
Biomimetic total syntheses of renifolin F and antiarone K†
Jarish
Ahamad
and
Faiz Ahmed
Khan
 *
*
Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, 502284, India. E-mail: faiz@chy.iith.ac.in
First published on 22nd May 2024
Abstract
The first biomimetic and concise racemic total syntheses of renifolin F and antiarone K, accomplished in 8 and 7 linear steps, respectively, are presented in this article. Our synthetic approach commences with substituted aldehydes to produce prenylated aldol products followed by ene-type intramolecular cyclization affording a five-member core ring. This key step mediated by InCl3·4H2O is a novel procedure first utilized in prenylated systems which directly culminates mainly into tertiary alcohols.
Introduction
For synthetic chemists, the goal of synthetic efficiency typically quantified by step count and overall yields provides a wealth of incentive and inspiration for developing novel tactics and techniques. Many of the pharmaceutical chemicals used today are derived from natural sources, once a major medication source. Flavonoids’ fascinating biological and therapeutic properties, along with their intricate structure, have attracted the attention of the synthetic organic community. Flavonoids are abundant in the kingdom of plants and are precious natural resources. Chalcone has been acknowledged as a preferred scaffold in medicinal chemistry.1 Naturally occurring Chalcones are categorized as phenolic compounds within the flavonoid class and have a five-membered ring assembled from β-carbon of chalcone and dimethylallyl carbon. They exhibit numerous pharmacological and biological properties such as antiparasitic, antitumor, cytotoxic, antifungal, anti-inflammatory, anti-allergic, antiviral, and antibacterial. Some of these compounds have the potential to treat neurodegenerative and vasodilatory disorders.2–4Renifolin D–F were isolated from entire Desmodium Renifolium plants by Yan-Ping Li et al. in 2014 (Fig. 1).5 Utilizing five tumor cell lines, the cytotoxicity of each isolate was assessed. Compared to the positive control drug paclitaxel renifolin E and F were 100 times less potent and showed only little cytotoxicity (IC50 values of 2.8 and 2.2 μM, respectively) against A549 human lung carcinoma cells.1 Furthermore, Feng Huang and co-workers isolated renifolin F (2) from another medicinal plant Shuteria Involucrate in 2022 and mentioned the therapeutic effect on allergic asthma. Approximately 300 million people worldwide suffer from allergic asthma, a chronic and diverse illness. Currently, corticosteroids, β-agonists, and leukotriene receptor antagonists are the major treatments for asthma. However, for 5–10% of individuals with severe asthma, the benefits of these therapies are insufficient, and long-term use of these medications might result in major side effects. Therefore, finding a safe anti-asthma medicine is essential.6 Given the biological significance and natural scarcity of renifolin F (2), it is imperative to establish a succinct strategy for its total synthesis. This would facilitate additional biological assessments and evaluations. Taro Nomura and co-workers extracted antiarone K (1) from the root bark of Antiaris Toxicaria in 1991, collected from Indonesia.7 Antiarone K (1) and renifolin F (2) are recognized as chalcone derivatives that possess an isoprenoid moiety.
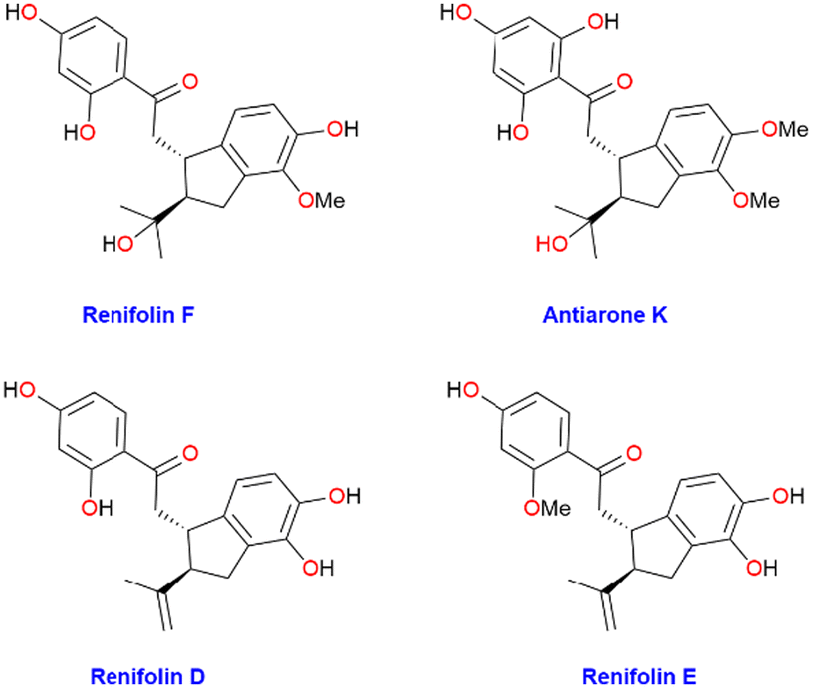 | ||
| Fig. 1 Structure of few chalcone natural products. | ||
We conceived a biomimetic synthesis as illustrated in retrosynthetic Scheme 1. The key step in this strategy is an ene-type cyclization based on proposed biosynthesis.5 Renifolin F (2) and antiarone K (1) could be synthesized from the deprotection of benzylated cyclized compound 3. Intermediate 3 was envisaged to be obtained by an intramolecular ene-type cyclization of compound 4 which in turn could be derived from prenylated aldehyde 5 through an aldol condensation reaction. Different substituted prenylated aldehydes 5 could be accessed from 7via prenylation. Compound 7 was synthesized using readily accessible and cost-effective starting material 8via regioselective bromination. (The blue circles in Scheme 1 serve as an indicator for the formation of a new C–C bond.)
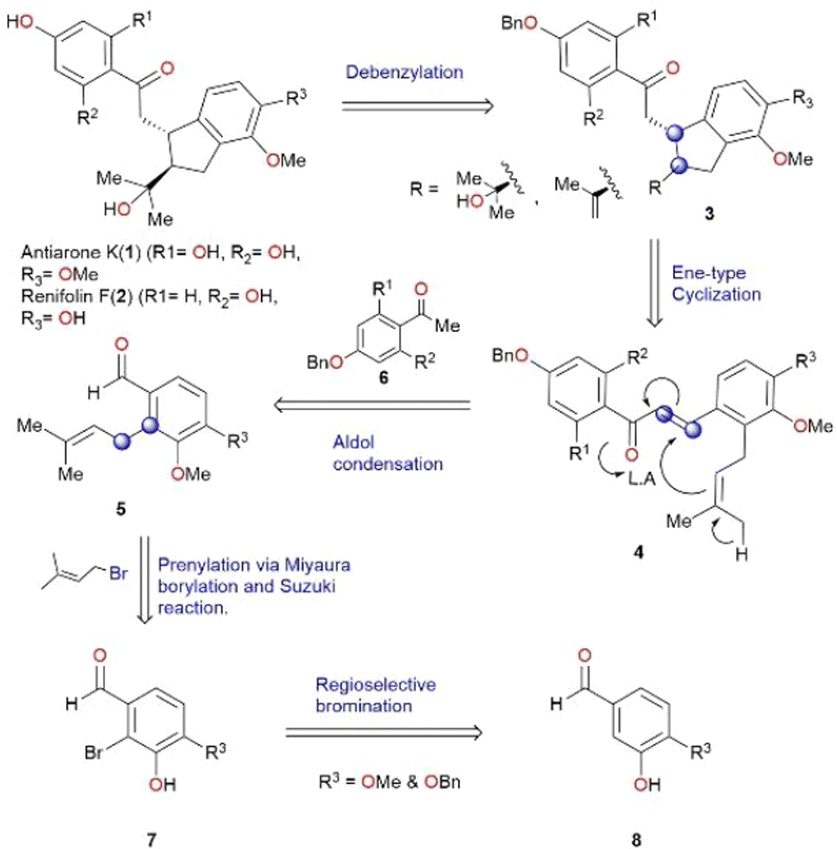 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Herein we disclose an effective and flexible approach for the total synthesis of renifolin F (2) and antiarone K (1) from commercially available starting materials. With a strong emphasis on our retrosynthetic plan, we aim to produce substituted prenylated aldehyde 13, a crucial intermediate in our synthesis process. The synthesis of compound 13 has been previously established through the direct prenylation of veratraldehyde 28, resulting only in 12% of the desired product, as well as other isomers by Irinel and co-authors.8 In order to achieve better results, we have implemented an alternative approach based on oxazoline directed metalation to attain the required regiochemistry and to avoid unwanted isomers (Scheme S1, ESI†).9–14 This approach has furnished an improved overall yield of 35%. The yield was further enhanced finally to an impressive overall 64% by a novel approach (Scheme 2). In this method for the synthesis of precursor 13, we began with commercially procurable and inexpensive isovanillin 9. Bromination15 of 9 in the presence of Br2 and iron powder furnished 10 in 94% yield and subsequent methylation15 of the 10 produced 11 in 95% yield. Bromo compound 11 was subjected to Miyaura borylation16 using a palladium catalyst, resulting in the formation of borylated product 12 in very good yield. Suzuki17 reaction was carried out with 12, resulting in the smooth formation of compound 13 in a satisfactory yield using tetrakis(triphenylphosphine) [Pd(PPh3)4] and prenyl bromide.
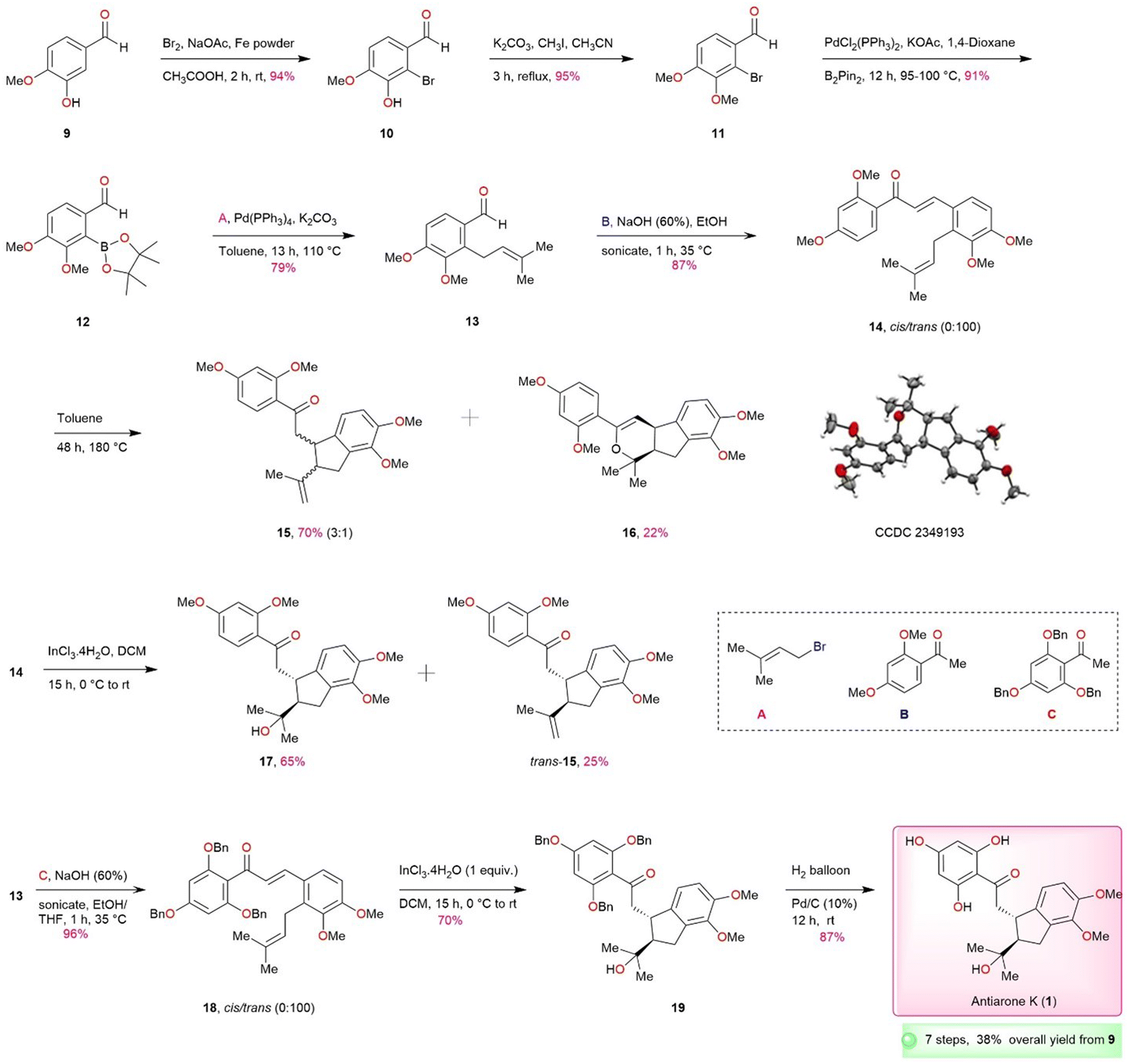 | ||
| Scheme 2 Total synthesis of antiarone K (1). | ||
Following the acquisition of requisite 13 through our improved prenylation strategy, an aldol condensation reaction was conducted using 13 and benzylated acetophenone derivative B under sonication.18 The outcome was the generation of prenylated aldol condensation product 14 in 87% yield. Subsequently, our attention is directed towards forming the five-membered core ring via intramolecular ene-type cyclization. Based on literature precedents of closely related systems,19,20 we initially subjected 14 to thermal conditions at high temperature (toluene, 180 °C) in a sealed tube. The reaction was non-selective and furnished an inseparable 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trans/cis mixture (1H and 13C NMR, see ESI†) along with a further cyclized product 16, originating from cis-15. The unambiguous confirmation of structure 16 was done by X-ray crystallography. Several standard demethylation protocols were attempted on 15 but none were successful (Table S1, ESI†). We first thought of addressing non-selective ene-type cyclization that gave trans/cis mixture under thermal conditions. It was planned to explore the intramolecular cyclization of the 14 in the presence of Lewis acids. To start our investigation, we conducted experiments under various reaction conditions using 14 as a model substrate and the results are depicted in Table 1. The best results were observed when 1 equiv. of InCl3·4H2O was used in the presence of DCM at 0 °C (Table 1, entry 11) which furnished the desired product 17 stereoselectively in 65% yield along with 25% of trans-15. Reducing the amount of Lewis acid to 0.5 equiv. gave diminished yields (Table 1, entry 15). This is a significant achievement as this type of cyclization is novel and not reported earlier with InCl3·4H2O to the best of our knowledge. The structural assignments of 17 and trans-15 were based on 1H and 13C NMR analysis. The presence of a tertiary alcohol moiety (2-hydroxy-2-propanyl group) in 17 was supported by the presence of a peak at 72.8 ppm (carbinol carbon) in the 13C NMR spectrum, consistent with the findings in the isolation report. The structure of 17 was further corroborated with 2D NMR (HMBC and HMQC) techniques. Demethylation of 17 also proved to be problematic as for 15. Therefore, it was imperative to opt for a protecting group that could be readily removed at the end after the cyclization. In this context, antiarone K (1) possessing a tertiary alcohol unit and two methyl ether moieties on the fused aromatic ring appeared to be a straightforward target. So, we commenced the total synthesis of antiarone K (1) (Scheme 2) starting with differentially protected precursor via this novel cyclization method utilizing InCl3·4H2O, which is expected to directly furnish the required tertiary alcohol derivative as the major product. With precursor 13 as the key component, differentially protected 18 was synthesized using aldol reaction of acetophenone derivative C under sonication15 employing NaOH (aq.) and EtOH as a solvent in 96% yield. The key intramolecular cyclization of 18 mediated by InCl3·4H2O gratifyingly produced tertiary alcohol derivative 19 stereoselectively in 70% yield. Alkene product, similar to 15, was not detected in this case. The final step for the completion involves debenzylation using 10% Pd/C under hydrogen atmosphere (balloon)21 leading to the successful production of 87% antiarone K (1) from 19. The overall yield achieved was 38%, and the entire process involved 7 consecutive steps. The NMR data pertaining to the synthetic antiarone K (1) aligns with the results documented in the isolation report.
:1 trans/cis mixture (1H and 13C NMR, see ESI†) along with a further cyclized product 16, originating from cis-15. The unambiguous confirmation of structure 16 was done by X-ray crystallography. Several standard demethylation protocols were attempted on 15 but none were successful (Table S1, ESI†). We first thought of addressing non-selective ene-type cyclization that gave trans/cis mixture under thermal conditions. It was planned to explore the intramolecular cyclization of the 14 in the presence of Lewis acids. To start our investigation, we conducted experiments under various reaction conditions using 14 as a model substrate and the results are depicted in Table 1. The best results were observed when 1 equiv. of InCl3·4H2O was used in the presence of DCM at 0 °C (Table 1, entry 11) which furnished the desired product 17 stereoselectively in 65% yield along with 25% of trans-15. Reducing the amount of Lewis acid to 0.5 equiv. gave diminished yields (Table 1, entry 15). This is a significant achievement as this type of cyclization is novel and not reported earlier with InCl3·4H2O to the best of our knowledge. The structural assignments of 17 and trans-15 were based on 1H and 13C NMR analysis. The presence of a tertiary alcohol moiety (2-hydroxy-2-propanyl group) in 17 was supported by the presence of a peak at 72.8 ppm (carbinol carbon) in the 13C NMR spectrum, consistent with the findings in the isolation report. The structure of 17 was further corroborated with 2D NMR (HMBC and HMQC) techniques. Demethylation of 17 also proved to be problematic as for 15. Therefore, it was imperative to opt for a protecting group that could be readily removed at the end after the cyclization. In this context, antiarone K (1) possessing a tertiary alcohol unit and two methyl ether moieties on the fused aromatic ring appeared to be a straightforward target. So, we commenced the total synthesis of antiarone K (1) (Scheme 2) starting with differentially protected precursor via this novel cyclization method utilizing InCl3·4H2O, which is expected to directly furnish the required tertiary alcohol derivative as the major product. With precursor 13 as the key component, differentially protected 18 was synthesized using aldol reaction of acetophenone derivative C under sonication15 employing NaOH (aq.) and EtOH as a solvent in 96% yield. The key intramolecular cyclization of 18 mediated by InCl3·4H2O gratifyingly produced tertiary alcohol derivative 19 stereoselectively in 70% yield. Alkene product, similar to 15, was not detected in this case. The final step for the completion involves debenzylation using 10% Pd/C under hydrogen atmosphere (balloon)21 leading to the successful production of 87% antiarone K (1) from 19. The overall yield achieved was 38%, and the entire process involved 7 consecutive steps. The NMR data pertaining to the synthetic antiarone K (1) aligns with the results documented in the isolation report.
a
| Entry | Lewis acid (equiv.) | Solvent | Time (h) | Yieldd (%) (15) | Yieldd (%) (17) |
|---|---|---|---|---|---|
|
a Reaction conditions: the reaction was carried out with 14 (0.1 mmol, 1 equiv.), reagents in different solvent (3 ml), temp (0 °C to r.t
b 180 °C.
c 80 °C).
d isolated yield.
e
trans/cis (3:1).
f
trans, ND = not detected, compound 16 (entry 1, 22%).
|
|||||
| 1b | None | Toluene | 48 | 70e | ND |
| 2 | In(OTf)3 (1) | DCM | 18 | 35e | ND |
| 3 | Bi(OTf)3 (1.6) | THF | 8 | 51e | 25 |
| 4 | Yb(OTf)3 (1) | THF | 20 | ND | ND |
| 5 | ZnCl2 (1) | DCM | 11 | 20f | 55 |
| 6 | ZnCl2 (1) | CH3CN | 15 | ND | ND |
| 7c | ZnBr2 (1) | Toluene | 22 | 33e | ND |
| 8 | BF3·Et2O (5) | THF | 18 | 22e | 50 |
| 9 | B(C6F5)3 (1) | CH3CN | 15 | ND | ND |
| 10 | Zn(OTf)2 | CH3CN | 48 | ND | ND |
| 11 | InCl 3 ·4H 2 O (1) | DCM | 20 | 25 | 65 |
| 12 | InCl3·4H2O (1) | THF | 24 | Trace | Trace |
| 13 | InCl3·4H2O (1) | CH3CN | 24 | ND | ND |
| 14 | InCl3·4H2O (1) | DMF | 24 | ND | ND |
| 15 | InCl3·4H2O (0.5) | DCM | 30 | 10f | 30 |
| 16 | InCl3·4H2O (1) | DCE | 15 | 20f | 60 |
| 17 | InCl3·4H2O (1) | Dioxane | 24 | ND | ND |
With the successful completion of total synthesis of antiarone K (1), we turned our attention to undertaking the total synthesis of renifolin F (2) (Scheme 3). Our objective was to synthesize prenylated aldehyde 25, a vital intermediate for this synthesis while considering the structure of renifolin F (2). In compound 25, the para and meta hydroxy are protected by the benzyl and methoxy groups respectively. This protective strategy is implemented to achieve the desired positioning of the hydroxy and methoxy groups following a targeted debenzylation step. The synthesis of Intermediate 23 was initially carried out in 5 steps using known protocols (Scheme S2, ESI†), yielding 36% overall.22–25 Through the adoption of an alternative route, we successfully produced 23 in just 3 steps (Scheme 3), with a remarkable overall yield of 77% – surpassing the previous route by more than double and reducing the number of steps required. To achieve this objective, established methods like benzylation,25 bromination, and methylation were employed on protocatechualdehyde 20 to obtain 23 through intermediates 21 and 22 in excellent yields. Compound 23 was further converted into key intermediate 25via24 using Miyaura borylation (93%) and Suzuki reaction (77%). The synthesis of renifolin F (2) was successfully achieved by utilizing key intermediate 25 in aldol condensation, InCl3·4H2O ene-type intramolecular cyclization, and selective debenzylation via26 and 27, yielding 91%, 86%, and 93% respectively (Scheme 3). We have accomplished the total synthesis of renifolin F (2) in 8 linear steps with an overall yield of 35%. The spectral data of synthetic renifolin F (2) corresponds with the findings outlined in the isolation report.
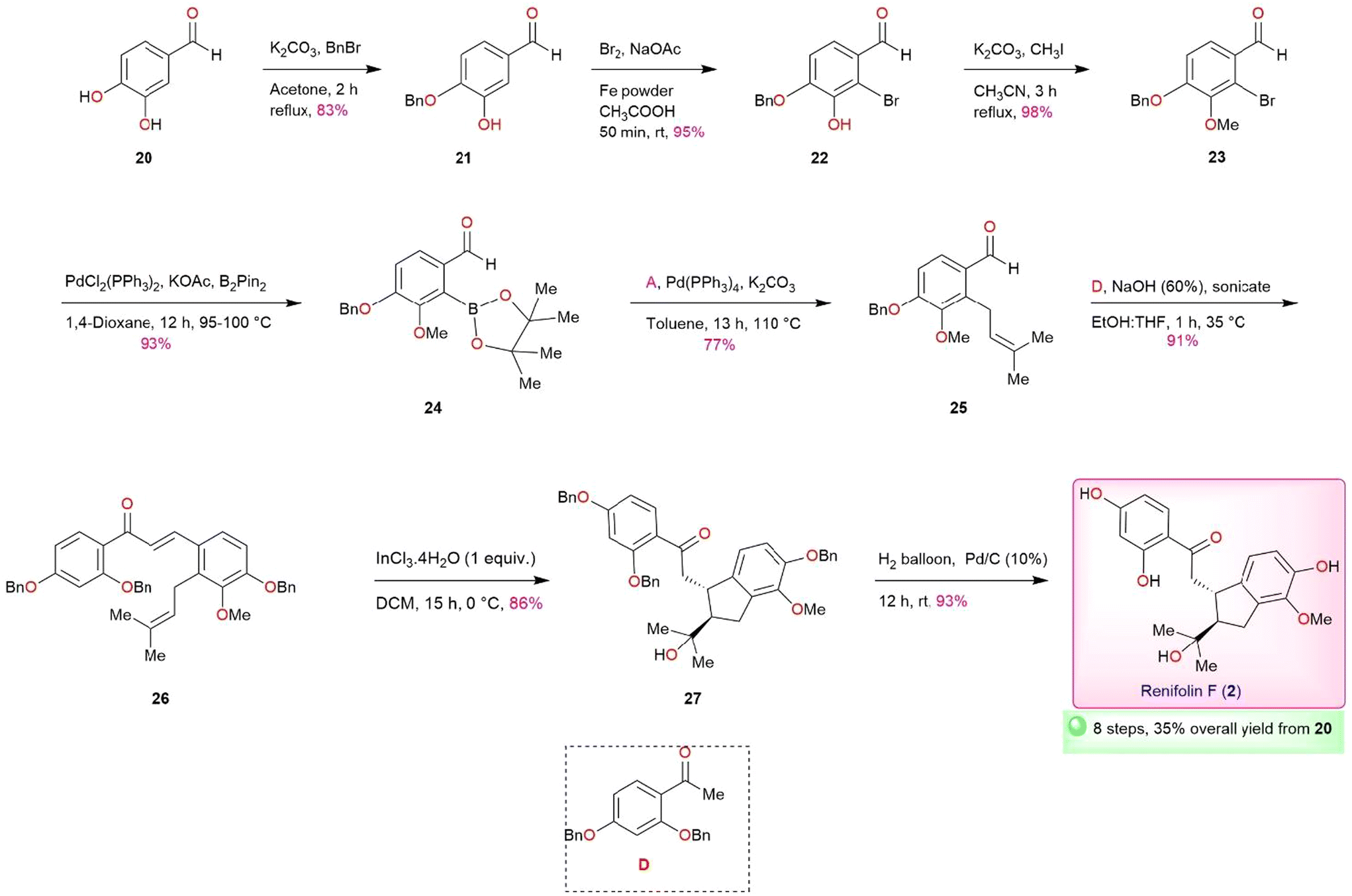 | ||
| Scheme 3 Total synthesis of renifolin F (2). | ||
Conclusions
To summarize, we have accomplished a concise and scalable first total synthesis of antiarone K (1) and renifolin F (2) in 7 and 8 steps with an overall yield of 38% and 35% respectively. The four crucial transformations facilitated the successful completion of the synthesis. These transformations include the synthesis of prenylated aldehyde, aldol condensation, In(III)-mediated cyclization to construct the five-member core ring, and finally palladium-catalyzed debenzylation to deliver phenolic compounds.Data availability
Data is available in the ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Science and Engineering Research Board (SERB), India, for financial support (CRG/2022/004763) and sincerely appreciate Indian Institute of Technology Hyderabad for providing facilities. J. A expresses gratitude to CSIR (09/1001(0099)/2021-EMR-I) New Delhi for offering a fellowship.References
- K. Zhou, S. Yang and S.-M. Li, Nat. Prod. Rep., 2021, 38, 2236–2260 RSC.
- N. A. A. Elkanzi, H. Hrichi, R. A. Alolayan, W. Derafa, F. M. Zahou and R. B. Bakr, ACS Omega, 2022, 7, 27769–27786 CrossRef CAS PubMed.
- H. A. Jasim, L. Nahar, M. A. Jasim, S. A. Moore, K. J. Ritchie and S. D. Sarker, Biomolecules, 2021, 11, 1203 CrossRef CAS PubMed.
- Z. Rozmer and P. Perjési, Phytochem. Rev., 2016, 15, 87–120 CrossRef CAS.
- Y.-P. Li, Y.-C. Yang, Y.-K. Li, Z.-Y. Jiang, X.-Z. Huang, W.-G. Wang, X.-M. Gao and Q.-F. Hu, Fitoterapia, 2014, 95, 214–219 CrossRef CAS PubMed.
- Z. Yang, X. Li, R. Fu, M. Hu, Y. Wei, X. Hu, W. Tan, X. Tong and F. Huang, Molecules, 2022, 27, 3789 CrossRef CAS PubMed.
- Y. Hano, P. Mitsui, T. Nomura, T. Kawai and Y. Yoshida, J. Nat. Prod., 1991, 54, 1049–1055 CrossRef CAS.
- I. Badea, P. Cotelle and J.-P. Catteau, South. Braz. J. Chem., 2001, 9(10), 5–8 CAS.
- L. Luo, Q. Song, Y. Li, Z. Cao, X. Qiang, Z. Tan and Y. Deng, Bioorg. Med. Chem., 2020, 28, 115400 CrossRef CAS PubMed.
- A. I. Meyers and J. J. Willemsen, Tetrahedron, 1998, 54, 10493–10511 CrossRef CAS.
- J. Nakano, K. Uchida and Y. Fujimoto, Heterocycles, 1989, 29, 427 CrossRef CAS.
- S. R. Wilson, D. T. Mao and H. N. Khatri, Synth. Commun., 2007, 10, 17–23 CrossRef.
- R. Mohan and J. A. Katzenellenbogen, J. Org. Chem., 1984, 49, 1238–1246 CrossRef CAS.
- H. Nate, Y. Sekine, Y. Honma, H. Nakai, H. Wada, M. Takeda, H. Yabana and T. Nagao, Chem. Pharm. Bull., 1987, 35, 1953–1968 CrossRef CAS PubMed.
- S. N. Momm and R. Brückner, J. Org. Chem., 2022, 87, 15415–15420 CrossRef CAS PubMed.
- Y. K. Zhang, J. J. Plattner, E. E. Easom, D. Waterson, M. Ge, Z. Li, L. Li and Y. Jian, Tetrahedron Lett., 2011, 52, 3909–3911 CrossRef CAS.
- Z. Zhuang, P. Shen, J. Li, J. Li, Z. Zhao and B. Z. Tang, CCS Chem., 2021, 4, 286–303 CrossRef.
- F. Kayamba, T. Malimabe, I. K. Ademola, O. J. Pooe, N. D. Kushwaha, M. Mahlalela, R. L. van Zyl, M. Gordon, P. T. Mudau, T. Zininga, A. Shonhai, V. O. Nyamori and R. Karpoormath, Eur. J. Med. Chem., 2021, 217, 113330 CrossRef CAS PubMed.
- K. Kato, K. Ikeuchi, T. Suzuki and K. Tanino, Org. Lett., 2022, 24, 6407–6411 CrossRef CAS PubMed.
- J. E. Resek, J. Org. Chem., 2008, 73, 9792–9794 CrossRef CAS PubMed.
- M. H. Keylor, B. S. Matsuura, M. Griesser, J. P. R. Chauvin, R. A. Harding, M. S. Kirillova, X. Zhu, O. J. Fischer, D. A. Pratt and C. R. J. Stephenson, Science, 2016, 354, 1260–1265 CrossRef CAS PubMed.
- H. Garcia, R. Martinez-Utrilla and M. A. Miranda, Tetrahedron, 1985, 41, 3131–3134 CrossRef CAS.
- A. Mrozek-Wilczkiewicz, D. S. Kalinowski, R. Musiol, J. Finster, A. Szurko, K. Serafin, M. Knas, S. K. Kamalapuram, Z. Kovacevic, J. Jampilek, A. Ratuszna, J. Rzeszowska-Wolny, D. R. Richardson and J. Polanski, Bioorg. Med. Chem., 2010, 18, 2664–2671 CrossRef CAS PubMed.
- M. Banerjee, R. Mukhopadhyay, B. Achari and A. K. Banerjee, J. Org. Chem., 2006, 71, 2787–2796 CrossRef CAS PubMed.
- S. Radix and R. Barret, Tetrahedron, 2007, 63, 12379–12387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2349193. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00651h |
| This journal is © The Royal Society of Chemistry 2024 |