
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sumanene–carbazole conjugate with push–pull structure and its chemoreceptor application†
Dominika
Ufnal‡
a,
Jakub S.
Cyniak‡
 a,
Maurycy
Krzyzanowski
a,
Krzysztof
Durka
a,
Hidehiro
Sakurai
bc and
Artur
Kasprzak
*a
a,
Maurycy
Krzyzanowski
a,
Krzysztof
Durka
a,
Hidehiro
Sakurai
bc and
Artur
Kasprzak
*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego Str. 3, 00-664 Warsaw, Poland. E-mail: artur.kasprzak@pw.edu.pl
bDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, 565-0871 Osaka, Japan
cInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICSOTRI), Osaka University, Suita 565-0871, Osaka, Japan
First published on 15th May 2024
Abstract
The first-of-its-kind tetra-substituted sumanene derivative, featuring the push–pull chromophore architecture, has been successfully designed. The inclusion of both strong electron-withdrawing (CF3) and electron-donating (carbazole) moieties in this buckybowl compound has enhanced the charge transfer characteristics of the molecule. This enhancement was supported by ultraviolet-visible (UV-Vis) and emission spectra analyses along with density functional theory (DFT) calculations. The application of the title sumanene–carbazole push–pull chromophore as a selective recognition material for cesium cations (Cs+) was also presented. The title compound exhibited effective and selective Cs+-trapping ability, characterized by a high apparent binding constant value (at the level of 105) and a low limit of detection (0.09–0.13 μM). Owing to the tuned optical properties of the title push–pull chromophore, this study marks the first time in sumanene-tethered chemoreceptor chemistry where efficient tracking of Cs+ binding was possible with both absorption and fluorescence spectroscopies. This work introduces a new approach toward tuning the structure of bowl-shaped optical chemoreceptors.
Introduction
Bowl-shaped and π-conjugated compounds, termed buckybowls, constitute an intriguing class of compounds featuring various interesting physicochemical properties.1–6 Sumanene buckybowl (1; Fig. 1a) serves as the subunit of fullerene C60, and was first successfully synthesized in 2003.7,8 The unique geometrical feature of sumanene is its bowl shape, with the identified concave and convex sites of bowl, see Fig. 1a.3,9,10 Over the past decade, sumanene chemistry has advanced significantly. Owing to the π-conjugated and bowl structures, the sumanene backbone became an attractive motif for designing effective strongly fluorescent compounds. Tuning the properties of sumanene-tethered chromophore architectures can be achieved through methods like expanding the π-electron network,11–16 heteroatom doping of the bowl,17–21 or the formation of metal complexes.22,23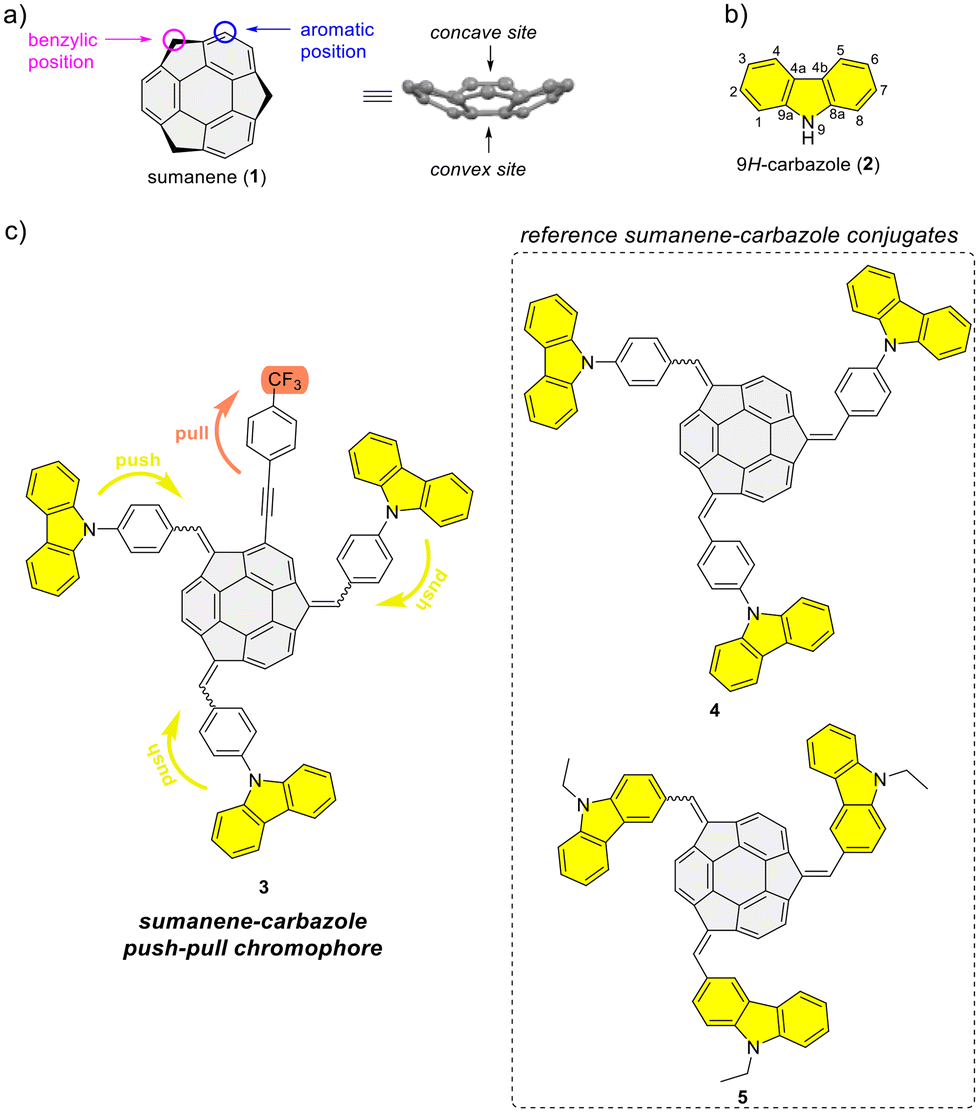 | ||
| Fig. 1 (a) Structure of sumanene (1), (b) structure of 9H-carbazole (2) together with atom numbering, and (c) structure of target sumanene–carbazole push–pull chromophore 3 together with the reference sumanene–carbazole conjugates. | ||
There is continuous interest in designing efficient light-emitting compounds,24–30 and one of the effective methods for tuning the optical properties of molecules is through the design of push–pull chromophores. These chromophores exhibit intramolecular charge-transfer properties through donor–acceptor interactions. As a result of promising optical properties, widespread applications of such systems have been investigated over the years.31–33 In light of the push–pull chromophore design, the 9H-carbazole backbone (2; Fig. 1b) is commonly used as an effective electron donor.34–38 The addition of a nitrogen atom to a π-conjugated carbon system not only alters the energy levels of the molecular orbitals of the aromatic compound but also crucially impacts its electronic structure, thereby profoundly affecting the electronic properties of the molecule. The 9H-carbazole backbone has demonstrated many important properties for designing effective light-emitting compounds.39–42 Incorporating this motif into various organic π-systems, including those with bowl-shaped corannulene,43–45 has resulted in improved light-emission properties.46–50 Importantly, tuning the optical and electronic properties of 9H-carbazole derivatives is feasible, given the numerous reported methods for functionalizing this heterocyclic compound.51–53
From 2019 to 2023, our study delved into the application of selected sumanene derivatives as chemoreceptors with high selectivity for cesium cations (Cs+).13,54–56 This research holds environmental importance in designing new Cs+ recognition materials results, e.g., from the need to monitor the presence of radioactive Cs+ in the environment, especially in the post-disaster areas after nuclear plant accidents.57–59 Despite yielding satisfactory binding parameters, we recognized a limitation of the sumanene-based chemoreceptors, specifically, the effective tracking of interactions in solution was achievable only through spectrofluorimetry. On the other hand, ultraviolet-visible (UV-Vis) absorption instruments are generally more cost-effective than spectrofluorometers and are widely available in environmental laboratories. Therefore, designing sumanene-based probes capable of efficiently tracking Cs+ binding using both UV-Vis spectroscopy and spectrofluorimetry is deemed desirable. Given the attractive optical properties of carbazole-containing push–pull chromophores,34–38 we identified an opportunity to merge the chemistry of carbazole-based chromophores with the science of sumanene-tethered Cs+-selective optical chemoreceptors. Resulting of these considerations, we report the successful design of the sumanene–carbazole conjugate 3 (Fig. 1c), featuring the push–pull chromophore architecture in sumanene science for the first time. Anticipating that this molecular architecture, enhancing internal charge-transfer characteristics within the buckybowl compound 3, would enable effective tracking of the Cs+-trapping phenomenon using both absorption and emission spectroscopies. The design of molecule 3 was based on the presence of both strong electron-withdrawing (CF3) and electron-donating moieties (carbazole units) attached to the sumanene buckybowl backbone. Furthermore, compound 3 was designed based on comparative studies on the photophysical properties of reference sumanene–carbazole conjugates 4–5 (Fig. 1c) and comprehensive density functional theory (DFT) calculations (Gaussian16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 software).
60 software).
Results and discussion
Synthesis
Refer to the ESI, Subsection S1,† for full experimental details on the synthesis of sumanene–carbazole push–pull chromophore 3, as well as compounds 4–5 and 10.The synthesis of reference compounds 4–5 (Scheme 1a) relied on the sodium hydroxide-mediated condensation-type reaction61 under the phase transfer catalysis (PTC) conditions between sumanene (1) and the respective aldehyde (6–7; yield 40%–53%). Building on the successful synthesis of compound 4 and leveraging the possibilities of modifying sumanene (1) at aromatic and benzylic positions, the design of compound 3, bearing both electron-withdrawing (CF3) and electron-donating groups (carbazole units), was accomplished. The synthesis of compound 3 (Scheme 1b) commenced with the production of 2-((4-(trifluoromethyl)phenyl)ethynyl)sumanene (compound 10) as the key-starting material. Initially, 2-iodosumanene (8) was obtained via the electrophilic aromatic substitution (SEAr) reaction55,62 using 1,3-diiodo-5,5-dimethylhydantoin as the iodinating agent, in the presence of catalytic amount of trifluoromethanesulfonic acid. Compound 10 was synthesized (72%) through the Sonogashira cross-coupling reaction between 8 and 1-ethynyl-4-(trifluoromethyl)benzene (9). This reaction was performed at 45 °C in triethylamine (TEA) acting both as the solvent and a base, with the usage of copper(I) iodide (CuI) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (Pd(PPh3)2Cl2) as the catalysts. The target compound 3 was then synthesized (48%) from 10 and aldehyde 6, using similar PTC condensation-type conditions as for compounds 4–5. The highest isolated yield of 3 was found for 8 molar equivalents of aldehyde 6 used per 1 molar equivalent of compound 10. Pure compound 3 was isolated as a red solid by means of the preparative thin layer chromatography (PTLC) using 50% CH2Cl2/hexane as an eluent. Importantly, to date, only native sumanene (1) was used in the discussed PTC condensation-type reaction for the synthesis of tris-substituted sumanenes.13,56,61,63,64 Therefore, from the synthetic viewpoint, the successful synthesis of compound 3 demonstrated the feasibility of using a 2-substituted sumanene derivative in this process, leading to the formation of a tetra-substituted sumanene-containing compound.
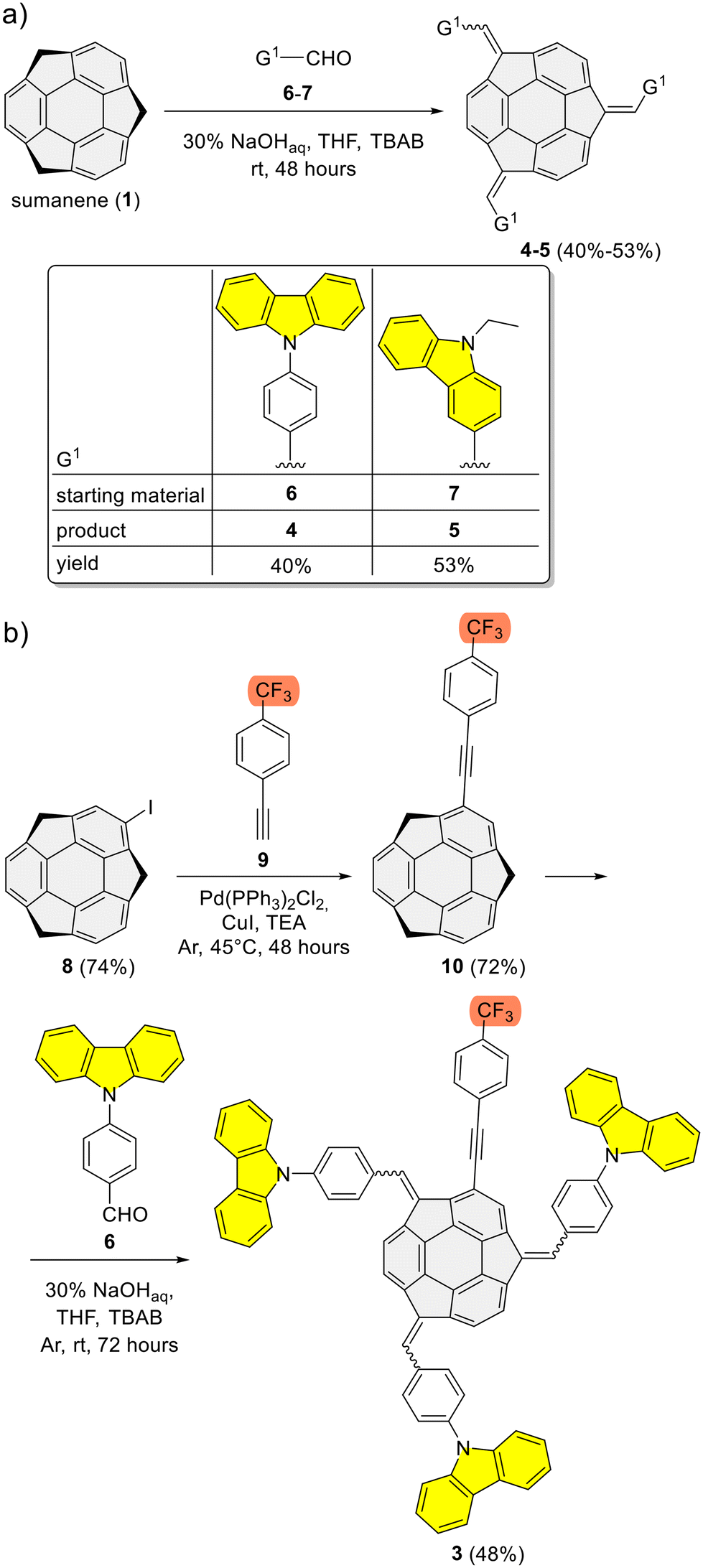 | ||
| Scheme 1 Synthesis of sumanene–carbazole conjugates: (a) reference compounds 4–5 and (b) push–pull chromophore 3. | ||
The formation of compounds 3–5 and 10 was confirmed using 1D (1H, {1H}13C), 2D (1H–1H COSY, 1H–13C HSQC, 19F–13C HSQC, 1H–13C HMBC) and pseudo-2D (1H DOSY, 19F DOSY) nuclear magnetic resonance (NMR) spectroscopy and electrospray ionization high-resolution mass spectrometry (ESI-HRMS), whereas their purity was further confirmed using elemental analysis. Refer to the ESI, subsections S1–S3,† for full characterization data for 3–5 and 10. Section S2 in ESI† contains the copies of NMR spectra of 3–5 and 10, as well as detailed discussion on the NMR spectra of sumanene–carbazole conjugates 3–5. Notably, similar to previous studies on sumanene derivatives synthesized with the PTC condensation-type reaction,13,56,61,63,64 the NMR spectra revealed the formation of diastereoisomers of compounds 3–5 (two and four possible diastereoisomers for 4–5 and 3, respectively). The relative stabilities of DFT-optimized diastereoisomers of compounds 3–5 are provided in the paragraph on “theoretical calculations” in the text. In brief regarding NMR characterization data for the target sumanene–carbazole push–pull chromophore 3, 1H NMR spectrum of 3 comprised two major groups of multiplets in the aromatic region (Fig. 2b). The signals from the multiplet group at 8.33–8.01 ppm (12H) were ascribed to the H-1, H-4, H-5 and H-8 protons within of the carbazole moiety (see atom numbering in Fig. 2a). The signals included the multiplet group at 7.89–7.06 ppm (36H) were ascribed to the protons of the following moieties: 2-substituted sumanene (5H), carbazole H-2, H-3, H-6, H-7 (12H), p-phenylene linkers (16H) and methidene linkers (3H). Notably, signals coming from the benzylic protons of sumanene backbone were not found in the 1H NMR spectrum of 3, what further supported tetra-substitution of the sumanene skeleton (in the spectrum of starting material 10 (in CDCl3), characteristic doublets originating from the presence of benzylic Hexo and Hendo protons of sumanene were found between 4.79–4.69 ppm and 3.64–3.42 ppm, respectively). The presence of 2-substituted sumanene backbone in 3 was also supported based on the comparative analyses with the 1H NMR spectrum of respective compound 4, which contained sumanene skeleton non-substituted at the aromatic position, see Fig. S30 in ESI together with the discussion in section S2.4.† The 19F NMR experiment (Fig. 2c) supported the presence of fluorine in the 3 sample in the form of trifluoromethyl (CF3) group (δF from −63.45 to −63.48 ppm), whereas 13C decoupled 19F–13C HSQC experiment enabled to distinguish chemical shift for CF3 group in the {1H}13C NMR spectrum of 3 (δC 125.3–125.2 ppm, see Fig. S35 in ESI†). The signals in the {1H}13C NMR spectrum of 3 were also tracked based on the 1H–13C HMBC NMR spectrum, see Fig. S36–S37 in ESI together with the discussion in section S2.4.† In particular in terms of the analysis of {1H}13C NMR spectrum of 3, 1H–13C HMBC NMR spectrum enabled the confirmation of the presence of low-intensity quaternary carbons of sumanene skeleton between 150–120 ppm and –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– moiety (93.4–92.3 ppm). Notably, 1H DOSY NMR experiment (Fig. 2d) confirmed that the 3 sample comprised a single type of molecule (diffusion coefficient for 3 was 4.78 × 10−10 m2 s−1), with an approximate hydrodynamic radius of ca. 0.96 nm. Importantly, similar outcomes were found from 19F DOSY NMR experiment (Fig. 2e), with measured diffusion coefficient (4.76 × 10−10 m2 s−1) consistent with the respective value from the 1H DOSY NMR experiment. Finally, ESI-HRMS spectrum (Fig. 2f) ultimately confirmed that 3 was obtained. Refer to section S2.4 in ESI† for further detailed discussion about the NMR spectra of push–pull chromophore 3.
C– moiety (93.4–92.3 ppm). Notably, 1H DOSY NMR experiment (Fig. 2d) confirmed that the 3 sample comprised a single type of molecule (diffusion coefficient for 3 was 4.78 × 10−10 m2 s−1), with an approximate hydrodynamic radius of ca. 0.96 nm. Importantly, similar outcomes were found from 19F DOSY NMR experiment (Fig. 2e), with measured diffusion coefficient (4.76 × 10−10 m2 s−1) consistent with the respective value from the 1H DOSY NMR experiment. Finally, ESI-HRMS spectrum (Fig. 2f) ultimately confirmed that 3 was obtained. Refer to section S2.4 in ESI† for further detailed discussion about the NMR spectra of push–pull chromophore 3.
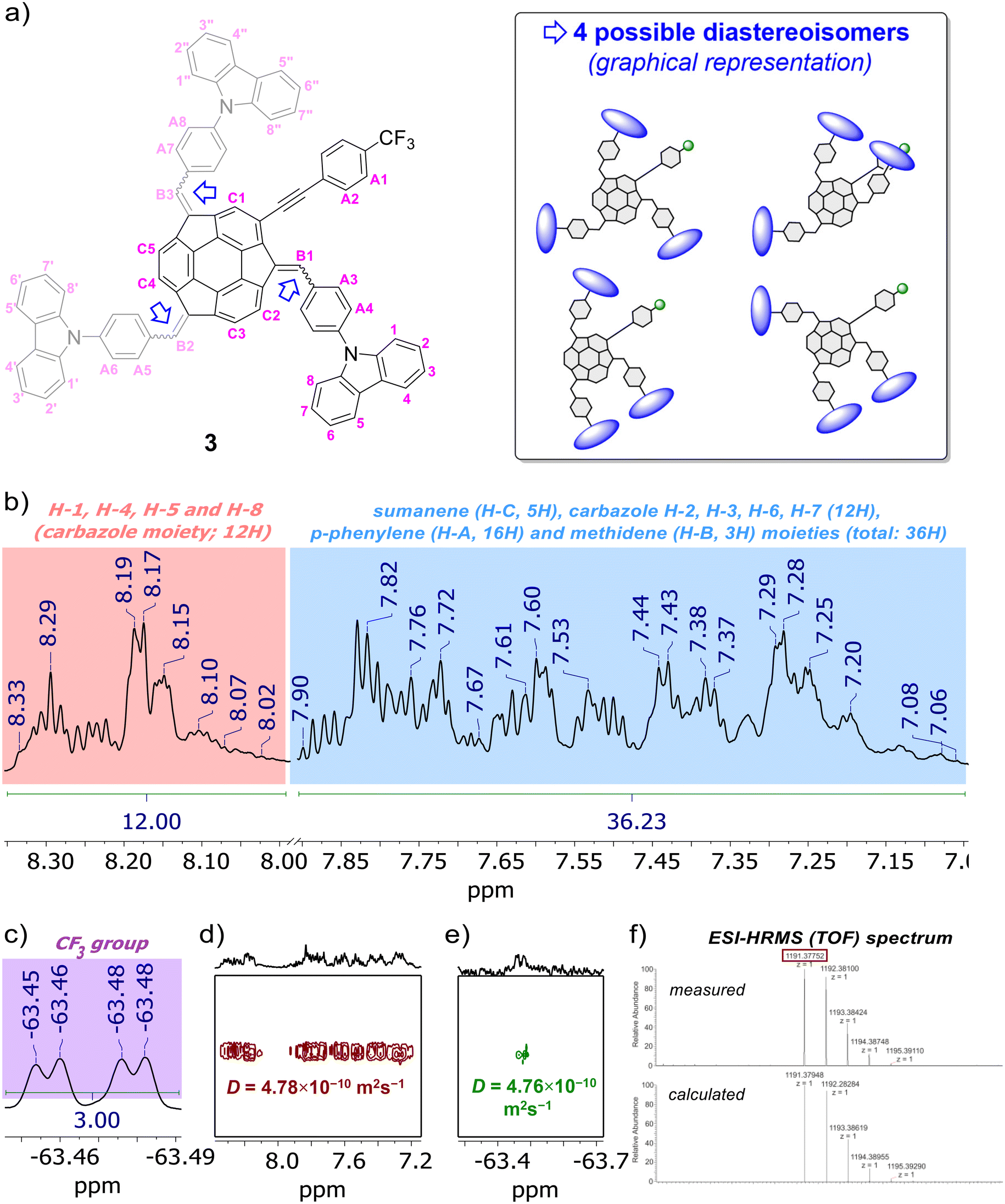 | ||
| Fig. 2 (a) Structure of push–pull chromophore 3 with atom numbering regarding NMR analyses, as well as graphical representation of the possible diastereoisomers (DFT-optimized diastereoisomers of 3 are presented and discussed in section S5, ESI†). Selected characterization data for push–pull chromophore 3: insets of (b) 1H NMR, (c) 19F NMR, (d) 1H DOSY NMR, (e) 19F DOSY NMR spectra (600 MHz, THF-d8) and (f) ESI-HRMS (TOF) spectrum. In the presented insets of NMR spectra, selected peaks are marked for the clarity of the images (for the full range spectra and full characterisation data on 3 with discussion, see sections S1–S3, ESI†). | ||
Photophysical properties
The spectral data of investigated compounds are summarized in Table 1, with detailed discussion provided in section S4 in ESI.†| Experimental data | TD-DFT calculations | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
λ
absa/nm |
λ
emb/nm |
Φ F |
λ
absc/nm |
λ
emc/nm |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Molar absorption coefficient values (ε; dm3 mol−1 cm−1) are given in parentheses. b For solid state samples values are provided in parentheses. c Excited state numbers and oscillator strength are given in parentheses. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
155 |
280 | 375 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 478 (1.0 × 105), 388 (2.2 × 105), 340 (2.5 × 105), 290 (3.3 × 105) | 552 (615) | 0.76 | 493 (S1, 0.056), 401 (S7, 0.286), 359 (S11, 0.308) | 552 (S1, 0.055) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 474 (2.0 × 104), 386 (4.7 × 104), 342 (4.7 × 104), 290 (6.7 × 104) | 549 (580) | 0.53 | 497 (S1, 0.327), 370 (S10, 0.432), 358 (S12, 0.358) | 547 (S1, 0.262) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 490 (3.1 × 104), 394 (6.4 × 104), 332 (3.4 × 104), 302 (5.0 × 104) | 558 (594) | 0.12 | 480 (S1, 0.356), 368 (S5, 0.227), 352 (S8, 0.648) | 570 (S1, 0.159) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 346 (2.6 × 104), 280 (7.0 × 104) | 400 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
1165 |
255 | 361 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 290 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The substitution of the sumanene skeleton with carbazole moieties led to a significant alternation in light absorption and emission properties of compounds 3–5 compared with native sumanene (1; one absorption maximum (λabs) at ca. 280 nm) and 1,3,5-tri(9H-carbazol-9-yl)benzene 11 (λabs = 255, 290, 346 nm),65 which served as the representative C3-symmetrical aromatic derivative comprising three carbazole units in the formula. Differences between the spectra of reference compounds 4–5 were concluded, what elucidated the effect the method of substitution of carbazole skeleton on the photophysical properties of the resultant conjugates with sumanene.
In the case of compounds 4–5, the tris-substitution of the sumanene skeleton with carbazole moieties resulted in the emergence of three major absorption bands ranging from 300 to 550 nm (refer to the spectra of 4–5 and respective discussions, including DFT calculations regarding experimental absorption spectra and calculated electronic transitions, in ESI, sections S4 and S5†). The UV-Vis spectrum of compound 3 (2 × 10−5 M; CHCl3) is presented in Fig. 3, along with a comparison with the spectrum of parent compound 10 and reference compound 4. Compound 3 exhibited several absorption bands with maxima at 250, 290, 340, 388, and 478 nm. For comparison, parent compound 10 exhibits the longest wavelength absorption band at 355 nm. Although the spectral profile of 3 was relatively similar to that of compound 4, notable changes were found in the absorption intensities and, thus, molar absorption coefficient (ε) values. Notably, ε values for the respective absorption bands for 3 were ca. 5-fold higher than for 4 (at respective λabs, Table 1). In particular, when the spectrum of 3 was measured at the same concentration as 4 (2 × 10−5 M), the absorption intensities of 3 were out of the range of the instrument. Notably, compound 3 featured higher absorption intensity values at the 10-fold lower concentration (2 × 10−6 M) than reference compound 4 (2 × 10−5 M).
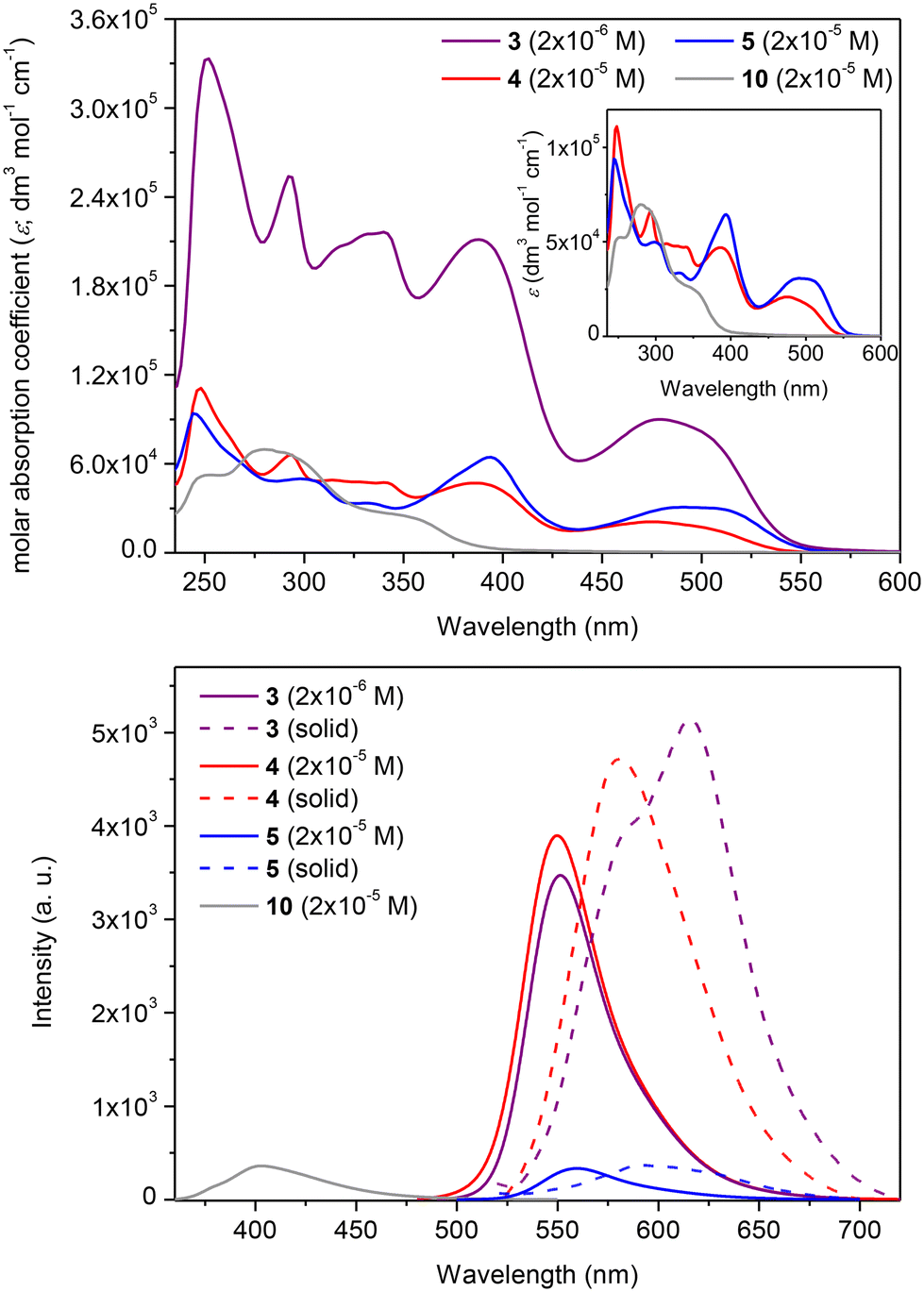 | ||
| Fig. 3 (top) UV-Vis spectra (CHCl3) of compounds 3–5 and 10 and (bottom) solution (CHCl3) and solid-state (dashed lines) emission spectra of compounds 3 (λex = 380 nm), 4 (λex = 380 nm), 5 (λex = 380 nm) and 10 (λex = 380 nm). | ||
The emission properties of 3 were also improved compared with the starting material 10 and reference sumanene–carbazole conjugate 4, which does not contain the electron-withdrawing –p(CF3)C6H4 group (Fig. 3; the 3-D emission spectrum of 3 is presented in ESI, section S4†). Compounds 4 and 5 were found to be yellow–orange light emitters with emission maxima (λem) at 549 and 558 nm, respectively. Emission intensity for 4 was ca. 12-fold higher in comparison to 5 (see spectra and discussion in ESI, section S4†). Thus, in general, the light-emission properties of 5 were not as satisfactory as those of 4, revealing the effect of the method of attachment of the carbazole moiety on the photophysical properties of the resultant conjugate with sumanene. Importantly, the λem for 3 (552 nm) was significantly red-shifted (ca. 152 nm; 6890 cm−1) compared with 10 (400 nm) and was similar to that of 4 (Fig. 3b). The intensity of emission for 3 was similar to the respective value for 4, although the spectrum of 3 was measured at the 10-fold lower concentration. In consequence, the fluorescence quantum yield (ΦF) for 3 (0.76) was ca. 1.5-fold higher than for 4 (0.53) and significantly higher than for 5 (0.12). Finally, the λem for a solid-state spectrum of 3 was red-shifted (ca. 35 nm; 980 cm−1) compared with 4.
Theoretical calculations
To get a deeper insight into the photophysical features of the studied compounds 3–5, we performed a series of DFT (B3LYP-D366–69) and time-dependent-DFT (TD-DFT; B3LYP66,67) theoretical calculations using the Gaussian16 software (basis set: 6-31+G(d,p)70). Our first goal was to define the optimal geometries of the molecules and compare the thermodynamic stability of their diastereoisomers. According to our computations, the unsymmetrical diastereoisomers of compounds 4 and 5 are by 10.0 and 14.4 kJ mol−1 more stable than corresponding symmetrical diastereoisomers (Fig. S51, ESI†). In the case of 3, four diastereoisomers should be considered (Fig. S50, ESI†). The most stable diastereoisomer comprises two neighboring carbazole units held by C–H⋯C(π) interactions, whereas the third carbazole group forms C–H⋯C(π) interaction with the p-(CF3)C6H4 group. Importantly, the fluorine atoms and the carbazole skeleton are sufficiently distant to avoid any repulsions. On the contrary, the lack of any such stabilizing interaction in the remaining isomers led to substantially lower thermodynamic stability.
In the subsequent step, the electronic transitions for the most stable diastereoisomers were explored. Evaluating these transitions could potentially explain the observed differences in the UV-Vis spectra. Specifically, we were interested in the investigation of electronic and spectral features of the push–pull system 3. Due to the complex nature of electronic transitions involving contributions from multiple HOMO and LUMO molecular orbitals, the natural transition orbitals (NTO)71 were calculated. NTO provides a more intuitive description of the electronic transitions from the highest-occupied NTO (HONTO) to the lowest-unoccupied NTO (LUNTO) representing the hole and electron, respectively.
Calculated absorption and emission wavelengths generally correspond to the observed experimental bands (Table 1). The differences in the positions of lmax observed in UV-Vis spectra between 3, 4, and 5, although present, were not substantial. These variations in the absorption spectra predominantly stem from changes in the molar absorption coefficient, whereas the positions of local maxima display only minor discrepancies. For all considered compounds, the lowest-energy absorption bands at ca. 480 nm can be attributed to the π–π* transitions from HONTO spread over sumanene and pendant phenyl-carbazole groups to LUNTO localized on the central sumanene unit (Fig. 4). On the contrary, the electronic transitions corresponding to the absorption bands around 340 and 390 nm have different natures between 3 and 4–5. Although respective HONTO orbitals are quite similar for these transitions, i.e., they are delocalized over the central sumanene and pendant phenyl-carbazole moieties, substantial differences are observed in the distribution of LUNTO demonstrating unique structural variances between molecules. Specifically, in the case of 3, LUNTO includes the contribution from the electron-withdrawing –p(CF3)C6H4 group. Thus, the electron excitations responsible for the formation of higher-energy bands at 340 and 390 nm can be interpreted in terms of intramolecular charge transfer from carbazole–sumanene moiety to 2-((4-(trifluoromethyl)phenyl)ethynyl)sumanene moiety, related to the push–pull chromophore architecture of 3. At the same time, the excitation energies remain similar between all considered derivatives. Due to the different nature of higher-energy transitions, it can be expected that the interaction with Cs+ cation will pose different effects on the intensities of these bands, affecting the sensitivity of detection.
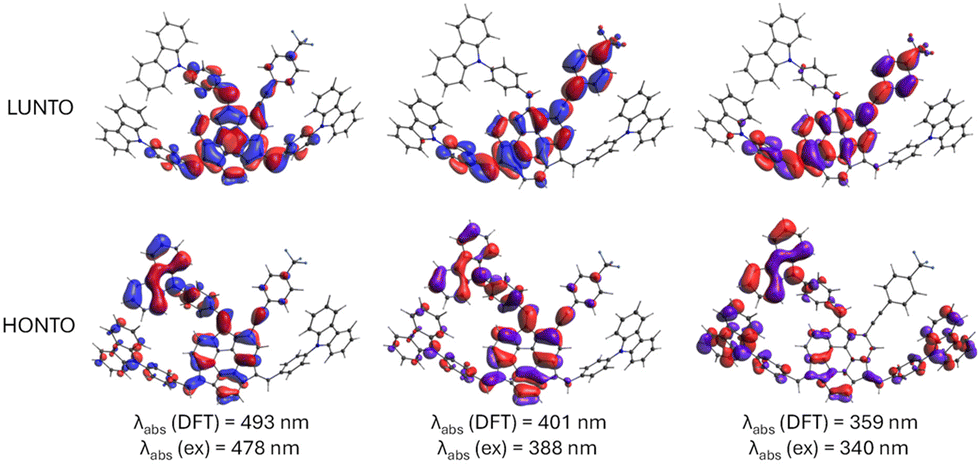 | ||
| Fig. 4 Graphical illustration of crucial highest-occupied and lowest unoccupied natural transition orbitals (HONTO and LUNTO, respectively) responsible for observed absorption bands in UV-Vis spectra of 3. Theory level: B3LYP/6-31+G(d,p). The NTO orbitals for 4 and 5 are provided in Fig. S52 in ESI.† | ||
Cesium cation (Cs+) recognition studies
Finally, the application of sumanene–carbazole conjugates 3–5 as optical chemoreceptors was investigated. In particular, the comprehensive studies on supramolecular interactions between sumanene–carbazole conjugates 3–5 and cesium cations (Cs+) in the form of hexafluorophosphate (PF6−) salt were performed in a THF:water (1:1 vol/vol) solvent system (see experimental details in ESI, section S6†). Specifically, Cs+ cations were selected for the chemoreceptor application studies based on recent reports on the action of sumanene derivatives toward binding these cations.13,54–56,63 Selective binding of lithium (Li+) by the sumanene-containing ruthenium(II)-bis(terpyridine) organometallic compounds was recently reported,22 therefore, Li+ was also included in our selectivity studies, whereas sodium (Na+) and potassium (K+) cations were selected as interferents commonly present in water and widely included in the selectivity studies for other chemoreceptors. Calcium (Ca2+) cations were selected as the representative divalent metal cations. All metal cations were tested in the form of their commercially available PF6− salts in order to exclude effects of the type of counteranion on the binding phenomena by compounds 3–5, if any.
At first, spectrofluorimetric titrations were performed. Our special attention was focused on the interactions between Cs+ and push–pull chromophore 3. To our delight, compound 3 exhibited exclusive emission quenching upon the addition of further portions (molar equivalents) of Cs+ (see Fig. 5a; the spectra for the interactions of reference compounds 4–5 are presented in ESI, section S6†). This experiment suggested that the Cs+ trapping abilities of derivative 3 were not excluded despite the structurally sophisticated structure of this sumanene derivative. No interfering effects of Na+ and K+ were found, and the changes after the addition of Li+ to the solutions were very slight as compared to the Cs+ titration experiments (see data in ESI, section S6†). No interfering effect was also found for the spectrofluorimetric selectivity studies with calcium hexafluorophosphate (Ca(PF6)2, see data in Fig. S66, ESI†). Therefore, satisfactory selectivity of the designed sumanene-tethered chemoreceptors toward the recognition of Cs+ can be concluded. Additionally, no changes in the emission intensity of reference 1,3,5-tri(9H-carbazol-9-yl)benzene 11 were found in the respective Cs+ binding experiments (see data in ESI, section S6†). This experiment supported the vital role of the presence of the sumanene skeleton in 3–5 toward providing cation-binding properties.
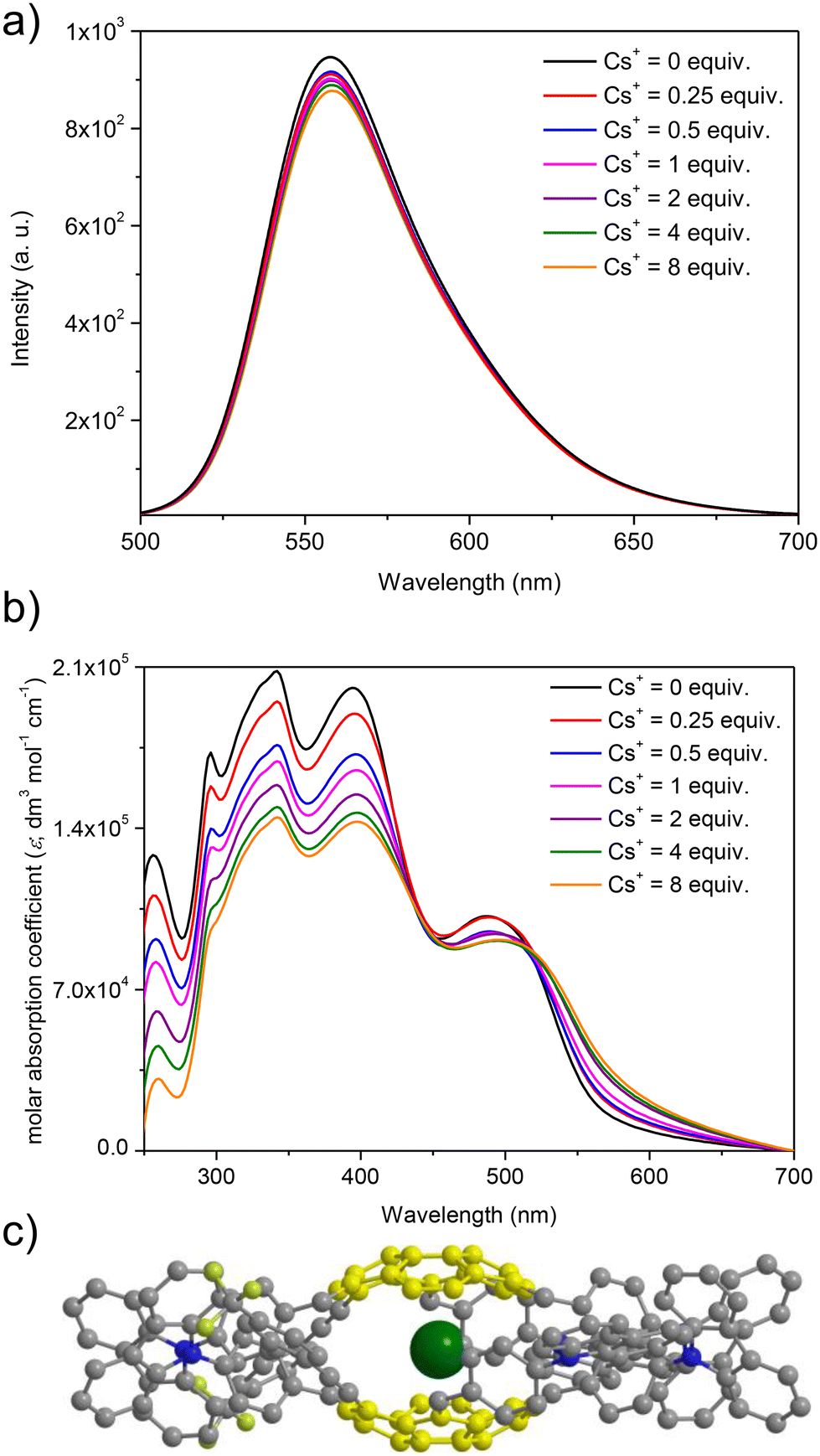 | ||
| Fig. 5 (a) Emission spectra of 3 in the presence of various molar equivalents of Cs+ (THF:water = 1:1 vol/vol, λex = 390 nm, C3 = 2 × 10−6 M); (b) UV–Vis spectra of 3 in the presence of various molar equivalents of Cs+ (THF:water = 1:1 vol/vol, C3 = 2 × 10−6 M); and (c) graphical representation on the possible structure of the formed non-covalent systems (for clarity sumanene bowl is marked yellow, whereas Cs+ is marked green). | ||
Taking into account significantly lower cost of instruments for UV-Vis absorption spectroscopy in comparison to spectrofluorometers, as well as common usage of UV-Vis spectrometers in almost every environmental laboratories, our attention was focused on investigating the possibility of applying UV-Vis absorption spectroscopy toward tracking the Cs+ binding by sumanene–carbazole conjugates 3–5. Importantly, while no significant changes in the UV-Vis spectra of 4–5 in the presence of Cs+ were observed, notable differences were found exclusively in the UV-Vis spectra of 3 (Fig. 5b). Notably, the changes in the UV-Vis spectra of 3 upon the addition of Cs+ were also more significant than in the case of emission spectra experiments, and the most essential among sumanene-based Cs+ optical chemoreceptors reported to date. The most essential changes were observed for λabs between 250 and 450 nm. Namely, a significant lowering of the absorption intensities, as well as slight red shifts (ca. 5 nm) for these bands was observed with the addition of further portions of Cs+. This possibility of applying UV-Vis spectroscopy toward effective tracking of the interactions between 3 and Cs+ was ascribed to the tuned structure of compound 3 featuring enhanced charge-transfer characteristics. Thus, among the reported sumanene-based optical chemoreceptors reported to date,13,22,54–56,63 compound 3 is the first molecule for which the application of both UV-Vis and emission spectroscopies toward effective tracking of the Cs+ recognition phenomenon is described.
The stoichiometry for each system, estimated with the Job's plot method (continuous variation method),72,73 was found to be 2:1, indicating 2 molecules of the sumanene derivative per one Cs+ (see data and Job's plots in ESI, section S6†). Thus, the observed changes might be ascribed to the dynamic formation of sandwich-type complexes (Fig. 5c), driven by the perfect size-match between concave sites of sumanene bowls and the van der Waals radius of Cs+, as well as improved cation–π interactions for this system.13,22,54–56,63,74,75 Notably, the system stoichiometries estimated from the emission and UV-Vis spectroscopy titrations for push–pull chromophore 3 were consistent.
The interactions between Cs+ and sumanene–carbazole conjugates 3–5 were characterized by the satisfactory apparent binding constant (Kapp) values (estimated with the Benesi–Hildebrand method76,77) at the level of 105 M−2 (see data in Table 2). Importantly, Kapp values estimated from emission spectroscopy experiments and respective UV-Vis assays (data collected for λabs = 394 nm) for the system consisting of the push–pull chromophore 3 were highly consistent, demonstrating that 3 is a versatile optical chemoreceptor of Cs+. The limit of detection (LOD) values for the system containing 3 estimated from emission and UV-Vis spectroscopy analyses were also highly consistent and equaled 0.09–0.13 μM. Notably, the LOD values for 3 are lower in comparison to the respective values for reference compounds 4–5 (LOD ranging from 0.30 to 2.09 μM).
Conclusions
In conclusion, we presented the successful design of a bowl-shaped and π-conjugated sumanene–carbazole push–pull chromophore (compound 3). Absorption and emission spectroscopy assays revealed improved photophysical properties of 3 compared with native sumanene, 9H-carbazole, and other reference sumanene–carbazole conjugates. Compound 3 featured exclusive, Cs+-selective trapping properties related to the dynamic formation of non-covalent sandwich-type complexes. For the first time, it was possible to apply both UV-Vis and emission spectroscopies toward effective tracking of the Cs+ recognition phenomenon with 3, due to the tuned optical properties of this molecule. The results of fluorescence and absorption spectroscopy titrations with Cs+ and 3 were highly consistent, revealing good apparent binding constant values (at the level of 105) for this system and low limits of detection (0.09–0.13 μM). Notably, compound 3 could be potentially applied in the analyses with cesium-contaminated real samples, as for example the concentration of cesium-137 in Japanese Mano River is about 0.06 mM.58,63 As a result of these studies, we demonstrated the benefits of using structurally sophisticated sumanene-containing push–push chromophore 3 in terms of designing versatile cation recognition materials. The findings of this work lay the basis for further progress in designing sumanene derivatives featuring beneficial optical properties toward applied sciences of this molecule as optical chemoreceptors.Author contributions
The manuscript was written through contributions of all authors and all authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial supports from the National Science Centre, Poland, OPUS grant no. 2021/43/B/ST4/00114 (A. K.) and Warsaw University of Technology (WUT; A. K.), as well as JSPS KAKENHI (grant no. JP19H00912 and JP21H05233; H. S.), are acknowledged. Computational facilities were provided by the Wrocław Centre for Networking and Supercomputing (grant no. 285). The authors would like to thank Dr Sergei Molchanov (WUT) for his assistance in the measurement of 2D NMR and DOSY NMR spectra.References
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635 RSC.
- T. Amaya, T. Nakata and T. Hirao, J. Am. Chem. Soc., 2009, 131, 10810 CrossRef CAS PubMed.
- T. Amaya and T. Hirao, Chem. Commun., 2011, 47, 10524 RSC.
- T. Amaya and T. Hirao, Chem. Rec., 2015, 15, 310 CrossRef CAS PubMed.
- W. Wang and X. Shao, Org. Biomol. Chem., 2021, 19, 101 RSC.
- X. Li, F. Kang and M. Inagaki, Small, 2016, 12, 3206 CrossRef CAS PubMed.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- H. Sakurai, Bull. Chem. Soc. Jpn., 2021, 94, 1579 CrossRef.
- T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, J. Am. Chem. Soc., 2009, 131, 408 CrossRef CAS PubMed.
- T. Amaya, W. Wang, H. Sakane, T. Moriuchi and T. Hirao, Angew. Chem., Int. Ed., 2010, 49, 403 CrossRef CAS PubMed.
- B. B. Shrestha, S. Higashibayashi and H. Sakurai, Beilstein J. Org. Chem., 2014, 10, 841 CrossRef PubMed.
- M. Nishimoto, Y. Uetake, Y. Yakiyama, A. Saeki, J. Freudenberg, U. H. F. Bunz and H. Sakurai, Chem. – Eur. J., 2023, 29, e202203461 CrossRef CAS PubMed.
- A. Kasprzak and H. Sakurai, Chem. Commun., 2021, 57, 343 RSC.
- H. Nakazawa, Y. Uetake, Y. Yakiyama and H. Sakurai, Synlett, 2023, 34, 374 CrossRef CAS.
- M. Nishimoto, Y. Uetake, Y. Yakiyama, F. Ishiwari, A. Saeki and H. Sakurai, J. Org. Chem., 2022, 87, 2508 CrossRef CAS PubMed.
- T. Amaya, T. Ito and T. Hirao, Angew. Chem., Int. Ed., 2015, 54, 5483 CrossRef CAS PubMed.
- J. Shang, R. Wang, C. Yuan, Z. Liu, H. Zhang and X. Shao, Angew. Chem., Int. Ed., 2022, 61, e202117504 CrossRef CAS PubMed.
- Q. Tan, P. Kaewmati, S. Higashibayashi, M. Kawano, Y. Yakiyama and H. Sakurai, Bull. Chem. Soc. Jpn., 2018, 91, 531 CrossRef CAS.
- P. Kaewmati, Y. Yakiyama, H. Ohtsu, M. Kawano, S. Haesuwannakij, S. Higashibayashi and H. Sakurai, Mater. Chem. Front., 2018, 2, 514 RSC.
- S. Wang, J. Shang, C. Yan, W. Wang, C. Yuan, H.-L. Zhang and X. Shao, Org. Chem. Front., 2019, 6, 263 RSC.
- Z. Liu, W. Song, C. Yan, Z. Liu, H.-L. Zhang and X. Shao, Org. Chem. Front., 2021, 8, 4767 RSC.
- J. Han, Y. Yakiyama, Y. Takeda and H. Sakurai, Inorg. Chem. Front., 2023, 10, 211–217 RSC.
- B. Topolinski, B. M. Schmidt, S. Higashibayashi, H. Sakurai and D. Lentz, Dalton Trans., 2013, 42, 13809 RSC.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49 CrossRef CAS.
- A. K. Singh, A. V. Nair, Sk. S. Shah, S. Ray and N. D. P. Singh, J. Med. Chem., 2023, 66, 3732 CrossRef CAS PubMed.
- J. Liu, W. Chen, C. Zheng, F. Hu, J. Zhai, Q. Bai, N. Sun, G. Qian, Y. Zhang, K. Dong and T. Lu, Eur. J. Med. Chem., 2022, 244, 114843 CrossRef CAS PubMed.
- G. Prabakaran, C. I. David and R. Nandhakumar, J. Environ. Chem. Eng., 2023, 11, 109701 CrossRef CAS.
- P. Vishnoi, D. Kaleeswaran and R. Murugavel, RSC Adv., 2018, 8, 17535 RSC.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105 RSC.
- F. Bureš, RSC Adv., 2014, 4, 58826 RSC.
- M. Klikar, P. Solanke, J. Tydlitát and F. Bureš, Chem. Rec., 2016, 16, 1886 CrossRef CAS PubMed.
- C. Pigot, G. Noirbent, D. Brunel and F. Dumur, Eur. Polym. J., 2020, 133, 109797 CrossRef CAS.
- S.-L. Hsu, C.-M. Chen and K.-H. Wei, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5126 CrossRef CAS.
- A. S. Steparuk, S. G. Tolshchina, N. A. Kazin, R. A. Irgashev, E. F. Zhilina, A. E. Aleksandrov, A. R. Tameev and G. L. Rusinov, Russ. Chem. Bull., 2021, 70, 1109 CrossRef CAS.
- P. K. M. Lokhande, D. S. Patil, M. Kadam and N. Sekar, J. Fluoresc., 2019, 29, 779 CrossRef CAS PubMed.
- R. D. Telore and N. Sekar, Dyes Pigm., 2016, 129, 1 CrossRef CAS.
- A. M. Asiri, O. I. Osman, S. H. Al-Thaqafy and S. A. Khan, RSC Adv., 2017, 7, 8402 RSC.
- G. Krucaite and S. Grigalevicius, Synth. Met., 2019, 247, 90 CrossRef CAS.
- J. Yin, Y. Ma, G. Li, M. Peng and W. Lin, Coord. Chem. Rev., 2020, 412, 213257 CrossRef CAS.
- S. Oner and M. R. Bryce, Mater. Chem. Front., 2023, 7, 4304 RSC.
- G. Kleinhans, A. J. Karhu, H. Boddaert, S. Tanweer, D. Wunderlin and D. I. Bezuidenhout, Chem. Rev., 2023, 123, 8781 CrossRef CAS PubMed.
- J.-J. Huang, H.-A. Lin and S.-C. Luo, ACS Appl. Polym. Mater., 2023, 5, 5727 CrossRef CAS.
- Y. Tokimaru, S. Ito and K. Nozaki, Angew. Chem., 2018, 130, 9966 CrossRef.
- A. Sacristán-Martín, D. Miguel, A. Diez-Varga, H. Barbero and C. M. Álvarez, J. Org. Chem., 2022, 87, 16691 CrossRef PubMed.
- A. Battal, S. B. Kassa, N. A. Gultekin, M. Tavasli and Y. Onganer, J. Fluoresc., 2023, 33, 1421 CrossRef CAS PubMed.
- F. Kolcu and İ. Kaya, Arabian J. Chem., 2022, 15, 103935 CrossRef CAS.
- X. J. Feng, P. Z. Tian, Z. Xu, S. F. Chen and M. S. Wong, J. Org. Chem., 2013, 78, 11318 CrossRef CAS PubMed.
- C. I. David, G. Prabakaran, A. Karuppasamy, J. C. Veetil, R. S. Kumar, A. I. Almansour, K. Perumal, C. Ramalingan and R. Nandhakumar, J. Photochem. Photobiol., A, 2022, 425, 113693 CrossRef.
- S. Qu, C. Zheng, G. Liao, C. Fan, G. Liu and S. Pu, RSC Adv., 2017, 7, 9833 RSC.
- T. Aggarwal, S. Sushmita and A. K. Verma, Org. Biomol. Chem., 2019, 17, 8330 RSC.
- I. A. Pocock, A. M. Alotaibi, K. Jagdev, C. Prior, G. R. Burgess, L. Male and R. S. Grainger, Chem. Commun., 2021, 57, 7252 RSC.
- M. Zhao, S. H. Pun, Q. Gong and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 24124 CrossRef CAS PubMed.
- A. Kasprzak and H. Sakurai, Dalton Trans., 2019, 48, 17147 RSC.
- A. Kasprzak, A. Gajda-Walczak, A. Kowalczyk, B. Wagner, A. M. Nowicka, M. Nishimoto, M. Koszytkowska-Stawińska and H. Sakurai, J. Org. Chem., 2023, 88, 4199 CrossRef CAS PubMed.
- J. S. Cyniak, Ł. Kocobolska, N. Bojdecka, A. Gajda-Walczak, A. Kowalczyk, B. Wagner, A. M. Nowicka, H. Sakurai and A. Kasprzak, Dalton Trans., 2023, 52, 3137 RSC.
- H. Kaeriyama, Fish. Oceanogr., 2017, 26, 99 CrossRef.
- T. Mizuno and H. Kubo, Sci. Rep., 2013, 3, 1742 CrossRef PubMed.
- Y. Morino, T. Ohara and M. Nishizawa, Geophys. Res. Lett., 2011, 38, L00G11 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- T. Amaya, K. Mori, H.-L. Wu, S. Ishida, J. Nakamura, K. Murata and T. Hirao, Chem. Commun., 2007, 1902 RSC.
- B. B. Shrestha, S. Karanjit, G. Panda, S. Higashibayashi and H. Sakurai, Chem. Lett., 2013, 42, 386 CrossRef CAS.
- A. Kasprzak, A. Kowalczyk, A. Jagielska, B. Wagner, A. M. Nowicka and H. Sakurai, Dalton Trans., 2020, 49, 9965 RSC.
- A. Kasprzak, A. Tobolska, H. Sakurai and W. Wróblewski, Dalton Trans., 2022, 51, 468 RSC.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998 CrossRef CAS PubMed.
- C. Y. Huang, in Methods in Enzymology, Elsevier, 1982, vol. 87, p. 509 Search PubMed.
- S. N. Spisak, Z. Wei, A. Yu. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, Angew. Chem., Int. Ed., 2017, 56, 2582 CrossRef CAS PubMed.
- D. Vijay, H. Sakurai, V. Subramanian and G. N. Sastry, Phys. Chem. Chem. Phys., 2012, 14, 3057 RSC.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS.
- S. Goswami, K. Aich, S. Das, A. K. Das, A. Manna and S. Halder, Analyst, 2013, 138, 1903 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, experimental section, compounds characterization data, additional UV-Vis and emission spectra, details on DFT calculations, details and additional data on Cs+ binding experiments. See DOI: https://doi.org/10.1039/d4ob00539b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |