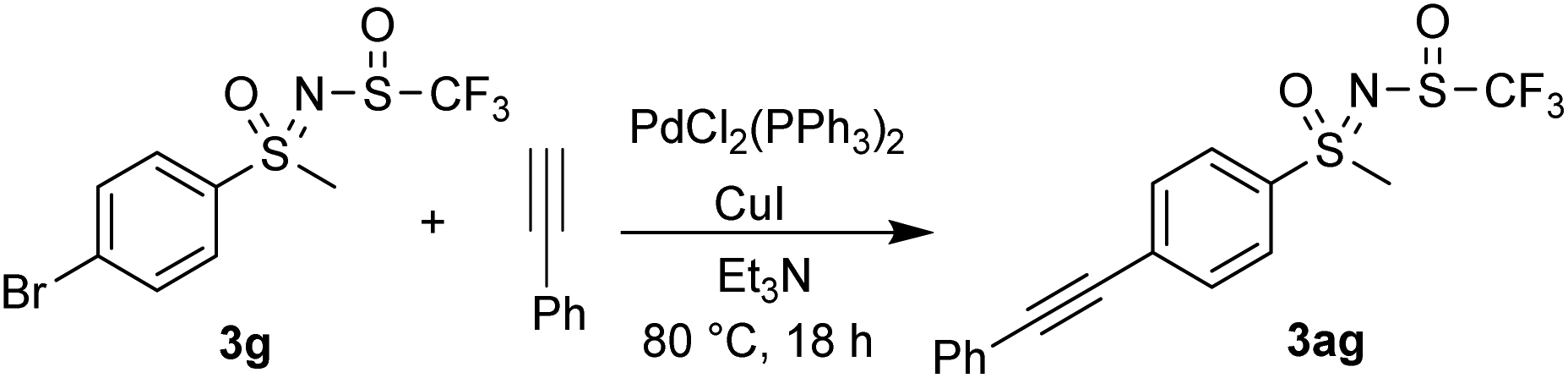 | ||
| Scheme 4 Sonogashira coupling reaction. | ||
Kumada-type coupling reactions with nickel and iron catalysts were also examined. The reaction of 3g with Fe(acac3), HMTA, TMEDA and phenylmagnesium bromide in THF failed to produce any product and only the substrate 3g was isolated. Reaction of 3g with methylmagnesium bromide in the presence of NiCl2(dppf) in diethyl ether led to deprotection of the substrate and the sulfoximine 1g was recovered (Scheme 5).
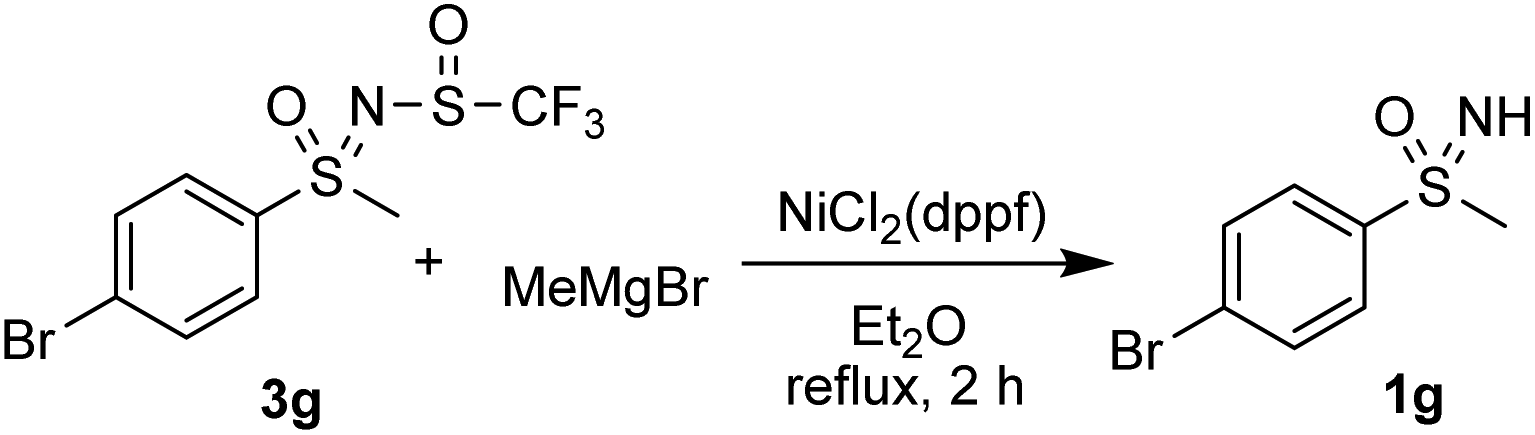 | ||
| Scheme 5 Attempted nickel-catalysed Kumada-type reaction. | ||
Next, electrophilic ring bromination was investigated. The use of NBS in hexafluoroisopropanol (HFIP) and MeCN did not lead to any reaction with 3b. Using bromine in acetic acid and in chloroform produced the sulfoximine 1b. Addition of FeBr3 to further promote ring bromination also produced the sulfoximine 1b (Scheme 6).
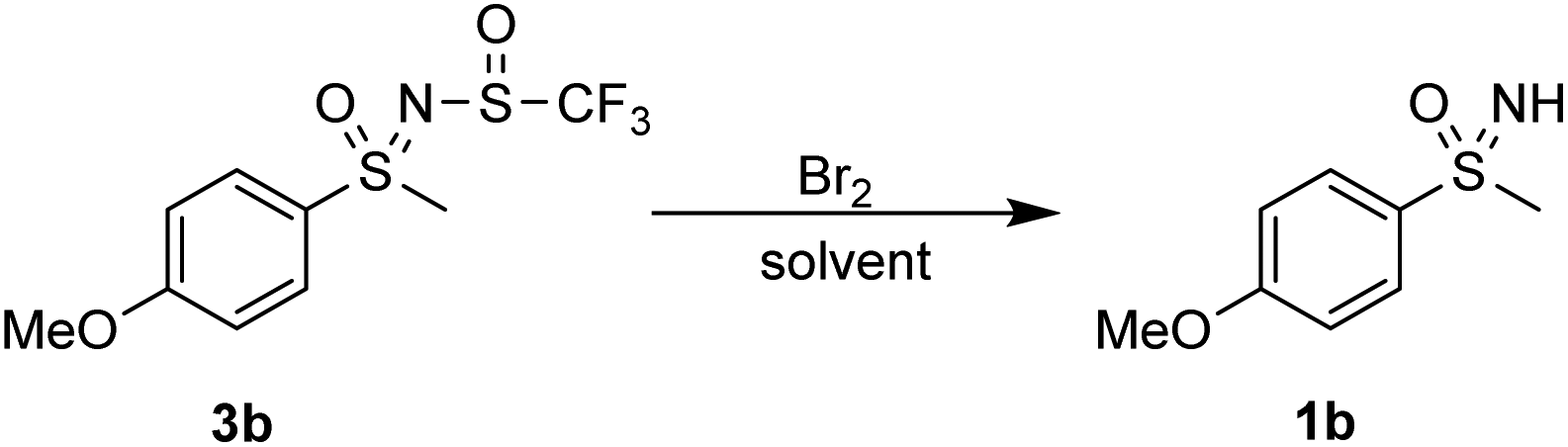 | ||
| Scheme 6 Attempted ring bromination. | ||
To further examine the stability of these compounds, which show no deterioration at room temperature or in air, compound 3g was subjected to elevated temperature (80 °C) and no changes were observed after 3 h (Scheme 7). Stability at different pH values was tested by stirring 3g in 2 M aqueous HCl and NaOH solutions for 3 h. Under basic conditions, degradation to sulfoximine 1g was observed, while no change occurred in an acidic medium.
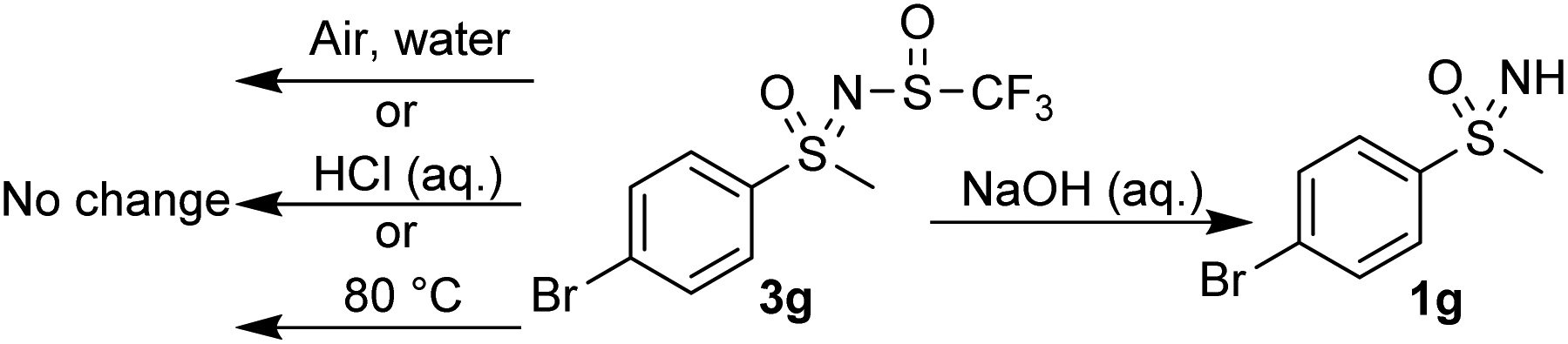 | ||
| Scheme 7 Stability tests for compound 3g. | ||
Conclusion
In conclusion, we report a sustainable, practical, cost-effective and highly selective oxidation of various aryl-, heteroaryl- and alkyl-substituted N-trifluoromethythio sulfoximines to N-trifluoromethylsulfaneylidene sulfoximines. These novel functionalized sulfoximines were prepared in high yields and with almost no need for purification using NaOCl·5H2O as a green oxidant, which produces ecologically benign waste. In competitive experiments, a general trend of reactivity that electron-rich substrates are more reactive than electron-poor substrates was observed. A scale-up experiment was performed with an atom economy of 82%, actual atom economy of 79.5%, a reaction mass efficiency of 79.7%, an E-factor of 16.48 and a total of 84.5 EcoScale points, denoting the method as a “great” synthesis from a green metric point of view. Further modifications of the functionalized sulfoximines are also possible and were demonstrated with Suzuki–Miyaura and Sonogashira coupling reactions. The stability of this new functional group was also investigated. While decomposition to sulfoximine 1 was observed under basic conditions, the compounds are stable in air, water, an acidic environment and at elevated temperatures.Author contributions
Ž. T.: investigation, validation, data curation, writing – original draft and writing – review & editing. M. J.: conceptualization, resources, validation, supervision, writing – original draft and writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (Research Core Funding Grant P1-0230 and Young Researcher Grant to Ž. T.). The authors thank Dr Damijana Urankar from the Research Infrastructure Center at the Faculty of Chemistry and Chemical Technology, the University of Ljubljana, for HRMS analyses of small molecules.References
- See for example:
(a) H. Kour, A. Sharma, J. Kour, M. Ahmed and S. D. Sawant, Eur. J. Org. Chem., 2022, e202201375 CrossRef CAS
; (b) D. Kong, D. Ma, P. Wu and C. Bolm, ACS Sustainable Chem. Eng., 2022, 10, 2863–2867 CrossRef CAS
- See for example:
(a) P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed
- V. N. Boiko, Beilstein J. Org. Chem., 2010, 6, 880–921 CrossRef PubMed
- C. A. Dannenberg, V. Bizet and C. Bolm, Synthesis, 2015, 47, 1951–1959 CrossRef CAS
- S. Y. Song, X. Zhou, Z. Ke and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202217130 CrossRef CAS PubMed
- P. Shi, Y. Tu, C. Wang, D. Ma and C. Bolm, J. Org. Chem., 2022, 87, 3817–3824 CrossRef CAS PubMed
- S. Greed, O. Symes and J. A. Bull, Chem. Commun., 2022, 58, 5387–5390 RSC
- A. Wimmer and B. König, Org. Lett., 2019, 21, 2740–2744 CrossRef CAS PubMed
- D. Liu, Z. R. Liu, C. Ma, K. J. Jiao, B. Sun, L. Wei, J. Lefranc, S. Herbert and T. S. Mei, Angew. Chem., Int. Ed., 2021, 60, 9444–9449 CrossRef CAS PubMed
- C. Wang, H. Zhang, L. A. Wells, T. Liu, T. Meng, Q. Liu, P. J. Walsh, M. C. Kozlowski and T. Jia, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed
- Z. P. Shultz, T. Scattolin, L. Wojtas and J. M. Lopchuk, Nat. Synth., 2022, 1, 170–179 CrossRef PubMed
- M. Song, L. Zhang, D. Wei, Y. He, J. Jia, H. Li and B. Yuan, Green Chem., 2022, 24, 6119–6124 RSC
- D. Ma, D. Kong, P. Wu, Y. Tu, P. Shi, X. Wang and C. Bolm, Org. Lett., 2022, 24, 2238–2241 CrossRef CAS PubMed
- Y. Maeda, S. Hamada, Y. Aota, K. Otsubo, T. Kano and K. Maruoka, J. Org. Chem., 2022, 87, 3652–3660 CrossRef CAS PubMed
- D. Zhang, H. Wang, C. Bolm and R. Li, Chem. Commun., 2018, 54, 5772–5775 RSC
- Y. Tu, P. Shi and C. Bolm, Org. Lett., 2022, 24, 907–911 CrossRef CAS PubMed
- P. Qiu, X. Duan, M. Li, Y. Zheng and W. Song, Org. Lett., 2022, 24, 2733–2737 CrossRef CAS PubMed
- K. K. Rajbongshi, S. Ambala, T. Govender, H. G. Kruger, P. I. Arvidsson and T. Naicker, Synthesis, 2020, 52, 1279–1286 CrossRef CAS
- Y. Tu, D. Zhang, P. Shi, C. Wang, D. Ma and C. Bolm, Org. Biomol. Chem., 2021, 19, 8096–8101 RSC
- See for example:
(a) H. Wang, M. Frings and C. Bolm, Org. Lett., 2016, 18, 2431–2434 CrossRef CAS PubMed
-
(a) F. Teng, J. Cheng and C. Bolm, Org. Lett., 2015, 17, 3166–3169 CrossRef CAS PubMed
- V. D. Romanenko, Curr. Org. Chem., 2023, 27, 411–434 CrossRef CAS
- N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6, 65 Search PubMed
- D. Wang, C. G. Carlton, M. Tayu, J. J. W. McDouall, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2020, 59, 15918–15922 CrossRef CAS PubMed
- R. P. Singh, G. Cao, R. L. Kirchmeier and J. M. Shreeve, J. Org. Chem., 1999, 64, 2873–2876 CrossRef CAS PubMed
- C. Wakselman, M. Tordeux, C. Freslon and L. Saint-Jalmes, Synlett, 2001, 550–552 CrossRef CAS
- T. Billard, A. Greiner and B. R. Langlois, Tetrahedron, 1999, 55, 7243–7250 CrossRef CAS
- H. Chachignon and D. Cahard, J. Fluorine Chem., 2017, 198, 82–88 CrossRef CAS
- D. W. Sun, X. Jiang, M. Jiang, Y. Lin and J. T. Liu, Eur. J. Org. Chem., 2017, 3505–3511 CrossRef CAS
- L. Yang, S. Wang, F. Liang, Y. Han, Y. Li, D. Shan, L. Liu, Q. Wang and D. Zhu, Org. Chem. Front., 2023, 10, 4368–4380 RSC
- See for example:
(a) K. J. Liu, J. H. Deng, J. Yang, S. F. Gong, Y. W. Lin, J. Y. He, Z. Cao and W. M. He, Green Chem., 2020, 22, 433–438 RSC
- See for example:
(a) B. Zhao, G. B. Hammond and B. Xu, Tetrahedron Lett., 2021, 82, 153376 CrossRef CAS
- C. M. M. Hendriks, P. Lamers, J. Engel and C. Bolm, Adv. Synth. Catal., 2013, 355, 3363–3368 CrossRef CAS
- R. Y. Tang, P. Zhong and Q. L. Lin, J. Fluorine Chem., 2007, 128, 636–640 CrossRef CAS
- See for example:
(a) H. Marom, S. Antonov, Y. Popowski and M. Gozin, J. Org. Chem., 2011, 76, 5240–5246 CrossRef CAS PubMed
- M. Kirihara, T. Okada, Y. Sugiyama, M. Akiyoshi, T. Matsunaga and Y. Kimura, Org. Process Res. Dev., 2017, 21, 1925–1937 CrossRef CAS
- See for example:
(a) M. Kirihara, R. Osugi, K. Saito, K. Adachi, K. Yamazaki, R. Matsushima and Y. Kimura, J. Org. Chem., 2019, 84, 8330–8336 CrossRef CAS PubMed
- J. Hachtel and H.-J. Gais, Eur. J. Org. Chem., 2000, 1457–1465 CrossRef CAS
- Y. Xie, B. Zhou, S. Zhou, S. Zhou, W. Wei, J. Liu, Y. Zhan, D. Cheng, M. Chen, Y. Li, B. Wang, X. S. Xue and Z. Li, ChemistrySelect, 2017, 2, 1620–1624 CrossRef CAS
- K. Van Aken, L. Strekowski, L. Patiny and L. Strekowski, Beilstein J. Org. Chem., 2006, 2, 3–17 Search PubMed
- A. K. Bachon, A. D. Steinkamp and C. Bolm, Adv. Synth. Catal., 2018, 360, 1088–1093 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and the copies of 1H, 19F and 13C NMR spectra. See DOI: https://doi.org/10.1039/d3ob02033a |
| This journal is © The Royal Society of Chemistry 2024 |