
PIDA-promoted metal-free [3 + 2] heteroannulation of β-ketothioamides with 4-hydroxy coumarins: chemo-/regioselective access to furo[3,2-c]chromen-4-ones at room temperature†
Anup
Kumar Yadav
,
Dhananjay
Yadav
,
Vipin
Kumar
,
Subhasish
Ray
 and
Maya Shankar
Singh
*
and
Maya Shankar
Singh
*
Department of Chemistry, Institute of Science, Banaras Hindu University, Varanasi 221005, India. E-mail: mssingh@bhu.ac.in
First published on 29th April 2024
Abstract
Herein, we report a viable protocol to access furo[3,2-c]chromen-4-ones by engaging easily accessible 4-hydroxy coumarins as a three-atom CCO unit and thioamides as a C2 coupling partner, mediated by phenyliodine(III) diacetate (PIDA) at room temperature in a highly efficient and pot-/step-economical manner. This strategy not only avoids potential toxicity and tiresome workup conditions, but also features operational simplicity, a broad substrate scope, good functional group tolerance, high yields, easy scalability and exclusive selectivity. A mechanistic study has shown that this metal-free reaction is triggered by PIDA via activation of the β-carbon of 4-hydroxy coumarin, followed by a nucleophilic addition/intramolecular cyclization/dethiohydration cascade. High-resolution mass spectra (HRMS) study confirms the key intermediates involved during the course of the reaction, elucidating the reaction pathways, and demonstrates the excellent regio- and chemoselectivity of this approach.
Introduction
Synthesis of fused heterocyclic motifs has always been an important task in organic synthesis, owing to their diverse range of properties, such as aromaticity, rigidity and electron delocalization, which make them ideal candidates for the development of therapeutic agents. Coumarins and their fused analogues represent a class of important heterocycles that have made outstanding contributions in the prevention and treatment of disease.1 Three-substituted coumarins and their derivatives have attracted a certain amount of attention due to their wide scope of pharmacological properties.2 Owing to their remarkable biological significance, the search for new bioactive coumarins is still of immense interest to scientists to reveal their medical applications. Hence, several approaches for functionalization of coumarins to achieve novel analogues with new biological properties have been described.3,4 Coumarin motifs are frequently found in many natural products and biologically active intermediates.5 In the scientific community, there is a growing interest in coumarin motifs due to their anti-HIV, antioxidant, antifungal, antihelminthic and antibacterial properties.6 Moreover, they are commonly used in the food and cosmetic industries as additives and are also found to act as insecticides, optical brighteners and as fluorescent and laser dyes.7A remarkable growth in hypervalent iodine-mediated transformation, as stoichiometric reagents as well as catalysts, has been well documented due to their excellent properties, such as mildness, ease of handling, good selectivity, environmental friendliness and high stability in the presence of atmospheric oxygen and moisture.8 The peculiar reactivity of hypervalent iodine reagents arises from the 3-center–4-electron bond (3c–4e), or hypervalent bond, which is longer, more polarized and weaker than a standard covalent bond, leading to higher electrophilic reactivity.9 In recent years, efforts have focused on cyclic hypervalent iodine reagents due to their higher stability.10 Moreover, confinement of the oxygen lone pairs out of the 3c–4e plane disfavours reductive elimination between the axial ligands.11 Over the past few years, hypervalent iodine reagents have emerged as efficient organocatalysts for oxidative transformation of a wide range of organic substrates.12
The development of new synthetic approaches to furo-fused chromen-4-ones is an important research area of organic synthesis. Various methods have been developed for the synthesis of furo[3,2-c]coumarin motifs, employing coupling and cyclization reactions involving suitable precursors.13 Mosslemin et al.14a reported a one-pot three-component reaction of 4-hydroxy coumarin with aldehydes and isocyanides (Scheme 1a). Phan and co-workers14b coupled 4-hydroxy coumarin with ketones in the presence of iodine to synthesize furo-fused coumarins (Scheme 1b). Vagh et al.14c reported an acyl transfer reaction of suitable starting material in the presence of acyl halides and base to construct furo-fused coumarin motifs (Scheme 1c). Although these reported methods are useful tools to access furo[3,2-c]coumarins, most of them suffer from significant limitations, such as the use of specific substrates and harsh conditions. Hence, it is highly desirable to explore a sustainable and efficient general strategy to construct furo[3,2-c]coumarins, especially those containing amino and aroyl groups, which can be further functionalized. Our careful literature survey revealed that 4-hydroxycoumarin and β-ketothioamide have not been explored as coupling partners for the synthesis of furo[3,2-c]coumarins. Recently, we devised a cascade metal-/catalyst-free strategy to construct furo-fused quinoxalines from β-ketothioamide and quinoxalin-2-ones at room temperature.15 To this end, our continuing efforts to synthesize diverse heterocycles16 prompted us to develop more innovative and sustainable methods for newer analogues. Herein, we report a convenient metal-free one-pot strategy to synthesize furo[3,2-c]coumarins employing 4-hydroxy coumarin and β-ketothioamide at room temperature (Scheme 1d).
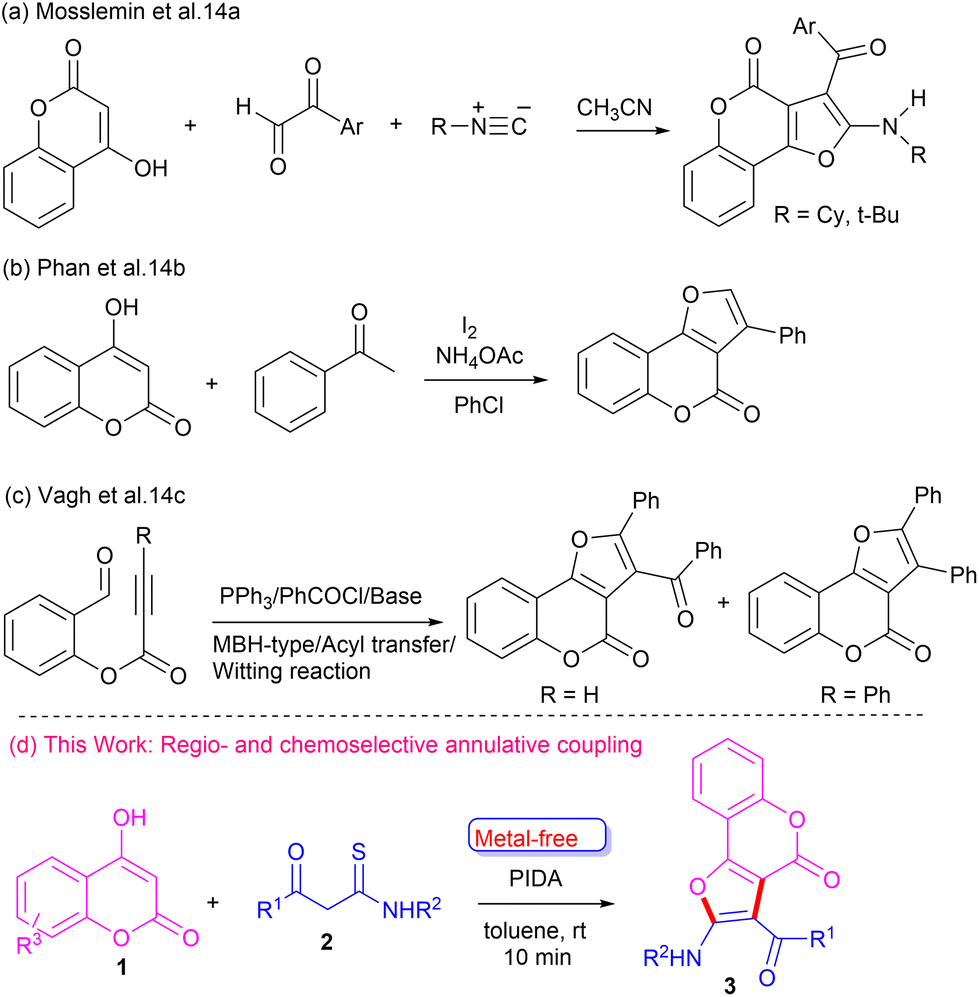 | ||
| Scheme 1 Previous reports and this work. | ||
Results and discussion
The efficient and selective synthesis of fused heterocyclic systems via cascade reactions remains an important pursuit in synthetic organic chemistry. Synthons containing both electrophilic and nucleophilic sites (ambiphilic synthons) have great potential in developing new reaction pathways. One such synthon are β-ketothioamides (KTAs), which have proven to be a versatile class of strategic precursors due to their intrinsic synthetic tunability with fitting partners in a chemo- and regioselective manner, for the construction of various heterocycles.17 Due to our constant efforts to develop new protocols towards heterocyclic frameworks utilizing KTA as a synthon,18 we envisioned further expanding their synthetic repertoire by reacting them with 4-hydroxy coumarin as a coupling partner. Herein, we report the synthesis of furo[3,2-c]coumarins from 4-hydroxy coumarins and β-ketothioamides via an intermolecular cross-coupling and intramolecular cyclization strategy mediated by the eco-friendly hypervalent iodine reagent phenyliodine(III) diacetate (PIDA), at room temperature under open air, in a highly efficient, rapid and pot-/step-economical manner (Scheme 1d). The reactions were completed within 10 minutes and the pure products were isolated in good yield.4-Hydroxycoumarin (1a) and β-ketothioamide (2a) were selected as test substrates to optimize the reaction conditions. Initially, the reaction of 1a (0.30 mmol) and 2a (0.25 mmol) in 2 mL of toluene at room temperature in an open atmosphere was carried out without any reagent. The reaction did not proceed at all, and the starting materials remained completely unreacted, even after 24 h (Table 1, entry 1). In an effort to trigger the reaction, this test reaction was performed in the presence of iodine, which enabled the formation of product in a trace amount after 1 h (Table 1, entry 2). The above result encouraged us to continue our investigations to obtain efficient condition for the formation of product in good yield.
|
|
||||
|---|---|---|---|---|
| Entry | Reagent (equiv.) | Solvent (mL) | Time | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.25 mmol), and solvent (2.0 mL), room temperature in an open atmosphere. b Isolated yield. PIDA = phenyliodine(III) diacetate; PIFA = phenyliodine(III) bis(trifluoroacetate); NIS = N-iodosuccinimide. | ||||
| 1 | None | Toluene | 24 h | NR |
| 2 | I2 (1.5) | Toluene | 60 min | Trace |
| 3 | PIDA (0.2) | Toluene | 30 min | 10 |
| 4 | PIDA (1.0) | Toluene | 30 min | 43 |
| 5 | PIDA (1.5) | Toluene | 10 min | 64 |
| 6 | PIDA (2.0) | Toluene | 10 min | 64 |
| 7 | PIDA (1.5) | Benzene | 10 min | 58 |
| 8 | PIDA (1.5) | CCl4 | 10 min | 50 |
| 9 | PIDA (1.5) | CHCl3 | 10 min | 45 |
| 10 | PIDA (1.5) | DCM | 10 min | 30 |
| 11 | PIDA (1.5) | DMF | 10 min | 40 |
| 12 | PIDA (1.5) | DCE | 10 min | 52 |
| 13 | PIFA (1.5) | Toluene | 10 min | 60 |
| 14 | NIS (1.5) | Toluene | 10 min | NR |
| 15 | ZnCl2 (1.5) | Toluene | 10 min | NR |
| 16 | Yb(OTf)3 (0.5) | Toluene | 10 min | NR |
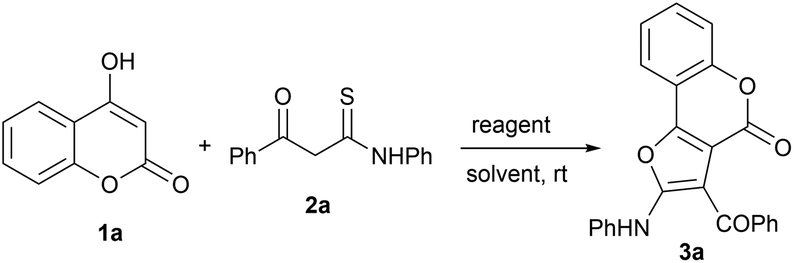
Next, we used phenyliodine(III) diacetate (0.2 equiv.) instead of iodine, and the product was obtained in 10% isolated yield after 30 min (Table 1, entry 3). With PIDA as a good promoter in hand, next we planned to optimize its loading, and it was found that the use of 1.5 equiv. of PIDA provided the best results (Table 1, entries 4–6). In order to achieve better efficiency, the model reaction was also assessed by varying reaction medium. Thus, the test reaction was performed in several non-polar and polar aprotic solvents, such as benzene, tetrachloromethane chloroform, dichloromethane, dimethylformamide and dichloroethane, but none of them provided better results than toluene (Table 1, entries 7–12). After optimizing toluene as the choice of solvent, we screened other promoters, such as PIFA (bis(trifluoroacetoxy)iodobenzene), NIS (N-iodosuccinimide), ZnCl2 and Yb(OTf)3 (Table 1, entries 13–16). Of these only PIFA triggered the reaction, providing the product in good yield (Table 1, entry 13). Thus, the best yield, cleanest reaction and most facile workup were achieved employing 1a (0.30 mmol), 2a (0.25 mmol) and PIDA (1.5 equiv.) in 2.0 mL of toluene at room temperature in an open atmosphere (Table 1, entry 5).
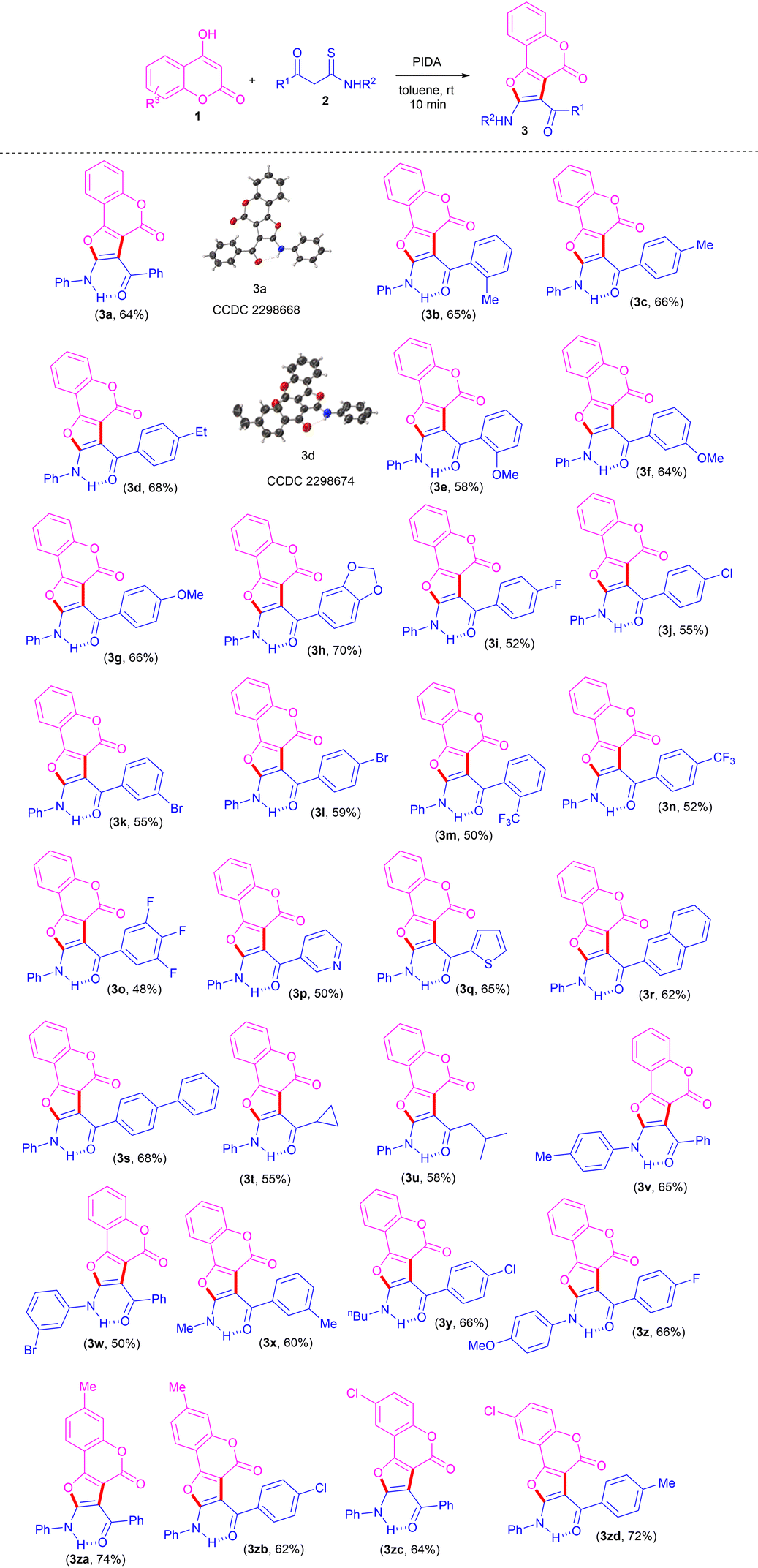
With the optimized conditions to hand, to delineate this approach, the scope and generality of this protocol was next examined by employing a broad range of β-ketothioamides and substituted 4-hydroxy coumarins. Notably, a wide range of β-ketothioamides bearing an R1 moiety as phenyl and aryl groups substituted with both electron-donating (Me, Et, OMe, methylenedioxy) as well as electron-withdrawing (F, Cl, Br, CF3) substituents, irrespective of their ortho-, meta- and para-positions and substitution pattern, were well tolerated, affording the products in moderate to good yield (Table 2, 3a–3l). It was noteworthy that the strong electron-withdrawing groups, such as trifluoromethyl and 3,4,5-trifluorobenzene, were also tolerated well and gave the desired products in good yield (Table 2, 3m–3o). After successful coupling of thioamides bearing the R1 moiety as diverse aryl groups, the R1 moiety as heteroaromatics, such as pyridyl and thienyl, were also tolerated well, furnishing the product in 50% and 65% yield, respectively (Table 2, 3p, 3q).
| Reaction conditions: 1a (0.3 mmol), 2a (0.25 mmol), PIDA (1.5 equiv.) and solvent (2.0 mL), room temperature in an open atmosphere. |
|---|
|
Notably, thioamides bearing not only aromatic and heteroaromatic groups at the R1 moiety, but also extended aromatics, such as naphthyl and biphenyl, were also found to be compatible, affording the products in good yields (Table 2, 3r, 3s). In order to gain further insight into this transformation and to show the versatility of this protocol, thioamides containing an R1 moiety as aliphatic substituents, such as cyclopropyl and iso-butyl, were also used and found to be tolerated well, giving the desired products in good yield (Table 2, 3t, 3u).
To expand the range of substrates for this reaction, the applicability of thioamides bearing different substituents at the R2 moiety was also investigated under standard conditions. The reaction proceeded well, with thioamides bearing the R2 moiety as 4-methylphenyl and 3-bromophenyl furnishing the desired products in good yield (Table 2, 3v, 3w). This protocol has also been found to be compatible with random variation in the moieties R1 and R2, resulting in the corresponding products in high yields (Table 2, 3x–3z). Next, we turned our attention to evaluating the scope of substituted coumarins. 7-Methyl and 6-choloro-coumarins also displayed good reactivity for this transformation, furnishing the corresponding products (Table 2, 3za–3zd) in 62–74% yields.
Furthermore, to check the practical efficacy of this protocol, we performed a large-scale reaction employing 4-hydroxy coumarin 1a (9.6 mmol, 1.55 g) and β-ketothioamide 2a (8 mmol, 2.04 g) in the presence of PIDA (12 mmol, 3.86 g) under optimized reaction conditions (Scheme 2). The desired product 3a was obtained in 61% yield (1.86 g), which was found to be comparable to the small-scale reaction. This observation indicated that this method could be easily implemented for large-scale preparation of furo[3,2-c] coumarins.
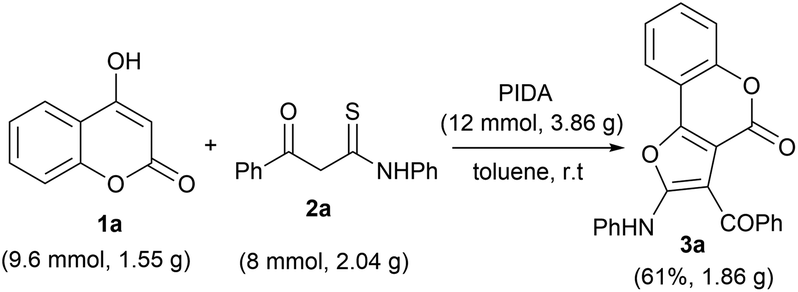 | ||
| Scheme 2 Gram-scale synthesis of 3a. Reaction conditions: 1a (0.3 mmol), 2a (0.25 mmol), PIDA (12 mmol) and solvent (2.0 mL), room temperature in an open atmosphere. | ||
The structures of all the newly synthesized furo[3,2-c]coumarins 3 were fully characterized by their satisfactory spectral (1H, 13C{1H} NMR and HRMS) studies. The mass spectra of these compounds displayed molecular ion peaks at the appropriate m/z values. Further structures of two representative compounds 3a and 3d were unambiguously established by single-crystal X-ray diffraction analysis (Table 2; see ESI for more details, Fig. S1 and S2†).
To devise a probable mechanism, we also carried out some control experiments using a simple coumarin moiety (1′) with β-ketothioamide (2a) under the standard reaction conditions (Scheme 3, eqn (1)). The reaction did not proceed at all and the starting materials remained unconsumed, indicating that the hydroxy group is necessary for the transformation to occur. Next, we performed the reaction of 1a with 2a in the presence of the radical scavenger BHT (butylated hydroxytoluene) (3.0 equiv.) to understand the reaction pathway. Workup of the reaction provided the desired product 3a exclusively, suggesting that the reaction does not involve the radical pathway (Scheme 3, eqn (2)).
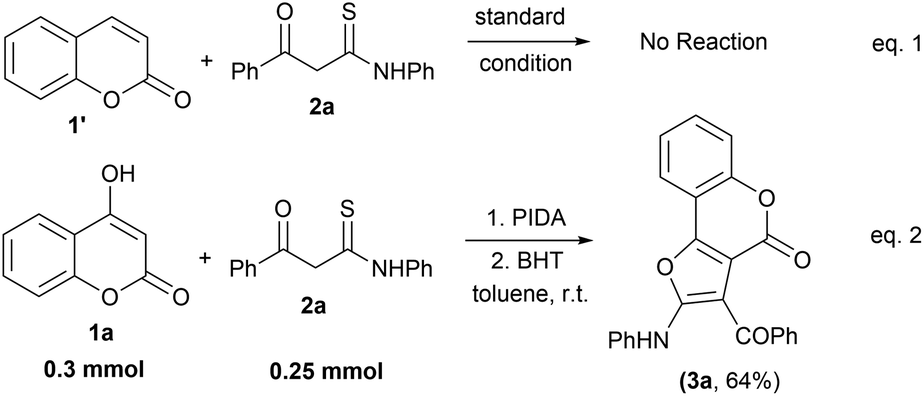 | ||
| Scheme 3 Control experiment. Reaction conditions: 1a (0.3 mmol), 2a (0.25 mmol), PIDA (1.5 equiv.), BHT (3 equiv.) and solvent (2.0 mL), room temperature in an open atmosphere. | ||
Taking into consideration all these reaction outcomes, a plausible pathway for this PIDA-initiated cascade sequence enabling the formation of compounds 3 is depicted in Scheme 4. Initially, PIDA reacts with the 3-position of 4-hydroxy coumarin, increasing its electrophilicity to form intermediate A, which subsequently undergoes nucleophilic attack by the α-carbon of thioamide to generate intermediate B, eliminating PhI and AcOH. Intermediates A and B have both been detected by HRMS analysis (see ESI for details, Fig. S3 and S4†). Intermediate B probably undergoes intramolecular O- or S-cyclization via path I or path II, to give intermediates C and D, respectively. Next, intermediate C suffers dethiohydration to furnish the desired furo[3,2-c]coumarin 3. Generation of H2S as the only by-product makes this process highly attractive19 (detected by the lead acetate test). The alternative path II, for intermediate B, could also be possible, enabling the formation of thiophene-fused coumarin 4via intermediate D, which was not observed, even in a trace, during our observations, with only coumarin 3 being obtained exclusively, suggesting O-cyclization through path I, and making the protocol highly chemo- and regioselective.
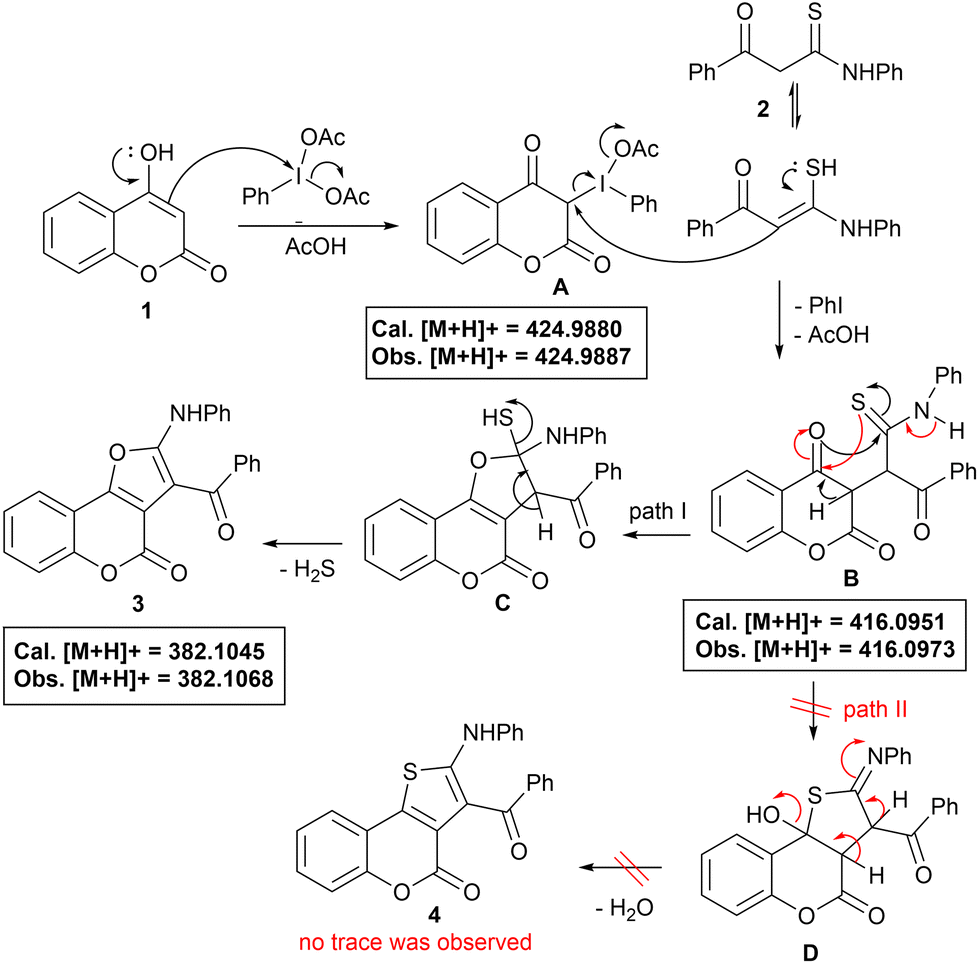 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have successfully synthesized diverse furo[3,2-c]coumarins in moderate to good yields featuring excellent chemo- and regioselectivity under mild conditions. The unique structure of the furo-fused coumarin ring not only imparts unique chemical reactivity, but also the ability to interact with various biological targets, which makes them attractive candidates for the development of novel therapeutic agents. Moreover, to the best of our knowledge, direct intermolecular cross-coupling employing β-ketothioamide to access furo[3,2-c]coumarin has not been described so far. The simple operating module at room temperature, metal-free conditions, protecting group-free, easy workup and gram scalability not only make this cascade strategy very promising for academic research and medicinal applications but also broadens the application of β-ketothioamides. Furo[3,2-c] coumarins are highly significant motifs in natural products and pharmaceutical applications. We anticipate that this approach will be useful for providing a valuable source of new physiologically active coumarin agents.Experimental
General information
The commercially available solvents and reagents were used as received without any further purification. The β-ketothioamides (2) were synthesized by the reported procedure.20 Toluene was purchased from Merck. PIDA and coumarins were purchased from Sigma Aldrich. All the reactions were monitored by analytical thin layer chromatography (TLC) using Merck pre-coated aluminium sheets, and visualized by a UV lamp. Flash column chromatography was performed on silica gel (230–400 mesh). The 1H and 13C{1H} NMR spectra were recorded on a JEOL 500 FT-NMR spectrometer operating at 500 and 125 MHz, respectively. Chemical shifts (δ) for 1H and 13C{1H} NMR are given in parts per million (ppm) using the residual solvent peaks as a reference relative to tetramethylsilane (TMS). Coupling constant (J) values are reported in hertz (Hz). High-resolution mass spectra (HRMS, m/z) were recorded in EI or ESI mode, on a Sciex X500R QTOF instrument. All the reactions were carried out using a single-neck round-bottom (25 mL) borosilicate flask. Melting points were determined with a Büchi B-540 melting point apparatus and are uncorrected. IUPAC names were obtained using the ChemDraw (version 19.1) software.General procedure for the synthesis of compounds 3
4-Hydroxy coumarins 1 (0.3 mmol), β-ketothioamides 2 (0.25 mmol) and PIDA (1.5 equiv.) were added into a 25 mL oven-dried, single-neck, round-bottom flask, followed by addition of 2.0 mL of toluene. The reaction mixture was stirred at room temperature for 10 min in an open atmosphere. After completion of the reaction (monitored by TLC), the reaction was quenched with water (10 mL), followed by extraction with ethyl acetate (15 mL) and water (2 × 10 mL). The combined organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude residue thus obtained was purified by silica gel flash column chromatography to give the pure products 3 (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane/ethyl acetate). The isolated product was dried under vacuum and then the analytical studies were performed.
:1 hexane/ethyl acetate). The isolated product was dried under vacuum and then the analytical studies were performed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Science and Engineering Research Board (CRG/2023/000021), IoE Incentive grants, BHU (Scheme No. 6031) and JC Bose National Fellowship (JCB/2020/000023), New Delhi. The authors (AKY, DY, VK and SR) are thankful to UGC, New Delhi for fellowships. We also acknowledge the support of DST-FIST for basic and high-end analytical facilities for the department.References
- (a) C. Kontogiorgis, A. Detsi and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat., 2012, 22, 437–454 CrossRef CAS PubMed; (b) K. N. Venugopala, V. Rashmi and B. Odhav, BioMed. Res. Int., 2013, 2013, 1–14 CrossRef PubMed; (c) D. Yu, M. Suzuki, L. Xie, S. L. Morris-Natschke and K. H. Lee, Med. Res. Rev., 2003, 23, 322–345 CrossRef CAS PubMed.
- (a) M. Musa, J. Cooperwood and M. O. Khan, Curr. Med. Chem., 2008, 15, 2664–2679 CrossRef CAS PubMed; (b) A. Kathuria, S. Jalal, R. Tiwari, A. Nasrolahi-Shirazi, S. Gupta, S. Kumar, K. Parang and S. K. Sharma, Chem. Biol. Interfaces, 2011, 1, 279–296 CAS; (c) G. Hertrampf, K. Kusserow, S. Vojnovic, A. Pavic, J. I. Müller, J. Nikodinovic-Runic and T. A. M. Gulder, Chem. – Eur. J., 2022, 28, e202200394 CrossRef CAS PubMed.
- (a) M. Golshani, M. Khoobi, N. Jalalimanesh, F. Jafarpour and A. Ariafard, Chem. Commun., 2017, 53, 10676–10679 RSC; (b) B. Niu, W. Zhao, Y. Ding, Z. Bian, C. U. Pittman, A. Zhou and H. Ge, J. Org. Chem., 2015, 80, 7251–7257 CrossRef CAS PubMed; (c) F. Jafarpour, M. Darvishmolla, N. Azaddoost and F. Mohaghegh, New J. Chem., 2019, 43, 9328–9332 RSC; (d) H. Stefani, K. Gueogjan, F. Manarin, S. Farsky, J. Zukerman-Schpector, I. Caracelli, S. Pizano Rodrigues, M. Muscará, S. Teixeira, J. Santin, I. Machado, S. Bolonheis, R. Curi and M. Vinolo, Eur. J. Med. Chem., 2012, 58, 117–127 CrossRef CAS PubMed; (e) M. Kumar, T. Kaur, V. K. Gupta and A. Sharma, RSC Adv., 2015, 5, 17087–17095 RSC.
- (a) M. Gupta, P. Kumar, V. Bahadur, K. Kumar, V. S. Parmar and B. K. Singh, Eur. J. Med. Chem., 2018, 2018, 896–900 CAS; (b) D. Kang, K. Ahn and S. Hong, Asian J. Org. Chem., 2018, 7, 1136–1150 CrossRef CAS; (c) M. Min, Y. Kim and S. Hong, Chem. Commun., 2013, 49, 196–198 RSC; (d) X. H. Cao, X. Pan, P. J. Zhou, J. P. Zou and O. T. Asekun, Chem. Commun., 2014, 50, 3359–3362 RSC.
- (a) Biology, Applications, and Mode of Action, ed. R. O'Kennedy and R. D. Thornes, John Wiley & Sons, Chichester, New York, 1997 Search PubMed; (b) Advances in Structure and Activity Relationship of Coumarin Derivatives, ed. S. Penta, Academic Press, 2015 Search PubMed; (c) Q. Yang, R. Guo and J. Wang, Asian J. Org. Chem., 2019, 8, 1742–1765 CrossRef CAS; (d) L. Wu, X. Wang, W. Xu, F. Farzaneh and R. Xu, Curr. Med. Chem., 2009, 16, 4236–4260 CrossRef CAS PubMed; (e) R. D. H. Murray, J. Mendey and S. A. Brown, The Natural Coumarins, Wiley, New York, 1982 Search PubMed.
- (a) L. Huang, X. Yuan, D. Yu, K. H. Lee and C. H. Chen, Virology, 2005, 332, 623–628 CrossRef CAS PubMed; (b) F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887–916 CrossRef CAS PubMed; (c) H. A. El-Wahab, M. A. El-Fattah, N. A. El-Khalik, H. S. Nassar and M. M. Abdelall, Prog. Org. Coat., 2014, 77, 1506–1511 CrossRef CAS; (d) G. Zhao, C. Peng, W. Du and S. Wang, Fitoterapia, 2013, 89, 250–256 CrossRef CAS PubMed; (e) T. Smyth, V. N. Ramachandran and W. F. Smyth, Int. J. Antimicrob. Agents, 2009, 33, 421–426 CrossRef CAS PubMed; (f) O. Galayev, Y. Garazd, M. Garazd and R. Lesyk, Eur. J. Med. Chem., 2015, 105, 171–181 CrossRef CAS PubMed; (g) N. Renuka and K. A. Kumar, Bioorg. Med. Chem. Lett., 2013, 23, 6406–6409 CrossRef CAS; (h) M. Z. Zhang, R. R. Zhang, J. Q. Wang, X. Yu, Y. L. Zhang, Q. Q. Wang and W. H. Zhang, Eur. J. Med. Chem., 2016, 124, 10–16 CrossRef CAS PubMed; (i) Y. Lu, D. Hu, S. Ma, X. Zhao, S. Wang, G. Wei, X. Wang, A. Wen and J. Wang, Int. Immunopharmacol., 2016, 34, 44–52 CrossRef CAS PubMed; (j) Y. G. Peng and L. Zhang, J. Dermatol. Treat., 2019, 30, 389–395 CrossRef CAS PubMed.
- (a) S. M. Razavi, H. Nazemiyeh, A. Delazar, R. Hajiboland, M. M. Rahman, S. Gibbons, L. Nahar and S. D. Sarker, Phytochem. Lett., 2008, 1, 159–162 CrossRef CAS; (b) N. Z. Ballin and A. T. Sørensen, Food Control, 2014, 38, 198–203 CrossRef CAS; (c) A. Fischer, C. Cremer and E. H. Stelzer, Appl. Opt., 1995, 34, 1989–2003 CrossRef CAS PubMed; (d) G. Cravotto, G. M. Nano, G. Palmisano and S. Tagliapietra, Tetrahedron: Asymmetry, 2001, 12, 707–709 CrossRef CAS; (e) R. Pratap and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed; (f) S. Sandhu, Y. Bansal, O. Silakari and G. Bansal, Bioorg. Med. Chem., 2014, 22, 3806–3814 CrossRef CAS PubMed; (g) M. Zahradink, The Production and Application of Fluorescent Brightening Agents, Wiley, New York, 1992 Search PubMed; (h) M. Maeda, Laser Dyes, Academic Press, New York, 1984 Search PubMed.
- (a) R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402–4404 CrossRef CAS PubMed; (b) M. Ochiai and K. Miyamoto, Eur. J. Org. Chem., 2008, 4429–4239 Search PubMed; (c) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC; (d) M. S. Yusubov and V. V. Zhdankin, Mendeleev Commun., 2010, 20, 185–191 CrossRef CAS; (e) F. V. Singh and T. Wirth, Chem. – Asian J., 2014, 9, 950–971 CrossRef CAS PubMed.
- (a) J. I. Musher, Angew. Chem., Int. Ed. Engl., 1969, 8, 54–68 CrossRef CAS; (b) J. C. Martin, Science, 1983, 221, 509–514 CrossRef CAS PubMed; (c) E. Le Du and J. Waser, Chem. Commun., 2023, 59, 1589–1604 RSC.
- (a) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef CAS PubMed; (b) R. L. Amey and J. C. Martin, J. Org. Chem., 1979, 44, 1779–1784 CrossRef CAS.
- T.-Y. Sun, X. Wang, H. Geng, Y. Xie, Y.-D. Wu, X. Zhang and H. F. Schaefer III, Chem. Commun., 2016, 52, 5371–5374 RSC.
- (a) V. V. Zhdankin, Hypervalent Iodine Chemistry; Preparation, Structure and Synthetic, Wiley, Chichester, vol. I, 2013 CrossRef; (b) T. Dohia and Y. Kita, Chem. Commun., 2009, 2073–2085 RSC; (c) F. V. Singh and T. Wirth, Chem. – Asian J., 2014, 9, 950 CrossRef CAS PubMed; (d) R. Narayan, S. Manna and A. P. Antonchick, Synlett, 2015, 26, 1785–1803 CrossRef CAS; (e) A. Maity and D. C. Powers, Synlett, 2019, 30, 257–262 CrossRef CAS; (f) F. V. Singh, S. E. Shetgaonkar, M. Krishnan and T. Wirth, Chem. Soc. Rev., 2022, 51, 8102–8139 RSC.
- (a) L. Santana, E. Uriarte, F. Roleira, N. Milhazes and F. Borges, Curr. Med. Chem., 2004, 11, 3239–3261 CrossRef CAS PubMed; (b) X. Zhu, X. P. Xu, C. Sun, T. Chen, Z. L. Shen and S. J. Ji, Tetrahedron, 2011, 67, 6375–6381 CrossRef CAS; (c) M. Adib, M. Mahdavi, S. Bagherzadeh and H. R. Bijanzadeh, Synlett, 2009, 2542–2544 CrossRef CAS; (d) M. Kumar, S. Bagchi and A. Sharma, RSC Adv., 2015, 5, 53592–53603 RSC; (e) N. Kerru, L. Gummidi, K. K. Gangu, S. Maddila and S. B. Jonnalagadda, ChemistrySelect, 2020, 5, 4104–4110 CrossRef CAS; (f) R. Lei, W. Lan, M. Li, Z. Qin and B. Fu, Tetrahedron Lett., 2023, 128, 154697 CrossRef CAS; (g) S. Battula, P. Kothuri, H. Bhumannagaria and K. Nayani, Org. Biomol. Chem., 2024, 22, 1671–1675 RSC; (h) L. Chen, Y. Li and M.-H. Xu, Org. Biomol. Chem., 2010, 8, 3073–3077 RSC; (i) S. M. Rajesh, S. Perumal, J. C. Menendez, S. Pandian and R. Murugesan, Tetrahedron, 2012, 68, 5631–5636 CrossRef CAS.
- (a) M. H. Mosslemin, M. Anary-Abbasinejad, A. F. Nia, S. Bakhtiari and H. Anaraki-Ardakani, J. Chem. Res., 2009, 2009, 599–601 CrossRef; (b) P. H. Pham, Q. T. Nguyen, N. K. Tran, V. H. Nguyen, S. H. Doan, H. Q. Ha, T. Truong and N. T. Phan, Eur. J. Org. Chem., 2018, 4431–4435 CrossRef CAS; (c) S. S. Vagh, B. J. Hou, A. Edukondalu, P. C. Wang and W. Lin, Org. Lett., 2021, 23, 842–846 CrossRef CAS PubMed.
- S. Ray, V. Kumar, S. Singh, K. Bandyopadhyay, S. Saha and M. S. Singh, Adv. Synth. Catal., 2024, 366, 262–268 CrossRef CAS.
- (a) P. Pali and M. S. Singh, Org. Lett., 2023, 25, 2258–2263 CrossRef CAS PubMed; (b) A. K. Yadav, V. Kumar, P. Pali, S. Ray, A. Verma and M. S. Singh, Adv. Synth. Catal., 2023, 365, 2943–2948 CrossRef CAS; (c) G. Shukla, K. Raghuvanshi and M. S. Singh, J. Org. Chem., 2022, 87, 13935–13944 CrossRef CAS PubMed; (d) S. Ray, S. Singh, G. Kumar, G. Shukla and M. S. Singh, Adv. Synth. Catal., 2022, 364, 3204–3210 CrossRef CAS; (e) S. Soni, G. Shukla and M. S. Singh, Org. Biomol. Chem., 2022, 20, 6784–6798 RSC.
- (a) C. H. Ding, J. L. Dong, L. S. H. Yu and J. W. Xie, Chin. Chem. Lett., 2018, 29, 517–520 CrossRef CAS; (b) L. Wen, N. Wang, W. Du, M. Zhu, C. Pan, L. Zhang and M. Li, Chin. J. Chem., 2021, 39, 1831–1837 CrossRef CAS; (c) J. Zheng, H. Gu, Q. Chen and W. Yang, Org. Biomol. Chem., 2023, 21, 2069–2080 RSC; (d) M. Li, X. J. Kong and L. R. Wen, J. Org. Chem., 2015, 80, 11999–12005 CrossRef CAS; (e) H. Lu, C. Y. Tan, H. X. Zhang, J. L. Zhang, J. Y. Liu, H. Y. Li and P. F. Xu, J. Org. Chem., 2018, 83, 15245–15255 CrossRef CAS PubMed; (f) X. Zhang, C. Liu, Y. Deng and S. Cao, Org. Biomol. Chem., 2020, 18, 7540–7544 RSC; (g) Q. Zeng, X. Huang, M. Liu, Z. Yu and Y. Xiao, Org. Lett., 2022, 24, 8186–8191 CrossRef CAS PubMed.
- (a) M. A. Ansari, D. Yadav, S. Soni, A. Srivastava and M. S. Singh, J. Org. Chem., 2019, 84, 5404–5412 CrossRef CAS; (b) M. A. Ansari, D. Yadav, S. Soni and M. S. Singh, Org. Biomol. Chem., 2019, 17, 9151–9162 RSC; (c) M. A. Ansari, D. Yadav and M. S. Singh, Chem. – Eur. J., 2020, 26, 8083–8089 CrossRef CAS PubMed; (d) P. Pali, G. Shukla, P. Saha and M. S. Singh, Org. Lett., 2021, 23, 3809–3813 CrossRef CAS PubMed; (e) G. Shukla, P. Saha, P. Pali, K. Raghuvanshi and M. S. Singh, J. Org. Chem., 2021, 86, 18004–18016 CrossRef CAS PubMed; (f) P. Pali, D. Yadav, S. C. Sahoo and M. S. Singh, J. Org. Chem., 2022, 87, 12342–11235 CrossRef CAS PubMed; (g) S. Singh, G. Shukla, S. Ray and M. S. Singh, Synthesis, 2023, 55, 1893–1903 CrossRef CAS; (h) S. Khan, M. A. Ansari and M. S. Singh, Org. Lett., 2023, 25, 642–646 CrossRef CAS PubMed; (i) G. Kumar, M. A. Ansari and M. S. Singh, Tetrahedron, 2023, 142, 133542 CrossRef CAS; (j) D. Yadav and M. S. Singh, Eur. J. Org. Chem., 2023, e202201335 CrossRef CAS.
- T. Ausma and L. J. De Kok, Front. Plant Sci., 2019, 10, 1–10 CrossRef PubMed.
- X. M. Zeng, C. Y. Meng, J. X. Bao, D. C. Xu, J. W. Xie and W. D. Zhu, J. Org. Chem., 2015, 80, 11521–11528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2298668 and 2298674. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00438h |
| This journal is © The Royal Society of Chemistry 2024 |