
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFriedel–Crafts reactions for biomolecular chemistry†
Jun
Ohata

Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, USA. E-mail: johata@ncsu.edu
First published on 9th April 2024
Abstract
Chemical tools and principles have become central to biological and medical research/applications by leveraging a range of classical organic chemistry reactions. Friedel–Crafts alkylation and acylation are arguably some of the most well-known and used synthetic methods for the preparation of small molecules but their use in biological and medical fields is relatively less frequent than the other reactions, possibly owing to the notion of their plausible incompatibility with biological systems. This review demonstrates advances in Friedel–Crafts alkylation and acylation reactions in a variety of biomolecular chemistry fields. With the discoveries and applications of numerous biomolecule-catalyzed or -assisted processes, these reactions have garnered considerable interest in biochemistry, enzymology, and biocatalysis. Despite the challenges of reactivity and selectivity of biomolecular reactions, the alkylation and acylation reactions demonstrated their utility for the construction and functionalization of all the four major biomolecules (i.e., nucleosides, carbohydrates/saccharides, lipids/fatty acids, and amino acids/peptides/proteins), and their diverse applications in biological, medical, and material fields are discussed. As the alkylation and acylation reactions are often fundamental educational components of organic chemistry courses, this review is intended for both experts and nonexperts by discussing their basic reaction patterns (with the depiction of each reaction mechanism in the ESI†) and relevant real-world impacts in order to enrich chemical research and education. The significant growth of biomolecular Friedel–Crafts reactions described here is a testament to their broad importance and utility, and further development and investigations of the reactions will surely be the focus in the organic biomolecular chemistry fields.
 Jun Ohata | Jun Ohata was born and raised in Japan. He received his BSc and MSc from Osaka Prefecture University, where he worked with Prof. Matsuzaka studying the carbon species in diruthenium complexes. He earned his PhD in the Ball group at Rice University, studying transition metal-catalyzed protein bioconjugation. He conducted his postdoctoral work with Prof. Chang at the University of California–Berkeley as a JSPS postdoctoral fellow, developing chemical probes for the detection of cellular metal ions. He then took up his current position at North Carolina State University as an assistant professor, developing novel strategies for bioconjugation. |
1. Introduction
Organic chemistry reactions are the core element for the advancement of fields at the interface of chemistry and biology, offering control over the bond formation and scission processes of polyfunctional biomolecules. Even if biomolecules and cellular systems differ substantially from small molecule substrates in many ways such as unique three-dimensional structures and the presence of multiple copies of certain functional groups, the principles of reaction development in synthetic organic chemistry are still applicable to reactions for biomolecular studies that address reactivity and selectivity challenges. Indeed, many of the state-of-the-art strategies for studying and utilizing biomolecules are predicated heavily on classical organic chemistry reactions (Scheme 1A).1–12 Many of these reactions follow basic mechanistic patterns often taught in undergraduate-level courses yet enable a wide range of powerful chemical strategies including activity-based profiling/sensing/imaging,13,14 enhancement of drug potency,15 drug delivery,16 and bioorthogonal chemistry/click chemistry.17 In other words, the translation of classical chemical reactions has been one of the major approaches for creating chemical tools for biological and medical applications.18 This review discusses classical organic chemistry reactions, Friedel–Crafts alkylation and acylation in the multifaceted areas of biomolecular chemistry (Scheme 1B).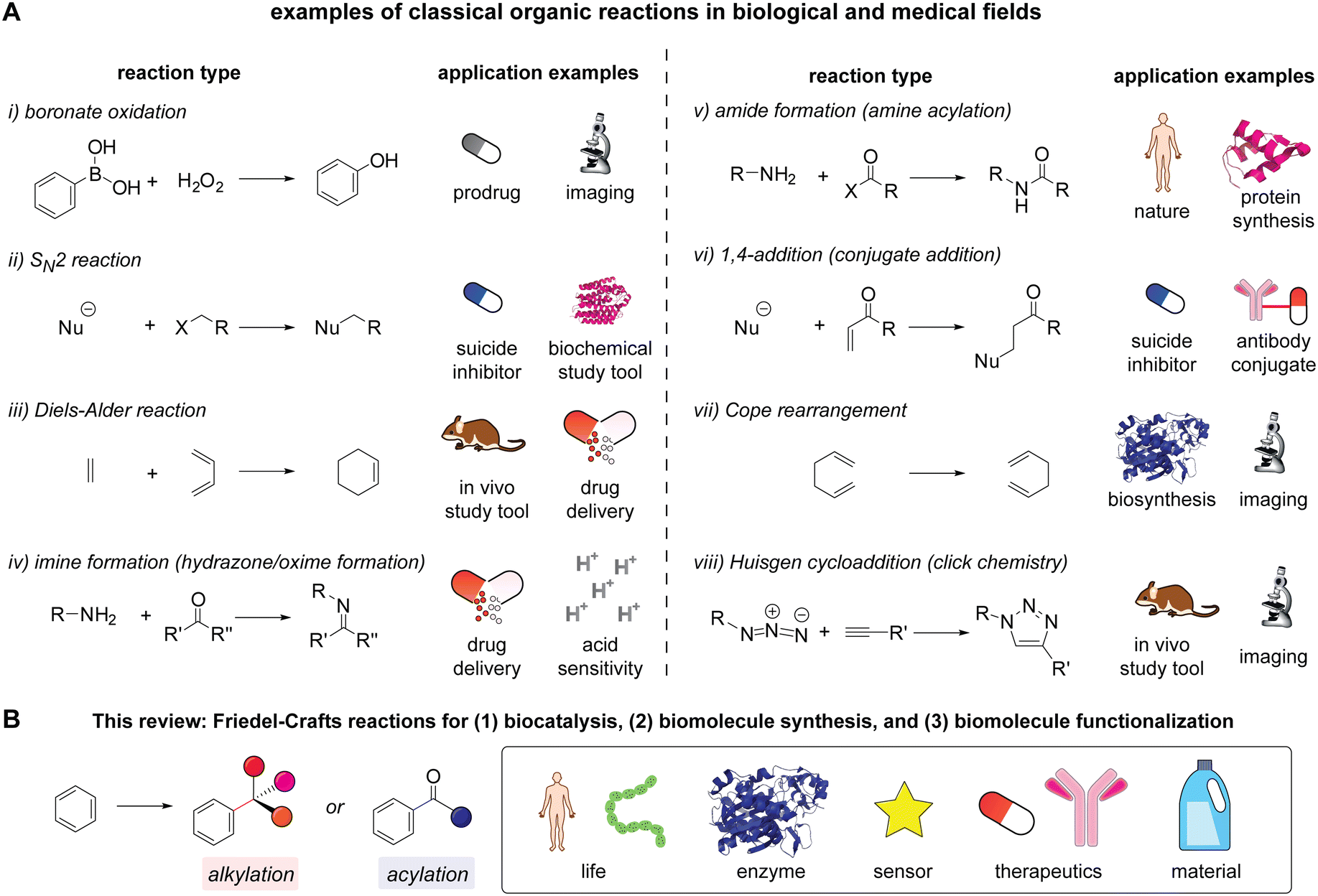 | ||
Scheme 1 Classical organic reactions in biological and medical fields. (A) Examples of the classical organic reactions including: (i) boronate oxidation,1,2 (ii) SN2 reactions with fluoro- and iodoacetamide,3,4 (iii) Diels–Alder reactions for bioorthogonal chemistry,5,6 (iv) imine and relevant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond formation such as hydrazone and oxime,7,8 (v) amide formation such as protein expression and native chemical ligation,9,10 (vi) 1,4-addition to a carbonyl group such as acrylamide and maleimide,11,12 (vii) Cope rearrangement,27–29 and (viii) Huisgen cycloaddition or azide–alkyne cycloaddition.18,30 The organization of the reactions is based on the appearance in an Organic Chemistry textbook (Marc Loudon and Jim Parise, sixth edition). (B) Friedel–Crafts alkylation and acylation for biomolecular chemistry as a theme of this review. N bond formation such as hydrazone and oxime,7,8 (v) amide formation such as protein expression and native chemical ligation,9,10 (vi) 1,4-addition to a carbonyl group such as acrylamide and maleimide,11,12 (vii) Cope rearrangement,27–29 and (viii) Huisgen cycloaddition or azide–alkyne cycloaddition.18,30 The organization of the reactions is based on the appearance in an Organic Chemistry textbook (Marc Loudon and Jim Parise, sixth edition). (B) Friedel–Crafts alkylation and acylation for biomolecular chemistry as a theme of this review. | ||
Friedel–Crafts reactions are alkylation and acylation processes that induce C–C bond formation. The original reaction was discovered by Friedel and Crafts in the late 19th century, as the historical account has been documented in previous literature.19,20 These reactions are often considered one of the basic organic chemistry reactions, as the reactions represent quintessential examples of electrophilic aromatic substitution covered in most of the undergraduate-level textbooks,21 and, therefore, have been an important subject in the chemical education community.22–24 Beyond their educational significance, the chemical industry has been relying on these reactions for the production of numerous carbon-based molecules for over a century.25,26
Diverse reaction types have become possible for Friedel–Crafts reactions through mechanisms different from traditional carbocation and acylium generation (Scheme 2 and Fig. S1†). Their simple and traditional examples involve alkyl or acyl chloride starting materials that lead to the generation of carbocation or acylium ions by aluminum chloride, which eventually react with an aromatic molecule to form a carbon–carbon bond (Scheme 2A). A broader definition of the alkylation and acylation reactions is also based on the process of carbon–carbon bond formation, often catalyzed/mediated by acids but with a wide collection of nucleophiles, electrophiles, and catalysts/additives (Scheme 2B).31 In addition to hetero-aromatic compounds (i.e., aromatic molecules with heteroatoms such as nitrogen, oxygen, and sulfur in the ring system), even nonaromatic alkenes can be considered possible nucleophiles.32 While separation of a distinct leaving group from an electrophile is a hallmark of the traditional reaction pattern, electrophiles such as carbonyl and epoxide derivatives can also be considered Friedel–Crafts reaction reagents. Aluminum chloride remains a common catalyst/additive but many other additives are now known to be able to induce alkylation and acylation reactions including Brønsted acids and enzymes.33 Notably, even additive- or catalyst-free reactions have been realized using fluorinated alcohol as a reaction solvent.34 Unsurprisingly, a number of review articles have been published regarding the growth of these reactions from a synthetic organic chemistry viewpoint.33,35–37
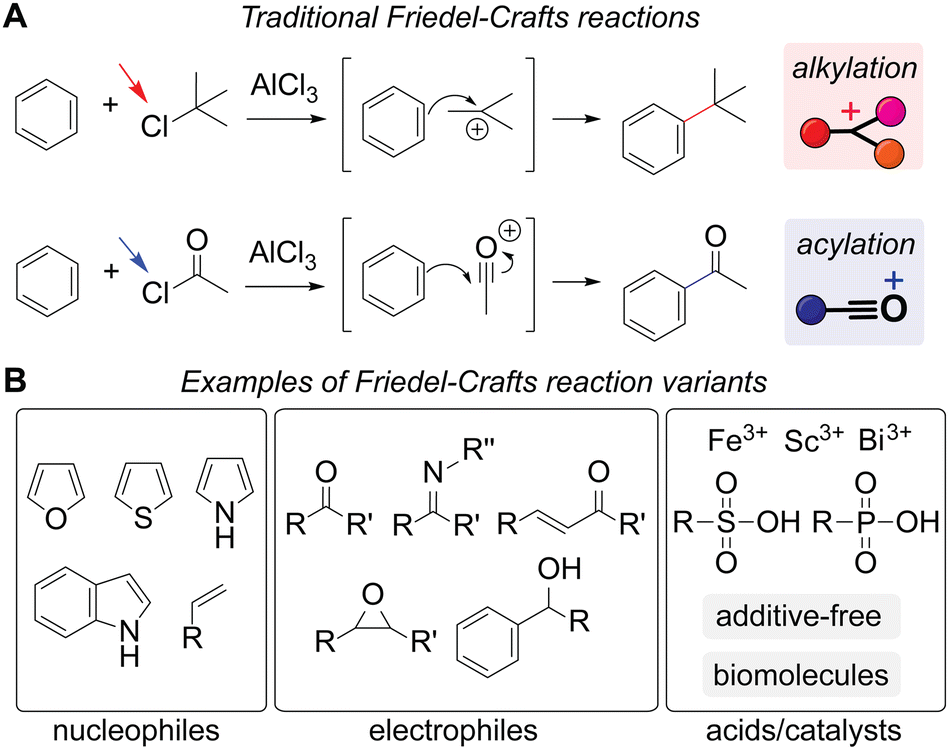 | ||
| Scheme 2 Friedel–Crafts reactions. (A) Traditional alkylation (top) and acylation (bottom) processes with aluminum chloride through carbocation and acylium ion generation, respectively. Even though reactions may not always proceed via such carbocation and acylium intermediates, all the reactions described below in the following sections are labeled as alkylation (red rectangle) and acylation (blue rectangle) as a visual aid for readers. Leaving groups are highlighted with arrows. The mechanisms are shown in Fig. S1 (ESI†). (B) Examples of more broadly defined reaction substrates and catalysts/additives than the traditional versions with alkyl and acyl chlorides with aluminum chloride. | ||
More recently, Friedel–Crafts alkylation and acylation have been increasingly used and studied for biomolecular chemistry. It is interesting that these alkylation and acylation reactions have not been as much studied as the other classical chemistries, perhaps due to the idea of the potential incompatibility of reactive intermediates and strong Lewis acids with biological samples.38 In fact, even though the toxicity of aluminum at high concentration is known,39 aluminum salts have been widely used as vaccine adjuvants since the early 20th century.40 In addition, the alkylation and acylation reactions do not always involve the generation of carbocation or acylium intermediates. These attributes probably enabled the study and development of biomolecular Friedel–Crafts reactions. This review is not intended just for experts in the chemistry fields but also for instructors and students, focusing on the basic reaction patterns and representative applications of each method. Thus, rather than a comprehensive collection of all the previous research publications, only selected examples of notable reactions and applications are discussed. Two major sections of the article are reactions catalyzed/mediated by biomolecules and synthesis/functionalization of biomolecules. Each reaction described below is labeled with alkylation (a red rectangle with the depiction of carbocation species) or acylation (a blue rectangle with the depiction of acylium species) as a visual aid for readers, even though these species are not always the active intermediates of a given reaction. In order to clarify the mechanistic details, arrow-pushing mechanisms of each reaction are shown in the ESI† (available as both pdf and ChemDraw files).
2. Friedel–Crafts reactions mediated by biomolecules (biocatalysis)
Similar to many classical organic chemistry reactions, a class of Friedel–Crafts reactions can also be mediated by enzymes (i.e., proteins) and other biomolecules. An enzyme acts as a catalyst to facilitate a reaction through a variety of mechanisms such as stabilization of reactive intermediates in a unique environment of biomolecule binding pockets.41,42 Examples of such biomolecule-catalyzed reactions include Diels–Alder reactions43 and cyclopropanation.44 Alkylation reactions of nucleotide bases (i.e., cytosine methylation) are also enzyme-mediated processes, and though their formal reaction patterns would appear to be Friedel–Crafts alkylation of a heteroaromatic group, the reactivity enhancement mechanism occurs through the formation of non-aromatic species which would not be considered an electrophilic aromatic substitution reaction (Fig. S2, ESI†).45 A class of enzymes can catalyze Friedel–Crafts reactions through various mechanisms such as activation of electrophiles without the generation of carbocation and acylium intermediates, as the proposed reaction mechanisms of the enzymatic Friedel–Crafts reactions are shown in Fig. S2–S6 (ESI†). Other types of biomolecules such as DNA,46 RNA,47 and carbohydrates (saccharides)48 are also known to catalyze a series of organic reactions. While such biomolecule-catalyzed or -assisted chemical reactions are known in living systems, the utilization of such biomolecule catalysts is also possible for reactions with unnatural reactants, allowing for synthetic applications of the catalytic transformation. There have been comprehensive review articles about such biocatalytic Friedel–Crafts reactions,49,50 and the following sections are representative examples of (1) natural and (2) artificial/unnatural Friedel–Crafts reactions mediated by biomolecules.2.1. Natural Friedel–Crafts reactions in living systems
Enzymatic modification of proteins with carbohydrate groups occurs in mammals and viruses through the Friedel–Crafts alkylation of a tryptophan residue (Scheme 3A). Proteins produced according to genomic sequences often undergo additional modification processes called post-translational modification,51 and this Friedel–Crafts reaction of tryptophan amino acid is such modification occurring through the attachment of a carbohydrate to a protein (known as C-mannosylation or more broadly as glycosylation).52 This alkylation process is found in diverse species including mammals and the Ebola virus and has relevance to several types of diseases and disorders such as developmental disease.53 Indole groups of tryptophan residues (1a) act as nucleophiles with the aid of electron donation from the nitrogen atom in the ring system, and the elimination of phosphate as a leaving group would occur when the carbohydrate electrophile (2a) is nucleophilically attacked, as suggested by a recent structural biology study (Fig. S3, ESI†).54 | ||
| Scheme 3 Friedel–Crafts reactions that occur in living systems catalyzed by a series of enzymes. Leaving groups are highlighted with arrows. Simplified mechanisms with selected amino acid functions of enzymes are shown in Fig. S3–S6 (ESI†). (A) Mannosylation (glycosylation) of proteins through an alkylation of a tryptophan residue with phosphorylated mannose in living organisms such as mammals and the Ebola virus.54 (B) Formation of a cytotoxin cylindrocyclophane (5) in cyanobacteria through intermolecular dimerization by alkylation reactions.56 (C) Formation of the antibiotic 2,4-diacetylphloroglucinol in bacteria Pseudomonas fluorescens.59 (D) Attachment of a fatty acid to an imidazole derivative in bacteria Streptomyces sp. OUCMDZ-944.61 | ||
A metabolite with electron-rich benzene rings and chloroalkanes is known to cause an enzymatic homo-dimerization process, generating a cytotoxin in cyanobacteria (Scheme 3B). Cylindrocyclophane derivatives are cytotoxins, and their biological importance is increasingly studied.55 With two hydroxyl groups and one alkyl group, the aromatic ring would be a great nucleophile that reacts with the secondary alkyl chloride in another molecule. Balskus and co-workers revealed the mechanism of action of enzymatic biosynthesis pathways, which showed activation of the nucleophiles through the deprotonation of the phenolic proton by the aspartate residue of the enzyme during the alkylation reaction (Fig. S4, ESI†).56,57
Plants utilize the formation of the antibiotic 2,4-diacetylphloroglucinol through enzymatic acylation reactions for their disease control against certain bacteria (Scheme 3C). The product of the acylation reaction 2,4-diacetylphloroglucinol (3b) shows antibiotic activity and is of agricultural importance. In this acylation reaction, two identical acetylated aromatic compounds react with each other, where formally one of the molecules acts as a nucleophile and the other as an electrophile/acetyl donor.58,59 Normally, ketone groups would not typically participate in the Friedel–Crafts acylation as an electrophile since the C–C bond would not be easily broken to liberate a leaving group; however, this reaction occurs in the enzymatic pocket activating the acetyl group to generate a thioester group linked to an enzyme (Fig. S5, ESI†). Afterward, nucleophilic attack by the other molecule on the thioester group causes the acylation reaction to produce the bis-acetylated product. Plants utilize certain bacteria (Pseudomonas fluorescens) that can induce the acylation reaction to produce antibiotics controlling the pathogens in soil, which highlights the interplay between plants and the types of bacteria mediated by the acylation reaction.60
Recently, a nucleotide derivative with an aminoimidazole group was found to undergo enzymatic acylation reactions with fatty acids in bacteria during fermentation (Scheme 3D).61 Imidazole derivatives can cause a nucleophilic attack often at the nitrogen atom in the ring.62,63 Likely, for this particular case of the nucleotide derivative, the nucleophilicity of the carbon center in the ring system is increased through electron donation by amine substitution, leading to the C–C bond forming reaction (Fig. S6†). Carboxylic acids are not a typical electrophile of Friedel–Crafts acylation reactions due to the modest leaving capability of the OH group as well as the poor electrophilicity of the deprotonated form of the COOH group, and this reaction proceeds through the activation of the carboxylic acid to its derivative by the action of the enzyme. The acylated imidazole product is further modified by enzymatic reactions to be connected with adenosine triphosphate (ATP), enabling the fermentation process in bacteria.
2.2. Biomolecule-catalyzed or -assisted Friedel–Crafts reactions of unnatural substrates
Inspired by natural reactions occurring in living systems, Friedel–Crafts alkylation and acylation reactions of unnatural reactants have been developed. Although some enzymes tend to show high substrate specificity and may not be able to host unnatural substrates, other classes of enzymes indeed would not recognize a slight structural modification of substrates, and such reactions of unnatural substrates can be facilitated by modulation of enzyme pockets/structures through mutation processes (i.e., change of an amino acid residue). For instance, alkylation reactions of phenol derivatives with unnatural allyl phosphate electrophiles (known as prenyl phosphates with trisubstituted alkene groups) have been realized using the natural enzyme with a slight modification to their amino acid composition (Scheme 4A and Fig. S7, ESI†).64 Similarly, indole derivatives can also be alkylated in an unnatural fashion.65 By employing the natural enzyme mentioned above (Scheme 3C), acylation reactions of aromatic compounds bearing a hydroxyl group(s) with several electrophiles such as esters and acyl-imidazoles have been demonstrated (Scheme 4B and Fig. S5, ESI†).66 While those enzymatic reactions are performed in a test tube with a pure enzyme and substrates, Roelfes and co-workers showed that enzymatic Friedel–Crafts reactions of unnatural substrates in vivo are also feasible.67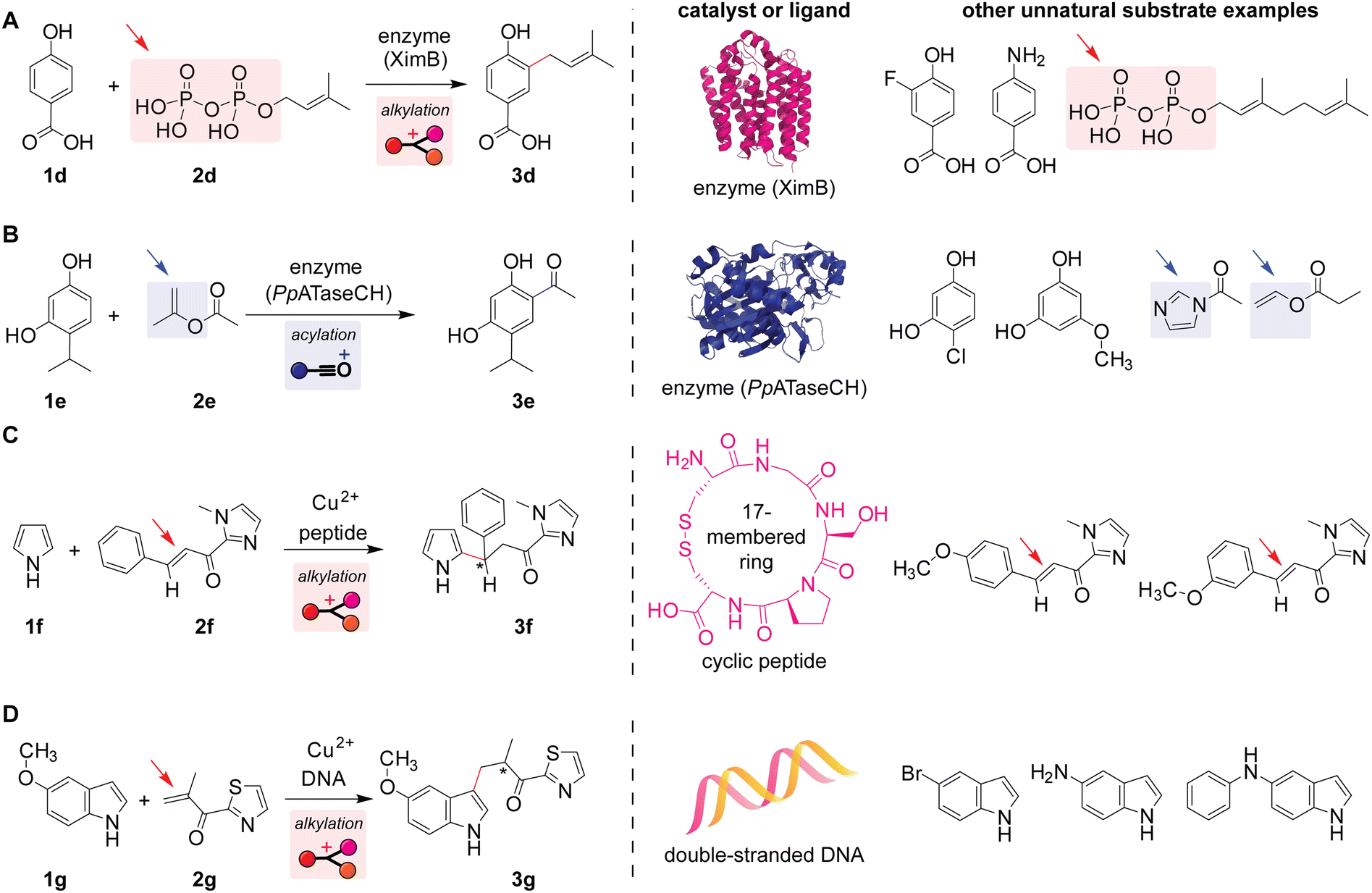 | ||
| Scheme 4 Biomolecule-catalyzed or -assisted Friedel–Crafts reactions of unnatural substrates in test tubes. Leaving groups or bonds that undergo cleavage are highlighted with arrows. Mechanisms of the reactions are shown in Fig. S7, S5, and S8 (ESI†). (A) Enzyme-catalyzed alkylation of benzoic acid derivatives with allylic phosphates.64 The predicted structure of enzyme XimB is obtained from the AlphaFold Protein Structure Database (uniprot code: Q96H96). (B) Enzyme-catalyzed acylation of phenol derivatives with esters.66 The partial structure of enzyme PpATaseCH is obtained from the Protein Data Bank (PDB ID: 5MG5). (C) Copper(II)-catalyzed alkylation reaction of pyrrole with α,β-unsaturated carbonyl compounds assisted by a peptide as a ligand to the metal catalyst.68 (D) Copper(II)-catalyzed alkylation reaction of indole derivatives with α,β-unsaturated carbonyl compounds assisted by double-stranded DNA as a ligand to the metal catalyst.73 | ||
Peptides and DNAs can act as auxiliary ligands for copper-catalyzed alkylation reactions with α,β-unsaturated carbonyl compound electrophiles. The aforementioned enzymatic reactions (i.e., protein-catalyzed reactions) offer high reaction efficiency and chemo-/regio-/enantio-selectivity. Without the large, intricate three-dimensional structures of enzymes, various types of smaller biomolecules such as peptides could also exert enantiocontrol (Scheme 4C) even in the presence of competing aromatic rings (e.g., a substrate with a methoxy-substituted benzene ring shown on the right of Scheme 4C).68,69 Unlike typical Friedel–Crafts reactions that involve activations of electrophiles through the elimination of leaving groups such as halides, α,β-unsaturated carbonyl compounds could act as electrophiles of the alkylation reaction through 1,4-addition, forming a new chiral center at the β position (Fig. S8, ESI†). Such a class of reactions generating a specific enantiomer over the other (i.e., asymmetric reactions) has been an important diversification of Friedel–Crafts alkylations.31,36,70–72 A similar asymmetric transformation is also possible with double-stranded DNA as a ligand that binds to the copper catalyst (Scheme 4D).73
3. Friedel–Crafts reactions for the synthesis and functionalization of biomolecules
Friedel–Crafts alkylation and acylation are commonly used for the synthesis of biomolecules/their analogues as well as the addition of unique functionalities to biomolecules through the formation of a stable carbon–carbon bond. The four basic biomolecules contain components for the Friedel–Crafts alkylation and acylation reactions including aromatic rings in nucleosides and amino acids/peptides/proteins, alkyl groups with a leaving group on carbohydrates, and acyl groups in fatty acids and amino acids/peptides/proteins. As nature takes advantage of these functional groups (e.g., tryptophan, mannose, and fatty acid in Scheme 3), numerous strategies were developed for various purposes of synthesis and functionalization of biomolecules. The following sections discuss the synthesis and functionalization of biomolecules and are organized based on biomolecule types: nucleosides, carbohydrates, fatty acids, and amino acids/peptides/proteins.3.1. Synthesis of nucleoside analogues through Friedel–Crafts alkylation
Friedel–Crafts reactions on the canonical aromatic nucleobases in ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) are not facile processes without protection strategies or enzymatic aids (Scheme 5A). Probably due to the presence of amine groups and the electron-deficient nature of nucleobases, Friedel–Crafts reactions of natural nucleotides are virtually unknown both in nature and for synthetic purposes. As discussed above, frequently occurring methylation of cytosine in nature is not considered an electrophilic aromatic substitution reaction (Fig. S2, ESI†),45 and the methylation of other nucleotides occurs on nitrogen atoms rather than carbon atoms.74 Indeed, the enzymatic acylation reaction of DNA analogues in living systems has not been reported until recently (Scheme 3D).61 Nucleobase acetylation is a well-known natural process among many species but amine acylation (i.e., amide bond formation) is a predominant pathway.75 Friedel–Crafts reactions for the synthesis or functionalization of natural nucleobases/nucleosides/nucleotides have not been reported,76 although the synthesis of RNA analogues through acylation is known (Scheme 5B and Fig. S9, ESI†).77 The challenges of nucleotide synthesis and functionalization may not only be because of the reactivity of amines and nucleobases, but the compatibility of acid catalysts/additives may also be relevant, as binding of nucleobases to metals has been commonly observed including an example of cisplatin, a platinum-based anti-tumor agent.78 Thus, the synthesis of nucleoside analogues often relies on functional group protection as shown below. | ||
| Scheme 5 Friedel–Crafts alkylation reactions for the construction and applications of synthetic nucleoside analogues. Leaving groups are highlighted with arrows. Mechanisms of the reactions are shown in Fig. S9–S11 (ESI†). (A) Chemical structures of natural (left) and synthetic (right) nucleosides. (B) Synthesis of a guanine analogue.77 (C) Synthesis of pyrene-substituted DNA.79 (D) Synthesis of sulfur-containing RNA (thienoguanosine).80 (E) An application of the pyrene-substituted nucleotide for the detection of DNA damage.81 (F) Complementarity of the sulfur-containing synthetic RNA analogue (thienoguanosine) with cytosine through Watson–Crick base pairing.83 (G) An application of sulfur-containing RNA (thienoguanosine) for the detection of a left-handed helical structure over a right-handed helical structure.85 The structural difference of Z-DNA and B-DNA is described in previous review papers.88,89 The images of the cyan fluorophore solutions were reproduced with permission.85 Copyright 2014, The Royal Society of Chemistry. | ||
Synthetic analogues of nucleosides with unique photophysical properties can be prepared through the Friedel–Crafts alkylation reaction with ribose derivatives (Scheme 5C, D and Fig. S9, S10, ESI†).79,80 As nucleosides are constructed with carbohydrate (i.e., ribose or deoxyribose) and nucleobase units, one of the general strategies for the synthesis of nucleoside analogues is to use nucleobase analogues as nucleophiles with carbohydrate electrophiles bearing a leaving group on the anomeric position—that is an acetal carbon in a cyclic ether. The OH and NH2 groups are normally protected as an ether, ester, and imine/amidine, likely to suppress side reactions on those elements and increase the solubility of the starting materials and products in organic solvents for synthetic ease. Protecting groups could enhance the stereoselectivity of the alkylation process as well (e.g., the silyl-Hilbert–Johnson type mechanism), as the actions of the groups during the reactions are depicted in Fig. S9–S11 (ESI†). It is noteworthy that the desired alkylation reactions by fluorescent aromatic groups in those examples occur in the presence of other arene groups, even though some substrates (e.g., pyrene 1i) may not be significantly more electron-rich compared to the alkoxy-substituted benzenes.
Synthetic analogues with fluorescent aromatic rings are useful for visualization of molecular events and behaviours of oligonucleotides such as DNA damage and conformational changes. Conjugated aromatic rings can offer fluorescence capabilities with sensitivity toward changes in local environments. One of the applications of pyrene nucleotides is the detection of DNA damage (Scheme 5E).81 The pyrene groups are known to display fluorescence color changes and increase upon a noncovalent dimerization process known as excimer formation,82 and the design by Kool and co-workers took advantage of the excimer formation when DNA undergoes deletion of a specific nucleotide.81 In addition to pyrene as a fluorescence reporter, thienoguanosine with a sulfur atom in the ring structure is also a fluorescent analogue with Watson–Crick base pairing ability (Scheme 5F).83 Thienoguanosine-based nucleotides possess unique photophysical features84 making the synthetic analogues useful for numerous applications including the detection of DNA conformational changes (Scheme 5F and G)85 and compatibility with gene-editing processes.86 Many other examples of applications of fluorescent nucleotides were described in a review by Kool and co-workers.87
3.2. Functionalization of carbohydrate derivatives through Friedel–Crafts alkylation
With carbon- and oxygen-based scaffolds, carbohydrate (or saccharide) units are often used as electrophiles of Friedel–Crafts alkylation reactions to construct intricate, functional molecules (Scheme 6A). Monomeric carbohydrates typically have no aromatic rings,90 and the presence of the acetal group at the anomeric position enables the generation of cationic intermediates from carbohydrate derivatives. The reaction patterns for the carbohydrate alkylation processes follow those in the DNA section of ribose where carbohydrates act as electrophiles (Scheme 5), and the alcohol groups are often protected in a similar fashion to the ribose system. Many biologically active small-molecule products are composed of carbohydrate groups with useful capabilities,91 and the alkylation reactions of carbohydrates with a leaving group in the acetal position can be a convenient approach. For instance, an intermediate molecule during the total synthesis of saptomycin B (15) with antitumor and antibacterial activities92 was facilitated by the scandium-mediated alkylation process with a carbohydrate group bearing an acetate moiety (Scheme 6B and Fig. S12, ESI†).93 Another example includes the formation of an intermediate species for the total synthesis of chafuroside B (16) by the silyl triflate (Me3Si-OTf)-mediated alkylation reaction;94 the synthesis may have been motivated by the light protection abilities of the natural product (Scheme 6C and Fig. S13, ESI†).95 It is noteworthy that these reactions allowed for the isolation of specific diastereomeric products. | ||
| Scheme 6 Friedel–Crafts alkylation reactions for the construction and applications of carbohydrate derivatives. Leaving groups are highlighted with arrows. Mechanisms of the reactions are shown in Fig. S12–S14 (ESI†). (A) Chemical structures of common, abundant (left) and complex (right) carbohydrates. (B) Formation of an intermediate for the total synthesis of saptomycin B (15) that may have the potential of anti-tumor and anti-bacteria applications.92,93 (C) Formation of an intermediate for the total synthesis of chafuroside B (16) that may have the potential of a light-protection application.94,95 (D) Fries-type rearrangement of a carbohydrate derivative causing C–C bond formation on the oxygen-rich aromatic ring.96 | ||
The Fries-type rearrangement that is mechanistically related to Friedel–Crafts reactions provides opportunities for the stereoselective functionalization of carbohydrate derivatives (Scheme 6D and Fig. S14, ESI†).96 Fries-type rearrangements are reactions of phenol derivatives that transfers a substitution group on the phenolic oxygen to its ortho position.97 The elimination of a phenolic anion as a leaving group generates cation intermediates on the carbohydrate group, assisted by electron donation from the oxygen in the ring system. An electrophilic aromatic substitution-type process occurs afterward to form an ortho-substituted product. Although the rearrangement can be considered an intermolecular reaction between the phenolic anion and oxonium cation intermediates, the characteristic inversion of the stereochemistry at the anomeric position is often observed. One of the rationalizations of such stereospecificity is that the generated cation and anion would have intimate charge–charge interaction (as an ion pair),98,99 which may spatially control the orientation of the two intermediates in a specific position to give the specificity. Many similar intramolecular chemistry examples have been reported too.100–102
3.3. Functionalization of fatty acids through Friedel–Crafts reactions
Long alkyl chains of fatty acids are useful for chemical and biological materials including detergents, and carboxylic acid moieties of fatty acids can be employed for the synthesis of such materials through Friedel–Crafts acylation (Scheme 7A). Fatty acids are essential components in life forms and also abundant, useful building blocks for organic synthesis. Furan-based detergents with enhanced stability in hard water can be synthesized through Friedel–Crafts acylation using the anhydride form of lauric acid (Scheme 7B).103 With the use of a solid acid catalyst, mesoporous aluminosilicate, the reaction of the furan nucleophile and the lauric anhydride electrophile produced the acylated product. A mechanistic study of the reaction with the alumino-silicate catalyst suggested that the activation of the acylating reagent would occur through the activation of the acyl group by an oxygen atom on the catalyst (Fig. S15, ESI†).104 The acylated furan was further reduced to alkyl product 22 that was tested in hard water, where micelle aggregation tends to be disrupted.103 Indeed, compared to frequently used detergent 23 that became turbid at a higher concentration of calcium ions, the solution of the alkyl-furan detergent (22) remained clear, demonstrating the practical utility of the furan detergent made from a fatty acid through the acylation and reduction reactions. | ||
| Scheme 7 Friedel–Crafts acylation reactions of fatty acids. Leaving groups are highlighted with arrows. Mechanisms of the reactions are shown in Fig. S15 and S16 (ESI†). (A) Chemical structures of natural fatty acids (left) and detergent molecules that contain fatty alkyl chains. Linear alkylbenzene sulfonates are often a mixture of isomers but a single isomer structure (23) is shown for brevity. (B) Formation of an intermediate for the synthesis of furan-based detergents that display higher tolerance to hard water (right). The image of hard water was reproduced with permission.103 Copyright 2016, American Chemical Society (https://pubs.acs.org/doi/10.1021/acscentsci.6b00208). (C) An acylation reaction of oleic acid as a nucleophile using its alkene group, rather than aromatic rings for typical Friedel–Crafts reactions.105 | ||
As the breadth of nucleophilic reactivity of alkene groups is well known, unsaturated fatty acids with an alkyl-substituted, nonaromatic alkene are also known to act as nucleophiles for Friedel–Crafts reactions (Scheme 7C).105 Friedel–Crafts reactions with nonaromatic alkene nucleophiles have been known for decades and actively studied to date.32 Even if the nonaromatic systems are a relatively rarer type of Friedel–Crafts reaction than their aromatic counterparts, a number of alkylation reactions with nonaromatic alkene nucleophiles are reported in synthetic organic chemistry fields.106–109 Such nonaromatic reactions often produce regioisomers, depending on which alkene carbon forms a new σ bond with an electrophile (Fig. S16, ESI†).
3.4. Synthesis and functionalization of amino acids, peptides, and proteins through Friedel–Crafts alkylation
Although Friedel–Crafts reactions can be useful strategies for constructing carbon–carbon bonds in amino acid-based biomolecules (Scheme 8A), the selectivity of the reactions in the presence of other nucleophilic functional groups is often a substantial challenge. Among the 20 canonical amino acids, four aromatic amino acid residues could be potential nucleophile substrates: phenylalanine with an alkylbenzene, tyrosine with a phenol, tryptophan with an indole, and histidine with an imidazole. In particular, phenol (tyrosine) and indole (tryptophan) are relatively electron-rich aromatic systems and often good nucleophiles for Friedel–Crafts reactions, as similar/relevant substrates are covered in the previous sections (e.g., Scheme 4) whereas an alkylbenzene (phenylalanine) with a mildly electron-donating alkyl group and imidazole (histidine) with the CN bond acting as an electron-withdrawing group are often challenging substrates. In the presence of the four potentially reactive aromatic amino acids, nature addresses reactivity and selectivity challenges through enzymatic processes as discussed above (e.g., acylation of imidazole derivatives shown in Scheme 3D). Theoretically, carboxylic acid and its derivatives could serve as electrophiles for acylation reactions, although such reactions can be challenging or require harsh conditions in practice.110 The following sections describe the putative synthesis of a type of amino acid in the early Earth (chemical evolution), the functionalization of amino acids/peptides/proteins, and their potential implications and applications.
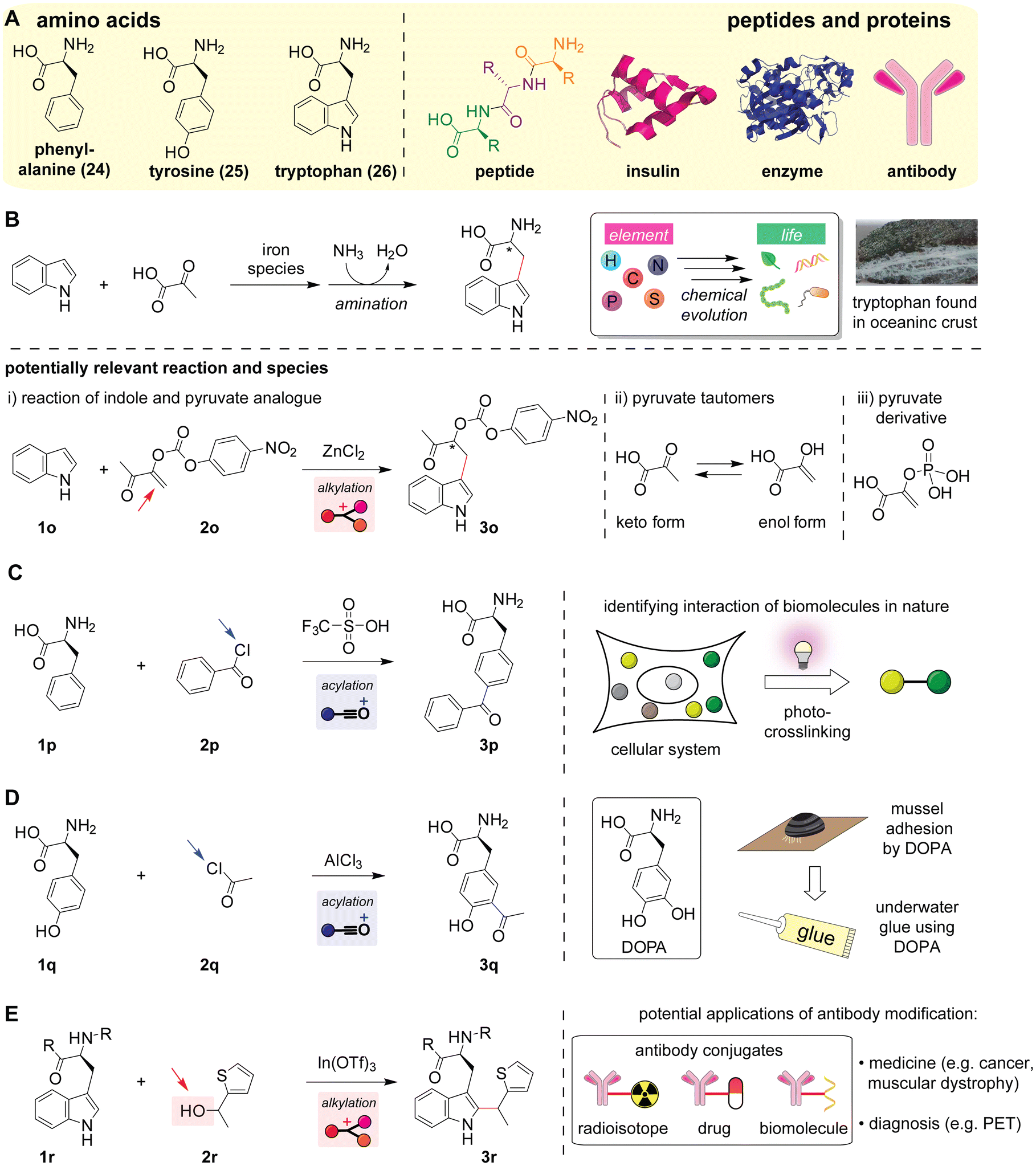 | ||
| Scheme 8 Friedel–Crafts reactions of amino acids, peptides, and proteins. Leaving groups are highlighted with arrows. Mechanisms of the reactions are shown in Fig. S17–S20 (ESI†). (A) Chemical structures of amino acids (left) and peptides/proteins (right). Structures of insulin (shown in magenta) and enzyme PpATaseCH (shown in blue) are obtained from the Protein Data Bank (PDB ID: 5CNY and 5MG5, respectively). (B) Formation of tryptophan from indole and pyruvate, proposed as one of the chemical evolution pathways in prebiotic eras.116 An image of a part of the oceanic crust (serpentinized harzburgite from the Atlantic Massif) is shown on the right. The crust image was reproduced with permission.115 Copyright 2011, the American Geophysical Union. Potentially relevant reactions to the proposed chemical evolution reaction are shown at the bottom: (i) reaction of indole and pyruvate analogues (oxygen-substituted α,β-unsaturated carbonyl compounds),120 (ii) tautomerization of pyruvate forming hydroxy-substituted α,β-unsaturated carboxylic acid as an enol form, and (iii) chemical structure of phosphoenolpyruvate (α,β-unsaturated carbonyl compound with a substitution on the oxygen of the enol unit) that is a known intermediate for modern glycolysis121 as well as a possible species in the prebiotic time.122 (C) Reaction of phenylalanine and benzoyl chloride forming benzophenone-substituted unnatural amino acid126 that is useful for the identification of interactions between biomolecules in cellular systems through irradiation of ultraviolet light (photo-crosslinking).124 (D) Acetylation of tyrosine with acetyl chloride forming acetyltyrosine as an intermediate for the chemical synthesis of 3,4-dihydroxyphenylalanine (DOPA).130,131 Schematic description of the utility of DOPA as a natural adherent is shown on the right. (E) Reaction of tryptophan residues in peptides or proteins with thiophene-ethanol, which can be used for labeling of antibodies.135 Examples of antibody conjugates are shown on the right. | ||
The proposed formation of tryptophan amino acid (Scheme 8B, top)116 may be relevant to an enol form of pyruvate derivatives (Scheme 8B, bottom). No literature precedents in synthetic chemistry fields exist about the reactions of non-substituted indole and pyruvate eventually producing tryptophan or even derivatives. Nonetheless, there was a report of a reaction of indole with a ketone analogue bearing an O-acylated enol group (Scheme 8B, (i) and Fig. S17, ESI†).120 Although the keto-acylated enol compound is not an exact pyruvate derivative, the chemical structure is akin to the enol form of pyruvate to a certain extent (Scheme 8B, (ii)), perhaps suggesting that an enol form of the pyruvate derivative might have been a reactant of the proposed tryptophan formation in the prebiotic time. Indeed, phosphoenolpyruvate (Scheme 8B, (iii)) is a ubiquitous pyruvate derivative with an enol unit as an intermediate species of glycolysis pathways in living systems.121 A recent report also suggested that phosphoenolpyruvate intermediates might have been present in the prebiotic time too,122 and even though further experimental investigations would be necessary to support those speculations, a set of evidence may be crucial to account for the prebiotic origin of tryptophan.
Friedel–Crafts acylation can be of use for the chemical synthesis of the natural, noncanonical amino acid 3,4-dihydroxyphenylalanine (DOPA) that can exert adhesive effects (Scheme 8D). DOPA is a naturally occurring amino acid with adherent properties (as mussel adhesion is enabled by the properties), although the natural amino acid is not one of the 20 canonical/proteinogenic amino acids. Since the adhesive properties are effective even under underwater conditions, synthetic glues often rely on DOPA compositions as a bioinspired material.127,128 Simply speaking, although direct hydroxylation/oxidation of tyrosine amino acid would provide DOPA by formally inserting an oxygen atom into the C–H bond, phenol derivatives tend to undergo the formation of various oxidation products including ortho-benzoquinone unless there is an enzymatic control of reactions,129 representing a synthetic challenge of DOPA. An approach originally reported by Boger and co-workers was a selective acetylation reaction toward the aromatic ring of the tyrosine starting material using excess AlCl3 at 100 °C (Scheme 8D and Fig. S19, ESI†), followed by a unique oxidative C–C bond cleavage of the acetyl group to form protected DOPA.130,131 The use of harsh reaction conditions with excess Lewis acid and high temperature is presumably key to achieving the selective acylation of aromatic rings over amines. However, such harsh conditions could induce loss of enantiopurity (i.e., racemization) through keto–enol tautomerization of the backbone carbonyl of the amino acid.132 In this context, asymmetric alkylation of amino acid derivatives/precursors could be a complementary strategy,133 or the development of milder reactions would be necessary as described in the next section.
4. Conclusions
Since their inception approximately 150 years ago, Friedel–Crafts reactions have proven their significance not only for synthetic methods of small molecule compounds but also for biomolecular chemistry. Even during the past decade, a series of natural Friedel–Crafts reactions in living systems have been revealed, and it would be reasonable to assume that there may be many other examples of undiscovered natural Friedel–Crafts reactions. The discovery of natural reactions would expand the field of enzymology in conjunction with bioengineering strategies such as directed evolution.139 Many synthesis and functionalization methods of biomolecules described above are dependent on protecting group strategies which require additional synthetic efforts and cost; hence, unique reaction control approaches for unprotected biomolecules would be an important focus for future development. In addition to the aforementioned relevance to chemical evolution (Scheme 8A), Friedel–Crafts alkylation has been employed for studies of autocatalysis, which is in part relevant to self-replication processes in the early Earth.140 As such, alkylation and acylation reactions remain essential for diverse aspects of chemistry and biology research fields.It is interesting that, despite the significant advance in biomolecular Friedel–Crafts reactions, there have not been any examples of bioorthogonal Friedel–Crafts reactions reported to date. Many classical reactions can be used to achieve bioorthogonal chemistry such as Diels–Alder reactions5 and even imine/hydrazone/oxime formation in certain circumstances7 (Scheme 1A, (iii), (iv), and (viii)). The challenges of such a bioorthogonal Friedel–Crafts reaction may be related to the general incompatibility of traditional Friedel–Crafts reagents (i.e., acyl chloride and aluminum chloride), although the generation of acyl chloride in cellular environments could be possible at a low concentration.141 However, reagents that are frequently considered too reactive/unstable in aqueous solutions (e.g., Wittig142 and diazo143 reagents) have been successfully used in cellular applications too, which indicates the potential feasibility of Friedel–Crafts reactions in cellular systems. The development of methods with unique media could potentially be an alternative approach, as discussed above (Scheme 8E).135 Even if intact small molecule reagents would not be usable for cellular contexts, there are other unique chemical strategies such as genetically encodable amino acids and peptides as bioorthogonal tags144 as well as encapsulation methods of reactive molecules in aqueous solutions.145 Biomolecular Friedel–Crafts reactions have rapidly grown in multiple ways, and alongside technological and strategic advances in chemical methods in various fields, the full potential of alkylation and acylation chemistry in biological systems may not have been seen yet.
Conflicts of interest
The author declares no conflict of interest.Acknowledgements
This work was financially supported by North Carolina State University.References
- A. R. Lippert, G. C. Van de Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed.
- J. P. Cadahía, V. Previtali, N. S. Troelsen and M. H. Clausen, Med. Chem. Commun., 2019, 10, 1531–1549 RSC.
- R. Lagoutte, R. Patouret and N. Winssinger, Curr. Opin. Chem. Biol., 2017, 39, 54–63 CrossRef CAS PubMed.
- E. S. Nakayasu, M. Gritsenko, P. D. Piehowski, Y. Gao, D. J. Orton, A. A. Schepmoes, T. L. Fillmore, B. I. Frohnert, M. Rewers, J. P. Krischer, C. Ansong, A. M. Suchy-Dicey, C. Evans-Molina, W.-J. Qian, B.-J. M. Webb-Robertson and T. O. Metz, Nat. Protoc., 2021, 16, 3737–3760 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- M. Peplow, Nat. Biotechnol., 2023, 41, 883–885 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- J. Wahbeh and S. Milkowski, SLAS Technol., 2019, 24, 161–168 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- M. J. Niphakis and B. F. Cravatt, Annu. Rev. Biochem., 2014, 83, 341–377 CrossRef CAS PubMed.
- K. J. Bruemmer, S. W. M. Crossley and C. J. Chang, Angew. Chem., Int. Ed., 2020, 59, 13734–13762 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- N. Diamantis and U. Banerji, Br. J. Cancer, 2016, 114, 362–367 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- A. A. Ashdown, Ind. Eng. Chem., 1927, 19, 1063–1065 CrossRef CAS.
- A.-M. Stadler and J. Harrowfield, Chem. Soc. Rev., 2011, 40, 2061–2108 RSC.
- K. L. Marsi and S. H. Wilen, J. Chem. Educ., 1963, 40, 214 CrossRef CAS.
- A. M. Reeve, J. Chem. Educ., 2004, 81, 1497 CrossRef CAS.
- M. R. Dintzner, J. J. Maresh, C. R. Kinzie, A. F. Arena and T. Speltz, J. Chem. Educ., 2012, 89, 265–267 CrossRef CAS.
- D. Elford, S. J. Lancaster and G. A. Jones, Comput. Educ.: X Reality, 2023, 2, 100021 Search PubMed.
- D. J. Macquarrie, Catalytic Asymmetric Friedel–Crafts Alkylations, John Wiley & Sons, Ltd, 2009, pp. 271–288 Search PubMed.
- P. H. Groggins and S. B. Detwiler, Ind. Eng. Chem., 1952, 44, 2012–2015 CrossRef CAS.
- L. Y. P. Luk, Q. Qian and M. E. Tanner, J. Am. Chem. Soc., 2011, 133, 12342–12345 CrossRef CAS PubMed.
- Z. Li, B. Xu, T. A. Alsup, X. Wei, W. Ning, D. G. Icenhour, M. A. Ehrenberger, I. Ghiviriga, B.-D. Giang and J. D. Rudolf, J. Am. Chem. Soc., 2023, 145, 22361–22365 CrossRef CAS PubMed.
- J. Ohata, K. J. Bruemmer and C. J. Chang, Acc. Chem. Res., 2019, 52, 2841–2848 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS PubMed.
- J. K. Groves, Chem. Soc. Rev., 1972, 1, 73–97 RSC.
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 Search PubMed.
- H. F. Motiwala, R. H. Vekariya and J. Aubé, Org. Lett., 2015, 17, 5484–5487 CrossRef CAS PubMed.
- S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190–2201 RSC.
- M. M. Heravi, V. Zadsirjan, M. Heydari and B. Masoumi, Chem. Rec., 2019, 19, 2236–2340 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan, P. Saedi and T. Momeni, RSC Adv., 2018, 8, 40061–40163 RSC.
- G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7202–7236 CrossRef CAS PubMed.
- I. O. Igbokwe, E. Igwenagu and N. A. Igbokwe, Interdiscip. Toxicol., 2019, 12, 45–70 CrossRef CAS PubMed.
- R. Danielsson and H. Eriksson, Semin. Cell Dev. Biol., 2021, 115, 3–9 CrossRef CAS PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC.
- M. J. Byrne, N. R. Lees, L.-C. Han, M. W. van der Kamp, A. J. Mulholland, J. E. M. Stach, C. L. Willis and P. R. Race, J. Am. Chem. Soc., 2016, 138, 6095–6098 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- Q. Du, Z. Wang and V. L. Schramm, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2916–2921 CrossRef CAS PubMed.
- A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083–2092 RSC.
- V. J. DeRose, Chem. Biol., 2002, 9, 961–969 CrossRef CAS PubMed.
- X. Liu, Y. Xiao, Z. Zhang, Z. You, J. Li, D. Ma and B. Li, Chin. J. Chem., 2021, 39, 3462–3480 CrossRef CAS.
- R. B. Leveson-Gower and G. Roelfes, ChemCatChem, 2022, 14, e202200636 CrossRef CAS PubMed.
- V. Kumar, W. B. Turnbull and A. Kumar, ACS Catal., 2022, 12, 10742–10763 CrossRef CAS.
- E. K. Keenan, D. K. Zachman and M. D. Hirschey, Mol. Cell, 2021, 81, 1868–1878 CrossRef CAS PubMed.
- S. L. Crine and K. R. Acharya, FEBS J., 2022, 289, 7670–7687 CrossRef CAS PubMed.
- S. Minakata, S. Manabe, Y. Inai, M. Ikezaki, K. Nishitsuji, Y. Ito and Y. Ihara, Molecules, 2021, 26, 5258 CrossRef CAS PubMed.
- J. S. Bloch, A. John, R. Mao, S. Mukherjee, J. Boilevin, R. N. Irobalieva, T. Darbre, N. E. Scott, J.-L. Reymond, A. A. Kossiakoff, E. D. Goddard-Borger and K. P. Locher, Nat. Chem. Biol., 2023, 19, 575–584 CrossRef CAS PubMed.
- G. E. Chlipala, M. Sturdy, A. Krunic, D. D. Lantvit, Q. Shen, K. Porter, S. M. Swanson and J. Orjala, J. Nat. Prod., 2010, 73, 1529–1537 CrossRef CAS PubMed.
- H. Nakamura, E. E. Schultz and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 916–921 CrossRef CAS PubMed.
- N. R. Braffman, T. B. Ruskoski, K. M. Davis, N. R. Glasser, C. Johnson, C. D. Okafor, A. K. Boal and E. P. Balskus, eLife, 2022, 11, e75761 CrossRef CAS PubMed.
- A. Hayashi, H. Saitou, T. Mori, I. Matano, H. Sugisaki and K. Maruyama, Biosci. Biotechnol. Biochem., 2012, 76, 559 CrossRef CAS PubMed.
- X. Sheng, M. Kazemi, A. Żądło-Dobrowolska, W. Kroutil and F. Himo, ACS Catal., 2020, 10, 570–577 CrossRef CAS PubMed.
- F. Yang and Y. Cao, Appl. Microbiol. Biotechnol., 2012, 93, 487–495 CrossRef CAS PubMed.
- Y. Xia, G. Zhu, X. Zhang, S. Li, L. Du and W. Zhu, J. Am. Chem. Soc., 2023, 145, 26308–26317 CrossRef CAS PubMed.
- S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS PubMed.
- K. Peciak, E. Laurine, R. Tommasi, J. Choi and S. Brocchini, Chem. Sci., 2019, 10, 427–439 RSC.
- B.-B. He, X.-L. Bu, T. Zhou, S.-M. Li, M.-J. Xu and J. Xu, ACS Synth. Biol., 2018, 7, 2094–2104 CrossRef CAS PubMed.
- S. Lund, R. Hall and G. J. Williams, ACS Synth. Biol., 2019, 8, 232–238 CrossRef CAS PubMed.
- N. G. Schmidt, T. Pavkov-Keller, N. Richter, B. Wiltschi, K. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2017, 56, 7615–7619 CrossRef CAS PubMed.
- S. Chordia, S. Narasimhan, A. Lucini Paioni, M. Baldus and G. Roelfes, Angew. Chem., Int. Ed., 2021, 60, 5913–5920 CrossRef CAS PubMed.
- L. Zheng, A. Marcozzi, J. Y. Gerasimov and A. Herrmann, Angew. Chem., Int. Ed., 2014, 53, 7599–7603 CrossRef CAS PubMed.
- A. J. Metrano, A. J. Chinn, C. R. Shugrue, E. A. Stone, B. Kim and S. J. Miller, Chem. Rev., 2020, 120, 11479–11615 CrossRef CAS PubMed.
- N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370–4371 CrossRef CAS PubMed.
- D. A. Evans, K. R. Fandrick, H.-J. Song, K. A. Scheidt and R. Xu, J. Am. Chem. Soc., 2007, 129, 10029–10041 CrossRef CAS PubMed.
- B. M. Trost and C. Müller, J. Am. Chem. Soc., 2008, 130, 2438–2439 CrossRef CAS PubMed.
- A. García-Fernández, R. P. Megens, L. Villarino and G. Roelfes, J. Am. Chem. Soc., 2016, 138, 16308–16314 CrossRef PubMed.
- K. Boulias and E. L. Greer, Nat. Rev. Genet., 2022, 23, 411–428 CrossRef CAS PubMed.
- W. R. Sinclair, D. Arango, J. H. Shrimp, T. T. Zengeya, J. M. Thomas, D. C. Montgomery, S. D. Fox, T. Andresson, S. Oberdoerffer and J. L. Meier, ACS Chem. Biol., 2017, 12, 2922–2926 CrossRef CAS PubMed.
- Preparation of the guanine nucleoside by a Friedel–Crafts catalyst is reported (W. W. Lee, A. P. Martinez and L. Goodman, J. Org. Chem., 1971, 36, 842) but this C–N bond forming reaction would not be considered a Friedel–Crafts alkylation reaction.
- N. S. Girgis, M. A. Michael, D. F. Smee, H. A. Alaghamandan, R. K. Robins and H. B. Cottam, J. Med. Chem., 1990, 33, 2750–2755 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- S. Hainke, S. Arndt and O. Seitz, Org. Biomol. Chem., 2005, 3, 4233–4238 RSC.
- D. Shin, R. W. Sinkeldam and Y. Tor, J. Am. Chem. Soc., 2011, 133, 14912–14915 CrossRef CAS PubMed.
- Y. W. Jun, D. L. Wilson, A. M. Kietrys, E. R. Lotsof, S. G. Conlon, S. S. David and E. T. Kool, Angew. Chem., Int. Ed., 2020, 59, 7450–7455 CrossRef CAS PubMed.
- C. Wu, C. Wang, L. Yan and C. J. Yang, J. Biomed. Nanotechnol., 2009, 5, 495–504 CrossRef CAS PubMed.
- Y. Li, A. Fin, L. McCoy and Y. Tor, Angew. Chem., Int. Ed., 2017, 56, 1303–1307 CrossRef CAS PubMed.
- J. Kuchlyan, L. Martinez-Fernandez, M. Mori, K. Gavvala, S. Ciaco, C. Boudier, L. Richert, P. Didier, Y. Tor, R. Improta and Y. Mély, J. Am. Chem. Soc., 2020, 142, 16999–17014 CrossRef CAS PubMed.
- S. Park, H. Otomo, L. Zheng and H. Sugiyama, Chem. Commun., 2014, 50, 1573–1575 RSC.
- H. Yang, E. Eremeeva, M. Abramov, M. Jacquemyn, E. Groaz, D. Daelemans and P. Herdewijn, Nucleic Acids Res., 2023, 51, 1501–1511 CrossRef CAS PubMed.
- W. Xu, K. M. Chan and E. T. Kool, Nat. Chem., 2017, 9, 1043–1055 CrossRef CAS PubMed.
- A. Rich, A. Nordheim and A. H. Wang, Annu. Rev. Biochem., 1984, 53, 791–846 CrossRef CAS PubMed.
- A. Rich and S. Zhang, Nat. Rev. Genet., 2003, 4, 566–572 CrossRef CAS PubMed.
- H. Dashti, W. M. Westler, J. R. Wedell, O. V. Demler, H. R. Eghbalnia, J. L. Markley and S. Mora, Sci. Data, 2020, 7, 210 CrossRef CAS PubMed.
- R. Mettu, C.-Y. Chen and C.-Y. Wu, J. Biomed. Sci., 2020, 27, 9 CrossRef CAS PubMed.
- N. Abe, Y. Nakakita, T. Nakamura, N. Enoki, H. Uchida and M. Munekata, J. Antibiot., 1993, 46, 1530–1535 CrossRef CAS PubMed.
- K. Kitamura, Y. Maezawa, Y. Ando, T. Kusumi, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2014, 53, 1262–1265 CrossRef CAS PubMed.
- T. Furuta, M. Nakayama, H. Suzuki, H. Tajimi, M. Inai, H. Nukaya, T. Wakimoto and T. Kan, Org. Lett., 2009, 11, 2233–2236 CrossRef CAS PubMed.
- T. Hasegawa, S. Shimada, H. Ishida and M. Nakashima, PLoS One, 2013, 8, e77308 CrossRef CAS PubMed.
- O. J. Plante, E. R. Palmacci, R. B. Andrade and P. H. Seeberger, J. Am. Chem. Soc., 2001, 123, 9545–9554 CrossRef CAS PubMed.
- M. Korb and H. Lang, Chem. Soc. Rev., 2019, 48, 2829–2882 RSC.
- M. J. S. Dewar and L. S. Hart, Tetrahedron, 1970, 26, 973–1000 CrossRef CAS.
- J. L. Gibson and L. S. Hart, J. Chem. Soc., Perkin Trans. 2, 1991, 1343–1348 RSC.
- H. Liu, M. Lang, D. Hazelard and P. Compain, J. Org. Chem., 2023, 88, 13847–13856 CrossRef CAS PubMed.
- M. Liu, B.-H. Li, T. Li, X. Wu, M. Liu, D.-C. Xiong and X.-S. Ye, Org. Biomol. Chem., 2020, 18, 3043–3046 RSC.
- E. R. Palmacci and P. H. Seeberger, Org. Lett., 2001, 3, 1547–1550 CrossRef CAS PubMed.
- D. S. Park, K. E. Joseph, M. Koehle, C. Krumm, L. Ren, J. N. Damen, M. H. Shete, H. S. Lee, X. Zuo, B. Lee, W. Fan, D. G. Vlachos, R. F. Lobo, M. Tsapatsis and P. J. Dauenhauer, ACS Cent. Sci., 2016, 2, 820–824 CrossRef CAS PubMed.
- A. V. Naik, K. E. Joseph, M. Shetty, M. A. Ardagh and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2020, 8, 18616–18625 CrossRef CAS.
- J. O. Metzger and U. Biermann, Fett/Lipid, 1998, 100, 2–6 CrossRef CAS.
- B. Xiang, T.-F. Xu, L. Wu, R.-R. Liu, J.-R. Gao and Y.-X. Jia, J. Org. Chem., 2016, 81, 3929–3935 CrossRef CAS PubMed.
- S. Tanaka, T. Kunisawa, Y. Yoshii and T. Hattori, Org. Lett., 2019, 21, 8509–8513 CrossRef CAS PubMed.
- A. Ishii, J. Kojima and K. Mikami, Org. Lett., 1999, 1, 2013–2016 CrossRef CAS.
- B. B. Snider and A. C. Jackson, J. Org. Chem., 1982, 47, 5393–5395 CrossRef CAS.
- A. Sumita and T. Ohwada, Molecules, 2022, 27, 5984 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- A. Eschenmoser, Tetrahedron, 2007, 63, 12821–12844 CrossRef CAS.
- L. M. Barge and R. E. Price, Nat. Geosci., 2022, 15, 976–981 CrossRef CAS.
- S. A. Sandford, M. Nuevo, P. P. Bera and T. J. Lee, Chem. Rev., 2020, 120, 4616–4659 CrossRef CAS PubMed.
- D. K. Blackman, B. Ildefonse, B. E. John, Y. Ohara, D. J. Miller, N. Abe, M. Abratis, E. S. Andal, M. Andreani, S. Awaji, J. S. Beard, D. Brunelli, A. B. Charney, D. M. Christie, J. Collins, A. G. Delacour, H. Delius, M. Drouin, F. Einaudi, J. Escartín, B. R. Frost, G. Früh-Green, P. B. Fryer, J. S. Gee, M. Godard, C. B. Grimes, A. Halfpenny, H.-E. Hansen, A. C. Harris, A. Tamura, N. W. Hayman, E. Hellebrand, T. Hirose, J. G. Hirth, S. Ishimaru, K. T. M. Johnson, G. D. Karner, M. Linek, C. J. MacLeod, J. Maeda, O. U. Mason, A. M. McCaig, K. Michibayashi, A. Morris, T. Nakagawa, T. Nozaka, M. Rosner, R. C. Searle, G. Suhr, M. Tominaga, A. von der Handt, T. Yamasaki and X. Zhao, J. Geophys. Res.: Solid Earth, 2011, 116, B07103 Search PubMed.
- B. Ménez, C. Pisapia, M. Andreani, F. Jamme, Q. P. Vanbellingen, A. Brunelle, L. Richard, P. Dumas and M. Réfrégiers, Nature, 2018, 564, 59–63 CrossRef PubMed.
- E. Camprubi, S. F. Jordan, R. Vasiliadou and N. Lane, IUBMB Life, 2017, 69, 373–381 CrossRef CAS PubMed.
- A. Sato, K. Soeno, R. Kikuchi, M. Narukawa-Nara, C. Yamazaki, Y. Kakei, A. Nakamura and Y. Shimada, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203633119 CrossRef CAS PubMed.
- A. Kirschning, Chem. – Eur. J., 2022, 28, e202201419 CrossRef CAS PubMed.
- D. Zárate-Zárate, R. Aguilar, R. I. Hernández-Benitez, E. M. Labarrios, F. Delgado and J. Tamariz, Tetrahedron, 2015, 71, 6961–6978 CrossRef.
- Y. Kondo, Y. Ishitsuka, D. Kadowaki, M. Kuroda, Y. Tanaka, M. Nagatome, M. Irikura, S. Hirata, K. Sato, T. Maruyama, N. Hamasaki and T. Irie, Biol. Pharm. Bull., 2012, 35, 606–611 CrossRef CAS PubMed.
- J. Zimmermann, R. J. Mayer and J. Moran, Chem. Sci., 2023, 14, 14100–14108 RSC.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- G. Dormán, H. Nakamura, A. Pulsipher and G. D. Prestwich, Chem. Rev., 2016, 116, 15284–15398 CrossRef PubMed.
- T.-A. Nguyen, M. Cigler and K. Lang, Angew. Chem., Int. Ed., 2018, 57, 14350–14361 CrossRef CAS PubMed.
- Y. Murai, L. Wang, Y. Muto, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and M. Hashimoto, Heterocycles, 2013, 87, 2119–2126 CrossRef CAS.
- D. Giuri, P. Ravarino and C. Tomasini, Org. Biomol. Chem., 2021, 19, 4622–4636 RSC.
- Y. Li, J. Cheng, P. Delparastan, H. Wang, S. J. Sigg, K. G. DeFrates, Y. Cao and P. B. Messersmith, Nat. Commun., 2020, 11, 3895 CrossRef CAS PubMed.
- C. Honisch, M. Gazziero, R. Dallocchio, A. Dessì, D. Fabbri, M. A. Dettori, G. Delogu and P. Ruzza, Int. J. Mol. Sci., 2022, 23, 6240 CrossRef CAS PubMed.
- D. L. Boger and D. Yohannes, J. Org. Chem., 1987, 52, 5283–5286 CrossRef CAS.
- C. Chen, Y.-F. Zhu and K. Wilcoxen, J. Org. Chem., 2000, 65, 2574–2576 CrossRef CAS PubMed.
- I. Urruzuno, P. Andrade-Sampedro and A. Correa, Org. Lett., 2021, 23, 7279–7284 CrossRef CAS PubMed.
- S. Brunen, B. Mitschke, M. Leutzsch and B. List, J. Am. Chem. Soc., 2023, 145, 15708–15713 CrossRef CAS PubMed.
- R. Rajan, S. Ahmed, N. Sharma, N. Kumar, A. Debas and K. Matsumura, Mater. Adv., 2021, 2, 1139–1176 RSC.
- M. Nuruzzaman, B. M. Colella, C. P. Uzoewulu, A. E. Meo, E. J. Gross, S. Ishizawa, S. Sana, H. Zhang, M. E. Hoff, B. T. W. Medlock, E. C. Joyner, S. Sato, E. A. Ison, Z. Li and J. Ohata, J. Am. Chem. Soc., 2024, 146, 6773–6783 CrossRef CAS PubMed.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef.
- P. J. Santangelo, K. A. Rogers, C. Zurla, E. L. Blanchard, S. Gumber, K. Strait, F. Connor-Stroud, D. M. Schuster, P. K. Amancha, J. J. Hong, S. N. Byrareddy, J. A. Hoxie, B. Vidakovic, A. A. Ansari, E. Hunter and F. Villinger, Nat. Methods, 2015, 12, 427–432 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184 CrossRef CAS PubMed.
- R. B. Leveson-Gower, Z. Zhou, I. Drienovská and G. Roelfes, ACS Catal., 2021, 11, 6763–6770 CrossRef CAS PubMed.
- S. Kamioka, D. Ajami and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 541–544 CrossRef CAS PubMed.
- S. Pani, T. Qiu, K. Kentala, S.-A. Azizi and B. Dickinson, Nat. Chem., 2024 DOI:10.1038/s41557-024-01493-1.
- Y. Shi, L. Fu, J. Yang and K. S. Carroll, Nat. Chem., 2021, 13, 1140–1150 CrossRef CAS PubMed.
- K. A. Andersen, M. R. Aronoff, N. A. McGrath and R. T. Raines, J. Am. Chem. Soc., 2015, 137, 2412–2415 CrossRef CAS PubMed.
- J. Ohata, Y. Zeng, L. Segatori and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 4015–4019 CrossRef CAS PubMed.
- P. La Manna, C. Talotta, G. Floresta, M. De Rosa, A. Soriente, A. Rescifina, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2018, 57, 5423–5428 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00406j |
| This journal is © The Royal Society of Chemistry 2024 |