
A divergent one-pot thiol-Michael strategy to create β-thiophene-fused porphyrins†
Jagmeet
Singh
,
Abhijeet
Singh
 and
Mahendra
Nath
*
and
Mahendra
Nath
*
Department of Chemistry, Faculty of Science, University of Delhi, Delhi 110 007, India. E-mail: mnath@chemistry.du.ac.in
First published on 8th May 2024
Abstract
A divergent one-pot domino strategy for the synthesis of nickel(II) and copper(II) β-thiophene-fused 5,10,15,20-tetraarylporphyrins was developed through a thiol-Michael addition of thioglycolic/thiolactic acid to the corresponding 2-iminoporphyrins, formed in situ after the reaction of nickel(II) and copper(II) 2-formyl-5,10,15,20-tetraarylporphyrins with sterically hindered tert-butylamine in 1,2-dichloroethane at 80 °C. Interestingly, the reaction of 2-formylporphyrins with comparatively less sterically hindered primary amines and thioglycolic acid afforded a mixture of β-substituted porphyrinic thiazolidinones and β-thiophene-fused porphyrins. A similar one-pot thiol-Michael protocol was applied to construct a novel free-base thieno[2,3-b]-meso-tetrakis(4-methoxyphenyl)porphyrin, which underwent zinc insertion by using zinc acetate in a CHCl3–MeOH mixture and afforded zinc(II) β-thiophene-fused meso-tetrakis(4-methoxyphenyl)porphyrin in an appreciable isolated yield. On photophysical evaluation, these new porphyrins displayed a modest bathochromically shifted electronic absorption in contrast to meso-tetraarylporphyrin building blocks.
Introduction
Porphyrins with extended π-conjugation have received considerable attention in recent years due to their numerous applications in diverse research areas such as catalysis,1 optoelectronics,2 supramolecular chemistry,3 chemical sensors,4 photovoltaic applications,5 electron transfer6 and photomedicine.7 Fascinatingly, these tetrapyrrolic macrocycles have played pivotal roles in several biomimetic processes,8 photodynamic therapy,9 and dye-sensitized solar cells.10a,b During the past decades, a large number of periphery modified π-extended porphyrins have been constructed by using meso-tetraarylporphyrins as easily accessible building blocks.11–27,28a–d Some of these macrocycles have been explored as light-harvesting materials in organic solar cells due to their strong light absorption properties.29 Similarly, thiophene-fused aromatic systems have demonstrated their potential usage in organic semiconductors,30 microelectronics,31 electroluminescent materials,32 conducting polymers33 and solar cells.34 Considering the biological and material significance of thiophene and porphyrin scaffolds, efforts were made to synthesize hybrid molecular frameworks by incorporating a thiophene moiety on the periphery of porphyrin through functionalization of β- and meso-positions, and some of these thiophene-tethered porphyrins have demonstrated interesting features such as antiaromaticity, red-shifted absorption and emission and narrow HOMO–LUMO band gaps.35–40,41a–c In view of the inspiring optical properties of β-thiophene-appended porphyrins, Li and co-workers synthesized β,β′-thiophene-fused meso-tetraphenylporphyrins in two steps.42 According to the reported methodology, a reaction of 2-nitro-meso-tetraphenylporphyrin with 1,4-dithiane-2,5-diol in dichloromethane in the presence of triethylamine affords a diastereomeric mixture of β-tetrahydrothiophene-fused meso-tetraphenylporphyrins, which on aromatization at 190 °C in DMSO provides β-thiophene-fused meso-tetraphenylporphyrin (Scheme 1A). However, this protocol involves two steps to synthesize targeted β-thiophene-fused porphyrins and requires harsh reaction conditions, particularly in the second step. Hence, we contemplated a domino strategy to construct these porphyrins by employing 2-formyl-meso-tetraarylporphyrins as building blocks under mild conditions. To this end, we report herein a new straightforward one-pot strategy for the synthesis of β-thiophene-fused 5,10,15,20-tetraarylporphyrins through a reaction of 2-formyl-5,10,15,20-tetraarylporphyrins with tert-butylamine and thioglycolic/thiolactic acid in 1,2-dichloroethane at 80 °C (Scheme 1B).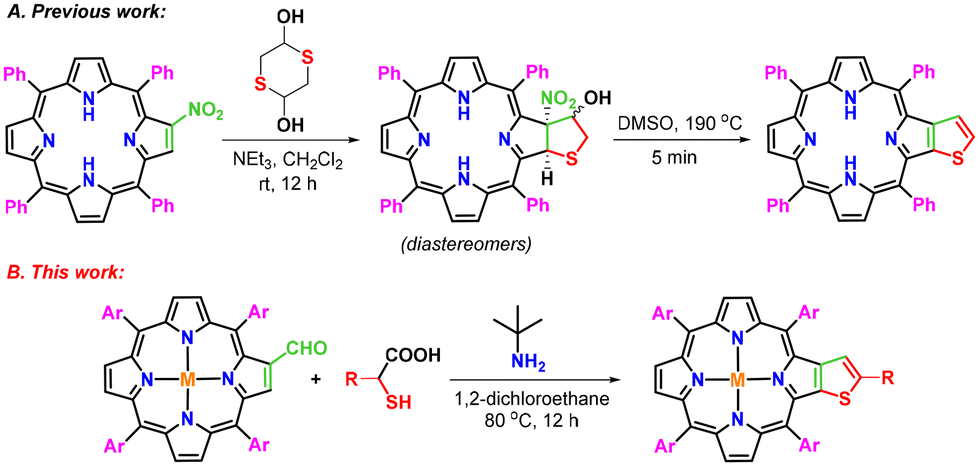 | ||
| Scheme 1 Methods for the synthesis of β-thiophene-fused meso-tetraarylporphyrins. | ||
Results and discussion
The present methodology involves a one-pot three-component synthesis of nickel(II) and copper(II) thieno[2,3-b]-5,10,15,20-tetraarylporphyrins (3a–e) from the corresponding 2-formyl-5,10,15,20-tetraarylporphyrins (1a–d). At first, the required precursors, 2-formyl-5,10,15,20-tetraarylporphyrins (1a–d), were synthesized from easily accessible meso-tetraarylporphyrins by following a Vilsmeier–Haack protocol as reported earlier.43 Then, these 2-formyl-5,10,15,20-tetraarylporphyrins were transformed into β-thiophene-fused meso-tetraarylporphyrins through a reaction with tert-butylamine and thioglycolic acid in 1,2-dichloroethane at 80 °C. For the optimization of reaction conditions, nickel(II) 2-formyl-5,10,15,20-tetraphenylporphyrin (1a), thioglycolic acid (2a) and tert-butylamine were selected as model substrates and experiments were first carried out in various chlorinated solvents such as dichloromethane, chloroform, 1,2-dichloroethane and 1,2-dichlorobenzene at different temperatures (Table 1, entries 1–5). The reaction of nickel(II) 2-formyl-5,10,15,20-tetraphenylporphyrin (1a) with tert-butylamine and thioglycolic acid (2a) in dichloromethane at room temperature was unsuccessful to produce a nickel(II) β-thiophene-fused 5,10,15,20-tetraphenylporphyrin (3a) (Table 1, entry 1); however, when the reaction temperature was elevated to 40 °C, only 13% product (3a) was formed (Table 1, entry 2). When a similar reaction was set up in chloroform at 60 °C, it generated the anticipated porphyrin (3a) in 35% isolated yield within 12 hours (Table 1, entry 3).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Yieldb (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 2-formylporphyrin (1a, 0.071 mmol), thioglycolic acid (2a, 0.142 mmol), and tert-butylamine (0.284 mmol) in 10 mL of solvent. b Isolated yields. c The starting material was recovered quantitatively. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1c | Dichloromethane | 25 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Dichloromethane | 40 | 13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Chloroform | 60 | 35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 1,2-Dichloroethane | 80 | 40 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 1,2-Dichlorobenzene | 100 | 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 1,4-Dioxane | 80 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Toluene | 80 | 9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8c | DMSO | 80 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | THF | 80 | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
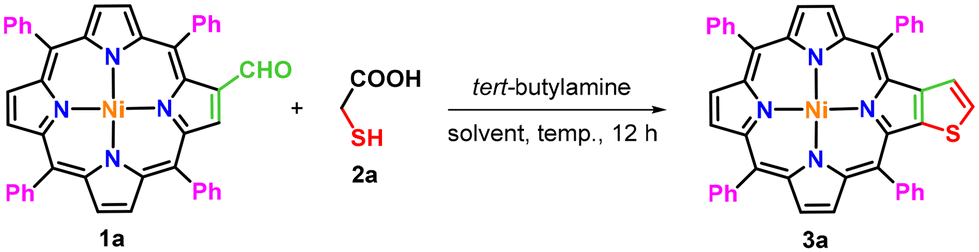
Furthermore, a reaction of porphyrin (1a) with tert-butylamine and thioglycolic acid (2a) in 1,2-dichloroethane at 80 °C afforded the desired nickel(II) β-thiophene-fused porphyrin (3a) in 40% isolated yield (Table 1, entry 4). To examine the role of temperature in enhancing the yield of porphyrin (3a), an experiment was conducted at 100 °C using 1,2-dichlorobenzene as a solvent. Surprisingly, the isolated yield of porphyrin (3a) significantly decreased to 10% (Table 1, entry 5). Hence, it was concluded that the 80 °C temperature is an ideal temperature for the synthesis of porphyrin (3a). Furthermore, various other solvents such as 1,4-dioxane, toluene, DMSO and THF have also been screened (Table 1, entries 6–9) under identical reaction conditions at 80 °C. The reaction in 1,4-dioxane and toluene afforded porphyrin (3a) in poor yields (14% and 9%, respectively; Table 1, entries 6 and 7). In contrast, the reaction did not proceed in DMSO (Table 1, entry 8) and only a trace amount of porphyrin (3a) was formed when THF was employed as a solvent (Table 1, entry 9). Therefore, the use of 1,2-dichloroethane at 80 °C for 12 hours was found to be the optimum reaction conditions for the construction of nickel(II) β-thiophene-fused 5,10,15,20-tetraphenylporphyrin (3a) from 2-formylporphyrin (1a).
After the standardization of the synthetic procedure, diverse nickel(II) and copper(II) β-thiophene-fused 5,10,15,20-tetraarylporphyrins (3b–e) were prepared in 28–46% isolated yields by treating nickel(II) or copper(II) 2-formyl-5,10,15,20-tetraarylporphyrins (1a–d) with tert-butylamine and thioglycolic/thiolactic acid (2a–b) in 1,2-dichloroethane at 80 °C for 12 hours under one-pot operation (Scheme 2). Surprisingly, the conversion of 2-formyl-5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin to a free-base β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3f) was found to be extremely sluggish under identical conditions. However, the desired free-base porphyrin (3f) was obtained in 30% isolated yield when the reaction of 2-formyl-5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin with tert-butylamine and thioglycolic acid was performed in xylene at 140 °C for 16 hours. Later, a traditional demetallation procedure was applied to synthesize the free-base β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3f) in appreciable yield from the corresponding copper(II) porphyrin (3e) using sulfuric acid in chloroform at 0 °C. For comparative photophysical studies, zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3g) was prepared in 77% yield through zinc insertion into the core of the free-base porphyrin (3f) by using Zn(OAc)2, as presented in Scheme 2.
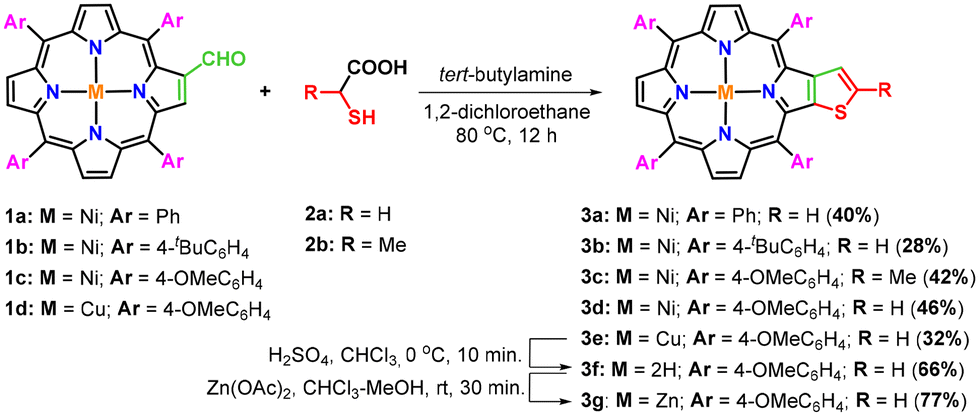 | ||
| Scheme 2 Synthesis of thieno[2,3-b]-5,10,15,20-tetraarylporphyrins (3a–g). | ||
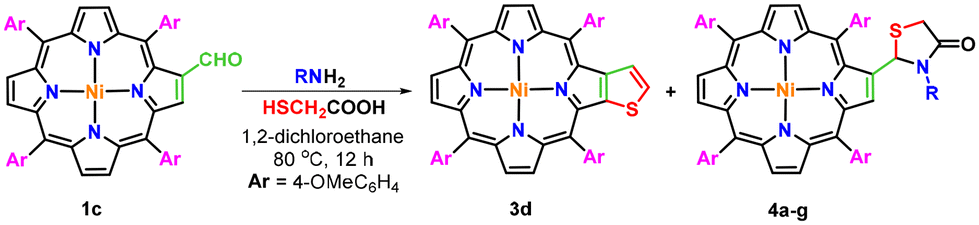
Furthermore, a series of experiments were also conducted by reacting nickel(II) 2-formyl-5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (1c) with ammonia and various aliphatic/aromatic primary amines and thioglycolic acid under optimized reaction conditions (Table 2). The results obtained from this study revealed that the use of a sterically hindered primary amine namely tert-butylamine selectively favoured the formation of β-thiophene-fused porphyrin (3d) in 46% isolated yield (Table 2, entry 1). In contrast, the reaction of 2-formylporphyrin (1c) with ammonia and thioglycolic acid under the same reaction conditions afforded exclusively nickel(II) 2-[2′-(5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin)]-thiazolidin-4-one (4a) in 63% isolated yield (Table 2, entry 2), whereas the reaction with other less bulky aliphatic and aromatic amines provided a mixture of β-thiophene-fused porphyrin (3d) and β-substituted porphyrinic thiazolidinones (4b–g) in unequal amounts (Table 2, entries 3–8).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Porphyrin (3d) | Porphyrins (4a–g) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 2-formylporphyrin (1c, 0.061 mmol), primary amine (0.244 mmol), and thioglycolic acid (0.122 mmol) in 10 mL of 1,2-dichloroethane. b Isolated yields. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | t Bu | 46 | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | H | 0 | 63 (4a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | n Bu | 13 | 57 (4b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Ph | 13 | 58 (4c) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 2-FC6H4 | 10 | 45 (4d) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 4-OMeC6H4 | 18 | 58 (4e) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 3-OMeC6H4 | 17 | 31 (4f) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 2-OMeC6H4 | 27 | 27 (4g) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Based on the results obtained from the present study and previous literature reports, the proposed mechanism for the formation of the desired β-thiophene-fused porphyrins (3a–e) and β-substituted porphyrinic thiazolidinones (4a–g) is depicted in Fig. 1. Firstly, 2-formyl-5,10,15,20-tetraarylporphyrins (1a–d) react with primary amines to generate iminoporphyrins (I) as key intermediates. Subsequently, these iminoporphyrins react with thioglycolic/thiolactic acid either in a 1,2- or 1,4-addition manner. Both these reaction pathways are found to be highly dependent on the presence of steric bulk44 around the nitrogen atom of primary amines. The 1,4-addition of thioglycolic/thiolactic acid to the iminoporphyrin intermediate (I) resulted in the formation of a zwitterionic intermediate (II), which afforded another zwitterionic intermediate (III) via successive proton transfer and tautomerization steps. On decarboxylation,45 intermediate III facilitates intramolecular cyclization possibly via the formation of a carbanion intermediate followed by aromatization to afford β-thiophene-fused porphyrins (3a–e) in moderate yields. However, the 1,2-addition of thioglycolic acid to the iminoporphyrins afforded zwitterionic species (IV), which upon proton transfer followed by successive intramolecular cyclization and dehydration produced β-substituted porphyrinic thiazolidinones (4a–g) in moderate to good yields.
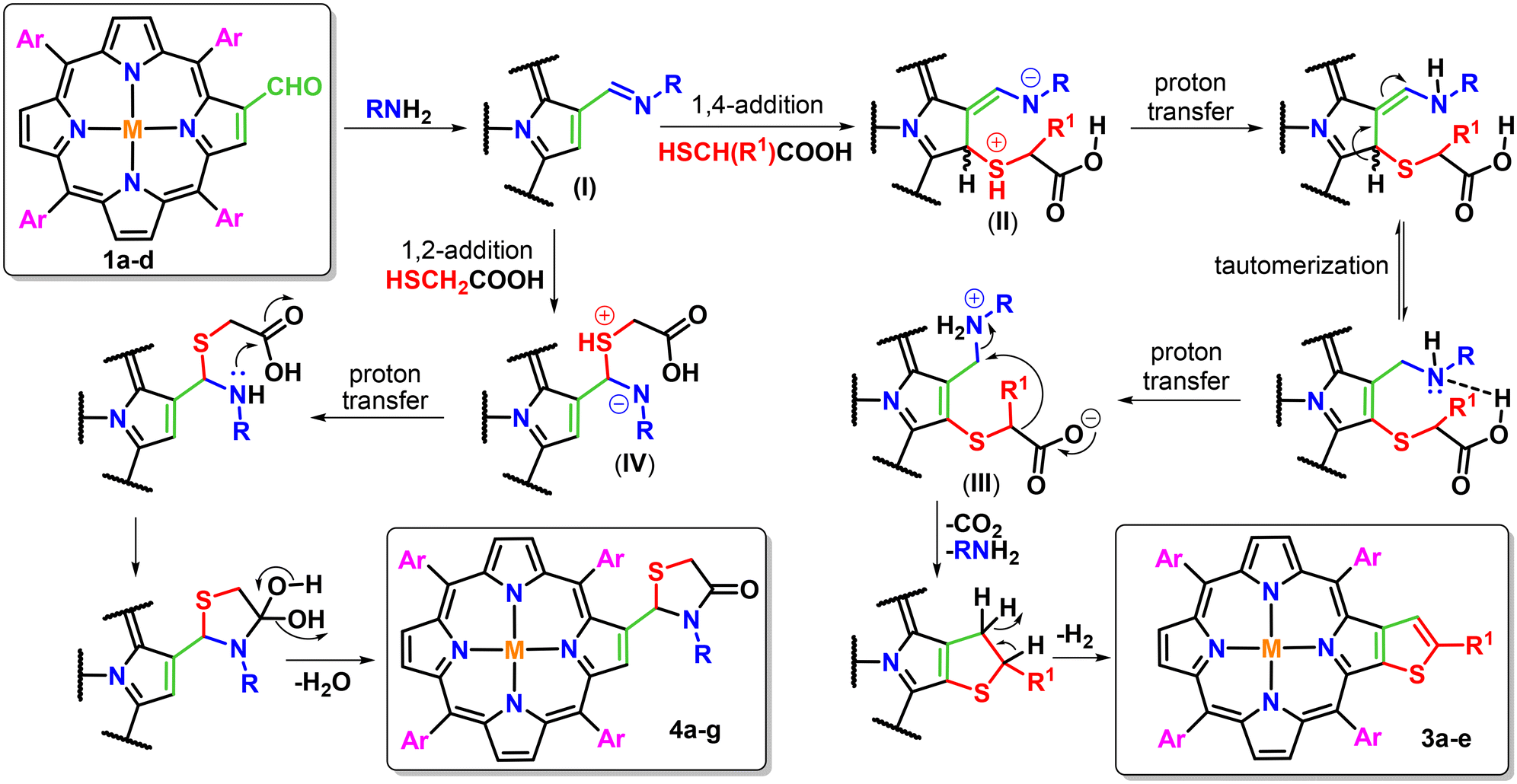 | ||
| Fig. 1 Plausible mechanistic pathway for the formation of β-thiophene-fused porphyrins (3a–e) and β-substituted porphyrinic thiazolidinones (4a–g). | ||
After chromatographic purification, the newly synthesized β-thiophene-fused 5,10,15,20-tetraarylporphyrins (3a–g) and nickel(II) β-substituted porphyrinic thiazolidinones (4a–g) were characterized on the basis of IR, 1H NMR, 13C NMR and high-resolution mass spectral data analysis. In the IR spectrum of a representative compound (3a), the absence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency in the carbonyl region indicates the formation of the desired porphyrin (3a). In the 1H NMR spectrum, the two characteristic doublets at δ 6.30 and δ 7.32 ppm with coupling constants J = 5.0 Hz and J = 5.1 Hz, respectively, confirmed the presence of a thiophene ring in the compound. In addition, a multiplet between δ 8.57 and 8.84 ppm was observed for six β-pyrrolic protons of the porphyrin ring. The three multiplets between δ 7.57 and 8.03 ppm were assigned to the twenty meso-phenyl protons in the set of four, four and twelve protons. In the 13C NMR spectrum, the absence of a carbonyl peak also supported the formation of the desired porphyrin (3a).
O stretching frequency in the carbonyl region indicates the formation of the desired porphyrin (3a). In the 1H NMR spectrum, the two characteristic doublets at δ 6.30 and δ 7.32 ppm with coupling constants J = 5.0 Hz and J = 5.1 Hz, respectively, confirmed the presence of a thiophene ring in the compound. In addition, a multiplet between δ 8.57 and 8.84 ppm was observed for six β-pyrrolic protons of the porphyrin ring. The three multiplets between δ 7.57 and 8.03 ppm were assigned to the twenty meso-phenyl protons in the set of four, four and twelve protons. In the 13C NMR spectrum, the absence of a carbonyl peak also supported the formation of the desired porphyrin (3a).
In the end, the high-resolution mass spectrum (HRMS) confirmed the formation of porphyrin (3a) by showing a molecular ion peak as [M + H]+ at m/z 727.1481 for the molecular formula C46H29N4NiS. Besides, the IR spectrum of nickel(II) 2-[2′-(5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin)]-thiazolidin-4-one (4a) showed a CO stretching frequency at 1690 cm−1, which confirmed the formation of a cyclic amide in porphyrin 4a. In the 1H NMR spectrum of porphyrin 4a, a singlet peak at δ 8.81 ppm corresponds to a β-pyrrolic proton, present adjacent to the thiazolidin-4-one ring, and remaining six β-pyrrolic protons appeared between δ 8.60 and 8.77 ppm as two multiplets of three protons each. A doublet of one proton at δ 7.97 ppm with a coupling constant J = 8.3 Hz was assigned to one of the meso-(4-methoxyphenyl) protons. The remaining fifteen meso-(4-methoxyphenyl) protons appeared between δ 7.10 and 7.92 ppm as multiplets of six, one and eight protons, and twelve methoxy (OCH3) protons appeared as two singlets at δ 4.03 and 4.05 ppm in a set of three and nine protons, respectively. The methine (C–H) and N–H protons of the thiazolidin-4-one ring appeared as sharp singlets at δ 6.32 and 5.84 ppm, respectively, and the two diastereotopic methylene (CH2) protons of the thiazolidine-4-one ring were observed as two doublets of one proton each at δ 3.39 and 3.60 ppm having coupling constants J = 15.7 Hz each. Finally, high-resolution mass spectral analysis confirmed the formation of porphyrin (4a) by showing a molecular ion peak as [M + H]+ at m/z 892.2106 for the molecular formula C51H40N5NiO5S. Similarly, other β-thiophene-fused porphyrins (3b–g) and β-substituted porphyrinic thiazolidinones (4b–g) were characterized and their spectral data are presented in the Experimental section.
The electronic absorption spectra of newly constructed β-thiophene-fused porphyrins (3a–g) and β-substituted porphyrinic thiazolidinones (4a–g) were recorded in CHCl3 (1.5 × 10−6 M) at 298 K. The UV-vis spectra of the representative nickel(II), copper(II), free-base and zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrins (3d–g) are presented in Fig. 2a. The nickel(II) and copper(II) porphyrins (3d and 3e) displayed a strong Soret band at 426 nm and 423 nm, respectively, along with two weak Q-bands between 539 and 582 nm. In contrast, the free-base β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3f) exhibited a slightly bathochromically shifted Soret band at 429 nm and four Q-bands between 521 and 661 nm. Besides, zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3g) exhibited a significantly red-shifted Soret band at 431 nm and two Q-bands at 556 and 592 nm as compared to the corresponding nickel(II) and copper(II) porphyrins (Fig. 2a). In comparison with nickel(II), copper(II), free-base and zinc(II) 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin precursors (Soret bands at 418 nm, 418 nm, 421 nm and 425 nm, respectively), the newly constructed porphyrins (3d–g) have demonstrated a modest red-shift (up to 8 nm) in their electronic absorption spectra.
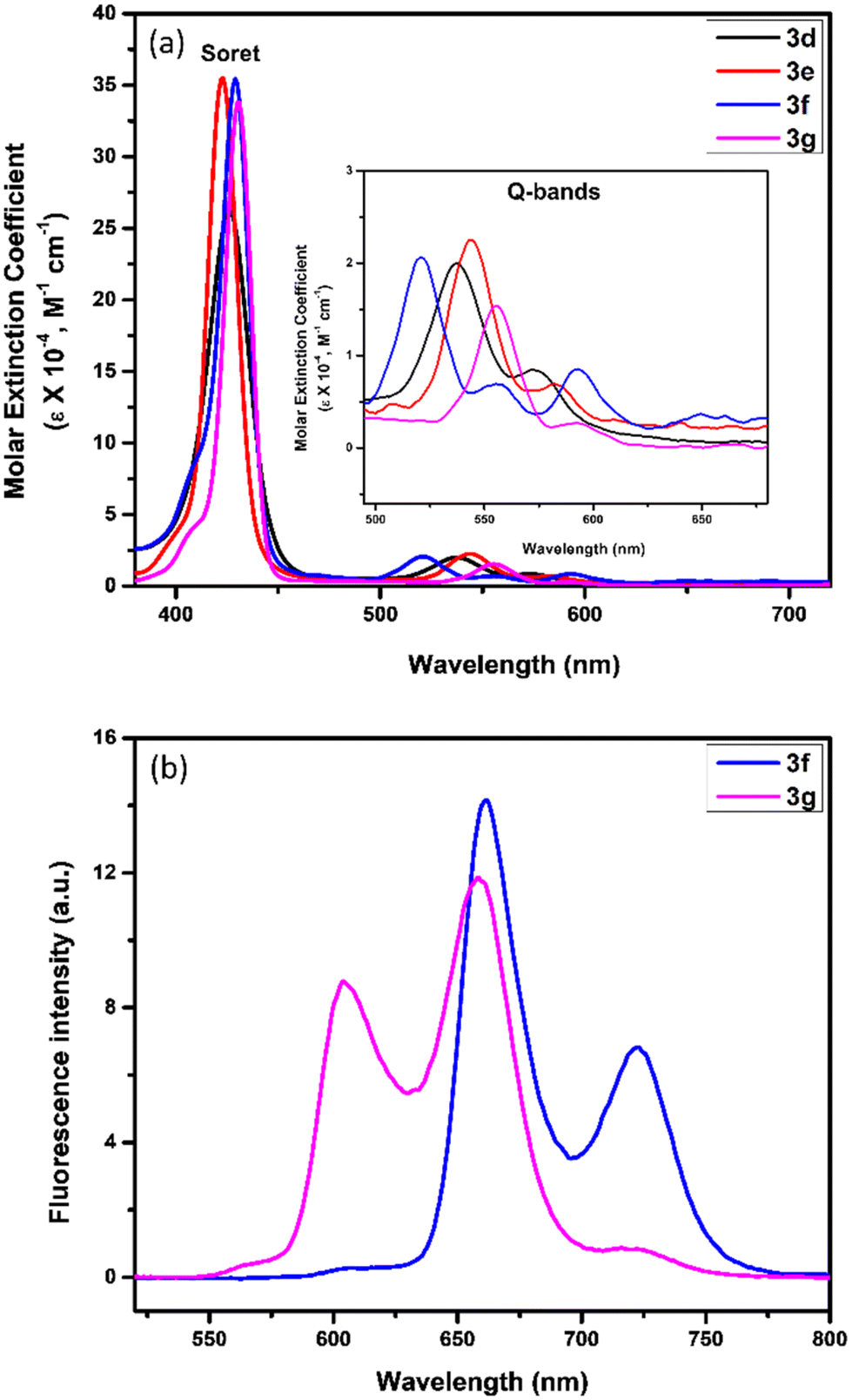 | ||
| Fig. 2 (a) Electronic absorption spectra of nickel(II), copper(II), free-base and zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrins (3d–g) in CHCl3 (1.5 × 10−6 M) at 298 K. The inset shows Q-bands. (b) Emission spectra of free-base and zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrins (3f and 3g) in CHCl3 (1.5 × 10−6 M) at 298 K (λEx = 425 nm). | ||
Furthermore, the emission spectra of free-base and zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrins (3f and 3g) are shown in Fig. 2b. The free-base β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3f) displayed two emission bands at 662 nm and 723 nm, whereas zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3g) exhibited two emission bands at 604 nm and 658 nm. The emission spectra of both the newly prepared free-base and zinc(II) porphyrins (3f and 3g) were found to be similar as reported in the case of 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (emission bands at 660 and 723 nm) and zinc(II) 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (emission bands at 605 and 654 nm), respectively.
Conclusions
In summary, we have developed a cascade thiol-Michael strategy for the construction of a series of nickel(II) and copper(II) β-thiophene-fused 5,10,15,20-tetraarylporphyrins via a reaction of 2-formyl-5,10,15,20-tetraarylporphyrins with tert-butylamine and thioglycolic/thiolactic acid in 1,2-dichloroethane at 80 °C. In addition, β-substituted porphyrinic thiazolidinones could also be obtained when ammonia and other less sterically hindered aliphatic/aromatic primary amines were reacted with 2-formylporphyrins and thioglycolic acid under standardized conditions. Furthermore, free-base and zinc(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrins were also obtained by using standard demetallation and zinc insertion procedures. On photophysical investigation, the synthesized β-thiophene-fused 5,10,15,20-tetraarylporphyrins and β-substituted porphyrinic thiazolidinones have shown a modest bathochromic shift in the electronic absorption as compared to their meso-tetraarylporphyrin building blocks. The results presented herein may be useful for the future development of π-conjugated molecules with diverse applications in materials science and medicine.Experimental section
General methods
All reagents and solvents used in the present study were purchased from Merck, Spectrochem, Thomas Baker (Chemicals) and Sisco Research Laboratories. Spectroscopic grade CHCl3 purchased from Spectrochem was used to record UV-visible absorption and emission spectra of the compounds. Thin-layer chromatography was performed on silica gel 60 F254 (pre-coated aluminium sheets) from Merck. All porphyrin products were purified on either activated neutral aluminium oxide (Brockmann grade I-II, Merck) or silica gel (60–120 mesh) columns. The NMR spectra were recorded in CDCl3 either on Jeol ECX 400P (400 MHz) or Bruker Ascend 400 NMR spectrometers by using TMS as an internal standard and chemical shifts are given in parts per million (ppm) relative to residual CHCl3 (δ = 7.26 for 1H NMR and δ = 77.00 for 13C NMR). The coupling constants (J) are reported in hertz (Hz). The IR spectra of products were recorded on a PerkinElmer IR spectrometer. Electronic absorption spectra were recorded on a Lab India Analytical UV-3092 UV-vis spectrophotometer and emission spectra were recorded on a Hitachi F-4700 fluorescence spectrometer using spectroscopic grade CHCl3 as a solvent. Mass spectra (ESI-HRMS) were recorded on an Agilent G6530AA (LC-HRMS-Q-TOF) and an LCMS-Waters SYNAPT G2 mass spectrometer. MALDI-TOF mass spectra were recorded using a Bruker UltrafleXtreme-TN MALDI-TOF/TOF spectrometer and dithranol as a matrix.General procedure for the synthesis of nickel(II) and copper(II) thieno[2,3-b]-5,10,15,20-tetraarylporphyrins (3a–e)
To a solution of nickel(II) and copper(II) 2-formyl-5,10,15,20-tetraarylporphyrins (1a–d, 0.071 mmol) in 1,2-dichloroethane (10 mL), tert-butylamine (0.284 mmol) and thioglycolic acid or thiolactic acid (0.142 mmol) were added and the reaction mixture was stirred at 80 °C for 12 hours. The reaction was monitored by using thin layer chromatography. After the completion of the reaction, the mixture was evaporated under reduced pressure. The crude solid was dissolved in CHCl3 (20 mL) and the resulting solution was washed with water (2 × 20 mL). The organic layer was dried over anhydrous sodium sulphate and evaporated under reduced pressure to obtain a red coloured solid. The crude products were purified on a silica gel column using 50–70% chloroform in hexane as an eluent (in the case of porphyrins 3a–b and 3d–e) and a neutral alumina column using 40% chloroform in hexane as an eluent (in the case of porphyrin 3c).Method B: To a solution of copper(II) β-thiophene-fused 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3e, 0.036 mmol) in CHCl3 (5 mL), conc. H2SO4 (0.5 mL) was added dropwise at 0 °C. The reaction mixture was allowed to stir at 0 °C for 5 minutes and the progress of the reaction was monitored by TLC. After the completion of the reaction, the mixture was neutralized by adding aqueous NaHCO3 solution. Then, the mixture was diluted with CHCl3 (10 mL) and washed with water (2 × 20 mL). The organic layer was dried over anhydrous sodium sulphate and evaporated under reduced pressure. The crude porphyrin (3f) was purified on a neutral alumina column by using 50% chloroform in hexane as an eluent. Red solid, yield: 20 mg (66%); UV λmax (ε × 10−4 M−1 cm−1): 429 (35.4), 521 (2.06), 556 (0.69), 593 (0.84), 661 (0.34) nm; λEm (CHCl3; λEx 425 nm): 662, 723 nm; IR (CHCl3) vmax: 3340, 2916, 2850, 1606, 1502, 1286, 1246, 1172, 1136, 1105, 1031, 991, 964, 804, 731 cm−1; 1H NMR (400 MHz, CDCl3) δH: 8.94–8.78 (m, 6H, β-pyrrolic H), 8.13–8.01 (m, 8H, meso-ArH), 7.48 (s, 1H, thiophene H), 7.39–7.26 (m, 8H, meso-ArH), 6.49 (s, 1H, thiophene H), 4.12 (s, 6H, CH3), 4.10 (s, 6H, CH3), −2.81 (s, 2H, internal N–H); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C50H39N4O4S 791.2692; found 791.2634.
General procedure for the synthesis of nickel(II) β-substituted porphyrinic thiazolidinones (4a–g)
To a solution of nickel(II) 2-formyl-5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (1c; 0.061 mmol) in 1,2-dichloroethane (10 mL), ammonia or aliphatic/aromatic primary amines (0.244 mmol) and thioglycolic acid (0.122 mmol) were added and the mixture was refluxed at 80 °C for 12 hours. After the completion of the reaction, the solvent was evaporated under reduced pressure. The crude solid was dissolved in CHCl3 (20 mL) and the solution was washed with water (2 × 20 mL) and dried over anhydrous sodium sulphate. The organic layer was evaporated under reduced pressure to obtain a mixture of two products, which on separation over a neutral alumina column using 50% chloroform in hexane afforded nickel(II) thieno[2,3-b]-5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (3d) as a minor product. However, β-substituted porphyrinic thiazolidinones (4a–g) were obtained as major products in 70–80% chloroform in hexane as a solvent and their characterization data are given below.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mahendra Nath is thankful to IoE, University of Delhi, India, for providing an FRP grant. Jagmeet Singh and Abhijeet Singh thank the CSIR, New Delhi, India, for the award of the Senior Research Fellowship.References
- X. Q. Yu, J. S. Huang, X. G. Zhou and C. M. Che, Org. Lett., 2000, 2, 2233–2236 CrossRef CAS PubMed.
- P. Liang, Z. Du, D. Wang, Z. Yang, H. Sheng, S. Liang, H. Cao, W. He and H. Yang, ChemPhysChem, 2014, 15, 3523–3529 CrossRef CAS PubMed.
- R. van der Weegen, A. J. P. Teunissen and E. W. Meijer, Chem. – Eur. J., 2017, 23, 3773–3783 CrossRef CAS PubMed.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS PubMed.
- M. M. Al Mogren, N. M. Ahmed and A. A. Hasanein, J. Saudi Chem. Soc., 2020, 24, 303–320 CrossRef CAS.
- K. Velappan, R. Rajalingam and A. Venkattappan, J. Fluoresc., 2021, 31, 1575–1585 CrossRef CAS PubMed.
- H. Huang, W. Song, J. Rieffel and J. F. Lovell, Front. Phys., 2015, 3, 127972 Search PubMed.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2010, 40, 340–362 RSC.
- (a) T. Higashino and H. Imahori, Dalton Trans., 2014, 44, 448–463 RSC; (b) J. Lu, S. Liu and M. Wang, Front. Chem., 2018, 6, 541 CrossRef CAS PubMed.
- L. Jaquinod, C. Gros, M. M. Olmstead, M. Antolovich and K. M. Smith, Chem. Commun., 1996, 1475–1476 RSC.
- P. Singh and M. Nath, Org. Lett., 2022, 24, 8586–8591 CrossRef CAS PubMed.
- A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Synlett, 2002, 1155–1157 CrossRef CAS.
- S. Sharma and M. Nath, New J. Chem., 2011, 35, 1630–1639 RSC.
- L. L. Yang, X. F. Li, X. L. Hu and X. Y. Yu, Tetrahedron Lett., 2016, 57, 1265–1267 CrossRef CAS.
- R. Tiwari and M. Nath, SynOpen, 2018, 2, 133–137 CrossRef CAS.
- M. Lo, J. F. Lefebvre, D. Leclercq, A. Van Der Lee and S. Richeter, Org. Lett., 2011, 13, 3110–3113 CrossRef CAS PubMed.
- P. Singh and M. Nath, SynOpen, 2020, 4, 44–50 CrossRef CAS.
- C. M. A. Alonso, M. G. P. M. S. Neves, A. C. Tome, A. M. S. Silva and J. A. S. Cavaleiro, Tetrahedron, 2005, 61, 11866–11872 CrossRef CAS.
- D. K. Singh and M. Nath, Org. Biomol. Chem., 2015, 13, 1836–1845 RSC.
- T. D. Lash and V. Gandhi, J. Org. Chem., 2000, 65, 8020–8026 CrossRef CAS PubMed.
- J. Singh, P. Singh and M. Nath, J. Org. Chem., 2023, 88, 7302–7310 CrossRef CAS PubMed.
- C. M. Cillo and T. D. Lash, Tetrahedron, 2005, 61, 11615–11627 CrossRef CAS.
- A. Singh, J. Singh and M. Nath, Eur. J. Org. Chem., 2023, e202201319 CrossRef CAS.
- C. S. Tekuri, P. Singh and M. Nath, Beilstein J. Org. Chem., 2023, 19, 1216–1224 CrossRef CAS PubMed.
- P. S. S. Lacerda, A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro and A. L. Llamas-Saiz, Angew. Chem., Int. Ed., 2006, 45, 5487–5491 CrossRef CAS PubMed.
- C. S. Tekuri, P. Singh and M. Nath, Org. Biomol. Chem., 2020, 18, 2516–2523 RSC.
- (a) N. Fukui, K. Fujimoto, H. Yorimitsu and A. Osuka, Dalton Trans., 2017, 46, 13322–13341 RSC; (b) I. A. Abdulaeva, K. P. Birin, A. B. Lemeune, A. Y. Tsivadze and Y. G. Gorbunova, Coord. Chem. Rev., 2020, 407, 213108 CrossRef CAS; (c) M. Umetani, K. Naoda, T. Tanaka, S. K. Lee, J. Oh, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 6305–6309 CrossRef CAS PubMed; (d) J. Singh, A. Singh and M. Nath, Tetrahedron, 2023, 148, 133684 CrossRef CAS.
- V. Piradi, F. Yan, X. Zhu and W. Y. Wong, Mater. Chem. Front., 2021, 5, 7119–7133 RSC.
- T. Higashino, S. Inoue, S. Arai, S. Tsuzuki, H. Matsui, R. Kumai, K. Takaba, S. Maki-Yonekura, H. Kurokawa, I. Inoue, K. Tono, K. Yonekura and T. Hasegawa, Chem. Mater., 2024, 36, 848–859 CrossRef CAS.
- N. Zhao, N. Ai, M. Cai, X. Wang, J. Pei and X. Wan, Polym. Chem., 2015, 7, 235–243 RSC.
- G. Turkoglu, M. E. Cinar, A. Buyruk, E. Tekin, S. P. Mucur, K. Kaya and T. Ozturk, J. Mater. Chem. C, 2016, 4, 6045–6053 RSC.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed.
- J. An, X. Yang, Z. Tian, B. Cai, L. Zhang, Z. Yu, X. Wang, A. Hagfeldt and L. Sun, Tetrahedron, 2021, 88, 132124 CrossRef CAS.
- T. Ghosh, L. Gerbig, M. Lambov, M. Dechant and M. Lehmann, J. Mater. Chem. C, 2020, 8, 5562–5571 RSC.
- Y. Mitsushige, S. Yamaguchi, B. S. Lee, Y. M. Sung, S. Kuhri, C. A. Schierl, D. M. Guldi, D. Kim and Y. Matsuo, J. Am. Chem. Soc., 2012, 134, 16540–16543 CrossRef CAS PubMed.
- N. Fukui, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6311–6314 CrossRef CAS PubMed.
- V. A. Nuay, D. H. Kim, S. H. Lee and J. Ko, Bull. Korean Chem. Soc., 2009, 30, 2871–2872 CrossRef CAS.
- R. Kumar and M. Sankar, Inorg. Chem., 2014, 53, 12706–12719 CrossRef CAS PubMed.
- J. Song, N. Aratani, H. Shinokubo and A. Osuka, Chem. Sci., 2011, 2, 748–751 RSC.
- (a) P. Bhyrappa and V. Velkannan, Inorg. Chim. Acta, 2012, 387, 64–73 CrossRef CAS; (b) X. L. Zheng, H. S. Lin, B. W. Zhang, S. Maruyama and Y. Matsuo, J. Org. Chem., 2022, 87, 5457–5463 CrossRef CAS PubMed; (c) Y. Kurumisawa, T. Higashino, S. Nimura, Y. Tsuji, H. Iiyama and H. Imahori, J. Am. Chem. Soc., 2019, 141, 9910–9919 CrossRef CAS PubMed.
- L. L. Yang, X. L. Hu, Z. Q. Tang and X. F. Li, Chem. Lett., 2015, 44, 1515–1517 CrossRef CAS.
- E. Annoni, M. Pizzotti, R. Ugo, S. Quici, T. Morotti, M. Bruschi and P. Mussini, Eur. J. Inorg. Chem., 2005, 3857–3874 CrossRef CAS.
- K. Tomioka, Y. Shioya, Y. Nagaoka and K. I. Yamada, J. Org. Chem., 2001, 66, 7051–7054 CrossRef CAS PubMed.
- N. K. Thalji, W. E. Crowe and G. L. Waldrop, J. Org. Chem., 2009, 74, 144–152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, HRMS and electronic absorption spectra of compounds. See DOI: https://doi.org/10.1039/d4ob00598h |
| This journal is © The Royal Society of Chemistry 2024 |