Asymmetric total synthesis of humulane sesquiterpenoids alashanoids B, C, E, and F and 2,9-humuladien-6-ol-8-one†
Received
10th March 2024
, Accepted 4th April 2024
First published on 10th April 2024
Abstract
Naturally occurring sesquiterpenes having humulane frameworks are structurally intriguing and possess significant biological profiles. Asymmetric synthesis of the alashanoids B, C, E, and F and 2,9-humuladien-6-ol-8-one is achieved for the first time through a linear synthetic strategy. Intramolecular late-stage Nozaki–Hiyama–Kishi (NHK) coupling is employed to access the eleven-membered macrocyclic core present in the target molecules. The NHK precursors are accessed using the Evans and non-Evans syn and anti-aldol reaction as a key transformation. X-ray and ECD analysis reconfirmed the synthesized compounds’ structures and chirotopical properties.
Introduction
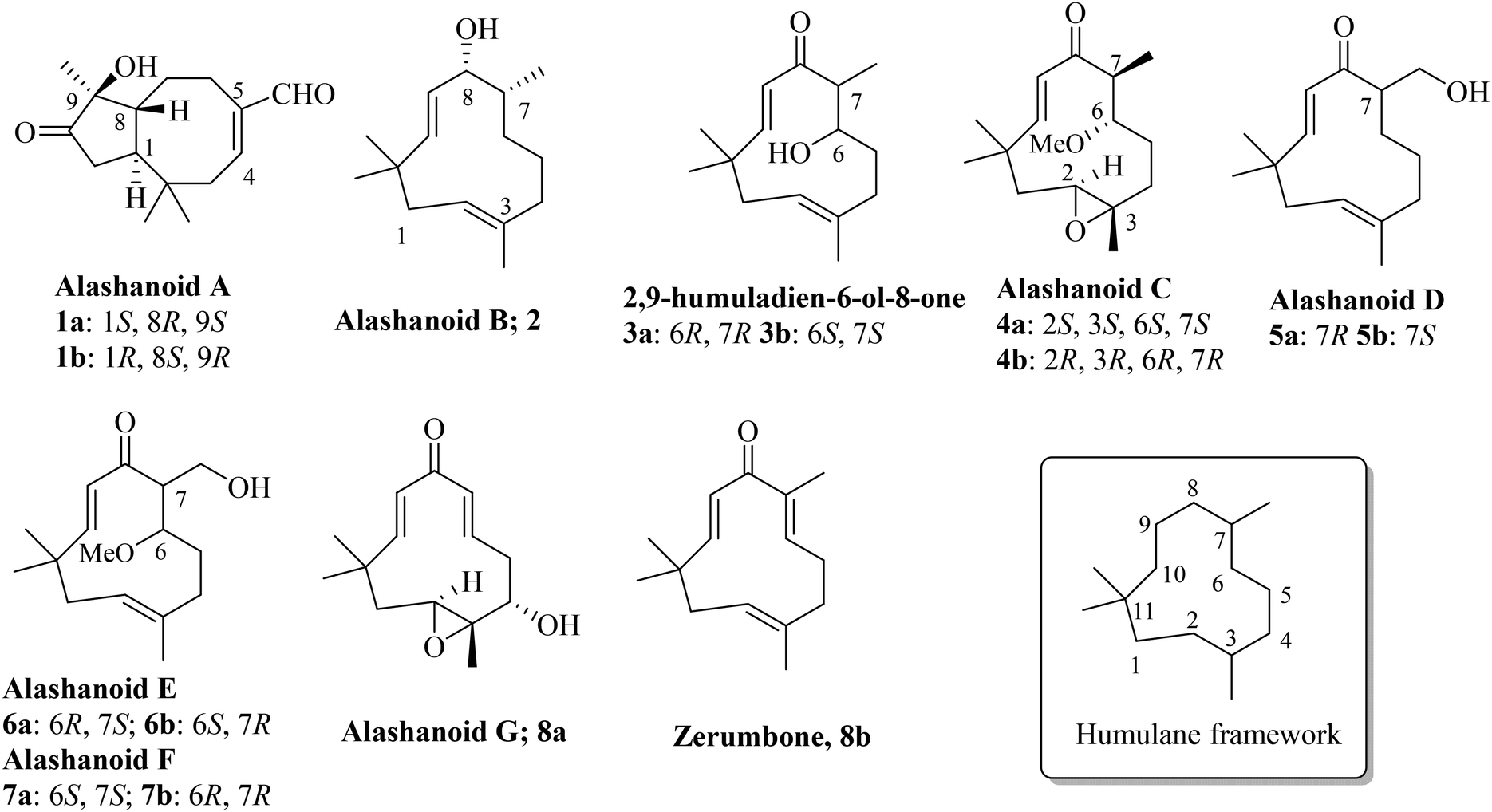
Syringa pinnatifolia, belonging to the Oleaceae family, is a deciduous shrub of height 8 to 12 feet and has an elegant bushy habit and is mainly found in western China. The extract from this plant's peeled roots, stems, and twigs have been used for a long time as a traditional folk medicine for the treatment of cardiovascular diseases and for pain management. The stems of this plant contain zerumbone, a humulane type sesquiterpenoid exhibiting a cardio protective effect against acute myocardial ischemia (AMI) in rats.1,2 Recently, a few structurally new sesquiterpenoids were isolated from the stem of Syringa pinnatifolia; among them are alashanoids A–G (1, 2, 4–7 and 8a) and 2,9-humuladien-6-ol-8-one (3), which are unique and have been isolated from the first time from this species.3 Alashanoid A (1) has a rare 2,2,5,9-tetramethylbicyclo[6.3.0]-undecane skeleton and both the enantiomers have been isolated from the same plant. Alashanoids B–G (2, 4–7 and 8a) and 2,9-humuladien-6-ol-8-one (3) have a humulane-type framework in their structure (Fig. 1). Zerumbone (8b) is another known humulane-type sesquiterpenoid which contains a di-enone functionality and is known for its cardioprotective effect, as shown in earlier studies.1,2 The structures of all the isolated natural products have been established through extensive NMR analysis and X-ray crystallographic analysis. Their absolute stereo-configuration was also assigned through theoretical and experimental ECD data analysis. Few of these compounds exhibit a wide range of bioactivities, including protective effects against hypoxia injury to H9c2 cells and the ability to inhibit NO production in LPS-induced RAW264.7 macrophage cells; hence such compounds might be of tremendous significance for clinical use in traditional medicines used to treat coronary diseases.
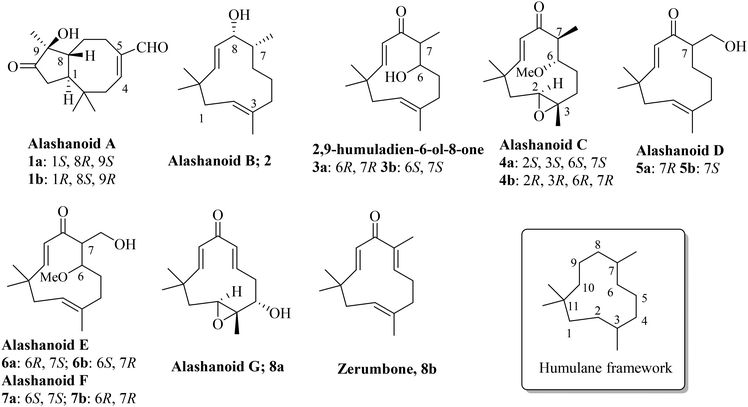 |
| | Fig. 1 Naturally occurring humulane-type sesquiterpenoids alashanoids and 2,9-humuladien-6-ol-8-one. | |
In the literature, it was reported that alashanoid A (1), 2,9-humuladien-6-ol-8-one (3), and alashanoids C–F (4–7) were obtained as a scalemic (racemic) mixture, but separation through chiral phase HPLC was performed and after CD measurement, the absolute configuration was assigned to the individual enantiomers (Fig. 1). Alashanoid B (2) and alashanoid G (8a) were obtained as enantiopure compounds from the stem of Syringa pinnatifolia, and their absolute configuration was assigned through theoretically calculated and experimentally measured ECD spectra. Recently, a comprehensive review of the synthesis of humulanolides (sesquiterpene lactones) has been reported.4 Synthetic studies towards naturally occurring humulanes, shown in Fig. 1, are not available in print yet.
Results and discussion
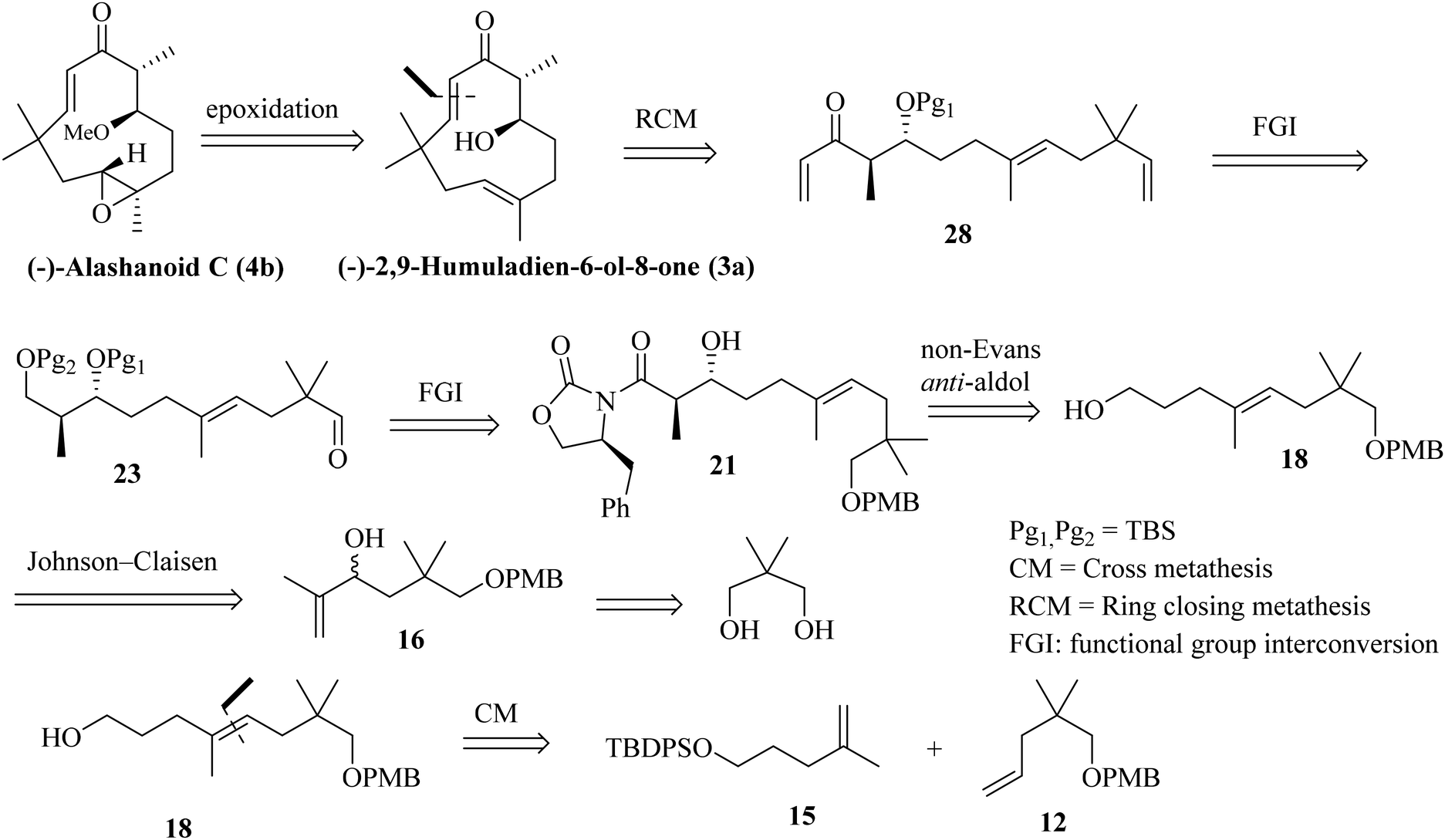
In this article, we report the first asymmetric total synthesis of (+)-alashanoid B, (−)-2,9-humuladien-6-ol-8-one, (−)-alashanoid C and (+)-alashanoids E and F and also establish their absolute configuration via comparison with those of reported natural products. Close structural visualization revealed that compounds 2, 3a, 4b, 6a, and 7a all contain eleven-membered humulane ring systems with two stereocenters and enone systems; in addition, they also contain two all-carbon-quaternary centers (C11 bearing the gem-dimethyl group and C3 with the E-olefinic geometry is stereogenic). (−)-Alashanoid C (4b) contains an oxirane ring at the C2–C3 carbon, and the C6 carbon contains a methoxy (–OMe) substitution. (−)-2,9-Humuladien-6-ol-8-one can be regarded as a precursor for (−)-alashanoid C. The retrosynthetic disconnection for alashanoid C and (−)-2,9-humuladien-6-ol-8-one is shown in Scheme 1. A late-stage RCM reaction5 is proposed to access 3a, and the subsequent regioselective epoxidation should furnish 4b. The RCM precursor can be conveniently prepared through an Evans auxiliary mediated anti-selective aldol reaction (non-Evans anti) through an open TS (transition state) model.6 The aldol precursor could be accessed through the Johnson–Claisen rearrangement of allylic alcohol,7 which could easily be accessed using neo-pentyl glycol as a starting precursor. A cross-metathesis (CM) reaction8 is also envisioned to access the aldol precursor, as shown in Scheme 1.
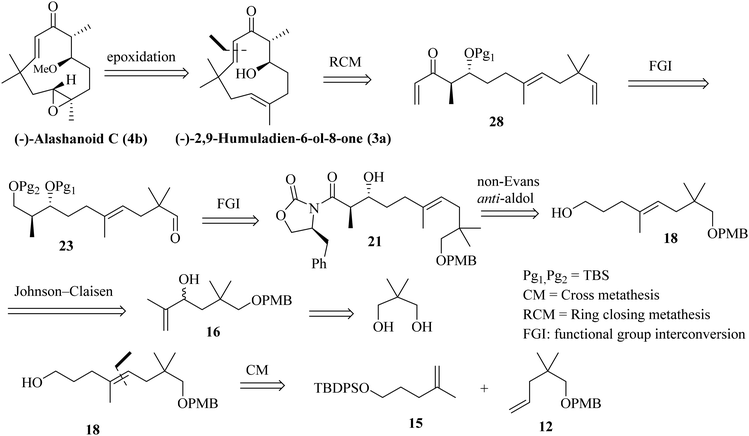 |
| | Scheme 1 Retrosynthetic disconnection for (−)-alashanoid C and (−)-2,9-humuladien-6-ol-8-one. | |
Synthesis of (−)-alashanoid C and (−)-2,9-humuladien-6-ol-8-one
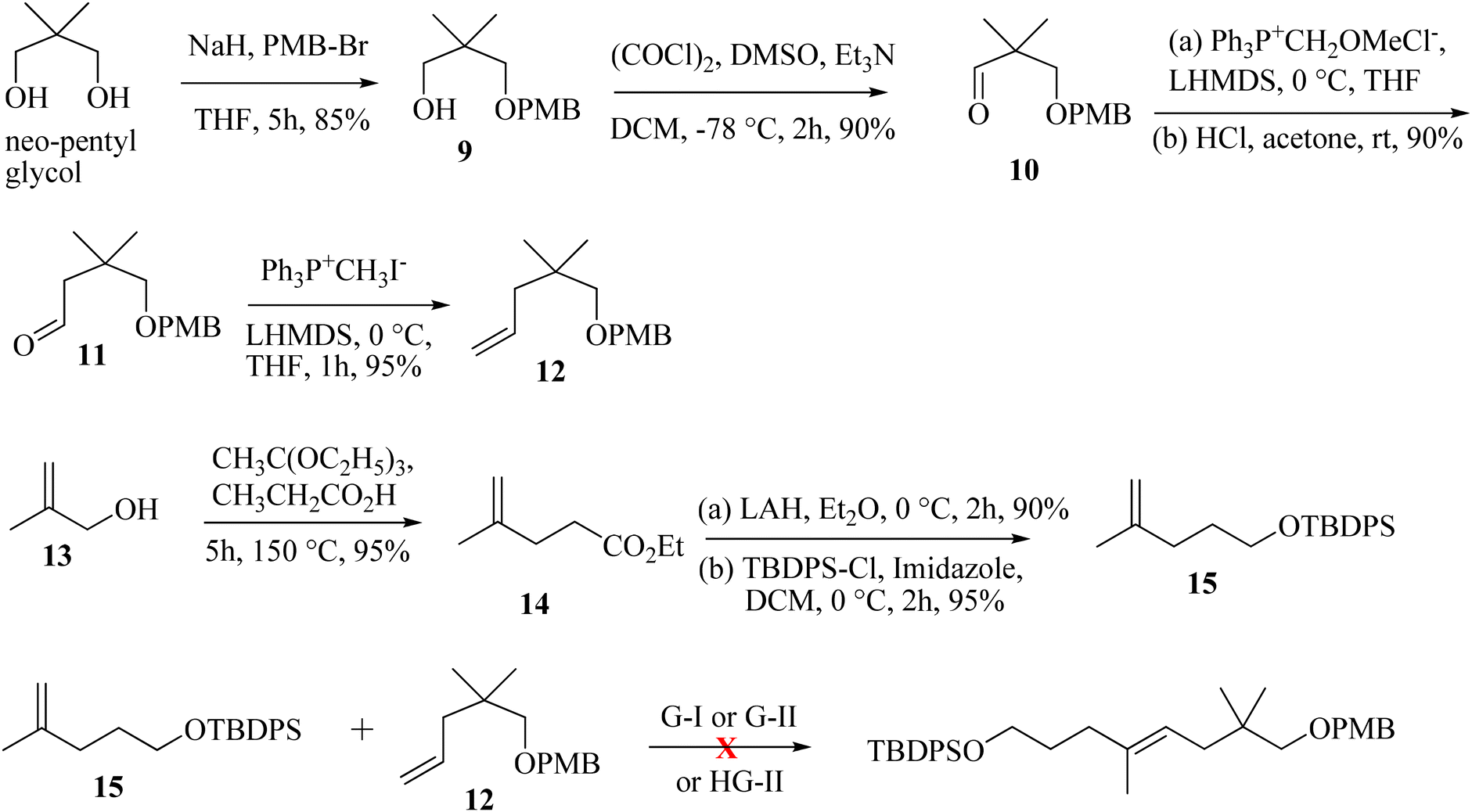
The synthesis is initiated from the known aldehyde 10 prepared from neo-pentylglycol.9 Aldehyde 10 on homologation through the Wittig protocol10 and the subsequent hydrolysis of the enol-ether furnished aldehyde 11 in 90% yield. The Wittig olefination of 11 provided olefin 12 in 95% yield. The other CM partner was synthesized from 2-methylprop-2-en-1-ol (13), which undergoes the Johnson–Claisen rearrangement and furnishes the γ,δ-unsaturated ester 14; subsequent reduction with LiAlH4 and treatment with TBDPS-Cl afforded compound 15 (81% yield in three steps). The proposed CM reaction was next attempted with the G-I or G-II or HG-II catalyst (5 mol%);8 unfortunately the CM product was not observed as anticipated. Unwanted self-dimerization occurred, and the desired CM product was not obtained (Scheme 2).
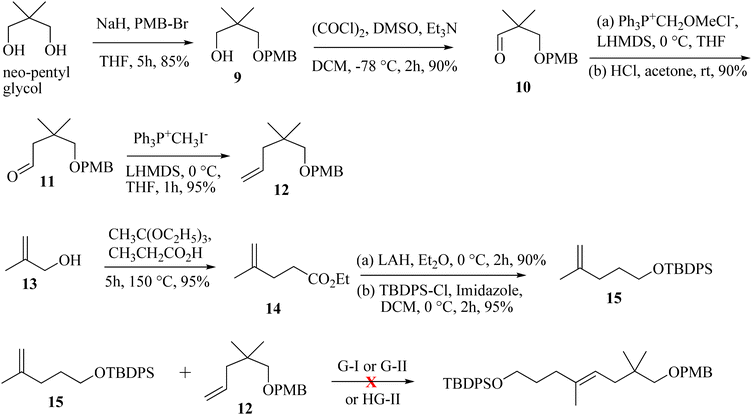 |
| | Scheme 2 Unsuccessful CM approach towards the advanced intermediate for (−)-2,9-humuladien-6-ol-8-one. | |
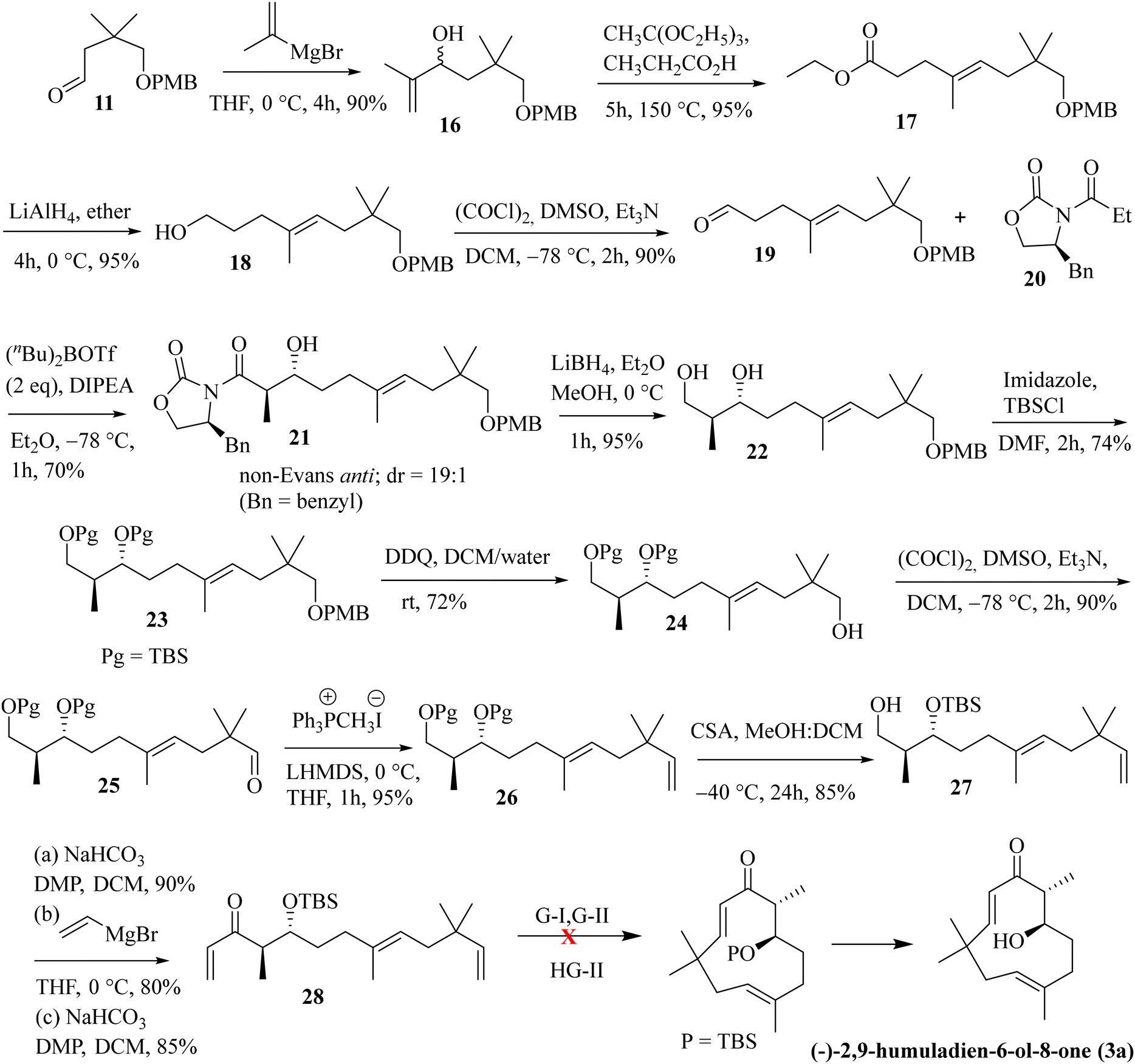
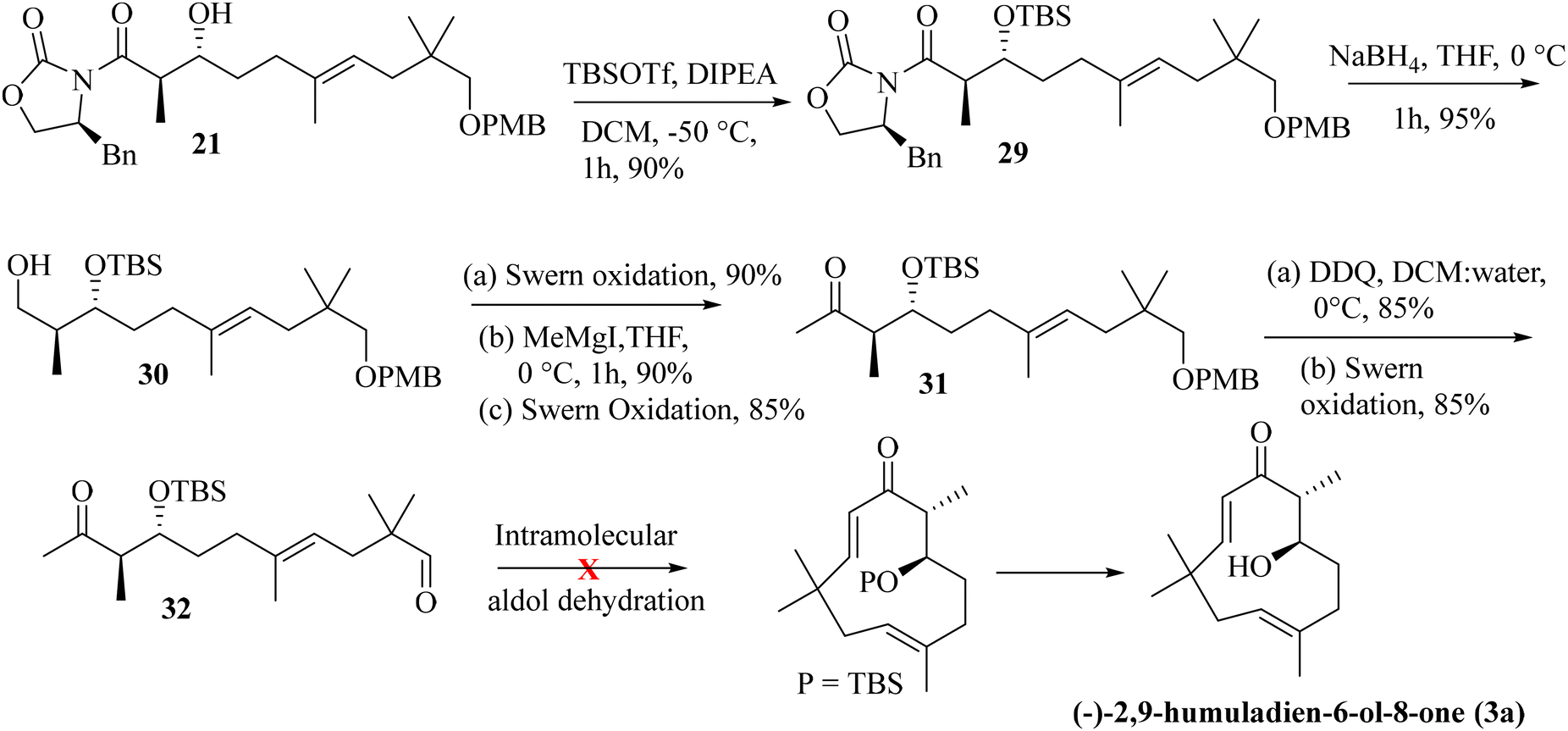
Next, we have explored the feasibility of preparing a suitably substituted γ,δ-unsaturated ester that will lead us to the aldol precursor (Scheme 1). Aldehyde 11 was reacted with freshly prepared Grignard reagent generated from 2-bromoprop-1-ene to furnish the allylic alcohol 16 in 90% yield. The Johnson–Claisen rearrangement of 16 afforded the desired γ,δ-unsaturated ester 17 in 95% yield, and the subsequent reduction11 with LiAlH4 then furnished alcohol 18 (95% yield). Oxidation of alcohol 18 under the Swern conditions then produced aldehyde 19 in 90% yield. The asymmetric aldol reaction of Evans oxazolidinone 20 with aldehyde 19 then afforded the anti-aldol product (non-Evans anti) 21 in 70% yield with excellent diastereoselectivity and enantioselectivity.12 The excellent stereoselectivity can be explained through the Heathcock open-chain transition state (via non-chelated acyclic stereocontrol) model.12a Reductive removal of the auxiliary with LiBH4 furnished the corresponding diol 22 (95% yield). Both the hydroxy groups were then protected as –TBS ether to provide compound 23 in a quantitative yield (98%). The –PMB ether group in 23 was oxidatively removed by DDQ treatment to furnish alcohol 24 in 72% yield. The Swern oxidation of 24 afforded aldehyde 25 in 90% yield. The Wittig olefination of aldehyde 25 gave olefin 26, as shown in Scheme 3. Chemoselective deprotection of the primary TBS-ether group13 was next achieved by treating 26 with CSA/MeOH at −40 °C for 24 h to afford compound 27 in 85% yield. Functional group manipulation with 27 then afforded the enone compound 28 (a three-step protocol involving DMP oxidation, vinyl Grignard addition, and further DMP oxidation; 60% yield in three steps), which served as the RCM precursor. The RCM reaction, mainly with the G-II and HG-II catalysts, was next attempted with compound 28. But to our disappointment, the desired ring closed product remained elusive (Scheme 3). We reasoned that the RCM precursor 28 bearing a terminal olefinic unsaturated group with two adjacent gem-dimethyl groups is creating enough steric crowding to inhibit the initial complexation with the Ru-based alkylidene catalyst. Such instances depicting the unsuccessful RCM reaction with the sterically crowded olefin and Ru-based metathesis catalysis have been reported.8e
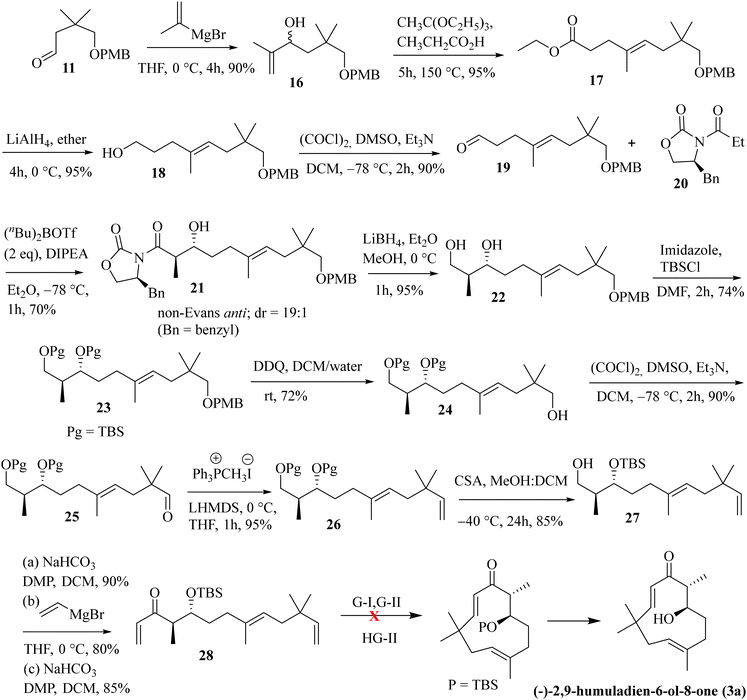 |
| | Scheme 3 Attempted RCM reaction for accessing the humulane core in the target molecule. | |
As the attempted RCM reaction was unsuccessful, we envisioned that an intramolecular aldol reaction14 might be suitable for achieving E-olefinic unsaturation (C9–C10) in the target structure. The aldol product 21 was protected as its TBS-ether by treatment with TBS-OTf and DIPEA at −50 °C (at a higher temperature, the protection becomes futile, and subsequent epimerization was observed at the α-carbon attached to the Evans-auxiliary) to furnish compound 29 in 90% yield. Removal of the Evans-auxiliary with NaBH4 reduction15 then produced alcohol 30 in 95% yield. Functional group manipulation (the Swern oxidation, MeMgI addition, and the Swern oxidation) led to the keto-methyl compound 31 in 70% yield (in three steps). Removal of the PMB group with DDQ and oxidation of the resulting alcohol under the Swern conditions then furnished the intramolecular aldol precursor 32. We attempted the intramolecular aldol dehydration under numerous base-mediated conditions, but it was unsuccessful as in most cases, intermolecular condensation seems to be the main product (Scheme 4).
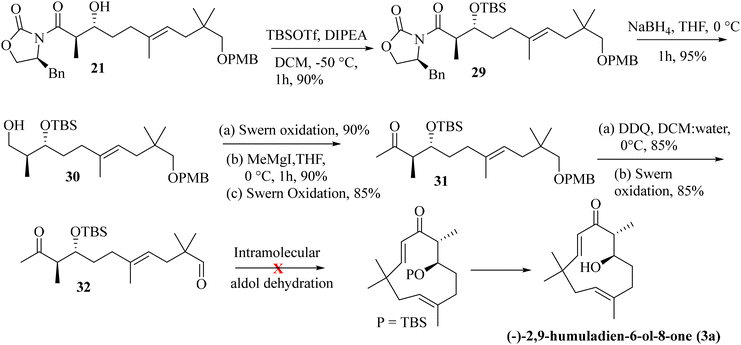 |
| | Scheme 4 Attempted intramolecular aldol reaction for accessing the humulane core in the target molecule. | |
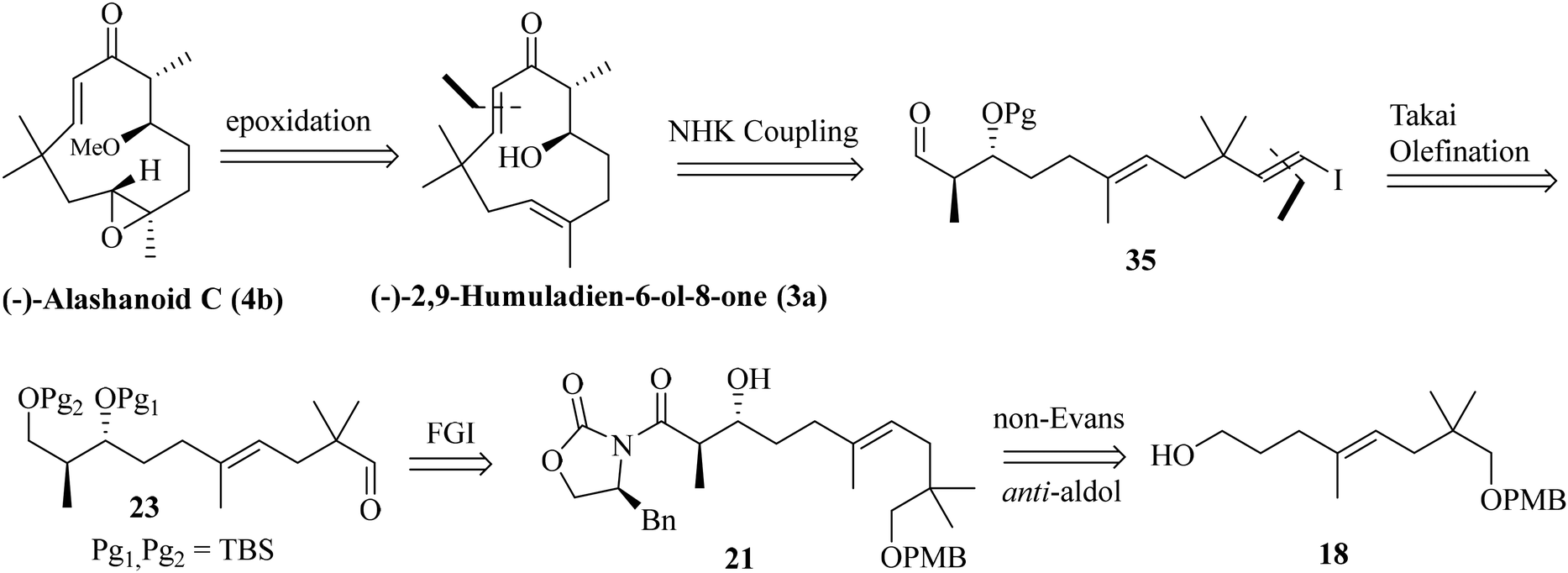
As both the RCM and intramolecular aldol routes did not help us our pursuit of the humulane core of the target natural products, we decided to explore the feasibility of an intramolecular NHK coupling with a suitably substituted E-vinylic iodide-aldehyde.16 A revised retrosynthetic disconnection is depicted in Scheme 5. As shown earlier, the required stereocenters were constructed through the non-Evans anti-aldol pathway (Scheme 5).
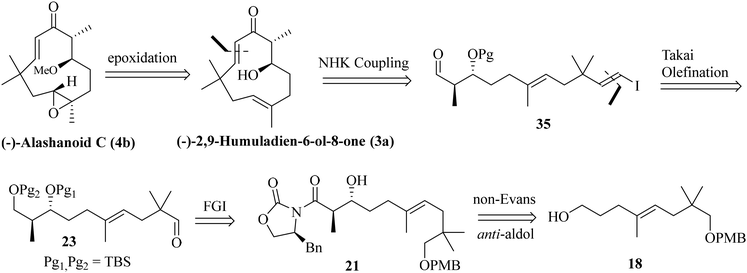 |
| | Scheme 5 A revised retrosynthetic route towards the humulane core through intramolecular NHK coupling. | |
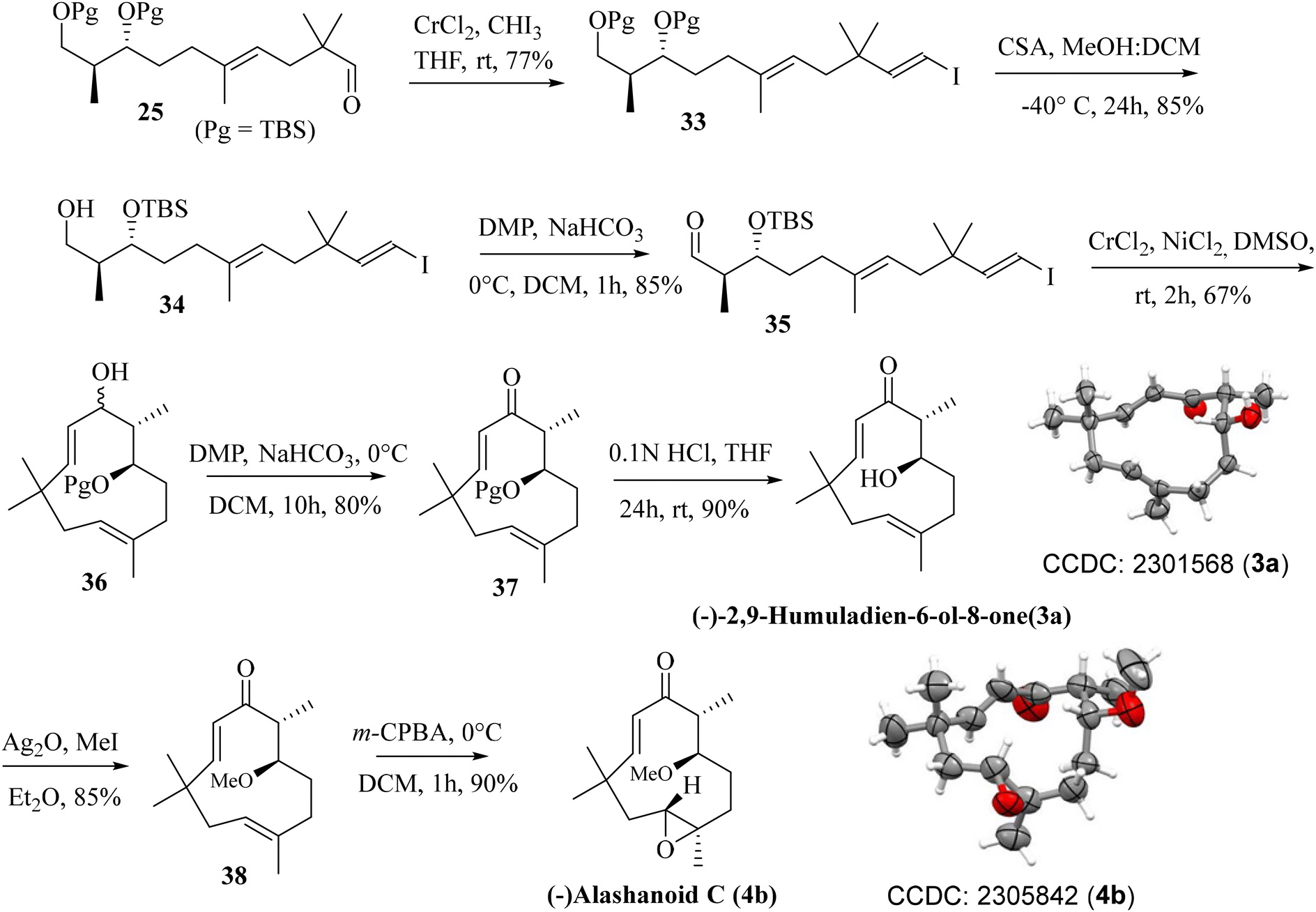
Previously synthesized aldehyde 25 was reacted with CrCl2/CHI3 under Takai olefination conditions17 to access the E-vinylic iodide 33 in a good yield and with excellent diastereoselectivity. Chemoselective deprotection of the primary TBS-ether group was next achieved by treating 33 with CSA/MeOH at −40 °C for 24 h to afford compound 34 in 85% yield. Oxidation of the primary hydroxy group in 34 was next achieved by treatment with DMP18 to afford the desired aldehyde 35 in 85% yield. The crucial intramolecular NHK coupling worked smoothly, and the ring-closed alcohol 36 (humulane framework) was obtained in a pleasing 67% yield. Subsequent oxidation of 36 under DMP conditions then afforded ketone 37 in 80% yield. Finally, removal of the –TBS group from 37 with 0.1 N HCl furnished (−)-2,9-humuladien-6-ol-8-one (yield 90%, overall yield 4.3% from neo-pentyl glycol in 18 steps). The secondary hydroxy group in 3a reacted with Ag2O and MeI19 to furnish the corresponding methoxy derivative of compound 3a. Chemoselective epoxidation of 32 with mCPBA20 then furnished (−)-alashanoid C (4b) (overall yield 3.3% from neo-pentyl glycol in 20 steps; Scheme 6). The spectroscopic (1H, 13C, 2D-NMR) and optical rotation values of the synthesized sample match well with those of the isolated natural product (see the ESI† for detailed information). The ECD spectra of the synthesized (−)-2,9-humuladien-6-ol-8-one (3a) and (−)-alashanoid C (4b) match perfectly with those of natural samples, as reported.3 The experimental ECD curve of 4b exhibited a positive Cotton effect (CE) at 235 nm and a negative CE at 308 nm, which match well with the reported ECD spectrum for the natural product. Hence, the absolute configuration of both the synthesized natural products has been reconfirmed as (6R,7R) for 3a and (2R,3R,6R,7R) for 4b. Single crystal X-ray analysis for 3a and 4b also revealed the structural integrity of both the natural products, as shown in Scheme 6.
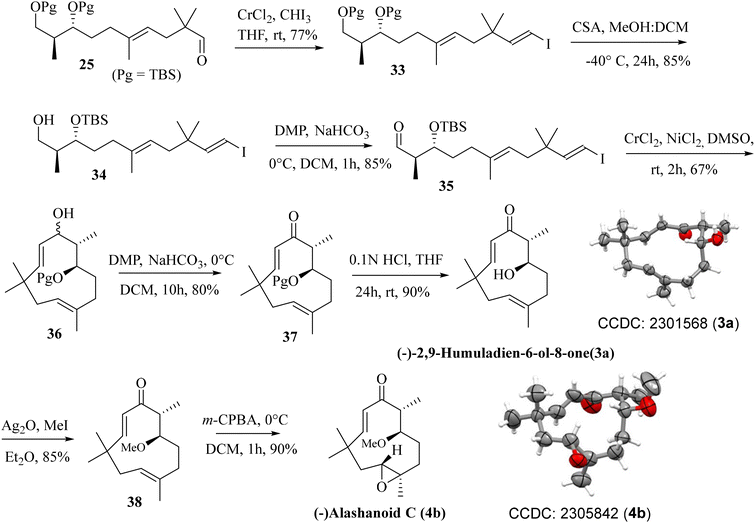 |
| | Scheme 6 Completion of the total synthesis of (−)-2,9-humuladien-6-ol-8-one and (−)-alashanoid C. | |
Total synthesis of alashanoids E and F
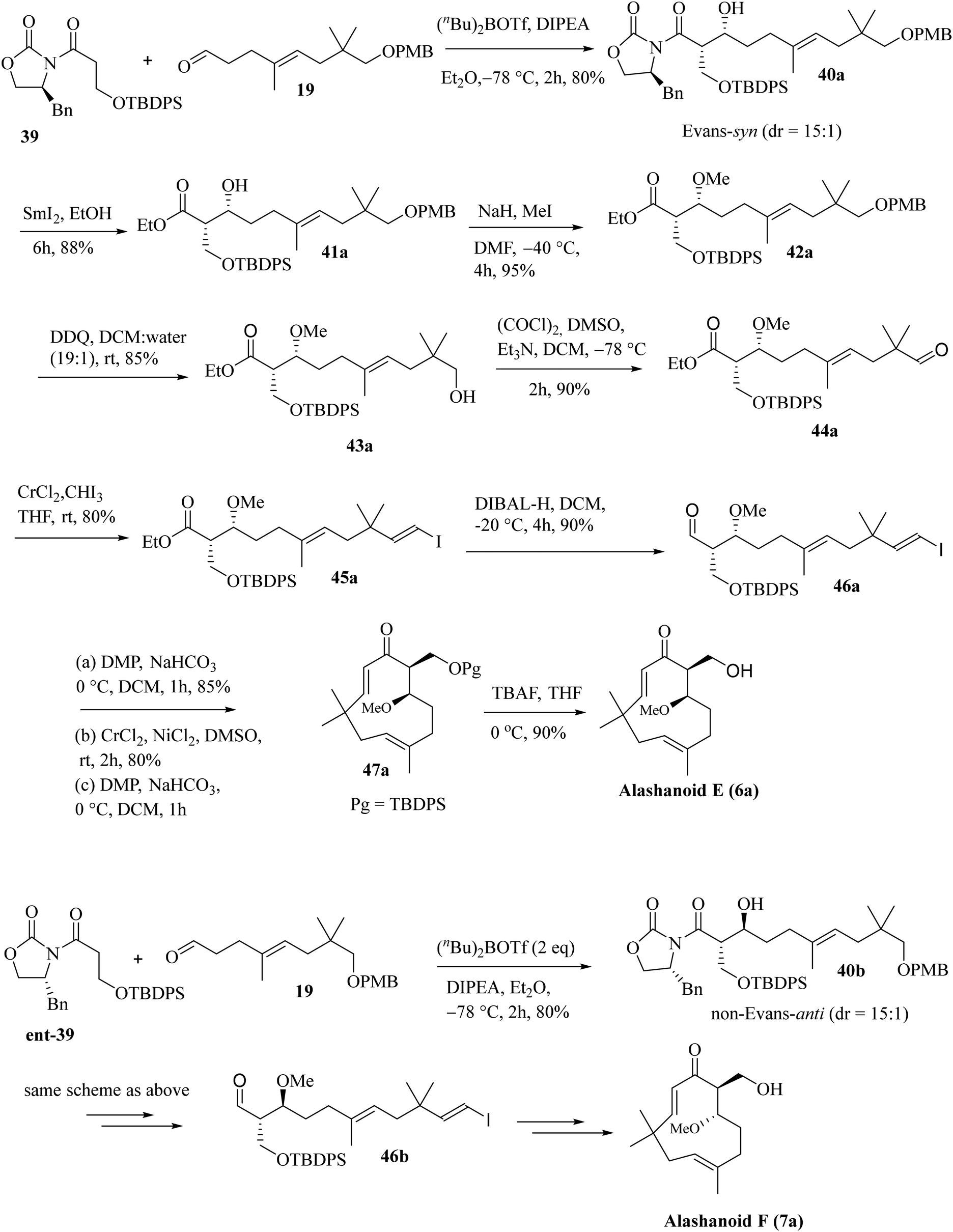
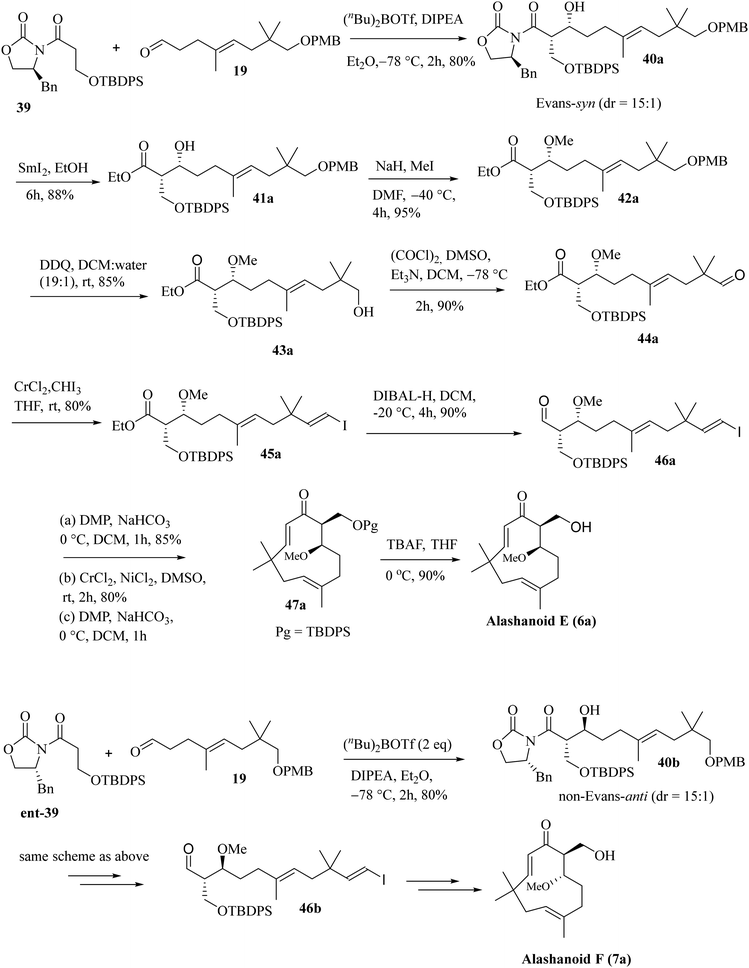
Next, we have focused on the total synthesis of alashanoids E and F. As the late-stage intramolecular NHK coupling reaction successfully constructed the humulane framework, we also intended to explore the same strategy here. Hence, the known aldehyde 19, when subjected to an asymmetric aldol reaction with auxiliary 39 (see the ESI† for its synthesis), afforded the Evans-syn-aldol21 product 40a in a reasonable yield (80%) with excellent diastereoselectivity (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The oxazolidinone auxiliary was removed on treatment with SmI2 and EtOH22 and under hydrolytic conditions afforded compound 41a in 88% yield. Treatment of compound 41a with excess NaH/MeI installed the –OMe group present at the C6 of alashanoids E and F to furnish 42a in 95% yield. Oxidative removal of the –PMB group with DDQ in a DCM buffer solution afforded alcohol 43a in 85% yield, which on Swern oxidation produced the corresponding aldehyde 44a in 90% yield. Stereoselective Takai olefination of aldehyde 44a with CrCl2/CHI3 delivered the corresponding E-vinylic iodide 45a in 80% yield. Reduction of the ester functionality with DIBAL-H furnished the corresponding alcohol 46a in 90% yield. The alcohol on oxidation under DMP conditions yielded the corresponding aldehyde. The intramolecular NHK coupling with CrCl2/NiCl2 (similar conditions as depicted earlier) of the resulting aldehyde afforded the ring closed product (humulane scaffold) as a diastereomeric mixture. Oxidation under DMP conditions furnished the corresponding ketone 47a in 80% yield. Deslilylation23 of 47a with TBAF, AcOH in THF at 0 °C afforded the desired target product alashanoid E (6a) (90% yield; overall yield 7% from neo-pentyl glycol in 18 steps; Scheme 7).
:1). The oxazolidinone auxiliary was removed on treatment with SmI2 and EtOH22 and under hydrolytic conditions afforded compound 41a in 88% yield. Treatment of compound 41a with excess NaH/MeI installed the –OMe group present at the C6 of alashanoids E and F to furnish 42a in 95% yield. Oxidative removal of the –PMB group with DDQ in a DCM buffer solution afforded alcohol 43a in 85% yield, which on Swern oxidation produced the corresponding aldehyde 44a in 90% yield. Stereoselective Takai olefination of aldehyde 44a with CrCl2/CHI3 delivered the corresponding E-vinylic iodide 45a in 80% yield. Reduction of the ester functionality with DIBAL-H furnished the corresponding alcohol 46a in 90% yield. The alcohol on oxidation under DMP conditions yielded the corresponding aldehyde. The intramolecular NHK coupling with CrCl2/NiCl2 (similar conditions as depicted earlier) of the resulting aldehyde afforded the ring closed product (humulane scaffold) as a diastereomeric mixture. Oxidation under DMP conditions furnished the corresponding ketone 47a in 80% yield. Deslilylation23 of 47a with TBAF, AcOH in THF at 0 °C afforded the desired target product alashanoid E (6a) (90% yield; overall yield 7% from neo-pentyl glycol in 18 steps; Scheme 7).
 |
| | Scheme 7 Total synthesis of alashanoids E and F. | |
The synthesis of another naturally occurring compound, alashanoid F (7a) (structurally similar to alasahnoid E, only differing in the absolute configuration at C6 and C7), was initiated with Evans auxiliary ent-35. Asymmetric aldol reaction through a non-Evans-anti selective TS model6 furnished the aldol adduct 40b in good yield (70%) and diastereoselectivity (19:1). The rest of the synthesis of alashanoid F is precisely similar to that of alashanoid E (Scheme 7). The spectroscopic (1H, 13C, and 2D-NMR) and optical rotation values of the synthesized sample match well with those of the isolated natural product (see the ESI† for detailed information). As reported, the recorded ECD spectra of the synthesized (+)-alashanoids E and F match perfectly with those of the natural samples.3,27 Hence, the absolute configuration of the synthesized (+)-alashanoid E has been reconfirmed as 6R,7S and that of (+)-alashanoid F as 6S,7S, as reported for the natural samples. HPLC chromatogram also unambiguously proves that both the synthesized compounds are enantiopure (see the ESI† for HPLC traces).
Total synthesis of (+)-alashanoid B
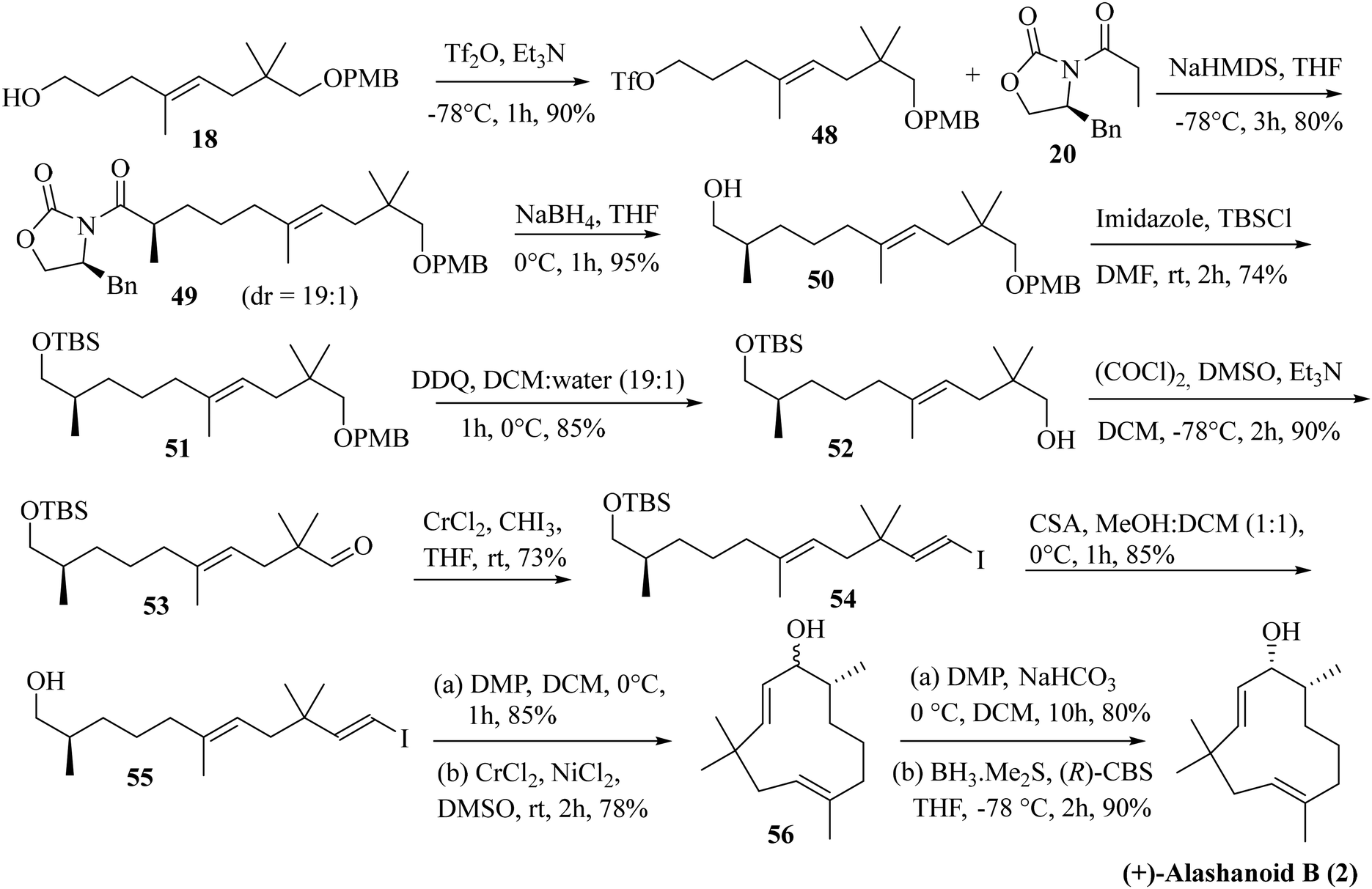
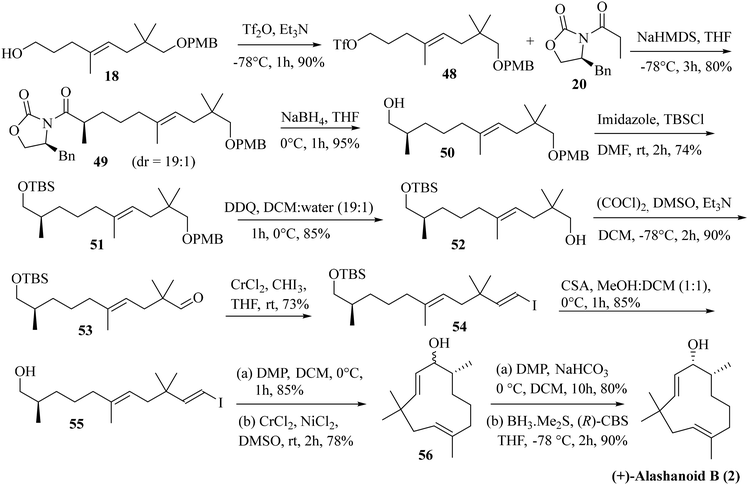
Next, we focused our attention on the enantioselective total synthesis of another structurally related humulane sesquiterpenoid alashanoid B. Close structural analysis revealed that alashanoid B has structural similarity with (−)-2,9-humuladien-6-ol-8-one (the difference being the presence of the –OH functionality at C8 and the absence of the –OMe group at C6). We have started our synthetic venture from the previously synthesized alcohol 18. The hydroxy group in compound 18 was converted to its triflate by treatment with Tf2O and Et3N. Asymmetric alkylation by employing an Evans oxazolidinone-based auxiliary (20) was next attempted with NHMDS as a base.24 The alkylated product 49 was obtained with good yield (80%) and excellent diastereoselectivity. Reductive elimination of the chiral auxiliary with NaBH4 then furnished alcohol 50 in 95% yield. Protection of the hydroxy group in 50 as its –TBS ether provided compound 51 in 74% yield. Removal of the –PMB group in 51 was next achieved by treatment with DDQ to afford alcohol 52 in 85% yield. The Swern oxidation of 52 afforded aldehyde 53, which was immediately subjected to Takai olefination to furnish the vinylic iodide 54 in 73% yield (exclusively E isomer was obtained). Removal of the –TBS group in 54 was achieved by treatment with CSA to furnish compound 55 in 85% yield. Oxidation of 55 with DMP followed by intramolecular NHK coupling of the resulting aldehyde proceeded smoothly as anticipated and yielded compound 56 in 66% yield (in two steps; dr is not determined). Oxidation of 56 under DMP conditions produced the cyclic ketone with the required humulane framework present in the target alashanoid B. Stereoselective reduction of the carbonyl group with (R)-CBS25 furnished (+)-alashanoid B (2) as a single diastereomer (95% yield; 6.4% overall yield from neo-pentyl glycol in 18 steps, Scheme 8). The spectroscopic (1H, 13C, and 2D-NMR) and optical rotation values of the synthesized sample match well with the isolated natural product (see the ESI† for detailed information). The absolute configuration of the C8 stereocenter in alashanoid B was further confirmed through the Mosher ester analysis26 (see the ESI† for details). The recorded ECD spectrum of the synthesized (+)-alashanoid B cannot be compared with those of natural samples as it was not reported in the literature.3,27 The optical rotation value confirms the absolute configuration of the synthesized alashanoid B as 7R,8R, the same as those reported for the natural samples.
 |
| | Scheme 8 Total synthesis of (+)-alashanoid B. | |
Conclusion
In conclusion, we have completed the first asymmetric synthesis of a few naturally occurring humulane-type sesquiterpenoids, alashanoids B, C, E, and F, and (−)-2,9-humuladien-6-ol-8-one through a late-stage intramolecular NHK coupling as a key reaction from simple building blocks. The non-Evans-anti aldol reaction was successfully explored to construct the required stereocenter present in (−)-2,9-humuladien-6-ol-8-one and then a stereoselective epoxidation completes the total synthesis of alashanoid C. A divergent and flexible asymmetric aldol reaction was adopted by employing an Evans oxazolidinone-based auxiliary through two independent pathways to access alashanoids E (Evans-syn) and F (non-Evans-anti). For alashanoid B, Evans oxazolidinone-based asymmetric enolate alkylation was used to construct the C7 stereocenter, and finally, stereoselective reduction through the CBS protocol installed the desired C8 stereocenter present in the target natural product. Further studies towards the total synthesis of the remaining humulane-based sesquiterpenoids in the series (Fig. 1) are underway in our laboratory.
Experimental section
Detailed experimental procedures and characterization data for all the synthesized compounds are provided in the ESI.† The characterization data of the final compounds are given in the following sections.
(−)-2,9-Humuladien-6-ol-8-one (3a)
[α]25D = −97.8 (c = 0.1, MeOH). 1H NMR (500 MHz, CDCl3) δ 6.15 (d, J = 16.0 Hz, 1H), 6.10 (d, J = 16.0 Hz, 1H), 5.08 (dd, J = 11.0, 3.5 Hz, 1H), 4.22 (m, 1H), 2.90 (m, 1H), 2.21 (t, J = 12.0 Hz, 1H), 2.07 (dd, J = 12.5, 3.5 Hz, 1H), 1.95 (t, J = 12.0 Hz, 1H), 1.86 (dd, J = 13.0, 3.5 Hz, 1H), 1.37 (s, 3H), 1.30 (m, 1H), 1.16 (s, 3H), 1.11 (m, 1H), 1.12 (s, 3H), 1.03 (d, J = 6.5 Hz, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 201.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 152.0 (C-10), 137.8 (C-3), 128.1 (C-9), 122.2 (C-2), 73.0 (C-6), 54.3 (C-7), 41.3 (C-1), 40.0 (C-11), 37.7 (C-4), 30.7 (C-5), 28.9 (C-15), 23.0 (C-14), 16.4 (C-12), 6.1 (C-13). IR: 3480, 2965, 2936, 2872, 2856, 1669, 1619, 1453, 1387, 1302, 1083, 1049, 1007, 999, 988 cm−1.
O), 152.0 (C-10), 137.8 (C-3), 128.1 (C-9), 122.2 (C-2), 73.0 (C-6), 54.3 (C-7), 41.3 (C-1), 40.0 (C-11), 37.7 (C-4), 30.7 (C-5), 28.9 (C-15), 23.0 (C-14), 16.4 (C-12), 6.1 (C-13). IR: 3480, 2965, 2936, 2872, 2856, 1669, 1619, 1453, 1387, 1302, 1083, 1049, 1007, 999, 988 cm−1.
HRMS (ESI) m/z: for C15H25O2 [M + H]+, calculated: 237.1855; found: 237.1853.
(−)-Alashanoid C (4b)
[α]25D = −140.2 (c = 0.1, MeOH). 1H NMR (500 MHz, CDCl3) δ 6.29 (s, 2H), 3.59 (dd, J = 8.5, 3.5 Hz, 1H), 3.38 (s, 3H), 3.10 (m, 1H), 2.71 (d, J = 11.0 Hz, 1H), 2.14 (dd, J = 14.0, 8.5 Hz, 1H), 1.92 (d, J = 14.0 Hz, 1H), 1.46 (m, 1H), 1.38 (dd, J = 14.0, 11.5 Hz, 1H), 1.28 (s, 3H), 1.16 (s, 3H), 1.10 (s, 3H), 1.06 (m, 1H), 0.97 (d, J = 6.5 Hz, 3H), 0.96 (overlap, 1H). 13C NMR {1H} (125 MHz, CDCl3) δ 202.3 (CO), 150.8 (C-10), 128.0 (C-9), 83.6 (C-6), 61.9 (C-3), 60.8 (C-2), 57.2 (C-16), 48.2 (C-7), 40.5 (C-1), 37.4 (C-4), 36.4 (C-11), 29.7 (C-15), 25.8 (C-5), 23.5 (C-14), 16.8 (C-12), 6.0 (C-13). IR: 2963, 2940, 2878, 1693, 1631, 1455, 1388, 1372, 1303, 1269, 1232, 1094, 1060, 1037, 1024, 1001, 983, 967, 913, 894, 677 cm−1. HRMS (ESI) m/z: for C16H27O3 [M + H]+, calculated: 267.1960; found: 267.1962.
(+)-Alashanoid E (6a)
[α]25D = +19.0 (c = 0.1, MeOH). 1H NMR (500 MHz, CDCl3) δ 6.23 (d, J = 16.0 Hz, 1H), 6.05 (d, J = 16.0 Hz, 1H), 5.08 (dd, J = 11.5, 4.0 Hz, 1H), 3.98 (dd, J = 12.0, 8.0 Hz, 1H), 3.77 (dd, J = 13.5, 4.5 Hz, 1H), 3.72 (dd, J = 5.5, 4.0 Hz, 1H), 3.45 (s, 3H), 3.26 (m, 1H), 2.22 (t, J = 14.0 Hz, 1H), 2.12 (dd, J = 14.0, 7.0 Hz, 1H), 1.86 (overlap, 2H), 1.48 (m, 1H), 1.37 (s, 3H), 1.21 (overlap, 1H), 1.18 (s, 3H), 1.14 (s, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 201.0 (CO), 152.9 (C-), 138.1 (C-3), 127.8 (C-9), 122.2 (C-2), 82.3 (C-6), 58.1 (C-13), 57.6 (C-7), 56.6 (C-16), 41.3 (C-1), 40.3 (C-11), 39.0 (C-4), 29.0 (C-15), 23.1 (C-14), 16.4 (C-12). IR: 3464, 2955, 2929, 2855, 1690, 1658, 1625, 1456, 1386, 1367, 1303, 1215, 1105, 1081, 1033, 1000 cm−1. HRMS (ESI) m/z: for C16H27O3 [M + H]+, calculated: 267.1960; found: 267.1961.
(+)-Alashanoid F (7a)
[α]25D = +18.4 (c = 0.1, MeOH). 1H NMR (500 MHz, CDCl3) δ 6.25 (d, J = 16.5 Hz, 1H), 5.92 (d, J = 16.5 Hz, 1H), 5.17 (dd, J = 11.5, 4.5 Hz, 1H), 4.02 (dd, J = 11.5, 7.0 Hz, 1H), 3.88 (dd, J = 12.0, 5.5 Hz, 1H), 3.54 (m, 1H), 3.30 (s, 3H), 2.82 (dd, J = 12.0, 6.0 Hz, 1H), 2.33–2.21 (overlap, 2H), 2.00 (m, 1H), 1.92 (dd, J = 13.5, 4.5 Hz, 1H), 1.75–1.72 (m, 2H), 1.56 (s, 3H), 1.17 (s, 3H), 1.16 (s, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 203.2 (CO), 155.5 (C-10), 137.0 (C-3), 126.3 (C-9), 123.7 (C-2), 82.4 (C-6), 60.8 (C-7), 60.4 (C-13), 57.5 (C-16), 41.6 (C-1), 39.8 (C-4), 39.2 (C-11), 32.5 (C-5), 28.9 (C-15), 23.8 (C-14), 16.5 (C-12). IR: 3437, 2957, 2932, 2872, 2855, 1686, 1621, 1461, 1386, 1366, 1216, 1175, 1105, 1083, 1059, 1001, 754 cm−1. HRMS (ESI) m/z: for C16H27O3 [M + H]+, calculated: 267.1960; found: 267.1964.
(+)-Alashanoid B (2)
[α]25D = +51.3 (c = 0.1, MeOH). 1H NMR (500 MHz, CDCl3) δ 5.35 (dd, J = 16.0, 6.0 Hz, 1H), 5.17 (d, J = 16.5 Hz, 1H), 5.05 (dd, J = 8.5, 7.0 Hz, 1H), 4.13 (d, J = 5.0 Hz, 1H), 2.10 (m, 1H), 1.99 (dd, J = 12.5, 10.5 Hz, 1H), 1.86 (dd, J = 3.0, 12.0 Hz, 1H), 1.81 (overlap, 1H), 1.62 (m, 1H), 1.50 (s, 3H), 1.42 (m, 1H), 1.24, 1.20 (overlap, 2H), 1.10 (s, 3H), 1.08 (s, 3H), 1.04 (m, 1H), 0.99 (d, J = 7.0 Hz, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 137.4 (C-10), 134.3 (C-3), 131.3 (C-9), 124.9 (C-2), 76.6 (C-8), 42.4 (C-7), 40.9 (C-4), 40.7 (C-1), 37.6 (C-11), 29.1 (C-15), 26.5 (C-6), 25.1 (C-14), 23.7 (C-5), 17.7 (C-13), 16.2 (C-12). IR: 3422, 2929, 2858, 1687, 1516, 1451, 1385, 1364, 1270, 1254, 1215, 1180, 1165, 1103, 1079, 1058, 997, 971, 751, 719, 697, 668 cm−1.
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. SN designed the project. RB did all the experimental work reported in this manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
Financial support from IIT-Kharagpur (SGIRG grant) is gratefully acknowledged. The authors are thankful to DST (India) for providing a research fellowship to one of the authors, RB. IIT-Kharagpur is acknowledged for providing all the instrumental facilities needed for this research.
References
- G. B. Zhang, L. J. Liu, M. K. Wang and Y. Z. Chen, Chin. Pharm. J., 1994, 29, 271–273 CAS.
- Y. Yan, W. L. J. Ao, X. J. Zhao, X. C. Ye, C. Zhang, J. J. Hao, J. J. He, X. Zhu, H. B. Xu and X. L. Yang, J. Ethnopharmacol., 2010, 131, 248–255 CrossRef CAS PubMed.
- R. Zhang, X. Feng, G. Su, Z. Mu, H. Zhang, Y. Zhao, S. Jiao, L. Cao, S. Chen, P. Tu and X. Chai, J. Nat. Prod., 2018, 81, 1711–1720 CrossRef CAS PubMed.
- J. Han, X. Liu, J. Zhao, S. Li and C. Li, Tetrahedron, 2017, 73, 3289–3303 CrossRef CAS.
-
(a) G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 3800–3801 CrossRef CAS;
(b) G. C. Fu, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 9856–9857 CrossRef CAS;
(c) G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 7324–7325 CrossRef CAS;
(d) G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 5426–5542 CrossRef CAS.
-
(a) M. A. Walker and C. H. Heathcock, J. Org. Chem., 1991, 56, 5747–5750 CrossRef CAS;
(b) J. Willwacher, N. K. Busies and A. Furstner, Angew. Chem., Int. Ed., 2012, 51, 12041–12046 CrossRef CAS PubMed;
(c) B. C. Raimundo and C. H. Heathcock, Synlett, 1995, 12, 1213–1214 CrossRef;
(d) D. A. Evans, J. M. Takacs, L. R. McGee, M. D. Ennis, D. J. Mathre and J. Bartroli, Pure Appl. Chem., 1981, 53, 1109–1127 CrossRef CAS.
-
(a) W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T. T. Li, D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741–743 CrossRef CAS;
(b) R. A. Fernandes, A. K. Chowdhury and P. Kattanguru, Eur. J. Org. Chem., 2014, 2833–2871 CAS.
-
(a) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed;
(b) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed;
(c) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef CAS;
(d) R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS;
(e) T. A. Kirkland and R. H. Grubbs, J. Org. Chem., 1997, 62, 7310–7318 CrossRef CAS PubMed.
-
(a) K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef CAS;
(b) A. J. Mancuso, S. L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480–2482 CrossRef CAS;
(c) T. Trieselmann, R. W. Hoffmann and K. Menzel, Eur. J. Org. Chem., 2002, 1292–1304 CrossRef CAS.
-
(a) A. Maercker, Org. React., 1965, 14, 270–490 CAS;
(b)
W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, UK, 1971, pp. 81–90. ISBN: 0-521-31117-9 Search PubMed;
(c) R. W. Hoffmann, Angew. Chem., Int. Ed., 2001, 40, 1411–1416 CrossRef CAS.
- A. E. Finholt, A. C. Bond
Jr. and H. I. Schlesinger, J. Am. Chem. Soc., 1947, 69, 1199–1203 CrossRef CAS.
-
(a) M. A. Walker and C. H. Heathcock, J. Org. Chem., 1991, 56, 5747–5750 CrossRef CAS;
(b) D. Sanchez, T. Andreou, A. M. Costa, K. G. Meyer, D. R. Williams, I. Barasoain, J. F. Diaz, D. L. Agell and J. Vilarrasa, J. Org. Chem., 2015, 80, 8511–8519 CrossRef CAS PubMed.
- S. W. Roberts and J. D. Rainier, Org. Lett., 2007, 9, 2227–2230 CrossRef CAS PubMed.
-
(a)
C. H. Heathcock, in The aldol reaction: Acid and general base catalysis, ed. B. M. Trost and I. Fleming, Comprehensive Organic Synthesis, 1991, vol. 2, pp. 133–179 Search PubMed;
(b) T. Wehlage, A. Krebs and T. Link, Tetrahedron Lett., 1990, 31, 6625–6628 CrossRef CAS;
(c) K. Toshima, K. Ohta, K. Yanagawa, T. Kano, M. Nakata, M. Kinoshita and S. Matsumura, J. Am. Chem. Soc., 1995, 117, 10825–10831 CrossRef CAS.
- T. Motozaki, K. Sawamura, A. Suzuki, K. Yoshida, T. Ueki, A. Ohara, R. Munakata, K. I. Takao and K. I. Tadano, Org. Lett., 2005, 7, 2265–2267 CrossRef CAS PubMed.
-
(a) Y. Okude, S. Hirano, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1977, 99, 3179–3181 CrossRef CAS;
(b) D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 1995, 117, 10391–10392 CrossRef CAS.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408–7410 CrossRef CAS.
-
(a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS;
(b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- J. N. Moorthy, S. Samanta, A. L. Koner, S. Saha and W. M. Nau, J. Am. Chem. Soc., 2008, 130, 13608–13617 CrossRef CAS PubMed.
- K. I. Takao, H. Kai, A. Yamada, Y. Fukushima, D. Komatsu, A. Ogura and K. Yoshida, Angew. Chem., Int. Ed., 2019, 58, 9851–9855 CrossRef CAS PubMed.
-
(a) D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS;
(b) C. Steinborn, R. E. Wildermuth, D. M. Barber and T. Magauer, Angew. Chem., Int. Ed., 2020, 59, 17282–17285 CrossRef CAS PubMed;
(c) M. H. Sahana, D. Saha and R. K. Goswami, J. Org. Chem., 2022, 87, 11805–11815 CrossRef CAS PubMed.
- C. M. Bouvier, I. Reboule, R. Gill and J. Collin, Synlett, 2008, 1211–1215 Search PubMed.
- S. Das, A. Panda and S. Pal, Carbohydr. Res., 2015, 416, 24–31 CrossRef CAS PubMed.
- G. Hofle, K. Gerth, H. Reichenbach, B. Kunze, F. Sasse, E. Forche and E. V. Prusov, Chem. – Eur. J., 2012, 18, 11362–11370 CrossRef PubMed.
-
(a) E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS;
(b) E. J. Corey, R. K. Bakshi, S. Shibata, C. P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925–7926 CrossRef CAS.
-
(a) J. A. Dale, D. L. Dull and H. S. Mosher, J. Org. Chem., 1969, 34, 2543–2549 CrossRef CAS;
(b) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS;
(c) D. L. Dull and H. S. Mosher, J. Am. Chem. Soc., 1967, 89, 4230 CrossRef CAS.
- The ECD spectrum of the (R)-MTPA ester of (+)-alashanoid B was recorded and it showed a positive Cotton effect, confirming its enantiopurity. HPLC analysis also confirms the enantiopurity (see the ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for all the synthesized compounds, spectral data (1H, 13C, and 2D NMR), ECD curve, and X-ray crystallographic data. CCDC 2301568 and 2305842. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00393d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*